
Abstract
Pharmacologic activation of the cystic fibrosis transmembrane conductance regulator (CFTR) has transformed cystic fibrosis (CF) therapy. Other, more common airway diseases can also be associated with CFTR deficiency. For example, individuals with one dysfunctional CFTR variant (i.e., CF carriers), as well as those with acquired CFTR deficiency, are predisposed to both non-CF bronchiectasis and chronic rhinosinusitis, raising the possibility that CFTR stimulation in these settings could provide clinical improvement. This study describes a new triazolo-thiadiazine-based compound series optimized to augment mutant and wildtype CFTR function when administered topically to airway epithelium. Mechanism of action appears attributable—at least in part—to phosphodiesterase 4 inhibition (PDE4i), with effects on other PDEs also noted. Together with a growing body of previous and emerging evidence, our results suggest a novel therapeutic strategy for treating people with CF (PwCF) who lack access to effective modulator therapy—and addressing common diseases such as chronic bronchiectasis and rhinosinusitis in the non-CF population.
Similar content being viewed by others

Introduction
Over the past several years, drug discovery has dramatically benefited people with cystic fibrosis1,2,3,4,5,6,7. Highly effective modulator treatment (HEMT) is available in the US to >90% of individuals with the disease, and the FDA label includes approvals for over 300 mutations in the cystic fibrosis transmembrane conductance regulator (CFTR), most of which have been established as disease-causing variants. “Triple combination therapy” (comprised of elexacaftor (VX-445), tezacaftor (VX-661), and ivacaftor (VX-770; ETI) or vanzacaftor (VX-121), tezacaftor, and deutivacaftor (VX-561)8) confer improvement in CF lung function, rhinosinusitis, quality of life, and other endpoints (including markers of CFTR activity such as sweat chloride) within days to a few weeks8,9,10. As with any new therapy, a subset of PwCF fail HEMT (e.g., ~ 5% of individuals encoding the common F508del variant may not benefit from ETI) or exhibit limiting toxicity (for example, hepatic impairment) that precludes use6,9,10,11. In addition, hundreds of disease-causing CFTR variants are not FDA-approved for modulator treatment, including refractory defects of folding and maturational processing1,12. Based on successful precedent achieved with ETI, several novel compounds with superior efficacy are being advanced by academic and pharmaceutical laboratories8,13,14,15. A recent survey examined over 650 CFTR variants associated with clinical disease for modulator responsiveness16 using cell systems considered predictive of pulmonary benefit in vivo1,4,5,17,18,19,20,21. That study revealed many CFTR mutations will require new and distinct activators specifically designed to improve lung function in patients with cystic fibrosis.
Individuals with CF experience tissue damage throughout the respiratory tract, including nasal passages, sinuses, lung parenchyma, and submucosal glands1,7,22,23,24,25,26,27,28,29,30,31. Involvement of the upper airways is very common ( > 90% of PwCF have radiographic evidence of chronic sinusitis). Impaired mucociliary clearance, airway obstruction, bioelectric abnormalities, immunologic hyperactivity, and chronic infection (by P. aeruginosa, H. influenza, S. aureus, B. cepacia and other pathogens) characterize both nasal and lower CF respiratory tissues6,7. HEMT strongly palliates—but does not resolve—existing CF lung and sinonasal pathology. While it has been suggested that CFTR modulators given at or before birth may prevent airway disease from occurring, longitudinal studies will be required to formally test that possibility. In either case, improved small molecules are actively being sought based on the view that a therapeutic ceiling has not yet been achieved with existing modulators14,15. Moreover, data in primary airway epithelial cells—model systems that correlate with clinical benefit—indicate potential for activation of the common F508del variant well above what can be achieved with HEMT alone14,15,32,33.
Respiratory failure due to bronchiectasis is the primary cause of morbidity and mortality in people with CF34. As above, pulmonary tissues exhibit many of the same clinical findings as the CF sinonasal tract, and pathogenic mechanisms are often viewed as comparable6,7. Epithelial monolayers from both nasal and lower airways have been used as tools for understanding and advancing access to HEMT, further supporting the relationship between nasal passages and modulator activity in lungs35,36,37,38. In addition, the importance of improved treatment for chronic rhinosinusitis in CF has been emphasized as a means to prevent “seeding” of lower pulmonary tissues and to improve outcomes post-lung transplantation39,40,41,42,43,44,45,46. Chronically infected nasal sinuses serve as a bacterial reservoir that infects the lower respiratory tract, impairs pulmonary graft viability, and causes failure of attempts to eradicate pulmonary pathogens in PwCF. Findings such as these indicate mechanistic similarities and strong interrelationships between CF lower and upper airway disease, with implications for other, more common illnesses.
A pulmonary condition that exhibits pathologic features with close similarity to CF lung disease—non-cystic fibrosis bronchiectasis (NCFB)—afflicts millions worldwide. Comparable clinical findings include abnormal airway dilation, diminished mucociliary clearance (MCC), and infection by similar (and recalcitrant) bacterial pathogens47. Endobronchial damage, associated inflammatory changes, and pulmonary remodeling in NCFB can lead to respiratory failure and death48,49,50. Although NCFB has traditionally been viewed as mechanistically unrelated to CFTR dysfunction, individuals who are silent carriers of CF (heterozygous with one pathogenic CFTR variant and one wildtype CFTR allele) are at higher risk of developing NCFB51,52,53. In addition, persistent inflammation, hypoxia, cigarette smoke exposure, and other toxins have been shown to contribute to an acquired loss of CFTR in conditions such as NCFB54,55. Abnormalities of the CFTR airway bioelectric phenotype54 and a variety of mutations in CFTR have been associated with NCFB. Moreover, recent case studies indicate potential usefulness of CFTR modulators for treating a subset of individuals with non-CF bronchiectasis56.
In the same context, approximately 1 in 8 Americans suffers from intermittent, non-CF chronic rhinosinusitis (CRS), which (like NCFB) has been increasingly linked to both mutant CFTR and diminished wildtype CFTR activity55,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72. Non-CF CRS accounts for >10 million coded office visits annually in the US73 and is a cause of significant morbidity, including persistent and/or severe facial pain, headache, rhinorrhea, anosmia, absenteeism from work, and substantial debility. From a clinical perspective, predisposition to non-CF CRS is associated with heterozygosity for CFTR mutations, indicating that relative CFTR deficiency and diminished mucociliary clearance are partly responsible for pathogenesis59,60,61,62. Moreover, in semblance to NCFB, chronic airway infection/inflammation impairs CFTR biogenesis and ion transport, further supporting a therapeutic role for CFTR modulators in the treatment of this disease55,57,58,63,69,70,71,74.
Clinical cystic fibrosis and “forme frustes” of CF such as NCFB and CRS are clearly caused—at least in part—by inherited or acquired CFTR deficiency. We are developing a new class of compounds that activate both WT and mutant CFTR through robust, PKA-dependent R-domain phosphorylation. The drugs are intended for use as monotherapy or in combination with available CFTR modulators for treating PwCF who exhibit suboptimal clinical response, poor modulator tolerance, or those encoding CFTR variants that remain “off label” for emerging modulator therapy. Because the new compounds strongly activate both mutant and wildtype CFTR, contributions to treatment of non-CF airway diseases attributable to CFTR deficiency and inadequate mucociliary clearance (for example, CRS and NCFB) are also anticipated.
Results
Overview
We previously screened 300,000 agents from a library of bioactive compounds as a means to rescue N1303K or W1282X CFTR, two relatively common variants with measurable (albeit low level) function75. The screen was conducted using Fischer rat thyroid (FRT) cells stably transduced with CFTR encoding an extracellularly tagged horseradish peroxidase (HRP) for monitoring cell surface localization. The FRT model has been successful in many CFTR pharmaceutical drug discovery programs from both academic and commercial laboratories4,17,21,37,76, leading to several FDA-registered compounds. Following confirmation and dose-dependence analysis, as well as in silico (modified ‘PAINS’) profiling to exclude candidate drugs with inherent liabilities77, active scaffolds were identified. Based on a high compound “dropout” rate associated with drug screening in FRT cells, candidate molecules were tested using complementary cell systems. The most promising agents were advanced to validation in primary airway epithelial monolayers grown on permeable supports—a model shown previously to predict in vivo clinical benefit and recognized by the FDA for the purpose of drug registration4,21.
HDCF drug analysis and CFTR ion transport
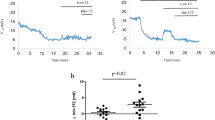
Lead agents resulting from the original compound library screen and subsequent medicinal chemistry optimization75 identified a drug series that strongly augments wildtype CFTR activity as judged by changes in short circuit current (Isc) in FRT cells (Fig. 1). Two examples of this series—termed HDCF104 and uHTS159—are reported in detail here. Functional assays in primary human bronchial epithelial monolayers encoding WT CFTR also showed robust enhancement of CFTR transepithelial transport (Fig. 2). Importantly, compounds in the newly identified class acutely stimulate many disease-causing CFTR variants. Unlike CFTR “corrector” agents, which rescue folding but do not augment channel gating, drugs described in this report block phosphodiesterase activity, enhance PKA-dependent phosphorylation of CFTR, and stimulate ion transport (see below). Data showing use of the new drug series against a number of CFTR variants are provided in the following sections.

Ussing chamber analysis of Fischer rat thyroid (FRT) cells shows stimulation of WT CFTR. a, b Escalating doses of HDCF104 demonstrate strong WT CFTR activity with EC50 = 101 μM (95% CI 91.7 μM to 112.3 μM) as judged by dose-dependence profile. In similar fashion, (c, d) uHTS159 displays robust enhancement of WT CFTR with EC50 = 14.4 μM (95% CI 12.5 μM−17.2 μM). Data is representative of 3 biological replicates and expressed as mean ± SD. Maximal stimulation of wildtype CFTR by saturating concentrations of forskolin in this model is 300−500 μA/cm2. Amiloride (100 µM) was added to block epithelial sodium channels; forskolin (5 µM) augments cellular cAMP and activates CFTR by PKA-dependent phosphorylation of the regulatory domain; inh-172 (10 µM) is a CFTR inhibitor. e Structures of the compounds. Statistics were by use of GraphPad Prism 10 software with the function of Sigmoidal, 4PL, with X as concentration. “HDCF” denotes the medicinal chemistry laboratory in which new compounds were synthesized (directed by Dr. Huw Davies). Panels (a, c) plotted with Microsoft Excel; (b, d) plotted (and compound EC50 calculated) using GraphPad Prism 10 software; Panel (e) created with ChemDraw; panels arranged and presented using Affinity Designer software.

a Escalating doses of HDCF104 provide robust WT CFTR activation with EC50 = 4.16 μM (95% CI 2.748 μM to 5.572 μM). b Data summarized by dose-activity profile. c, d In similar fashion, escalating doses of uHTS159 demonstrate activation of WT CFTR with EC50 = 0.71 μM (95% CI 0.558 μM to 0.864 μM). Data is representative of 4 biological replicates and expressed as mean ± SD. Addition of amiloride (100 µM), forskolin (10 µM), inh-172 (10 µM), and UTP (stimulates non-CFTR chloride channels as a control for monolayer integrity; 100 µM) are shown. HBEC = primary human bronchial epithelial cells. Statistics were by use of GraphPad Prism 10 software with the function of Sigmoidal, 4PL, with X as concentration. Panels (a, c) plotted with Microsoft Excel; (b, d) plotted (and compound EC50 calculated) using GraphPad Prism 10 software; panels arranged and presented using Affinity Designer.
G551D CFTR activation by uHTS159
An FDA-approved CFTR modulator (VX-770) was specifically developed by the pharmaceutical industry as a “potentiator” to rescue the G551D CFTR channel gating defect37,78. Figure 3 shows uHTS159 activates G551D CFTR independent of VX-770 and can strongly enhance VX-770 potentiation. In terms of efficacy as a single agent, uHTS159 is roughly equivalent to VX-770. When saturating concentrations of the clinically approved potentiator are applied, uHTS159 promotes further short circuit current, significantly surpassing the effects of VX-770 alone.

Activation of G551D CFTR in the FRT model by uHTS159 (10 μM) is shown, including substantial potentiation of Isc beyond VX-770 alone irrespective of the order in which compounds are added. a Representative tracings; b summary of 8 biological replicates per condition expressed as mean ± SEM. p < 0.01 for maximal activation of G551D CFTR by forskolin and VX-770 vs addition of uHTS159 with forskolin and VX-770. Statistics were by Excel software, ANOVA, single factor analysis function. Figures plotted with Microsoft Excel; plots arranged and presented using Affinity Designer software.
HDCF stimulation of F508del CFTR following ETI
The FRT model has been viewed by pharmaceutical groups and FDA as predictive of in vivo clinical response—particularly when a test compound augments mutant CFTR function to >10% wildtype activity levels (e.g., Fig. 3)4,16,17,21,37. As described above, primary CF airway epithelial cells are also well-established for predicting patient benefit4,19,20,21. Figure 4 provides an example of significant (10−20 μA/cm2) CFTR-dependent anion current increase conferred by HDCF104 following pharmaco-correction of the common F508del CFTR protein processing defect (present in ~90% of PwCF), indicating substantial and clinically meaningful enhancement of CFTR well above what can be accomplished with ETI alone. Importantly, HDCF104 elevates Isc to levels that are nearly comparable to wildtype CFTR activity in this model system. In the same context, note that although VX-770 activates F508del CFTR even in the absence of forskolin, total ion transport is significantly lower than what can be achieved by combination treatment of VX-770 with HDCF104. While levels of constitutively active PKA in primary human airway cells (Fig. 4) are therefore sufficient for stimulation of F508del CFTR by VX-770, augmenting PKA-dependent R-domain phosphorylation provides much stronger enhancement of protein function.

Activity in homozygous F508del human bronchial epithelial monolayers is shown. a, b Representative Ussing assay tracings. c Summary data using saturating concentrations of either VX-770 or HDCF104 added first. n = 4 biological replicates per condition; bars depict standard deviation (SD). Acute treatments shown are amiloride (100 µM), VX-770 (5 µM), HDCF104 (20 µM), and inh-172 (10 µM). p = 6.8 × 10-6 for HDCF104 plus VX-770 versus VX-770 alone (HDCF compound added first). Statistics were by Excel Data Analysis ToolPak, T-test tool. Figures plotted with Microsoft Excel; plots arranged and presented using Affinity Designer software.
Findings regarding N1303K CFTR
N1303K is the second most common class 2 CFTR defect, and a variant for which ETI has recently been FDA-approved based on clinical findings showing favorable—albeit less than maximal—lung function improvement79. Figure 5 shows activation of N1303K CFTR by HDCF104 in primary airway epithelial monolayers beyond what can be accomplished with ETI alone. In Fig. 5a–h, nasal airway epithelial cells from an individual with genotype N1303K/N1303K exhibited an additional 4- 5 µA/cm2 stimulation when corrector treatment was followed by HDCF104. Nasal airway epithelium from a patient with N1303K/G542X genotype (Fig. 5i, j) also responded to the HDCF compound. G542X is expected to be “null,” and findings indicate ~ 4 µA/cm2 stimulated CFTR current in the sample being tested. This level of rescue when combined with ETI amounts to ~30% of wildtype CFTR in primary nasal airway epithelium, a value seen as indicative of strong clinical respiratory improvement4,19,20,21,79.

a−h HDCF104 activates short circuit current in N1303K/N1303K homozygous and (i, j) N1303K/G542X heterozygous nasal airway epithelial monolayers. Drug additions include amiloride (100 µM), forskolin (5 µM), HDCF104 (10 µM), curcumin (40 µM), Inh-172 (10 µM), and VX-770 (5 µM). UTP, which activates non-CFTR dependent anion transport, was tested at 100 µM. Corrector 4a has been shown to stabilize CFTR. Curcumin potentiates certain CFTR variants such as W1282X (Wang et al., PLoS One, 2016). n = 3 biological replicates per condition. ETI (48 h) = elexacaftor (3 μM)—tezacaftor (3 μM)—ivacaftor (5 μM). p < 0.03 for HDCF104 plus ETI versus previous addition. Statistics were by Excel Data Analysis ToolPak, T-test tool. Figures plotted with Microsoft Excel; plots arranged and presented using Affinity Designer software.
HDCF104 and treatment of other CFTR mutations
With regard to levels of CFTR activation shown in Figs. 4 and 5, it is reasonable to expect that combination drug treatment may ultimately be required to maximally benefit many refractory CF variants. For example, as introduced above, although ETI has been FDA-approved for N1303K CFTR, a recent clinical trial demonstrated substantive but only partial clinical benefit in vivo among patients with this abnormality79. In other words, the magnitude of FEV1 rescue in human subjects with N1303K was less than what has been observed for several CFTR variants following triple combination therapy, and there is no question that further improvements in CFTR activation would be desirable79. Drugs in the HDCF series appear well suited to achieve that objective. In the same context, we evaluated a series of common CFTR mutations, many of which have recently been approved for ETI despite comparatively modest in vitro CFTR activation. The magnitude of lung function improvement for these variants in human subjects is not yet known. However, based on preclinical findings from predictive model systems it is likely that—in semblance to N1303K—much greater pulmonary improvement will be possible than with triple combination therapy alone. Our findings indicate that HDCF104 confers a robust increase in CFTR function when combined with ETI for many of these CFTR abnormalities, and may be useful to improve clinical outcomes (Table 1).
HDCF mechanism of action
Because the HDCF agents were identified by unbiased compound library screening, their mechanism of action was not initially known. Wildtype CFTR and many variants, including less responsive mutations such as N1303K (Fig. 5), as well as CFTR proteins exhibiting severe gating or other defects (Fig. 3 and Table 1) are activated by this drug class. Based on such findings, we initially anticipated a mechanism that involved direct binding of drug to the CFTR protein itself. However, the broad spectrum of activity observed for HDCF agents prompted us to explore a variety of possible contributing pathways. In one series of experiments, a specific PKA inhibitor (H89), which diminishes PKA-induced CFTR phosphorylation and activation, was used to investigate mechanism of action. Electrophysiologic (Ussing) tracings (Fig. 6a, b) demonstrated strong stimulation of WT CFTR by uHTS159 or HDCF104 in a CF bronchial epithelial cell line (CFBE41o-). Unexpectedly, a brief (1 h) pretreatment with H89 had minimal impact on VX-770 potentiation, whereas CFTR activation by HDCF analogs was abolished, suggesting a mechanism distinct from that of VX-770 (a drug that directly binds and improves gating of certain CFTR variants but with modest effects on wildtype CFTR in the absence of forskolin/cAMP pre-stimulation) (ref. 7, Fig. 6c), and unpublished observations. Acute addition of H89 partially blocked VX-770-stimulated current, which then recovered spontaneously (Fig. 6c, red tracings). These findings suggest VX-770 opens CFTR by virtue of conformational change that does not confer robust R-domain phosphorylation, and operates in a manner partially independent of the classical CFTR activation mechanism. The results using VX-770 also contrast studies of acute H89 addition followed by forskolin (Fig. 6a–c, red and black tracings), in which forskolin-stimulated CFTR chloride current is abrogated.

a, b CFTR-dependent chloride currents stimulated by saturating concentrations of uHTS159 or HDCF104 in CFBE41o- cells were inhibited by acute or more chronic addition of H89, a specific PKA blocker. c VX-770 displays a distinct activation pattern that was only transiently inhibitable by acute H89. d Western blot analysis demonstrates dose-dependent phosphorylation of WT CFTR by uHTS159 utilizing a phospho-sensitive CFTR antibody (UNC217) in comparison with nonphospho-sensitive CFTR detection (UNC596; an antibody recognizing an epitope in NBD2). Phosphorylation diminishes UNC217 affinity at a specific CFTR R-domain epitope (aa 807- 819). One additional PKA phosphorylation site assayed by this approach showed evidence of uHTS159 dependent phosphorylation (UNC450, aa 696 - 705). e Density of protein bands was quantified with Image Lab software (Bio-Rad) and plotted as % phosphorylation normalized to UNC596 and vehicle control (DMSO) (Methods). n = 3 biological replicates per condition. Significance levels determined by one-way ANOVA. *p ≤ 0.006; **p ≤ 0.04; NS not significant compared with H89 alone. Statistics were by Excel software, ANOVA single factor function. Panels (a−c) plotted using Microsoft Excel; (d) blot images generated with Image Lab 5.2 software, edited with Adobe Photoshop 2025, and labeled using Microsoft PowerPoint; (e) plotted with Microsoft Excel. All plots were arranged and presented using Affinity Design software.
To further investigate the role of phosphorylation during CFTR activation by the HDCF class, we conducted biochemical measurements of the CFTR regulatory domain (R-domain). PKA-dependent R-domain phosphorylation represents a key first step during CFTR ion channel gating80. In particular, CFBE cells expressing WT CFTR were treated with uHTS159 and surface CFTR biotinylated, followed by immunoprecipitation. CFTR was then probed with four phospho-sensitive anti-CFTR antibodies, as well as an anti-CFTR antibody raised against a nonphospho-epitope. Western blot and percent R-domain phosphorylation by PKA were determined (Fig. 6d, e), and showed enhanced phosphate addition to serine at R-domain position 813, visualized by diminished binding of UNC217 antibody. The same CFTR position (serine 813) was recently studied using Cryo-EM by Fiedorczuk K et al. and shown to be in close proximity to a PKA-C binding site, suggesting this location may be particularly important for initiating R-domain phosphorylation and CFTR channel activity80. H89 treatment led to decreased CFTR phosphorylation as judged by increased antibody binding at the same epitope. A comparable alteration was noted at serine 700 after uHTS159 treatment. Historically, certain phospho-epitopes (serines 737 and 768) have been identified as inhibitory81 of CFTR gating. These two positions appeared less sensitive to uHTS159-induced phosphate addition. In contrast, saturating concentrations of forskolin strongly enhanced phosphorylation of the two inhibitory sites (not shown), indicating the mechanistically complex process of CFTR domain rearrangement and channel opening elicited by forskolin as opposed to HDCF104 or uHTS159. Together, the findings point to CFTR stimulation by HDCF compounds as dependent on PKA phosphorylation at specific positions within the regulatory domain.
We next examined whether PDE inhibitors would influence WT CFTR activation by HDCF compounds in primary human bronchial epithelial cells (HBEC). Administration of IBMX (a broad-range PDE inhibitor) prior to HDCF104 (10 µM) completely eliminated CFTR ion transport elicited by HDCF104 (Fig. 7a, b). Rolipram, a specific PDE4 blocker, also abolished HDCF104 stimulation (Fig. 7c). Similar findings were obtained at higher concentrations of HDCF104 (100 µM, Fig. 7d–f). Cilostamide (a specific PDE3 inhibitor) did not activate CFTR either prior to or after HDCF104 administration (Fig. 7g, h). These data indicate that compounds in the HDCF104 class75, which have been specifically optimized for CFTR activation, act as PDE inhibitors to elevate cAMP intracellularly and compartmentally82,83,84,85 to phosphorylate the CFTR regulatory domain.

a−h Loss of HDCF104 activity demonstrated following pretreatment with IBMX or rolipram, but not a PDE3 inhibitor (cilostamide). Pretreatment with HDCF104 attenuated the activity of rolipram and IBMX. Compounds added acutely include amiloride (100 µM), forskolin (5 µM), Inh-172 (10 µM), and UTP (100 µM). To assure saturating drug concentrations, IBMX (broad spectrum PDE inhibitor) was added at 200 µM, whereas rolipram (PDE4 specific inhibitor) and cilostamide (PDE3 inhibitor) were tested at 10 µM and 1 µM, respectively. HDCF104 was added at 10 µM in Panels (a) through (c), and 100 µM in Panels (d) through (h). The studies were conducted in non-CF primary nasal airway epithelial cells and show robust wildtype CFTR activation mediated by HDCF104. All panels were plotted using Microsoft Excel; plots were arranged and presented using Affinity Design software.
We also evaluated the HDCF series by a conventional cell-free enzymatic assay using purified PDE protein in which luciferase serves as an ATP “sink” over the linear range of luminescence. Because ATP is a substrate for PKA, inhibition of PDE4 (with a resultant rise in cAMP and PKA activity) elicits a decrease in ATP available to luciferase and decreased assay luminescence (Fig. 8). The inhibitory profile of HDCF104 showed IC50 of 7.5 µM for purified PDE4B—a concentration that agrees well with half-maximal activation levels of HDCF104 needed to stimulate wildtype CFTR in primary airway cells (Fig. 2). We note that drug concentrations of this magnitude are commonly used for mucosal administration to the human respiratory tract (unlike oral agents, where greater potency may be required but with an associated risk of systemic toxicity). For example, levels of approved antibiotics or other compounds administered by aerosol to cystic fibrosis lungs are often at a concentration range of 100−200 µM in CF airway secretions in vivo86,87.

a Dose-response curve of HDCF104 as a blocker of PDE4B (IC50 of HDCF104 = 7.5 ± 0.32 μM). b Shows approximately two orders of magnitude lower IC50 of uHTS159 compared to HDCF104. c More modest inhibition of PDE3A was observed (in the μM range). (IC50 of uHTS159 = 0.06 ± 0.0021 μM as an inhibitor of PDE4B and 14.9 ± 1.57 μM for PDE3A). HDCF104 did not demonstrate inhibition of PDE3A at concentrations up to 100 μM (data not shown). IC50 of compounds calculated using GraphPad Prism 10 software, which was also employed for plotting all panels. Panels were arranged and presented with Affinity Designer software.
Finally, for many other PDE inhibitors—including those approved for clinical use—cross-reactivity with more than one PDE isoform is commonly observed. As case in point, in addition to strong blockade of PDE4s, modest inhibition of PDE5 by HDCF104 has also been identified. A plan for nebulized airway delivery of HDCF104 is intended to minimize PDEi-related off-target toxicities. Extensive safety analysis of the compound in two mammalian species has been presented to FDA as part of pre-IND interactions. The findings to date support future use of aerosolized HDCF104, including absence of PDE-related or other safety concerns.
Discussion
This report describes a novel drug scaffold developed specifically for mucosal activation of CFTR and well suited for testing as innovative therapy in airway diseases caused by deficient CFTR. Compounds in this class are intended to promote mucus clearance in people with CF or non-CF lung and sinonasal conditions. For engineered FRT cells and primary airway epithelial monolayers (two models recognized by FDA as relevant to registration of CFTR activators), drugs in the HDCF class markedly augment mutant CFTR activity beyond what can be achieved with HEMT alone (e.g., variants such as F508del, G551D, and many others), while also providing a means to strongly stimulate wildtype CFTR. Importantly, the compounds also increase short circuit current in cell monolayers encoding variants for which CFTR modulators are not considered “highly active” (e.g., N1303K79; also Table 1). Agents in the HDCF series exhibit several advantages relevant to treatment of CF and non-CF respiratory conditions including: (1) novel mechanism of action that does not overlap with currently approved CFTR modulators, (2) topical (mucosal) route of administration intended to minimize systemic exposure, (3) the ability to activate mutant CFTR (including “corrected” F508del CFTR) to levels that exceed what can be achieved with modulators alone, and (4) expected amelioration of immune-mediated pulmonary injury (see below).
In our studies, we used H89, a selective PKA inhibitor, to determine whether the new compound class acts through a mechanism involving PKA and CFTR phosphorylation. Administration of H89 following either HDCF104 or uHTS159 abolished CFTR stimulation, supporting a mechanism dependent upon R-domain phosphorylation (Fig. 6a, b). Moreover, we show that the compound series directly inhibits purified PDE4 enzyme (Fig. 8) and biochemically phosphorylates the CFTR R-domain (Fig. 6). In addition, HDCF agents stimulate CFTR activity in a dose-dependent manner using relevant cell-based systems (an effect expected to increase cAMP/PKA and promote R-domain phosphorylation) (Figs. 1 and 2) and exhibit a cross-reactive mechanism of action with other PDE inhibitors, including rolipram, a specific PDE4i (Fig. 7). In unpublished studies, we have shown that HDCF compounds elevate cellular cAMP in epithelial cells and fail to activate ∆R CFTR – findings that provide additional support for dependence on R-domain phosphorylation. Future studies using CFTR constructs encoding amino acid substitutions at consensus PKA sites may offer a means to characterize the R-domain conformational changes that underlie CFTR folding and gating after treatment with HDCF compounds.
A functional threshold in primary airway epithelial cells of 10−30% wild-type CFTR activity is often viewed in the field as predictive of respiratory benefit among PwCF4,20,21,37,88,89. In vitro studies of sinonasal epithelia show that establishing a benchmark can be somewhat imprecise, particularly due to significant variability of wildtype CFTR within cell monolayers obtained from nasal airways. In our laboratory, 10−15 µA/cm2 is frequently observed in epithelial monolayers derived from non-CF nasal tissue samples, although much larger CFTR currents can be obtained in cells from certain individuals4,21,36,37. As described above, model systems such as FRT lines have also been very useful for predicting respiratory improvement, and are recognized by the FDA for this purpose, particularly at CFTR activation levels above 10% of wildtype4,16,17,20,21. These thresholds suggest that for commercial and academic groups developing CFTR activators for rare variants, the impact of a molecule like HDCF104 could be transformational as a means to maximally stimulate mutant CFTR. Among gating mutations such as G551D, we show that HDCF104 can function either as monotherapy or provide a strong complement to FDA-approved modulators, leading to activity far above 10% of wildtype when combined with ETI (Fig. 3). For other variants, including N1303K (Fig. 5 and Table 1), a combination of drug treatments (similar to what has been required for F508del rescue) will likely be needed to achieve maximal benefit. Data from several laboratories, including recent clinical findings from our group79, indicate that N1303K stimulation by ETI occurs at levels that provide incomplete lung function improvement. Our current results demonstrate that HDCF104 confers activation of the N1303K variant to significantly above 30% of wildtype when combined with available modulators (Fig. 5). For PwCF who fail, are ineligible, or cannot gain access to HEMT, compounds in the HDCF104 series are anticipated to provide considerable value.
Enzymatically and pharmacologically distinct members of the PDE family—including PDE subtypes 1-11—are found in nearly all mammalian cells or tissues, and hydrolyze/inactivate cyclic nucleotides. Blockade of certain PDEs elicits compartmental elevation of cyclic AMP, which (in a manner dependent on PDE category, tissue, cell type, etc.) can strongly enhance protein kinase A activity and stimulate CFTR through phosphorylation of specific positions within the regulatory or other protein domains90,91. PDE4is have been previously recognized as activators of wildtype CFTR in airway epithelium90,92,93,94,95,96,97. Even within the same PDE4 subclass, however, differences in airway epithelial partitioning, PDE4i affinity, subcellular compartmentalization, plasma membrane permeability, cellular compound clearance, and other features may influence the magnitude and duration of effects on CFTR gating – including distinctions from use of forskolin to stimulate cAMP/PKA. We believe the application of high throughput assays and discovery of new PDE4is specifically tailored for CFTR activation in primary airway epithelium, as described here, will be essential for optimizing interventions that address CFTR deficiency in a number of human disease states. A large body of clinical, biophysical, biochemical, and genetic evidence supports that contention51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,68,69,70,71,72,82,83,84,85. As indicated above, emerging data establishes that several common muco-obstructive respiratory conditions, including non-CF bronchiectasis (NCFB) and CRS, share key pathogenic and mechanistic features with CF lung disease51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,68,69,70,72. For patients with these illnesses, stimulation of CFTR and improved mucociliary clearance are likely to result from treatment with agents such as those described here.
PDE4 blockade has been established as a clinically effective means to diminish immune hyper-responsiveness within human lungs98,99,100,101,102,103,104,105,106,107. In recent preclinical studies, PDE4 inhibitors were shown to blunt inflammation in models of asthma, mediate bronchodilation and suppress inflammatory response associated with COPD, diminish immune activity of eosinophils in human bronchial specimens from subjects with bronchitis, repress inflammation in human airway tissue, and mediate numerous additional anti-inflammatory effects of this sort98,99,100,101,102,103,104,105,106,107. From the standpoint of the current findings, anti-inflammatory effects attributable to PDE4i blockade would be in addition to ways in which CFTR activation, itself, may diminish cytokine release from airway cells108. AstraZeneca markets the oral PDE4 inhibitor roflumilast for treatment of COPD101,109,110,111 as a broad-spectrum compound that blunts several inflammatory pathways. The oral drug has been significantly limited by gastrointestinal and other systemic sequelae, including pronounced diarrhea. Side effects due to oral (systemic) PDE4is have led commercial developers to advance aerosolized PDE3 and PDE4 blockers for treating inflammation in COPD. Such drugs have shown considerable promise—without significant GI or other limiting complications98,108,112,113,114,115,116,117,118,119.
Mucosal administration of CFTR activators represents an emerging area of interest for treatment of chronic respiratory conditions such as CF. Although wildtype CFTR stimulation by PDE4is is well described, activation of disease-causing CFTR variants has been less well studied in the CF modulator era. Recent work from Hanrahan and colleagues demonstrated that RPL554 (a PDE3»PDE4 inhibitor) enhanced G551D and a number of other mutant CFTRs in the FRT model120,121,122. Based on high-throughput compound analysis, Beekman et al. recently concluded that certain PDE4i molecules rank among the most promising for stimulating mutant CFTR123. Findings presented in the current study highlight the effectiveness of PDE4i compounds for activating both wildtype and variant CFTR at levels expected to provide clinical improvement.
In summary, we describe a novel class of CFTR gating enhancers discovered using unbiased cell-based screening. The compounds exhibit robust activity in both FRT and primary airway cell models with a molecular target (CFTR) already well validated by an extensive body of genetic, biophysical, and clinical literature. Drugs described here are expected to have a therapeutic role not only in cystic fibrosis pulmonary and sinonasal disease, but also much more common illnesses associated with CFTR deficiency such as non-CF bronchiectasis and chronic sinusitis. The mechanism of action for the new drug series has been identified as phosphodiesterase blockade, and lead agents have been optimized specifically for topical use in human airway epithelium to activate CFTR-dependent mucociliary clearance. Importantly, since both mutant and wildtype CFTR are strongly enhanced, both CF and non-CF muco-obstructive illnesses are appropriate targets for the new drug class. Compounds in the same mechanistic category have already been shown to provide clinically beneficial anti-inflammatory properties in diseases such as COPD, but have not been well tested in CF, NCFB, or CRS. Future clinical development is merited to determine the extent to which mucosal CFTR activators can be used to improve treatment of inherited and acquired CFTR deficiency in human airways.
Methods
Cell culture
Fischer rat thyroid (FRT) cells expressing diverse CFTR variants were cultured in Coon’s modification of Nutrient Mixture F-12 Ham media (Sigma, F6636) supplemented with 2.68 g/l sodium-bicarbonate in the presence of 5% fetal bovine serum (Gibco). The cells were maintained at 37 C° under humidified, 5% CO2 – 95% air. For electrophysiology studies, polarized cell cultures were established, with FRT cells seeded onto Transwell permeable supports (Corning, 3470). Cells were cultured for 5 days until a mature monolayer was established (with transepithelial resistance of at least 400 Ω × cm2).
Primary nasal or bronchial epithelial cells carrying diverse CFTR variants (hNEC N1303K/N1303K patient K and hNEC N1303K/G542X patient H, SickKids, Toronto, Canada; hBEC F508del/F508del product code EP57AB, Geneva Switzerland; hBEC healthy donor FC0035, Lifeline Cell Technology, Frederick, MD; hNEC healthy donor patient 21.A2, University of Alabama, Birmingham) were expanded using PneumaCult-Ex Plus medium (StemCell Technology; 05040) on collagen-coated flasks, followed by seeding onto collagen-treated Transwell inserts. Resulting monolayers were cultured for 2–4 days under submerged conditions, and subsequently transitioned to air-liquid interface (ALI) utilizing PneumaCult ALI medium (StemCell Technology; 05001). Cells were then maintained at 37 C° under humidified, 5% CO2—95% air until well-differentiated epithelium was established, typically within 3 weeks of seeding.
CFBE41o- cells with stable expression of WT CFTR124 were cultured in MEM medium supplemented with L-glutamine and 10% FBS. Cells for surface CFTR biotinylation studies or electrophysiology assays were seeded onto Transwell permeable supports (Corning, 3450 or Corning, 3470) and grown until well polarized under submerged conditions.
FRT clonal cell lines with stable expression of CFTR variants
Fischer Rat Thyroid cells (FRT) were used to establish an FRT Flip-In host cell line utilizing pFRT/lacZeo vector. WT or mutant human CFTR cDNAs were incorporated at a single insertion site according to the manufacturer’s protocol (Thermo Fisher Scientific). Cell clones demonstrating comparable levels of CFTR mRNA expression (within 0.5–1.5 fold of wildtype) were selected from each CFTR variant pool to minimize differences in response after modulator treatment (a confounder that may otherwise result from distinct CFTR protein expression levels).
Transient expression of CFTR variants in FRT cells
Rare CFTR missense variants were selected from among those listed in the CFTR2 mutation database (https://cftr2.org). CFTR constructs encoding these molecular defects contained nucleotide changes identical to those found in patients (including methionine at position 470) and were cloned into the pcDNA5/FRT mammalian expression vector (Thermo), as previously described17. Expression was under regulatory control of the CMV promoter. Variants were transiently expressed and studied by short-circuit current (Ussing chamber) assays to investigate CFTR function.
Electrophysiology
A total of 1.2 × 10^5 FRT cells were seeded and cultured on each Transwell permeable support (Corning 3470). One day after seeding, cells on each filter were transfected with 0.5 μg of CFTR plasmid using Lipofectamine 3000 (Thermo). The media was changed 72 h post-transfection, and cells were treated with the indicated CFTR modulators for 24 h. CFTR functional assays were performed on day 5 post-seeding in the Ussing chamber17. Eight filter supports were analyzed for each CFTR variant by Ussing chamber experiments, and each experiment included wildtype CFTR-expressing cells as positive controls.
Ussing chamber analysis
Monolayers were mounted onto an EasyMount Ussing Chamber System (Physiologic Instruments) and bathed apically in low chloride Ringer’s solution containing: 140 mM Na-gluconate, 1.2 mM NaCl, 25 mM NaHCO3, 3.33 mM KH2PO4, 0.83 mM K2HPO4, 1.2 mM CaCl2, 1.2 mM MgCl2, and 10 mM D-glucose (pH 7.4). Basolateral solutions contained 120 mM NaCl, 25 mM NaHCO3, 3.33 mM KH2PO4, 0.83 mM K2HPO4, 1.2 mM CaCl2, 1.2 mM MgCl2, and 10 mM D-glucose (pH 7.4) to establish a chloride gradient. Temperature of bathing solutions was maintained at 37 °C and stirred by bubbling through 5% CO2/95% O2. Test compounds were dissolved in DMSO at 1000X concentration and diluted as described. For bioelectric measurements, once a baseline had been established, 100 μM amiloride (MilliporeSigma, A7410) was applied to both the apical and basolateral surfaces to inhibit epithelial sodium channel (ENaC) activity. CFTR protein was activated by applying forskolin (MilliporeSigma, F3917), typically 5 μM (FRT cells) or 10 μM (primary cells), followed by addition of test compound. In some experiments, VX-770 (SelleckChem, S1144) was added apically at 5 μM. At the end of each recording, CFTRinh-172 (10 μM; MilliporeSigma C2992) was applied at the mucosal surface to inhibit CFTR currents. In primary cell experiments, UTP (stimulates non-CFTR chloride channels; VWR, 0145) was added to confirm monolayer integrity. Rolipram (Selleck S1430), Cilostamide (Enzo BML-PD125) and IBMX (MilliporeSigma, I7018) were used to determine importance of phosphodiesterase activity. Across all experiments, short circuit current was recorded under voltage clamp conditions with change in Isc following acute treatment calculated and expressed as mean ± SD.
CFTR phosphorylation
CFBE41o- cells expressing WT CFTR were seeded onto permeable supports to generate polarized monolayers grown under immersed conditions (typically ~6 days until mature). Test compounds (e.g., uHTS159, ChemSpace US Inc., CSSS00160668684) and/or PKA inhibitor (H89 2HCl, Selleckchem, S1582) were applied bilaterally for 15 min. At the end of each incubation, monolayers were transferred to ice-cold PBS and oxidation of glycan chains followed by biotinylation with Hydrazide-LC-Biotin (Thermo Scientific, 21340)125. Cells were next lysed in RIPA buffer supplemented with protease and phosphatase inhibitor, and equal amounts of total protein immunoprecipitated using Streptavidin magnetic beads (Thermo Scientific (Pierce), 88817). Immunoprecipitated proteins were resolved on 8-16% Novex, Tris-Glycine SDS-PAGE (Thermo Fischer Scientific, XP08165BOX) and transferred to nitrocellulose membrane (Bio-Rad, 1620112). CFTR proteins were blotted with specific antibodies UNC596, UNC217, UNC432, UNC570, UNC450 (CFTR Antibody Distribution Program, UNC-Chapel Hill, Dr. Martina Gentzsch), followed by detection with HRP-goat-anti-mouse antibody (Abcam, ab205719). Specific bands were visualized with SuperSignal ELISA Femto Maximum Sensitivity Substrate (Thermo Scientific, 37074). The ChemiDoc MP System was used for imaging. Band densities were quantified with ImageLab 5.2.1 software, and normalized to corresponding bands from UNC596 (as loading control) followed by normalization to DMSO-treated conditions. Results were plotted and expressed as mean ± SEM. Single-factor ANOVA was used to determine significance.
IC50 determination and in vitro PDE enzyme activity assay
PDE-Glo Phosphodiesterase Assay (Promega, V1361), recombinant human PDE4B (Abcam, ab125582), and recombinant human PDE3A (Abcam, ab125582) were used in a 384-well plate format to determine IC50 of test compounds according to the manufacturer’s protocol. Recombinant PDE enzymes were titrated to establish optimal concentrations (determined for PDE4B or PDE3A as 3 ng/reaction) and reaction times (identified as 20 min). Test compound was added to each plate in serial dilution and relative luminescent signal was detected using a BioTek Synergy Neo2 plate reader. For calculating IC50 of test compounds, relative luminescent signal was plotted against concentration with GraphPad Prism 10 software (Sigmoidal, 4PL, X as concentration function).
Statistical methods
Descriptive statistics (mean, median, standard deviation and standard error of mean) were applied to in vitro continuous data (e.g., Isc). All in vitro studies were conducted in paired fashion (same day, same cell passage). A p-value (α) of less than or equal to 0.05 was considered statistically significant, based on single-factor ANOVA, paired t-tests and/or Mann-Whitney analysis.
Data availability
All data is provided within the manuscript.
References
Manfredi, C., Tindall, J. M., Hong, J. S. & Sorscher, E. J. Making precision medicine personal for cystic fibrosis. Science 365, 220–221 (2019).
Bell, S. C. et al. The future of cystic fibrosis care: a global perspective. Lancet Respir. Med. 8, 65–124 (2020).
Shteinberg, M. and Taylor-Cousar, J. L. Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. Eur. Respir. Rev. 29, 190112 (2020).
Clancy, J. P. et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros 18, 22–34 (2019).
McCague, A. F. et al. Correlating cystic fibrosis transmembrane conductance regulator function with clinical features to inform precision treatment of cystic fibrosis. Am. J. Respir. Crit. Care Med. 199, 1116–1126 (2019).
Grasemann, H. & Ratjen, F. Cystic fibrosis. N. Engl. J. Med. 389, 1693–1707 (2023).
Sorscher, E. J. Cystic fibrosis. In Harrison’s Principles of Internal Medicine(ed. Jameson, J. L. et al.) (McGraw-Hill Education/Medical, New York, NY, 2022).
Drugs.com. FDA approves Alyftrek. https://www.drugs.com (2024).
Keating, D. et al. VX-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 379, 1612–1620 (2018).
Middleton, P. G. et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N. Engl. J. Med. 381, 1809–1819 (2019).
TRIKAFTA [prescribing information]. (Vertex Pharmaceuticals Incorporated, Boston, MA, 2020).
CFF. FDA Approves Trikafta for 94 Additional Rare CFTR Mutations. https://news.vrtx.com/news-releases/news-release-details/vertex-announces-us-fda-approval-trikafta (2024).
CFF. Drug Development Pipeline. https://www.pfizer.com/science/drug-product-pipeline (2021).
Sionna. Sionna Therapeutics Launches With $ 111 Million Series B Financing To Advance Pipelinee Of Novel Small Molecules With The Potential To Fully Restore Cftr Function In Cystic Fibrosis. https://www.prnewswire.com/news-releases (2022).
Marchesin, V. et al. A uniquely efficacious type of CFTR corrector with complementary mode of action. SciAdv. 10, eadk1814 (2024).
Bihler, H. et al. In vitro modulator responsiveness of 655 CFTR variants found in people with cystic fibrosis. J. Cyst. Fibros 23, 664–675 (2024).
Han, S. T. et al. Residual function of cystic fibrosis mutants predicts response to small molecule CFTR modulators. JCI Insight 3, e121159 (2018).
Sabusap, C. M. et al. Analysis of cystic fibrosis-associated P67L CFTR illustrates barriers to personalized therapeutics for orphan diseases. JCI Insight 1, e86581 (2016).
Sosnay, P. R. et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet 45, 1160–7 (2013).
Millen, L. et al. Theratyping 2019. Pediatr. Pulmonol. 54, 2300110 (2019).
Durmowicz, A. G., Lim, R., Rogers, H., Rosebraugh, C. J. & Chowdhury, B. A. The U.S. Food and Drug administration’s experience with ivacaftor in cystic fibrosis. establishing efficacy using in vitro data in lieu of a clinical trial. Ann. Am. Thorac. Soc. 15, 1–2 (2018).
Le, C., McCrary, H. C. & Chang, E. Cystic fibrosis sinusitis. Adv. Otorhinolaryngol. 79, 29–37 (2016).
Hamilos, D. L. Chronic rhinosinusitis in patients with cystic fibrosis. J. Allergy Clin. Immunol. Pract. 4, 605–12 (2016).
Lee, S. E. et al. Cystic fibrosis transmembrane conductance regulator modulator therapy: a review for the otolaryngologist. Am. J. Rhinol. Allergy 34, 573–580 (2020).
Lowery, A. S. et al. Chronic rhino-sinusitis treatment in children with cystic fibrosis: a cross-sectional survey of pediatric pulmonologists and otolaryngologists. Int. J. Pediatr. Otorhinolaryngo.l 124, 139–142 (2019).
Chang, E. H. New insights into the pathogenesis of cystic fibrosis sinusitis. Int. Forum Allergy Rhinol. 4, 132–7 (2014).
Loebinger, M. R., Bilton, D. & Wilson, R. Upper airway 2: Bronchiectasis, cystic fibrosis and sinusitis. Thorax 64, 1096–101 (2009).
Chang, M. T. & Patel, Z. M. Update on long-term outcomes for chronic rhinosinusitis in cystic fibrosis. Curr. Opin. Otolaryngol. Head Neck Surg. 28, 46–51 (2020).
Zemke, A. C. et al. Clinical predictors of cystic fibrosis chronic rhinosinusitis severity. Int. Forum. Allergy Rhinol. 9, 759–765 (2019).
Gostelie, R. et al. The impact of ivacaftor on sinonasal pathology in S1251N-mediated cystic fibrosis patients. PLoS One 15, e0235638 (2020).
Henig, N. Sinusitis and cystic fibrosis. Vol. 2021 (Stanford Medicine, The Cystic Fibrosis Center at Stanford).
Sorscher, E. Cystic fibrosis: embracing risk and inspiration. In Plenary Address, CF Foundation Invitational Research Conference (Stowe, VT, 2024).
Della Sala, A. et al. A nonnatural peptide targeting the A-kinase anchoring function of PI3Kγ for therapeutic cAMP modulation in pulmonary cells. J. Biol. Chem. 300, 107873 (2024).
Garcia, B. & Flume, P. A. Pulmonary complications of cystic f.ibrosis. Semin. Respir. Crit. Care Med. 40, 804–809 (2019).
Pranke, I. M. et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 7, 7375 (2017).
Keegan, D. E. & Brewington, J. J. Nasal epithelial cell-based models for individualized study in cystic fibrosis. Int. J. Mol. Sci. 22, 4448 (2021).
Van Goor, F. et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 106, 18825–30 (2009).
Neuberger, T., Burton, B., Clark, H. & Van Goor, F. Use of primary cultures of human bronchial epithelial cells isolated from cystic fibrosis patients for the pre-clinical testing of CFTR modulators. Methods Mol. Biol. 741, 39–54 (2011).
Safi, C., Zheng, Z., Dimango, E., Keating, C. & Gudis, D. A. Chronic rhinosinusitis in cystic fibrosis: diagnosis and medical management. Med. Sci. (Basel) 7, 32 (2019).
Aanæs, K. Bacterial sinusitis can be a focus for initial lung colonisation and chronic lung infection in patients with cystic fibrosis. J. Cyst. Fibros 12, S1–20 (2013).
Johnson, B. J., Choby, G. W. & O’Brien, E. K. Chronic rhinosinusitis in patients with cystic fibrosis-Current management and new treatments. Laryngoscope Investig. Otolaryngol. 5, 368–374 (2020).
Fothergill, J. L., Neill, D. R., Loman, N., Winstanley, C. & Kadioglu, A. Pseudomonas aeruginosa adaptation in the nasopharyngeal reservoir leads to migration and persistence in the lungs. Nat. Commun. 5, 4780 (2014).
Armbruster, C. R. et al. Evolution and adaptation of Pseudomonas aeruginosa in the paranasal sinuses of people with cystic fibrosis. bioRxiv https://doi.org/10.1101/2020.10.29.359844 (2020).
Rosbe, K. W., Jones, D. T., Rahbar, R., Lahiri, T. & Auerbach, A. D. Endoscopic sinus surgery in cystic fibrosis: do patients benefit from surgery?. Int. J. Pediatr. Otorhinolaryngol. 61, 113–9 (2001).
Nelson, J., Karempelis, P., Dunitz, J., Hunter, R. & Boyer, H. Pulmonary aspiration of sinus secretions in patients with cystic fibrosis. Int Forum. Allergy Rhinol. 8, 385–388 (2018).
Luparello, P. et al. Outcomes of endoscopic sinus surgery in adult lung transplant patients with cystic fibrosis. Eur. Arch. Otorhinolaryngol. 276, 1341–1347 (2019).
Bopaka, R. G., El Khattabi, W., Janah, H., Jabri, H. & Afif, H. Bronchiectasis: a bacteriological profile. Pan. Afr. Med. J. 22, 378 (2015).
Maselli, D. J., Amalakuhan, B., Keyt, H. & Diaz, A. A. Suspecting non-cystic fibrosis bronchiectasis: What the busy primary care clinician needs to know. Int. J. Clin. Pract. 71, e12924 (2017).
Chalmers, J. D., Chang, A. B., Chotirmall, S. H., Dhar, R. & McShane, P. J. Bronchiectasis. Nat. Rev. Dis. Primers 4, 45 (2018).
Flume, P. A., Chalmers, J. D. & Olivier, K. N. Advances in bronchiectasis: endotyping, genetics, microbiome, and disease heterogeneity. Lancet 392, 880–890 (2018).
Miller, A. C. et al. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc. Natl. Acad. Sci. USA 117, 1621–1627 (2020).
Pignatti, P. F., Bombieri, C., Marigo, C., Benetazzo, M. & Luisetti, M. Increased incidence of cystic fibrosis gene mutations in adults with disseminated bronchiectasis. Hum. Mol. Genet 4, 635–9 (1995).
Casals, T. et al. Bronchiectasis in adult patients: an expression of heterozygosity for CFTR gene mutations?. Clin. Genet 65, 490–5 (2004).
Bienvenu, T. et al. Cystic fibrosis transmembrane conductance regulator channel dysfunction in non-cystic fibrosis bronchiectasis. Am. J. Respir. Crit. Care Med. 181, 1078–84 (2010).
Teerapuncharoen, K. et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction and radiographic bronchiectasis in current and former smokers: a cross-sectional study. Ann. Am. Thorac. Soc. 16, 150–153 (2019).
Swenson CE, H. W. et al. Treating non-cystic fibrosis bronchiectasis with CFTR modulators: early case reports. Ann. Case Rep. 9, 101910 (2021).
Banks, C., Freeman, L., Cho, D. Y. & Woodworth, B. A. Acquired cystic fibrosis transmembrane conductance regulator dysfunction. World J. Otorhinolaryngol. Head Neck Surg. 4, 193–199 (2018).
Cho, D. Y. & Woodworth, B. A. Acquired cystic fibrosis transmembrane conductance regulator deficiency. Adv. Otorhinolaryngol. 79, 78–85 (2016).
Wang, X. & Cutting, G. R. Chronic rhinosinusitis. Adv. Otorhinolaryngol. 70, 114–121 (2011).
Wang, X., Kim, J., McWilliams, R. & Cutting, G. R. Increased prevalence of chronic rhinosinusitis in carriers of a cystic fibrosis mutation. Arch. Otolaryngol. Head Neck Surg. 131, 237–40 (2005).
Wang, X. et al. Mutation in the gene responsible for cystic fibrosis and predisposition to chronic rhinosinusitis in the general population. JAMA284, 1814–9 (2000).
Raman, V., Clary, R., Siegrist, K. L., Zehnbauer, B. & Chatila, T. A. Increased prevalence of mutations in the cystic fibrosis transmembrane conductance regulator in children with chronic rhinosinusitis. Pediatrics 109, E13 (2002).
Woodworth, B. A. Resveratrol ameliorates abnormalities of fluid and electrolyte secretion in a hypoxia-Induced model of acquired CFTR deficiency. Laryngoscope 125(Suppl 7), S1–s13 (2015).
Tipirneni, K. E. et al. Assessment of acquired mucociliary clearance defects using micro-optical coherence tomography. Int. Forum. Allergy Rhinol. 7, 920–925 (2017).
Alexander, N. S. et al. Cystic fibrosis transmembrane conductance regulator modulation by the tobacco smoke toxin acrolein. Laryngoscope 122, 1193–7 (2012).
Zhang, S. et al. Quercetin increases cystic fibrosis transmembrane conductance regulator-mediated chloride transport and ciliary beat frequency: therapeutic implications for chronic rhinosinusitis. Am. J. Rhinol. Allergy 25, 307–12 (2011).
Cho, D. Y. et al. LPS decreases CFTR open probability and mucociliary transport through generation of reactive oxygen species. Redox. Biol. 43, 101998 (2021).
Clunes, L. A. et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. Faseb J. 26, 533–45 (2012).
Dransfield, M. T. et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest 144, 498–506 (2013).
Virgin, F. W. et al. Exposure to cigarette smoke condensate reduces calcium activated chloride channel transport in primary sinonasal epithelial cultures. Laryngoscope 120, 1465–9 (2010).
Mall, M. A. & Hartl, D. CFTR: cystic fibrosis and beyond. Eur. Respir. J. 44, 1042–54 (2014).
Sloane, P. A. et al. A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS One 7, e39809 (2012).
Palmer, J. N. et al. Efficacy of EDS-FLU for Chronic rhinosinusitis: two randomized controlled trials (ReOpen1 and ReOpen2). J. Allergy Clin. Immunol. Pract. 12, 1049–1061 (2024).
Marklew, A. J. et al. Cigarette smoke exposure induces retrograde trafficking of CFTR to the endoplasmic reticulum. Sci. Rep. 9, 13655 (2019).
Rab, A. et al. A Novel 7H-[1,2,4]Triazolo[3,4-b]thiadiazine-based cystic fibrosis transmembrane conductance regulator potentiator directed toward treatment of cystic fibrosis. ACS Med. Chem. Lett 14, 1338–1343 (2023).
Cell_Model_Resources. Cell Model Resources. https://www.cff.org/researchers/cell-model-resources (2021).
Baell, J. B. & Nissink, J. W. M. Seven year itch: pan-assay interference compounds (PAINS) in 2017-utility and limitations. ACS Chem. Biol. 13, 36–44 (2018).
Ramsey, B. W. et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 365, 1663–72 (2011).
Solomon, G. M. et al. Elexacaftor-Tezacaftor-Ivacaftor improves clinical outcomes in individuals with CF encoding N1303K CFTR. Lancet Respir. Med. 27, s2213−2600 (2024).
Fiedorczuk, K. et al. The structures of protein kinase A in complex with CFTR: Mechanisms of phosphorylation and noncatalytic activation. Proc. Natl. Acad. Sci. USA 121, e2409049121 (2024).
Hegedus, T. et al. Role of individual R domain phosphorylation sites in CFTR regulation by protein kinase A. Biochim. Biophys. Acta.1788, 1341–9 (2009).
Murabito, A., Bhatt, J. & Ghigo, A. It Takes Two to Tango! Protein-Protein Interactions behind cAMP-Mediated CFTR Regulation. Int. J. Mol. Sci. 24, 10538 (2023).
Monterisi, S. et al. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J. Cell Sci. 125, 1106–17 (2012).
Huang, P. et al. Local regulation of cystic fibrosis transmembrane regulator and epithelial sodium channel in airway epithelium. Proc. Am. Thorac. Soc. 1, 33–7 (2004).
Barnes, A. P. et al. Phosphodiesterase 4D forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J. Biol. Chem. 280, 7997–8003 (2005).
Ruddy, J. et al. Sputum tobramycin concentrations in cystic fibrosis patients with repeated administration of inhaled tobramycin. J. Aerosol. Med. Pulm Drug Deliv. 26, 69–75 (2013).
Hubert, D. et al. Pharmacokinetics and safety of tobramycin administered by the PARI eFlow rapid nebulizer in cystic fibrosis. J. Cyst. Fibros 8, 332–7 (2009).
van Goor, F., Hadida S., Grootenhuis, P. Pharmacological rescue of mutant CFTR function for the treatment of cystic fibrosis. In Ion Channels. Topics in Medicinal Chemistry, (ed. Fermini, B., Priest, B. T.) 3 (Springer, Berlin, Heidelberg, 2008).
Van Goor, F., Yu, H., Burton, B. & Hoffman, B. J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros 13, 29–36 (2014).
Kelley, T. J., Al-Nakkash, L., Cotton, C. U. & Drumm, M. L. Activation of endogenous deltaF508 cystic fibrosis transmembrane conductance regulator by phosphodiesterase inhibition. J. Clin. Invest. 98, 513–20 (1996).
Penmatsa, H. et al. Compartmentalized cyclic adenosine 3’,5’-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol. Biol. Cell 21, 1097–110 (2010).
Liu, S. et al. Dynamic activation of cystic fibrosis transmembrane conductance regulator by type 3 and type 4D phosphodiesterase inhibitors. J. Pharmacol. Exp. Ther. 314, 846–54 (2005).
Schultz, B. D., Singh, A. K., Devor, D. C. & Bridges, R. J. Pharmacology of CFTR chloride channel activity. Physiol. Rev. 79, S109–44 (1999).
Cobb, B. R. et al. A(2) adenosine receptors regulate CFTR through PKA and PLA(2). Am. J. Physiol. Lung Cell Mol. Physiol. 282, L12–25 (2002).
Blanchard, E. et al. Anchored PDE4 regulates chloride conductance in wild-type and ΔF508-CFTR human airway epithelia. Faseb J. 28, 791–801 (2014).
Rowe, S. M. et al. DeltaF508 CFTR processing correction and activity in polarized airway and non-airway cell monolayers. Pulm Pharmacol. Ther. 23, 268–78 (2010).
Cobb, B. R., Fan, L., Kovacs, T. E., Sorscher, E. J. & Clancy, J. P. Adenosine receptors and phosphodiesterase inhibitors stimulate Cl- secretion in Calu-3 cells. Am. J. Respir. Cell Mol. Biol. 29, 410–8 (2003).
Phillips, J. E. Inhaled phosphodiesterase 4 (PDE4) inhibitors for inflammatory respiratory diseases. Front Pharmacol. 11, 259 (2020).
Zuo, H., Cattani-Cavalieri, I., Musheshe, N., Nikolaev, V. O. & Schmidt, M. Phosphodiesterases as therapeutic targets for respiratory diseases. Pharmacol. Ther. 197, 225–242 (2019).
Turner, M. J., Dauletbaev, N., Lands, L. C. & Hanrahan, J. W. The phosphodiesterase inhibitor ensifentrine reduces production of proinflammatory mediators in well differentiated bronchial epithelial cells by inhibiting PDE4. J. Pharmacol. Exp. Ther. 375, 414–429 (2020).
Rabe, K. F. et al. Anti-inflammatory effects of roflumilast in chronic obstructive pulmonary disease (ROBERT): a 16 week, randomised, placebo-controlled trial. Lancet Respir. Med. 6, 827–836 (2018).
Cazzola, M., Calzetta, L., Rogliani, P. & Matera, M. G. Ensifentrine (RPL554): an investigational PDE3/4 inhibitor for the treatment of COPD. Expert. Opin. Investig. Drugs 28, 827–833 (2019).
Southworth, T. et al. Anti-inflammatory effects of the phosphodiesterase type 4 inhibitor CHF6001 on bronchoalveolar lavage lymphocytes from asthma patients. Cytokine 113, 68–73 (2019).
Govoni, M. et al. Sputum and blood transcriptomics characterisation of the inhaled PDE4 inhibitor CHF6001 on top of triple therapy in patients with chronic bronchitis. Respir. Res. 21, 72 (2020).
Yougbare, I. et al. NCS 613, a potent PDE4 inhibitor, displays anti-inflammatory and anti-proliferative properties on A549 lung epithelial cells and human lung adenocarcinoma explants. Front Pharmacol. 11, 1266 (2020).
Yang, J. X. et al. Synergistic effect of phosphodiesterase 4 inhibitor and serum on migration of endotoxin-stimulated macrophages. Innate. Immun. 24, 501–512 (2018).
Lea, S. et al. The modulatory effects of the PDE4 inhibitors CHF6001 and roflumilast in alveolar macrophages and lung tissue from COPD patients. Cytokine 123, 154739 (2019).
Veit, G. et al. Proinflammatory cytokine secretion is suppressed by TMEM16A or CFTR channel activity in human cystic fibrosis bronchial epithelia. Mol. Biol. Cell 23, 4188–202 (2012).
Rhee, C. K. & Kim, D. K. Role of phosphodiesterase-4 inhibitors in chronic obstructive pulmonary disease. Korean J. Intern. Med. 35, 276–283 (2020).
Wedzicha, J. A., Calverley, P. M. & Rabe, K. F. Roflumilast: a review of its use in the treatment of COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 11, 81–90 (2016).
Lennon, K. FDA Approves Starting Dose of Roflumilast. https://www.mdedge.com/content/fda-approves-starting-dose-roflumilast (2018).
Singh, D. et al. Efficacy and safety of CHF6001, a novel inhaled PDE4 inhibitor in COPD: the PIONEER study. Respir. Res. 21, 246 (2020).
Study to evaluate the efficacy and safety of two doses of CHF6001 DPI (Tanimilast) as add-on to maintenance triple therapy in subjects with COPD and chronic bronchitis (PILLAR). (Good Clinical Practice Network).
Singh, D. et al. COPD patients with chronic bronchitis and higher sputum eosinophil counts show increased type-2 and PDE4 gene expression in sputum. J. Cell Mol. Med. 25, 905–918 (2021).
Singh, D. et al. Effect of the inhaled PDE4 inhibitor CHF6001 on biomarkers of inflammation in COPD. Respir. Res. 20, 180 (2019).
Mariotti, F., Govoni, M., Lucci, G., Santoro, D. & Nandeuil, M. A. Safety, tolerability, and pharmacokinetics of single and repeat ascending doses of CHF6001, a novel inhaled phosphodiesterase-4 inhibitor: two randomized trials in healthy volunteers. Int. J. Chron. Obstruct. Pulmon. Dis. 13, 3399–3410 (2018).
Lucci, G. et al. Safety, tolerability and pharmacokinetics of CHF 6001, a novel selective inhaled PDE4 inhibitor, in healthy volunteers. Eur. Resp. J. 48, PA4086 (2016).
PracticeUpdate. ATS 2019: the novel inhaled PDE4 inhibitor CHF6001 shows promise in moderate to severe COPD and chronic bronchitis. (Respiratory Medicine, 2019).
GlobeNewswire. Verona Pharma announces US FDA approval of Ohtuvayre (ensifentrine). https://www.globenewswire.com/newsrelease/2024/06/26/2904839/0/en/Verona-Pharma-Announces-US-FDA-Approval-of-Ohtuvayre-ensifentrine.html (2024).
Turner, M. J., Abbott-Banner, K., Thomas, D. Y. & Hanrahan, J. W. Cyclic nucleotide phosphodiesterase inhibitors as therapeutic interventions for cystic fibrosis. Pharmacol. Ther. 224, 107826 (2021).
Nguyen, J. P. et al. Modulation of cAMP metabolism for CFTR potentiation in human airway epithelial cells. Sci. Rep.11, 904 (2021).
Turner, M. J., Luo, Y., Thomas, D. Y. & Hanrahan, J. W. The dual phosphodiesterase 3/4 inhibitor RPL554 stimulates rare class III and IV CFTR mutants. Am. J. Physiol. Lung Cell Mol. Physiol. 318, L908–l920 (2020).
de Poel, E. et al. FDA-approved drug screening in patient-derived organoids demonstrates potential of drug repurposing for rare cystic fibrosis genotypes. J. Cyst. Fibros 22, 548–559 (2023).
Bebok, Z. et al. Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. J. Physiol. 569, 601–15 (2005).
Prince, L. S., Workman, R. B. Jr & Marchase, R. B. Rapid endocytosis of the cystic fibrosis transmembrane conductance regulator chloride channel. Proc. Natl. Acad. Sci. USA 91, 5192–6 (1994).
Acknowledgements
Funding for this study was provided by the Cystic Fibrosis Foundation (SORSCH21XX0) and Georgia Research Alliance (GRA VL24.C10).
Author information
Authors and Affiliations
Contributions
CRediT author statementAndras Rab: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Writing—original draft, Writing—review and editing. Jeong S. Hong: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Resources, Data curation, Writing—review and editing, Visualization, Supervision. Candela Manfredi: Investigation, Data curation, Writing—review and editing, Visualization. Disha Joshi: Investigation, Writing—review and editing, Visualization. Sadhana Ponnaluri: Investigation, Writing—review and editing. William F. Tracy: Investigation, Writing—review and editing. Yujie Luo: Investigation. Xun Yang: Investigation. Alexander A. Kolykhalov: Conceptualization, Methodology. Huw M.L. Davies: Conceptualization, Methodology, Writing—review and editing. Eric J. Sorscher: Conceptualization, Methodology, Formal analysis, Writing—original draft, Writing—review and editing, Supervision, Project administration, Funding acquisition.
Corresponding author
Ethics declarations
Competing interests
Dr. Sorscher serves as a non-voting member of the Cystic Fibrosis Foundation Board of Trustees and is Chair of the Foundation’s Medical Advisory Council, both unpaid positions. The Foundation had no participation in the preparation of this manuscript or the research reported herein. All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rab, A., Hong, J.S., Manfredi, C. et al. A novel drug series optimized to address cystic fibrosis and other CFTR deficiency diseases of human airways. npj Drug Discov. 2, 27 (2025). https://doi.org/10.1038/s44386-025-00026-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44386-025-00026-1
