
Abstract
Spatial organization of the disease microenvironment informs patient prognosis. Key modalities for studying spatial biology include H&E images (WSIs) for tissue structure and spatial transcriptomics (ST) for transcriptome-level programs. Spatial analysis aims to (1) identify markers linked to clinical outcome, (2) understand functional programs driving these associations, and (3) guide targeted therapies. Current research addresses these topics but offers limited explainability across the full morphology - molecular mechanism - outcome axis. Further, given the abundance of WSIs and limited availability of ST, there is a need for analyses integrating these complementary datasets. We present an AI-driven framework combining foundation-model features, multiple-instance learning, unsupervised clustering, and molecular analyses to identify mechanisms underlying outcome associated patterns. Applied to HER2+ breast cancer, we identify CCND1 and PTK6 signaling in tumor regions linked to trastuzumab resistance, consistent with prior studies. Our approach offers interpretable insights for multi-level resistance mechanisms, tissue-specific drug targeting, and precision medicine.
Similar content being viewed by others
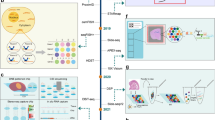

Introduction
The tissue microenvironment in any disease is a complex ecosystem of cell types and tissue structures. Spatial context of these components is fundamental to understanding biological behavior. In cancer, such spatial biomarkers are predictive of disease progression; for example, high presence of M2 tumor associated macrophages (TAMs) at the invasive edge of gastric tumors has been found correlated to recurrence1. With rapid advancements in spatial data generation, the ultimate goal is to decode spatial biology across different modalities and resolutions to (1) Identify spatial markers associated with clinical outcome, (2) Interpret these associations, and (3) Leverage this deeper understanding to develop effective targeted therapies. 3 areas of research currently address different parts of this challenge: tissue morphology analysis, cell neighborhood analysis, and spatial transcriptomics (ST) analysis including spatial domain identification and mechanistic discovery. We first review existing methodologies in each of these areas before presenting our novel framework.
Hematoxylin and Eosin (H&E) tissue staining, which highlight nuclei and cytoplasm to reveal spatial structures, is fundamental to cancer diagnostic imaging. Recent morphological profiling methods leverage digitized H&E whole slide images (WSIs) with image features derived from foundation models, to predict both prognostic and molecular data directly from WSIs with high accuracy2. Due to the large size of WSIs, multiple instance learning (MIL) is commonly used, treating each WSI as a ‘bag of instances’ with instances corresponding to image tiles3,4. Understanding model decision-making is challenging, making attention-MIL and transformer models popular for their ability to score tiles by importance to the clinical outcome or molecular profile5.
Multiplex imaging identifies tissue-wide single cell (sc) phenotypes, while ST identifies gene expression at spot or sc resolution depending on the technology. Cell type proximity measures can be quantified from both these modalities through point pattern statistics or window clustering to identify cell type neighborhoods6. Correlation between these metrics and clinical outcome can then provide spatial markers6. Several studies have integrated morphology pattern discovery with cell type enrichment. Namely, in 2024 Quiros et al. presented a workflow to cluster representative H&E image features into morphology phenotypes, associate these with patient survival, and calculate cell type enrichment in each phenotype from cell segmentation7.
The rapid advancement of spatial technology has led to the explosion of computational techniques for ST, including spatial domain identification, regional differential expression, cell-type deconvolution, and cell communication analysis8,9,10,11,12,13. While studies associating ST domains and underlying molecular mechanisms with clinical outcomes are still limited, this field is gaining momentum—such as Zheng et al., which identified AC-like subclusters linked to poor prognosis in GBM14. Recent efforts further integrate H&E with ST to detect spatial domains using paired datasets, but this approach offers limited explainability of how specific morphological patterns relate to molecular programs and outcomes15,16.
Although ST technologies are becoming increasingly accessible, H&E remains the standard modality in pathological prognosis. Given the abundance and availability of WSIs, there is significant room to develop analytical pipelines leveraging WSIs to discover clinical outcome associations and integrate this knowledge with ST and other modalities downstream17. The closest literature that develops such a workflow is by Laury et al.18. In this paper, they performed microarray ST on high confidence (HC) and background regions of H&E slides, previously obtained by training a convolutional neural network (CNN) to predict ovarian carcinoma therapeutic response. They discovered several genes upregulated in the HC regions between short and long term patient groups. However, their methodology is not designed to integrate analysis of separate complementary WSI and ST datasets, is not fully computational as it employs ST after HC region identification, does not obtain or characterize clinically relevant fine-grain morphology features, and does not perform diverse molecular analyses. In this paper, we present a novel framework to identify clinically-associated H&E patterns by analyzing high-attention regions from a predictive MIL model, and link these patterns to molecular mechanisms including cell neighborhood and gene program enrichment. Importantly, we design our framework so that H&E pattern analyses and molecular mechanism analyses can be performed on separate complementary WSI and ST datasets. To the best of our knowledge, no such framework exists to map disease morphology to underlying molecular programs. Our framework is interpretable and addresses all 3 goals listed above. Since our novelty lies not in one component, but in the integrative workflow, our framework is highly generalizable, allowing the choice of any high performance predictive model or molecular analyses based on biological question and further enables the easy integration of additional data modalities. We apply proof of concept (PoC) on separate complementary WSI and ST HER2 + BRCA datasets to identify mechanisms driving imaging patterns associated with trastuzumab resistance. Our methodology can answer the following main questions for any disease application: 1. What morphological patterns may be associated with my outcome of interest? 2. Are these associated with cell neighborhoods on any scale? 3. What cell-type specific pathways are activated in morphology patterns from 1? 4. Are there any upstream cell- cell interactions that could be driving these pathways? 5. What potential drug targets can we nominate based on these identified mechanisms?
In this PoC, a top result from our methodology identifies the CCND1 gene and PTK6 signaling pathway activated in tumor cells in a resistance-associated pattern compared to the rest of the tissue, indicating hierarchical resistance mechanisms and potential drug targets. Future direction of our framework focuses on agentic representation for workflow efficiency, with key implications for designing tissue-specific drug delivery, clinical trial optimization, and interpretable precision medicine.
Results
Multi-modal framework identifies mechanisms underlying outcome-associated morphology patterns
We developed a novel, 4-module framework to identify morphology patterns associated with clinical outcome and elucidate their distinct molecular characteristics. This methodology is outlined in Fig. 1. Here we present a PoC using 2 datasets - the first is a cohort of 82 HER2 + BRCA patients with matched (FFPE) H&E, tumor ROIs, and trastuzumab response outcome19. The second is a cohort of 29 HER2 + BRCA samples with matched (frozen) H&E and ST20. Throughout this paper, we refer to these as the “outcomes” dataset and the “spatial” dataset respectively. We interchangeably use “resistant” and “nonresponder” to refer to patients who do not respond to trastuzumab treatment. Additionally, we interchangeably use “cluster” and “pattern” to indicate the results of image tile clustering. We start with module 1: pre-processing both datasets to obtain comparable tile-level feature vectors, including style transfer for frozen to FFPE in the spatial dataset. We then identify outcome-associated morphology patterns in module 2 by (1) training an attention-MIL classifier to predict outcome, (2) clustering high-attention tile features, and (3) performing statistical tests to validate pattern compositional differences between groups. Once we identify patterns associated with the resistant group from the outcomes dataset, we map all patterns to the spatial dataset in module 3. We also perform cell type deconvolution in module 3. Finally, in module 4 we perform molecular analysis on the ST spots underlying group-associated patterns, including cell neighborhood enrichment, cell type specific differential expression (DE), and ligand receptor (LR) analysis. Integrating the results of these molecular analyses, we generate hypotheses on therapy resistance mechanisms and potential drug targets. Table 1 shows parameter decisions that can be made by the user in each module, to guide both understanding and implementation. If the user chooses, the pipeline can also be fully run with all default values and no changes (excluding directory paths) on any other dataset.

Overview of AI-driven framework.
Preprocessing produces high quality real and pseudo-FFPE images
We first processed all WSIs from both datasets using Otsu’s background thresholding and Reinhard stain normalization. We extracted tiles of size 224 × 224 pixels with 50% overlap, using available invasive tumor ROIs for the outcomes dataset. Example tiled images for 2 samples from the outcomes dataset are shown in part a of Fig. 2. We then input the outcomes dataset tiles into the Convolutional Neural Network (CNN) backbone of the RetCCL foundation model encoder.

a Example of tiles cut from annotated invasive ROIs. b UMAP of RetCCL features before and after AI-FFPE style transfer on frozen spatial dataset tiles. c Pathologist tile classification performance and example tiles for QC. d Comparison of attention-MIL average AUC across 3-fold CV, without and with self-attention function before model training. e UMAP representation of high-attention features colored by leiden cluster, and all features colored by leiden clusters from separate high and low attention clustering. f Example tile pattern assignments in high attention regions.
Before passing the normalized spatial dataset tiles to RetCCL, we performed style transfer using AI-FFPE21. FFPE is the diagnostic gold standard for H&E staining and is used in our outcomes dataset, while our spatial dataset uses fresh frozen tissue. AI-FFPE is a generative adversarial network (GAN) that converts an input image in the reference space (frozen) to the target space (FFPE). When training this model, we observed the total loss function remaining steady after an initial decrease in the first 3 epochs, while the discriminator loss decreased and generator loss increased. This trend indicates that the discriminator is likely improving faster than the generator; thus the generator cannot further improve without removing biological context. To validate this, we trained the model for 15 epochs, ran the trained model per epoch on each frozen spatial dataset tile to obtain FFPE style images, and used the RetCCL encoder to obtain final feature embeddings. We then calculated the Frechet Inception Distance (FID) score between the original outcomes dataset and pseudo-FFPE spatial dataset RetCCL features. We observe that the FID score plateaued by epoch 4 (Supplementary Fig. 1), indicating early convergence of the generator-discriminator dynamics as predicted from our loss function trends. Thus, we limited training to 15 epochs and selected the last epoch for the final pseudo-FFPE tile embeddings.
We created UMAP representations of all outcomes dataset tile features, original frozen spatial dataset features, and final pseudo-FFPE spatial dataset features. From the 2 plots in part b of Fig. 2, we can identify the clear domain shift in feature representation, noting that the final pseudo-FFPE spatial dataset features are well mixed with the outcomes dataset features.
To qualitatively evaluate pseudo-FFPE tiles, we obtained pathologist (S.B.) annotations across real, frozen, and pseudo-FFPE tile images. To assess whether the pseudo-FFPE tiles appeared convincingly similar to FFPE tiles, 75 tiles were randomly selected from FFPE, frozen, and pseudo-FFPE WSIs and the pathologist was tasked with assigning the appropriate label for each tile in a blinded fashion. The pathologist was able to classify pseudo-FFPE tiles with an F1-score of 0.62, lower than both the FFPE and frozen tile classification scores (F1 = 0.72 and 0.69 respectively). While the classification was performed in a manner highly sensitive to pseudo-FFPE (recall: pseudo-FFPE = 0.72, FFPE = 0.64, frozen = 0.65), leading to a high-detection rate, low specificity (pseudo-FFPE = 0.71, FFPE = 0.93, frozen = 0.87) and precision (pseudo-FFPE = 0.55, FFPE = 0.81, frozen=0.72) highlight poor discrimination of pseudo-FFPE from FFPE. In a follow up pathologist inspection of the pseudo-FFPE tiles, only 1 tile contained artifacts that would significantly negatively impact histopathological assessment. These statistics and example tiles from FFPE, frozen, and pseudo-FFPE are shown in Fig. 2c. We can validate quality improvement after style transfer, and the similarity between real and psuedo-FFPE images from these examples. Altogether, these demonstrate the frozen to pseudo-FFPE transformation performed convincingly well at the level of visual histopathological examination.
Attention classifier and leiden clustering highlight resistance associated morphology pattern 13
Our framework uses the attention-MIL classifier to predict outcome from input H&E tile features. We note in the reference publication for our outcomes dataset, they predicted both HER2 status and Trastuzumab resistance directly from WSIs using weakly supervised learning through convolutional neural networks (CNNs)19. Their study revealed, after using tumor ROIs, the average slide-level AUC across 5 CV folds for HER2 prediction was 0.90 and for treatment response prediction was 0.80. Thus in our framework, we repeat this prediction of the Trastuzumab response, but with an attention-MIL methodology that allows downstream mechanistic mapping, reaffirming that our novelty lies in the integrative pipeline and not in one single component. We performed 3-fold cross validation (CV) and calculated slide-level AUC scores for each fold. We start with the attention-MIL model because it has proven high accuracy; however, a key assumption is tile independence, ignoring inter-tile relationships (refs. 22,23). Recent methods to address this include TransMIL, inspired by a transformer encoder. Encoders use self-attention to embed each tile with contextual information, capturing similarities with all other tiles while considering positional encoding. However, transformers are inherently computationally expensive; thus we trained 2 versions of the attention-MIL - one with a single self-attention layer prior to model training, and one without. In our self-attention layer, we calculate similarities between the center tile and only its nearest k neighbors. Then, a weighted aggregate of the neighboring tile embeddings is calculated to represent the new center tile embedding, which is input into the attention network of the classifier. This is inspired by existing neighborhood applications, including cell neighborhood identification through “windows”, and cell graph modeling using graph neural networks (GNN’s) to aggregate over node neighbors6,24,25. We see from part d of Fig. 2 that while the average AUC across 3 folds remains steady from 0.71 to 0.72 after incorporating self-attention, the standard deviation across 3 folds reduces from 0.11 to 0.06, stabilizing the model. Thus, we ran our classifier with a self-attention step on all samples in the outcomes dataset.
To calculate a final attention score per tile, we took the weighted sum of attention scores across the 3 folds using the normalized AUC as the weight per fold. We then aggregated the original embeddings from the top 20% (per sample) attention tiles to perform Leiden community clustering. We created a UMAP representation of the resulting optimal 17 clusters from the high attention tile embeddings as part e of Fig. 2. We also clustered the original embeddings from the low attention (remaining tiles) across all samples, resulting in 23 clusters, to later allow downstream mapping of non-relevant invasive regions to the spatial dataset (Fig. 2e). We note that although the silhouette score is close to 0 for both high and low attention clustering, indicating fuzzy boundaries, the UMAP visualizations reveal enough group distinction for PoC. We show spatial tile pattern assignments for the top attention regions of 2 nonresponder samples in Fig. 2f. However, high-attention regions from such an MIL approach often have false positives even if the classifier has high AUC, i.e, regions that the classifier claims are relevant to outcome prediction but may actually be noise or random image artifacts. To account for this, we performed two statistical tests to validate pattern differences between outcomes.
First, for each pattern, we ran a nonparametric rank-sum test between the patient group pattern composition values across high-attention regions. Patterns 7, 13, and 14 were found significantly different after p-value adjustment, using a threshold of 0.1 and noting that the p-values of patterns 13 and 14 are close to 0.05; both at 0.069 (Fig. 3a). We only consider patterns 13 and 14 to be of interest as these comprise a greater composition of high-attention regions in resistant patients than responders. Second, we ran a Fisher’s test to identify the enrichment of the resistant group in each pattern region. From part b of Fig. 3, this test identifies non-responder enrichment in 8 patterns. We focus on evaluating the intersection of both tests to account for inter-sample pattern heterogeneity, which is pattern 13. The box plot in Fig. 3 reveals pattern compositions in high-attention regions across samples in each patient group. From this, we first note the heterogeneity of pattern composition across patients’ high attention regions, which is expected in any cancer. Looking at pattern 13, we notice while the median difference in composition is small, the compositional variance and 3rd quartile are much higher in resistant patients. From the sample-specific compositional heatmap in part d of Fig. 3, we see that all responders have less than 20% composition of pattern 13 among high attention tiles, but nonresponders have either more than 40% or none of pattern 13. The presence of nonresponder samples with no pattern 13 explains the similarity in median composition between groups, but the heatmap clearly reveals high variance across nonresponders. This suggests that when present, pattern 13 is likely a resistance-associated signal and our downstream molecular analysis thus focuses on regions belonging to this pattern in detail.

a Rank-sum test results comparing non-responder and responder group pattern compositions. b Fisher’s test non-responder results; * reveals enriched patterns in high attention regions of non-responders. c Boxplot of pattern composition from high-attention regions of all samples grouped by treatment response. d Heatmap of individual patterns compositions from high-attention regionsacross all samples with pattern 13 column highlighted.
Mapping morphology patterns from outcomes to spatial dataset reveals 5 samples with pattern 13 regions
To discover molecular trends underlying pattern 13, we mapped all morphology patterns, both from high and low attention regions across outcomes dataset WSIs, to the spatial dataset. We used K-Nearest Neighbors (KNN) on the tile feature embeddings, resulting in 16 of the 17 high attention patterns and all 23 of the low attention patterns identified as shown in part a of Fig. 4. An example of spatial tiles overlaid on the H&E image with assigned patterns is also shown in part b of Fig. 4. We then assigned each underlying ST spot to a pattern, noting that there are a total of 200 pattern 13 spots across 9051 spots over all samples. Specifically, 5 samples from the spatial dataset have more than 10 pattern 13 spots. These are: SPA125 with 74, SPA120 with 22, SPA121 with 21, SPA139 with 20, and SPA140 with 11 spots. Figure 4c shows spot-level pattern assignments of several samples.

a UMAP of spatial dataset RetCCL features colored by outcomes dataset pattern assignments. b Example overlays of tile-level pattern assignments in spatial dataset. c Example mapping of tile-level pattern to spot-level pattern assignments in spatial dataset. d Enrichment testing to determine if any histopathological features are pattern 13-specific. e Example tiles from pattern 13 and other patterns.
Pathologist interpretation of patterns reveals distinct pattern 13 morphology
To assess pattern 13 for unique histopathological features, a pathologist (S.B) analyzed 277 tiles sampled across all patterns, including 50 tiles from pattern 13. 45 of 50 tiles in pattern 13 contained tumor, a significant enrichment compared to all other patterns (O.R = 5.88, P = 2.81e-05) (Fig. 4d). When comparing tumor tiles in pattern 13 to tumor tiles in all other patterns, pattern 13 was enriched for tiles with high pleomorphism (O.R. = 3.90, P = 0.02) and dense stroma (O.R. = 31.86, P = 7.34e-08) (Fig. 4d), while no difference was found in tissue-type composition, lymphocyte infiltration, vascularity, mitotic activity, apocrine features, plasmacytoid morphology or tumor necrosis (Supplementary Fig. 1, part b). These features can be visually validated in the tile examples shown in Fig. 4e across pattern 13 tiles and other patterns from both low and high attention regions. Specifically, in non-pattern 13 tiles, we observe tumor or normal epithelial cells among loose stroma, or in the bottom right corner tile we identify dense stroma with necrosis present. Cases with pleomorphic tumor cells among dense stroma are prevalent only in pattern 13, indicating distinct biological signal.
Molecular analysis underlying pattern 13 identifies tumor-specific PTK6 signaling and downstream CCND1 enrichment as a mediator of tumor cell resistance
First, to identify cell type organization across space and allow cell type specific DE for multi-cellular resolution spots, we performed deconvolution using BayesPrism and a HER2+ sc reference (Supplementary Fig. 3)11,26. The spatial pie maps for deconvolution results of samples SPA125 and SPA121 are shown in part a of Fig. 5. We also show spatial maps for the remaining 2 samples with more than 20 pattern 13 spots, and 3 samples with no pattern 13 spots in Supplementary Fig. 2. As a sanity check, we note there is a low presence of regular epithelial cells across samples, but a high proportion of tumor cells as expected.

a Cell type deconvolution. b Pattern 13 vs rest of tissue compositions. c Tumor-specific pattern 13 differential expression. d CCND1 expression spatial map alongside pattern 13. e IPA top pathways by p value. f Drug-gene set enrichment results from DSigDB and DGIdb. g Filtered CellChat results for tumor-specific interactions.
We performed both individual cell type and neighborhood enrichment in pattern 13 regions after deconvolution, as described in Supplementary Figs. 3 and 4. Both did not reveal any cell type enrichment in pattern 13, as can be validated in part b of Fig. 5. Thus, we infer that the resistance-associated signal identified in pattern 13 is not due to cell type composition, but likely due to more complex changes in tissue structure, molecular behavior, and cellular interactions.
To gain spatial molecular insights into resistance, we performed cell type-specific DE between pattern 13 spots and the rest of the tissue using C-SIDE. In the context of trastuzumab response, we are especially interested in why tumor cells in pattern 13 might be resistant to anti-HER2 therapy, and therefore continue to present cell proliferation and anti-apoptosis behavior. Thus, we focus on tumor cell-specific genes here, shown in part c of Fig. 5. The remaining cell type results are in Supplementary Fig. 4. Among the top 10 upregulated tumor DE genes ranked by log fold change, several have been previously implicated in HER2 + BRCA including CCND1 and HOXB7.
First, we visually assess the correspondence of CCND1 high spots with pattern 13 spots of sample SPA125, which has the largest number of pattern 13 spots. Figure 5 part d suggests that CCND1 expression is low-moderate, ranging from 0 to 3 log normalized counts (2–7 raw counts) and we confirm the pattern correspondence. CCND1 expression is directly regulated by HER2 through several downstream pathways such as PI3K/AKT, and codes for cyclin D127. Cyclin D1 forms a subunit with CDK4/6 and is thus required for cell cycle G1/S transition28. The cyclin-CDK4 axis has been shown to mediate targeted treatment resistance in mouse models of HER2-positive BRCA, supporting CCND1 expression even without an upstream HER2 stimulus27. A recent study investigated the role of dual HER2 blockades in HER2E (Her2-enriched) tumors, and observed that tumor cells that are sensitive to anti-HER2 therapy such as trastuzumab, but do not die, become luminal A type29. This opens opportunities to use existing anti-luminal A treatments including FDA-approved CDK4/6 inhibitors. Using EnrichR, we identified drug-gene sets enriched in our tumor-specific DE genes from DGIdb, a repository of known drugs with specific gene targets. We see from the top table in part f of Fig. 5 that CCND1 is a key recurring target and 4 of the top 10 drugs are kinase inhibitors, of which LY2835219 (Amebaciclib) and Ribociclib are specifically built for CDK4/6 inhibition. Thus, in our study, the upregulation of CCND1 in tumor cells in resistance-associated pattern 13 across samples supports the potential efficacy of CDK4/6 inhibitors, either through a sequential maintenance program or directly in conjunction with anti-HER2 therapies.
HOXB7 codes for a transcription factor regulating cell proliferation and differentiation30. Several studies have shown the overexpression of HOXB7 in BRCA, supporting epithelial-to-mesenchymal transition and therefore tumor metastasis31. The MYC-HOXB7-HER2 signaling pathway has been proposed as a target in endocrine-resistant BRCA32. While HOXB7 has not been identified as a specific target in HER2 + BRCA resistance, the clear role of this gene in BRCA metastasis further supports the validity of our results.
We ran Ingenuity Pathway Analysis (IPA) to perform pathway enrichment on each cell type specific set of DE genes, focusing first on the 39 identified genes in tumor cells. From part e of Fig. 5, PTK6 signaling is the most significantly enriched pathway, including the DE genes: ARHGAP35, CCND1, and UBC. PTK6 is an oncogenic non-receptor kinase that typically binds to HER2 heterodimers, resulting in the activation of several downstream mechanisms that can regulate CCND1 expression (Reactome database). In normal tissue, these mechanisms regulate tissue homeostasis, but in tumor cells, PTK6 signaling supports proliferation and inhibition of cell death. PTK6 signaling inhibition has been shown to promote apoptosis in tumors resistant to targeted Lapatinib therapy, and simultaneous knockdown of HER2 and PTK6 in HER2 over-expressing BRCA tumors was shown to significantly reduce cell proliferation33,34. These studies indicate persistent PTK6 activity even after HER2 has been blocked through other stimuli. We then used EnrichR to identify drug-gene sets enriched in tumor specific genes in the DSigDB database, a diverse repository of known drug-gene interactions using drug influence on gene expression. This database is more broad than DGIdb, including both direct and indirect drug-gene set interactions, making it a good resource for functional analysis. From the bottom table in part f of Fig. 5, we see the top drug is arsenenous acid, or arsenic trioxide, also found in the top 10 drugs from DGIdb. Integrating results from both databases, this compound directly targets CCND1 and influences the expression of 10 other DE genes, including those involved downstream of PTK6 signaling. While this drug is not currently part of a clinical trial for HER2 + BRCA, it has been approved for acute promyelocytic leukemia (APL) and is being investigated in other cancers35. Thus, our framework identifies both the PTK6 pathway and Cyclin D1-CDK4/6 complex in conjunction with anti-HER2 therapy as strong potential trastuzumab resistance targets, with enrichment analysis proposing the investigation of Amebaciclib/Ribociclib kinase inhibitors and arsenic trioxide with anti-HER2 therapy. A high-level overview of HER2, PTK6 and CCND1 regulation mechanism is shown in Fig. 6, revealing the direct HER2-CCND1 regulation pathway, indirect PTK6-CCND1 regulation pathway, and protein complex activation.

High-level overview of HER2, PTK6, and CCND1 mechanism.
To infer cell communication through spatial gene expression profiling at spot resolution and still consider likely interacting cell types, we used an integrative approach with both sc and spatial data. We first used CellChat to identify top LR pairs of interest from the same public sc reference used for deconvolution. In parallel, we used spatialDM to identify significant spatially co-expressed LR pairs in pattern 13 from at least one sample with more than 10 pattern 13 spots. We note that in this dataset, we did not find LR pairs uniquely co-expressed only in pattern 13 vs rest of the tissue; however, LR interactions often drive differential downstream pathways resulting from tissue microenvironment characteristics. Thus, we proceed with our PoC analysis, but in future applications, pattern-specific significance of LR co-expression would be ideal. We then identified the intersection of spatially proximal pairs and interacting pairs from sc expression, resulting in 10 LR pairs. CellChat results for these are shown in Fig. 5g. We further found the intersection of these ligand and receptor molecules with upstream regulators of cell-type specific DE genes indicated by IPA, with the goal of discovering potential intercellular signaling mechanisms of gene expression regulation and pathway activation specific to pattern 13.
First, IPA analysis found that the ligands and receptors FN1, CDH1, CDH2, MIF, and PECAM1 were upstream regulators of tumor-specific genes differentially expressed in pattern 13. Interestingly, CCND1 was found downstream of all these ligands and receptors. We focus on 2 identified pairs here based on literature - from the CellChat results, FN1 most likely interacts with the SDC4 receptor between CAF and tumor cells, and CDH1-CDH1 interactions are most likely between tumor cells. However, from our spatial co-expression analysis, the CDH1-CDH1 interaction is significant in 3 out of 4 samples’ pattern 13 regions, while FN1-SDC4 is significant in 1. Thus, we further investigate CDH1-CDH1. First, CDH1 codes for the E-cadherin protein, a cell-cell adhesion molecule that promotes strong cell connections (NIH gene report). Yamauchi et al. showed that HER2 overexpressing breast tumor cells presenting E-cadherin were less sensitive to antibody-dependent cellular cytoxicity (ADCC) mediated by trastuzumab as compared to those without E-cadherin (by knocking down CDH1)36. Additionally, Bajrami et al. used perturbation screening in diverse breast cancer cell lines to identify synthetic lethality between E-cadherin/ROS1 tyrosine kinase inhibition, supporting the role of E-cadherin in tumor sensitivity to treatment37. While there is no literature revealing the direct influence of CDH1-CDH1 on downstream CCND1 regulation in HER2 + BRCA, Fournier et al. show that blocking of both E-cadherin and integrin adhesion is required to reduce CCND1 expression in normal mammalian epithelial cells38. From our integrative analysis and literature, we can hypothesize that E-cadherin interactions in pattern 13 could be indirectly regulating CCND1 expression, and maintaining connected tumor cell structures across the tissue, thus potentially disrupting antibody access.
Discussion
The spatial organization of the microenvironment significantly influences patient prognosis in many diseases. A well-established example is that high levels of infiltrating lymphocytes (TILs) in the tumor microenvironment (TME) are often associated with improved patient outcome39. Current methods to discover spatial TME patterns include correlating tissue morphology with clinical outcome, identifying cell proximity patterns, and finding molecular domains through gene expression and morpho-transcriptomic analysis. With advancements in spatial omics and imaging, there is an urgent need to connect across these themes in an interpretable manner to support a structured understanding of clinical outcome. In this study, we present a multi-modal integrative framework to discover mechanisms underlying outcome-associated tissue patterns, with the ultimate goal of identifying regional therapy targets. We deliver PoC on HER2 + BRCA using 2 datasets - the first from a study of matched FFPE H&E with trastuzumab response outcome as patient group, and the second from a study investigating general spatial molecular patterns with matched frozen H&E and ST.
Our methodology consists of 4 main modules - (1) Preprocessing and Style Transfer, (2) Identifying group-associated morphology patterns from outcomes dataset, (3) Map morphology patterns to spatial dataset, and (4) Molecular analysis to identify mechanisms driving tissue regions associated with patient group. In the first module, we used a foundation model encoder to extract features from the processed tile images from both datasets, using style transfer as needed. In module 2, we discovered outcome-associated morphology patterns by clustering tile-level feature embeddings across high-attention WSI regions obtained from our trained attention-MIL classifier. We performed two statistical tests to validate enrichment in the resistant patient group, from which we identified pattern 13 of interest. We further clustered low-attention regions to account for non-relevant tissue patterns. In module 3, we assigned outcomes dataset morphology patterns to the spatial dataset using KNN on tile feature embeddings, and mapped the tile-level pattern assignments to ST spot level. We also deconvolved each sample to get cell type proportions across the tissue using a pre-treatment sc reference. This allowed us to perform comprehensive molecular analysis in module 4, including cell type composition enrichment, neighborhood discovery, and cell type specific DE to investigate mechanisms underlying pattern 13 compared to the rest of the tissue.
There are 3 key results of our methodology applied to HER2 + BRCA. First, we identify morphology pattern 13 associated with trastuzumab resistance, corresponding to pathologist annotations of pleomorphic tumor cells within dense stroma. Second, at the gene level, we discover CCND1 is a top upregulated gene (by log FC: fold change) in tumor cells in pattern 13 compared to other regions. CCND1 sustains tumor growth post-anti-HER2 therapy, and its role in CCND1-CDK4/6 complexes driving cell cycle progression supports the potential efficacy of combining CDK4/6 inhibitors, such as Amebaciclib, with anti-HER2 treatment27,28. Third, at the pathway level, PTK6 signaling is activated in tumor cells in pattern 13 compared to other regions. PTK6 signaling supports cell proliferation and inhibition of cell death in tumor cells, as it is upstream of CCND1 expression regulation. This proposes PTK6 as a candidate target path along with CDK4/6 inhibitors, for which arsenic trioxide could be a candidate compound from drug-gene enrichment. PTK6 signaling has been directly implicated in targeted-therapy resistance in HER2 + BRCA, validating our results34. A fourth result is from our integrative LR analysis, as we identified CDH1-CDH1 interaction between tumor cells as a potential upstream regulator of tumor-specific differential genes. While IPA identifies CDH1 upstream of CCND1, the E-cadherin interaction is not found to be directly involved in CCND1 regulation in breast cancer literature. However, CDH1-CDH1 has been shown to promote trastuzumab resistance and influences CCND1 expression in a perturbation experiment on normal mammalian epithelial cells; thus we can hypothesize indirect regulation of CCND1 expression and parallel mechanisms of therapy resistance through disrupting antibody access. In other disease applications, this novel step in our methodology can present a strong connection between cell-type specific genes and/or pathways, giving additional/synergistic targets in a communication cascade of interest.
While our study presents a framework with significant potential for treatment evaluation, there are several limitations inherent in the datasets used for PoC. First, the sample sizes for both the outcomes and spatial datasets are limited. In typical predictive modeling, sample sizes in the hundreds are preferred; here we only have 82 images. In the spatial dataset, only 5 samples out of 29 express pattern 13. This observation aligns with our findings that this pattern is predominantly associated with resistance; however, molecular analyses such as DE typical benefit from larger datasets. Although these low sample sizes do not preclude analysis and have helped identify markers of interest for PoC, we acknowledge that larger datasets will be important for future application and validation.
Second, the morphology patterns from the outcomes dataset are derived solely from invasive tumor, while the spatial dataset lacks ROIs and may include several tissue regions including invasive, in situ carcinoma, and adipose. We chose to use invasive tumor ROIs in the outcomes dataset because Farahmand et al. demonstrated a 0.12 decrease in slide-level AUC when predicting resistance using the full tissue compared to using only invasive ROIs19. To obtain the most accurate predictive model in Module 2, we therefore focused exclusively on invasive tumor regions. This decision introduces a discrepancy; there will be no direct mapping from non-invasive regions of the spatial dataset to patterns identified from the outcomes dataset. However, based on pathologist input, pattern 13 has a particularly prominent morphology - highly pleomorphic tumor cells in dense stromal tissue - that is typically not expressed in non-invasive regions, and has limited presence in boundary zones where invasive front tend to have looser stroma. Given this, we expect our current KNN method is unlikely to assign non-invasive regions from the spatial dataset to pattern 13, and instead be mapped to low attention clusters, reducing the risk of false positives. To further mitigate misclassification, we use majority voting when assigning spot-level pattern labels from overlapping tiles. While we believe our PoC mechanistic results are reliable- given methodological rigor and integration of biological context- false positives are still possible due to smaller ROIs in the outcomes dataset. In the future, to validate similarity in pattern assignments between datasets, H&E based cell segmentation, phenotyping, and cell type enrichment between the 2 datasets for each pattern could be implemented in module 3. Additionally, since ROI selection is user-defined, the framework could be extended to include larger regions—such as invasive fronts—which are often clinically relevant.
As ST data becomes increasingly available at cellular/subcellular resolution (such as Xenium and Visium HD), our methodology can be adapted to operate on cell level features rather than tile level features. Specifically, our framework could leverage new architectures such as cell graph networks when deriving WSI features, as this would lead to more fine-grained pattern identification and mapping25. Further, our methodology could be modularized through agentic modeling to increase framework efficiency and automate internal reasoning to identify potential spatial targets. We propose a multi-agentic system that is a combination of sequential modeling to identify patterns, and an LLM-based agent to perform all molecular analyses.
Downstream implications of our AI-based methodology could be transformative for the way candidate drug discovery, clinical trials, and precision medicine are approached. The following are potential key applications: 1. Discovering regional targets and proposing studies on region-specific drug delivery, including exploring synergistic target interactions if multiple proposed. 2. If identified targets are already in clinical trials, the next step is prioritizing those trials over others for a given disease or condition. 3. Once trained on diverse datasets for a disease, our framework aids personalized clinical decisions by: (1) deploying a classifier/model to predict patient outcomes, and (2) identifying outcome-associated tissue patterns to enhance predictions and optimize drug delivery.
Methods
Preprocessing, style transfer, and tile feature representation
The first stage in our framework is preprocessing and creating tile feature representations. After performing Otsu’s thresholding followed by Reinhard color normalization, we extract tiles of 224 × 224 pixel size with 50% overlap from each dataset. We specify a resolution of 0.7 µm/pixel for both datasets to maintain the lowest resolution available; thus, the outcomes dataset is downsampled when extracting tiles. This tile size was initially chosen as it is commonly used for downstream pre-trained CNN models. We then used the ResNet50 pre-trained backbone from RetCCL, a histopathology foundation model, to extract feature vectors of length 2048 for each outcomes dataset tile. Although RetCCL is a well-trained model, there are other foundation models that could also be used in this step such as Virchow40. Additionally, we used a popular tile size for ResNet50 backbones, but this could be changed in a parameter sweep to determine the best downstream classifier result. We applied the AI-FFPE model from Mahmood laboratory for frozen-to-FFPE style transfer on the spatial dataset before also using the RetCCL backbone on these tiles. Specifically, AI-FFPE uses the CUT GAN model for image-to-image translation with unpaired training data. We split the spatial dataset into 21 train and 8 test samples, and utilized the full FFPE outcomes dataset tiles for both training and evaluation. Since we are using a separate input dataset for testing and the ‘real FFPE’ tiles of the outcomes dataset make up a diverse set of tissue patterns, data leakage should not be a point of concern. During evaluation, we used the FID score, commonly used to determine the quality of images from a generative model. The equation for this distance metric between two multivariate gaussians X1 ∼ N(µ1,C1) and X2 ∼ N(μ2, C2) is,
We calculated this score between 2048 dimensional tile embedding distributions from the pseudo-FFPE output tiles of the test spatial dataset (X1) and from the real FFPE tiles of the outcomes dataset (X2). If the spatial dataset has FFPE tissue, there is no need for style transfer. However, the tile embedding distributions should be well mixed from a UMAP. If they are separated distributions, a batch normalization method should be used such as ComBat, which is frequently used for gene expression counts41. We note that ComBat batch correction requires each feature to follow a normal distribution.
Identify group-associated morphology patterns from outcomes H&E dataset
First, we trained and evaluated an attention-MIL classifier to predict outcome directly from tile features. This model is a two step process as described in Ilse et al. (ref. 42). First, each tile embedding is passed through a network to determine its relative importance weight. Then, the weighted aggregate of all tile embeddings for a given WSI is calculated and passed as input to a classifier network. In our study, we incorporated one “self-attention” layer on the tile-level embeddings before passing the embeddings into the attention-MIL model. Specifically, we calculated the similarity between the center tile embedding and its k = 10 nearest neighbors, then took the weighted aggregate of neighboring embeddings to assign a new, context-informed, embedding to the center tile.
For model training and evaluation, we used a 3-fold stratified cross-validation approach and calculated the average AUC/ROC curve across the 3 iterations per class. We used 3-fold CV as opposed to LOOCV or 5-fold CV to reduce computational time and complexity, considering the downstream ensemble modeling approach in obtaining the final attention scores. Finally, we ran the trained model from each fold across all samples and used an ensemble approach to assign one attention score per tile for downstream analysis. Specifically, we took the weighted average of attention scores for a given tile across the 3 folds, with each weight representing the normalized average AUC for that fold. This allows greater trust in assigned attention scores.
We then used a 2-step approach to identify group-associated morphology patterns. First, we identified tiles from each sample belonging to the top 20% attention scores. Subsets of these regions are most likely to correlate with patient group for a well-trained model. We performed leiden community clustering on the tile embeddings from these regions across all samples to identify diverse morphology patterns. Since leiden clustering requires choosing k nearest neighbors when constructing the point graph, we selected k = 1200. We chose this by performing a sweep from k = 400 to 2700 and observing the silhouette score. The silhouette score typically ranges between −1 and 1, where 1 indicates ideal cluster separation. Here, for all of our k values the silhouette score was close to 0, indicating a fuzzy boundary between clusters. Thus, we chose a value of k that had the highest positive score of 0.023 and created UMAPs of the final pattern assignments over tiles to determine level of separation as described in results. We further clustered tile embeddings corresponding to the remaining ’low-attention’ regions across all samples using the same k to account for non-relevant patterns. Second, once we assigned tile patterns, we used 2 complementary statistical tests to investigate pattern differences between patient groups. We first used the Wilcoxon rank-sum test with Benjamini-Hochberg (BH) multiple testing correction to compare the composition of each pattern between the 2 groups. In parallel, we performed a one-sided Fisher’s test for each pattern to determine pattern enrichment in high attention regions between groups. We can think of this multi-step approach as applying multiple filters to remove false positives, and consider only morphology patterns that are very likely associated with patient group.
All parts of this module up to statistical testing used the python programming language (v3.9). The slideflow package, built for creating and testing deep learning (DL) models for histopathology, was used for preprocessing, tile extraction, and MIL model training. The AI-FFPE model was trained and tested using the Mahmood lab Github repository. The scanpy package was used for leiden clustering. The final result of module 2 is an anndata object of tile embeddings across all samples with corresponding (low and high attention) pattern assignments, as well as a list of outcome-associated patterns.
Pattern mapping and preparing ST data for molecular analysis
To discover mechanisms underlying tissue patterns identified from the outcomes dataset, we needed to (1) Map outcomes patterns to spatial dataset tiles, and (2) Map tile level patterns to the spot level. First, we used KNN to assign a pattern to a spatial tile using k = 50 nearest neighbors. We chose 50 to account for fuzzy decision boundaries. The difference in ROIs between the outcomes and spatial datasets is addressed in the discussion section. Once patterns are assigned to each tile, we mapped these assignments to the spot level using a majority vote. This allows us to prioritize reducing false positives, since a spot can have more than one overlaid tile due to 50% tile overlap. However, if we want to prioritize reducing false negatives, in the future we could assign a spot to a pattern if at least one tile of that pattern overlays the spot. We implemented this module in Python; thus, the output is an anndata object of spot gene expression with saved pattern assignments.
To determine spatial cell type organization from ST, we performed deconvolution using InstaPrism, a fast, computationally efficient version of BayesPrism implemented in R. BayesPrism uses a sc reference from the same tissue to determine cell type gene signatures and fit a bayesian model of cell type proportions µns and cell type-specific gene expression vectors Uns for each spot/sample n and cell type s.
Spatial molecular analysis
To discover the molecular bases behind pattern(s) of interest, we performed cell type neighborhood enrichment, cell type specific DE, and LR analysis, followed by drug-gene and pathway enrichment for deeper insight. First, we performed both individual and neighborhood cell type enrichment to discover any associations between cell types and patterns. We fit a linear mixed-effect model to represent the composition of a cell type c as a function of binary pattern assignment p for each (c,p) at a time while accounting for sample s as fixed effect. We then evaluated the significance of the pattern coefficient for each combination of variables. Thus, the model was c = β∗p + z∗s. In parallel, we clustered cell type compositions to identify neighborhoods across samples followed by enrichment analysis in pattern(s) of interest. To do this, we used a 4 step process: (1) Since compositional data is not in the euclidean space, we ran the centralized log-ratio (CLR) transform on each compositional vector across spots. If our spots were high resolution, we would identify a window around each spot to create new window compositions before taking the CLR transform. (2) We then clustered transformed spot compositions across all samples and identified an ideal cluster number k using a penalized k-means method, similar to the BIC metric that is often used for probabilistic clustering. (3) Once we identified cluster centroids, we assigned a cluster (neighborhood) to each spot (4) Finally, we performed a one-sided Fisher’s test per neighborhood on pattern regions of interest to identify neighborhoods enriched in the pattern. While our enrichment analyses did not identify any associations between cell types and our pattern of interest, this methodology is important in future disease applications as it can both explain morphology trends and prioritize downstream DE.
In order to perform DE, we fit a generalized linear mixture model using CSIDE by Cable et al. to determine cell type-specific differential expression as a function of the pattern region8.This tool models gene counts as a function of cell-type specific expression, in turn dependent on specified covariates(s). In our study, we have 1 discrete covariate; we assigned a binary value of 1 or 0 to each spot across all samples based on whether the spot belonged to the pattern region or not. Since statistical power of C-SIDE is hindered with a small sample size for each value of the discrete covariate on a per sample basis, we aggregated all spots across all samples. We then input raw counts with assigned covariates to the model, since C-SIDE internally normalizes across spots. C-SIDE has several parameters that can be set by the user to improve model fit, including cell type threshold t. This value is the minimum required sum of proportions for a given cell type in all spots belonging to each value of the covariate, and can thus filter out cell types with too little presence.
To identify likely interacting LR pairs in pattern region(s) of interest, we used an integrative workflow with both the public sc reference and spatial dataset. We first ran CellChat, a mass-action model, on the sc reference to calculate communication probabilities and p-values across all LR pairs in CellChatDB (database). In parallel, we ran SpatialDM only on the pattern region(s). This tool uses bivariate Moran’s statistic to calculate spatial co-expression of the ligand and receptor genes. Ideally, we would like to consider only LR pairs with significant co-expression unique to the pattern region(s), but can still continue analysis otherwise. We then filtered the list of all significant pairs from CellChat by the pairs that were significantly spatially co-expressed in pattern(s) of interest. The final LR pairs represent the likely interactions in pattern(s) of interest between cell types defined by the sc reference. If the previous cell-type neighborhood or individual cell type enrichment gave significant results, we would further filter our LR pair list by combinations of these significant cell types.
Alternative LR analysis pipelines for spot-resolution ST include SpaTalk and Tangram with CellChat. These methods rely on mapping the sc reference onto spatial coordinates by reducing the difference in gene expression between the ST dataset and mapping single cells. While mathematically rigorous, these methods (1) assume similar expression profiles between sc and spatial and (2) are computationally inefficient. Thus, we utilized the above integrative workflow for our methodology.
We used the EnrichR tool from the Ma’ayan laboratory (https://maayanlab.cloud/Enrichr/) to input our list of tumor-specific DE genes. We then used the Appyter extension to get the table of top drug targets and overlapping genes from both DSigDB and DGIdb.
We used the IPA tool from Qiagen to perform pathway analysis for our PoC because it has a large consolidated set of internal studies and directly provides upstream regulators to link with our LR analysis. Though not used in our PoC, similar pathway and term enrichment can be performed using open source alternatives including fGSEA43 and enrichR44,45 which are compatible with the wide array of publicly available datasets for gene ontology, disease pathways, drug targets, and more.
All downstream molecular analysis methods except pathway analysis and drug-gene set enrichment were implemented and run in the R language. Identifying cell type neighborhoods involves calculating the CLR transform and performing a k-means sweep to identify an ideal k, for which we used the compositions and coseq package respectively. InstaPrism and C-SIDE can both be used through the instaprism and spacexr R packages respectively.
Data availability
For this PoC study, we used two public datasets - the first “outcomes” dataset is from a Yale study that predicts HER2 status and trastuzumab resistance from H&E FFPE images, with 82 tumor-ROI annotated WSIs (taken pre-treatment)19. The image magnification is 20X at a resolution of 0.5 µm/pixel. The “spatial” dataset is a HER2 + BRCA cohort of 29 samples across 8 patients with matched ST and fresh frozen H&E (taken pre-treatment), from the HEST library of matched samples gathered from public and private cohorts46. While these H&E images are also at 20X magnification, the resolution of these images is close to 0.7 µm/pixel. The spot resolution of this data is 100 µm in diameter with 200 µm distance between spot centers. We used a public sc HER2 + BRCA dataset from the Broad Institute as a reference for deconvolution and starting point for LR analysis26. To avoid treatment as a confounder, we removed one post-treatment sample as indicated in an integrative sc BRCA study47. Additionally, we plotted ERBB2 (gene name for HER2) count histograms to confirm low expression in normal epithelial and high expression in cancer epithelial cells (Supplementary Fig. 2).
Code availability
The code for this study will be available at the following github repo: https://github.com/Revak007/Multimodal_Imaging_Mechanisms.
References
Chu, X., Tian, Y. U. & Lv, C. Decoding the spatiotemporal heterogeneity of tumor-associated macrophages. Mol. Cancer 23, 150 (2024).
Heather, D. Couture. Deep learning-based prediction of molecular tumor biomarkers from h&e: A practical review. J. Personal. Med. 12, 2022 (2022).
Ahn, B. et al. Histopathologic image–based deep learning classifier for predicting platinum-based treatment responses in high-grade serous ovarian cancer. Nat. Commun. 15, 4253 (2024).
Shamai, G. et al. Clinical utility of receptor status prediction in breast cancer and misdiagnosis identification using deep learning on hematoxylin and eosin-stained slides. Commun. Med. 4, 276 (2024).
M Ilse, J Tomczak, & M Welling. Attention-based deep multiple instance learning. In Proc. International conference on machine learning, 2127–2136 (PMLR, 2018).
Schu¨rch, C. M. et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 182, 1341–1359 (2020).
Claudio Quiros, A. et al. Mapping the landscape of histomorphological cancer phenotypes using self-supervised learning on unannotated pathology slides. Nat. Commun. 15, 4596 (2024).
Cable, D. M. et al. Cell type-specific inference of differential expression in spatial transcriptomics. Nat. Methods 19, 1076–1087 (2022).
Cable, D. M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 40, 517–526 (2022).
Cang, Z. et al. Screening cell–cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods 20, 218–228 (2023).
Chu, T., Wang, Z., Pe’er, D. & Danko, C. G. Cell type and gene expression deconvolution with bayesprism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat. Cancer 3, 505–517 (2022).
Dries, R. et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 1–31 (2021).
Svensson, V., Teichmann, S. A. & Stegle, O. Spatialde: identification of spatially variable genes. Nat. Methods 15, 343–346 (2018).
Zheng, Y., Carrillo-Perez, F., Pizurica, M., Heiland, D. H. & Gevaert, O. Spatial cellular architecture predicts prognosis in glioblastoma. Nat. Commun. 14, 4122 (2023).
Hu, J. et al. Spagcn: integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 18, 1342–1351 (2021).
Pham, D. et al. stlearn: integrating spatial location, tissue morphology and gene expression to find cell types, cell-cell interactions and spatial trajectories within undissociated tissues. Preprint at https://doi.org/10.1101/2020.05.31.125658 (2020).
Walker, B. L., Cang, Z., Ren, H., Bourgain-Chang, E. & Nie, Q. Deciphering tissue structure and function using spatial transcriptomics. Commun. Biol. 5, 220 (2022).
Laury, A. R. et al. Opening the black box: spatial transcriptomics and the relevance of artificial intelligence–detected prognostic regions in high-grade serous carcinoma. Mod. Pathol. 37, 100508 (2024).
Farahmand, S. et al. Deep learning trained on hematoxylin and eosin tumor region of interest predicts her2 status and trastuzumab treatment response in her2+ breast cancer. Mod. Pathol. 35, 44–51 (2022).
Jaume, G. et al. Hest-1k: a dataset for spatial transcriptomics and histology image analysis. Adv. Neural Inform. Process. Syst. 37, 53798–53833 (2024).
Ozyoruk, K. B. et al. A deep-learning model for transforming the style of tissue images from cryosectioned to formalin-fixed and paraffin-embedded. Nat. Biomed. Eng. 6, 1407–1419 (2022).
Tomita, N. et al. Attention-based deep neural networks for detection of cancerous and precancerous esophagus tissue on histopathological slides. JAMA Netw. open 2, e1914645 (2019).
Hashimoto, N. et al. Multi-scale domain-adversarial multiple-instance CNN for cancer subtype classification with unannotated histopathological images. In Proc. IEEE/CVF conference on computer vision and pattern recognition, 3852–3861 (IEEE, 2020).
Mi, H. et al. Spatial architecture of single-cell and vasculature in tumor microenvironment predicts clinical outcomes in triple-negative breast cancer. Mod. Pathol. 38, 100652 (2025).
Zhang, B., Li, C., Wu, J., Zhang, J. & Cheng, C. Deepcg: A cell graph model for predicting prognosis in lung adenocarcinoma. Int. J. Cancer 154, 2151–2161 (2024).
Wu, S. Z. et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 53, 1334–1347 (2021).
Goel, S. et al. Overcoming therapeutic resistance in her2-positive breast cancers with cdk4/6 inhibitors. Cancer Cell. 29, 255–269 (2016).
Gao, X., Leone, G. W. & Wang, H. Cyclin d-cdk4/6 functions in cancer. Adv. Cancer Res. 148, 147–169 (2020).
Fara Bras´O-Maristany et al. Phenotypic changes of her2-positive breast cancer during and after dual her2 blockade. Nat. Commun. 11, 385 (2020).
Shah, N. & Sukumar, S. The hox genes and their roles in oncogenesis. Nat. Rev. Cancer 10, 361–371 (2010).
Wu, X. et al. Hoxb7, a homeodomain protein, is overexpressed in breast cancer and confers epithelial-mesenchymal transition. Cancer Res. 66, 9527–9534 (2006).
Jin, K. et al. Hoxb7 is an erα cofactor in the activation of her2 and multiple er target genes leading to endocrine resistance. Cancer Discov. 5, 944–959 (2015).
Ludyga, N. et al. Effects of simultaneous knockdown of her2 and ptk6 on malignancy and tumor progression in human breast cancer cells. Mol. Cancer Res. 11, 381–392 (2013).
Park, S. H., Ito, K., Olcott, W. & Katsyv, I. Gwyneth HalsteadNussloch, and Hanna Y Irie. Ptk6 inhibition promotes apoptosis of lapatinib-resistant her2+ breast cancer cells by inducing bim. Breast Cancer Res. 17, 1–13 (2015).
National Center for Biotechnology Information. Arsenic trioxide (cid 14888). PubChem [Internet]. U.S. National Library of Medicine, 2025. Accessed March 20 2025.
Yamauchi, C. et al. E-cadherin expression on human carcinoma cell affects trastuzumab-mediated antibody-dependent cellular cytotoxicity through killer cell lectin-like receptor g1 on natural killer cells. Int. J. Cancer 128, 2125–2137 (2011).
Bajrami, I. et al. E-cadherin/ros1 inhibitor synthetic lethality in breast cancer. Cancer Discov. 8, 498–515 (2018).
Fournier, A. K. et al. Rac-dependent cyclin d1 gene expression regulated by cadherinand integrin-mediated adhesion. J. Cell Sci. 121, 226–233 (2008).
Jochems, C. & Schlom, J. Tumor-infiltrating immune cells and prognosis: the potential link between conventional cancer therapy and immunity. Exp. Biol. Med. 236, 567–579 (2011).
Vorontsov, E. et al. A foundation model for clinical-grade computational pathology and rare cancers detection. Nat. med. 30, 2924–2935 (2024).
Zhang, Y., Parmigiani, G. & Johnson, W. E. Combat-seq: batch effect adjustment for rna-seq count data. NAR Genom. Bioinform. 2, lqaa078 (2020).
Maximilian Ilse, Jakub Tomczak, & Max Welling. Attention-based deep multiple instance learning. In Proc. 35th International Conference on Machine Learning (eds Jennifer, D and Andreas, K) 2127–2136 (PMLR, 2018).
Korotkevich, G. et al. Fast gene set enrichment analysis, 60012 Section: New Results. (2021).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Xie, Z., et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 1, e90 (2021).
Andersson, A. et al. Spatial deconvolution of her2-positive breast cancer delineates tumor-associated cell type interactions. Nat. Commun. 12, 6012 (2021).
Pang, L. et al. Single-cell integrative analysis reveals consensus cancer cell states and clinical relevance in breast cancer. Sci. Data 11, 289 (2024).
Acknowledgements
We acknowledge several funding sources for the support. R.K. was funded by the Biomedical Informatics and Data Science Training Program (T32-GM141746). A.M. was funded by the University of Michigan Medical Scientist Training Program and Department of Computational Medicine and Bioinformatics through F074041 and F072372 NIH grants. The following grants also supported this project: R37CA214955-01A1 and award number U01CA287852 (Saleh M, Sayed S, Waljee AK, Balis U, and Rao A). The second award (U01CA287852) is under the Office Of The Director, National Institutes Of Health (OD), the National Institute Of Biomedical Imaging And Bioengineering (NIBIB), the National Institute Of Mental Health (NIMH), and the Fogarty International Center (FIC) of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
A.R. and R.K. devised the project and developed the framework methodology. R.K. and A.M. implemented the methodology with A.M. identifying public data sources. S.B. provided pathologist annotations for both QC and pattern interpretation. R.K. and A.M. wrote the manuscript, and all authors reviewed/edited.
Corresponding authors
Ethics declarations
Competing interests
A.R serves as consultant to Tempus and Telperian, member for Voxel Analytics, LLC, and faculty advisor for TCS Ltd. Additionally, A.R. serves as Satish Dhawan Visting Professor at the Indian Institute of Science and Affiliate Investigator at Fred Hutchinson Cancer Research Center (FHCRC) in Seattle.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kulkarni, R., Maddox, A., Bailey, S. et al. A multimodal framework to identify molecular mechanisms driving patient group-associated morphology through the integration of spatial transcriptomics and whole slide imaging. npj Artif. Intell. 2, 9 (2026). https://doi.org/10.1038/s44387-025-00050-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44387-025-00050-6
