
Abstract
Latin America’s diverse genetic landscape provides a unique opportunity to study Alzheimer’s disease (AD) and frontotemporal dementia (FTD). The Multi-Partner Consortium to Expand Dementia Research in Latin America (ReDLat) recruited 2162 participants with AD, FTD, or healthy controls from six countries: Argentina, Brazil, Chile, Colombia, Mexico, and Peru. Participants underwent genomic sequencing, and population structure analyses were conducted using Principal Component Analysis and ADMIXTURE. The study revealed a predominant mix of American, African, and European ancestries, with an additional East Asian component in Brazil. Variant curation identified 17 pathogenic variants, a pathogenic C9orf72 expansion, and 44 variants of uncertain significance. Seventy families showed autosomal dominant inheritance, with 48 affected by AD and 22 by FTD. This represents the first large-scale genetic study of AD and FTD in Latin America, highlighting the need to consider diverse ancestries, social determinants of health, and cultural factors when assessing genetic risk for neurodegenerative diseases.
Similar content being viewed by others

Introduction
Latin America is home to a unique blend of Indigenous American, European, and African ancestries, resulting in a diverse admixed genetic landscape that emerged in the 1500s from the conquest of the Americas and the transatlantic slave trade1. Significant East Asian immigration during the early 20th century has further contributed to the continent’s population diversity observed today2,3. This admixture harbors a spectrum of novel genetic variants, including some that may modulate susceptibility to neurodegenerative diseases like Alzheimer’s disease (AD) and frontotemporal dementia (FTD) or may result in a higher allelic frequency of known risk-conveying variants for neurodegeneration4. Studying these populations, with their complex genetic architecture, offers an invaluable resource for understanding neurodegenerative diseases.
Assessing admixed populations is particularly interesting because admixture introduces a rich heterogeneity of alleles, which can be crucial for understanding genetic risk. For instance, in the Colombian population, researchers from a single center in Medellín have identified 13 different pathogenic PSEN1 variants from different ancestral backgrounds4. This contrasts with the nine independent variants described in a screening study from nine centers throughout the Iberian Peninsula5. The presumptive excess of PSEN1 variants may have become fixed in a relatively small effective population due to the antimicrobial benefits of high beta-amyloid levels4. Additionally, in the Peruvian population, a recent genome-wide association study and functional analysis suggested that the NFASC gene, located on chromosome 1, is associated with AD. The NFASC locus showed significant contributions from both European and African ancestries6. This finding emphasizes the crucial role of relatively recent founder effects among diverse ancestral backgrounds in uncovering novel variation and understanding the complex genetic foundations of AD.
Due to cultural, religious, and historical factors, Latin American families have traditionally been large. Government policies during the 1960s and 1970s further encouraged population growth, resulting in an average family size of six children7,8. Additionally, geographic and socioeconomic conditions often led to extended families residing in proximity7,9. This unique demographic structure provides an exceptional opportunity to study founder effects in large families by tracing the lineage and impact of specific genetic variants across generations. For example, the PSEN1 Glu280Alavariant (canonically known as PSEN1 E280A) in Colombia can be traced back approximately 500 years to the time of the Spanish invasion. In the large families that descend from the original carrier, the variant became fixed and spread widely among numerous distant relatives. This historical and genetic tracing provides valuable insights into how genetic drift occurs over time in small populations, increasing the likelihood of identifying both risk and protective genetic variants10,11,12.
The scientific community from the region recognized the immense potential of this approach and formed the Multi-Partner Consortium to Expand Dementia Research in Latin America (ReDLat)13. This consortium fosters collaboration among researchers, clinicians, and institutions across six Latin American countries and the United States to leverage the unique genetic diversity and demographic characteristics that influence AD and FTD in Latin American populations. ReDLat aims to highlight the unique genetic diversity and demographic features of these populations associated with neurodegenerative diseases of cognition14. Ultimately, the consortium’s work seeks to refine diagnostic approaches, develop targeted therapeutic interventions, and significantly enhance our understanding of dementia across Latin America13.
This paper is the first genetic report from this consortium, focusing on AD and FTD in admixed Latin American participants, with a particular emphasis on families. Our research efforts aim to identify genetic variants associated with these neurodegenerative diseases in the region and provide insights into some of the clinical presentations observed within the studied families, thereby enhancing our understanding of AD and FTD across this vast and diverse population. By expanding the genomic dataset in the coming years, we aim to deepen our understanding of the genetic underpinnings and clinical presentations of these neurodegenerative diseases across diverse Latin American populations, enhancing the potential for targeted interventions and therapies.
Results
Characterization of the population
To investigate the genetic landscape of neurodegeneration in Latin America, we recruited patients with mild to moderate AD or FTD, along with healthy controls from ten memory clinics across six Latin American countries: Argentina, Brazil, Chile, Colombia, Mexico, and Peru. Recruitment occurred in two phases due to the COVID-19 pandemic, which delayed the initiation of ReDLat’s prospective enrollment, originally planned for 2020. Despite differences in timing, the inclusion criteria for both the retrospective and prospective cohorts were identical. As of August 17, 2024, a total of 5834 participants had been enrolled in the study (Fig. 1).

WES whole exome sequencing, WGS whole genome sequencing, SNP single-nucleotide polymorphism.
By the time of manuscript submission, PCR-free whole genome sequencing (WGS), whole exome sequencing (WES), and/or single-nucleotide polymorphism (SNP) array genotyping had been performed in 2254 participants from both cohorts. After thorough quality control, genomic data from 2162 individuals were retained for analysis. The final dataset included 658 participants with SNP array data, of whom 174 also had WES; 1495 participants with WGS; and 9 individuals with only WES [Fig. 1]. All participants underwent medical and neuropsychological evaluation. Among those with high-quality genomic data, 999 were diagnosed with AD, 381 with FTD, and 755 were classified as cognitively healthy at the time of assessment. The dataset also included eight participants with mild cognitive impairment and 19 individuals with other neuropsychiatric diagnoses, such as Parkinson’s disease, Lewy body dementia, atypical parkinsonism, neurodevelopmental disorders, cerebellar ataxia, brain tumor, cognitive impairment associated with non-brain cancer, vascular dementia, severe depressive disorder, bipolar disorder, obstructive sleep apnea, and chronic traumatic encephalopathy. These individuals were members of recruited families and had a relative enrolled in the study with AD- or FTD-related dementia.
As expected, a higher percentage of participants with FTD were under 65 years of age compared to those with AD (22.3% vs. 13.9%). Furthermore, the percentage of female participants was higher in the AD group (66.6%) compared to the FTD group (52.8%). Those in the AD group also showed a higher proportion of individuals who are heterozygous (41.5%) and homozygous (8.6%) for the APOE ε4 allele (Table 1). The distribution of APOE alleles of unrelated individuals varies per country, as shown in Supplementary Table 1. Homozygous APOE ε2 carriers were observed only in Colombia (0.2%) and Brazil (1%). Conversely, the highest numbers of APOE ε4 alleles were found in Argentina and Colombia (39% in both), although Colombia has 1% more homozygous carriers than Argentina. It is worth noting that these numbers may not be fully representative due to the Colombian sample size being considerably larger. As larger numbers of samples from all regions are collected, allele frequency estimates will be further refined.
Genetic ancestry
To assess genetic ancestry similarity among our samples, we initially generated a merged ReDLat dataset that included 2153 participants with WGS or SNP array data that passed the concordance analysis. We then used WGS from the 1000 Genomes Project (1000GP) as reference populations to estimate the global ancestry of the participants, employing Principal Component Analysis (PCA) and ADMIXTURE software to estimate global ancestry. We used the 1000GP cohort to identify variants with allelic frequency >10% and in linkage equilibrium that were present in both the 1000GP and the merged ReDLat dataset, resulting in a total of 226,524 variants used for ancestry analyses.
PCA (Fig. 2, and Supplementary Fig. 1) reveals that the ReDLat dataset shows substantial overlap with the American populations (AMR) sampled by the 1000GP, which also included Colombian, Mexican, and Peruvian participants. Though a substantial number of participants overlap with European (EUR) cohorts, there were 21 individuals clustering with the East Asian (EAS) population, suggesting recent EAS descent subgroups within ReDLat.

Panels represent the main principal component (PC) comparisons. a, d, g: PC1 vs. PC2. b, e, h: PC1 vs. PC3. c, f, i PC2 vs. PC3. Different color schemes were used to differentiate sample origin. The 1000GP includes African (AFR), European (EUR), South Asian (SAS), East Asian (EAS) and Admixed American (AMR) individuals. ReDLat subcohorts include individuals from Argentina (ARG), Brazil (BRA), Chile (CHI), Colombia (COL), Mexico (MEX), and Peru (PER). Panels a, b, c show color-coded ReDLat cohort participants projected onto the 1000GP reference samples PCA (gray). ReDLat cohort genomes are colored according to their country of origin. d, e, f show the ReDLat cohort (magenta) plotted against the color-coded 1000GP subpopulations. Panels g, h, i show ReDLat cohort (magenta) overlaid on 1000GP cohort (gray).
When analyzing the data by country, participants from Peru, Mexico, Chile, and Argentina exhibit minimal variation along Principal Component 1 (PC1), which is associated with African ancestry. In contrast, there is greater variation along Principal Component 2 (PC2), which is associated with Amerindian ancestry. This variation is particularly pronounced among participants from Mexico and Peru, who have individuals with a majority of their ancestry being Amerindian. These findings suggest a predominant two-way ancestry pattern in these populations. There is a clear overlap between Argentinian and Brazilian samples with the European (EUR) cohort; however, the Brazilian samples show significant variation along PC1, highlighting the African component of the sample. While most Brazilian samples are distributed primarily between African (AFR) and EUR populations, a small subset clusters with EAS, indicating a distinct ancestral subgroup within Brazil. Colombia displays a clear three-way admixture pattern, as evidenced by its wide distribution along both PC1 and PC2. Additionally, Peruvian and Mexican samples from ReDLat exhibit clear overlap with their 1000GP counterparts, while ReDLat Colombian samples showed greater diversity than those in 1000GP (Fig. 2). This increased diversity is likely due to ReDLat’s broader sampling across multiple regions of Colombia. Overall, the PCA analysis confirms that the ReDLat cohort accurately represents the different historical admixture patterns previously described in the corresponding countries1.
To calculate global ancestry proportions (Q-values), which are adjusted p-values accounting for multiple testing and controlling the false discovery rate, we projected the ReDLat samples onto the 1000GP dataset ADMIXTURE results at multiple clustering values (Supplementary Fig. 2). At K = 5, where K represents the number of ancestral populations in the clustering analysis, we observed a continental separation of ancestral origins and were able to differentiate the Amerindian component (Fig. 3). Peru is the only country where Amerindian ancestry exceeds European ancestry, followed by Mexico, where these two ancestries show similar distribution. In Argentina, Brazil, Colombia, and Chile, European ancestry is the most prevalent among the participants, with a median value of 86.9% (mean value of 79.7%, standard deviation 17.5) for Argentina and 84.2% (mean value of 73.8%, standard deviation 26.9) for Brazil. African ancestry is present in Colombia and Brazil at lower levels; we observe a continuum of this ancestry, with individuals having over 90% and 75% African descent, down to the mean levels for both countries (around 10%), suggesting ongoing admixture over generations. In contrast, this continuum is not observed in the EAS component of the Brazilian samples, suggesting a recent diaspora without intercontinental admixture (Supplementary Fig. 3).

The 1000 Genomes project includes African (AFR): GWD Gambian in Western Divisions in the Gambia, LWK Luhya in Webuye, MSL Mende in Sierra Leone, YRI Yoruba in Ibadan, Nigeria, ACB African Caribbean in Barbados, ASW African Ancestry in the Southwest US. European (EUR): CEU Utah Residents (CEPH) with Northern and Western European Ancestry, FIN Finnish in Finland, GBR British in England and Scotland, IBS Iberian Population in Spain, TSI Tuscany in Italia. South Asian (SAS): BEB Bengali in Bangladesh, GHI Gujarati Indians in Houston, ITU Indian Telugu in the UK, PJL Punjabi in Lahore, STU Sri Lankan Tamil in the UK. East Asian (EAS): CDX Chinese Dai in Xishuangbanna, China, CHB Han Chinese in Beijing, CHS Han Chinese South, JPT Japanese, Kyushu, KHV Kinh Vietnamese. Admixed American (AMR): CLM Colombian from Medellin, PUR Puerto Rican from Puerto Rico. ReDLat subcohorts: ARG Argentina, BRA Brazil, CHI Chile, COL Colombia, MEX Mexico, PER Peru.
Variant pathogenicity analysis
To identify Mendelian forms of neurodegeneration in our cohort, we analyzed data from 1678 participants who had high-quality WGS or WES data to detect pathogenic variants. Following standard practices in complex systems analysis, we employed both “bottom-up” and “top-down” approaches.
Our bottom-up approach was a “gene-to-family” search, in which we initially assessed the genes most commonly associated with adult-onset neurodegeneration for pathogenic variants (see “Methods”). We identified a total of 17 pathogenic variants, a pathogenic C9orf72 expansion, and 44 variants of uncertain significance (VUS) (Table 2 and Supplementary Table 2). We note that the C9orf72 expansions were identified using ExpansionHunter software from PCR-free WGS but were not explicitly confirmed by Southern blot, although this predictive tool is very accurate15.
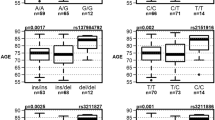
In families with AD, we identified several previously described PSEN1 variants: c.356 C > T (p.Thr119Ile), c.415 A > G (p.Met139Val), c.428 T > C (p.Ile143Thr), and c.519 G > T (p.Leu173Phe)4,16,17 (Supplementary Fig. 4). Notably, Thr119Ile and Ile143Thr are of European origin and exhibit identity by descent among Colombian carriers for Ile143Thr and between Colombian and Argentine carriers for Thr119Ile4. In contrast, the PSEN1 c.415 A > G (p.Met139Val) variant, identified in a large family from Argentina, was determined to be of Amerindian origin using the local ancestry inference software RFMix (see “Methods”). This haplotype was also present in two individuals from the 1000 Genomes Project, originating from Peru and Colombia (Supplementary Fig. 5). The PSEN1 Met139Val variant has been reported across diverse ancestral backgrounds18,19,20 and is associated with either AD or atypical dementia, characterized by amnestic and behavioral symptoms. These symptoms may also include spastic paraparesis, psychosis, seizures, and myoclonus21 (see Supplementary Note 1 for the clinical description of this family). APP duplications were assessed using the methods described by Bowling et al.22. No duplication detected by multiple copy number variation callers overlapped the APP gene.
In families with FTD, the gene most commonly associated with hereditary forms of the illness in our cohort was GRN, followed by MAPT [See Supplementary Notes 2 and 3 for the clinical description of these families]. Pathogenic C9orf72 repeat expansions were observed in several families from geographically non-adjacent areas. The variants in GRN, MAPT, C9orf72, and TARDBP that were present in more than one individual were all of European descent (Table 3). The pedigrees of families with pathogenic variants are shown in the Supplementary Figs. 6–9. Allele frequencies of the pathogenic variants identified in the ReDLat cohort, compared with their respective frequencies in gnomAD v4.1.0, are reported in Supplementary Table 3.
After the initial analysis of these primary genes, we expanded our search to include secondary genes associated with adult-onset neurodegeneration. Utilizing the OMIM database, we identified a set of genes where single-nucleotide variants or short insertions/deletions could be disease-causing (Supplementary Table 4)19,23,24,25. In our analysis, we found four additional pathogenic variants (Supplementary Tables 5 and 6). Three of these variants are present in families with autosomal dominant disease and are located in the PRNP and NOTCH3 genes. Most notably, we identified an FTD patient without motor symptoms carrying a pathogenic variant in SOD1 c.388 G > A (p.Phe21Leu), which was previously reported in another FTD patient from the same geographical region and is believed to have originated from an Amerindian haplotype4.
In contrast, our top-to-bottom approach consisted of a “family-to-gene” search, where we analyzed the pedigrees of all recruited participants with WES or WGS data available (766 individuals grouped in 592 families). After excluding individuals recruited as “healthy”, we identified 426 independent families and classified them based on the presence of affected individuals (Fig. 4). In this cohort, 70 families exhibited autosomal dominant inheritance of neurodegenerative diseases, as evidenced by the presence of three affected individuals in two consecutive generations. The families were later classified according to the diagnosis of the proband, with 48 families identified as having AD and 22 as FTD. These families were subsequently categorized based on the age at disease onset: ‘late onset’ was assigned to families where all affected members presented dementia at ages older than 65 years; ‘early onset’ applied to those where all affected individuals were 65 years or younger at the time of dementia onset; and ‘mixed onset’ described families that included members with both early and late-onset disease (Supplementary Table 7). Though many of the carriers of pathogenic variants belonged to the ReDLat retrospective cohort, 14 of the recruited families were carriers of pathogenic variants, and the majority had a positive family history of neurodegeneration.

Cosegregation analysis of APOE ε4 and neurodegeneration in families with multiple affected individuals.Footnote
✠ Deceased: Francisco Lopera.
In a further analysis of families exhibiting autosomal dominant patterns, we assessed for the presence of at least one allele of APOE ε4. Among the families diagnosed with AD, nine were negative for APOE ε4 alleles, while 32 had at least one APOE ε4 carrier. We were able to determine the APOE ε4 allele status for more than one participant in seven families, and only four families showed co-segregation of the APOE ε4 allele with the illness. In contrast, when assessing the families with FTD, six were negative for APOE ε4 alleles, and in the one family where we could determine the APOE ε4 allele status for more than one participant, no co-segregation was observed (Fig. 4).
Discussion
This initial release of genomic data from the ReDLat cohort provides early insights into the genetic underpinnings of neurodegeneration within a Latin American population, supported by genomic analyses of established variants associated with AD and FTD. Our genetic ancestry analysis, leveraging data from the 1000 Genomes Project, revealed tricontinental admixture patterns across most regions and an East Asian component in Brazil, reflecting historical migration and admixture events. The study notably identifies a significant prevalence of autosomal dominant inheritance patterns in AD and FTD, characterized by distinct age-of-onset categorizations, geographic distribution of genetic variants, and a stronger presence of the APOE ε4 allele in AD families. These patterns also include newly discovered variants in the PSEN1 and APP genes for AD, which play critical roles in the disease’s pathogenesis and the recurrence of a SOD1 variant presenting as FTD without motor symptoms, suggesting a novel disease phenotype association. Families with as-yet unidentified variation remain strong candidates for future novel gene discovery as additional family members are recruited for gene-mapping linkage studies.
Indeed, there is considerable potential for novel genetic discovery in diverse cohorts such as ReDLat, both in terms of risk for AD and related dementias (ADRD) and resilience against it, in both families and sporadic cases. Previous work in the region has unveiled more than 13 PSEN1 pathogenic variants in Colombia, including the E280A kindred that spans more than 5000 descendants of a founder couple11. Leveraging larger, diverse cohorts—as well as genetic families with substantial clinical heterogeneity—represents a unique opportunity for the discovery of resilience factors for ADRD, which may serve as strong targets for disease intervention. Notably, two genetic variants previously described as protective for AD, APOE Christchurch Arg154Ser (rs121918393)26 and RELN His3447Arg27 (rs201731543), were observed in our cohort. APOE Arg154Ser was found in one patient with FTD and another with AD, with ages at onset of 66 and 80 years, respectively. RELN His3447Arg was observed in three individuals with AD, all with dementia onset in their early 70 s. The lack of replication for RELN His3447Arg as a protective variant was also observed in the TANGL cohort4, where we identified four carriers who had early-onset dementia; three with FTD (onset at 50, 60, and 65 years) and one with AD (onset at 59 years). All carriers of the APOE and RELN variants described here were heterozygous and did not carry pathogenic mutations in PSEN1 or other known disease-causing genes. This finding suggests that protective effects conferred by the RELN variants may be restricted to individuals carrying the PSEN1 E280A. Although APOE Arg154Ser heterozygosity was not associated with a protective effect in a patient with FTD and an elderly individual with clinical AD, ongoing evaluation in non-PSEN1 E280A carriers is necessary to elucidate its potential role in resilience.
As previously noted by Browning et al.28, historical factors like colonization, migration, and bottlenecks have significantly shaped the genetic landscape of Latin American populations. During and after the period of colonization, many Latin Americans lived in small, often isolated villages. This created a population structure characterized by multiple mini-bottlenecks as descendants of a small number of founders in each village tended to remain in the same location for many generations. As families expanded, the specific rare alleles in each place became common, even surpassing the allelic frequencies of the same variant in the ancestral population, as is the case for the SQSTM1 FTD risk-conferring variant Pro392Leu (rs104893941)29,30,31. This phenomenon resulted in a genetic map of the region that closely corresponds to the geographic map, where multiple individuals share the same deleterious variants and are identical by descent. The long stretches of identical haplotypes created by the bottlenecks and increased ancestral diversity within isolated populations are advantageous for researchers seeking to identify rare variants associated with diseases like AD and FTD or the interaction between genetic variation and ancestral haplotypes32. These factors underscore the value of family studies in Latin America, offering unique insights into genetic patterns and the potential for discovering new genetic contributions to disease28.
The first-wave study cohort reported here has several limitations. First, we have chosen not to conduct unbiased discovery efforts, such as genome-wide association studies and burden analyses, in this cohort due to the extensive family structure and relatively small sample size of the cross-sectional cohort collected to date. Second, despite the tangible advancement in global representation offered by this cohort, participants are still enriched for higher socioeconomic status due to the urban-centric recruitment. This drawback is being actively addressed through ongoing enrollments and community outreach efforts in more rural areas. Third, as with any clinic-based enrollment cohort, there is a possibility of ascertainment bias among recruited participants because the study recruits from clinical practices specializing in cognitive disorders, which may lead to an overrepresentation of more extreme clinical phenotypes. The findings to date represent significant advances in understanding the etiology of Alzheimer’s and Frontotemporal dementia in this region. Continued enrollment in this project will provide additional valuable insights through future studies that map the genetic underpinnings of disease risk in large families, genetic risk burden in cases, and offer well-powered cohorts for case-control studies to identify common risk variants. Moreover, the robust family structure already observed in ReDLat provides a unique opportunity to map genetic modifiers and assess the impact of local genomic ancestry. As global population representation continues to expand, it will be critical to evaluate the generalizability of genetic risk factors for AD and FTD across diverse ancestral backgrounds, within the context of distinct social determinants of health, and accounting for modifiable risk factors that may influence disease risk and resilience across distinct cultures.
Methods
Participant recruitment and inclusion criteria
Clinical diagnosis was determined by site investigators through consensus conferences at each site, adhering to the current diagnostic criteria for AD and FTD33,34,35. Healthy controls were recruited at the same locations, meeting the following criteria: clinical dementia rating (CDR)36 of 0, a mini-mental state examination (MMSE)37 score greater than 25, or having been evaluated by a neuropsychologist who confirmed normal cognition in participants with few years of formal education. Family members of participants with AD or FTD, aged 18 years or older, were included if there were two or more individuals with neurodegenerative illnesses in the family, or were related to a study participant with a known dementia-associated genetic mutation and had undergone genetic counseling. All participants (diagnosed patients, healthy controls, and family members) demonstrated minimum fluency in the language of assessment (Spanish or Portuguese), had adequate vision and hearing for cognitive testing as determined by the investigator, and were required to have a study partner (informant) with at least 6 months of knowledge about their daily activities and cognitive/functional status. All participants (prospective and retrospective) had to be capable of providing informed consent or have a legally authorized representative.
Written informed consent was obtained from all participants or their legally authorized proxies for all evaluations and assessments conducted, following a detailed explanation of the procedures, associated risks, and potential benefits. This process adhered to the ethical guidelines of each participating country, the Code of Ethics of the World Medical Association, the Declaration of Helsinki, and the Belmont Report. Assent was also secured from participants themselves, ensuring their willingness to participate. The consent process included explicit permission to publish the findings. The study and informed consent procedures were approved by the Institutional Review Board of each participating medical institution.
Participating institutions and their respective Federalwide Assurance (FWA) numbers included the following: Argentina—INECO-Centro de Psicología Médica San Martín de Tours (FWA00028264); Brazil—Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (FWA00001035); Chile—Hospital Clínico Universidad de Chile (FWA00029089) and Universidad Adolfo Ibáñez (FWA00030846); Colombia—Comité de Bioética del Instituto de Investigaciones Médicas, Facultad de Medicina, Universidad de Antioquia (FWA00028864), Pontificia Universidad Javeriana—Hospital Universitario San Ignacio (FWA00001113), and Fundación Valle de Lili (FWA00029865); Mexico—Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (FWA00014416); Peru— Hospital Nacional Docente Madre Niño San Bartolomé (FWA00010121); and the United States—University of California, San Francisco—Memory and Aging Center (FWA00000068).
Clinical characterization
As part of the recruitment process, participants were interviewed about their family history of neurodegeneration. Information was collected via self-report from both patients and their study partners using the Genetic Pedigree Software-Progeny®38. A positive family history was defined as having at least one first- or second-degree relative with dementia or another neurodegenerative disorder. Families with three or more affected individuals in two consecutive generations were then labeled as ‘strong family aggregation’. Medical history and a full neuropsychological examination were conducted as described in Ibañez et al.13 Retrospective participants were assessed based on a re-evaluation of the available clinical data for each individual. The cognitive tests were harmonized as described in Maito et al.39.
Genetic sequencing and data processing
Sample acquisition and processing
Standardized phlebotomy with EDTA tubes was used for sample collection. Genomic DNA was extracted using Wizard® Genomic DNA Purification Kit (Promega), QIAamp® DNA Mini Kit (Qiagen), or similar salting-out methods. Samples were shipped quarterly from the various participating sites in Latin America to the United States. HudsonAlpha Institute for Biotechnology (Alabama, USA) performed Single Nucleotide Polymorphism (SNP) Arrays, WES, and/or WGS of the samples. Additional whole genome sequencing was performed at Psomagen, Inc. (Maryland, USA)
SNP arrays
Variants were genotyped using the NeuroBooster array from Illumina, designed to capture variants relevant to neurological conditions40. Quality control (QC) of the SNP Array data was conducted using Genotools v1 default settings41. Prior to imputation, the QC’ed output files from Genotools were processed with the no_qc_imputation_prep.sh script. This script ran the datasets through the Wrayner script to compare them against all TOPMed freeze 8 variants. Excluded variants were then flipped to rescue additional variants, after which the dataset was processed again through the Wrayner script to compare against the PASS TOPMed freeze 8 variants. Data was subsequently imputed using the TOPMed Imputation Panel and Server v1.3.3, following a previously developed pipeline for multi-ancestral sample sets as described in Vitale et al.41.
Whole exome sequencing
DNA was processed using Integrated DNA Technologies xGen Exome Hyb Panel v2, and sequenced on the NovaSeq 6000 platform using paired-end 100-base pair reads to a target depth of 100×.
Whole genome sequencing
Samples at Psomagen were prepared using the TruSeq DNA PCR-Free library prep method to avoid PCR amplification bias. Samples at HudsonAlpha (64 of the pass-QC genomes) underwent a custom PCR-free preparation involving Covaris shearing (fragmenting the DNA), end repair (preparing the DNA fragments for ligation), and adapter ligation, all without PCR amplification. All libraries from both sites were then normalized using KAPA qPCR and sequenced on the Illumina NovaSeq 6000 platform to a target depth of 30×. The sequencing was paired-end with a read length of 150 bp (Illumina 150bpPE).
Alignment and variant calling
The raw sequence data (fastq files) were aligned to the hg38 reference genome using the Sentieon v202112.05 implementation of the BWA MEM algorithm at HudsonAlpha. Sentieon v202112.05 utilities were used to sort the reads, mark duplicate sequences, and recalibrate the base quality scores. Variant calling, which identifies differences between the sample DNA and the reference genome, was performed using GATK4 tools implemented by Sentieon v202112.05. This step was conducted across all samples in a batch for exomes and one for genomes to maintain consistency. Finally, variant quality score recalibration (VQSR) was applied to filter out false positive variant calls, ensuring high-quality data. This comprehensive approach achieved an average recall rate of 99.22% when compared to the Genome in a Bottle high-confidence truth set, indicating a high level of accuracy in detecting genetic variants.
Genomic analyses
Genomic data quality control
-
a.
Variant Call Format (VCF) files were filtered according to established criteria to ensure high-quality data. For whole genome and exome sequences, variants with genotype quality greater than 20 and read depth scores above 10 were retained. The filtered VCF was then annotated with gene names, variant types, and amino acid changes for all exonic variants using GRCh38.99 with SnpEff, dbSNP release 156, CADD 1.6, TOPMed Bravo Freeze 8 allele frequencies, and ClinVar through BCFtools and Annovar42,43,44,45,46. Variants with genotyping rates below 95% by individual and 95% by variant were removed. Chromosomal sex was further validated via genetic data by splitting the pseudoautosomal regions of the X chromosome and analyzing the heterozygosity of the X chromosome, as well as the count of variants present on the Y chromosome. A detailed pipeline and scripts are available at https://github.com/TauConsortium/redlat-genetics.
-
b.
Relatedness: Family history was documented through elaboration of detailed pedigrees for each recruited participant. Disclosed relatedness was compared to expected genetic relatedness using KING47. Individuals with cryptic relatedness (kinship coefficient <0.125 without a familial relationship documented on the pedigrees) or discrepancies between disclosed and genetic relatedness were removed.
-
c.
Combining Datasets: To combine the arrays and WGS data, all variant IDs in both datasets were first annotated with “chrom:pos:ref:alt”. The VCFs were then intersected using BCFtools v1.9 isec, producing a list of intersecting variants between the two datasets. Both VCFs were filtered to include only these intersecting variants and then merged using BCFtools v1.9 merge48. After merging, the VCF was annotated with dbSNP 15649.
-
d.
Concordance Check: To ensure concordance between the imputed arrays and WGS, concordance was checked for individuals with data from both methods. The imputed array data was filtered for varying allele frequencies (AF) using BCFtools v1.948. Concordance was assessed for each AF-filtered VCF using SnpSift concordance. The final concordance for each individual was determined by dividing the number of correct variant calls by the total number of intersecting variant calls. The complete script for the concordance check is available at the project’s GitHub repository (see data sharing). Samples with concordance below 0.95 at sites with AF less than 0.0001 were excluded.
Population stratification
To capture the ancestral diversity of the cohort and represent all participating countries, we merged the WGS data with imputed SNP array data for individuals lacking WGS data. The ReDLat dataset was then combined with high-depth WGS data from the 1000 Genomes Project (1000GP)50 and filtered to include only biallelic variants with a genotyping rate of >95%. The resulting ReDLat-1000GP dataset was used for the following analyses:
-
a.
Principal component analysis (PCA) was conducted using the smartpca package from EIGENSOFT (version 8.0.0)51 PCA was performed on samples from 1000GP, and the ReDLat samples were subsequently projected onto the principal components.
-
b.
Global ancestry was estimated using ADMIXTURE (version 1.3)52. We performed an unsupervised ancestry analysis on the 1000GP data, modeling ancestry from two to eight populations (K). The ancestry fractions for the ReDLat samples were calculated using the allelic ancestry proportions derived from the 1000GP analysis.
Variant pathogenicity analysis
Data from WGS and WES were merged for a joint analysis of pathogenic variation. We used the Online Mendelian In Man (OMIM) database to search for genes associated with autosomal dominant, autosomal recessive, or X-linked forms of adult-onset dementia53.
We manually curated protein-altering variants in the ten genes most commonly associated with adult-onset neurodegeneration: APP, CHMP2B, FUS, GRN, MAPT, PSEN1, PSEN2, TARDBP, TBK1, and VCP. Expansions in C9orf72 were identified using ExpansionHunter v5.0.054,55, copy number variations in APP were called using DELLY, ERDS56,57, MANTA58, and CNVnator59, following the methodology used in Bowling et al.22. Variants located in introns, the 3′ untranslated region (3′ UTR), the 5′ untranslated region (5′ UTR), and synonymous variants within exons were included if their in silico splice-predicting scores (dbscSNV_RF_SCORE and dbscSNV_ADA_SCORE) were both greater than 0.6, since these variants were considered likely to have an impact on splicing, making them relevant for further study60. Exonic non-synonymous variants (missense, nonsense, and frameshift) were analyzed following guidelines from the American College of Medical Genetics and Genomics (ACMG)55 and the Guerreiro algorithm for PSEN1 and PSEN2 genes5. The variants identified in the remaining genes listed in Supplementary Table 4 were queried in the ClinVar61 database and reported if previously classified as pathogenic or likely pathogenic. Variants identified through this process were then tested by hand for familial segregation to confirm their association with the disease within families. ExAC and gnomAD v.4.1.0 databases were used to assess the allelic frequency of variants of interest in global populations.
Local ancestry of pathogenic variants
To determine the haplotypic origin of pathogenic variants, we first phased the ReDLat WGS using SHAPEIT5 (v5.1.1)62. Pedigree information and 1000GP phased genomes were included as family and population references to improve phase accuracy. Since heterozygous variants in a single individual cannot be reliably assigned to a specific haplotype, we restricted our assessment of local ancestry to pathogenic variants present in two or more individuals.
After phasing, we estimated the local ancestry of pathogenic variants using RFMix (v2.03-r0)63. We constructed the reference population panel by merging high-coverage sequence data from the Human Genome Diversity Project (HGDP)64 and 1000GP. Due to the absence of Amerindian individuals in the 1000GP dataset, we used admixed American samples from 1000GP that were over 99.9% Amerindian in the ADMIXTURE analysis at K = 3 as Amerindian reference samples. We then extracted representative individuals of African, Amerindian, European, and East Asian ancestry to build reference cohorts of similar size (100–150 individuals). Finally, we divided the ReDLat WGS into subgroups of individuals with similar global ancestry based on their ADMIXTURE results and ran RFMix with the following settings: terminal node size of five, five expectation-maximization iterations, and both with and without the —reanalyze-reference option.
Data availability
The data used in this publication have been deposited in the FAIR Data Portal within the AD Workbench, supported by the Alzheimer’s Disease Data Initiative, and are accessible at https://www.alzheimersdata.org/ad-workbench. Allelic counts for variants in twenty genes associated with neurodegeneration can be explored through the Tau Consortium Variant Visualizer65, an interactive tool designed to plot and compare variant frequencies across subcohorts of the ReDLat dataset. https://doi.org/10.5062/F4BR8QFB.
Code availability
All the scripts used for the study have been compiled into a single GitHub repository: https://github.com/TauConsortium/redlat-genetics. The scripts for the Tau Consortium Variant Visualizer65 can be found at https://github.com/TauConsortium/variant-visualizer.
Notes
✠ Deceased: Francisco Lopera.
References
Adhikari, K., Chacón-Duque, J. C., Mendoza-Revilla, J., Fuentes-Guajardo, M. & Ruiz-Linares, A. The Genetic Diversity of the Americas. Annu. Rev. Genomics Hum. Genet. 18, 277–296 (2017).
Kunimoto, I. Japanese Migration to Latin America. in Japan, the United States, and Latin America: Toward a Trilateral Relationship in the Western Hemisphere? (eds Stallings, B. & Székely, G.) 99–121 (Palgrave Macmillan UK, London, 1993). https://doi.org/10.1007/978-1-349-13128-0_4.
Hassan, W. S. Arabs and the Americas: a multilingual and multigenerational legacy. Rev. Lit. Arts Am. 52, 166–169 (2019).
Acosta-Uribe, J. et al. A neurodegenerative disease landscape of rare mutations in Colombia due to founder effects. Genome Med. 14, 27 (2022).
Guerreiro, R. J. et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol. Aging 31, 725–731 (2010).
Akgun, B. et al. A genome-wide association study in Peruvians suggests new risk loci for Alzheimer disease. Preprint at medRxiv https://doi.org/10.1101/2023.11.29.23299201 (2023).
Arriagada, I. Familias latinoamericanas: diagnóstico y políticas públicas en los inicios del nuevo siglo. (Naciones Unidas, CEPAL, Div. de Desarrollo Social, Santiago de Chile, 2001).
Van Bavel, J. The world population explosion: causes, backgrounds and projections for the future. Facts Views Vis. Obgyn 5, 281–291 (2013).
Arriagada, I. Familias latinoamericanas: cambiantes, diversas y desiguales. Papeles Población 13, 9–22 (2007).
Acosta-Baena, N. et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: a retrospective cohort study. Lancet Neurol. 10, 213–220 (2011).
Lalli, M. A. et al. Origin of the PSEN1 E280A mutation causing early-onset Alzheimer’s disease. Alzheimers Dement 10, S277–S283.e10 (2014).
Cochran, J. N. et al. Genetic associations with age at dementia onset in the PSEN1 E280A Colombian kindred. Alzheimers Dement 19, 3835–3847 (2023).
Ibanez, A. et al. The multi-partner consortium to expand dementia research in Latin America (ReDLat): driving multicentric research and implementation science. Front. Neurol. 12, 631722 (2021).
Moguilner, S. et al. Brain clocks capture diversity and disparities in aging and dementia across geographically diverse populations. Nat. Med. https://doi.org/10.1038/s41591-024-03209-x (2024).
Dolzhenko, E. et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 27, 1895–1903 (2017).
Arango, D. et al. Systematic genetic study of Alzheimer disease in Latin America: mutation frequencies of the amyloid β precursor protein and presenilin genes in Colombia. Am. J. Med. Genet. 103, 138–143 (2001).
Itzcovich, T. et al. A novel mutation in PSEN1 (p.T119I) in an Argentine family with early- and late-onset Alzheimer’s disease. Neurobiol. Aging 85, 155.e9–155.e12 (2020).
Alzheimer’s Disease, C. ollaborative Group. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat. Genet. 11, 219–222 (1995).
Rippon, G. A. et al. Presenilin 1 mutation in an African American family presenting with atypical Alzheimer dementia. Arch. Neurol. 60, 884–888 (2003).
Jia, L. et al. PSEN1, PSEN2, and APP mutations in 404 Chinese pedigrees with familial Alzheimer’s disease. Alzheimer’s. Dement. 16, 178–191 (2020).
Ryan, N. S. et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: a case series. Lancet Neurol. 15, 1326–1335 (2016).
Bowling, K. M. et al. Genome sequencing as a first-line diagnostic test for hospitalized infants. Genet. Med. 24, 851–861 (2022).
Clark, R. F. et al. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat. Genet. 11, 219–222 (1995).
Sandbrink, R. et al. Missense mutations of the PS-1/S182 gene in german early-onset Alzheimer’s disease patients. Ann. Neurol. 40, 265–266 (1996).
Islam, S. et al. Presenilin is essential for ApoE Secretion, a novel role of presenilin involved in Alzheimer’s disease pathogenesis. J. Neurosci. 42, 1574–1586 (2022).
Arboleda-Velasquez, J. F. et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat. Med. 25, 1680–1683 (2019).
Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man—PubMed. https://pubmed.ncbi.nlm.nih.gov/37188781/.
Browning, S. R. et al. Ancestry-specific recent effective population size in the Americas. PLOS Genet. 14, e1007385 (2018).
Karczewski, K. J. et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv https://doi.org/10.1101/531210 (2019).
van der Zee, J. et al. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 128, 397–410 (2014).
gnomAD. Variant: 5-179836445-C-T. gnomAD (2024).
Blue, E. E., Horimoto, A. R. V. R., Mukherjee, S., Wijsman, E. M. & Thornton, T. A. Local ancestry at APOE modifies Alzheimer’s disease risk in Caribbean Hispanics. Alzheimers Dement 15, 1524–1532 (2019).
Jack Jr, C. R. et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup. Alzheimer’s Dementia. 20, 5143–5169 (2024).
Rascovsky, K. et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477 (2011).
Gorno-Tempini, M. L. et al. Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014 (2011).
Arnold, B. R., Cuellar, I. & Guzman, N. Statistical and clinical evaluation of the Mattis Dementia Rating Scale-Spanish adaptation: an initial investigation. J. Gerontol. B Psychol. Sci. Soc. Sci. 53, 364–369 (1998).
Folstein, M. F., Folstein, S. E. & McHugh, P. R. Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198 (1975).
Progeny Genetics, L. Pedigrees Were Constructed and Drawn Using the Genetic Data Management System, Progeny Clinical – Version N from Progeny Genetics. (Copyright 2019. Reprinted with permission of Progeny Genetics LLC, Delray Beach, FL).
Maito, M. A. et al. Classification of Alzheimer’s disease and frontotemporal dementia using routine clinical and cognitive measures across multicentric underrepresented samples: a cross sectional observational study. Lancet Reg. Health Am. 17, 100387 (2023).
Bandres-Ciga, S. et al. NeuroBooster array: a genome-wide genotyping platform to study neurological disorders across diverse populations. medRxiv https://doi.org/10.1101/2023.11.06.23298176.
Vitale, D. et al. GenoTools: an open-source Python package for efficient genotype data quality control and analysis. G3 Genes|Genomes|Genetics 520, jkae268 (2025).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly ((Austin)) 6, 80–92 (2012).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
ClinVar. https://www.ncbi.nlm.nih.gov/clinvar/.
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001).
Byrska-Bishop, M. et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426–3440.e19 (2022).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLOS Genet. 2, e190 (2006).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Home—OMIM. https://www.omim.org/.
Dolzhenko, E. et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35, 4754–4756 (2019).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med.17, 405–423 (2015).
Zhu, M. et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am. J. Hum. Genet. 91, 408–421 (2012).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016).
Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984 (2011).
Li, Q. & Wang, K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 100, 267–280 (2017).
ALZFORUM | NETWORKING FOR A CURE. https://www.alzforum.org/.
Hofmeister, R. J., Ribeiro, D. M., Rubinacci, S. & Delaneau, O. Accurate rare variant phasing of whole-genome and whole-exome sequencing data in the UK Biobank. Nat. Genet. 55, 1243–1249 (2023).
Maples, B. K., Gravel, S., Kenny, E. E. & Bustamante, C. D. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 93, 278–288 (2013).
Bergström, A. et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 367, eaay5012 (2020).
Lu, D. & Acosta-Uribe, J. Tau Consortium Variant Visualizer. https://doi.org/10.5281/zenodo.15652736 (2025).
Acknowledgements
We dedicate this manuscript to Professor Francisco Lopera, a pioneer in the study of neurodegeneration and Alzheimer’s disease in Latin America. We thank the individuals and the families who participated in this study. We are grateful to Dr. Sharon R. Browning for her assistance with determining local ancestry, Dylan Lu for developing a variant visualization tool for our data and to all the researchers in the participating centers, whose collaboration made ReDLat possible. The Multi-Partner Consortium to Expand Dementia Research in Latin America (ReDLat) was supported in part by the National Institutes of Health; Intramural Research Program of the NIH; National Institute on Aging (NIA)[R01 AG057234, R01 AG075775, R01 AG021051, R01 AG083799]; NIH Department of Health and Human Services [ZO1 AG000534 and AG000548]; National Institute of Neurological Disorders and Stroke (NINDS); Fogarty International Center; Alzheimer’s Association (SG-20–725707); Rainwater Charitable Foundation–Tau Consortium; Bluefield Project to Cure Frontotemporal Dementia; and the Global Brain Health Institute. In addition to the grants reported above, A.S. is supported by ANID/FONDAP [15150012] and ANID/FONDECYT [Regular 1231839]. C.D.A. is supported by ANID/FONDECYT [Regular 1210622] and ANID/PIA/ANILLOS [ACT210096]. D.L.M. received funding from Bluefield Project to Cure Frontotemporal Dementia and Alector. A.I. is supported by ReDLat grants mentioned above and by ANID/FONDECYT Regular [1210195, 1210176, and 1220995]; ANID/FONDAP [15150012]; ANID/PIA/ANILLOS [ACT210096]; and FONDEF [ID20I10152 and ID22I10029]. J.SY. receives funding from NIH-NIA [R01AG062588, P30AG062422, P01AG019724, U19AG079774]; NIH-NINDS [U54NS123985]; NIH-NIDA [75N95022C00031]; AFTD Susan Marcus Memorial Fund; Larry L. Hillblom Foundation; Bluefield Project to Cure Frontotemporal Dementia; French Foundation; Mary Oakley Foundation; Alector and Transposon Therapeutics. This work utilized the computational resources of the NIH STRIDES Initiative (https://cloud.nih.gov) through the Other Transaction agreement [Azure: OT2OD032100, Google Cloud Platform: OT2OD027060, Amazon Web Services: OT2OD027852] and the NIH high-performance computing Biowulf cluster (https://hpc.nih.gov). M.A.N., H.L., C.J., D.V. and M.J.K.’s participation in this project was part of a competitive contract awarded to DataTecnica LLC by the National Institutes of Health to support open science research.
Author information
Authors and Affiliations
Consortia
Contributions
J.A.-U., S.D.P.-E., J.S.Y., and K.S.K. conceived and designed the project, with contributions from J.N.C., J.W.T., P.A.C., C.J., D.L.M., and M.A.N. A.R.L. and D.S.G. managed pedigree information and conducted the heritability assessment. E.A.B. handled sample processing, while K.R. generated SNP-array data at HudsonAlpha.J.A.-F., M.I.B., M.A.B., L.I.B., N.C., C.D-A., F.L., A.S., L.T.T., L.M.Z-R., D.E.D-R., E.P.F.R., L.F.G., N.G., M.B.B., M.E.G., M.A.M., S.J., B.L.M., C.B.P., A.B.S., V.V., and A.I. contributed to participant recruitment, evaluation, sample acquisition, genomic data generation, as well as project management and coordination. H.L., D.V., S.B-C., and M.J.K. assisted with genetic data analyses. F.L. passed away in September 2024; although he was an active collaborator in data acquisition, analyses, and interpretation, he was not able to review the final version of the paper. All remaining authors read and approved the final paper. J.A.-U. and S.D.P.-E. contributed equally to this work, as did J.S.Y. and K.S.K.
Corresponding authors
Ethics declarations
Competing interests
M.A.N. owns stock from Character Bio Inc. and Neuron23 Inc. K.S.K. and J.S.Y. are members of the scientific advisory board of the Epstein Family Alzheimer’s Research Collaboration. K.S.K. is a member of the board of directors of the Tau Consortium and consults with Expansion Therapeutics and ADRx Pharma. J.S.Y. is Editor-in-Chief for npj Dementia. J.S.Y. was not involved in the journal’s review of, or decisions related to, this paper.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Acosta-Uribe, J., Piña-Escudero, S.D., Cochran, J.N. et al. Genetic contributions to Alzheimer’s disease and frontotemporal dementia in admixed Latin American populations. npj Dement. 2, 5 (2026). https://doi.org/10.1038/s44400-025-00025-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44400-025-00025-z
