
Abstract
Despite decades of mechanistic investigation of Alzheimer’s disease (AD), wide gaps exist in disease modeling, particularly the pathobiological arm of tau pathology. Relying on transgenic models expressing mutated forms of tau has contributed much knowledge about primary tauopathy, yet with limited relevance to human AD. To eliminate blind spots for basic and translational research, we review recent developments in this area and discuss key refinements toward next-generation AD-relevant tauopathy modeling.
Similar content being viewed by others
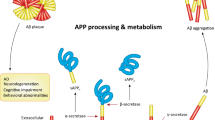


The importance of accurately modeling tauopathy for AD research
Tau, encoded in humans by MAPT, is a neuronal protein that stabilizes microtubules and controls dynamic cytoskeletal remodeling in a phosphorylation-dependent manner in homeostasis, playing important roles in axonal transport and synaptic function1,2,3. In a class of neurodegenerative disorders known as tauopathies, tau protein becomes dysregulated through abnormal processing, causing its aberrant aggregation inside of cells4. In Alzheimer’s disease (AD), tau pathology plays a central role in neurodegeneration5, becoming abnormally hyperphosphorylated and forming fibrillar aggregates called neurofibrillary tangles (NFTs), which disrupt cellular function6,7. In addition, AD is marked by increased overall levels of both total and phosphorylated tau in the brain parenchyma8 as well as in the cerebrospinal fluid9 and plasma10, serving as key imaging and fluid biomarkers of disease progression, respectively.
Histologically, AD is also characterized by amyloid-β plaque deposition, which precedes tau aggregation. Moreover, because of the strong association of familial early-onset AD with genetic mutations involving the enzymatic processing of amyloid precursor protein, amyloid-β plaques have historically received more attention; however, tau pathology shows a closer clinical correlation with symptom severity and disease stage11. Tau pathology in AD is both a marker and a driver of neurodegeneration, making it crucial for diagnosis, mechanistic interrogation, and potential intervention strategies.
Tauopathies are broadly classified into “primary” and “secondary” tauopathies, a distinction based on the underlying cause(s) of the tau pathology12. Primary tauopathies, such as frontotemporal dementia (FTD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and Pick’s disease, are diseases where tau protein abnormalities (most often mutations13) are the defining and originating etiological feature. In these disorders, tau aggregates are the primary driver of subsequent neurodegeneration and other related pathological sequelae14. Secondary tauopathies, on the other hand, possess tau pathology as a consequence of other disease processes. While traumatic brain injuries15 and certain viral infections16 can lead to tau aggregation, by far the most common secondary tauopathy is AD.
Numerous attempts have been made to model the detrimental effects of tau aggregation in living systems17,18, but as we shall see, much previous work has relied on non-AD relevant modes of tau aggregation, which has muddied the waters for accurate mechanistic understanding of AD. Such understandings are necessary if we are to target this complex pathway therapeutically, hence the dire need for more relevant modeling of tau pathology for investigation19.
Modeling of primary tauopathy
Experimental modeling of tau aggregation and tauopathy has traditionally relied on mutated forms of tau, which are, in general, more prone to aberrant processing such as hyperphosphorylation, leading to more rapid appearance of aggregates20. Mutations in the MAPT gene, which encodes the tau protein, are causative in several inherited primary tauopathies13, particularly FTD21,22. Among the most studied are the P301L and P301S mutations, both located in exon 10 within the microtubule-binding domain of tau. These mutations substitute proline with leucine (P301L) or serine (P301S), disrupting tau’s ability to stabilize microtubules and promoting pathological aggregation21. Both mutations increase the propensity for tau hyperphosphorylation and fibrillization, contributing to neurofibrillary tangle (NFT) formation and neurodegeneration. These two mutations altering the P301 residue have been instrumental in understanding tau’s toxic gain-of-function mechanisms in human primary tauopathy.
Several transgenic mouse models have had a profound impact on our understanding of tau toxicity. The JNPL3 mouse expresses human 4-repeat (4R) tau containing the P301L mutation under the control of the mouse prion promoter (PrP) and displays tau hyperphosphorylation and inclusions, NFTs (particularly in the spinal cord and brainstem), and finally age-dependent motor deficits23. The rTg4510 model combines P301L expression with a tetracycline-regulatable system, leading to rapid forebrain atrophy, NFTs, and memory deficits24. A P301L-based model, TgTauP301S (Line 2541), displays early-onset tau aggregation and gliosis, mainly in the brainstem and spinal cord25. The widely used PS19 mouse model expresses human P301S tau, also driven by the PrP promoter, and exhibits robust forebrain tau pathology, synaptic loss, and gliosis beginning around 6 months of age, with region-specific brain atrophy appearing later in progression26. All of these models recapitulate key aspects of human tauopathies, including tau aggregation and neurodegeneration, making them valuable tools for studying disease mechanisms and testing therapeutics. Differences in expression patterns and regional vulnerability offer complementary insights into tau-mediated toxicity. However, transgenic approaches to modeling carry several drawbacks27, such as off-target gene disruption due to transgene insertion, supraphysiological transgene levels, leaky promoter activation in non-target cell types, and splicing differences, leading to observation of non-hTau (human Tau) driven phenotypes28,29. Importantly, many of these FTD models suffer from fatal degeneration in the brain stem and spinal cord, pathologies that do not manifest in human clinical diseases. These caveats merit the advent of alternative strategies.
Humanized knock-in mouse lines are being developed to avoid several of these caveats. Two recent studies reported MAPT knock-ins bearing multiple FTD-related mutations, one of which exhibited tau hyperphosphorylation, synapse and behavior alterations despite a lack of aggregated forms of tau30, and the other showing robust tau pathology and extensive brain alterations such as atrophy, synaptic impairment, and behavioral abnormalities31. These mice may be more useful for studying mechanisms of tau-mediated disease than transgenic overexpression models due to their more physiological expression of tau, though the mutations in tau still distance them from being relevant models of AD.
Viral-mediated human tau overexpression, particularly using adeno-associated virus (AAV), provides a more direct approach to study tau-induced neurodegeneration. Several studies have been published demonstrating robust tau expression in murine brain via AAV-mediated transgene delivery, including both non-mutated wild-type (WT) human tau32,33,34,35, or mutated forms32,33,35,36,37,38. Generally, unlike transgenic mouse lines such as PS1926 and rTg451024, AAV-driven human tau overexpression has not been shown to instigate overt neurodegeneration36,37, with a few exceptions32,35. Because neuronal loss is a defining characteristic of neurodegeneration, there is still a strong preference for using the established transgenic lines that consistently recapitulate this phenotype. While the potential off-target effects of AAV infection in the CNS39 should always be taken into account and controlled for experimentally, viral-mediated approaches avoid many drawbacks of using genetically engineered mouse strains and are becoming more commonly used due to their versatility.
Non-mutated and mutated tau have differing effects in the brain
To model AD brains that build up 3–8 times more non-mutated tau protein than non-diseased controls5,8,9,40,41, a strategy to overexpress WT human tau was adopted early on. A transgenic mouse line of WT human tau overexpression generated by the Davies laboratory42 displayed tau hyperphosphorylation and filamentous aggregation, neuronal cell death43, and synaptic and cognitive dysfunction44. However, transgenic models engineered to overexpress WT MAPT typically lack the rapid and extensive tangle formation seen in P301L or P301S models45. The underlying reason for this was baffling. However, it became clear that transgene expression in vivo is grossly affected by the promoter of choice as well as transgene copy number and insertion sites, making it difficult to directly compare different transgenic lines. By constructing genetically matched transgenic mice overexpressing WT or P301L 0N4R hTau, Gamache et al. observed a marked increase in pathogenicity and cognitive impairment in mice overexpressing WT hTau compared to genetically-matched animals overexpressing P301L hTau instead29. In this study, WT tau exhibited more significant hyperphosphorylation than mutant tau, but conversely did not form insoluble aggregates29, indicating that aggregation is not a requisite for cognitive impairment or other aspects of neurotoxicity, in agreement with earlier studies43. The disparities in pathological outcomes from WT tau expression versus mutated tau in these transgenics may be influenced by distinct interactions of the two with neurodevelopment29, enhanced spreading of wild-type tau46, or differing protein-protein interactions47,48. Regardless, other studies have further demonstrated the sufficiency of WT human tau in causing CNS dysfunction49, thus highlighting their utility in studying AD-related neurodegeneration.
Recent advances and insights obtained from AD-relevant tau pathology models
Instead of a transgenic approach, Jaworski et al. demonstrated that AAV-mediated delivery and overexpression of WT tau in adult brain resulted in profound hippocampal atrophy, which was more deleterious than P301L and occurring in the absence of overt NFT formation32. More recently, Tetlow et al. demonstrated that AAV-mediated overexpression of WT tau, but not P301L or R406W tau, induced localized atrophy in brain tissue after stereotaxic administration to older animals, despite a lesser degree of tau aggregation35. These findings, in agreement with the transgenic studies, highlight a distinctive ability of elevated WT human tau to cause brain pathology, which has been largely overlooked for a long time.
Using the AAV vectors expressing either WT or P301L human tau from Jaworski et al.32, our group has observed key differences between the two forms of tau in both cellular and animal models. In cultured mouse primary neurons, WT tau induced greater toxicity than P301L, as measured by axonal degeneration and cell death50. Interestingly, WT tau was more prone to pathogenic phosphorylation than mutant tau, in agreement with previous studies29. Accompanying enhanced neurotoxicity of WT tau was the presence of phosphorylated tau in axons and dendrites and a more dramatic upregulation of markers of cellular stress, phenotypes that were absent or blunted with mutant tau overexpression in vitro50. Notably, DLK-MAPK signaling is likely a unique driver for axon degeneration associated with tauopathy in the absence of genetic mutations, thus a novel pathway with therapeutic potential.
We further examined the effects of both tau forms in living brains recently, and saw very similar phenotypes in young mice51. P301L-mutated and wild-type human tau overexpression selectively in neurons resulted in abundant phosphorylated tau accumulation, but only WT tau was able to induce significant loss of brain weight as early as 6 weeks post-infection. Further characterization of WT tau pathogenicity revealed its sufficiency in causing brain atrophy, neuronal cell loss in various brain regions, and elevation of both genomic damage and cell stress markers51. Single-nuclei RNA sequencing revealed a selective loss of hippocampal excitatory neurons by WT tau, accompanied by the upregulation of neurodegeneration-related pathways in the affected neuronal populations. Furthermore, expression of WT tau elicited reactive astrogliosis and microgliosis, indicative of neuroinflammatory activation in the brain51. Remarkably, the extent of neurodegeneration in the hippocampus and thalamus was differentially affected by the lifelong absence of microglia, signifying a major influence of neuron-extrinsic responses in the diseased brain.
These cumulative results further underline the exceptional ability of non-mutated human tau, when at an elevated dose, to disrupt homeostasis and cause neurodegenerative outcomes in multiple model systems. Because these changes were most prominently induced by non-mutated rather than mutated tau, we believe that such experimental systems may allow for more accurate modeling of AD in the absence of MAPT mutations associated with primary tauopathy.
Bridging the gaps in next-generation secondary tauopathy modeling
There is no perfect tauopathy model. Several pitfalls still exist in various model systems that attempt to create an artificial but disease-relevant experimental tauopathy, thus warranting further adaptation and refinement19.
One technical pitfall in many models of tauopathy is the possibility of effects on neurodevelopment29. For example, in numerous transgenic lines, and indeed in AAV-driven overexpression paradigms that introduce transgenes at neonatal stages, there are potentially unwanted effects on the neurodevelopmental process that need to be taken into account. These could be avoided if overexpression occurred in adult animals, after the brain has fully developed. This would far more accurately recapitulate the true tauopathy disease course, where accumulation of tau protein does not occur until later in life. In addition, AAV vectors may be delivered to adult mice at distal sites within the CNS, such as intracerebroventricular52 or intrathecal spaces53, avoiding mechanical injury to parenchymal sites of interest. Alternatively, different AAVs such as AAV-PHP.B, AAV-PHP.eB, or AAV.CAP-B22 can be used to bypass the blood-brain barrier of the adult hosts after indirect administration of viral particles54.
Tau expression, the molecular basis for models of tauopathy, would ideally match human patterns of expression, splicing, processing, localization, and potential for pathogenicity as closely as possible. Due to the great differences in mouse and human tau, complete humanization of MAPT sequences is most likely the best approach for studying human disease in mice. For example, a knock-in mouse line generated by Saito et al. replaced the entire murine Mapt gene with a non-mutated humanized copy under the control of the endogenous Mapt promoter55. Notably, these mice express all six isoforms of tau known to exist in the human brain55, but show no signs of cognitive impairment, overt pathology, or changes in neurons in old age56, thus modeling a “healthy aged” human phenotype upon which stressors may be added experimentally in future studies. Another recent initiative reported a series of even more extensive MAPT humanizations (both H1 and H2.1 wild-type haplotypes and several mutated variants), which included the entire 190 kb genomic locus of the human tau sequence57. Scientifically, these humanized knock-in lines will provide a much more disease-relevant background upon which to study tauopathy.
AD is histopathologically complex, manifesting as both tau and amyloid pathology (along with secondary hallmarks), but most murine models of tauopathy only focus on tau alone, limiting relevance to human disease. Models engineered to introduce more than one hallmark of the disease simultaneously may have added merit. Several recent studies have crossed humanized APP and mutant MAPT knock-in strains and demonstrated enhanced tau pathology in the presence of β-amyloid, one with P290S tau58 and one with three FTD-related mutations31. Another group performed intravenous AAV-mediated mutant human tau expression in the CNS of adult amyloid-bearing mice and saw synergistic effects of amyloid on the accumulation of pathogenic tau59. More recently, Desai et al. reported an age- and amyloid-dependent accumulation of tau pathology in double knock-in mice expressing humanized APP and WT MAPT60. The tauopathy phenotype of all these models invariably develops in a delayed manner, after 14 months of age, but more accurately models human AD, thus raising the bar for the next generation of tauopathy models.
Despite an improved relevance to AD, a noticeable limitation of these double knock-in models is the lack of fully mature tau tangle pathology, which is far more commonly observed in models of mutant human tau and may reflect differences in post-translational modification or interactions with glial or other cell types. In order to drive mature tangle pathology in wild-type human tau models, further adaptation may be necessary, such as altering environmental factors (e.g. diet, injury, and infection), optimizing the neuroimmune landscape with xenotransplanted humanized glial cell types61,62, aging the animals either naturally or mimicking enhanced aging genetically63,64, or perhaps combining human APP and MAPT knock-in strains with further humanized alleles in AD-relevant risk genes like APOE, TREM2, and/or others57.
One critical aspect of tau pathology is its spread via the transmission of seeds of pathogenic tau65. Therefore, another important approach for modeling AD tau pathology in mice is the introduction of exogenous tau-containing particles, such as recombinant human tau packaged into pre-formed fibrils66, or aggregated tau isolated from post-mortem human brain tissues67,68,69. These agents, in contrast to native mouse tau, which lacks the ability to form true de novo aggregates, have been demonstrated to initiate tau pathology formation, and even seeding aggregation of native mouse tau67,68, in murine brain. Tau humanization has been shown to significantly accelerate cell-to-cell propagation of AD brain-derived pathological tau, even in the absence of β-amyloid55, allowing experimental modeling of this aspect of AD tau pathology.
It is worth noting that, despite a focus on insoluble NFT formation when modeling tauopathy, neuronal loss is more closely associated with tau expression than with tangle formation, as tangle-bearing neurons paradoxically have a reduced risk of cell death compared to neurons without NFTs in AD brain70. Soluble, high-molecular-weight human tau has emerged as a pathological species that can impair synaptic health, disrupt network function, and predispose neuronal death71,72. In the absence of NFT formation, we have observed a unique capacity of WT tau to induce the formation of high-molecular-weight tau, together with tau hyperphosphorylation, both in the soma and axon/dendrites of neurons that undergo neurodegeneration50. Therefore, neurodegeneration can be biologically disconnected from tangle formation, a crucial point to consider when modeling tauopathy.
Lastly, non-human primates have been used in a limited number of studies to model tauopathy in hopes of overcoming species differences between mice and humans. Though primates are known to display age-related cognitive impairment and amyloid beta pathology, there is little evidence that spontaneous tau pathology can influence neurodegeneration in these animals73,74, calling into question their relevance to human disease75. Nevertheless, AAV-mediated human tau expression has been shown to instigate neurodegeneration in primates76, and even initiate other AD-relevant phenotypes, including hippocampal neuronal loss and cognitive deficits77. Therefore, despite a relative lack of genetic tools and added ethical considerations with non-human primates, they likely still hold a place of value in the investigation of AD-related tau pathological mechanisms or therapeutics74.
Conclusion
The AD field is in dire need of more relevant animal models of the secondary tauopathy seen in human disease. Although the use of transgenic mice and mutated forms of tau has yielded a great deal of information about the regulation and dysregulation of tau, not all findings appear to be strictly relevant to AD, which has made their utility as pre-clinical models far less effective. Alternative approaches, such as using wild-type sequences of human tau, avoiding developmental artifacts, allowing significant periods of time for aging and pathological maturation, and combining with other AD pathogenic or risk factors, will be needed to reach new levels of refinement.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Chang, C. W., Shao, E. & Mucke, L. Tau: enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 371, https://doi.org/10.1126/science.abb8255 (2021).
Wang, Y. & Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21 (2016).
Guo, T., Noble, W. & Hanger, D. P. Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704 (2017).
Holtzman, D. M. et al. Tau: from research to clinical development. Alzheimers Dement. 12, 1033–1039 (2016).
Knopman, D. S. et al. Alzheimer disease. Nat. Rev. Dis. Prim. 7, 33 (2021).
Parra Bravo, C., Naguib, S. A. & Gan, L. Cellular and pathological functions of tau. Nat. Rev. Mol. Cell Biol. 25, 845–864 (2024).
Tracy, T. E. & Gan, L. Tau-mediated synaptic and neuronal dysfunction in neurodegenerative disease. Curr. Opin. Neurobiol. 51, 134–138 (2018).
Khatoon, S., Grundke-Iqbal, I. & Iqbal, K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer’s disease: a radioimmuno-slot-blot assay for nanograms of the protein. J. Neurochem. 59, 750–753 (1992).
Hu, Y. Y. et al. Levels of nonphosphorylated and phosphorylated tau in cerebrospinal fluid of Alzheimer’s disease patients: an ultrasensitive bienzyme-substrate-recycle enzyme-linked immunosorbent assay. Am. J. Pathol. 160, 1269–1278 (2002).
Pillai, J. A. et al. Highly elevated cerebrospinal fluid total tau level reflects higher likelihood of non-amnestic subtype of Alzheimer’s disease. J. Alzheimers Dis. 70, 1051–1058 (2019).
Malpas, C. B., Sharmin, S. & Kalincik, T. The histopathological staging of tau, but not amyloid, corresponds to antemortem cognitive status, dementia stage, functional abilities and neuropsychiatric symptoms. Int. J. Neurosci. 131, 800–809 (2021).
Orr, M. E., Sullivan, A. C. & Frost, B. A brief overview of tauopathy: causes, consequences, and therapeutic strategies. Trends Pharm. Sci. 38, 637–648 (2017).
Goedert, M. & Jakes, R. Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta 1739, 240–250 (2005).
Johnson, A. M. & Lukens, J. R. The innate immune response in tauopathies. Eur. J. Immunol. 53, e2250266 (2023).
McKee, A. C. et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64 (2013).
Di Primio, C. et al. Severe acute respiratory syndrome coronavirus 2 infection leads to Tau pathological signature in neurons. PNAS Nexus 2, pgad282 (2023).
Gotz, J. et al. Transgenic animal models of Alzheimer’s disease and related disorders: histopathology, behavior and therapy. Mol. Psychiatry 9, 664–683 (2004).
Sanchez-Varo, R. et al. Transgenic mouse models of Alzheimer’s disease: an integrative analysis. Int. J. Mol. Sci. 23, 5404 (2022).
Samudra, N., Lane-Donovan, C., VandeVrede, L. & Boxer, A. L. Tau pathology in neurodegenerative disease: disease mechanisms and therapeutic avenues. J. Clin. Invest. 133, https://doi.org/10.1172/JCI168553 (2023).
Alonso, A. D. et al. Hyperphosphorylation of tau associates with changes in its function beyond microtubule stability. Front. Cell Neurosci. 12, 338 (2018).
Hutton, M. et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705 (1998).
Strang, K. H., Golde, T. E. & Giasson, B. I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab Invest. 99, 912–928 (2019).
Lewis, J. et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491 (2001).
Santacruz, K. et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481 (2005).
Allen, B. et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci. 22, 9340–9351 (2002).
Yoshiyama, Y. et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351 (2007).
Goodwin, L. O. et al. Large-scale discovery of mouse transgenic integration sites reveals frequent structural variation and insertional mutagenesis. Genome Res. 29, 494–505 (2019).
Gamache, J. et al. Factors other than hTau overexpression that contribute to tauopathy-like phenotype in rTg4510 mice. Nat. Commun. 10, 2479 (2019).
Gamache, J. E. et al. Developmental pathogenicity of 4-repeat human tau is lost with the P301L mutation in genetically matched tau-transgenic mice. J. Neurosci. 40, 220–236 (2020).
Watamura, N. et al. In vivo hyperphosphorylation of tau is associated with synaptic loss and behavioral abnormalities in the absence of tau seeds. Nat. Neurosci. 28, 293–307 (2025).
Morito, T. et al. Human MAPT knockin mouse models of frontotemporal dementia for the neurodegenerative research community. Cell Rep. Methods 5, 101024 (2025).
Jaworski, T. et al. AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in wild-type mice. PLoS ONE 4, e7280 (2009).
Koller, E. J. et al. Combining P301L and S320F tau variants produces a novel accelerated model of tauopathy. Hum. Mol. Genet. 28, 3255–3269 (2019).
Vogels, T., Vargova, G., Brezovakova, V., Quint, W. H. & Hromadka, T. Viral delivery of non-mutated human truncated tau to neurons recapitulates key features of human tauopathy in wild-type mice. J. Alzheimers Dis. 77, 551–568 (2020).
Tetlow, A. M. et al. Neural atrophy produced by AAV tau injections into hippocampus and anterior cortex of middle-aged mice. Neurobiol. Aging 124, 39–50 (2023).
Cook, C. et al. Tau deposition drives neuropathological, inflammatory and behavioral abnormalities independently of neuronal loss in a novel mouse model. Hum. Mol. Genet. 24, 6198–6212 (2015).
Liu, X. et al. Expression of P301L-hTau in mouse MEC induces hippocampus-dependent memory deficit. Sci. Rep. 7, 3914 (2017).
Mustroph, M. L., King, M. A., Klein, R. L. & Ramirez, J. J. Adult-onset focal expression of mutated human tau in the hippocampus impairs spatial working memory of rats. Behav. Brain Res. 233, 141–148 (2012).
Costa-Verdera, H. et al. AAV vectors trigger DNA damage response-dependent pro-inflammatory signalling in human iPSC-derived CNS models and mouse brain. Nat. Commun. 16, 3694 (2025).
Scheltens, P. et al. Alzheimer’s disease. Lancet 397, 1577–1590 (2021).
Yamamori, H. et al. Tau in cerebrospinal fluid: a sensitive sandwich enzyme-linked immunosorbent assay using tyramide signal amplification. Neurosci. Lett. 418, 186–189 (2007).
Andorfer, C. et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 86, 582–590 (2003).
Andorfer, C. et al. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454 (2005).
Polydoro, M., Acker, C. M., Duff, K., Castillo, P. E. & Davies, P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J. Neurosci. 29, 10741–10749 (2009).
Sahara, N. & Yanai, R. Limitations of human tau-expressing mouse models and novel approaches of mouse modeling for tauopathy. Front. Neurosci. 17, 1149761 (2023).
Kruger, L. & Mandelkow, E. M. Tau neurotoxicity and rescue in animal models of human Tauopathies. Curr. Opin. Neurobiol. 36, 52–58 (2016).
Lim, J. et al. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J. Clin. Invest. 118, 1877–1889 (2008).
Tracy, T. E. et al. Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell 185, 712–728.e714 (2022).
Khan, K. M. et al. Human tau-overexpressing mice recapitulate brainstem involvement and neuropsychiatric features of early Alzheimer’s disease. Acta Neuropathol. Commun. 11, 57 (2023).
Li, S., Roy, E. R., Wang, Y., Watkins, T. & Cao, W. DLK-MAPK signaling coupled with DNA damage promotes intrinsic neurotoxicity associated with non-mutated tau. Mol. Neurobiol. 61, 2978–2995 (2024).
Roy, E. R. et al. Non-mutated human tau stimulates Alzheimer’s disease-relevant neurodegeneration in a microglia-dependent manner. Sci. Rep. 15, 27664 (2025).
Passini, M. A. et al. Intraventricular brain injection of adeno-associated virus type 1 (AAV1) in neonatal mice results in complementary patterns of neuronal transduction to AAV2 and total long-term correction of storage lesions in the brains of beta-glucuronidase-deficient mice. J. Virol. 77, 7034–7040 (2003).
Puhl, D. L., D’Amato, A. R. & Gilbert, R. J. Challenges of gene delivery to the central nervous system and the growing use of biomaterial vectors. Brain Res. Bull. 150, 216–230 (2019).
Natale, D. & Holt, M. Retro-orbital delivery of AAVs for CNS wide astrocyte targeting. Methods Mol. Biol. 2896, 13–31 (2025).
Saito, T. et al. Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J. Biol. Chem. 294, 12754–12765 (2019).
Benskey, M. J. et al. Behavioral and neuropathological characterization over the adult lifespan of the human tau knock-in mouse. Front. Aging Neurosci. 15, 1265151 (2023).
Benzow, K. et al. Gene replacement-Alzheimer’s disease (GR-AD): modeling the genetics of human dementias in mice. Alzheimers Dement. 20, 3080–3087 (2024).
Huang, M. et al. Increase in tau pathology in P290S Mapt knock-in mice crossed with app (NL-G-F) mice. eNeuro 9, https://doi.org/10.1523/ENEURO.0247-22.2022 (2022).
Finneran, D. J. et al. Induction of tauopathy in a mouse model of amyloidosis using intravenous administration of adeno-associated virus vectors expressing human P301L tau. Alzheimers Dement. 10, e12470 (2024).
Desai, S. et al. Age- and amyloid-beta-dependent initiation of neurofibrillary tau tangles: an improved mouse model of Alzheimer’s disease without mutations in MAPT. bioRxiv https://doi.org/10.1101/2024.11.04.621900 (2024).
Mancuso, R. et al. Xenografted human microglia display diverse transcriptomic states in response to Alzheimer’s disease-related amyloid-beta pathology. Nat. Neurosci. 27, 886–900 (2024).
Preman, P. et al. Human iPSC-derived astrocytes transplanted into the mouse brain undergo morphological changes in response to amyloid-beta plaques. Mol. Neurodegener. 16, 68 (2021).
Cai, N., Wu, Y. & Huang, Y. Induction of accelerated aging in a mouse model. Cells 11, https://doi.org/10.3390/cells11091418 (2022).
Ong, J. et al. Senescence accelerated mouse-prone 8: a model of neuroinflammation and aging with features of sporadic Alzheimer’s disease. Stem Cells 43, https://doi.org/10.1093/stmcls/sxae091 (2025).
Gibbons, G. S., Lee, V. M. Y. & Trojanowski, J. Q. Mechanisms of cell-to-cell transmission of pathological tau: a review. JAMA Neurol. 76, 101–108 (2019).
Peeraer, E. et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol. Dis. 73, 83–95 (2015).
Guo, J. L. et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J. Exp. Med. 213, 2635–2654 (2016).
Narasimhan, S. et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci. 37, 11406–11423 (2017).
Mate De Gerando, A. et al. Tau seeding and spreading in vivo is supported by both AD-derived fibrillar and oligomeric tau. Acta Neuropathol. 146, 191–210 (2023).
Zwang, T. J. et al. Neurofibrillary tangle-bearing neurons have reduced risk of cell death in mice with Alzheimer’s pathology. Cell Rep. 43, 114574 (2024).
Takeda, S. et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 6, 8490 (2015).
Harris, S. S. et al. Alzheimer’s disease patient-derived high-molecular-weight tau impairs bursting in hippocampal neurons. Cell 188, 3775–3788.e3721 (2025).
Heuer, E., Rosen, R. F., Cintron, A. & Walker, L. C. Nonhuman primate models of Alzheimer-like cerebral proteopathy. Curr. Pharm. Des. 18, 1159–1169 (2012).
Colwell, J. C. & Emborg, M. E. Tau and tauopathies across primate species: implications for modeling neurodegenerative disorders. Front. Aging Neurosci. 17, 1598245 (2025).
Jucker, M. The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat. Med. 16, 1210–1214 (2010).
Tu, Z. et al. Tauopathy promotes spinal cord-dependent production of toxic amyloid-beta in transgenic monkeys. Signal Transduct. Target Ther. 8, 358 (2023).
Jiang, Z. et al. A nonhuman primate model with Alzheimer’s disease-like pathology induced by hippocampal overexpression of human tau. Alzheimers Res. Ther. 16, 22 (2024).
Acknowledgements
Writing was funded by the National Institute on Aging, National Institutes of Health, grant numbers AG057587 and AG074283; BrightFocus ADR A20183775; Brown Foundation 2020 Healthy Aging Initiative; and Pilot Grant support from UTHealth McGovern Medical School (W.C.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
E.R.R.: manuscript writing, revision of the final draft. W.C.: conception of the original idea, revision of the final draft, and approval.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Roy, E.R., Cao, W. Advancing Alzheimer’s research by improving disease modeling of secondary tauopathy. npj Dement. 1, 38 (2025). https://doi.org/10.1038/s44400-025-00044-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44400-025-00044-w
