
Abstract
Agricultural and urban wastewater acts as a primary vehicle for transporting human pathogens and antimicrobial resistance genes (ARGs) into aquatic ecosystems. This study presents an integrated framework combining metagenomic sequencing, quantitative PCR (qPCR), microbial source tracking (MST), meta-MLST genotyping, and quantitative microbial risk assessment (QMRA) to systematically assess the occurrence, sources, and public health risks of microbial contaminants in the Sanjiangyuan watershed from 2023 to 2024. Metagenomic profiling identified over 30 high-risk bacterial and viral pathogens, with Salmonella enterica, Campylobacter jejuni, Vibrio cholerae, and Adenovirus exhibiting clear seasonal peaks. Zoonotic pathogens, including Brucella and Leptospira, were exclusively detected in agricultural effluents. Meta-MLST analysis revealed epidemic sequence types of V. cholerae (e.g., ST69) and V. parahaemolyticus (e.g., ST925, ST2141) in agricultural effluents. MST markers (BacHum, BacCow, qC160F) revealed dual contributions of human and livestock fecal pollution, and were significantly correlated with nutrient indicators such as nitrate and COD. Meta-MLST confirmed epidemic sequence types of V. cholerae and V. parahaemolyticus, indicating environmental circulation of clinically relevant strains. Resistome analysis revealed 772 to 972 ARG subtypes across sites, dominated by multidrug, β-lactam, and tetracycline resistance genes. Notably, mobile ARGs were frequently co-localized with virulence factors (VFs) in opportunistic pathogens such as Pseudomonas aeruginosa and Acinetobacter baumannii, indicating elevated mobility and pathogenicity. Seasonal variation showed the highest resistome risk in October, with 55% of ARGs classified as high-risk and associated with mobile genetic elements. Our findings underscore the value of integrating resistome analysis into risk-based frameworks to enhance early warning of microbial threats and health-relevant ARGs. This approach supports targeted mitigation strategies in highland watersheds facing mixed-source pollution.

Similar content being viewed by others


Introduction
The SanjiangYuan watershed, covering 39,500 km² with an annual water storage of 1.32 billion m³, is the largest natural reserve in China and is considered the country’s “Water Tower.” This region plays a crucial role in ecological protection and water supply, with various government initiatives, such as the “Returning Pasture to Grassland” and “Ecological Protection Zones,” aimed at preserving its ecological integrity1,2. While sparsely populated, the watershed faces significant environmental challenges, particularly from agricultural wastewater discharge, which is a major contributor to microbial contamination in water systems. Wastewater, especially from animal husbandry, introduces a wide range of microorganisms, including pathogens, antibiotic resistance genes (ARGs), and virulence factors (VFs), into aquatic environments3,4. Moreover, the lack of wastewater treatment infrastructure in rural areas exacerbates this issue, with untreated effluents flowing directly into natural water systems, further spreading ARGs and pathogens5,6. Agricultural wastewater has been shown to harbor a variety of opportunistic pathogens, including Pseudomonas aeruginosa, Legionella pneumophila, and Staphylococcus aureus7. These pathogens, along with the ARGs they carry, can pose significant public health risks, as they may spread to human populations through contaminated water sources, exacerbating the global challenge of antimicrobial resistance8. The overuse of antibiotics in agriculture contributes to the proliferation of antibiotic-resistant bacteria in wastewater, which acts as a vector for the environmental dissemination of resistance genes, thus amplifying the antimicrobial resistance crisis, complicating the management of microbial risks9.
To evaluate health risks from microbial contamination in water, Quantitative Microbial Risk Assessment (QMRA) is a widely used framework10. However, traditional QMRA approaches, which rely on culturable organisms and pathogen enumeration, may underestimate microbial risks due to the limited detection sensitivity of conventional microbiological methods11. Recent advancements in molecular techniques, such as quantitative PCR (qPCR) and metagenomic sequencing, have significantly enhanced pathogen detection sensitivity, allowing for more accurate risk assessments by detecting a broader spectrum of pathogens, including viruses, bacteria, and protozoa12. By integrating these molecular tools into QMRA frameworks, we can obtain a better understanding of the health risks posed by waterborne pathogens13.
Despite the advancements, evaluating health risks associated with ARGs remains a challenge. Because antibiotic resistance genes are non-infectious and lack an established dose–response, quantitative risk assessment for ARGs cannot yet be performed within the standard QMRA paradigm14. This gap in risk assessment methodologies highlights the need for further research into the transfer dynamics of ARGs and their interaction with pathogens in environmental settings. Previous attempts to integrate ARGs into risk assessment frameworks have oversimplified the complexity of genetic transfer, environmental persistence, and pathogen variability, leading to limited practical applicability15. Recent advances have already addressed several complexities in ARG risk assessment. In particular, omics-based and global frameworks now incorporate genetic mobility via mobile genetic elements, environmental persistence, and host/pathogen variability when prioritizing ARGs and estimating potential health risks16,17.
Although integrating ARGs into QMRA frameworks could substantially enhance the assessment of health risks associated with antibiotic-resistant pathogens, this integration remains underdeveloped. The complexity of accurately quantifying risks linked to ARG dissemination, horizontal gene transfer, and antibiotic-resistant pathogen dynamics in environmental waters necessitates further research. Specifically, integration can be approached by: (1) identifying and quantifying ARG presence through metagenomic sequencing; (2) associating detected ARGs with specific bacterial hosts, particularly clinically relevant pathogens; (3) determining ARG transfer potentials by examining mobile genetic elements (MGEs)18. Although integrating ARGs into QMRA frameworks is still in its early stages, ARG dissemination is closely linked to fecal contamination, which serves as an important source of antibiotic-resistant bacteria and associated genetic elements in aquatic environments. Thus, accurately assessing ARG-related health risks necessitates reliable identification and tracking of fecal contamination sources. Clarifying the sources of fecal contamination in SanjiangYuan watershed is paramount19. Fecal contamination is closely associated with ARG dissemination and serves as a significant source of antibiotic-resistant bacteria in aquatic environments. Therefore, reliable identification and tracking of fecal contamination sources are essential for accurately assessing ARG-related health risks20. In the SanjiangYuan watershed, the identification of fecal contamination sources is particularly crucial for water quality management21. Microbial Source Tracking (MST) has become an indispensable tool for pinpointing fecal pollution sources in aquatic systems, enabling effective water quality monitoring22.
To address these knowledge gaps, we systematically investigated microbial communities, ARGs profiles, pathogen occurrences, microbial source tracking (MST) markers, and associated public health risks at agricultural wastewater discharge points situated in rural and urban regions over two consecutive years (2023–2024). Utilizing an integrated approach that combined high-throughput metagenomic sequencing, qPCR validation, MST marker quantification, resistome analysis, and quantitative microbial risk assessment (QMRA), our study aimed to: (1) elucidate bacterial and viral community structures, pathogen distributions, and temporal dynamics across various discharge points, with an emphasis on clinically relevant pathogens and geographical differentiation; (2) employ MST markers to accurately trace fecal contamination sources in surface waters impacted by wastewater discharge; (3) quantify ARG diversity and abundance, examining their associations with pathogen hosts; and (4) employ a resistome-informed QMRA framework to quantitatively evaluate the public health risks posed by ARG-carrying pathogens in aquatic environments.
Results
Screening of potential pathogens
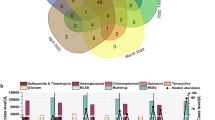
Samples were collected from agricultural wastewater annually during 2023–2024. Two sample types were collected—receiving surface water and effluent at four local sewage discharge point. In the metagenomic data, 92.78% of reads were assigned into the bacteria. At the phylum level, the sequenced reads from all samples were assigned to 254 different phyla (Fig. 1A). Among the overall 254 phyla, the Proteobacteria dominated all samples. The second-largest phylum in the samples was Bacteroidota (17.56%), while Actinobacteria was the third-dominant phylum (13.88%). The top 20 most abundant genera represented between 27 and 43% of the microbial community at each sampling time. Species of the genus Limnohabitans were found in all samples at high levels, whereas the genera Acidovorax and Acinetobacter peaked in samples taken after October 2023 (0.73 and 3.7%, respectively) (Fig. 1B). However, no significant differences in microbial composition were observed among the different discharge outlets. This phenomenon may be attributed to the overflow of septic tanks in residential sewage systems and contamination from livestock manure in agricultural areas. At the phylum level, the sequenced reads from all samples differed markedly in their patterns of bacterial composition across seasons, as demonstrated by principal coordinate analysis (PCoA) (Fig. 1C). Across regions, community profiles were broadly similar and showed no discernible separation by region.

A Relative abundance of bacterialcommunities at the phylum level from 2023 to 2024. B Relative abundance of bacterial communities at the genus level. C Principal coordinate analysis (PCoA) based on Bray–Curtis dissimilarity of bacterial community composition of different samples in discharge point. D Dynamics of bacterial pathogens in wastewater the size of circle indicates the relative abundance PHI database. Quality-filtered reads were taxonomically assigned by alignment to the bacterial PHI database; abundances are reported as the percentage of classified reads per sample
We further investigated the potential pathogens in the metagenomic dataset of waste water samples. Kraken2 was used for taxonomic classification of each sample by PHI database. A total of 120 bacterial pathogens that infect Homo sapiens (human), as listed in the Pathogen-Host Interaction (PHI) database, were selected for screening. Among these, 30 bacterial pathogens were abundantly detected in the metagenomic dataset, including both waterborne pathogens (e.g., Pseudomonas aeruginosa, Shigella flexneri, Campylobacter jejuni, Salmonella enterica, Clostridium perfringens) and respiratory pathogens (e.g., Legionella pneumophila, Streptococcus mutans, Staphylococcus aureus). Afterwards, in rural discharge point the relative abundance of Staphylococcus aureus decreased to 2.56% in May 2023, before becoming dominant (4.42%) in October 2023. Salmonella enterica reached high relative abundance in 2023 and 2024, while Vibrio spp. (0.23%) and Helicobacter pylori (0.083%) increaseed simultaneously in May 2023. Bacillus anthracis and Listeria monocytogenes were notably more frequent in October 2023. In contrast, in urban discharge point the relative abundance of other pathogens varied seasonally (Fig. 1D). The relative abundance of Shigella flexneri, Campylobacter jejuni, and Mycobacterium tuberculosisassociated with clinical cases of dysentery, enteritis, and tuberculosis, respectively (Fig. S1). The results demonstrate the significant potential of wastewater-based epidemiology in serving as a sensitive indicator for tracking and predicting local epidemic trends.
In addition, a comparative analysis between wastewater and surface water revealed clear differences in pathogen composition. Wastewater samples showed a higher diversity and relative abundance of human-associated pathogens, including Escherichia coli, Salmonella enterica, and Staphylococcus aureus. In contrast, surface water samples exhibited lower pathogen abundances overall, with the community dominated by environmental such as Pseudomonas spp.
Quantification and genotype of the abundance of pathogens in wastewater
This study screened pathogenic DNA viruses of public-health concern by mapping metagenomic reads to reference genomes (NR database), identifying eight DNA viruses (Fig. S1). In total, 2.1% of reads were classified as DNA viruses, 96.24% of which were bacteriophages. African swine fever virus and Adenovirus 6 appeared in May 2023 and persisted at low abundance thereafter. Norovirus was not detected by qPCR, likely reflecting low copy numbers. qPCR assays confirmed eight frequently detected viruses; signals for Variola virus (smallpox virus) and Monkeypox virus were relatively high at rural discharge points, with peaks in October 2023, indicating seasonal amplification. Adenovirus and Fowlpox virus also showed seasonal variation, and viral indicators were higher near discharge points and during wet months.
To quantify bacterial risks, species-specific qPCR targeted Cryptococcus neoformans, Vibrio cholerae, Salmonella enterica, Neisseria meningitidis, Staphylococcus aureus, Pseudomonas aeruginosa, Escherichia coli, Klebsiella pneumoniae, Streptococcus pneumoniae, Campylobacter jejuni, Shigella flexneri, Enterococcus faecalis, and Mycobacterium tuberculosis in surface water and at discharge points. E. coli was detected at all sampling times, peaking in May 2024 (0.00013% of reads). Salmonella, Staphylococcus, and Enterococcus also peaked at this time. Metagenomic data showed Neisseria meningitidis at discharge points more than 100-fold above the dry season, and Vibrio cholerae increased by more than tenfold in May 2023. Intestinal pathogens (e.g., Salmonella, Shigella, E. coli) were ubiquitous across agricultural and urban wastewater, whereas zoonotic pathogens were detected exclusively in agricultural effluents; Brucella and Leptospira were identified by metagenomics and subsequently confirmed by qPCR.
Across all samples, Legionella pneumophila (20.3%), Pseudomonas aeruginosa (19.5%), and Shigella flexneri (17.2%) were the most frequently detected potential pathogens in surface water, with median abundances of 4.6 × 10⁴, 4.5 × 10⁴, and 4.3 × 10⁴ copies/L, respectively. Viral targets in surface water showed lower detection frequencies, with rotavirus A (16.4%) and adenovirus 41 (14.1%) as the most prevalent. In wastewater, Enterovirus and P. aeruginosa exhibited the highest detection frequencies (31.3%), followed by Mycobacterium tuberculosis (29.7%) and rotavirus A (26.6%), with median abundances exceeding 5.0 × 10⁵ copies/L for most dominant taxa. Compared to surface water, wastewater consistently harbored higher detection frequencies and abundances for both bacterial pathogens and viruses, indicating greater pathogen loads and potential public health risks.
Genotyping using Meta-MLST verified pathogenic lineages. For Vibrio parahaemolyticus, five and six sequence types (STs) were detected in May and October, respectively, in farming wastewater, with ST925 and ST2141 present in waste samples. In Zhanjiang, four V. cholerae STs (ST2665, ST2830, ST2413, ST69) were observed, and ST69 belongs to the pandemic O1 serogroup. Additionally, ST1577 and ST7648 were detected in urban and farming wastewaters in May. Most identified STs correspond to epidemic genotypes according to PubMLST (https://pubmlst.org).
Detection and quantification of MST markers in surface water samples
The qPCR assays were tested in 146 surface water samples with general and host-associated markers. The three human-associated markers displayed similar levels. All sites exhibited the highest detection frequencies for the B.theta Hum-assay (Fig. 2A). The three general markers showed an overall strong correlation between BacUni, E. coli and Ent, with the correlations between Entero1 and BacUni and Entero1 and E. coli. B.theta were slightly higher for the other hum-marker at all sites. BacUni has been reckoned as a generalized marker sequence for the quantitative detection of all fecal germs. Thus, higher levels of this marker are also expected. As well as significantly higher correlations between the two markers in sites. Studies comparing host specificities among different fecal sources for the BacHum and B.theta markers have shown that both assays were equally sensitive. Among the host-associated markers, it showed the higher detections for B. theta and BacCow, indicating the human and cow are the main sources in this watershed (Fig. 2B).

A Comparison between seasons for host-associated markers using qPCR assays. Bubble size represent averages concentration and error bars represent standard deviations. Error bars show the 95% confidence interval computed on log10(copies) using a two-sided Student’s t approach. B Heat map of Spearman’s rank correlation coefficients matrix for qPCR markers. C Correlation Network Spearman’s rank correlation coefficients matrix for qPCR markers and water quality parameters. Coefficients are colored based on the following scale of absolute value of the coefficients: no correlation (0.0–0.19), weak correlation (0.20–0.39), moderate correlation (0.40–0.59), strong correlation (0.60–0.79), and very strong correlation (0.80–1.0). Significance thresholds are denoted by asterisks: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
Among the host-associated markers, all samples represent the highest detection rates for qC160F and BacCow, suggesting that a larger percentage of fecal contamination comes from poultry and cow sources in SanjiangYuan watershed. Relevance analysis showed significant correlations for both the human markers and general germ markers BacUni and there were no significant correlations between MST markers. The host-associated markers, three human markers, and livestock markers showed a strong correlation, as all markers have been developed specifically for human fecal waste. Only the porcine marker showed significant relationships with V. cholerae. There were no significant relationships found between the Salmonella spp. with other markers. Correlations among the other host-associated markers varied from weak to moderate (Fig. 2C).
Temporal variation in the average concentrations of general indicators, FIBs, human-associated markers, and other host-associated markers over the course of the study at SanjiangYuan was also assessed. The results from the discharge points confirm that higher concentrations were observed during the early winter. Bac32, BacUni, and BacCow all exhibited elevated concentrations during the early summer (October 2018) in surface water. Surface water temperature was measured for each sampling event throughout the study, with an average of 13 °C during the winter and 19 °C during the spring and fall months. Thus, seasonal variability and the effects of water temperature likely contribute to the rapid decay of Bacteroidales during the summer months, resulting in lower marker concentrations. The findings suggest that the high snowmelt from the mountains may influence the source of fecal contamination. High surface permeability, characterized by a low percentage of impermeable surfaces from animal husbandry and wildlife manure, likely contributes to this contamination. The dilution effect brought on by large volumes of meltwater cannot be overlooked. The concentration of human fecal pollutants remains stable at the discharge outlet but becomes diluted in surface water during the summer, confirming this effect.
The presence of host-associated indicators, such as B.theta and QmiHu, exhibited significant correlations with nitrate concentrations, with Spearman’s rank correlation coefficients of 0.43 and 0.36, respectively. In contrast, BacCow showed a stronger correlation with nitrite (0.44), exceeding the associations observed for general indicators. These findings suggest that nitrate serves as a strong predictor of human-associated fecal pollution. Furthermore, cow fecal contamination is also likely a major contributor to nitrogen pollution in creek systems. This is supported by the positive correlation (0.52) between BacCan marker levels and COD concentrations, suggesting that cow manure, with its high organic content, contributes to elevated chemical oxygen demand (COD) in surface water. Together, these observations emphasize the distinct roles of human and cow fecal pollution in driving nitrogen and organic matter contamination in aquatic environments.
Our redundancy analysis (RDA) revealed significant relationships between specific environmental parameters and the distribution of microbial markers across various river sampling points, with the first canonical axis (RDA1) accounting for a substantial 12.34% of the variability in microbial communities (F = 2.1969, p = 0.001) (Fig. 3). Notably, dissolved organic phosphorus (DOP) and specific measures of rainfall emerged as significant predictors of microbial distribution patterns. The presence of DOP was closely linked to increased concentrations of general fecal indicators such as E.coli and BacCow, indicating that nutrient-rich environments could facilitate the proliferation of these microorganisms, which are often indicative of bovine and other fecal contaminations. Conversely, prolonged rainfall correlated with the presence of human-associated markers such as HF183 and qC160F-HU, highlighting the potential for extended wet conditions to enhance runoff and leaching effects, thus increasing the detectability of human-related fecal contamination. Interestingly, factors such as water temperature and turbidity did not significantly contribute to the variance in microbial distributions, as evidenced by their respective F-values (temperature: F = 0.7604, p = 0.617; turbidity: F = 0.9993, p = 0.459). This implies that although these parameters are commonly monitored, their roles in microbial dynamics within this specific river ecosystem may be limited under the conditions studied. In contrast, the Impervious Surface, serving as a proxy for urban runoff, aligned closely with the spatial distribution of total and fecal coliforms, further reinforcing the notion that urbanization and the resultant non-porous surfaces significantly impact water quality. Heavy rainfall often results in increased runoff and saturation of soils, which can cause leakage from septic-leach field systems. Such leakage may increase pathogen concentrations in surface waters, particularly those pathogens that are usually removed by more stable environmental conditions. This scenario underscores the importance of robust waste management systems and highlights the potential public health risks associated with inadequate infrastructure, especially during periods of intense precipitation.

The first axis significantly described all the variability (p = 0.001). Significant predictor environmental variables are identified with a ∗ (α = 0.05).
QMRA quantified human health risks due to exposure to human pathogens
Exposure to Vibrio cholerae poses a significant health risk, as illustrated by the probability of illness associated with each of the seven human pathogens, along with the cumulative illness risk when all pathogens are considered collectively (Fig. 4A). These pathogens are associated with a wide range of adverse health outcomes, including gastrointestinal, respiratory, ocular, auditory, and dermatological conditions. Among them, Vibrio cholerae, an opportunistic pathogen, typically contributes the most to the overall illness risk, followed by Campylobacter jejuni and Legionella pneumophila.

A Predicted probability of illness from Drinking untreated water ingestion exposure event by 7 different pathogens and the cumulative risk due to exposure to all these pathogens. Red dashed lines were benchmarks to the USEPA. B A risk radar plot demonstrating metagenomics-based resistome risk assessment is presented. Bar plots illustrate specific resistome assessment criteria, including risk rank, host pathogenicity, and gene mobility. C A holistic assessment of beach water microbial risk based on E. coli abundance, QMRA, and resistome.
Levels of traditional FIBs are shown in through a culture method. The four general indicators bacteria, E. coli (EC23S857), Enterococci (Entero1), total coliforms and fecal coliforms also showed the high levels of detections among the culture methods (detection frequency >84%) in waste water discharge point (Fig. S3). General marker levels were similar across and showed the similar concentrations among the qPCR markersIn general. The targeted fecal bacterial groups were frequently detected in surface water samples. This study assessed the health risks associated with fecal contamination in recreational waters by analyzing concentrations of key fecal indicator bacteria (FIB), namely Escherichia coli and Enterococci. The findings revealed that in several sampling locations and time periods, FIB levels exceeded the U.S. Environmental Protection Agency’s (EPA) recommended thresholds for primary contact recreation 126 MPN/100 mL for E. coli and 35 MPN/100 mL for Enterococci. Exceeding these thresholds is associated with an increased risk of gastrointestinal illnesses among swimmers, estimated at approximately 36 cases per 1000 individuals.Furthermore, when evaluating the recommended Fecal Indicator Bacteria (FIB) for water quality monitoring Enterococci all four discharge points complied with regulatory standards, which limit illness rates to a maximum of 36 per 1000 people (USEPA, 2022). However, the QMRA results for water quality monitoring revealed that data from May 2023 and May 2024 (averaging 23.4 illnesses per 1000 people) exceeded the benchmark in four areas, while the October data remained within the marginally acceptable range (1.4–1.8 illnesses per 1000 people) as per guidelines. QMRA predicted a high annual risk of gastrointestinal diseases, including shigellosis, campylobacteriosis, and salmonellosis, due to exposure to bacterial contaminants in surface water within the informal settlement.
This risk applied to all farm members except those who consistently consumed treated water. The elevated risks were attributed to the high frequency and volume of water consumption. Additionally, QMRA identified season-specific patterns of illness risk, each linked to different potential contamination sources. For instance, in May, a significant presence of Campylobacter D. jejuni was detected in wastewater, leading to a heightened risk of illness associated with this pathogen. Overall, the discharge point for direct drinking water sources used by local residents fall within the permissible range and exhibit no significant seasonal variations (Fig. S4). The estimated per event infection risk for human norovirus (HuNoV) ranged from 2.5 × 10−4 to 3.8 × 10−2 exceeding the 10⁻⁴ benchmark in 41.7% combinations. HuNoV, despite moderate environmental concentrations, emerged as one of the top three pathogens in median risk owing to its high infectivity.
Host distribution and correspondence with ARGs and VFs in the waste water
Resistome profiles diverged between farm and urban wastewaters, encompassing ARG richness, class composition, and clinically relevant hosts. ARG composition also differed by source. In farm wastewater, we detected 772 ARG subtypes across 22 types, with 673–753 subtypes per site. Among these, 79 subtypes (10.23%) conferred resistance to β-lactams and 234 (30.30%) to multiple drug classes; tetracycline and glycopeptide resistance genes predominated. In urban wastewater, 972 subtypes across 34 types were identified at two discharge points, with 792–862 subtypes per site representing 23–31 types. Of these, 245 subtypes (25.20%) conferred β-lactam resistance and 102 (10.49%) were multidrug-resistance subtypes; sulfonamide resistance genes were rare (0.0002% of detected subtypes) (Fig. S6). Similar to farm wastewater, tetracycline (23.14%) and glycopeptide (14.92%) resistance genes were predominant. Human-associated pathogens detected in wastewater, such as A. baumannii and P. aeruginosa, carried both ARGs and virulence factors commonly reported in clinical settings. Among ARGs, bacA showed the highest abundance and is generally considered intrinsic; sulfonamide genes (sul1, sul2) and the MLS gene macB were also frequently detected across matrices.
In the metagenomic co-occurrence analysis, several species carried multiple ARGs linked to multidrug resistance. Acinetobacter baumannii, Klebsiella pneumoniae, and Escherichia coli harbored ARGs such as adeB, emrB, and acrB. Notably, E. coli contained the most diverse ARG repertoire, conferring resistance to 13 antibiotic classes, consistent with a multidrug-resistant profile. Many pathogens also carried MGEs; twelve taxa carried more than one ARG, including Pseudomonas aeruginosa, E. coli, Enterobacter cloacae, Campylobacter concisus, Enterococcus faecium, and Bifidobacterium bifidum (Fig. S5). ARG hosts differed between agricultural and urban settings. In agricultural environments, dominant hosts included Corynebacterium diphtheriae and Elizabethkingia anophelis, whereas K. pneumoniae and P. aeruginosa dominated in urban systems. In wastewater, P. aeruginosa was a key ARG host carrying four subtypes—tet(33), tet(G) (tetracycline), Erm(35) (MLS), and bacA (peptide) (Fig. S6); Acinetobacter and Staphylococcus were also major hosts, carrying four and six ARG subtypes, respectively. Host diversity was higher in urban wastewater (113 distinct hosts) than in farm wastewater (87 hosts).
Potential health risk was inferred when both a pathogen and its key virulence genes were detected. Metagenomic reads were mapped to VFDB. At the functional-category level (Fig. S5C), virulence assignments were dominated by Offensive functions (48.56%), followed by Defensive (25.85%) and Non-specific functions (21.03%); regulation accounted for 4.56%. At the gene-family level (Fig. S5D), unclassified families (“others”) comprised 80.34% of reads, whereas annotated families accounted for 19.66%. The most abundant determinants included LOS (CVF494, 3.81%), pdhB (CVF227, 3.07%), polar flagella (2.62%), LPS (CVF66, 2.26%), β-hemolysin (CVF171, 2.09%), Type IV pilus (CVF082/268, 1.82%), pyoverdine biosynthesis (A001, 1.67%), and HABC family members (e.g., CVF268, 1.33%) (Fig. S6).
Metagenomics-enabled risk assessment of ARGs
Wastewater was identified as the dominant source of ARGs. Pathogens carrying ARGs pose a significant threat to human health. In discharge point, 13 bacterial genera were identified, with Enterococcus faecium and Campylobacter jejuni serving as the primary hosts of ARGs. Host mapping (Fig. S5B) links the five most abundant ARG classes to representative species-level hosts spanning environmental and clinically associated taxa, including opportunistic pathogens (e.g., Acinetobacter, Pseudomonas, Enterobacteriaceae) and commensal/environmental carriers (e.g., Bacteroides). Additionally, we identified specific ARGs in pathogenic bacteria at the discharge point, which may have clinical implications. For instance, the vanXA gene, associated with glycopeptide antibiotics such as vancomycin, was carried by a Bacteroidetes bacterium in the groundwater of the working swine feedlot. This gene exhibited high homology with those present in clinical pathogens, including Klebsiella pneumoniae, Staphylococcus aureus, and Streptococcus gallolyticus, indicating the potential transmission of ARGs from wastewater to humans. The presence of ARGs and MGEs on the same contig was employed as a proxy to evaluate the potential horizontal transferability of ARGs. Notably, pathogens harboring ARGs associated with MGEs posed the highest resistome risk, emphasizing their potential for horizontal gene transfer and the amplification of resistance traits. In October 2023, a significant proportion of ARGs were classified as high-risk. Specifically, 55% of these ARGs were ranked within the top two risk categories, which primarily included mobile ARGs enriched in human-impacted environments. This led to the highest observed absolute abundance of active, high-risk ARGs (Fig. 4B). Additionally, host tracking and genetic context analysis (Fig. 4B) revealed that ARGs present in viable bacterial cells from discharge point in October 2023 were associated with the highest number of potentially pathogenic hosts and exhibited the greatest mobility capacity. In contrast, while May 2023 showed the highest total number of ARGs, only a quarter of these posed a significant risk to human health. Furthermore, the risk posed by ARGs at the sewage discharge outlet was lower during the summer compared to the winter, highlighting a seasonal variation in ARG risk. A comprehensive assessment of human health risks associated with swimming requires the integration of QMRA and resistome risk evaluation. In this context, surface water quality is primarily assessed by measuring E. coli levels. Although all surface water samples met the local statutory water quality standards (≤50 MPN/100 mL, GB 3838-2002), those influenced by sewage effluent exhibited the highest E. coli counts, indicating an elevated potential risk to human health. Regarding human illness risks from both enteric and opportunistic pathogens, as assessed by QMRA (Fig. 4B), the May seasons exhibited marginally acceptable illness risks, with illness probabilities surpassing the USEPA’s recommended thresholds, potentially influenced by both aquaculture and wastewater treatment plants (WWTPs). Health risks associated with the resistome were highest in October 2024 and lowest in October 2023. In summary, October 2023 exhibited the lowest overall human health risks, while May 2023 presented the highest risks in terms of both illness probability and resistome-related risks.
Discussion
The workflow consists of three key stages: pathogen screening and metagenotyping, microbial source tracking, and risk assessment. First, metagenomic surveillance was employed to identify bacterial pathogens and DNA viruses, followed by multiplex qPCR for RNA virus detection. In addition, fine-resolution epidemiological typing using Meta-MLST was employed to study the presence and epidemiology of specific bacterial pathogens, with results compared to clinical isolates. Overall, most pathogen genotypes were detected in both wastewater and clinical samples, suggesting that wastewater surveillance may serve as an effective tool for public health risk assessment. Subsequently, a traceability analysis of fecal contamination was conducted using MST markers, and the relationship between physical and chemical water parameters was examined. Finally, pathogen abundance data were integrated into QMRA to evaluate drinking water risks, with the resistome included in the assessment to address challenges posed by antibiotic-resistant pathogens.
Metagenomic surveillance robustly detects novel and underreported pathogens in environmentally complex samples. Their emergence often coincides with local epidemics. Untreated wastewater, which serves as a reservoir for both pathogens and multidrug-resistant organisms, poses a significant health risk to humans upon its release into natural aquatic systems. This risk encompasses activities such as drinking water consumption, swimming, irrigation, and various other recreational activities. The primary advantage of wastewater surveillance lies in its ability to represent both the entire population and the pollution sources within the catchment area. In our study, the relative abundance trend of Shigella flexneri, Campylobacter jejuni, and Mycobacterium tuberculosis—pathogens linked to dysentery, enteritis, and tuberculosis, respectively remained consistent with clinical case data throughout the study period.
An inherent limitation of metagenomic sequencing is that a substantial number of detected species may not necessarily be human pathogens or pathogenic types. Additionally, fine-resolution epidemiological typing methods, such as MLST or WGS, have been employed to understand the presence and epidemiology of specific bacterial pathogens, often through comparisons with clinical isolates23. However, the full implementation of public health risk assessment tools presents notable challenges, particularly due to the significant time investments required for the comprehensive steps involved in the assessment process. Additional procedures, including bacterial isolation, MLST, or PCR, are conducted for pathogens of particular concern, adding several months to the genotyping timeline24. Culture assays are also performed to obtain pure cultures of bacterial pathogens for subsequent genotyping. Meta-MLST offers considerable time savings. By leveraging metagenomics, Meta-MLST enables the simultaneous identification of pathogens and their allelic profiles in a single sequencing run. This approach not only reduces the time and resources required for pathogen identification but also enhances overall efficiency, making it particularly advantageous for large-scale surveillance and monitoring studies25. In this study, Meta-MLST was utilized to investigate the epidemiology of bacterial pathogens in wastewater, specifically targeting pathogens with potential public health implications such as Vibrio cholerae, Escherichia coli, and Pseudomonas aeruginosa. Through high-resolution sequencing, we were able to identify epidemic genotypes and track their prevalence across different discharge points and seasons. For instance, specific sequence types of V. cholerae (e.g., ST69) were identified in rural wastewater and confirmed to be associated with pandemic strains. This high level of detail enables a deeper understanding of the genetic diversity of pathogens in the environment and their potential to contribute to local epidemics. Moreover, Meta-MLST enhances pathogen source tracking by linking pathogen strains in wastewater to those found in clinical samples, thereby providing insights into potential cross-contamination and the movement of pathogens between the environment and human populations. This is crucial for early detection of epidemic risks, especially in areas where traditional surveillance systems may not have the capacity to detect emerging strains26.
Implementing routine metagenomic surveillance constitutes an effective biosecurity measure with the potential to prevent large-scale epidemics. This approach proves particularly valuable in detecting underestimated pathogens and is optimally suited for tracking spatial and temporal trends in disease incidence on local farms prior to outbreak occurrence. One limitation of our research is that, while many studies have successfully assembled pathogen genomes from metagenomic data, the quality of these genomes is often compromised when pathogens are present at low abundances27,28. Our study demonstrates that only bacterial species with a relative abundance greater than 0.537% can be reliably recovered. Consequently, pathogens at lower abundances remain undetected, thereby hindering the accurate assessment of health risks. This suggests that the qPCR method can complement metagenomics in detecting microorganisms.
The adoption of QMRA enabled the identification of region-specific health risks undetectable through FIBs measurements alone. Vibrio cholerae was identified as the primary contributor to cumulative illness risks in aquatic environments. This finding highlights the need to consider both pathogens originating from fecal contamination and indigenous marine pathogens that pose significant health risks. Additionally, a notably high illness risk was observed for Legionella pneumophila, which can be partially attributed to two favorable conditions promoting its persistence in aquatic environments: wastewater discharge and protozoan hosts, which act as reservoirs and facilitate bacterial survival. Anthropogenic activities have dramatically intensified the spread of antibiotic resistance in the environment, complicating the treatment of resistant pathogens. To address this issue, we incorporated the analysis of viable pathogenic hosts carrying ARGs into our risk assessment. This integrative approach offered a broader perspective on potential human health impacts. ARGs were the most prevalent in wastewater samples, primarily carried by Pseudomonas aeruginosa. This phenomenon is linked to the extensive use of oxytetracycline in aquaculture operations near farming zones. Previous studies suggest that antibiotic use in farming practices may result in significant environmental impacts21. In our analysis, tetracycline ARGs were dominant resistance markers, with Vibrio species emerging as the predominant pathogenic hosts of ARGs, consistent with the findings of Jo et al.29. This demonstrates the potential of ARG-host associations as tools for source tracking, offering valuable insights into pollution sources, including agricultural runoff and sewage effluents in aquatic environments. These findings underscore the complex interplay between environmental pollution and public health risks, highlighting the necessity of comprehensive monitoring and mitigation strategies. To address these gaps, future research should focus on elucidating the mechanisms by which environmental factors such as agricultural runoff and wastewater effluents influence the transmission of pathogens and the spread of antibiotic resistance.
Importantly, the SanjiangYuan watershed, a unique high-altitude region exhibits distinct hydrogeological and sociocultural features that may amplify these risks. Seasonal snowmelt and high runoff during spring accelerate the diffusion of both pathogens and ARGs from agricultural lands into surface waters. Simultaneously, the coexistence of rural settlements, livestock enclosures, and aquaculture ponds within short distances creates dense point sources of contamination, compounding the burden on aquatic ecosystems. These geographic and hydrological conditions must be accounted for in risk prediction frameworks, as they differ fundamentally from those in plains or urban regions. Our results highlight that ARG-host associations can be leveraged not only for pollution source tracking but also as early indicators of emerging resistant pathogens. For instance, the frequent detection of Acinetobacter baumannii and Klebsiella pneumoniae both known clinical threats suggests that pathogenic strains in the environment may already resemble those circulating in healthcare settings. This convergence underscores the urgency of monitoring both environmental and clinical microbial resistomes in an integrated manner. We propose a new perspective: that the dynamic interaction between environmental niches (e.g., sediment, water column, biofilms) and anthropogenic inputs (e.g., antibiotics, organic load, nutrient enrichment) creates microenvironments that favor the selection and persistence of ARG-carrying pathogens. Future QMRA frameworks should incorporate such micro-scale ecological heterogeneity to improve their predictive accuracy and risk stratification capacity30.
Despite the comprehensive approach employed in this study, several limitations warrant attention. First, the metagenomic sequencing applied here, while effective in broad-spectrum pathogen detection, may have limited sensitivity in identifying low-abundance pathogens or viral RNA genomes without prior enrichment or targeted amplification. This is particularly important in high-altitude regions such as the SanjiangYuan watershed, where environmental dilution effects due to extensive snowmelt and low human density may further obscure pathogen signals31. While Meta-MLST enabled high-resolution genotyping of dominant bacterial species, the method remains constrained by database completeness and read coverage. Pathogens present below a critical relative abundance threshold (0.5%) could not be confidently genotyped, which is a notable limitation when tracking emerging or rare strains in the region.
By integrating QMRA with analyses of ARG-host associations, researchers can gain deeper insights into the relationship between environmental pathogens and antibiotic resistance genes, potentially identifying key sources of health risks. Furthermore, developing region-specific risk assessment frameworks that account for local variations in water quality and pollution sources is critical for more accurate and effective public health risk evaluations. These efforts will not only enhance our understanding of the risks associated with aquatic pathogens but also inform the development of more targeted water quality monitoring and pollution control strategies.
Methods
Sampling sites and pretreatment
Samples were collected from SanjiangYuan region of the Qinghai-Tibet Plateau (Fig. 5). The sample collection was divided into two components: one from surface water and the other from four local sewage discharge points. Specifically, 72 surface water samples were collected from the Yellow River Watershed, and 90 from the Yangtze River Watershed. Two farm discharge points were located from the Yellow River Watershed, and two urban discharge point from the Yangtze River Watershed. These served as sampling points for natural water bodies around each discharge point. Samples were selected from national surface water assessment sections to represent a variety of environmental conditions and human influences, with sampling conducted in and, 2023 October, 2024 May Two-liter wastewater samples were collected from both the surface (0.5 m) and bottom (2–3 m) depths. Sampling was performed at 10:00 AM to minimize sunlight exposure and its potential impact on microbial viability. For microbiological analysis, 4 to 6 liters of water were collected from 1 m below the surface in sterile containers. Additionally, 2 liters were collected from 1.5 m below the surface for physicochemical analysis. Each sample was collected in duplicate to ensure the reliability of the data. Simultaneously, at each watershed, samples were collected from three local sewage discharge outlets, with three untreated wastewater outlets within a 3 km radius selected from each watershed, representing primary discharge points in the respective agricultural regions. For microbiological analysis, 4 to 6 liters of water were collected from 1 m below the surface in sterile containers. Additionally, 2 liters were collected from 80 cm below the surface for physicochemical analysis. Each sample was collected in duplicate to ensure the reliability of the data.

Sewage discharge points included two from agricultural areas in the Yellow River watershed, and two from an urban area in the Yangtze River watershed. Samples were taken in October 2023 and May 2024.
Fecal coliforms were enumerated using the five-tube most probable number (MPN) technique, following the standard methods of the American Public Health Association. Enterococci were enumerated using membrane filtration and mEI agar, in accordance with US Environmental Protection Agency (US EPA) Method 1600 (US EPA, 2005a). Coliphages and somatic coliphages were cultured following US EPA Method 1601 (US EPA, 2001), while Salmonella spp. were cultured using US EPA Method 1682 (US EPA, 2006). Clostridium perfringens was cultured using Standard Method ASTM D5916-96(2002) (American Public Health Association, 1996).
Sample collection and DNA extraction
All water samples were transported on ice to the laboratory and processed within 24 h of collection. Each 1-liter sample was filtered in duplicate through 0.22-μm-pore-size filters and immediately stored at −80 °C until DNA extraction. DNA extraction was performed using the DNeasy PowerLyzer PowerSoil Kit (MP, USA) following the manufacturer’s protocol. To monitor potential contamination during DNA extraction, each batch included a blank control. DNA purity and concentration were assessed using a Nanodrop One spectrophotometer, and extracts were stored at −20 °C until further analysis.
For microbiological analyses, water samples were kept at 4 °C for no longer than 1 day before measuring total coliforms and fecal coliforms using the multiple tube fermentation technique, as outlined by the Ministry of Ecology and Environment, China (2023). Enterococcus counts were determined using the membrane filtration technique, followed by incubation at 36 ± 2 °C for 44 ± 4 h. Colonies were then transferred to preheated Enterococcus agar plates and further incubated at 44 ± 0.5 °C for 2 h to confirm their identity, characterized by black/brown colonies on agar plates.
qPCR analyses
Using the extracted DNA as templates (Table. S1), six microbial source tracking (MST) markers were measured via TaqMan qPCR assays (SanGon Biotech, China). These markers targeted various bacterial groups, including Bacteroidales 16S rRNA genes: Universal Bacteroidales (BacUni), human-associated Bacteroidales (HF183 and BacHum), Chicken/Duck Bacteroidales (qC160F-HU), and Cow Bacteroidales (BacCow). Additionally, conventional fecal bacterial groups (E.coli EC23S857) and (Enterococcus spp.) were assessed using qPCR-based assays. The qPCR assays were categorized into general indicators (BacUni, EC23S857, Entero1), host-associated markers (HF183, BacHum, Chicken/DuckBac, BacCan, BacCow), and pathogen detection. The abundance of eight opportunistic pathogens, including E. coli, Legionella pneumophila, Vibrio cholerae, and Shigella spp., was also detected. The range of quantification for most qPCR assays in each sample was between 10−1 and 106 copies per reaction. According to the standard curve R2 values were all greater than 0.9219. PCR inhibition tests were done for one set of samples for each site (12.5% of total samples) and resulted in a Ct value proportional to a 10-fold dilution. It suggests that PCR inhibition did not interfere with the amplification efficiency. DNA extraction controls and no template controls show that there is no contamination in the qPCR experiments as shown in table S1.
All qPCR assays were conducted using the 7500 StepOne Plus QuantStudio 3 ViiA 7 Detection Real-Time qPCR System (Thermo, USA). The 20 μL reaction mixtures contained SYBR Green (Tiangen, China), 10 μL supermix, 0.6 μL of each forward and reverse primer, 7.4 μL H2O, 0.4 μL dye, and 1 μL of DNA template. Standard curves were generated in duplicate for each qPCR plate using serially diluted plasmid standards purchased from Tsingke (China), which contained the target gene sequences. Each standard curve comprised at least eight 10-fold dilutions of plasmid, and percent amplification efficiencies were calculated. No-template controls were employed to monitor for potential cross-contamination, while sample-added controls were used to detect PCR inhibition. To assess qPCR inhibition, plasmid standards were included in PCR reactions without the sample of interest, and the mean Cycle Threshold (Ct) value served as a reference for comparison with sample Ct values. All qPCR reactions were performed in triplicate. Detailed information on qPCR primer can be found in Supplementary Materials Table S1. This structured approach enhances clarity and coherence, ensuring transparency in the experimental design and procedures employed.
Metagenomics sequencing and Meta-MLST
DNA libraries (350 bp) were prepared using Covaris M220 and NEXTFLEX Rapid DNA-Seq, then sequenced (paired-end) on an Illumina NovaSeq™ X Plus platform. Raw reads were trimmed and quality-filtered using fastp (v0.20.0), then assembled using MEGAHIT (v1.1.2). DNA extract was fragmented to an average size of about 350 bp using Covaris M220 (Gene Company Limited, China) for paired-end library construction. Paired-end library was constructed using NEXTFLEX Rapid DNA-Seq (Bioo Scientific, Austin, TX, USA). Paired-end sequencing was performed on Illumina NovaSeq™ X Plus (Illumina Inc., San Diego, CA, USA) at Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China) using NovaSeq X Series 25B Reagent Kit according to the manufacturer’s instructions. the raw sequencing reads were trimmed of adapters, and low-quality reads (length < 50 bp or with average quality value < 20) were removed by fastp (https://github.com/OpenGene/fastp, version 0.20.0). The quality-filtered data were assembled using MEGAHIT (https://github.com/voutcn/megahit, version 1.1.2) to obtain MAGs.
Contigs with a length ≥300 bp were selected as the final assembling result. Open reading frames (ORFs) from each assembled contigs were predicted using Prodigal[7] (https://github.com/hyattpd/Prodigal, version2.6.3) and a length ≥100 bp ORFs were retrieved. A non-redundant gene catalog was constructed using CD-HIT (http://weizhongli-lab.org/cd-hit/, version 4.7) with 90% sequence identity and 90% coverage. Gene abundance for a certain sample was eatimated by SOAPaligner (https://github.com/ShujiaHuang/SOAPaligner, version soap2.21release) with 95% identity. The best-hit taxonomy of non-redundant genes was obtained by aligning them against the NCBI NR database by DIAMOND (http://ab.inf.uni-tuebingen.de/software/diamond/, version 2.0.13) with an e-value cutoff of 1e-5. Similarly, the functional annotation (VFDB, CARD, PHI) of non-redundant genes was obtained. Based on the taxonomic and functional annotation and the abundance profile of non-redundant genes, the differential analysis was carried out at each taxonomic, functional, or gene-wise level by Kruskal-Wallis test. MetaMLST was employed to characterize the microbial composition of the samples at the strain level and to construct phylogenetic trees. MEGAHIT was employed for metagenome assembly.
Filtered reads were assembled using SPAdes, specifying k-mer size values of 21, 33, 55, and 77, with contigs longer than 1000 base pairs (bps) retained. Filtered contigs were processed using the Binning Across a Series of AssembLies Toolkit (BASALT) to obtain metagenome-assembled genomes (MAGs), with binning performed using MetaBAT version 2.12.1, MaxBin version 2.2.4, and CONCOCT version 0.4.2. The completeness and contamination of the bins were then estimated using CheckM version 1.0.13, with lineage-specific marker genes and default parameters. Bins with completeness greater than 50% and contamination below 10% were retained as MAGs.
High-risk ARGs were identified as the top two arg_ranker risk ranks, which encompass mobile ARGs highly enriched in human-impacted environments; ARGs were ranked using arg_ranker, and mobility was evaluated by mapping ARG-carrying reads to the MobileGeneticElementDatabase to detect integrases/transposases/plasmid signatures17. This study employed Meta-MLST to perform in-silico MLST on metagenomic samples. Genomic DNA was extracted from wastewater samples collected from farming and urban sources in May and October. The Meta-MLST tool was used to perform the MLST analysis by identifying sequence types (STs) from the genomic data. The Meta-MLST repository was cloned from GitHub, and Bowtie2 index files were created for sequence alignment. The FASTQ files were then mapped to the index, generating BAM files, which were subsequently analyzed using the metamlst.py to identify strain typing. Escherichia and Pseudomonas had incomplete MLST locus recovery and could not be reliably typed (non-typable). Accordingly, strain-level results are reported only for Vibrio.
Quantitative microbial risk assessment
Using results generated by our QMRA was conducted to evaluate the probability of human health illnesses associated with drinking per exposure event. The typical QMRA paradigm was adopted to determine human health risks: problem formulation, exposure assessment, health-effects assessment, and risk characterization (WHO, 2016).
To evaluate human health risks posed by exposure to wastewater-contaminated surface water, we applied a QMRA framework encompassing pathogen selection, dose estimation, and risk characterization. Seven reference pathogens were included to represent common waterborne infection routes. Exposure doses were calculated by integrating measured pathogen concentrations with swimmer ingestion volumes, modeled via a triangular distribution (min: 20 mL, mode: 35 mL, max: 50 mL). Dose–response relationships were fitted using established beta-Poisson and exponential models, with illness probability derived accordingly. Cumulative illness risk was calculated by aggregating single-pathogen probabilities under a single exposure scenario.
Problem formulation
At the problem formulation stage, our primary focus was on human health risks associated with the direct consumption of water polluted by wastewater discharge. Seven reference pathogenic bacteria were included in the QMRA analysis to comprehensively account for various waterborne infections (e.g., gastrointestinal, skin, and pulmonary).
Exposure Assessment: To assess the exposure level, concentrations of the reference pathogens were presented as point estimates. The volume of water accidentally ingested by swimmers was modeled using a triangular distribution, defined by a minimum of 20 mL, a most likely value (mode) of 35 mL, and a maximum of 50 mL32. The dose of pathogen i per exposure event during activity j (D_{i,j}) was estimated as: IV_j is the ingestion volume (mL) for activity j, C_{j,k} is the concentration of faecal indicator k for activity j (colony-forming units or genes/mL), and f_{i,k} is the ratio of viable pathogens i (cells) per faecal indicator k (colony-forming units or genes). For the conversion of genes to viable pathogen cells, we considered the number of gene copies per genome of the faecal indicator organisms, along with an estimated ratio of derived genomes to viable cells, based on previously reported flow cytometry and qPCR experiments using sand from water biofilters33,34. To assessment the exposure level (step 2), concentrations of the reference pathogens were presented as point estimates calculated by in-silico rapid viable cell enumeration workflow described by Yu Yang (Yang, 2023).
Health-effects assessment
Epidemiological dose-response models for each reference pathogen were taken from published studies (Table S2) with infection or illness as the endpoints. Popularly, beta-Poisson (Eq.2) and exponential (Eq. 3) dose-response models were used to for fittings of bacterial pathogens. For dose-response models with infection as the endpoint response, probability of illness was obtained using illness-infection ratio (Eq. 4).
where P_response(dose) denotes the probability of response (infection or illness) resulting from a single dose of a pathogen in an individual; dose refers to the quantity of pathogen exposure; k represents the pathogen-specific survival constant; N50 represents the median infectious dose required to infect 50% of the test population; α and β are pathogen-specific constants that optimize model fitting; P_ill(dose) denotes the probability of illness resulting from a single dose of a pathogen in an individual; a represents the pathogen-specific illness-to-infection ratio.
Risk characterization
To characterize the risk, the cumulative risk from a single exposure event contributed by all seven reference pathogenic bacteria was calculated and compared to the specified health outcome levels (Eq. 5). In this study, we estimated the cumulative probability of illness per exposure event related to drinking water for all relevant reference pathogens, rather than calculating daily or annual risks, which could be computed by incorporating additional parameters.
where CumP_ill denotes the cumulative probability of illness resulting from exposure to i pathogens; P_ill(patho_i) represents the probability of illness for pathogen i per exposure event.
Water quality parameters
Water quality parameters, including resistance, conductivity, salinity, oxygen partial pressure (OPP), dissolved oxygen (DO), potential value, pH, temperature (TEMP), chemical oxygen demand (COD), biochemical oxygen demand (BOD), permanganate index, ammonia nitrogen, total phosphorus, total nitrogen, turbidity, total suspended solids (TSS), total dissolved solids (TDS), and suspended solids (SS), were measured using handheld portable devices Hach DR 900 Series (America) and Lachat Quikchem QC8500 Automated Ion Analyser (LACHAT Instruments, USA). Samples were transported to the laboratory at 4 °C within 24 h of collection for further analysis. Chemical oxygen demand (COD) was determined using the potassium dichromate (K₂Cr₂O₇) method. Water quality parameters were continuously monitored at all sites throughout the study period to analyze their spatial-temporal distribution and assess overall water quality. Values below the detection limit were recorded as “below detection limit” (BDL), and non-detect (ND) data points were treated as zero for statistical analysis.
An assessment of study sites was conducted based on proximal land-use information obtained from Geographic Science Information Center of Chinese Academy of Sciences. A promotable analysis of each site was performed as factors influencing fecal contamination sources and levels differ from land use characterization. Precipitation data within 1 and 7 days were downloaded from the National Meteorological Scientific Data (China Meteorological Administration land surface data assimilation system, cldas-v2.0) and applied to all sites (which are between 20 and 50 km from the weather station). The total precipitation amount (mm) during the 24 h preceding sample collection was calculated based on hourly rainfall. Two or more millimeters of rainfall at the time of sample collection was classified as “wet” weather, between 0 and 2 mm as “damp” weather, no precipitation was categorized as “dry” weather. Additionally, land use parameters were also estimated for each individual site and correlation between the MST markers and land use variables was calculated. Average human population for each site was obtained from LandScan Global. The percentage of developed or undeveloped land was obtained from Google Earth Engine (ClLC dataset).
Statistical analyses
All statistical analyses were performed on R. Targeted marker copy numbers per 100 mL of water were calculated for all samples based on standard curves. Raw data from each assay were log-transformed (log10) prior to statistical analysis to elucidate the levels of bacterial indicators of fecal contamination, including Microbial Source Tracking (MST) markers and Fecal Indicator Bacteria (FIB), across five watersheds. Geometric means of the log-transformed technical replicates were used as response variables in all analyses. The nonparametric Wilcoxon signed-rank test was employed to assess whether differences in marker concentrations across sampling sites were statistically significant. The correlation between rainfall in the previous 1 and 7 days and the concentrations of general and host-associated markers at all sites was analyzed using Spearman’s rank correlation in Microsoft Excel. Correlation strength was interpreted using an established scale for biological statistics (McDonald, 2009). Spearman’s rank correlation coefficients (r) between marker concentrations and water quality parameters were also computed. For comparison, the coefficients were characterized using a previously published scale (Stachler et al.): 0.2–0.39 (weak correlation), 0.4–0.59 (moderate correlation), 0.6–0.79 (strong correlation), and 0.8–1.0 (very strong correlation). Differences and correlations were considered statistically significant if p < 0.05.
Data availability
The data that support the findings of this study are available from the corresponding author Fengchang Wu upon request.
References
Cao, W., Wu, D., Huang, L. & Liu, L. Spatial and temporal variations and significance identification of ecosystem services in the Sanjiangyuan National Park, China. Sci. Rep. 10, 6151 (2020).
Wu, G., Li, X. & Gao, J. The evolution of hummock-depression micro-topography in an alpine marshy wetland in Sanjiangyuan as inferred from vegetation and soil characteristics. Ecol. Evol. 11, 3901–3916 (2021).
Yao, Z. et al. Distribution, source identification, and output flux of barium in surface waters in the Sanjiangyuan region and Qilian mountain region of Tibetan Plateau. Bull. Environ. Contam Toxicol. 111, 7 (2023).
Li, H. et al. Atmospheric water vapor and soil moisture jointly determine the spatiotemporal variations of CO(2) fluxes and evapotranspiration across the Qinghai-Tibetan Plateau grasslands. Sci. Total Environ. 791, 148379 (2021).
Nienie, A. B. et al. Microbiological quality of water in a city with persistent and recurrent waterborne diseases under tropical sub-rural conditions: the case of Kikwit City, Democratic Republic of the Congo. Int. J. Hyg. Environ. Health 220, 820–828 (2017).
Foggin, P. M. et al. Assessment of the health status and risk factors of Kham Tibetan pastoralists in the alpine grasslands of the Tibetan plateau. Soc. Sci. Med. 63, 2512–2532 (2006).
Hong, P.-Y., Mantilla-Calderon, D. & Wang, C. Metagenomics as a tool to monitor reclaimed-water quality. Appl. Environ. Microbiol. 86, e00724–00720 (2020).
Wu, X., Nawaz, S., Li, Y. & Zhang, H. Environmental health hazards of untreated livestock wastewater: potential risks and future perspectives. Environ. Sci. Pollut. Res. Int. 31, 24745–24767 (2024).
Liu, Y. J., Li, Z. H., He, Y. T., Yuan, L. & Sheng, G. P. Antibiotic resistomes in face-mask biofilm along an urban river: multiple drivers and co-occurrence with human opportunistic pathogens. J. Hazard Mater. 455, 131587 (2023).
Beaudequin, D. et al. Beyond QMRA: modelling microbial health risk as a complex system using Bayesian networks. Environ. Int. 80, 8–18 (2015).
Petterson, S. R., Stenström, T. A. & Ottoson, J. A theoretical approach to using faecal indicator data to model norovirus concentration in surface water for QMRA: Glomma River, Norway. Water Res. 91, 31–37 (2016).
Ahmed, W. et al. Quantitative microbial risk assessment of microbial source tracking markers in recreational water contaminated with fresh untreated and secondary treated sewage. Environ. Int. 117, 243–249 (2018).
Ryan, U., Hill, K. & Deere, D. Review of generic screening level assumptions for quantitative microbial risk assessment (QMRA) for estimating public health risks from Australian drinking water sources contaminated with Cryptosporidium by recreational activities. Water Res. 220, 118659 (2022).
Leonard, A. F. C., Higgins, S. L., Hui, M. & Gaze, W. H. Investigating landscape-scale variables impacting human exposure to antibiotic resistant bacteria using a targeted metagenome approach. Environ. Pollut. 372, 126015 (2025).
Ahmed, Y., Lu, J., Yuan, Z., Bond, P. L. & Guo, J. Efficient inactivation of antibiotic resistant bacteria and antibiotic resistance genes by photo-Fenton process under visible LED light and neutral pH. Water Res. 179, 115878 (2020).
Zhang, Z. et al. Assessment of global health risk of antibiotic resistance genes. Nat. Commun. 13, 1553 (2022).
Zhang, A. N. et al. An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat. Commun. 12, 4765 (2021).
Yang, T. et al. Antibiotic resistance and resistome risks of inhalable bioaerosols at aeration tank of a full-scale wastewater treatment plant. J. Hazard Mater. 480, 136253 (2024).
Masciopinto, C., Vurro, M., Lorusso, N., Santoro, D. & Haas, C. N. Application of QMRA to MAR operations for safe agricultural water reuses in coastal areas. Water Res. X 8, 100062 (2020).
Stokdyk, J. et al. Sources and risk factors for nitrate, pathogens, and fecal contamination of private wells in rural southwestern Wisconsin, USA. Water Res. 275, 123202 (2025).
Burch, T. R., Stokdyk, J. P., Durso, L. M. & Borchardt, M. A. Quantitative microbial risk assessment for ingestion of antibiotic resistance genes from private wells contaminated by human and livestock fecal sources. Appl. Environ. Microbiol. 90, e0162923 (2024).
Derx, J. et al. Genetic microbial source tracking support QMRA modeling for a riverine wetland drinking water resource. Front. Microbiol. 12, 668778 (2021).
Álvarez-Narváez, S., Logue, C. M., Barbieri, N. L., Berghaus, L. J. & Giguère, S. Comparing PFGE, MLST, and WGS in monitoring the spread of macrolide and rifampin resistant Rhodococcus equi in horse production. Vet. Microbiol. 242, 108571 (2020).
Wareth, G. et al. Molecular characterization of german acinetobacter baumannii isolates and Multilocus Sequence Typing (MLST) analysis based on WGS reveals novel STs. Pathogens 10, https://doi.org/10.3390/pathogens10060690 (2021).
Soliman, E. A. et al. Exploring AMR and virulence in Klebsiella pneumoniae isolated from humans and pet animals: a complement of phenotype by WGS-derived profiles in a One Health study in Egypt. One Health 19, 100904 (2024).
Tang, S. et al. Assessment and comparison of molecular subtyping and characterization methods for Salmonella. Front. Microbiol. 10, 1591 (2019).
Ho, H. et al. Integrating chromatin conformation information in a self-supervised learning model improves metagenome binning. PeerJ 11, e16129 (2023).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Jo, H., Raza, S., Farooq, A., Kim, J. & Unno, T. Fish farm effluents as a source of antibiotic resistance gene dissemination on Jeju Island, South Korea. Environ. Pollut. 276, 116764 https://doi.org/10.1016/j.envpol.2021.116764 (2021).
Nguyen, A. Q. et al. Monitoring antibiotic resistance genes in wastewater treatment: Current strategies and future challenges. Sci. Total Environ. 783, 146964 (2021).
Deng, C. et al. Protecting water sources from animal and fecal pollution for preventing ARGs contamination. Ecotoxicol. Environ. Saf. 303, 118815 (2025).
Federigi, I. et al. Quantitative microbial risk assessment as support for bathing waters profiling. Mar. Pollut. Bull. 157, 111318 (2020).
Hansson, I., Persson, M., Svensson, L., Engvall, E. O. & Johansson, K. E. Identification of nine sequence types of the 16S rRNA genes of Campylobacter jejuni subsp. jejuni isolated from broilers. Acta Vet. Scand. 50, 10 (2008).
Yang, F. et al. Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res. 33, 6445–6458 (2005).
Acknowledgements
This work was supported by the National Key Research and Development Program of China [grant number 2022YFC3204703, 2022YFC3205400] and Young Elite Scientists Sponsorship Program by CAST [grant number 2022QNRC001].
Author information
Authors and Affiliations
Contributions
Zixuan Zhang contributed to conceptualization, methodology, investigation, data curation, formal analysis, and drafting of the manuscript. Miaomiao Teng was responsible for conceptualization, supervision, project administration, and critical review and editing of the manuscript. Wen Li contributed to methodology, data analysis, visualization, and manuscript revision. Hongbo Ma provided resources, validation, and assisted with manuscript editing. Wanting Zhou contributed to formal analysis, data curation, and validation. Fengchang Wu was responsible for conceptualization, funding acquisition, supervision, and critical review and editing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Z., Teng, M., Li, W. et al. Metagenomics with comprehensive validation as a supplementary tool for QMRA in SanjiangYuan watershed. npj Emerg. Contam. 1, 17 (2025). https://doi.org/10.1038/s44454-025-00017-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44454-025-00017-x
