
Abstract
Drought is the most serious abiotic stress that hinders rice production under rainfed conditions. Breeding for deep rooting is a promising strategy to improve the root system architecture in shallow-rooting rice cultivars to avoid drought stress. We analysed the quantitative trait loci (QTLs) for the ratio of deep rooting (RDR) in three F2 mapping populations derived from crosses between each of three shallow-rooting varieties (‘ARC5955’, ‘Pinulupot1’, and ‘Tupa729’) and a deep-rooting variety, ‘Kinandang Patong’. In total, we detected five RDR QTLs on chromosomes 2, 4, and 6. In all three populations, QTLs on chromosome 4 were found to be located at similar positions; they explained from 32.0% to 56.6% of the total RDR phenotypic variance. This suggests that one or more key genetic factors controlling the root growth angle in rice is located in this region of chromosome 4.
Similar content being viewed by others


Introduction
Global climate change in recent years has caused serious drought damage to crop production, resulting in higher food prices, lower farm income, and more food insecurity. Therefore, strategies for the genetic enhancement of drought resistance in crop plants are essential for a stable and adequate food supply. Rice (Oryza sativa L.), a staple food for nearly half of the world's population, is susceptible to drought stress because of its shallow rooting compared with other cereal crops and therefore a limited capacity to extract water from deep soil layers1,2. Nevertheless, rice is frequently exposed to drought in 20% of its total growing area in Asia3. Many studies have suggested that a deep root system helps plants to avoid drought stress by extracting water from deep soil layers4,5,6. Typical upland rice has deeper rooting than lowland rice7. The deep root system in upland rice may contribute to its drought avoidance through enhanced water uptake8. Therefore, to ensure drought avoidance in shallow-rooting rice, introduction of genes responsible for deep rooting into such rice cultivars is considered as one of the most promising breeding strategies6.
Deep rooting is a complex trait because the vertical root distribution is determined by a combination of the root growth angle and maximum root length in seminal and nodal roots of cereal crops9,10. Thus, the development of deep roots requires multiple genes that confer near-vertical root growth axis and increase root length along this axis. According to Courtois et al.11, a total of 103 quantitative trait loci (QTLs) for root length have been reported in rice. Recently, Obara et al.12 fine-mapped qRL6.1, a QTL located on chromosome 6 and associated with root length in hydroponically grown rice seedlings. However, there have been few reports of mapped QTLs associated with root growth angle in rice. DRO1, a QTL for deep rooting on chromosome 9 13, and qSOR1, a QTL for soil-surface rooting on chromosome 7 14 have been mapped. Both QTLs contribute considerably to the root growth angle. Cloning and characterization of DRO1 showed that it functions downstream of the auxin signalling pathway and controls the gravitropic curvature in rice roots15. Under upland conditions with drought stress, a near-isogenic line (NIL) with a functional allele of DRO1 introduced from the deep-rooting cultivar ‘Kinandang Patong’ has deeper roots and a significantly higher grain yield than the shallow-rooting parent variety ‘IR64’, which has a non-functional allele of DRO1. Thus, DRO1 is the first gene associated with root system architecture (RSA) that has been shown to improve the ability to avoid drought.
Sequence analysis of the DRO1 transcribed regions in 64 rice cultivars revealed six haplotypes15. Interestingly, some shallow-rooting cultivars grouped together with the cultivars carrying a functional allele of DRO1 such as ‘Kinandang Patong’, suggesting the existence of undetected QTL(s) associated with root growth angle distinct from DRO1. Availability of multiple genes that affect the root growth angle would be useful for breeding strategies using marker-assisted selection (MAS) to develop rice cultivars with high adaptability to drought.
To identify new QTLs for root growth angle in rice, we selected three accessions (other than ‘IR64’) with the shallowest rooting from different DRO1 haplotype groups based on our previous study15. ‘Tupa729’ belongs to Group I together with ‘Kinandang Patong’. ‘ARC5955’ and ‘Pinulupot1’ belong to Groups II and IV, respectively. We conducted QTL analyses for the root growth angle in three F2 mapping populations derived from a cross between each shallow-rooting variety and ‘Kinandang Patong’ as a donor line with deep roots.
Results
Phenotypic variations of the ratio of deep rooting (RDR) in the three mapping populations
Comparison of the RDR among the parental lines confirmed shallow rooting in ‘ARC5955’, ‘Pinulupot1’, and ‘Tupa729’ (ranging from 16.5 to 27.4%), whereas ‘Kinandang Patong’ showed deep rooting (approximately 80%) ( Fig. 1 ). The RDR values in the three F2 mapping populations from the crosses between ‘Kinandang Patong’ and ‘ARC5955’ (AK-F2), ‘Pinulupot1’ (PK-F2), and ‘Tupa729’ (TK-F2) were distributed between the values of the respective parental lines without considerable transgressive segregation ( Fig. 1 ). The broad-sense heritability (h2B) of RDR was 79.9% in AK-F2, 73.6% in PK-F2, and 88.9% in TK-F2.

Vertical and horizontal lines above the bars indicate the mean and SD of each parental line.
Detection of QTLs for deeper rooting in AK-F2
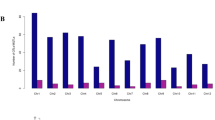
Among 768 single-nucleotide polymorphism (SNP) markers, 346 (45.1%) were polymorphic between ‘ARC5955’ and ‘Kinandang Patong’. To resolve the linkage gaps and to saturate the regions around the LOD support intervals for detected QTLs with DNA markers, we added 16 polymorphic simple sequence repeat (SSR) markers in these regions. The AK-F2 linkage map, composed of 362 markers, covered almost the entire rice genome ( Supplementary Fig. 1 ). The total map length was 1533.5 cM, and the average distance between markers was 4.38 cM. Two QTLs for RDR on chromosomes 2 and 4 were detected based on an LOD threshold of 5.96 ( Figs. 2 and 3a ; Supplementary Fig. 1 ). The QTL on chromosome 4 had a large contribution to the phenotypic variance, explaining 32.0% of the total ( Table 1 ). The mean RDRs of the lines homozygous for the ‘Kinandang Patong’ allele at the marker (RM6089) closest to the detected QTL were significantly higher than those of the lines homozygous for the ‘ARC5955’ allele ( Fig. 3b ). The ‘Kinandang Patong’ allele at this QTL increased the RDR. The minor QTL on chromosome 2 explained 9.5% of total phenotypic variance ( Fig. 2 ; Table 1 ; Supplementary Fig. 1 ). A two-dimensional scan revealed no significant epistatic interaction in the whole genome in this population ( Supplementary Fig. 2 ).

Rectangles represent linkage maps with DNA marker positions shown as vertical lines. Chromosome numbers are indicated under each linkage map (short arms are on the left). Dotted lines indicate LOD thresholds (AK-F2, 5.96; PK-F2, 5.72; TK-F2, 5.46).

(a) The peaks of the LOD curves indicate the putative positions of RDR QTLs. Vertical lines in each linkage map indicate the genetic positions (cM) of DNA markers. Black bars on the linkage maps indicate 1.8-LOD support intervals calculated by using the lodint function in R/qtl36. DNA markers are shown under the linkage maps; the numbers in parentheses indicate their physical map positions (Mb) on chromosome 4 of ‘Nipponbare’. (b) Frequency distribution of RDR in three F2 populations showing three genotype classes of the DNA markers closest to the RDR QTLs (RM6089 in AK-F2 and PK-F2; RM3916 in TK-F2). The inverted triangles indicate the mean, and the horizontal bars indicate SD of each allele. The same shading is used for triangles and corresponding bars. The means labelled with different letters differ significantly among the three alleles (P <0.05, Tukey's multiple comparison test).
Detection of QTLs for deeper rooting in PK-F2
Among 768 SNP markers, 184 (24.0%) were polymorphic between ‘Tupa729’ and ‘Kinandang Patong’. We also mapped 53 polymorphic SSR markers. The PK-F2 linkage map, composed of 237 markers, covered almost the entire rice genome ( Supplementary Fig. 3 ). The total map length was 1560.1 cM, and the average distance between markers was 6.93 cM. Only one QTL (on chromosome 4) was found based on a LOD threshold of 5.72 ( Figs. 2 and 3a ; Supplementary Fig. 3 ). This QTL had a large contribution to the phenotypic variance, explaining 36.2% of the total ( Table 1 ). The mean RDRs of lines homozygous for the ‘Kinandang Patong’ allele at the marker (RM6089) closest to the detected QTL were significantly higher than those of the lines homozygous for the ‘Pinulupot1’ allele ( Fig. 3b ). The Kinandang Patong allele at this QTL increased the RDR. A two-dimensional scan revealed no significant epistatic interaction in the whole genome in this population ( Supplementary Fig. 4 ).
Detection of QTLs for deeper rooting in TK-F2
Among 768 SNP markers, 160 (20.8%) were polymorphic between ‘Pinulupot1’ and ‘Kinandang Patong’. We also mapped 64 polymorphic SSR markers. The TK-F2 linkage map, composed of 224 markers, covered almost the entire rice genome ( Supplementary Fig. 5 ). The total map length was 1434.1 cM, and the average distance between markers was 6.76 cM. Two QTLs were detected on chromosomes 4 and 6 based on an LOD threshold of 5.46 ( Figs. 2 and 3a ; Supplementary Fig. 5 ). The QTL on chromosome 4 had a very large contribution to the phenotypic variance, explaining 56.6% of the total ( Table 1 ). The mean RDRs of lines homozygous for the ‘Kinandang Patong’ allele at the marker (RM3916) closest to the detected QTL were significantly higher than those of lines homozygous for the ‘Tupa729’ allele ( Fig. 3b ). The ‘Kinandang Patong’ allele at this QTL increased the RDR. A QTL detected on chromosome 6 showed a small genetic effect, explaining only 6.3% of total phenotypic variance ( Fig. 2 ; Table 1 ; Supplementary Fig. 5 ). A two-dimensional scan revealed no significant epistatic interaction in the whole genome in this population ( Supplementary Fig. 6 ).
Discussion
To identify new QTLs for root growth angle in rice, we selected three accessions with shallow rooting from different varietal groups of cultivated rice, and produced F2 mapping populations derived from their crosses with ‘Kinandang Patong’. We detected two QTLs for RDR in AK-F2, two in TK-F2, and one in PK-F2. These are novel QTLs for root growth angle because their chromosomal positions differed from those reported in our previous studies13,14. The three QTLs on chromosome 4, detected in the three populations, showed very high LOD values and a large contribution to the total phenotypic variance ( Figs. 2 and 3 ; Table 1 ).The physical positions of the markers most closely linked to the LOD peaks of these QTLs indicated that they may correspond to the same locus. Therefore, we assume that one or more key genes responsible for root growth angle may be located in this chromosome region, and we designate this QTL as DEEPER ROOTING 2 (DRO2). The ‘Kinandang Patong’ allele of DRO2 increased the RDR in all three populations compared with the alleles from the three shallow-rooting parents. The ‘Kinandang Patong’ allele of DRO1, detected in the ‘IR64’ × ‘Kinandang Patong’ recombinant inbred lines (IK-RILs), also resulted in deeper roots than the ‘IR64’ allele13. These results suggest that deep rooting of ‘Kinandang Patong’ is largely controlled by DRO1 and DRO2, although it is unknown whether these QTLs interact epistatically. To clarify the genetic interaction between them, further analysis using NILs for DRO1 and DRO2 will be needed.
We compared the relationship between the positions of DRO2 and other root QTLs by using the QTL Annotation Rice Online Database (Q-TARO, http://qtaro.abr.affrc.go.jp/)16 and the data summarized by Courtois et al.11. We found only one QTL for deep root ratio (percentage of the volume of deep roots [below 30 cm] relative to the total root volume), a parameter related to RDR, in the LOD support interval for DRO2 ( Fig. 3a ; 27.74–30.69 Mb based on the ‘Nipponbare’ genome sequence in the RAP database; http://rapdb.dna.affrc.go.jp/). This QTL was located between the SSR markers RM470 (28.28 Mb) and RM317 (29.25 Mb) in RILs derived from a cross between a lowland rice cultivar ‘Zhenshan97’ (indica) and an upland cultivar ‘IRAT109’ (tropical japonica)17, but comparison of the marker positions alone is not sufficient to determine whether DRO2 is related to this QTL. We also found that a QTL for root volume (qFSR4)18 had been fine-mapped near the candidate region of DRO2. However, the candidate region of qFSR4 is located in the long arm at >31 Mb18, and thus is outside of the LOD support interval for DRO2; this excludes qFSR4 as a candidate for DRO2. According to the report of Courtois et al.11, many QTLs for other root traits (but not root growth angle) have also been detected in the putative region of DRO2. Further study using a NIL for DRO2 should be conducted to clarify whether DRO2 has pleiotropic effects on other root morphology traits. For this purpose, we are currently developing a NIL for DRO2 in each of the shallow-rooting varieties.
We found no genes homologous to DRO1 in the DRO2 LOD support interval. This suggests that DRO1 and DRO2 are not structurally related. Many auxin-related genes such as CROWN ROOTLESS1 (CRL1)/ADVENTITIOUS ROOTLESS119,20, OsPID121, CRL4/OsGNOM122,23, and CRL524 are associated with root gravitropism, which determines the root growth angle. Therefore, we checked the DRO2 LOD support interval for the presence of auxin-related genes, but did not find any candidate genes in this region. We also compared the positions of DRO2 and known root genes characterized in mutants by using the Overview of functionally characterized Genes in Rice Online database (OGRO, http://qtaro.abr.affrc.go.jp/ogro)25. We found three root-related genes, DWARF17626, Oryza sativa indeterminate domain 1027, and GLR3.128, in the DRO2 LOD support interval, but none of them has been reported to affect the root growth angle. Therefore, DRO2 might be distinct from these known genes.
Sequences of the DRO1 transcribed regions of 64 rice cultivars have been grouped into six haplotypes15. The three shallow-rooting varieties used in this study belong to different haplotype groups: Group I (‘Tupa729’ together with deep-rooting ‘Kinandang Patong’), Group II (‘ARC5955’) and Group IV (‘Pinulupot1’). The variety of Group I haplotype showed functional allele of DRO1 in the previous study15. Therefore, we expected to detect QTLs other than DRO1 in TK-F2. Indeed, we detected two new QTLs (but not DRO1) accounting for 68.3% of the total RDR phenotypic variance in TK-F2 ( Table 1 ). This suggests that the differences in RDR between ‘Tupa729’ and ‘Kinandang Patong’ are mostly explained by these two new QTLs. On the other hand, we expected that DRO1 might be detected in AK-F2 and PK-F2 because ‘ARC5955’ and ‘Pinulupot1’ belong to different DRO1 haplotype groups than ‘Kinandang Patong’. However, DRO1 was not found in these mapping populations. In primary mapping populations such as F2, segregation of major QTLs often masks the presence of minor QTLs and hampers their statistical detection29. We assume that DRO1 could not be detected in the present study because of a small difference in the genetic effects of the ‘Kinandang Patong’ DRO1 allele and those of ‘ARC5955’ or ‘Pinulupot1’. This result also suggests that Group II and IV have functional allele of DRO1. The detected QTLs in AK-F2 and PK-F2 contributed to only 39.8% and 36.2% of the total RDR phenotypic variance, respectively. The h2B of RDR in IK-RILs was 77.7%13. In this study, AK-F2 and PK-F2 showed similar h2B; no significant epistatic interaction could be detected in either population. Therefore, we believe that other QTLs in addition to DRO2 were associated with the RDR in these two mapping populations but were not detected in this analysis. For instance, we observed two additional LOD peaks in AK-F2 at lower LOD threshold (LOD = 3.40 between ah03001245 and ah03001697 on chromosome 3; LOD = 3.78 at AD06014592 on chromosome 6) ( Fig. 2 ; Supplementary Fig. 1 ). In PK-F2, we found four additional LOD peaks at lower LOD threshold (LOD = 4.80 at P0634_1 and LOD = 4.09 at ah01003209 on chromosome 1; LOD = 4.24 between AE02000044 and P0221_3 on chromosome 2; LOD = 5.08 between ad07001335 and ad07001571 on chromosome 7) ( Fig. 2 ; Supplementary Fig. 3 ). Chromosome segment substitution lines are very powerful tools for detection of minor QTLs because they enable separation of minor QTLs from major QTLs located in other chromosome regions29. Such advanced materials will be useful to elucidate whether favorable alleles of DRO1 and other QTLs are present in AK-F2 and PK-F2.
We conclude that DRO2 is a promising QTL that can be used to enhance drought avoidance of shallow-rooting rice cultivars by controlling the RSA, because the ‘Kinandang Patong’ allele of DRO2 improves the RDR of cultivars in three different genetic backgrounds: aus, indica, and japonica. DRO2 cloning will accelerate elucidation of its molecular function, and DRO2 haplotype analysis using many rice accessions will enable breeding for a better RSA through MAS. Map-based cloning of DRO2 is currently underway in our laboratory.
Methods
Plant materials
For QTL analysis, we developed three F2 populations from crosses between ‘Kinandang Patong’ and (i) ‘ARC5955’ (138 plants; AK-F2), (ii) ‘Pinulupot1’ (134 plants; PK-F2), and (iii) ‘Tupa729’ (133 plants; TK-F2). ‘ARC5955’ (NIAS Acc. No. WRC35) is a landrace (aus) of Indian origin. ‘Pinulupot1’ (NIAS Acc. No. WRC24) is a landrace (indica) of Philippine origin. ‘Tupa729’ (NIAS Acc. No. WRC55) is a landrace (tropical japonica) of Bangladeshi origin. ‘Kinandang Patong’ (International Rice Research Institute Acc. No. IRGC23364) is a landrace (tropical japonica) of Philippine origin.
RDR measurement
RDR was evaluated quantitatively (as an index for root growth angle) based on a basket method that uses open stainless-steel mesh baskets (top diameter of 7.5 cm and depth of 5.0 cm; PROUD, Ushiku, Japan), as described previously13. We defined the RDR as the number of roots that penetrated the lower part of the mesh (i.e., ≥50° from the horizontal, centred on the stem of the rice plant) divided by the total number of roots that penetrated the whole mesh. Larger RDR values correspond to greater root growth angles.
DNA marker analysis
We determined the genotypes of F2 populations by using SNP markers selected from genome-wide SNP marker data30,31, and SSR markers selected on the basis of the data from the International Rice Genome Sequencing Project32. Total DNA was extracted from leaves by using the CTAB method33. The SNPs were detected from a set of 768 custom SNP panels by using a GoldenGate Genotyping Universal-32 768-plex Assay Kit and the BeadStation 500 G system (both from Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. PCR amplification was performed in 5-μl reaction mixtures containing 0.5 μl (20 ng) DNA, 1.0 μl 5× PCR buffer, 0.1 μl 10 mM dNTPs, 0.025 μl (5 units) KAPA2G Fast DNA Polymerase (Kapa Biosystems, Boston, MA, USA), 0.125 μl of a mixture of forward and reverse primers (20 pM each), and 3.25 μl H2O. The PCR programme consisted of an initial denaturation for 1 min at 95°C; followed by 35 cycles of 10 s at 95°C, 10 s at 55°C, and 1 s at 72°C; followed by a final extension for 30 s at 72°C. PCR products were separated by electrophoresis in 3% agarose gels (Type I agarose; Sigma-Aldrich, St. Louis, MO, USA) at 150 V for 120 min.
Statistical and QTL analyses
h2B of RDR was estimated from the variances of the parental plants and the variance among the F2 lines, and calculated as

where Vp[F2] is the phenotypic variance among F2 lines, and Vp[P1] and Vp[P2] are the phenotypic variances of the parental plants.
Linkage maps were constructed from the genotype data in MAPMAKER/EXP 3.0 software34. Genetic distances were estimated by using the software’s Kosambi map function35. QTL analysis was performed by using the composite interval mapping function of the R/qtl software36. The composite interval mapping threshold was based on the results of 1000 permutations at a 5% significance level37. The confidence intervals around each significant QTL peak were determined by 1.8-LOD support intervals36. The additive and dominant effects and the percentage of phenotypic variance explained by each QTL (R2) at the maximum LOD score were estimated by using the sim.geno, makeqtl, and fitqtl functions in R/qtl36. Two-dimensional scans with a two-QTL model were conducted in R/qtl with the thresholds based on the results of 1000 permutations at a 5% significance level36.
References
Kondo, M., Murty, M. V. R. & Aragones, D. V. Characteristics of root growth and water uptake from soil in upland rice and maize under water stress. Soil Sci. Plant Nutr. 46, 721–732 (2000).
Kondo, M. et al. Genotypic and environmental variations in root morphology in rice genotypes under upland field conditions. Plant Soil 255, 189–200 (2003).
Pandey, S. & Bhandari, H. Drought: economic costs and research implications. In: Drought Frontiers in Rice: Crop Improvement for Increased Rainfed Production. 3–17 (World Scientific Publishing, Singapore, 2008).
Yoshida, S. & Hasegawa, S. The rice root system: its development and function. In: Drought Resistance in Crops with Emphasis on Rice. 97–114 (International Rice Research Institute, Manila, Philippines, 1982).
Fukai, S. & Cooper, M. Development of drought-resistant cultivars using physio-morphological traits in rice. Field Crops Res. 40, 67–86 (1995).
Gowda, V. R. P. et al. Root biology and genetic improvement for drought avoidance in rice. Field Crops Res. 122, 1–13 (2011).
O’Toole, J. C. & Bland, W. L. Genotypic variation in crop plant root systems. Adv. Agr. 41, 91–143 (1987).
Price, A. H. et al. Mapping root and shoot traits in rice: experience in UK, IRRI and WARDA. In: Genetic improvement of rice for water-limited environments. 257–273 (International Rice Research Institute, Manila, Philippines, 1999).
Abe, J. & Morita, S. Growth direction of nodal roots in rice: its variation and contribution to root system formation. Plant Soil 165, 333–337 (1994).
Araki, H., Morita, S., Tatsumi, J. & Iijima, M. Physio-morphological analysis on axile root growth in upland rice. Plant Prod. Sci. 5, 286–293 (2002).
Courtois, B. et al. Rice root genetic architecture: meta-analysis from a drought QTL database. Rice 2, 115–128 (2009).
Obara, M. et al. Fine-mapping of qRL6.1, a major QTL for root length of rice seedlings grown under a wide range of NH4+ concentrations in hydroponic conditions. Theor. Appl. Genet. 121, 535–547 (2010).
Uga, Y., Okuno, K. & Yano, M. Dro1, a major QTL involved in deep rooting of rice under upland field conditions. J. Exp. Bot. 62, 2485–2494 (2011).
Uga, Y. et al. Identification of qSOR1, a major rice QTL involved in soil-surface rooting in paddy fields. Theor. Appl. Genet. 124, 5–86 (2012).
Uga, Y. et al. Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nature Genet. 45, 1097–1102 (2013).
Yonemaru, J. et al. Q-TARO: QTL Annotation Rice Online Database. Rice 3, 194–203 (2010).
Yue, B. et al. Genetic basis of drought resistance at reproductive stage in rice: separation of drought tolerance from drought avoidance. Genetics 172, 1213–1228 (2006).
Ding, X., Li, X. & Xiong, L. Evaluation of near-isogenic lines for drought resistance QTL and fine mapping of a locus affecting flag leaf width, spikelet number, and root volume in rice. Theor. Appl. Genet. 123, 815–826 (2011).
Inukai, Y. et al. Crown Rootless1, which is essential for crown root formation in rice, is a target of an AUXIN RESPONSE FACTOR in auxin signaling. Plant Cell 17, 1387–1396 (2005).
Liu, H. et al. ARL1, a LOB domain protein required for adventitious root formation in rice. Plant J. 43, 47–56 (2005).
Morita, Y. & Kyozuka, J. Characterization of OsPID, the rice ortholog of PINOID, and its possible involvement in the control of polar auxin transport. Plant Cell Physiol. 48, 540–549 (2007).
Kitomi, Y., Ogawa, A., Kitano, H. & Inukai, Y. CRL4 regulates crown root formation through auxin transport in rice. Plant Root 2, 19–28 (2008).
Liu, S. et al. Adventitious root formation in rice requires OsGNOM1 and is mediated by the OsPINs family. Cell Res. 19, 1110–1119 (2009).
Kitomi, Y. et al. The auxin responsive AP2/ERF transcription factor CROWN ROOTLESS5 is involved in crown root initiation in rice through the induction of OsRR1, a type-A response regulator of cytokinin signaling. Plant J. 67, 472–484 (2011).
Yamamoto, E., Yonemaru, J., Yamamoto, T. & Yano, M. OGRO: The Overview of functionally characterized Genes in Rice online database. Rice 5, 26 (2012).
Arite, T., Kameoka, H. & Kyozuka, J. Strigolactone positively controls crown root elongation in rice. J. Plant Growth Regul. 31, 165–172 (2012).
Xuan, Y. H. et al. Indeterminate domain 10 regulates ammonium-mediated gene expression in rice roots. New Phytol. 197, 791–804 (2013).
Li, J. et al. A rice glutamate receptor-like gene is critical for the division and survival of individual cells in the root apical meristem. Plant Cell 18, 340–349 (2006).
Fukuoka, S., Nonoue, Y. & Yano, M. Germplasm enhancement by developing advanced plant materials from diverse rice accessions. Breed. Sci. 60, 509–517 (2010).
Ebana, K. et al. Genetic structure revealed by a whole-genome single-nucleotide polymorphism survey of diverse accessions of cultivated Asian rice (Oryza sativa L.). Breed. Sci. 60, 390–397 (2010).
Ebana, K. et al. Uncovering of major genetic factors generating naturally occurring variation in heading date among Asian rice cultivars. Theor. Appl. Genet. 122, 1199–1210 (2011).
International Rice Genome Sequencing Project. The map based sequence of the rice genome. Nature 436, 793–800 (2005).
Murray, M. G. & Thompson, W. F. Rapid isolation of high molecular weight plant DNA. Nucl. Acids Res. 8, 4321–4325 (1980).
Lander, E. S. et al. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1, 174–181 (1987).
Kosambi, D. D. The estimation of map distance from recombination values. Ann. Eugen. 12, 172–175 (1944).
Broman, K. W., Wu, H. & Sen, S. R/qtl: QTL mapping in experimental crosses. Bioinformatics 19, 889–890 (2003).
Churchill, G. A. & Doerge, R. W. Empirical threshold values for quantitative trait mapping. Genetics 138, 963–971 (1994).
Acknowledgements
The rice accessions were provided by the Genebank of the National Institute of Agrobiological Sciences (Japan) and the International Rice Research Institute (Philippines). This work was supported by grants from the Ministry of Agriculture, Forestry and Fisheries of Japan (Genomics for Agricultural Innovation QTL-4003, Genomics-based Technology for Agricultural Improvement RBS2009).
Author information
Authors and Affiliations
Contributions
Y.U. designed the experiments and wrote the manuscript. Y.U. and N.K. evaluated RDR. N.K. and S.K. performed SSR analysis. T.M. and S.F. performed SNP analysis. Y.U. and E.Y. performed QTL analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplemental information (PDF 1177 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Uga, Y., Yamamoto, E., Kanno, N. et al. A major QTL controlling deep rooting on rice chromosome 4. Sci Rep 3, 3040 (2013). https://doi.org/10.1038/srep03040
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03040
This article is cited by
-
Root system architecture of historical spring wheat cultivars is associated with alleles and transcripts of major functional genes
BMC Plant Biology (2022)
-
Stable and Novel Quantitative Trait Loci (QTL) Confer Narrow Root Cone Angle in an Aerobic Rice (Oryza sativa L.) Production System
Rice (2021)
-
Meta-QTL and ortho-MQTL analyses identified genomic regions controlling rice yield, yield-related traits and root architecture under water deficit conditions
Scientific Reports (2021)
-
Intricate genetic variation networks control the adventitious root growth angle in apple
BMC Genomics (2020)
-
Pattern of alternative splicing different associated with difference in rooting depth in rice
Plant and Soil (2020)
