
Abstract
The role of membrane-bound complement regulatory proteins (mCRP) in the protection of tumor cells in vivo against elimination by the immune system is still unknown. In this study the effect of expression of these mCRP by cervical cancer cells was investigated. In situ expression of mCRP was observed on cervical carcinomas, normal cervical epithelial cells, and the surrounding stroma. Deposition of C3 and C5b-9 was sporadically found on the tumor cells and the surrounding stroma. A low expression of CD46 was statistically significantly associated with deposition of C3. Comparable expression patterns were shown on primary cervical tumor cell suspensions. A relatively high deposition of C4c was found on these tumor cells, indicating classical pathway activation. Furthermore, it was demonstrated that CD55 and CD59 were the most potent inhibitors of C3 deposition and classical pathway-mediated lysis, respectively, on cervical cancer cell lines. The feasibility of increasing complement activation at the tumor cell membrane surface was demonstrated with an anti-HLA Class I*anti-CD55 bispecific mAb. The potential immunotherapeutic applicability was investigated with both anti-G250*anti-CD55 and anti-Ep-CAM*anti-CD55 bispecific mAbs. An approximate 2-fold increase in C3 deposition, compared with the parental anti-Ep-CAM mAb, was attained with an anti-Ep-CAM*anti-CD55 bispecific mAb when the tumor-associated antigen was expressed in sufficient amounts. These results demonstrate that when tumor-associated antigens are expressed in adequate amounts, bispecific mAbs in vivo may be potent immunotherapeutic agents to enhance an inflammatory reaction at the tumor site.
Similar content being viewed by others


Introduction
Cervical cancer is the second leading cause of cancer death in women worldwide. It has an incidence of 500,000 newly diagnosed cases each year (Boring et al, 1994). The etiology of cervical cancer involves infection with a small epitheliotropic DNA virus, the human papilloma virus (HPV). Infection with HPV and integration of HPV in the genome are regarded as essential steps in the multistep process of cervical carcinogenesis. The E5, E6, and E7 genes encode viral oncogenes that have been shown to possess the capacity to induce cell proliferation, immortalization, and transformation (Herrington, 1994). Viral proteins, presented on the tumor cell surface, might induce an immune response. A cellular immune response directed against viral antigens indeed has often been described (Bontkes et al, 2000; Van der Burg et al, 2001). In addition, the humoral immune system is activated (de Gruijl et al, 1999). Antibodies induced against tumor cell surface proteins or immunotherapeutic antibodies might also be used to eradicate these tumors.
Binding of antibodies to tumor cells may induce complement activation, resulting in the generation of C3a and C5a, the initiation of an inflammatory response, and thus the attraction of leukocytes to the tumor site. Furthermore, complement activation may result in direct lysis of tumor cells through formation of the membrane attack complex (C5b-9) or promote cellular cytotoxicity through opsonization of tumor cells with C3 fragments. The level of complement activation on cell membranes is regulated by the expression of membrane-bound complement regulatory proteins (mCRP), which protect normal and tumor cells from uncontrolled complement-mediated injury (Gorter and Meri, 1999). mCRP comprise complement receptor 1 (CD35), membrane cofactor protein (CD46), decay-accelerating factor (CD55), and homologous restriction factor 20 (CD59) (Liszewski et al, 1996). CD35, CD46, and CD55 inhibit the deposition of C3 fragments on the cell surface and thereby limit complement-dependent cellular cytotoxicity. CD59 prevents the formation of membrane attack complexes and the subsequent osmotic lysis of the target cell. The expression of mCRP has been demonstrated on cervical epithelium, premalignant cervical tissue, and cervical carcinomas (Simpson et al, 1997). Several studies have shown that in situ tumor cells overexpress CD46, CD55, and CD59 (Koretz et al, 1992; Li et al, 2001; Mäenpää et al, 1996; Niehans et al, 1996; Yamakawa et al, 1994). The overexpression of these mCRP on tumor cells may prevent an efficient local immune response.
To investigate the role of mCRP on cervical carcinoma cells, we have determined the expression levels of CD46, CD55, and CD59 on cervical carcinoma cells in situ, the adjacent stroma, freshly isolated cervical tumor cells, and primary epithelial cervical cancer cell lines (CCCL). The relative importance of CD46, CD55, and CD59 on cell lysis and C3 deposition was investigated on CCCL using mCRP blocking mAbs. To determine the feasibility of specifically blocking the most important C3 regulatory protein on cervical tumor cells, bispecific antibodies directed against CD55 and HLA Class I, as a model for a tumor-associated antigen with a high expression level, were used. In addition, to determine the immunotherapeutic applicability, 17-1A/Ep-CAM*anti-CD55 and G250/MN/CAIX*anti-CD55 bispecific mAbs were developed.
Results
Expression of CD46, CD55, and CD59 on Cervical Carcinomas
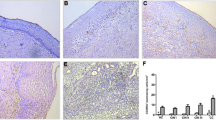
To compare the expression levels of mCRP on cervical carcinoma cells and the surrounding stroma, both the number of cells expressing mCRP and the observed intensity of this expression were scored semiquantitatively, using immunohistochemistry (Table 1). In general, the expression of CD46 was higher on carcinoma cells as compared with stromal cells (Fig. 1a). In contrast, both CD55 and CD59 were expressed in higher amounts on stroma than on tumor cells (Fig. 1, b and c). As reported previously, CD55 was also detected in the extracellular matrix, presumably bound to stromal fibers resulting in a fibrillate pattern (Medof et al, 1987; Sayama et al, 1992). To measure the efficiency of mCRP, the sections were also stained for deposition of C3d and C5b-9. C3d deposition, as well as C5b-9 deposition, was low or absent on tumor cells (Fig. 1, e and f). If present, C3d deposition was associated with a low expression of CD46 on the tumor cells (p = 0.016). C3d was detected in the stroma, but C5b-9 generally was not present. Both C3d and C5b-9 deposits were detected with a high intensity on blood vessels. This absence of C3d and C5b-9 on tumor cells suggests a role for mCRP in the protection of cervical tumor cells from complement.

Expression of membrane-bound complement regulatory proteins (mCRP) and tumor-associated antigens and deposition of complement proteins on cervical carcinoma. Immunohistochemical staining was performed as described in “Materials and Methods.” Nuclei were counterstained with Mayers hematoxylin. a, CD46 (J4-48 mAb); b, CD55 (Bric216 mAb); c, CD59 (Bric229 mAb); d, negative control (omission of the primary mAb); e, C3d; f, C5b-9; g, G250 (G250 mAb); h, Ep-CAM (323/A3 mAb). Magnification ×65.
Tissue sections were also stained for expression of the tumor-associated antigens G250/MN/CAIX (G250) and 17-1A/Ep-CAM (Ep-CAM) (Fig. 1, g and h; Table 1). G250 was absent in 2 of 10 cases or expressed heterogeneously by the tumor cells in the other 8 cases. Ep-CAM was moderately to highly expressed by all tumors. Stromal cells and normal cervical epithelial cells (NCE) were negative for Ep-CAM.
Expression of CD46, CD55, and CD59 on Primary Cervical Tumor Cell Suspensions
To substantiate the in situ immunohistochemical observations, the expression of mCRP, C3, C4, and C5b-9 on four freshly isolated primary cervical tumor cell suspensions was measured by flow cytometry (Fig. 2). Tumor cells were discriminated from stromal or inflammatory cells by selecting cells that were positive for the tumor-associated Ep-CAM antigen using the 323/A3 mAb. CD46 and CD55 were detected in similar amounts on the tumor cells. As observed on the tissue sections, CD59 was highly expressed on the tumor cells. In addition, iC3b, C4c, and to a lesser extent C5b-9 were detected on tumor cell membranes. Deposition of C4c suggests that the complement system had been activated via the classical route of complement activation.

Expression levels of mCRP and amounts of iC3b, C4c, and C5b-9 deposited on three cervical tumor cell suspensions. Expression levels were determined with flow cytometry and are depicted in MESF values (×104). The results of three experiments are shown (mean ± sd).
Expression of mCRP on Cervical Carcinoma Cell Lines
To determine whether the expression levels of mCRP are similar on CCCL compared with freshly isolated cervical tumor cells and NCE, the expression levels of CD46, CD55, and CD59 were quantified by flow cytometry. Three commercially available CCCL and six newly generated CCCL (primary CCCL) were investigated (Table 2). In general, both the expression levels of CD46 and CD55 were higher on the commercial CCCL than on the primary CCCL. The freshly isolated primary cervical tumor cells had a similar expression of CD46 and CD55 as the primary CCCL. The expression of CD59 was comparable for all CCCL. The expression of CD59 on the freshly isolated cervical tumor cells was approximately half the expression level of CD59 on CCCL. To examine whether mCRP were overexpressed on malignant cells, the mCRP expression level of normal NCE was compared with CCCL (Table 2). Interestingly, the expression of CD46 was slightly increased, and especially CD55 showed a 4- to 40-fold increase in expression on CCCL and the freshly isolated cervical tumor cells, when compared with NCE cultures. In contrast, the expression of CD59 was higher on NCE cultures as compared with both CCCL and freshly isolated tumor cells.
Relative Importance of mCRP
Because of the similarities in expression profiles of mCRP on primary CCCL and freshly isolated cervical tumor cells, CCCL were used to unravel the role of mCRP on cervical carcinoma. To study the relative importance of each of the mCRP, two representative CCCL, a commercial CCCL (HeLa) and a primary CCCL (CC8) were selected for further study. These CCCL were selected on the basis of their differences in expression patterns of CD46, CD55, CD59, and HLA Class I. None of the selected CCCL was lysed in the presence of anti-HLA Class I mAb and normal human serum (NHS). In contrast, in the presence of baby rabbit complement (BRC), which is not inhibited by human mCRP, both HeLa and CC8 demonstrated a significant amount of lysis. This illustrates that mCRP acting on homologous complement components restrict complement activation.
To investigate the role of the different mCRP, HeLa (high expression of HLA Class I) and CC8 (low expression of HLA Class I) were treated with different combinations of blocking mAbs (Fig. 3). It is clear that blocking of CD46 has no effect on lysis of HeLa cells. In contrast, blocking of especially CD59 and, to a minor extent, CD55 induces lysis of HeLa. To exclude that the effect of CD46 is masked by the effect of CD59, we also tested combinations of anti-CD55 and anti-CD46 and the combination of anti-CD59 and anti-CD46. In both cases no additional increase in lysis was observed. Only the combination of anti-CD55 and anti-CD59 resulted in an increase in lysis, and the effect was more additive than synergistic. These experiments suggest that on HeLa cells, CD55 and CD59 are indeed involved in the protection of cervical tumor cells from complement-mediated damage. The blocking mAbs themselves did not activate the complement system (Fig. 3). These data were confirmed using phosphatidylinositol-specific phospholipase C (PIPLC)-treated HeLa cells. PIPLC removes the phosphatidyl-inositol-anchored CD55 and CD59 from the cell membrane. Of the PIPLC-treated HeLa cells, 57% were lysed as measured by a 51Cr release assay, whereas only 4% of the untreated cells were killed by complement activation, under comparable conditions (data not shown). To substantiate the role of mCRP on CC8 cells, these cells were also incubated with anti-mCRP mAb. Only anti-CD59 or combinations of anti-mCRP mAb containing anti-CD59 mAb induced lysis of CC8 cells.

Complement-mediated lysis of HeLa and CC8 cells in the presence of anti-mCRP mAb. Experiments were performed as described in “Materials and Methods.” A representative experiment is shown. Amount of lysis is determined as the percentage 51Cr released, after complement activation by anti-HLA Class I mAb (W6/32). Baby rabbit complement (BRC) was used as a positive control for complement activation. Under the x axis, the blocked mCRP are depicted. *Negative control, complement activation in the absence of anti-HLA Class I and in the presence of blocking mAb against all three mCRP.
Subsequently, we have investigated the effect of CD46 and CD55 on C3 deposition. C3 deposition on the cell membrane of HeLa and CC8 cells was measured using flow cytometry after incubation with the anti-HLA Class I mAb W6/32 and NHS (Fig. 4). On HeLa cells the amount of deposited C3 was increased when CD55 was blocked with anti-CD55 mAb, whereas an anti-CD46 mAb had no effect. This suggests that in addition to CD59, CD55 is important to limit classical pathway–mediated complement activation. However, on CC8 cells neither anti-CD46, anti-CD55, nor a combination of both mAbs had any effect on C3 deposition. This supports the previous observation that on CC8 cells the expression of HLA Class I is too low to induce lysis of these cells.

The effect of anti-CD46, anti-CD55, and anti-CD59 on C3c deposition on HeLa and CC8 cells. Complement is activated by anti-HLA Class I mAb (W6/32). Experiments were performed as described in “Materials and Methods.” A representative experiment is shown. C3c deposition is determined with flow cytometry and expressed in MESF values (×104). Under the x axis, the blocked mCRP are depicted.
Effect of Model Bispecific Antibody Containing Anti-CD55 on C3 Deposition and Lysis
To investigate the feasibility of inhibiting CD55, the most efficient in vitro membrane-bound C3 regulatory protein, on the tumor cell surface, bispecific mAbs were generated. HLA Class I was initially used as a model for a tumor-associated antigen because of its high expression levels. The effect of these model bispecific mAbs on C3 deposition was investigated on HeLa cells (Table 3). Anti-HLA Class I*anti-CD55 bispecific mAb resulted in a 6-fold increase in C3c deposition as compared with anti-HLA Class I mAb. The combination of the parental anti-HLA Class I and anti-CD55 (MBC1) mAbs resulted in a plateau value of 286 × 104 molecules of equivalent soluble fluorescence (MESF), which was 3-fold the plateau value reached with anti-HLA Class I mAb alone. Interestingly anti-HLA Class I*anti-CD55 bispecific mAbs were able to increase this latter plateau value with an additional 100% to 567 × 104 MESF.
Effect of Potential Immunotherapeutic Anti-Ep-CAM mAb, Anti-G250 mAb, or Anti-Ep-CAM*Anti-CD55 and Anti-G250*Anti-CD55 Bispecific mAbs on C3 Deposition and Tumor Cell Lysis
The Ep-CAM antigen and the G250 antigen (recently detected on cervical carcinoma cells) may be candidate antigens for adjuvant mAb immunotherapy for cervical carcinoma. Tumor cells in situ indeed showed a high expression of these antigens (Fig. 1, g and h). Therefore, their expression levels on the CCCL and NCE were measured. Ep-CAM and G250 were expressed in low amounts on both CCCL and NCE cells (Table 2). Neither anti-Ep-CAM mAb, anti-G250 mAb, nor bispecific mAbs (anti-Ep-CAM*anti-CD55 or anti-G250*anti-CD55) induced C3 deposition or lysis (not shown). This might be a result of the low expression of both Ep-CAM and G250 in vitro (Table 2). To test this hypothesis, CaSki cells, which endogenously express a low amount of Ep-CAM, were transiently transfected with Ep-CAM. The transfection resulted in a doubled expression level of this antigen on the CaSki cell membrane surface (MESF value 183 × 104 compared with MESF value 92 × 104 of the untransfected cells). Incubation of the transfected cells with the anti-Ep-CAM*anti-CD55 bispecific mAb resulted in a clear increase in C3 deposition of MESF 44 × 104, as compared with MESF 27×104 in the presence of the anti-Ep-CAM mAb alone (Figs. 5 and 6). In addition, in the presence of bispecific mAb, complement-mediated lysis was also increased (13% versus 7%). Addition of anti-CD59 mAb was necessary to induce up to 80% of lysis (not shown). In that case no difference was seen between anti-Ep-CAM mAb and bispecific mAb.

C3 deposition caused by anti-Ep-CAM*anti-CD55 (323A3*MBC1) on Ep-CAM transfected CaSki cells. Experiments were performed as described in “Materials and Methods.” C3c deposition is determined with flow cytometry and expressed in MESF values (×104). Under the x axis, the mAb or bispecific mAb used is depicted.

Complement-mediated lysis caused by anti-Ep-CAM*anti-CD55 (323A3*MBC1) of Ep-CAM–transfected CaSki cells. Experiments were performed as described in “Materials and Methods.” Amount of lysis is determined as the percentage 51Cr released, after complement activation by the mAb or bispecific mAb that is depicted under the x axis. # BRC is added as a positive control for complement activation.
Discussion
The viral etiology of cervical carcinoma makes this tumor potentially suitable for immunotherapeutic treatment. Both the humoral and the cellular arm of the immune system are involved in the defense against cervical tumor cells (Bontkes et al, 2000; de Gruijl et al, 1999). Unfortunately this is apparently not sufficient to eliminate the tumor cells. The clinical importance of antibodies directed against HPV or tumor-associated antigens expressed on cervical tumor cells is limited and thus far attempts to improve the treatment of cervical carcinoma have been directed at enhancing the cellular immune response (Liu et al, 2000; Thornburg et al, 2000). In the present study, we have attempted to determine the role of CD46, CD55, and CD59 on cervical tumor cells in the protection against mAb-induced complement-mediated injury. In addition, we have investigated whether mAb immunotherapy for cervical carcinoma with the potentially immunotherapeutic anti-G250 mAb (Uemura et al, 1999), anti-Ep-CAM mAb (Riethmuller et al, 1998), or bispecific mAbs directed against G250 or Ep-CAM and CD55 is feasible.
To allow immune surveillance, complement undergoes a continuous low-level activation. A stringent regulation is therefore necessary to prevent nonspecific damage to host tissues. As on normal cells, the level of complement activation on the tumor cell surface is regulated by mCRP. The expression of these regulators is increased in several carcinoma types (Koretz et al, 1992; Kumar et al, 1993; Li et al, 2001; Niehans et al, 1996; Seya et al, 1990). The increased expression may limit the extent of complement-mediated injury. This latter observation may also explain the limited success of this type of immunotherapy (Gorter and Meri, 1999). Previously, expression of mCRP has been demonstrated on cervical epithelium, premalignant cervical tissue, and cervical carcinomas (Oglesby et al, 1996; Simpson et al, 1997). We have also observed mCRP expression in situ on cervical cancer cells. Knowledge of the in vivo role of mCRP can be obtained by assessing the association between mCRP expression levels on tumor cells and deposition of complement fragments on their cell surface, as has been done before for renal carcinoma, chronic lymphocytic leukemia, and thyroid cancer cells (Magyarlaki et al, 1996; Marquart et al, 1995; Yamakawa et al, 1994). However, it is unclear which mCRP is important to protect human tumor cells in vivo from complement-mediated damage. in situ, where a low expression of CD55 is usually observed on the tumor cells, CD46 may play a prominent role. This finding is supported by the observation that for renal tissue a correlation is found between the presence of C3 and low expression of CD46 (Blok et al, 2000). In the present study, we also observed an association between C3 deposition and a relatively low expression of CD46. These observations are compatible with the observed low expression of CD55 in situ and the inhibitory effect of anti-CD46 mAb on PC7 and PC9 lung adenocarcinoma cell lines (Azuma et al, 1995) and support the proposed view that only a low amount of CD55 is necessary to make C3 available for cleavage by CD46 (Brodbeck et al, 2000).
To substantiate these findings, we have quantified the mCRP expression on freshly isolated cervical tumor cells in situ, primary CCCL and normal NCE cells. As reported for breast, renal, and lung carcinomas, a low expression of CD55, a somewhat higher expression of CD46, and a high expression of CD59 were observed (Niehans et al, 1996). With regard to the role of CD46, in agreement with the majority of other in vitro studies, under these experimental conditions we could not find support for the proposed role of CD46 in vivo (Hakulinen and Meri, 1994; Juhl et al, 1997; Yamakawa et al, 1994; Yu et al, 1999). Our data also demonstrate that CD55 and CD59 are the most important regulators in vitro in regulating complement-mediated injury on cervical cancer cells.
CD55 may play a dual role in vivo. Overexpression of CD55 may increase the protection of tumor cells against complement-mediated injury. On the other hand, overexpression of CD55 may result in the presentation of CD55 peptides by major histocompatibility complex proteins. These MHC-peptide complexes have been shown to initiate a cellular immune response (Durrant et al, 2000) and may promote the elimination of tumor cells by cytotoxic T cells. In addition, highly expressed CD55 can act as a tumor antigen (Durrant et al 2000; Hensel et al 2001; Maxwell-Armstrong et al 2001) and can be exploited as a target for (bispecific) mAb immunotherapy. Humoral immunotherapy with bispecific mAb might, by inhibiting the function of CD55 (Blok et al, 1998), increase a local inflammatory reaction.
Adjuvant mAb or bispecific mAb immunotherapy may be used to stimulate a local inflammatory reaction (generation of C5a and C3b deposition), resulting in eradication of the tumor cells (Gorter and Meri, 1999; Riethmuller et al, 1998). The presence of bound mAb and C3 to tumor cells may enhance antibody-dependent cellular cytotoxicity and complement-dependent cellular cytotoxicity by FcYR or CR-positive leukocytes, leading to an increased antigen presentation and a potentially more efficient T-cell response to the viral antigens presented by the tumor cells. Importantly, it may render HLA Class I–negative tumor cells accessible to antibody-dependent cellular cytotoxicity and complement-dependent cellular cytotoxicity.
We have investigated the potential therapeutic effect of mAb and bispecific mAb directed against the G250 antigen and the Ep-CAM antigen on CCCL. High expression of Ep-CAM was observed in all cervical tumors and of G250 in some tumors (Litvinov et al, 1996). In contrast, low expression of these antigens was measured on the CCCL. Because of this low antigen expression and the presence of mCRP, no complement activation was observed on CCCL by either complement-activating anti-G250 or anti-Ep-CAM mAbs.
Because complement activation may be limited by the presence of mCRP at the tumor cell membrane (Gorter et al, 1996; Gorter and Meri, 1999), we have investigated the effect of bispecific mAb containing anti-CD55. To demonstrate the effectiveness of these types of bispecific mAbs to increase C3 deposition, we initially used anti-HLA Class I (a highly expressed antigen)*anti-CD55 bispecific mAb. Indeed in the presence of these bispecific mAbs, increased C3 deposition at the tumor cell surface was observed. In addition, the immunotherapeutic applicability of anti-G250*anti-CD55 and anti-Ep-CAM*anti-CD55 bispecific mAbs was investigated. In the presence of the latter bispecific mAb, initially no complement activation occurred on the CCCL. When the Ep-CAM expression on cervical cancer cells was increased by transfection with 17-1A/Ep-CAM, incubation with anti-Ep-CAM*anti-CD55 bispecific mAb resulted in both increased C3 deposition and lysis. Also, the potential applicability of this type of bispecific mAb has been demonstrated for other tumor types, renal cancer cells, and leukemic cells (Blok et al, 1998; Harris et al, 1997). Its use in cancer patients awaits proper animal models to test the feasibility of this approach in vivo.
Materials and Methods
Cell Lines
CCCL used were as follows: SiHa, HeLa, CaSki (American Type Tissue Collection, Rockville, Maryland) and CSCC-1, CSCC-7, CC-8, CC-10A, CC-10B, and CC-11+, generated from five primary squamous and adenosquamous tumor specimens obtained from hysterectomized FIGO Stage IB/IIB cervical carcinoma patients as previously described (Koopman et al, 1999). The normal primary cervical epithelial cell cultures NCE1 and NCE2 were derived from patients undergoing hysterectomy for nononcologic reasons. NCE were tested for HPV infection and were demonstrated not to be infected.
The cervical carcinoma cell lines were grown in serum-free keratinocyte medium (GIBCO BRL, Grand Island, New York) supplemented with 5 μg/L recombinant human epidermal growth factor and 50 μg/L bovine pituitary extract (GIBCO BRL). SiHa, HeLa, and CaSki were cultured in DMEM (Life Technologies, Rockville, Maryland) containing 22.5 mm HEPES supplemented with 10% heat-inactivated FCS, 2 mm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin (complete medium). Cell lines were passaged, once or twice a week, or used for experiments when 90% confluency was reached, using 0.25% trypsin (Flow Laboratories, Irving, United Kingdom) and 0.1% EDTA (Merck, Darmstadt, FRG) in calcium and magnesium-free HBSS (pharmacy of the Leiden University Medical Center, Leiden, The Netherlands).
Tissue Specimens and Processing
Human cervical tumor cell suspensions were acquired from three patients who underwent radical hysterectomy because of cervical carcinoma. Tumor specimens were obtained immediately after surgery. Tumor specimens were cut into 1-mm3 pieces and washed twice in DMEM. Subsequently, per 1 g of tissue, 5 ml of DMEM medium containing 4 mg/ml collagenase Ia (Sigma Chemicals, St. Louis, Missouri) and 0.002% DNAse (Sigma) was added and the tissue was incubated for 1 hour at 37° C. The acquired cell suspensions were filtered over a 100-μm nylon gauze (Verseidag-Industrietextilien GmbH, Kempen, Germany) and washed twice with DMEM complete medium. Erythrocytes were removed from the cell suspension by treatment (5 minutes, 4° C) with a solution of 0.16 m NH4Cl, 10 mm KHCO3, and 0.13 mm EDTA. Cell suspensions were stored at −70° C in 10% dimethyl sulfoxide in heat-inactivated fetal bovine serum until use. Both tumor and normal tissue were snap-frozen in liquid isopentane on dry ice and stored at −70° C until use.
Antibodies
The following antibodies were used: J4-48 (IgG1; CLB, Amsterdam, The Netherlands) and GB24 (Hsi et al, 1988) (IgG1; a gift from Prof. Dr. Atkinson, St. Louis, Missouri) directed against CD46; MBC1 (IgG1; a gift from Prof. Dr. B. P. Morgan, Cardiff, United Kingdom) and BRIC216 (IgG1; International Blood Group Reference Laboratories [IBGRL], Bristol, United Kingdom) directed against CD55; BRIC229 (IgG2b; IBGRL) and MEM43 (IgG2a; Sanbio, Uden, The Netherlands) directed against CD59; G250 (Oosterwijk et al, 1986) (IgG2a) directed against the G250 antigen; 323/A3 (Edwards et al, 1986) (IgG2a) directed against the Ep-CAM antigen; W6/32 (IgG2a, anti-HLA ABC) obtained from the American Type Culture Collec-tion; G250*MBC1, 323/A3*MBC1, and W6/32*MBC1 bispecific mAbs (all IgG2a*IgG1), developed in our own department (Blok et al, 1998); anti-human C3d and iC3b (both IgG1) (Quidel Corporation, San Diego, California); aE11 (IgG2a), directed against human C5b-9 (DAKO A/S, Glostrup, Denmark); FITC-labeled goat F(ab)2 anti-mouse IgG/M (DAKO); FITC-labeled goat F(ab)2 anti-mouse IgG1, FITC-labeled goat F(ab)2 anti-mouse IgG2b, and PE-labeled goat F(ab)2 anti-mouse IgG2a (Biotechnology Associates, Birmingham, Alabama); and horseradish peroxidase (HRP) rabbit-anti-mouse Ig EnVision+ kit (DAKO).
Complement Source
NHS was prepared from freshly collected human AB serum, aliquoted, and stored at −70° C. Rabbit serum (BRC) was purchased from Pel-Freeze (Brown Deere, Wisconsin) and stored at −70° C until use.
Generation of Quadroma Cells
Quadroma cells were generated as described by van Dijk et al (1989). Briefly, the MBC1 hybridoma was incubated with increasing amounts (10−7 m to 10−4 m) of 8-azaguanine (GIBCO BRL) to induce hypoxanthine-guanine phosphoribosyl transferase deficiency. The 323/A3 hybridoma was incubated with increasing amounts (10−7 m to 10−4 m) of 5-bromodeoxyuridine (Sigma) to induce thymidine kinase deficiency. Quadroma cells were generated by adding PEG-1500 to equal numbers (5 × 106) of either hypoxanthine-guanine phosphoribosyl transferase–deficient MBC1 cells or thymidine kinase–deficient 323/A3/G250/W6/32 cells. The generated quadroma cells were cultured in medium, containing 0.1 mm hypoxanthine, 7.7 μm aminopterin, and 16 μm thymidine (HAT medium). Cell cultures were cloned in 96-well microtiter plates (Greiner B.V., Alphen aan de Rijn, The Netherlands), with each well statistically containing 0.3 cells. Quadroma clones producing bi-isotypic mAbs with functional binding sites were subcloned. Quadroma clones were selected for further study, and bi-isotypic mAbs were purified from the supernatant, using affinity chromatography on a protein A column. The percentage of bispecific mAbs present in the bi-isotypic fraction was determined by HPLC on a Mono S column (Blok et al, 1998). The bi-isotypic fractions consisted of 15% to 25% of bispecific mAb.
Isotype-Specific ELISA
Microtiter plates (96 well) were coated for 2 hours at 37° C with rat anti-mouse IgG1 (Sanbio) at a concentration of 2.5 μg/ml in 0.1 m NaHCO3, pH 9.6. Serial dilutions of culture medium from quadroma clones, in phosphate-buffered saline containing 0.05% Tween- 20 (PBS/T) and 1% BSA were incubated in the coated microtiter plates for 1 hour at room temperature. Bi-isotypic mAb was detected using goat anti-mouse IgG2a conjugated with HRP (Southern Biotechnology Associates, Inc., Birmingham, Alabama). Cross-reactivity with coated rat Ig was blocked with 10% rat serum in PBS/T. The assay was developed with 1% 3,5,3′,5′-tetramethyl-benzidine in 0.11 m sodium acetate (pH 5.5, 100 μg/ml) containing 0.1% freshly added H2O2. The concentration of monospecific IgG1 (parental mAb) was measured with a goat anti-mouse–HRP kappa chain conjugation (Sanbio). To measure the concentration of monospecific IgG2a (parental mAb), the procedure was repeated with a goat anti-mouse–IgG2a coating (Southern Biotechnology Associates, Inc.). For determination of the mAb concentration, IgG1- and IgG2a standards (obtained from Centocor, Leiden, The Netherlands) were used.
Treatment of Tissue Specimens
Immunohistochemistry.
Cryostat sections of 5 μm were dried (1 hour, 50° C) and subsequently placed overnight in a 60° C oven. Sections were fixed in acetone (10 minutes). Sections were washed twice with PBS (5 minutes) and then incubated with predetermined concentrations of primary antibody diluted in PBS containing 1% BSA (PBS/1% BSA) (1 hour). Sections were washed three times for 5 minutes with PBS. Subsequently sections were incubated with HRP and rabbit anti-mouse Ig both coupled to the DAKO EnVision+ system (30 minutes). After three washes with PBS, sections were washed once with 0.05 m Tris-HCl, pH 7.6. Sections were developed with diamino-benzidine-tetrahydrochloride in Tris-HCl (10 minutes). The reaction was stopped with PBS. Sections were counterstained with Mayer’s hematoxylin. The intensity of the staining was scored as negative (0), weak (1) moderate (2), or strong (3). The percentage positive cells was scored as sporadic (1; 1% to 5%), local (2; 6% to 25%), occasional (3; 26% to 50%), majority (4; 51% to 75%), or large majority (5; 76% to 100%), according to the method described by Ruiter et al (1998). Sections in which the primary mAb was omitted were included in all experiments as negative controls. As a positive control for the presence of C3d and C5b-9 mAbs, renal tissue from a patient with glomerulonephritis was used.
Flow Cytometry.
Cells (2.5 × 105) were incubated with a 100-μl mixture of primary antibodies diluted in PBS/0.5% BSA (30 minutes, 4° C). Cells were washed twice with PBS/0.5% BSA and incubated with 100 μl of a mixture of secondary antibodies (30 minutes, 4° C). After washing, the cells were resuspended in 250 μl of PBS/0.5% BSA containing 1 μg/ml propidium iodide (PI) to stain dead cells. Specimens were measured with the FACSCalibur (Becton Dickinson, San Jose, California). Ten thousand events of positive tumor cells were counted. FITC-positive cells were measured on FL1: BP530/30 nm (green fluorescence). PE-positive cells were measured on FL2: BP585/42 nm (orange fluorescence). PI-positive (dead) cells were measured on FL3: LP 650 nm (red fluorescence). Fluorescence compensation was used to correct for spectral cross talk between the fluorescent signals. Data are expressed in MESF values. MESF values were calculated on the basis of a flow cytometry standardization kit (Quantum 25 FITC; Flow Cytometry Standards Europe, Leiden, The Netherlands). Tumor cells were distinguished from inflammatory and stromal cells by selecting only cells positive for expression of Ep-CAM. The fraction of PI-negative cells in the cell suspension consisted of Ep-CAM–positive tumor cells (10% to 25%) and leukocytes (approximately 50%), and the remaining cell population (25% to 40%) contained most probably stromal cells (fibroblasts) and endothelial cells. Differences in subclass of the CD59 mAb did not influence the mean fluorescence intensity, because both MEM43 (IgG2a) and BRIC229 (IgG2b) gave similar results (not shown).
C3c Deposition
C3c deposition was assessed by flow cytometric analysis as described above. After incubation with primary mAb (30 minutes, 4° C), cells were incubated with 10% NHS diluted in DMEM containing 1 mm Ca2+ and 0.5 mm Mg2+ (20 minutes, 37° C). C3c deposition was detected using GaH-C3c-FITC (30 minutes, 4o). Ten thousand living cells were counted. C3 deposition was expressed in MESF values.
PIPLC Treatment
Tumor cells were suspended in 50% DMEM/50% RPMI 1640 containing 1% BSA (4 × 106/ml). PIPLC (Bacillus Thurigiensis; ICN Biomedicals B.V., Zoetermeer, The Netherlands) was added (final concentration 100 mU/ml). The cells were incubated for 1 hour at 37° C, washed twice with 50% DMEM/50% RPMI 1640 containing 1% BSA, and either used in 51Cr release assays or flow cytometry experiments.
51Cr Release Assay
The total reaction volume of the 51Cr release assay was 250 μl, and all dilutions were performed in DMEM/RPMI/1% BSA medium. Fifty microliters of 51Cr-labeled target cells (2000 cells/well) were mixed with 100 μl of mAb in round-bottomed microtiter plates and incubated for 30 minutes at 37° C. As a homologous complement source, 100 μl of NHS was added (final concentration of 10%). As a control for complement activation, BRC (final concentration of 2.5%) was used as a heterologous complement source. Wells with NHS were incubated for 4 hours and wells with BRC were incubated for 2 hours at 37° C, after which 100 μl of supernatant was counted for 51Cr release. Maximal release was defined as the release obtained by the addition of 10% (v/v) Triton X-100 to the target cells. Spontaneous release was obtained by incubating the target cells with medium. The percentage of specific release was calculated as 100 × (counts experimental release − spontaneous release)/(maximal release − spontaneous release). All tests were performed in triplicate.
Transient Transfection
CaSki cells were grown in 10-cm Petri dishes until 30% confluency. DMEM (500 μl) without FCS, containing 30 μl of fugene-6 (Roche, Almere, The Netherlands) and 10 μl of pCep4 vector (Invitrogen BV, Leek, The Netherlands) containing Ep-CAM cDNA diluted in glycerol, was added and incubated at 37° C. After 4 days, again 500 μl DMEM without FCS, containing 30 μl of fugene-6 and 10 μl of vector, was added and incubated for another day. Then cells were harvested and used for flow cytometry and 51Cr release assays as described above.
Statistical Analysis
Associations between mCRP expression and complement deposition were evaluated by Pearson’s χ2 test. A p value less than 0.05 was considered statistically significant.
References
Azuma A, Yamano Y, Yoshimura A, Hibino T, Nishida T, Yagita H, Okumura K, Seya T, Kannagi R, Shibuya M, and Kudoh S (1995). Augmented lung adenocarcinoma cytotoxicity by the combination of a genetically modified anti-Lewis Y antibody and antibodies to complement regulatory proteins. Scand J Immunol 42: 202–208.
Blok VT, Daha MR, Tijsma O, Harris CL, Morgan BP, Fleuren GJ, and Gorter A (1998). Bispecific monoclonal antibody directed against both the membrane-bound complement regulator CD55 and the renal tumor-associated antigen G250 enhances C3 deposition and tumor cell lysis by complement. J Immunol 160: 3437–3443.
Blok VT, Daha MR, Tijsma OMH, Weissglas MG, Van den Broek LJCM, and Gorter A (2000). A possible role of CD46 for the protection in vivo of human renal tumor cells from complement-mediated damage. Lab Invest 80: 335–344.
Bontkes HJ, de Gruijl TD, van den Muysenberg AJ, Verheijen RH, Stukart MJ, Meijer CJ, Scheper RJ, Stacey SN, Duggan Keen MF, Stern PL, Man S, Borysiewicz LK, and Walboomers JM (2000). Human papillomavirus type 16 E6/E7-specific cytotoxic T lymphocytes in women with cervical neoplasia. Int J Cancer 88: 92–98.
Boring CC, Squires TS, Tong T, and Montgomery S (1994). Cancer statistics, 1994. CA Cancer J Clin 44: 7–26.
Brodbeck WG, Mold C, Atkinson JP, and Medof ME (2000). Cooperation between decay-accelerating factor and membrane cofactor protein in protecting cells from autologous complement attack. J Immunol 165: 3999–4006.
de Gruijl TD, Bontkes HJ, Walboomers JM, Coursaget P, Stukart MJ, Dupuy C, Kueter E, Verheijen RH, Helmerhorst TJ, Duggan-Keen MF, Stern PL, Meijer CJ, and Scheper RJ (1999). Immune responses against human papillomavirus (HPV) type 16 virus-like particles in a cohort study of women with cervical intraepithelial neoplasia. I. Differential T-helper and IgG responses in relation to HPV infection and disease outcome. J Gen Virol 80 (Pt 2):399–408.
Durrant LG, Maxwell Armstrong C, Buckley D, Amin S, Robins RA, Carmichael J, and Scholefield JH (2000). A neoadjuvant clinical trial in colorectal cancer patients of the human anti-idiotypic antibody 105AD7, which mimics CD55. Clin Cancer Res 6: 422–430.
Edwards DP, Grzyb KT, Dressler LG, Mansel RE, Zava DT, Sledge GW Jr, and McGuire WL (1986). Monoclonal antibody identification and characterization of a Mr 43,000 membrane glycoprotein associated with human breast cancer. Cancer Res 46: 1306–1317.
Gorter A, Blok VT, Haasnoot WHB, Ensink NG, Daha MR, and Fleuren GJ (1996). Expression of CD46, CD55, and CD59 on renal tumor cell lines and their role in preventing complemented-mediated tumor cell lysis. Lab Invest 74: 1039–1049.
Gorter A and Meri S (1999). Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol Today 20: 576–582.
Hakulinen J and Meri S (1994). Expression and function of the complement membrane attack complex inhibitor protectin (CD59) on human breast cancer cells. Lab Invest 71: 820–827.
Harris CL, Kan KS, Stevenson GT, and Morgan BP (1997). Tumour cell killing using chemically engineered antibody constructs specific for tumour cells and the complement inhibitor CD59. Clin Exp Immunol 107: 364–371.
Hensel F, Hermann R, Brandlein S, Krenn V, Schmausser B, Geis S, Muller-Hermelink HK, and Vollmers HP (2001). Regulation of the new coexpressed CD55 (decay-accelerating factor) receptor on stomach carcinoma cells involved in antibody SC-1-induced apoptosis. Lab Invest 81: 1553–1563.
Herrington CS (1994). Human papillomaviruses and cervical neoplasia. I. Classification, virology, pathology, and epidemiology. J Clin Pathol 47: 1066–1072.
Hsi BL, Yeh C, Fenichel P, Samson M, and Grivaux C (1988). Monoclonal antibody GB24 recognizes a trophoblast-lymphocyte cross-reactive antigen. Am J Reprod Immunol Microbiol 18: 21–27.
Juhl H, Helmig F, Baltzer K, Kalthoff H, Henne Bruns D, and Kremer B (1997). Frequent expression of complement resistance factors CD46, CD55, and CD59 on gastrointestinal cancer cells limits the therapeutic potential of monoclonal antibody 17-1A. J Surg Oncol 64: 222–230.
Koopman LA, Szuhai K, van Eendenburg JD, Bezrookove V, Kenter GG, Schuuring E, Tanke H, and Fleuren GJ (1999). Recurrent integration of human papillomaviruses 16, 45, and 67 near translocation breakpoints in new cervical cancer cell lines. Cancer Res 59: 5615–5624.
Koretz K, Brüderlein S, Henne C, and Möller P (1992). Decay-accelerating factor (DAF, CD55) in normal colorectal mucosa, adenomas and carcinomas. Br J Cancer 66: 810–814.
Kumar S, Vinci JM, Pytel BA, and Baglioni C (1993). Expression of messenger RNAs for complement inhibitors in human tissues and tumors. Cancer Res 53: 348–353.
Li L, Spendlove I, Morgan J, and Durrant LG (2001). CD55 is over-expressed in the tumor environment. Br J Cancer 84: 80–86.
Liszewski MK, Farries TC, Lublin DM, Rooney IA, and Atkinson JP (1996). Control of the complement system. Adv Immunol 61: 201–283.
Litvinov SV, van Driel W, Van Rhijn CM, Bakker HA, van Krieken H, Fleuren GJ, and Warnaar SO (1996). Expression of Ep-CAM in cervical squamous epithelia correlates with an increased proliferation and the disappearance of markers for terminal differentiation. Am J Pathol 148: 865–875.
Liu DW, Tsao YP, Hsieh CH, Hsieh JT, Kung JT, Chiang CL, Huang SJ, and Chen SL (2000). Induction of CD8 T cells by vaccination with recombinant adenovirus expressing human papillomavirus type 16 E5 gene reduces tumor growth. J Virol 74: 9083–9089.
Magyarlaki T, Mosolits S, Baranyay F, and Buzogany I (1996). Immunohistochemistry of complement response on human renal cell carcinoma biopsies. Tumori 82: 473–479.
Marquart HV, Gronbaek K, Christensen BE, Svehag SE, and Leslie RG (1995). Complement activation by malignant B cells from patients with chronic lymphocytic leukaemia (CLL). Clin Exp Immunol 102: 575–581.
Mäenpää A, Junnikkala S, Hakulinen J, Timonen T, and Meri S (1996). Expression of complement membrane regulators membrane cofactor protein (CD46), decay accelerating factor (CD55), and protectin (CD59) in human malignant gliomas. Am J Pathol 148: 1139–1152.
Maxwell-Armstrong CA, Durrant LG, Buckley TJ, Scholefield JH, Robins RA, Fielding K, Monson JR, Guillou P, Calvert H, Carmichael J, and Hardcastle JD (2001). Randomized double-blind phase II survival study comparing immunization with the anti-idiotypic monoclonal antibody 105AD7 against placebo in advanced colorectal cancer. Br J Cancer 84: 1443–1446.
Medof ME, Walter EI, Rutgers JL, Knowles DM, and Nussenzweig V (1987). Identification of the complement decay-accelerating factor (DAF) on epithelium and glandular cells and in body fluids. J Exp Med 165: 848–864.
Niehans GA, Cherwitz DL, Staley NA, Knapp DJ, and Dalmasso AP (1996). Human carcinomas variably express the complement inhibitory proteins CD46 (membrane cofactor protein), CD55 (decay-accelerating factor), and CD59 (protectin). Am J Pathol 149: 129–142.
Oglesby TJ, Longwith JE, and Huettner PC (1996). Human complement regulator expression by the normal female reproductive tract. Anat Rec 246: 78–86.
Oosterwijk E, Ruiter DJ, Hoedemaeker PHJ, Pauwels EKJ, Jonas U, Zwartendijk J, and Warnaar SO (1986). Monoclonal antibody G250 recognizes a determinant present in renal cell carcinoma and absent from normal kidney. Int J Cancer 38: 489–494.
Riethmuller G, Holz E, Schlimok G, Schmiegel W, Raab R, Hoffken K, Gruber R, Funke I, Pichlmaier H, Hirche H, Buggisch P, Witte J, and Pichlmayr R (1998). Monoclonal antibody therapy for resected Dukes’ C colorectal cancer: Seven-year outcome of a multicenter randomized trial. J Clin Oncol 16: 1788–1794.
Ruiter DJ, Ferrier CM, Van Muijen GNP, Henzen-Logmans SC, Kennedy S, Kramer MD, Nielsen BS, and Schmitt M (1998). Quality control of immunohistochemical evaluation of tumour-associated plasminogen activators and related components. European BIOMED-1 Concerted Action on Clinical Relevance of Proteases in Tumour Invasion and Metastasis. Eur J Cancer 34: 1334–1340.
Sayama K, Shiraishi S, and Miki Y (1992). Distribution of complement regulators (CD46, CD55 and CD59) in skin appendages, and in benign and malignant skin neoplasms. Br J Dermatol 127: 1–4.
Seya T, Matsumoto M, and Akedo H (1990). Quantitative analysis of membrane cofactor protein (MCP) of complement: High expression of MCP on human leukemia cell lines, which is down-regulated during cell differentiation. J Immunol 145: 238–245.
Simpson KL, Jones A, Norman S, and Holmes CH (1997). Expression of the complement regulatory proteins decay accelerating factor (DAF, CD55), membrane cofactor protein (MCP, CD46) and CD59 in the normal human uterine cervix and in premalignant and malignant cervical disease. Am J Pathol 151: 1455–1467.
Thornburg C, Boczkowski D, Gilboa E, and Nair SK (2000). Induction of cytotoxic T lymphocytes with dendritic cells transfected with human papillomavirus E6 and E7 RNA: Implications for cervical cancer immunotherapy. J Immunother 23: 412–418.
Uemura H, Nakagawa Y, Yoshida K, Saga S, Yoshikawa K, Hirao Y, and Oosterwijk E (1999). MN/CA IX/G250 as a potential target for immunotherapy of renal cell carcinomas. Br J Cancer 4: 741–746.
Van der Burg SH, Ressing ME, Kwappenberg KM, De Jong A, Straathof K, De Jong J, Geluk A, van Meijgaarden KE, Franken KL, Ottenhoff TH, Fleuren GJ, Kenter G, Melief CJ, and Offringa R (2001). Natural T-helper immunity against human papillomavirus type 16 (HPV16) E7-derived peptide epitopes in patients with HPV16-positive cervical lesions: Identification of 3 human leukocyte antigen class II-restricted epitopes. Int J Cancer 91: 612–618.
Van Dijk J, Warnaar SO, Van Eendenburg JDH, Thienpont M, Braakman E, Boot JHA, Fleuren GJ, and Bolhuis RLH (1989). Induction of tumor cell lysis by bispecific monoclonal antibodies recognizing renal cell carcinoma and CD3 antigen. Int J Cancer 43: 344–349.
Yamakawa M, Yamada K, Tsuge T, Ohrui H, Ogata T, Dobashi M, and Imai Y (1994). Protection of thyroid cancer cells by complement-regulatory factors. Cancer 73: 2808–2817.
Yu J, Caragine T, Chen S, Morgan BP, Frey AB, and Tomlinson S (1999). Protection of human breast cancer cells from complement-mediated lysis by expression of heterologous CD59. Clin Exp Immunol 115: 13–18.
Acknowledgements
We thank Mr. J. D. H. van Eendenburg for generating the bispecific mAb.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gelderman, K., Blok, V., Fleuren, G. et al. The Inhibitory Effect of CD46, CD55, and CD59 on Complement Activation After Immunotherapeutic Treatment of Cervical Carcinoma Cells with Monoclonal Antibodies or Bispecific Monoclonal Antibodies. Lab Invest 82, 483–493 (2002). https://doi.org/10.1038/labinvest.3780441
Received:
Published:
Issue date:
DOI: https://doi.org/10.1038/labinvest.3780441
This article is cited by
-
Unlocking the Power of Complement-Dependent Cytotoxicity: Engineering Strategies for the Development of Potent Therapeutic Antibodies for Cancer Treatments
BioDrugs (2023)
-
Bispecific antibodies targeting tumor-associated antigens and neutralizing complement regulators increase the efficacy of antibody-based immunotherapy in mice
Leukemia (2015)
-
Role of C5b-9 complement complex and response gene to complement-32 (RGC-32) in cancer
Immunologic Research (2013)
-
HER2 Expression Beyond Breast Cancer: Therapeutic Implications for Gynecologic Malignancies
Molecular Diagnosis & Therapy (2013)
-
Complement in Cancer and Cancer Immunotherapy
Archivum Immunologiae et Therapiae Experimentalis (2011)
