
Abstract
Patients with germline mutation of succinate dehydrogenase (SDH) subunit genes are prone to develop paraganglioma, gastrointestinal stromal tumor, and rarely renal cell carcinoma (RCC). However, SDH-deficient RCC is not yet widely recognized. We identified such tumors by distinctive morphology and confirmed absence of immunohistochemical staining for SDHB. Immunohistochemical features were evaluated using a panel of antibodies to renal tumor antigens. Targeted next-generation sequencing was performed on DNA extracted from paraffin-embedded tissue. Eleven tumors were identified from 10 patients, 22–72 years of age (median 40). Two patients had paragangliomas, 1 bilateral SDH-deficient RCC, and 1 contralateral oncocytoma. Grossly, tumors were tan or red–brown, 2–20 cm in diameter (median 4.25 cm). Fuhrman grade was 2 (n=10) or 3 (n=1). Stage was pT1a–pT2b. One patient developed widespread metastases 16 years after nephrectomy and died of disease 6 years later. All tumors were composed of uniform eosinophilic cells containing vacuoles or flocculent cytoplasmic inclusions. Architecture was primarily solid; entrapped renal tubules and intratumoral mast cells were common. By immunohistochemistry, tumor cells were negative for SDHB (11/11) and rarely SDHA (1/11). Labeling was uniformly positive for PAX8 and kidney-specific cadherin and absent for KIT, RCC, and carbonic anhydrase IX. Staining for broad-spectrum epithelial markers was often negative or focal (positive staining for AE1/AE3 in 4/10, CAM5.2 3/7, CK7 1/11, EMA 10/10). By sequencing, SDHB mutation and loss of the second allele were present in 5/6 tumors; the SDHA-deficient tumor showed no SDHB abnormality. SDH-deficient RCC is a unique neoplasm that is capable of progression, often harboring SDHB mutation. A monomorphic oncocytic renal tumor with solid architecture, cytoplasmic inclusions of flocculent material, and intratumoral mast cells should prompt evaluation of SDH status, as it may have implications for screening the patient and relatives. Negative immunohistochemistry for KIT and heterogeneous labeling for epithelial antigens are other supportive features.
Similar content being viewed by others
Main
Succinate dehydrogenase (SDH) is a mitochondrial enzyme complex composed of four protein subunits (SDHA, SDHB, SDHC, and SDHD) that functions as a member of the Krebs cycle and electron transport chain. Germline mutations of the genes that encode the SDH subunits result in hereditary paraganglioma-pheochromocytoma syndromes.1, 2 Patients with such mutations also develop gastrointestinal stromal tumors (GISTs) that can be recognized by their distinctive multinodular architecture, predominantly epithelioid morphology, and predilection for lymph node metastasis.1, 2, 3
In addition to paraganglioma and GIST, there is increasing evidence that patients with germline mutation of SDH subunit genes also develop renal tumors,4 which, similar to paragangliomas and GISTs in such patients, lack immunohistochemical labeling for SDHB in the neoplastic cells.5, 6 Many of the renal tumors that have been recognized to date in these patients exhibit distinctive morphology, characterized by sheets of uniform cells with eosinophilic or oncocytic cytoplasm that contain cytoplasmic vacuoles or flocculent inclusions.5, 6, 7, 8 However, renal cell carcinomas (RCCs) with other histologic appearances have been reported in patients with germline mutations of SDH subunit genes,4, 9 and a few RCCs of other histologic types have been found to be SDH-deficient in the absence of known germline gene mutation.8, 10 Nonetheless, experience with SDH-deficient RCC demonstrating this unique oncocytic histology remains very limited.11 Detailed pathologic features and immunohistochemical staining results for renal tumor antigens have been reported in only four tumors with this unique morphology.5, 6 Therefore, the precise constellation of features that differentiates these neoplasms from other subtypes of renal tumors remains poorly understood. To facilitate discrimination of these SDH-deficient oncocytic renal tumors from other benign and malignant renal neoplasms and to elaborate the spectrum of their pathologic features, we performed a detailed analysis of clinicopathologic, immunohistochemical, and genetic features of SDH-deficient RCCs.
Materials and methods
Patients and Tumors
Renal tumors that exhibited morphologic similarity to the previously reported SDH-deficient oncocytic renal neoplasms were retrieved from the institutional and consultation files of the contributing authors. Tumors with a spectrum of suspicious features were screened by immunohistochemistry for the absence of immunohistochemical staining for SDHB (as described below), as loss of any of the four SDH subunits results in destabilization of the enzyme complex and absence of detectable SDHB protein by immunohistochemistry.1 Tumors were included in the study cohort if they (1) demonstrated the distinctive morphologic features previously recognized in patients with germline SDH subunit gene mutations,5, 6 including a uniform population of cells with eosinophilic cytoplasm and cytoplasmic vacuoles or flocculent cytoplasmic inclusions, and (2) were confirmed to have an absence of immunohistochemical labeling for SDHB in the neoplastic cells, with appropriate internal control staining of adjacent renal tissue and intratumoral non-neoplastic cells. A total of 37 tumors were evaluated with SDHB immunohistochemistry, of which 11 were confirmed to be SDH-deficient and 27 exhibited strong, positive cytoplasmic staining for SDHB; findings in the latter group are summarized below in Excluded Tumors with Positive SDHB Immunohistochemistry. Clinicopathologic features of the confirmed SDH-deficient neoplasms were collected, including: patient age, patient gender, greatest tumor diameter, number of tumors, surgical procedure, pathologic stage, sites involved, follow-up status, other tumors (including paraganglioma, pheochromocytoma, and GIST), family history, and gross tumor appearance. Histopathologic features assessed included: Fuhrman nuclear grade, presence of cytoplasmic vacuoles or flocculent cytoplasmic inclusions, hyalinized or edematous stroma, entrapped non-neoplastic renal tubules, tumor pseudocapsule, foamy macrophages, degenerative cytologic atypia, multinucleated tumor cells, intratumoral mast cells, tumor necrosis, tumor calcification or metaplastic bone formation, lymphoid aggregates, and other lesions in the uninvolved renal parenchyma. Percentages of solid or nested, tubular, and cystic architecture were estimated based on the available gross and microscopic findings.
Immunohistochemistry and Histochemistry
Immunohistochemical staining was performed on the 11 tumors confirmed to be SDH-deficient with sufficient available material (paraffin blocks or unstained slides) with appropriate positive and negative internal and external controls. Antibodies and technical specifications are listed in Table 1. For a subset of cases, immunohistochemical stains performed as part of the original diagnostic evaluation were retrospectively reviewed. Staining reactions were estimated microscopically as a percentage of positively staining cells, and distinct patterns of reactivity were separately noted. Histochemical staining using a modified colloidal iron technique12 was also performed.
Next-Generation DNA Sequencing
For next-generation DNA sequencing, hematoxylin and eosin-stained slides were reviewed to evaluate tumor content and viability and areas enriched for tumor were circled. The entire slides from paired 5 micron-thick formalin-fixed paraffin-embedded tissue sections were scraped in five cases (because they contained 80% tumor or greater), and 5 to 10 × 0.5 mm diameter cores were obtained directly from the fixed tissue block in two cases. Samples were digested in proteinase K overnight, and DNA was isolated using according to the manufacturer’s protocol (QIAamp DNA Mini Kit, Qiagen, Gaithersburg, MD, USA). DNA concentration was assessed using PicoGreen ds DNA detection (Life Technologies, Carlsbad, CA, USA). Samples with at least 50 ng total DNA concentration were subjected to targeted next-generation sequencing using a cancer genomic assay to detect sequence and copy number variations in 275 oncogenes and tumor suppressor genes. The entire exonic sequence of the target genes was captured using a solution-phase Agilent SureSelect hybrid capture kit (Agilent Technologies, Santa Clara, CA, USA) and massively parallel sequencing was performed on an Illumina HiSeq 2500 sequencer (Illumina, San Diego, CA, USA). Mutation calls were made using Mutect and GATK software (Broad Institute, Cambridge, MA, USA) and copy number alterations were assessed using VisCap Copy (Dana Farber Cancer Institute, Boston, MA, USA).
Results
Clinicopathologic Features
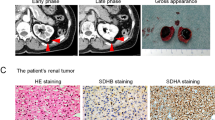
Eleven tumors from 10 patients were confirmed to be SDH deficient by immunohistochemistry and were classified or reclassified as SDH-deficient RCC (Table 2). These tumors arose in seven men and three women, who ranged from 22 to 72 years in age (mean 41, median 40 years). Greatest tumor diameter ranged from 2.0 to 20 cm (median 4.25 cm). One tumor was sampled only by core biopsy (patient 8) and measured 2.4 cm in greatest dimension based on computed tomography findings. Bilateral, morphologically identical SDH-deficient RCCs were resected from one patient (patient 1), 2 months apart, measuring 4.0 and 9.0 cm in greatest diameter. No tumor demonstrated invasion of the perinephric fat, renal vein branches, or renal sinus, and therefore, pathologic stages were pT1a (n=5), pT1b (n=2), pT2a (n=2), and pT2b (n=1). A regional lymph node was present in only one specimen and did not contain metastatic carcinoma. Two patients were known to have paragangliomas, which were also confirmed to lack immunohistochemical labeling for SDHB. In patient 2, a paraaortic paraganglioma was resected synchronously with the SDH-deficient RCC; in patient 10, a submandibular lymph node was excised 9 months after the RCC and found to contain metastatic paraganglioma (Figures 1a and b). In the latter patient, subsequent imaging revealed a large left carotid body tumor. No patient was known to have GIST. For patient 2, after concurrent resection of the SDH-deficient RCC and paraganglioma, a small contralateral renal mass was identified by imaging and removed via partial nephrectomy. In contrast to the SDH-deficient RCC, this tumor exhibited characteristic morphology of renal oncocytoma (large cell nests and tubular structures dispersed in hyalinized stroma) and was confirmed by immunohistochemistry to have strongly positive immunohistochemical staining for SDHB (Figures 1c and d). One patient (patient 4) had a brother with RCC that metastasized to the liver (not available for review), and another (patient 9) had a brother with known SDHB and FH germline mutations and, by report, cancers related to these mutations (details unknown). One patient (patient 5) was also noted to have BRCA2 mutation. None of the other patients had known family history of SDH-related neoplasia. Follow-up information was available for six patients, as detailed in Table 2. Of these, one patient developed widespread metastases 16 years after nephrectomy involving lungs, liver, rib, and vertebrae (patient 5). A small contralateral renal tumor measuring 2.2 cm in greatest diameter was also identified by imaging at that time but never resected. A lung metastasis was sampled by fine-needle aspiration and confirmed to represent a metastasis from the prior SDH-deficient RCC, based on identical histologic features in a cell block preparation, diffuse nuclear immunoreactivity for PAX8, and absence of labeling for SDHB (Figure 2). The patient was treated with sunitinib and remained stable for several additional years but experienced progression and died of disease 6 years later. A small ipsilateral renal mass (1.0 cm) was identified in patient 2, 4 years after resection of the SDH-deficient RCC. This tumor was treated with cryoablation and no tissue was retrieved for histologic analysis. At most recent follow-up (4 years), he remained free of disease. The remaining patients with follow-up information were alive without evidence of residual, recurrent, or metastatic disease at most recent follow-up, although for some patients follow-up duration was short.

A paraganglioma from patient 10 (a) was composed of a nested to whorled arrangement of cells with amphophilic cytoplasm. The paraganglioma cells exhibited absence of immunohistochemical staining for SDHB (b), whereas endothelial cells and other stromal cells exhibited normal positive labeling for SDHB. An oncocytoma from patient 2 (c) was resected after the SDH-deficient RCC. In contrast to the SDH-deficient RCC, normal SDHB labeling was present in the oncocytoma (d).

Sixteen years after resection of the SDH-deficient RCC, patient 5 developed widespread metastases involving lungs, liver, rib, and vertebrae. A lung metastasis was sampled by fine-needle aspiration. Diff-Quik-stained direct smears (a) revealed clusters of monotonous epithelioid cells with granular cytoplasm. A cell block preparation was composed of a similar population of cells with eosinophilic cytoplasm and occasional cytoplasmic vacuoles (b), identical to that of the primary tumor. By immunohistochemistry, the neoplastic cells were positive for PAX8 (c) and negative for SDHB (d), supporting the interpretation of metastatic SDH-deficient RCC. Rare inflammatory cells show weak staining for SDHB (d).
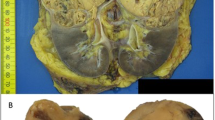
Grossly, tumors had a variegated, tan–brown or red–brown, sometimes hemorrhagic cut surface (Figure 3a). Three tumors were noted grossly to be partly cystic and one was grossly surrounded by a pseudocapsule. Microscopically, architecture was predominantly solid or nested, comprising 50–95% of tumor volume (Figure 3b). All tumors were composed of uniform neoplastic cells with eosinophilic, variably granular cytoplasm that contained cytoplasmic vacuoles or inclusions of flocculent eosinophilic material (Figure 3c). Nuclei were typically uniform and round, with smooth nuclear contours, finely clumped chromatin, and small or absent nucleoli. Architecturally, formation of tubules and microcystic structures with open lumina was typically less conspicuous (Figure 3d), comprising the remaining 5–50% of tumor volume. A partial tumor pseudocapsule was observed microscopically in four tumors. Six tumors had areas of edematous stroma. One tumor had hyalinized stroma and three had foci of metaplastic bone formation containing paucicellular bone marrow. Entrapped non-neoplastic renal tubules were present in nine tumors, five conspicuously (Figure 3e) and four focally. Necrosis was encountered in one tumor. Neither degenerative nuclear atypia with smudged chromatin nor perinuclear cytoplasmic clearing was present in any tumor. Foamy macrophages within lumina of microcystic structures were focally encountered in two tumors. Another two tumors had rare binucleated cells. Intratumoral mast cells could be identified histologically in nine tumors (Figure 3c), and they were conspicuous in both tumors from patient 1. Scattered aggregates of intratumoral lymphocytes were present in one tumor. The available slides demonstrating non-neoplastic renal tissue did not contain microscopic incipient tumors in any specimen, although in some, the non-neoplastic renal tissue was scant.

Grossly, tumor 5 (from patient 4) formed a round, circumscribed but unencapsulated tan–brown mass with areas of congestion or hemorrhage that bulged from the contour of the kidney (a). At low magnification (b), tumor 2 (from patient 1) was partly surrounded by peritumoral pseudocapsule (upper left). Architecture was predominantly solid, and entrapped non-neoplastic tubules were present (upper center). At high magnification (c), the same tumor contained scattered intratumoral mast cells (white arrow) and large flocculent cytoplasmic inclusions in the neoplastic cells (black arrows). In addition to solid architecture, tumors typically exhibited less conspicuous formation of tubules (d, tumor 5). Entrapped non-neoplastic renal tubules were commonly present (e, tumor 4). These tubules and other non-neoplastic cells were confirmed to have retained immunohistochemical labeling for SDHB (f, same tumor), whereas SDHB protein was not detected in the neoplastic cells.
Immunohistochemistry and Histochemistry
Results of immunohistochemical staining in SDH-deficient RCCs are summarized in Table 3. By criteria for inclusion in the study, SDHB was not detected in the neoplastic cells (Figure 3f). In one tumor (patient 8), a negative staining reaction was also obtained for SDHA. In the remaining 10 tumors, staining for SDHA was strongly positive in the cytoplasm and intensely labeled cytoplasmic inclusions (Figure 4a, inset). Immunohistochemistry with antibodies to broad-spectrum epithelial antigens revealed a wide spectrum of positive reactivity. Labeling for EMA was present in all tumors, often with a delicate membranous pattern, but varied from focal to diffuse in extent (Figure 4b). Similarly, the labeling for cytokeratin CAM5.2 and AE1/AE3 ranged from entirely absent (Figure 4c) to diffuse. Reactivity for cytokeratin 7 was absent in the majority of tumors, with the exception of one that exhibited positive staining in small clusters of cells (∼5%). Nuclear labeling for PAX8 (Figure 4d) was consistently uniform and diffuse, whereas staining for the RCC antigen was entirely absent. Carbonic anhydrase IX was not detected in any tumor, and similar to broad-spectrum epithelial markers, staining for CD10 varied from absent to diffuse. The neoplastic cells were entirely negative for vimentin, with the exception of one tumor that exhibited patchy reactivity. However, staining for vimentin reliably highlighted the intratumoral mast cells. Similarly, staining for KIT was consistently negative in the neoplastic cells of all tumors studied, although this antibody likewise labeled mast cells (Figure 4e). Kidney-specific cadherin and HNF1B (Figure 4f) both yielded diffuse positive reactions (membranous and nuclear staining patterns, respectively). Extent of labeling for S100A1 varied widely and cytoplasmic staining with the colloidal iron technique was absent (four tumors evaluated). Labeling for the other studied antigens was entirely negative, including chromogranin, synaptophysin, cathepsin K, TFE3, and HMB45.

Immunohistochemical staining of SDH-deficient RCCs revealed normal positive labeling for SDHA in the neoplastic cells of all tumors (a, inset, tumor 11 from patient 10 depicted), with the exception of tumor 9 from patient 8 (absence of SDHA staining not pictured). Intense labeling of the cytoplasmic inclusions supports their interpretation as abnormal mitochondria rather than true vacuoles. In this tumor, many of the cells were reactive for EMA (b), although an entirely negative reaction was observed for cytokeratin AE1/AE3 (c). Reactivity for other common renal epithelial antigens was highly variable. Entrapped non-neoplastic tubules show labeling for keratin AE1/AE3 as a positive internal control (C, upper center). Antibody to PAX8 uniformly labeled the nuclei of all tumors (d). The neoplastic cells were negative for KIT (CD117) in all tumors (e); however, this antibody characteristically highlighted many intratumoral mast cells. The pattern of nuclear labeling for HNF1B expected of normal cells was uniformly retained (f), in contrast to the lack of labeling for this marker that has been described in chromophobe RCC.
Next-Generation DNA Sequencing
Material was available from seven tumors for targeted exome capture and next-generation DNA sequencing of 275 oncogenes and tumor suppressors, including SDHB. One tumor that was resected in 1991 generated quantitatively adequate DNA but failed sequencing, likely due to DNA fragmentation. The remaining six met minimum sequencing quality standards. The mean target coverage across the specimens was 159 reads (range 52–274). Following filtration for common polymorphisms [NHLBI Exome Sequencing Project (ESP), Washington University, St Louis, MO] and noncoding or silent variants, each sample contained an average of six single-nucleotide variants or small insertion-deletion mutations (range 4–9). Four of the six tumors contained single-nucleotide variants in SDHB. An additional tumor contained an exon 3 deletion in SDHB detectable on manual review of the sequencing data on Integrated Genome Viewer (IGV, Broad Institute) and with VisCap (Figure 5). All five tumors with SDHB alterations (Table 4) also demonstrated one copy loss involving part of or the entire short arm of chromosome 1 including the SDHB gene, consistent with a second deleterious hit. The sixth evaluated tumor was the single case with negative SDHA immunohistochemistry (patient 8), which was not found to have an alteration of SDHB by sequencing. The targeted sequencing panel did not evaluate the SDHA gene.

Copy number analysis (one representative sample with SDH deficiency) shows isochromosome 1 with one copy loss of the entirety of 1p and relative gain of 1q. SDHB, located on 1p, shows one copy loss across the entire gene and two copy loss at exon 3 (arrow) suggestive of an intragenic deletion or splicing event with subsequent loss of heterozygosity in the tumor. Of note, SDHC is located on 1q, and demonstrates no mutational events and low copy gain.
The remaining sequence alterations largely consisted of rare single-nucleotide polymorphisms (present in less than 1% of American populations represented in ESP). Other detected variants of unknown significance included a REL p.R108* mutation and a MYB p.N241fs mutation, both expected to lead to loss of function of their respective genes. Of note, none of the cases contained variants in other genes recognized to contribute to RCC pathogenesis, including VHL, PIK3CA, AKT, MTOR, MET, and TP53.
Excluded Tumors with Positive SDHB Immunohistochemistry
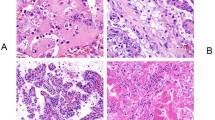
Twenty-seven tumors from 26 patients that exhibited a spectrum of morphologic features raising the possibility of SDH-deficient RCC showed strong positive cytoplasmic staining for SDHB, supporting exclusion from the category of SDH-deficient RCC (Figure 6). In brief, the possibility of SDH-deficient RCC was considered for these tumors due to the following reasons: features not fitting well into the categories of oncocytoma or eosinophilic variant of chromophobe RCC (n=10), an unclassified RCC composed of eosinophilic cells (n=7), prominent cytoplasmic vacuoles (n=5), multiple oncocytic renal neoplasms (n=3), and flocculent-appearing pale cytoplasm with prominent entrapped non-neoplastic renal tubules (n=2). Fifteen tumors in this group were noted to have at least focal cytoplasmic vacuoles (Figures 6a and b) and 10 had a prominent component of entrapped renal tubules (Figures 6c and d). Two tumors were composed of a mixture of epithelioid eosinophilic cells with cytoplasmic vacuoles and eosinophilic spindle-shaped cells, as has been rarely reported in SDH-deficient RCC.6 Nonetheless, all retained diffuse, strong, granular cytoplasmic immunohistochemical labeling for SDHB. Notably, none of these tumors exhibited a homogeneous constellation of histologic features identical to the SDH-deficient RCCs reported in this study. Often, cytoplasmic vacuoles had a predominantly empty appearance, and the granular cytoplasmic labeling for SDHB uniformly spared the vacuoles, indicating that they are unlikely to be large, abnormal mitochondria, as in SDH-deficient RCC.5 For 11 of these tumors, immunohistochemical staining for KIT was available for review, which highlighted at least scattered intratumoral mast cells in six and labeled the neoplastic cells with variable intensity.

Twenty-seven tumors with a range of morphologic features raising the possibility of SDH-deficient RCC were found to have positive immunohistochemical staining for SDHB and were excluded from the study group. Prominent cytoplasmic vacuoles were noted in 15 of these tumors. In one such example (a), abrupt transitions from areas of eosinophilic tumor cells to areas with numerous cytoplasmic vacuoles were present. Intense granular cytoplasmic labeling for SDHB was retained in this tumor (b), supporting exclusion from the category of SDH-deficient RCC. The cytoplasmic vacuoles were uniformly negative for SDHB, indicating that they are unlikely to be enlarged, abnormal mitochondria. Another unclassified RCC that was excluded from the SDH-deficient study group was unencapsulated and composed of cells with pale to flocculent cytoplasm with areas of cytoplasmic clearing (c). Numerous entrapped non-neoplastic renal tubules were present (center). Despite the pale-staining cytoplasm, positive staining for SDHB was readily recognizable by immunohistochemistry in both the neoplasm and entrapped tubules (d).
Discussion
Patients with germline mutations of SDH subunit genes are prone to develop paragangliomas and GISTs.1, 2 In addition to these tumor types, there is increasing evidence that some patients also develop renal neoplasms,5, 6, 7, 9 a subgroup of which have been reported to exhibit distinctive oncocytic morphology with cytoplasmic vacuoles or inclusions of flocculent material.5, 6 In such tumors, the neoplastic cells have been found to lack immunohistochemical staining for SDHB protein, in contrast to adjacent and intratumoral non-neoplastic tissues, which exhibit positive staining,6 likely due to germline mutation with initiation of tumorigenesis after a ‘second hit’ to the remaining SDH subunit gene allele. Nonetheless, experience with these renal neoplasms remains very limited,11 and few have been comprehensively evaluated by immunohistochemistry for commonly used markers of renal neoplasms.5, 6 Therefore, in this study, we sought to perform a detailed characterization of clinicopathologic, immunohistochemical, and genetic features of these oncocytic SDH-deficient RCCs, to facilitate their discrimination from other subtypes of renal neoplasia. This is the first study to focus on identification of these SDH-deficient RCCs based on their unique morphology rather than by a known history of hereditary syndrome or germline mutation,4, 6, 10 or comprehensive screening of unselected tumors.8, 10
An association between germline mutations of SDH genes and hereditary renal neoplasia has been recognized for a number of years;7, 13, 14, 15, 16, 17, 18 however, the studies by Housley et al.5 and Gill et al.6 first documented that at least a subgroup of the renal neoplasms from these patients have unique morphology that can be distinguished from that of other subtypes of renal tumors.5, 6 These tumors were observed to be composed of uniform populations of cuboidal cells with finely granular eosinophilic cytoplasm, centrally located round nuclei, and cytoplasmic inclusions of pale eosinophilic fluid-like material or flocculent-appearing areas of cytoplasmic clearing.6 By electron microscopy, these cytoplasmic inclusions have been found in one tumor to represent highly abnormal mitochondria with an excess of mitochondrial matrix and few cristae.5 In this study, we found SDH-deficient RCCs to reliably recapitulate this unique morphologic pattern. Supporting the hypothesis that these cytoplasmic inclusions represent abnormal mitochondria, we found immunohistochemical staining for SDHA, a mitochondrial protein, to strongly label the inclusions, with the exception of the single tumor that was entirely negative for SDHA. In contrast, an excluded group of other renal tumors with varied patterns of eosinophilic cells and cytoplasmic vacuoles were positive for SDHB by immunohistochemistry, typically demonstrating some contrasting morphologic features, such as empty-appearing cytoplasmic vacuoles (Figure 6), predominant or pure tubular architecture, or multinucleated tumor giant cells. Similarly, in a large study by Ricketts et al,4 14 patients from 12 families with germline SDHB mutations were reported to have RCCs, often with oncocytic morphology, although the pathologic characteristics and histologic appearances of these tumors were not separately described.
Discriminating SDH-deficient RCCs from other subtypes of renal neoplasms, especially renal oncocytoma, which they may closely resemble, is of substantial importance. One patient in our study experienced late recurrence with widespread metastases and ultimately died, supporting the ability of SDH-deficient RCC to progress over a protracted course. In addition, accurate classification of these tumors is important for prompting appropriate clinical follow-up, screening for paraganglioma, and evaluation of other family members for a hereditary tumor predisposition syndrome associated with germline SDH subunit gene mutation.
Several features identified in this study may be helpful in discriminating SDH-deficient RCC from other subtypes of renal neoplasms that have overlapping morphologic features. Oncocytoma and SDH-deficient RCC are both composed of cells with eosinophilic cytoplasm and round, centrally located uniform nuclei, arranged in variable admixtures of solid or nested, tubular, and cystic architectural patterns. Both of these tumors may or may not possess a peritumoral pseudocapsule, and entrapped non-neoplastic tubules are a common finding in both lesions. In contrast to renal oncocytoma, however, none of the tumors in this study demonstrated discrete, large rounded nests of tumor cells dispersed in hyalinized or edematous stroma. Rather, formation of tubules was typically a minor tumor component and solid architecture often included ill-defined, sheet-like growth. Similarly, none of the SDH-deficient tumors exhibited zones of degenerative nuclear atypia with enlarged nuclei and smudged nuclear chromatin, as are sometimes observed in renal oncocytoma. The characteristic cytoplasmic vacuoles or flocculent inclusions of eosinophilic material in SDH-deficient RCCs are a helpful diagnostic feature, although it is important to keep in mind that other renal tumors that are not SDH-deficient can also occasionally contain cytoplasmic vacuoles, often with an empty appearance. In this study, 27 renal tumors with a spectrum of morphologic features suspicious for SDH-deficient RCC, including 15 with cytoplasmic vacuoles, were evaluated, all of which retained positive immunohistochemical staining for SDHB. This labeling characteristically spared the cytoplasmic vacuoles, indicating that they are unlikely to be enlarged, abnormal mitochondria, as in SDH-deficient RCC. The absence of KIT (CD117) by immunohistochemistry is supportive of a diagnosis of SDH-deficient RCC when compared with oncocytoma, as the latter often shows reactivity with this antibody.19, 20 Of note, prominent intratumoral mast cells are a novel feature of SDH-deficient RCC identified in the current study that may be a helpful diagnostic clue, recognizable either by light microscopy or highlighted by immunohistochemistry for KIT or vimentin. Nonetheless, several of the tumors excluded from this study due to positive SDHB immunohistochemistry also contained scattered intratumoral mast cells, limiting the diagnostic usefulness of this feature alone. Oncocytomas also often have a distinctive pattern of labeling for CK7 wherein only scattered single cells and small clusters of cells show positive labeling.21, 22, 23 Notably, this pattern was shared only by 1 of the 11 SDH-deficient RCCs in this study, whereas the remaining tumors were entirely negative for CK7. None of the other evaluated immunohistochemical stains were useful to distinguish SDH-deficient RCC and oncocytoma (Table 3), including positive nuclear staining for hepatocyte nuclear factor-1β (HNF1B), an emerging marker that exhibits an absence of reactivity in chromophobe RCC,24 and diffuse labeling for kidney-specific cadherin, similar to both oncocytoma and chromophobe RCC.25
Although the morphology of SDH-deficient RCC does not closely resemble that of the eosinophilic variant of chromophobe RCC, this diagnostic possibility might also be considered for a renal epithelial neoplasm composed of eosinophilic cells. The immunohistochemical results for SDH-deficient RCC in this study appear relatively distinct from those of chromophobe RCC, which labels for KIT and CK7 more diffusely in ∼50–60% of cases and has been found to be negative for HNF1B.21, 22, 23, 24 Like chromophobe RCC, SDH-deficient RCC is usually negative for vimentin. Staining for S100A1, which has proposed utility in differentiating oncocytoma from chromophobe RCC, was highly variable in SDH-deficient RCCs and unlikely to be of diagnostic value. Reactivity for this antigen ranged from 5% of cells showing weak labeling (closer to the negative reactivity expected of chromophobe RCC) up to 80–90% of cells showing moderate to strong reactivity (similar to the positive reaction expected of oncocytoma).26 Key pathologic and immunohistochemical features helpful in discriminating SDH-deficient RCC from oncocytoma and the eosinophilic variant of chromophobe RCC are summarized in Table 5.
An unusual feature of SDH-deficient RCC is that tumors exhibit relatively limited and variable immunohistochemical reactivity for some renal epithelial antigens, other than PAX8 and kidney-specific cadherin, which were uniformly diffuse and strong in all studied tumors. The uniform positivity for kidney-specific cadherin, in particular, suggests origin from or differentiation toward the distal nephron, given the pattern of positivity in the distal nephron of the non-neoplastic kidney observed previously for this marker. Immunohistochemical labeling for CD10, cytokeratin AE1/AE3, CAM5.2, and EMA varied widely. Some tumors exhibited limited to no reactivity for these markers, whereas others demonstrated diffuse immunohistochemical labeling. Staining for RCC antigen was entirely negative in all tumors, and labeling for AMACR was often minimal. Taken together with the unique morphology of these tumors, the limited labeling for common renal tumor antigens might also raise a differential diagnosis that includes translocation-associated RCC (tumors associated with MITF gene family translocations); however, the morphologic pattern of uniform tumor cells with eosinophilic cytoplasm is not the prototypical appearance of Xp11 translocation-associated RCC. Nevertheless, tumors with oncocytoma-like morphology have been occasionally reported, and there is increasing evidence that the morphologic spectrum of these tumors is broader than originally described.27, 28 In this study, the absence of TFE3 protein, cathepsin K, and HMB45 was supportive in differentiating SDH-deficient RCC from translocation-associated RCC. Similarly, these staining patterns (negative cathepsin K and HMB45 and positivity for at least some epithelial antigens) can be helpful in differentiating these tumors from epithelioid angiomyolipoma or perivascular epithelioid cell tumor (PEComa).29, 30, 31
Other notable clinicopathologic features of SDH-deficient RCCs identified in this study include the occurrence of bilateral tumors with identical morphology in one patient and the occurrence of paragangliomas in two patients. Interestingly, one patient with concurrent paraganglioma and SDH-deficient RCC (patient 2) underwent subsequent resection of a small contralateral renal tumor, which demonstrated characteristic features of oncocytoma (rounded nests and tubular structures lined by eosinophilic cells, dispersed in hyalinized stroma) and showed strong positive reactivity for SDHB. The occurrence of oncocytoma has been previously reported in a patient with germline SDHB mutation;17 however, as the pathologic characteristics of the tumor in this prior report were not specifically described or illustrated, it is uncertain whether it represented a true oncocytoma or SDH-deficient RCC with oncocytic morphology. Nonetheless, our findings suggest that true oncocytoma may indeed occur in these patients.
Another tumor identified in this study (sampled only by core needle biopsy, patient 8), which demonstrated similar morphology to the other SDH-deficient RCCs, exhibited absence of labeling for both SDHB and SDHA by immunohistochemistry. Recent studies of SDH-deficient paragangliomas and GISTs have demonstrated that only tumors with SDHA mutations show negative SDHA immunohistochemistry (in addition to SDHB).32, 33, 34 As expected, there was no abnormality of the SDHB gene detected by next-generation sequencing in this case. Unfortunately, SDHA was not included in the targeted sequencing panel, and therefore, the presence of an inactivating SDHA mutation could not be confirmed. To our knowledge, SDHA-deficient RCC has not been previously described; however, our findings support the occurrence of this distinctive oncocytic morphology in such tumors as well. Similarly, Gill et al35 recently reported the occurrence of an SDH-deficient RCC showing this characteristic morphology in a patient with germline SDHC gene mutation and multiple SDH-deficient GISTs.
The precise malignant potential of SDH-deficient RCCs is not entirely understood; however, tumor progression and death from disease over a protracted course in one patient in our study (patient 5) indicate that such tumors do carry a risk for metastasis and death. The significance of an additional BRCA2 mutation in this patient in not known. Another patient recently reported by Papathomas et al10 likewise developed multifocal abdominal metastases from a tumor with similar oncocytic morphology. This patient was noted to be alive with disease 11 years after nephrectomy, suggesting that these tumors may in some cases behave in a relatively indolent manner, even in the setting of metastatic disease. The brother of another patient in our study (patient 4) reportedly developed liver metastasis from RCC, although full clinicopathologic details and tissue material from this tumor were not available for evaluation. Several patients with kidney cancer attributable to germline SDH subunit gene mutations in the study of Ricketts et al4 died of metastatic disease, at least one of which was specifically noted to have SDHB mutation and an oncocytic renal tumor. This patient had bone metastasis at presentation and died 7 months after nephrectomy, suggesting that more rapid progression is also possible. In some families, kidney tumors were the only manifestation of their hereditary tumor syndrome, and in some metastatic disease developed despite small, localized primary tumors. Two patients with germline SDHC mutations and one with germline SDHD mutation in their study developed metastatic disease, of which two died of kidney cancer, although interestingly, these were noted to be clear cell RCCs histologically, in contrast to the patients with SDHB gene mutations, whose tumors were noted to be predominantly oncocytic in appearance.4
Similarly, a few prior reports have identified RCCs with complete or partial absence of SDHB immunohistochemical staining8, 10 or RCCs in families with known SDH subunit gene mutations4, 9 that demonstrate other histologic appearances, including tumors resembling clear cell RCC, papillary RCC, sarcomatoid and unclassified RCCs.4, 8, 9, 10 The significance of this occurrence remains incompletely understood. In at least some circumstances, renal tumors9 and other neoplasms10, 35 occurring in patients with known germline SDH subunit mutations have been found to have normal immunohistochemical staining for SDHB, suggesting that they develop through mechanisms other than biallelic SDH subunit inactivation. In rare circumstances, it appears that absence of SDHB labeling can also occur in RCCs unrelated to SDH subunit gene germline mutation. As an illustration of this possibility, Papathomas et al10 recently identified one clear cell RCC with partial absence of immunohistochemical staining for SDHB from a group of 348 neoplasms (including 130 renal tumors) occurring in patients without known germline mutation of SDH subunit genes. This tumor exhibited positive reactivity for SDHB in areas with low-grade clear cell RCC morphology, transitioning to a high-grade sarcomatoid component with absence of reactivity, suggesting that SDHB protein loss was the result of additional genetic alterations that developed during tumor progression. In favor of this hypothesis, the authors found large intragenic deletions of SDHAF2 and SDHD in the tumor, without germline mutation of an SDH subunit gene. In another recent study, Miettinen et al8 screened 711 renal neoplasms with SDHB and SDHA immunohistochemistry and found 4 (0.6%) to be SDH-deficient. Of these, two tumors had oncocytic morphology and occurred in patients with a family history of early-onset RCC and a personal history of SDH-deficient GIST, respectively, suggesting that these tumors represent SDH-deficient RCCs associated with germline gene mutations, similar to those described in this study. In contrast, the remaining two SDH-deficient renal tumors histologically appeared to be a high-grade clear cell RCC and a type 2 papillary RCC. From a group of 1547 other neoplasms, a single prostatic adenocarcinoma, gastric adenocarcinoma, and testicular seminoma each were found to be SDH deficient. The significance of negative SDH immunohistochemistry in such tumors (RCCs with other morphologies and neoplasms of other organs) remains incompletely understood and a subject of potential interest for further study.
In this study, we also performed targeted next-generation DNA sequencing on formalin-fixed, paraffin-embedded tissue samples from seven tumors to further assess abnormalities of the SDHB gene and other genes involved in RCC pathogenesis. Four of the six tumors with SDHB protein loss by immunohistochemistry revealed single-nucleotide variants in the SDHB gene, and one tumor demonstrated an exon 3 deletion of SDHB; the sixth case, which demonstrated loss of SDHA protein, was negative for abnormalities in the SDHB gene. Similar sequence-level alterations were also observed by Papathomas et al10 and Neumann et al14 in the setting of hereditary pheochromocytoma–paraganglioma syndromes. In all of these five tumors, SDHB alterations were associated with losses of chromosome 1p containing the SDHB gene, in keeping with a second deleterious hit. Although we did not specifically assess for germline gene mutations, this pattern of genetic abnormalities would be in line with that of germline gene mutation followed by a ‘second hit’ initiating tumorigenesis. The targeted next-generation sequencing panel utilized in this study did not reveal alterations of other key genes involved in RCC pathogenesis, such as VHL, PIK3CA, AKT, MTOR, MET, or TP53.
Clinicopathologic features for six of the patients in this study are also suggestive of germline mutation and a hereditary tumor predisposition syndrome, including personal history of multiple renal tumors (three patients), synchronous or metachronous paraganglioma (two patients), other hereditary gene mutations (two patients, FH and BRCA2, respectively), and family history of syndrome-associated tumors (two patients). Whether two somatic alterations to SDH subunit genes in the absence of germline mutation can result in formation of a tumor with the unique morphology and negative SDHB immunohistochemistry described in this study remains unknown.
In summary, our findings support the classification of SDH-deficient RCC as a distinct type of renal neoplasm. These tumors are composed of a uniform population of cells with eosinophilic cytoplasm, cytoplasmic inclusions of flocculent or eosinophilic material (previously reported to be abnormal mitochondria), and predominantly solid or nested architecture. Intratumoral mast cells, entrapped non-neoplastic renal tubules, and a variable tumor pseudocapsule are other characteristic features, although in isolation, these findings can be encountered in renal epithelial neoplasms that are not SDH deficient. Other than diffuse nuclear labeling for PAX8 and diffuse membranous labeling for kidney-specific cadherin, these tumors often show limited and highly variable labeling for renal epithelial markers by immunohistochemistry. Tumor progression over a protracted course in some patients supports the classification of these neoplasms as carcinomas. Careful attention to subtle morphologic and immunohistochemical clues is helpful in triggering recognition of these tumors via screening for SDHB protein and clinical evaluation for a familial tumor predisposition syndrome.
References
Barletta JA, Hornick JL . Succinate dehydrogenase-deficient tumors: diagnostic advances and clinical implications. Adv Anat Pathol 2012;19:193–203.
Gill AJ . Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology 2012;44:285–292.
Doyle LA, Hornick JL . Gastrointestinal stromal tumours: from KIT to succinate dehydrogenase. Histopathology 2014;64:53–67.
Ricketts CJ, Shuch B, Vocke CD et al. Succinate dehydrogenase kidney cancer: an aggressive example of the Warburg effect in cancer. J Urol 2012;188:2063–2071.
Housley SL, Lindsay RS, Young B et al. Renal carcinoma with giant mitochondria associated with germ-line mutation and somatic loss of the succinate dehydrogenase B gene. Histopathology 2010;56:405–408.
Gill AJ, Pachter NS, Chou A et al. Renal tumors associated with germline SDHB mutation show distinctive morphology. Am J Surg Pathol 2011;35:1578–1585.
Ricketts C, Woodward ER, Killick P et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 2008;100:1260–1262.
Miettinen M, Sarlomo-Rikala M, Cue PM et al. Mapping of succinate dehydrogenase losses in 2258 epithelial neoplasms. Appl Immunohistochem Mol Morphol 2014;22:31–36.
Malinoc A, Sullivan M, Wiech T et al. Biallelic inactivation of the SDHC gene in renal carcinoma associated with paraganglioma syndrome type 3. Endocr Relat Cancer 2012;19:283–290.
Papathomas TG, Gaal J, Corssmit EP et al. Non-pheochromocytoma (PCC)/paraganglioma (PGL) tumors in patients with succinate dehydrogenase-related PCC-PGL syndromes: a clinicopathological and molecular analysis. Eur J Endocrinol 2014;170:1–12.
Srigley JR, Delahunt B, Eble JN et al. The International Society of Urological Pathology (ISUP) Vancouver Classification of renal neoplasia. Am J Surg Pathol 2013;37:1469–1489.
Tickoo SK, Amin MB, Zarbo RJ . Colloidal iron staining in renal epithelial neoplasms, including chromophobe renal cell carcinoma: emphasis on technique and patterns of staining. Am J Surg Pathol 1998;22:419–424.
Vanharanta S, Buchta M, McWhinney SR et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet 2004;74:153–159.
Neumann HP, Pawlu C, Peczkowska M et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004;292:943–951.
Srirangalingam U, Walker L, Khoo B et al. Clinical manifestations of familial paraganglioma and phaeochromocytomas in succinate dehydrogenase B (SDH-B) gene mutation carriers. Clin Endocrinol (Oxf) 2008;69:587–596.
Schimke RN, Collins DL, Stolle CA . Paraganglioma, neuroblastoma, and a SDHB mutation: resolution of a 30-year-old mystery. Am J Med Genet A 2010;152A:1531–1535.
Henderson A, Douglas F, Perros P et al. SDHB-associated renal oncocytoma suggests a broadening of the renal phenotype in hereditary paragangliomatosis. Fam Cancer 2009;8:257–260.
Ricketts CJ, Forman JR, Rattenberry E et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat 2010;31:41–51.
Al-Ahmadie HA, Alden D, Fine SW et al. Role of immunohistochemistry in the evaluation of needle core biopsies in adult renal cortical tumors: an ex vivo study. Am J Surg Pathol 2011;35:949–961.
Liu L, Qian J, Singh H et al. Immunohistochemical analysis of chromophobe renal cell carcinoma, renal oncocytoma, and clear cell carcinoma: an optimal and practical panel for differential diagnosis. Arch Pathol Lab Med 2007;131:1290–1297.
Leroy X, Moukassa D, Copin MC et al. Utility of cytokeratin 7 for distinguishing chromophobe renal cell carcinoma from renal oncocytoma. Eur Urol 2000;37:484–487.
Skinnider BF, Amin MB . An immunohistochemical approach to the differential diagnosis of renal tumors. Semin Diagn Pathol 2005;22:51–68.
Skinnider BF, Folpe AL, Hennigar RA et al. Distribution of cytokeratins and vimentin in adult renal neoplasms and normal renal tissue: potential utility of a cytokeratin antibody panel in the differential diagnosis of renal tumors. Am J Surg Pathol 2005;29:747–754.
Wang CC, Mao TL, Yang WC et al. Underexpression of hepatocyte nuclear factor-1beta in chromophobe renal cell carcinoma. Histopathology 2013;62:589–594.
Kuehn A, Paner GP, Skinnider BF et al. Expression analysis of kidney-specific cadherin in a wide spectrum of traditional and newly recognized renal epithelial neoplasms: diagnostic and histogenetic implications. Am J Surg Pathol 2007;31:1528–1533.
Tan PH, Cheng L, Rioux-Leclercq N et al. Renal tumors: diagnostic and prognostic biomarkers. Am J Surg Pathol 2013;37:1518–1531.
Rao Q, Williamson SR, Zhang S et al. TFE3 break-apart FISH has a higher sensitivity for Xp11.2 translocation-associated renal cell carcinoma compared with TFE3 or cathepsin K immunohistochemical staining alone: expanding the morphologic spectrum. Am J Surg Pathol 2013;37:804–815.
Green WM, Yonescu R, Morsberger L et al. Utilization of a TFE3 break-apart FISH assay in a renal tumor consultation service. Am J Surg Pathol 2013;37:1150–1163.
Nese N, Martignoni G, Fletcher CD et al. Pure epithelioid PEComas (so-called epithelioid angiomyolipoma) of the kidney: a clinicopathologic study of 41 cases: detailed assessment of morphology and risk stratification. Am J Surg Pathol 2011;35:161–176.
He W, Cheville JC, Sadow PM et al. Epithelioid angiomyolipoma of the kidney: pathological features and clinical outcome in a series of consecutively resected tumors. Mod Pathol 2013;26:1355–1364.
Eble JN, Amin MB, Young RH . Epithelioid angiomyolipoma of the kidney: a report of five cases with a prominent and diagnostically confusing epithelioid smooth muscle component. Am J Surg Pathol 1997;21:1123–1130.
Korpershoek E, Favier J, Gaal J et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab 2011;96:E1472–E1476.
Wagner AJ, Remillard SP, Zhang YX et al. Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod Pathol 2013;26:289–294.
Miettinen M, Killian JK, Wang ZF et al. Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. Am J Surg Pathol 2013;37:234–240.
Gill AJ, Lipton L, Taylor J et al. Germline SDHC mutation presenting as recurrent SDH deficient GIST and renal carcinoma. Pathology 2013;45:689–691.
Acknowledgements
We would like to thank Drs Alison Koehler and Edgar Hartle (Mooresville, North Carolina), Dr Tomas Slavic (Pretoria, South Africa) and Dr Ali Amin (Providence, Rhode Island) for contributing cases in consultation and providing additional materials and clinicopathologic information for this study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Williamson, S., Eble, J., Amin, M. et al. Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol 28, 80–94 (2015). https://doi.org/10.1038/modpathol.2014.86
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/modpathol.2014.86
This article is cited by
-
Expanding the clinicopathological spectrum of succinate dehydrogenase-deficient renal cell carcinoma with a focus on variant morphologies: a study of 62 new tumors in 59 patients
Modern Pathology (2022)
-
Classification of renal cell tumors – current concepts and use of ancillary tests: recommendations of the Brazilian Society of Pathology
Surgical and Experimental Pathology (2021)
-
Charakterisierung der unterschiedlichen Entitäten beim Nierenzellkarzinom
Der Urologe (2020)
-
Positive/retained SDHB immunostaining in renal cell carcinomas associated to germline SDHB-deficiency: case report
Diagnostic Pathology (2019)
-
Clinical and morphologic review of 60 hereditary renal tumors from 30 hereditary renal cell carcinoma syndrome patients: lessons from a contemporary single institution series
Medical Oncology (2019)
