
Abstract
Myeloid neoplasms occasionally occur in patients with sickle cell disease, and the underlying connection between the two diseases is unclear. Herein, we retrospectively analyzed four cases of sickle cell disease patients who developed myeloid neoplasm. Age at time of diagnosis ranged from 27 to 59 years with a median of 35.5 years. Two patients were treated with hydroxyurea and the other two with supportive care alone, with one out of the four patients receiving additional treatment with hematopoietic stem cell transplant. Three patients presented with leukocytosis, and the remaining patient presented with pancytopenia. Two patients were diagnosed with myelodysplastic syndrome/myeloproliferative neoplasm, one with myelodysplastic syndrome, and the other with acute myeloid leukemia. All four cases demonstrated certain degrees of myelodysplasia and complex cytogenetic abnormalities with − 7/7q- and/or − 5/5q- or with 11q23 (KMT2A) rearrangement. This cytogenetic profile resembles that seen in therapy-related myeloid neoplasm, suggesting that myeloid neoplasm in the setting of sickle cell disease may represent a subcategory of the disease distinct from de novo myeloid neoplasm in general. Extensive literature review further demonstrates this similarity in cytogenetic profile, as well as in other associated pathologic features. Potential etiology includes therapy for sickle cell disease, disease-related immunomodulation, or disease-related chronic inflammation. We hypothesize that constant hematopoietic hyperplasia, stimulated by a hemolysis-induced cytokine storm, may increase the chance of somatic mutations/cytogenetic aberrations, resulting in transformation of myeloid precursors. This group of myeloid neoplasms seems to herald a dismal clinical outcome, with median survival <1 year, although the exact pathogenesis and biology of the disease remain to be investigated by large cohorts in future studies.
Similar content being viewed by others

Introduction
Sickle cell disease is an autosomal recessive hereditary hemoglobinopathy. The genetic defect involves a point mutation that occurs on the sixth codon of the hemoglobin β subunit gene, resulting in a substitution of glutamic acid with valine [1, 2]. When a patient inherits a mutated allele from each parent, he or she has a homozygous hemoglobin S (hemoglobin SS) that often manifests with severe clinical signs and symptoms, whereas patients with a heterozygous mutant allele (hemoglobin SA), so-called carriers, usually demonstrate minimal clinical symptoms. When a carrier of hemoglobin S is compounded with another specific type of β subunit gene mutation, such as hemoglobin C or β thalassemia, it forms a compound heterozygous (hemoglobin SC or hemoglobin Sβ0) hemoglobinopathy that has a similar clinical course to hemoglobin SS2. With the exception of carriers, all these patients usually manifest with chronic hemolysis owing to unstable hemoglobin, and thus receive similar clinical treatment. The mainstay of clinical management involves conservative treatment with blood transfusion, antibiotic prophylaxis against opportunistic infections, hydration, or other supportive care [1]. Although a hematopoietic stem cell transplant may provide a potential chance of cure, it risks other medical complications such as graft versus host disease, graft rejection, post-transplant infection, and post-transplant neoplasms [3,4,5]. Therefore, the vast majority of sickle cell disease patients initially receive conservative therapy, with hematopoietic stem cell transplant reserved for patients that are refractory to conventional therapy. In addition to supportive care, the disease is often treated with hydroxyurea (hydroxycarbamide) to increase expression of hemoglobin F, and decrease the episode of sickling crisis by diluting hemoglobin S within individual red blood cells [1]. This therapy has greatly improved the survival of patients with sickle cell disease or other equivalent hemoglobinopathies in developed countries, with the majority of the patients now living to adulthood. However, along with this improvement of survival has come the risk of sickle cell disease patients developing secondary diseases, particularly malignancy [6]. The increased risk of sickle cell disease patients developing myeloid neoplasms remains controversial, despite sporadic reports [6, 7], and pathogenesis of myeloid leukemia in this setting is unclear at the present time. Herein, we report a retrospective analysis of four cases of myeloid neoplasm in the setting of sickle cell disease, focusing on pathological features, with extensive review of the literature.
Materials and methods
Case selection
Following approval of the study by the institutional review board of Duke University Medical Center, we retrieved four cases of myeloid neoplasm in the setting of sickle cell disease or equivalent hemoglobinopathy from four different patients (referred to as patients 1–4) within our pathology database during the last 2 years. These four cases (cases 1–4) were collected from a pool of 2206 total patients with sickle cell disease (1048 adults and 1158 children). All the cases were retrospectively evaluated by four hematopathologists (JLN, CMM, ASL, and EW), and the diagnosis of each individual case was confirmed according to the World Health Organization classification of tumors of haematopoietic and lymphoid tissues [8,9,10,11]. Clinical histories and laboratory data, including flow cytometric analyses, cytogenetic studies, and molecular diagnostic tests, were collected, and retrospectively reviewed and analyzed. In addition, we retrieved cases of bone marrow biopsies from five control patients (referred to as control patients 5–9) without evidence of myeloid neoplasm or other neoplasms from the total pool of sickle cell disease patients. All these five cases (control cases 5–9) were retrospectively evaluated in comparison with studied cases 1–4 described above.
Literature review
A PubMed literature search was performed using relevant title words, such as “sickle cell disease” or “sickle cell anemia”, combined with “leukemia”, “myelodysplastic”, “myeloproliferative”, “neoplasm”, or “malignancies” for the case reports of leukemia in the setting of sickle cell disease or equivalent hemoglobinopathy. Previously published cases of myeloid neoplasm in the setting of sickle cell disease or equivalent hemoglobinopathy were reviewed, and the relevant information was collected and analyzed.
Statistical analysis
Statistical analyses were performed with IBM SPSS Statistics, version 24 (www.ibm.com). Overall survival was calculated using Kaplan–Meier survival analysis, generating the estimated median survival and a 95% confidence interval.
Results
Clinical presentation, pathological features, diagnoses, treatments, and follow-up of the four cases of myeloid neoplasm in the setting of sickle cell disease or equivalent hemoglobinopathy are summarized in Table 1.
Clinical presentation
Of the four patients (patients 1–4) with myeloid neoplasm in the setting of sickle cell disease, two were male and two were female. The ages ranged from 27 to 59 years, with a median of 35.5 years, at the time of diagnosis of the myeloid neoplasm. The types of hemoglobinopathy included hemoglobin SS (two patients), hemoglobin SC (one patient), and hemoglobin Sβ0 (one patient). All four patients had a history of pain crises, and had received blood transfusions and antibiotic prophylaxis, in addition to other supportive care, as part of their treatment plan. Two patients (patients 1 and 3) began treatment with hydroxyurea at 9 and 23 years, respectively, before the diagnosis of myeloid neoplasm. The dose of hydroxyurea was started at 400 or 500 mg daily, respectively, with dose escalation to 1500 mg daily for each patient. One patient (patient 4) was found to have human immunodeficiency virus infection 3 years before the development of myeloid neoplasm. Patient 1 developed severe clinical symptoms of sickle cell disease, despite 2 prior years of therapy with hydroxyurea, and subsequently received non-myeloablative-conditioning regimens plus total body irradiation, followed by hematopoietic stem cell transplant with a matched sibling donor, 7 years before the identification of myelodysplastic syndrome. The donor hematopoietic stem cells were completely engrafted 1 month later, and blood hemoglobin electrophoresis demonstrated 98% hemoglobin A without detectable hemoglobin S. However, the patient’s blood cell origin became chimeric 1 year after donor stem cell infusion, the graft was completely lost 4 years later (2 years before the diagnosis of myelodysplastic syndrome), and the patient subsequently received conventional supportive therapy. Patient 3 was noted to have cervical lymphadenopathy, 1 year before the diagnosis of myeloid neoplasm, the biopsy of which demonstrated evidence of a human herpes virus 8-positive B-cell lymphoproliferative disorder. However, because this was a localized lesion and was completely excised, the patient received no further treatment, and remained free of lymphoma at the time of leukemic presentation. At the time of myeloid neoplasm diagnosis, three (patients 2–4) out of four patients presented with leukocytosis with associated moderate to severe anemia; patient 3 also demonstrated moderate thrombocytopenia, and the remaining patient (patient 1) manifested with moderate to severe pancytopenia.
Bone marrow biopsies from the five control patients (patients 5–9) with sickle cell disease but no evidence of myeloid neoplasm were retrospectively reviewed, and the features are summarized in Table 2. The patient age in this group ranged from 9 to 61 years with a median age of 22 years. The types of hemoglobinopathy included hemoglobin SS (two patients), hemoglobin SC (two patients), and hemoglobin Sβ+ (one patient). Of the five patients, four had bone marrow examination performed to rule out myeloid neoplasm, and the remaining patient (patient 9) was found to have serum paraprotein, which prompted a bone marrow examination to exclude plasma cell myeloma. At the time of biopsy, all the patients demonstrated moderate to severe microcytic anemia consistent with sickle hemoglobinopathy. In addition, patients 6 and 8 exhibited leukocytosis and thrombocytosis, respectively.
Morphologic evaluation and immunohistochemical analysis
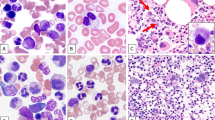
Of the four studied cases, three cases (cases 2–4) showed leukocytosis on examination of the peripheral blood film, and the remaining case (case 1) exhibited pancytopenia. In addition, all the peripheral blood films demonstrated morphologic distortion of red blood cells with anisopoikilocytosis including a few to many target cells, rare sickle cells, and various circulating erythroid precursors. These features of peripheral blood examination were consistent with an underlying sickle hemoglobinopathy, similar to the features seen in control patients with sickle cell disease but without myeloid neoplasm (Table 2). In bone marrow examination, all the cases (cases 1–4) exhibited hyperplastic hematopoiesis and various degrees of myelodysplasia. In case 1, peripheral blood film showed pancytopenia in addition to morphologic distortion of red blood cells with many target cells, increased polychromasia, and a few circulating nucleated red blood cells; circulating immature granulocytes were also noted, but blasts were not identified (Fig. 1a). Despite peripheral pancytopenia, the bone marrow biopsy for this case showed hypercellularity with erythroid and megakaryocytic hyperplasia and mild to moderate dysplasia (Fig. 1b–d); and blasts were not increased. Immunohistochemistry revealed scattered hematopoietic progenitor cells that were positive for p53 (Fig. 1d, inset). Case 2 demonstrated similar morphologic features to those seen in case 1, except for more severe megakaryocytic dysplasia. Peripheral blood smear demonstrated marked erythroblastosis, circulating micromegakaryocytes and a few blasts (Fig. 2a). The bone marrow core section showed hypercellularity with erythroid hyperplasia and increased micromegakaryocytes (Fig. 2c). Blasts were slightly increased comprising ~5% of the total bone marrow cellularity. A proliferation of micromegakaryocytes is highlighted by CD61 immunohistochemical stain (Fig. 2c). p53 stain was positive in many immature hematopoietic progenitors including those reminiscent of small megakaryocytes (Fig. 2d). The peripheral blood examination in case 3 showed marked leukocytosis with absolute monocytosis, a few circulating immature granulocytes and rare small megakaryocytes, in addition to morphologic distortion of red blood cells (Fig. 3a). Monocytes exhibited atypical morphology including a few immature forms and rare promonocytes. Examination of the bone marrow aspirate smear showed hyperplastic hematopoiesis with moderate dysplasia in granulopoiesis and megakaryopoiesis (Fig. 3b). Blasts were slightly increased comprising 5% of the total nucleated cells. Bone marrow core biopsy demonstrated hypercellularity with increased small megakaryocytes (Fig. 3c) and many immature hematopoietic cells with strong p53 staining (Fig. 3d). In case 4, markedly increased blasts were seen on the peripheral blood film and bone marrow aspirate smear, comprising 37 and 72% of the nucleated cells, respectively. A significant fraction of the blasts showed indented to folded nuclei and moderate to abundant light basophilic cytoplasm, features suggestive of monocytoid differentiation (Fig. 4a–c). In addition, erythropoiesis displayed megaloblastoid changes with occasional nuclear irregularity. The section of bone marrow core biopsy demonstrated hypercellularity with replacement of bone marrow elements by blasts (Fig. 4d), many of which were positive for p53 (Fig. 4d, inset).

Peripheral blood and bone marrow examination in case 1. a Peripheral blood smear shows red blood cell distortion with increased central pallor and many target cells, one immature granulocyte and nucleated red blood cell (inset). Wright–Giemsa stain, ×1000. b and c Wright–Giemsa-stained bone marrow aspirate smear shows erythroid hyperplasia and left-shifted myelopoiesis, and dysplasia in erythropoiesis (arrows) and megakaryopoiesis (arrow heads), ×1000. d Bone marrow core biopsy shows a hypercellular bone marrow with erythroid and megakaryocytic hyperplasia. Note many megakaryocytes display their small size and hypolobate nuclei. Hematotoxilin and eosin (H&E) stain, ×200. Inset shows a few p53-postive cells. Note weak to moderate nuclear staining in hematopoietic progenitor cells. p53 stain, ×400

Peripheral blood and bone marrow examination in case 2. a Peripheral blood smear shows red cell distortion, nucleated red blood cells and circulating micromegakaryocytes (arrow heads). Inset shows one circulating blast. Wright–Giemsa stain, ×1000. b Bone marrow core biopsy shows a hypercellular bone marrow with erythroid hyperplasia and increased micromegakaryocytes (arrow heads). Micromegakaryocytes are identified by their eosinophilic cytoplasm and eccentric hyperchromatic nuclei, which can only be recognized after comparing with CD61 stain (c). H&E stain, ×400. c Many micromegakaryocytes are highlighted by CD61 stain on the section of bone marrow biopsy, ×400. d p53 stain on the bone marrow biopsy. Note an islet of early normoblasts that are weakly positive in their nuclei at the top center, in addition to strong nuclear staining in scattered or clustered hematopoietic progenitor cells, ×400

Peripheral blood and bone marrow examination in case 3. a Peripheral blood smear shows circulating immature granulocytes, atypical monocytes and one atypical neutrophil in a background of hypochromic red blood cells with anisopoikilocytosis and increased target cells. The inset shows a circulating small megakaryocyte with naked nucleus. Wright–Giemsa stain, ×1000. b Bone marrow aspirate smears show a few dysplastic megakaryocytes (arrow heads) and many hypogranular neutrophils (arrows). Wright–Giemsa stain, ×1000. c Bone marrow core biopsy demonstrates hypercellularity with hyperplastic hematopoiesis. Note the increased immature precursors and a few small megakaryocytes. H&E stain, ×400. d p53 stain on the bone marrow biopsy showing many positive hematopoietic progenitor cells, ×400

Morphologic evaluation of bone marrow biopsy in case 4. a Peripheral blood smear shows circulating blasts and rare nucleated red blood cells (inset). Note one atypical monocyte with cytoplasmic vacuoles, suggesting a possible monocytic differentiation of the leukemic cells. Wright–Giemsa stain, ×1000. b and c Bone marrow aspirate smears show markedly increased blasts with morphologic features consistent with monoblasts or promonocytes. Note an erythroid precursor undergoing karyorrhexis (arrow) in image b. Wright–Giemsa stain, ×1000. d Bone marrow core biopsy demonstrates hypercellularity and replacement of bone marrow by leukemic blasts. H&E stain, ×200. Inset shows p53 staining in mononucleated cells, probably blasts. The stain was performed on a post-transplant bone marrow biopsy with relapsed acute myeloid leukemia instead of the pretreatment tissue block that was not available
Similarly, peripheral blood smears in the control patients (cases 5–9) with sickle cell disease but without myeloid neoplasm showed anisopoikilocytosis with many target cells and increased polychromasia (Fig. 5a). However, in contrast to the sickle cell disease patients with myeloid neoplasm, none of the control cases demonstrated circulating blasts, circulating small megakaryocytes, significant dysplasia other than erythroid dyspoiesis owing to hemolysis-induced stress or increase in myeloid blasts in the bone marrow biopsy. Of interest, the control patient 8 manifested with worsening microcytic anemia and eventual development of thrombocytosis. Although significant myelodysplasia or increase in blasts was not identified, the bone marrow aspirate smear showed active trilineage hematopoiesis (Fig. 5b) with increased sea blue histiocytes (Fig. 5c), a feature that has been previously described in bone marrow biopsies of sickle cell disease patients, besides those of myeloproliferative neoplasm. In all control cases (cases 5–9), bone marrow biopsy exhibited active hematopoiesis with erythroid hyperplasia and increased hemosiderin pigments (Fig. 5d). p53 immunohistochemical stain was negative in all five control cases (Fig. 5d, inset).

Peripheral blood smear and bone marrow biopsies in control cases (cases 5–9). a Peripheral blood smear from case 6 demonstrates red blood cells with anisopoikilocytosis including many with enlarged central pallor and increased target cells as well as single immature neutrophil with coarse azurophilic granules. Wright–Giemsa stain, ×1000. b Bone marrow aspirate smear from case 8 shows active hematopoiesis with erythroid hyperplasia. Wright–Giemsa stain, ×1000. c The same aspirate smear shows two sea blue histiocytes. Wright–Giemsa stain, ×1000. Inset exhibits a low magnification field with increased sea blue histiocytes. Wright–Giemsa stain, ×100. Arrows in images b and c indicate erythroid precursors with irregular nuclear contours or karyorrhexis. e Bone marrow core biopsy from case 9 demonstrates active hematopoiesis with erythroid hyperplasia. Note increased hemosiderin pigments throughout the hematopoietic elements. H&E stain, ×400. e, p53 stain on bone marrow core biopsy of case 9 showing negative staining, ×400
Flow cytometric analysis
Flow cytometric analysis was performed on the bone marrow aspirate of all the cases in our series (Table 1). Of these, two cases (cases 1 and 2) demonstrated no significant increase in myeloid blasts, one case (case 3) showed a borderline increase in myeloid blasts and the remaining case (case 4) detected a marked increase in blasts with monocytic differentiation. In those cases without a significant increase in blasts, an atypical immunophenotypic profile in hematopoietic elements was noted in flow cytometric analysis. Case 1 showed diminished CD10 and CD16 expression in neutrophils and their precursors, and both granulocytes and monocytes had aberrant expression of CD56. Flow cytometry in case 2 showed a suboptimal result with a significant amount of cell debris and nonspecific binding. Nonetheless, a small but discrete population of myeloid blasts was detected, which expressed aberrant CD7. In case 3, increased atypical myelomonocytic cells were detected, in addition to 3% myeloid blasts. These myelomonocytic cells exhibited reduced side scatter, and expression of CD11C, but diminished expression of CD10 and CD16.
Flow cytometric analysis of bone marrow biopsies from all control cases (cases 5–9) demonstrated no phenotypic abnormalities in each analysis.
Cytogenetic studies
Chromosomal analysis was performed on the bone marrow biopsies of each case, and demonstrated clonal abnormalities with complex karyotypic abnormalities in all four studied cases (cases 1–4; Table 1). Two of the cases (case 1 and 3) showed presence of subclones suggestive of clonal evolutions and genomic instability. In case 2, only four metaphase cells were collected, three of which demonstrated complex cytogenetic abnormalities. The karyotypic abnormalities in these cases suggested loss or partial loss of long arm in chromosome 7 in two cases (cases 1 and 3), loss or partial loss of long arm in chromosome 5 in two cases (cases 2 and 3) and rearrangement of KMT2A gene at 11q23 in one case (case 4). These specific changes were confirmed with interphase fluorescence in situ hybridization (FISH). Of note, the karyotypic abnormalities in case 3 contained both del (5q) and del (7q) in >90% of the total interphase nuclei. In this case, a specific FISH analysis for BCR/ABL fusion was also performed to rule out chronic myeloid leukemia, and it turned out to be negative.
Cytogenetic analysis of bone marrow biopsies from control cases 5–9 demonstrated no acquired karyotypic abnormalities. Of note, case 6 and case 8 showed inversion of chromosome 9 and t (7;11) translocation, respectively. Both changes have been considered constitutional rather than acquired abnormalities. Interphase FISH analysis demonstrated no myelodysplastic syndrome-related abnormalities in cases 5–8 and no myeloma-related abnormalities in case 9.
Molecular diagnostic analysis
Molecular diagnostic tests were performed on cases 1–4 at the time of bone marrow biopsy and subsequent diagnosis of myeloid neoplasm. Of these, cases 2 and 3 had next-generation sequencing analysis for myeloid panel performed, which detected somatic point mutations of the TP53 gene, occurring on exon 5 and exon 6 of the gene, respectively. The variant allele frequency was determined as 0.224 and 0.907, respectively. Both these point mutations were missense and predicted to be deleterious to the function of p53 protein. Other pathogenic mutations or variants, including mutations on JAK2, CALR, and MPL, were not detected by our myeloid panel in these two cases. In addition, case 2 had specific PCR assays performed for JAK2 and CALR mutations, which were negative as seen in next generation sequencing analysis. Quantitative RT-PCR for BCR/ABL fusion transcripts were also performed to rule out chronic myeloid leukemia in this case, and it turned out to be negative. In case 4, specific PCR assays were performed for somatic FLT3, IDH1, and IDH2 mutations, which were negative in this biopsy with acute myeloid leukemia. Case 4 had next-generation sequencing analysis performed on a post-transplant bone marrow biopsy with leukemic relapse, and demonstrated no pathogenic mutations including a wild type TP53 gene. Periodic PCR based analysis of short tandem repeats were performed on blood samples of patient 1 after allogenic hematopoietic stem cell transplant (7 years before the diagnosis of myeloid neoplasm). Initial test results demonstrated that this patient’s donor hematopoietic stem cells remained well engrafted until one year later, when the donor’s cell fraction started decreasing. Two years before the diagnosis of myeloid neoplasm, the analysis of short tandem repeats showed a complete loss of donor’s graft, including an assay performed on the bone marrow sample upon which the diagnosis of myelodysplastic syndrome with multilineage dysplasia was rendered.
Molecular tests were not performed on bone marrow biopsies from the five control cases without evidence of myeloid neoplasm.
Integrated diagnosis and classification for the four cases with myeloid neoplasm
The final diagnosis for each individual case was rendered based on its clinical presentation, morphologic features, flow cytometric analysis, and cytogenetic studies. Of three cases without significant increase in blasts, two (cases 2 and 3) were diagnosed as myelodysplastic/myeloproliferative neoplasm and the remaining case (case 1) was diagnosed as myelodysplastic syndrome with multilineage dysplasia. Case 3 was further classified as chronic myelomonocytic leukemia-1, proliferative type, and case 2 as myelodysplastic/myeloproliferative neoplasm, unclassifiable. Case 4 was diagnosed as acute myeloid leukemia with myelodysplasia-related changes according to the World Health Organization Classification (acute myeloid leukemia M5b per French, American, and British classification). The classification of case 4 was based primarily on the cytogenetic results, even though the leukemia cells displayed apparently monocytoid differentiation.
Clinical course
All four patients in our series (patients 1–4) had treatment and follow-up information available (Table 1), and all were treated for myeloid neoplasm with chemotherapy followed by hematopoietic stem cell transplantation (or were intended to have this treatment plan). The clinical follow-up period was relatively short, ranging from 2 to 21 months, with a median of 3.5 months. Two patients (patients 3 and 4) died of disease progression despite aggressive treatment. Patient 3 died of multiorgan failure owing to systemic leukemic infiltration, 2 months following diagnosis. Patient 4 received high dose chemotherapy followed by dual umbilical cord blood stem cell transplantation. In the case of patient 4, a bone marrow biopsy after chemotherapy showed no evidence of leukemia, with a cytogenetic result converted to normal karyotype, and negative FISH for KMT2A rearrangement. The donor stem cells were never engrafted in patient 4, and acute myeloid leukemia relapsed in a subsequent biopsy (1 month after the transplant, and 6 months after the diagnosis of acute myeloid leukemia). The patient then began a new chemotherapy regime with addition of oral venetoclax, with a plan of another dual umbilical cord blood stem cell infusion. Unfortunately, this patient later died of disease progression and treatment complications. The other two patients remained alive, but their follow-up was too short to make conclusive assessment of the clinical outcome. Of these two patients, one (patient 1) had received high-dose chemotherapy followed by allogenic hematopoietic stem cell transplantation, and the remaining patient (patient 2) was initially treated with immunomodulated therapy with Lenalidomide, intravenous immunoglobulin, and prednisone, but the myeloid neoplasm persisted with dysplastic neutrophils and rare circulating small megakaryocytes identified in the peripheral blood film. Patient 2 anticipated to receive allogenic hematopoietic stem cell transplantation once correct donor was identified. Patient 1 received conditioning regimens including Busulfen, fludarabine, and anti-thymocyte globulin before allogenic hematopoietic stem cell transplantation with a matched sibling donor. A complete donor cell engraftment was achieved 1 month after the procedure, and remained engrafted up to 21 months after the diagnosis of myelodysplastic syndrome with multilineage dysplasia, the time when this manuscript was prepared. Laboratory evaluation demonstrated normalized hemoglobin components in this patient.
Reviews of literature and analysis of the pooled data
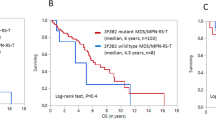
In this literature review, 11 additional cases of myeloid neoplasm in the setting of sickle cell disease were identified to have adequate description of pathologic features and/or some clinical follow-up [12,13,14,15,16,17,18,19,20,21,22]. Cases of myeloid neoplasm in the setting of sickle cell disease without description of pathologic features or clinical course were excluded from the review and subsequent analysis. The clinical and pathologic information for these cases were summarized in Table 3, together with the cases in the current series. Of 15 cases in total, seven patients were female and eight were male. Median age at diagnosis was 37 years with a range of 17–59 years. Approximately half of the patients were treated with hydroxyurea with a median interval of 8 years from treatment to the diagnosis of myeloid neoplasm. The types of myeloid neoplasm included myelodysplastic syndrome in five (33%) cases, acute myeloid leukemia in four (27%), chronic myeloid leukemia in four (27%), and myelodysplastic/myeloproliferative neoplasm in two (13%) cases. Ten non-chronic myeloid leukemia cases had cytogenetic information available, and all demonstrated complex abnormalities, including −5/5q-, as a component, in eight (80%) cases and −7/7q- in seven (70%) cases. All the patients with chronic myeloid leukemia received treatment with imatinib, but only one had adequate clinical follow-up, who was alive 72 months after the treatment. All the other patients (non-chronic myeloid leukemia) who had treatment information available were treated with combination chemotherapy, with or without hematopoietic stem cell transplant. Excluding the cases of chronic myeloid leukemia, 10 cases had clinical follow-up with median follow-up of 7 months (range = 1–24 months). Eight of the 10 patients died of the disease during the period of follow-up. The median overall survival was estimated at 7 months with 95% confidence interval of 3–12 months (Fig. 6).

Kaplan–Meier survival analysis of 10 cases of myeloid neoplasm in the setting of sickle cell disease. These include six reported cases with clinical follow-up, and four cases in our series (Table 2). Two cases are censored at 5 and 21 months as indicated by tick marks
Discussion
In this manuscript, we describe the development of a myeloid neoplasm in the setting of sickle cell disease, a rare event, in four patients. Could this myeloid neoplasm be coincidental with sickle cell disease or otherwise, is there an intrinsic linkage between myeloid neoplasm and hemoglobinopathy in this particular patient population? In other words, does a patient with sickle cell disease or equivalent hemoglobinopathy have an increased risk of myeloid leukemia? It has been debated whether or not patients with sickle cell disease have an increased risk of developing malignant neoplasms, especially hematolymphoid malignancies. In the past, the risk of malignancies associated with sickle cell disease had not been well defined, despite sporadic case reports and case series studies published [12,13,14,15,16,17,18,19,20,21,22]. Seminog et al. [6] have, via an epidemiology study in England, reported 2.1-fold increased risk of combined malignancies in patients with sickle cell disease, and the risk was higher for hematolymphoid neoplasms, particularly for myeloid neoplasm (~10-fold). Confounding factors such as ethnicity was excluded by using a black population without sickle cell disease as a control cohort in this retrospective analysis. Brunson et al. [7] calculated standard incidence ratios among patients with sickle cell disease in California, and reported an increased risk for hematolymphoid neoplasm, which is lower than that reported by Seminog et al., and no increase in the risk of combined malignancies. Both studies collected >6000 sickle cell disease patients and much more for control cohorts. In this series, the rate of myeloid leukemia in the setting of sickle cell disease is calculated as 0.18% (4/2206), which is similar to that reported by Seminog et al. (14/7516 or 0.19%), and higher than reported by Brunson et al. (6/6423 or 0.09%). An increased risk of myeloid neoplasm in patients with sickle cell disease is also suggested by the significantly younger age of our patients and reported cases (Table 3) compared with counterparts in the general population. It remains uncertain how the clinical presentation and pathologic features of myeloid neoplasm in the setting of sickle cell disease differ from those seen within the general population, and exactly what the possible underlying mechanisms are for the development of leukemia in this particular context. Besides the epidemiology studies already discussed, sporadic case reports and small series have been published (12–22), although the analyses of clinicopathologic features, particularly cytogenetic profile and molecular landscape, have never been reported in MN occurring in the setting of sickle cell disease.
Theoretically, if myeloid neoplasm is simply coincidental with sickle cell disease, its pathologic features should resemble de novo myeloid neoplasm in the general population. Instead, all four cases of myeloid neoplasm in our series demonstrated features suggestive of underlying myelodysplasia by morphologic evaluation. In addition, all four cases in our series showed complex cytogenetic abnormalities including loss or partial loss of chromosome 5 and/or chromosome 7 or KMT2A rearrangement. Two cases demonstrated subclone heterogeneity suggestive of genomic instability in the leukemia clone. A similar cytogenetic profile is noted in the reported cases of myeloid neoplasm in the setting of sickle cell disease (Table 3) [12,13,14,15,16,17,18,19,20,21,22]. Of interest, these morphologic features and cytogenetic profile are often associated with secondary myeloid neoplasm per the literature [23,24,25,26,27], prompting consideration of therapy as a cause for the development of myeloid neoplasm in this particular patient population. According to previous studies, myeloid neoplasm associated with alkylating agents and/or ionizing radiation tends to have a much higher proportion of unbalanced karyotypic abnormalities, including complex cytogenetic abnormalities with loss or partial loss of chromosomes 7q and/or 5q, than its de novo counterpart [23, 25,26,27,28]. Concordantly, two cases in our series demonstrated pathogenic mutations in the TP53 gene, which has been associated with therapy-related myeloid neoplasm via a possibly selective enrichment of the mutant clone [29, 30]. The mutant burden in each case correlates with upregulated p53 protein in our series, and the pattern is consistent with that observed in myeloid neoplasm with complex karyotype abnormalities reported in the recent literature [31, 32]. Of four patients in our series, only patient 1 was exposed to alkylating agents and ionizing radiation as a conditioning regimen before allogenic hematopoietic stem cell transplantation, and the interval between the procedure and diagnosis of myelodysplastic syndrome falls in the range of latency reported in the literature [23, 27, 31]. However, studies also suggest that hematopoietic stem cell transplantation for sickle cell disease does not increase the risk of developing myeloid neoplasm, compared with patients who have not undergone stem cell transplant [3]. Other patients in our series and the majority of reported cases did not involve stem cell transplantation before development of myeloid neoplasm, suggesting causes other than hematopoietic stem cell transplantation. In our series, two patients (patients 1 and 3) had been treated with hydroxyurea, starting 9 and 23 years, respectively, before the diagnosis of myeloid neoplasm. As the Food and Drug Administration’s approval of hydroxyurea for the treatment of sickle cell disease, its safety has been questioned for long-term use. Despite these safety concerns, of all the patients who develop myeloid neoplasm, only a minority have had a history of hydroxyurea treatment, and population based studies demonstrate no significant increase in the risk of myeloid neoplasm in the patients with long-term hydroxyurea use at the therapeutic dose level for sickle cell disease [7]. Alternatively, owing to chronic hemolysis and frequent blood transfusions required by sickle cell disease patients, there is often increased chronic inflammation and iron overload in hematopoietic tissue. Theoretically, these factors could possibly increase the risk of myeloid neoplasm by excessive production of reactive oxygen species that induce genomic damage and somatic mutations [33]. On the other hand, chronic hemolysis in sickle cell disease could lead to persistent stress on the bone marrow, forcing a disordered replication of hematopoietic elements via a cytokine-mediated compensative response [1, 2]. Somatic mutations or cytogenetic aberrations are generally increased in cells with high turnover rate, such as those undergoing constant hematopoietic hyperplasia in patients with sickle cell disease [34, 35]. Finally, patients with sickle cell disease may be associated with other genetic defects that cause genomic instability and thus hereditary predisposition to somatic mutations that lead to the development of myeloid neoplasm. Such a possibility of genetic predisposition is less likely, given the relatively low risk and sporadic cases reported in the literature. Nonetheless, the increased risk of hematolymphoid neoplasms in sickle cell disease could be owing to a complex mechanism involving multiple factors that influence leukemogenesis. The recent advance in high throughput sequencing techniques may provide an opportunity for a comprehensive study of myeloid neoplasm in the setting of sickle cell disease, in comparison with therapy-related myeloid neoplasm and its de novo counterpart, which may shed light on the genomic profile and pathogenesis of myeloid neoplasm in this particular patient population.
The exact role played by sickle cell disease as a factor in determining the clinical outcome of myeloid neoplasm is unclear at the present time. The clinical management of sickle cell disease patients is definitely complicated by the dual comorbidities of intrinsic hemoglobinopathy versus potential sickling episodes triggered by certain chemotherapeutic drugs. Given its association with high-risk cytogenetic abnormalities and possible secondary pathogenesis, myeloid neoplasm in the setting of sickle cell disease might be equivalent to its therapy-related counterpart and herald a dismal clinical outcome. This seems to be in keeping with the aggressive clinical course observed in our patients and the cases reported in the literature (Fig. 6). Although the case number is small and follow-up is short in our series, two out of four patients died of the disease progression despite aggressive treatment, and another was apparently refractory to the treatment. One patient (patient 1) received allogenic hematopoietic stem cell transplantation, and responded well with complete remission of myeloid neoplasm and disappearance of abnormal hemoglobin. Therefore, hematopoietic stem cell transplantation seems to be an optimal therapeutic modality for myeloid neoplasm in this clinical setting, given its additional therapeutic effect in the treatment of sickle cell disease. Interestingly, patients receiving hematopoietic stem cell transplant for sickle cell disease may have the increased risk of myeloid neoplasm normalized as long as the graft is successfully maintained. In fact, patient 1 in our series started losing the donor graft 1 year after the transplant for sickle cell disease, and developed myeloid neoplasm 2 years after completely losing it, raising a hypothesis of loss of protective effect of the donor graft and resuming the risk of myeloid neoplasm. Nonetheless, the exact biological behavior of myeloid neoplasm in this particular clinical setting and proper therapeutic approach remain to be determined with large cohorts in the future.
In conclusion, myeloid neoplasm in the setting of sickle cell disease likely represents a subgroup of the disease that exhibits clinicopathologic distinction from de novo myeloid neoplasm in general. It demonstrates cytogenetic and molecular profiles, as well as biological behavior, that resemble those seen in therapy-related myeloid neoplasm. The potential etiologies include therapy for sickle cell disease, disease-related immunomodulation, or disease-related chronic inflammation. We hypothesize that constant hematopoietic hyperplasia stimulated by a hemolysis-induced cytokine storm may increase the chance of somatic mutations and chromosomal aberrations, resulting in transformation of myeloid precursors, though the exact pathogenesis of the disease needs to be elucidated in future studies.
References
Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376:1561–73.
Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nat Rev Dis Prim. 2018;4:18010.
Gluckman E, Cappelli B, Bernaudin F, Labopin M, Volt F, Carreras J, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2017;129:1548–56.
Arnold SD, Bhatia M, Horan J, Krishnamurti L. Haematopoietic stem cell transplantation for sickle cell disease – current practice and new approaches. Br J Haematol. 2016;174:515–25.
Shenoy S, Angelucci E, Arnold SD, Baker KS, Bhatia M, Bresters D, et al. Current results and future research priorities in late effects after hematopoietic stem cell transplantation for children with sickle cell disease and thalassemia: a consensus statement from the second pediatric blood and marrow transplant consortium international conference on late effects after pediatric hematopoietic stem cell transplantation. Biol Blood Marrow Transpl. 2017;23:552–61.
Seminog OO, Ogunlaja OI, Yeates D, Goldacre MJ. Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J R Soc Med. 2016;109:303–9.
Brunson A, Keegan THM, Bang H, Mahajan A, Paulukonis S, Wun T. Increased risk of leukemia among sickle cell disease patients in California. Blood. 2017;130:1597–9.
Brunning RD, Bennett JM, Matutes E, et al. Myelodysplastic syndrome with multilineage dysplasia. In: Swerdlow SH, Campo E, Harris NL, et al. (eds) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. revised. 4th edn. France: IARC Press; 2017. p. 111–3.
Orazi A, Bennett JM, Bain BJ, et al. Myelodysplastic/mueloproliferative neoplasm, unclassifiable. In: Swerdlow SH, Campo E, Harris NL, et al. (eds) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. revised. 4th edn. France: IARC Press; 2017. p. 95–96.
Orazi A, Bain BJ, Bennett JM, et al. Chronic myelomonocytic leukaemia. In: Swerdlow SH, Campo E, Harris NL, et al. (eds) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. revised. 4th edn. France: IARC Press; 2017. p. 82–86.
Arber DA, Porwit A, Brunning RD, et al. Acute myeloid leukaemia with myelodysplasia-related changes. In: Swerdlow SH, Campo E, Harris NL, et al. (eds) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. revised. 4th edn. France: IARC Press; 2017. p. 150–2.
Aumont C, Driss F, Lazure T, Picard V, Creidy R, De Botton S, et al. Myelodysplastic syndrome with clonal cytogenetic abnormalities followed by fatal erythroid leukemia after 14 years of exposure to hydroxyurea for sickle cell anemia. Am J Hematol. 2015;90:E131–E132.
Zemenides S, Erblich T, Luqmani A, Bain BJ. Peripheral blood features of acute myeloid leukemia with myelodysplasia-related changes developing in a patient with sickle cell anemia. Am J Hematol. 2014;89:1010.
Wilson S. Acute leukemia in a patient with sickle-cell anemia treated with hydroxyurea. Ann Intern Med. 2000;133:925–6.
Baz W, Najfeld V, Yotsuya M, Talwar J, Terjanian T, Forte F. Development of myelodysplastic syndrome and acute myeloid leukemia 15 years after hydroxyurea use in a patient with sickle cell anemia. Clin Med Insights Oncol. 2012;6:149–52.
Taylor JG, Darbari DS, Maric I, McIver Z, Arthur DC. Therapy-related acute myelogenous leukemia in a hydroxyurea-treated patient with sickle cell anemia. Ann Intern Med. 2011;155:722–4.
Chen L, Zhuang M, Shah HQ, Lin JH. Chronic myelogenous leukemia in sickle cell anemia. Arch Pathol Lab Med. 2005;129:423–4.
Murphy M, Close J, Lottenberg R, Rajasekhar A. Effectiveness of imatinib therapy for sickle cell sanemia and chronic myeloid leukemia. Am J Med Sci. 2014;347:254–5.
Sallam M, Alsuliman A, Alahmed H, Alabdulaali M. Chronic myelogenous leukemia in sickle cell/beta 0 -thalassemia. Indian J Pathol Microbiol. 2011;54:597–8.
Stankovic Stojanovic K, Thiolière B, Garandeau E, Lecomte I, Bachmeyer C, Lionnet F. Chronic myeloid leukaemia and sickle cell disease: could imatinib prevent vaso-occlusive crisis? Br J Haematol. 2011;155:271–2.
Janakiram M, Verma A, Wang Y, Budhathoki A, Suarez Londono J, Murakhovskaya I, et al. Accelerated leukemic transformation after haplo-identical transplantation for hydroxyurea-treated sickle cell disease. Leuk Lymphoma. 2018;59:241–4.
Stricker RB, Linker CA, Crowley TJ, Embury SH. Hematologic malignancy in sickle cell disease: Report of four cases and review of the literature. Am J Hematol. 1986;21:223–30.
Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102:43.
Fianchi L, Pagano L, Piciocchi A, Candoni A, Gaidano G, Breccia M, et al. Characteristics and outcome of therapy-related myeloid neoplasms: Report from the Italian network on secondary leukemias. Am J Hematol. 2015;90:E80–E85.
Claerhout H, Lierman E, Michaux L, Verhoef G, Boeckx N. A monocentric retrospective study of 138 therapy-related myeloid neoplasms. Ann Hematol. 2018;97:2319–24.
McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer. 2017;17:513–27.
Vardiman JW, Matutes E, Arber DA, et al. Therapy-related myeloid neoplasms. In: Swerdlow SH, Campo E, Harris NL, et al. eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. revised. 4th edn. France: IARC Press; 2017. p. 153–5.
Schoch C, Kern W, Schnittger S, Hiddemann W, Haferlach T. Karyotype is an independent prognostic parameter in therapy-related acute myeloid leukemia (t-AML): an analysis of 93 patients with t-AML in comparison to 1091 patients with de novo AML. Leukemia. 2003;18:120.
Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552–5.
Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res. 2015;39:348–54.
Saft L, Karimi M, Ghaderi M, Matolcsy A, Mufti GJ, Kulasekararaj A, et al. p53 protein expression independently predicts outcome in patients with lower-risk myelodysplastic syndromes with del(5q). Haematologica. 2014;99:1041–9.
McGraw KL, Nguyen J, Komrokji RS, Sallman D, Al Ali NH, Padron E, et al. Immunohistochemical pattern of p53 is a measure of TP53 mutation burden and adverse clinical outcome in myelodysplastic syndromes and secondary acute myeloid leukemia. Haematologica. 2016;101:e320–23.
Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol. 2015;12:584.
Alves PM, Martins PRJ, Dias FdL, Burbano RM, Bianchi Mde L, Antunes LM. Sensitivity to cisplatin-induced mutations and elevated chromosomal aberrations in lymphocytes from sickle cell disease patients. Clin Exp Med. 2008;8:31–35.
Zhang J, MacLennan ICM, Liu Y-J, Lane PJL. Is rapid proliferation in B centroblasts linked to somatic mutation in memory B cell clones? Immunol Lett. 1988;18:297–9.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Li, Y., Maule, J., Neff, J.L. et al. Myeloid neoplasms in the setting of sickle cell disease: an intrinsic association with the underlying condition rather than a coincidence; report of 4 cases and review of the literature. Mod Pathol 32, 1712–1726 (2019). https://doi.org/10.1038/s41379-019-0325-6
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41379-019-0325-6
This article is cited by
-
Clonal selection of hematopoietic stem cells after gene therapy for sickle cell disease
Nature Medicine (2023)
-
Sickle cell disease and acute leukemia: one case report and an extensive review
Annals of Hematology (2023)
-
Fertility-preserving myeloablative conditioning using single-dose CD117 antibody-drug conjugate in a rhesus gene therapy model
Nature Communications (2023)
-
Effects of Erythrocytapheresis Procedures on Delayed Bone Marrow Conversion in Sickle Cell Disease
Clinical Hematology International (2022)
-
Lentiviral globin gene therapy with reduced-intensity conditioning in adults with β-thalassemia: a phase 1 trial
Nature Medicine (2022)
