
Abstract
Epigenetics governs a chromatin state regulatory system through five key mechanisms: DNA modification, histone modification, RNA modification, chromatin remodeling, and non-coding RNA regulation. These mechanisms and their associated enzymes convey genetic information independently of DNA base sequences, playing essential roles in organismal development and homeostasis. Conversely, disruptions in epigenetic landscapes critically influence the pathogenesis of various human diseases. This understanding has laid a robust theoretical groundwork for developing drugs that target epigenetics-modifying enzymes in pathological conditions. Over the past two decades, a growing array of small molecule drugs targeting epigenetic enzymes such as DNA methyltransferase, histone deacetylase, isocitrate dehydrogenase, and enhancer of zeste homolog 2, have been thoroughly investigated and implemented as therapeutic options, particularly in oncology. Additionally, numerous epigenetics-targeted drugs are undergoing clinical trials, offering promising prospects for clinical benefits. This review delineates the roles of epigenetics in physiological and pathological contexts and underscores pioneering studies on the discovery and clinical implementation of epigenetics-targeted drugs. These include inhibitors, agonists, degraders, and multitarget agents, aiming to identify practical challenges and promising avenues for future research. Ultimately, this review aims to deepen the understanding of epigenetics-oriented therapeutic strategies and their further application in clinical settings.
Similar content being viewed by others

Introduction
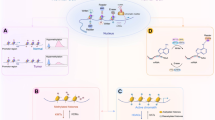
From a historical perspective, the term “epigenetics” was first introduced by Conrad Waddington in 1942 to describe heritable changes in gene function that do not involve alterations to the DNA sequence, leading to changes in biological phenotypes. Following nearly a century of rigorous research, a diverse array of epigenetic-modifying enzymes has been identified, and the elucidation of distinct molecular mechanisms has established epigenetics as a robust discipline.1 Presently, epigenetics is defined as a chromatin state regulatory system comprised of five principal mechanisms: DNA modifications,2 histone modifications,3 RNA modifications,4 chromatin remodeling,5 and the regulation based on non-coding RNA (ncRNA).6 These mechanisms independently transmit genetic information from the DNA sequence, enabling the activation or repression of specific genome regions in response to physiological or pathological signals (Fig. 1).

Epigenetic mechanisms and key examples of widely studied modifications and their modifying enzymes. a DNA modifications, histone modifications, RNA modifications, chromatin remodeling, and the regulation based on non-coding RNA constitute the core content of epigenetics, being responsible for passing on heritable variations of genetic information independently of the DNA sequence. b Epigenetic modifications are reversible progress catalyzed by functionally complementary modifying enzymes, which provide targets for disease therapeutics
Enzymes that regulate epigenetic modifications are categorized into “writers,” “erasers,” “readers,” and “remodelers” based on their functions.7,8,9 Writers modify specific bases or amino acids, whereas erasers remove these modifications, exerting reciprocal effects on gene expression. For instance, DNA methyltransferase (DNMT) catalyzes the addition of methyl groups to form 5-methylcytosine (m5C) in DNA bases,10 whereas the ten-eleven translocation (TET) enzymes initiate DNA demethylation, converting m5C into derivatives, such as 5-hydroxymethylcytosine (5hmC), 5-formylcytosine, and 5-carboxycytosine.11 Typically, genes expressed at higher levels exhibit lower methylation, whereas genes with lower expression levels tend to be more heavily methylated.12 Readers are proteins that contain specific motifs to recognize and bind these modifications, such as the methyl-CpG-binding domain (MBD) responsible for recognizing 5mC.13 These proteins influence chromatin status and recruit or collaborate with other enzymes to regulate gene expression.13,14 Remodelers are crucial in chromatin remodeling, moving or removing nucleosomes at vital regulatory elements like enhancers and promoters to modify chromatin accessibility.15 Furthermore, as unique epigenetic regulators distinct from epigenetic-modifying enzymes, ncRNAs directly bind to various genomic regions or specific RNA sequences to modulate gene expression.16 Variations in the given ncRNA may regulate the interactions or functions of its interactor partners, including proteins, RNAs, DNAs, and lipids, thereby influencing various biological cellular processes or pathological phenotypes.17
The discovery of functionally complementary epigenetic-modifying enzymes has underscored the reversibility of most known epigenetic modifications. This insight supports the development of strategies to modulate gene expression via targeted regulation of these enzymes, providing a strong theoretical basis for creating novel therapeutic approaches from an epigenetic perspective. To date, four categories of epigenetics-targeted drugs have received the Food and Drug Administration (FDA) approval for clinical use, with numerous clinical trials ongoing to refine their applications. A timeline of significant milestones in epigenetic research is depicted in Fig. 2.

Timeline of major discoveries and advances in epigenetic research. The significant discoveries and advances are depicted in the illustrator and displayed as primarily “Early foundations” (yellow boxes) on the top, “Improving safety and efficacy” (purple boxes) in the middle, and “Breakthroughs in the clinical practice” (pink boxes) at the bottom
Over the past few decades, numerous studies have underscored that abnormalities in the expression and function of epigenetic-regulating enzymes are crucial in the onset and progression of various diseases. Epigenetics-targeted drugs, therefore, have emerged as pivotal topics due to their significant physiological and pathological implications. The development of drug screening models rooted in epigenetic principles is anticipated to substantially expand therapeutic options in clinical settings. Moreover, advancements in epigenetic analysis and molecular modification techniques have accelerated the clinical adoption of these targeted drugs. Despite these developments, there remains a gap in comprehensive reviews that address epigenetic regulations in physiological and disease contexts and detail the latest advancements in drug development targeting these mechanisms. This review aims to fill that void by summarizing the current understanding of epigenetic regulations and clinical trials of targeted drugs, thereby outlining the future application of these promising agents. We begin with an overview of epigenetic mechanisms and their crucial roles in health and disease, followed by an in-depth discussion on the exploration and application of marketed epigenetic drugs. We then provide a systematic account of recent progress in developing potential therapeutic agents targeting various epigenetic enzymes, highlighting emerging research trends. Finally, we present the breakthroughs and challenges in epigenetic drugs, particularly the benefits of combining them with traditional therapies such as radiotherapy, chemotherapy, and targeted therapy, to underscore their potential in translational medicine.
Biological and pathological roles of epigenetics
Epigenetic modifications are a fundamental mechanism regulating gene expression, crucial for various cellular functions. Dysregulated epigenetic regulators, whether overexpressed or underactive, compromise normal functions and contribute to disease onset. Thus, epigenetic modifications hold significant potential for disease treatment and biotechnological applications, driving the development of targeted therapeutic drugs.
Epigenetics and early embryonic development
Epigenetic landscapes undergo substantial changes to ensure the coordinated progression of embryogenesis and subsequent development throughout an individual’s life.18 Mutations in epigenetic-modifying enzymes, whether heterozygous or hemizygous, are commonly associated with congenital conditions, such as Rubinstein-Taybi syndrome, linked to mutations in the cyclic adenosine monophosphate-responsive element-binding protein (CREB)-binding protein (CBP) and its paralog, E1A-binding protein (P300),19 immunodeficiency-centromeric instability-facial anomalies syndrome related to DNMT3B mutations,20 and Kabuki syndrome due to mutations in lysine methyltransferase 2D (KMT2D).21 DNA methylation reprogramming, a pivotal aspect of epigenetic modification in early embryonic stages, involves genome-wide removal of epigenetic marks through extensive DNA demethylation, followed by remethylation.22 This process, integral to mammalian development, has only been fully understood with the advent of whole-genome bisulfite sequencing, which allows for single-base resolution analysis of DNA methylation kinetics.23,24 Advances in precise assays for assessing DNA methylation at specific genetic loci have led to significant insights into these epigenomic reprogramming processes. This reprogramming results in global hypomethylation and significant loss of genetic memory, which is foundational for acquiring pluripotency and redetermining cell fate.25 Following fertilization, methylation patterns evolve progressively, enabling cells to differentiate and contribute to the development of various biological systems. The dynamic regulation of DNA methylation, including reprogramming, is indispensable for mammalian development and differentiation. Another vital mechanism, histone modification, plays a critical role during zygotic genome activation (ZGA), which involves the transition of the zygotic genome from a state of silence to active transcription.26 Notably, the de novo establishment of histone 3 lysine 14 acetylation (H3K14ac) and histone 3 lysine 9 trimethylation (H3K9me3) following fertilization is crucial for the timely activation of ZGA genes during development.27,28 The SWItch/Sucrose NonFermentable (SWI/SNF) complex also plays a significant role in the precise activation and repression of tissue-specific transcription factors, functioning as a chromatin remodeler that orchestrates the coordinated differentiation of multiple cell lineages during development.29,30 Additionally, the role of RNA modifications in embryo development is increasingly recognized and summarized.31,32 Recent studies indicate that deficiencies in methyltransferase-like proteins (METTLs) and their associated RNA N6-methyladenosine (m6A) levels can induce G1/S cell cycle arrest in hematopoietic stem and progenitor cells in model organisms.33 Furthermore, aberrant RNA modification patterns are integrated into the regulatory networks of other epigenetic mechanisms, such as histone deubiquitylation and DNA methylation, playing critical roles in nuclear reprogramming.34,35 Recently, preliminary evidence of ncRNAs being engaged in embryo development has been proposed according to the reported variation among ncRNAs contents during different stages of early embryonic development in mouse models.36,37,38 As an indicator of developmental competence, ncRNA plays an irreplaceable role in the continuous stages of pre-implantation development, embryo implantation, and post-implantation development.39 Aberrant levels of certain ncRNAs may disturb the transition of fertilized oocytes to pluripotent blastocysts, and may even affect the differentiation of epiblast stem cells.40,41 Notably, ncRNAs may also act as regulatory factors for other epigenetic mechanisms. For example, during mouse ZGA, the negative regulation of Dnmt3a/3b expression by microRNA-29b (miR-29b) helps maintain proper DNA demethylation to establish the imprinting of genes.42 However, considering that most of the current understanding has only been validated in animal models, much work is still required to explore the role of ncRNAs during embryonic and fetal development in humans.
Epigenetics and aging
Since the late 1990s and early 21st century, researchers have observed that epigenetic changes accompany aging based on data derived from cellular experiments.43 Initially, it was unclear whether these epigenetic alterations were a cause or a consequence of aging. Recent work by Yang et al.44 has successfully dissociated epigenetic dysregulation from genetic changes, confirming that the collapse of epigenetic modifications is a potent driver of aging. DNA methylation, a central epigenetic mechanism, regulates both development and aging. Notably, global DNA methylation levels in most regeneratively capable tissues tend to decrease with age.45 Beyond global changes, studies increasingly report high frequencies of age-related alterations in DNA methylation accumulated in specific cellular regions. These differentially methylated regions associated with aging lead to either the upregulation or repression of downstream genes. For example, age-related hypermethylation within the promoter regions of tumor suppressor and metabolic genes may partially explain the increased susceptibility of the elderly to tumors and various metabolic disorders.46 Conversely, DNMT inhibitor decitabine can reverse hypermethylation in tumor suppressors, enhancing their expression and inducing a senescence-like phenotype in tumor cell lines.47 Other mechanisms, such as histone modifications and chromatin remodeling, are also strongly linked to aging. A deficiency in sirtuin 7 (SIRT7) and histone methylation patterns like H3K9me2 and H3K27me3, for example, can activate the cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS)-stimulator of interferon genes pathway, a well-recognized aging-associated signaling pathway, thus exacerbating the aging process.48,49 Moreover, the importance of RNA modifications—particularly m6A and m5C—and ncRNA regulation is increasingly studied in aging research.50,51,52
Epigenetics and cancer
Abnormal epigenetic mechanisms play crucial roles at various stages of tumor development, including initiation, progression, invasion, migration, and the development of chemotherapy resistance (Fig. 3). DNA methylation was the first discovered epigenetic mechanism associated with tumors, initially implicated in the hypermethylation of specific gene promoter regions, which drives tumor development by silencing gene transcription.53 This silencing leads to the dysfunction of critical genes such as tumor suppressor and DNA repair genes, disrupting normal cell proliferation and differentiation and fostering the malignant phenotype of tumor cells.54,55 Moreover, methylation loss at specific sites in tumor cell genomes, particularly in oncogene promoter regions, and extensive demethylation in DNA repeat sequences, undermines chromosome stability, facilitating tumor development.56,57 Changes in histone modifications are also prevalent in tumors. The roles of histone methylation and acetylation in tumor progression have been extensively explored, with numerous reviews summarizing therapeutic strategies targeting these histone modifications or their associated epigenetic-modifying enzymes, underscoring their pathological significance and therapeutic potential.58,59 Noticeably, bromodomain (BD) and extra-terminal (BET) family member proteins, including BRD2, BRD3, BRD4, and BRDT, serving as interpreters of histone acetylation modification, have recently been found to facilitate tumorigenesis when overexpressed.60,61 Upregulated BET proteins can function as oncogenic transcriptional factors in tissue cells, driving a unique transcriptional program and controlling cell phenotype.62,63 Therefore, potent inhibitors targeting BET proteins may be considered potential agents for tumor treatment. Research into metabolic reprogramming and the Warburg effect in tumor cells has recently highlighted histone lactylation’s function in pathological processes.64,65 Histone lactylation, induced by glycolysis, has been studied extensively in various malignancies such as endometrial cancer, pancreatic ductal adenocarcinoma, and glioblastoma, where it plays roles in tumor progression and the suppression of the immune microenvironment.66,67,68 Additionally, dysregulation in RNA modifications, particularly m6A, is linked to the malignant potential and resistance of tumor cells,69,70 affecting multiple pathways that ensure tumor cell survival, including the maintenance of stemness,71 the establishment of vascular networks,72 and the formation of an immunosuppressive tumor microenvironment (TME).73 Thus, targeting aberrant RNA modifications could effectively disrupt the survival mechanisms of tumor cells, offering new avenues for cancer treatment.74 Changes in ncRNA families have also been observed in various tumors, first noted in chronic lymphocytic leukemia with chromosome 13q14 deletion, characterized by decreased levels of miR-15 and miR-16.75 Among these, various ncRNA molecules with antitumor effects have been identified and are commonly suppressed in various tumor diseases, representing promising targets for therapeutic intervention.76 Furthermore, ncRNAs can participate in the post-translational regulation of other epigenetic-modifying enzymes, integrating into broader epigenetic networks.77 In addition to functioning as pathogenic triggers in different tumors, ncRNAs present in extracellular vesicles in the TME also hold promise for assessing therapeutic response.78,79,80 According to clinical data from well-organized observational studies, unique plasma exosomal miRNA profiles are associated with predicting the efficacy of antitumor therapies in various tumor diseases, such as advanced non-small cell lung cancer,81 colorectal cancer,82,83 and breast cancer.84

Epigenetic mechanisms in cancer. Epigenetic alterations in cancer cells affect various cellular responses, such as cell proliferation, invasion, apoptosis, and drug resistance. These modifications, which include DNA modification, histone modification, RNA modification, chromatin remodeling, and non-coding RNAs, significantly affect the pathogenesis and progression of tumors. By targeting these epigenetic mechanisms, novel therapeutic strategies for combating cancer can be developed. The primary roles of epigenetic mechanisms in tumorigenesis and their further development are presented in the illustrator
Epigenetics and metabolic syndrome and related disorders
Metabolic syndrome encompasses a constellation of pathological conditions characterized by abnormal aggregation of metabolic components, notably abdominal obesity or overweight, dyslipidemia, insulin resistance and/or glucose tolerance abnormalities, and hypertension.85 These metabolic dysfunctions significantly elevate the risk of developing diseases such as type 2 diabetes mellitus (T2DM), nonalcoholic fatty liver disease (NAFLD), and cardiovascular diseases.86 Epigenetic modifications play a crucial role in nutrient metabolism under physiological conditions and also bridge the genetic and environmental factors contributing to metabolic disorders.87 For instance, dietary patterns significantly influence epigenetic markers; studies have shown that a high-fat diet in mice leads to hypermethylation in the promoter regions of genes like Rac family small guanosine triphosphate hydrolase (GTPase) 1, which promotes the progression of diabetic retinopathy.88 Dietary-induced epigenetic changes can impact subsequent generations, increasing their risk of glucose intolerance and diabetes.89 Moreover, epigenetic alterations linked to diet are implicated in developing gout and NAFLD.90,91
Additionally, the activity of epigenetics-modifying enzymes and their cofactors, such as TET and α-ketoglutarate (α-KG) from the tricarboxylic acid cycle (TCA), can be influenced by abnormal metabolite levels in patients with metabolic diseases, further disrupting epigenetic regulation and exacerbating disease progression.92 Epigenetic markers, especially DNA methylation landscapes, also provide diagnostic tools;93 in T2DM, for example, differential methylation in genes such as thioredoxin interacting protein, adenosine triphosphate (ATP)-binding cassette subfamily G member 1, peroxisome proliferator-activated receptor gamma-coactivator 1 alpha, and protein tyrosine phosphatase receptor type N2, can elucidate pathophysiological mechanisms.94 Understanding these epigenetic mechanisms in metabolic diseases is thus pivotal for developing innovative prevention, diagnosis, and treatment strategies.
Epigenetics and immune system disease
Epigenetic modifications are integral to the development and differentiation of immune cells and the regulation of immune functions. These modifications influence the differentiation of functional B and T cell subpopulations and maintain the homeostasis of innate immune cells by controlling specific gene expressions.95,96,97 Epigenetic dysregulation is closely linked to immune system diseases, including allergic reactions and autoimmune diseases, which have been extensively studied.98,99 For instance, allergic bronchial asthma involves reduced TET2 expression in regulatory T cells, leading to hypermethylation in the promoter region of forkhead box protein P3 and impaired immune function in controlling inflammatory responses.100 Additionally, low expression of METTL3 in monocyte-derived macrophages in allergy patients exacerbates airway inflammation through M2 macrophage polarization.97 Histone modification also plays a critical role in sustaining the therapeutic effects of glucocorticoids in asthma; oxidative stress in severe asthma cases leads to reduced histone deacetylase (HDAC) levels in alveolar macrophages, contributing to glucocorticoid resistance.101 Consequently, elevating HDAC levels in patients with severe, steroid-insensitive asthma could be a viable strategy to reduce airway hyperresponsiveness and restore steroid sensitivity.102
Epigenetic mechanisms play a significant role in the pathogenesis and progression of autoimmune diseases. For instance, hypomethylation mediated by TET2 within the promoter region of absent in melanoma 2, a critical component of the inflammasome, influences T follicular helper cell-dependent humoral immune responses in systemic lupus erythematosus (SLE).103 Additionally, altered patterns of miRNA in serum exosomes and immune cells have been identified, promising potential as biomarkers for diagnosis and indicators of disease severity.104,105 Histone modifications also play a pivotal role in SLE, where the administration of HDAC inhibitors has been shown to reduce cytokine profiles and improve pathogenesis in SLE and other inflammatory conditions.106 Moreover, the therapeutic potential of BET proteins in antibody-mediated diseases (e.g., SLE) has recently been evaluated. BET inhibitors alter the pro-inflammatory phenotypes of mononuclear phagocytes and impair the recruitment of dendritic cells in vitro.107 Beyond SLE, epigenetic mechanisms are implicated in the progression of other autoimmune diseases such as rheumatoid arthritis,108 autoimmune thyroid diseases,109 multiple sclerosis,110 T1DM,111 and severe aplastic anemia,112 highlighting the potential for epigenetics-modifying drugs in treatment strategies.
Epigenetics and neurodegenerative disease
Epigenetic modifications significantly influence learning, memory, and cognition, which are essential in maintaining synaptic plasticity.113,114 Disruptions in epigenetic regulation lead to the abnormal expression of genes involved in protein aggregation, neuroinflammation, and neuronal apoptosis, contributing to the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD).115 The deposits of extracellular Aβ plaques and tau phosphorylation, as well as the loss of plasticity, are basic pathogenesis of AD. In AD, aberrant histone modification patterns, particularly histone acetylation, have been observed in hippocampal neurons of AD mouse models, potentially driving cognitive decline and inadequate removal of Aβ plaques.116 Lactylation modifications of histones H4K12 and H3K18 affect the metabolic activity of various glial cells, influencing the progression of the AD phenotype.117,118 In addition, aberrant DNA-methylation patterns in the promoter regions of functional genes are linked to the accumulation of toxic peptides and the development of memory deficiency.119,120 Recent studies also consider RNA modifications and ncRNAs as potential therapeutic targets and diagnostic biomarkers for AD.121,122 PD is characterized by the misfolding and aggregation of α-synuclein, leading to the formation of Lewy bodies. Altered DNA methylation patterns have been observed in brain and blood samples from individuals with PD.123,124 TET2 may play a critical pathogenic role in PD, where its inactivation has shown a neuroprotective effect on dopaminergic neurons.125 Histone acetylation dysregulation is extensively studied in PD, associated with the accumulation of phosphorylated α-synuclein and mitochondrial respiratory dysfunction.126,127 Dysregulation in ncRNAs, particularly long non-coding RNAs (lncRNAs) and miRNAs affects the mRNA levels of pathogenic factors post-transcriptionally and is linked with clinical symptoms such as non-motor symptoms, cognitive deficits, and inflammation, presenting potential targets for PD treatment.128 In HD, epigenetic modification alterations are vital markers of its pathogenesis. Studies have shown the positive effects of using DNMT inhibitors, HDAC inhibitors, and extracellular vesicles loaded with miRNAs in preventing mutant huntingtin-induced neurotoxicity, emphasizing the potential roles of epigenetic dysregulations in HD.129,130,131 Recently, the impact of aberrant m6A RNA methylation on the progression of HD has been increasingly recognized. Hyper-methylation of m6A in genes related to HD and synaptic function has been linked to memory deficits. Conversely, inhibition of the fat mass and obesity associated protein (FTO) in the hippocampal regions of HD mouse models has shown promise in reversing cognitive symptoms, suggesting a potential therapeutic target.132
In summary, the dynamic nature of epigenetic modifications plays a crucial role in maintaining physiological functions and life cycle processes. During embryonic development, precise epigenetic regulation is crucial to cell differentiation and ensures proper tissue specialization by activating or suppressing specific genes. Furthermore, epigenetic modifications are closely linked to an individual’s adaptation to environmental influences such as nutritional status, stress, and toxin exposure, which can alter epigenetic landscapes and impact health and disease risk. On the other hand, understanding epigenetics offers a new perspective for disease prevention and treatment. The development and progression of many diseases, including cancer, metabolic disorders, immune system diseases, and neurodegenerative disorders, are closely associated with aberrant epigenetic modifications. A deeper understanding of epigenetic modulators could lead to novel therapeutic strategies, laying the groundwork for drug interventions targeting epigenetic processes.
Epigenetics-targeted drugs approved for clinical use
Epigenetic modifications and the enzymes involved can either activate or suppress the expression of specific genes at different levels (Table 1). Therefore, in contrast to traditional therapies, drugs targeting epigenetic-modifying enzymes have been developed with a focus on gene regulation. This unique mechanism provides epigenetic-targeted drugs with an advantage over other traditional treatments, especially for the treatment of tumors. More specifically, epigenetics-targeted drugs specifically target the abnormal epigenetic hallmarks of cancer cells, restoring their normal cellular function or enhancing the immune system’s recognition of tumor cells.133,134 Compared to traditional radiotherapy and chemotherapy, which directly kill cancer cells or prevent their proliferation, or immunotherapy, which activates or enhances the patient’s own immune system, the administration of epigenetic agents achieves maximized damage to tumor cells with usually fewer side effects.135,136,137 The application of these novel agents helps to reverse the progression of drug resistance caused by altered epigenetic characteristics in traditional antitumor therapies.138,139,140 Hence, these features endow epigenetic agents with importance and possibilities as monotherapy or adjuvants in combination with other therapeutic methods.141 Currently, some of these drugs have been approved for marketing, primarily for cancer treatmentt, and they exhibit exciting clinical potential. These drugs are categorized into four main types based on their mechanisms: DNMT inhibitors, HDAC inhibitors, isocitrate dehydrogenase (IDH) inhibitors, and enhancer of zeste homolog 2 (EZH2) inhibitors (Table 2).
DNMT inhibitors
Azacitidine and decitabine, known as hypomethylating agents (HMAs), target DNMT1 and were among the first epigenetics-targeted drugs approved for clinical use. The US FDA approved azacitidine in May 2004 and decitabine in June 2006 for treating myelodysplastic syndrome (MDS).142,143 The clinical success of these HMAs has led to a focus on optimizing their dosing schedules and administration methods. Early studies involving azacitidine and decitabine assessed their therapeutic potential through continuous and/or frequent intravenous or subcutaneous injections, establishing standard doses and delivery methods in clinical settings.144,145,146 Recent advancements have explored reduced dosages for patients at lower risk. In recent phase II clinical trials, azacitidine or decitabine was applied for three or five consecutive days in a 28-day cycle, exhibiting satisfactory therapeutic efficacy and tolerable safety.147,148 Some patients who received decitabine experienced myelosuppression, and future efforts are required to take steps to avoid this.147
Additionally, the development of oral formulations of azacitidine and decitabine, such as oral azacitidine (CC-486), approved in September 2020 for patients with acute myeloid leukemia (AML) who are not candidates for intensive curative therapy, has improved patient convenience and treatment adherence.149 In a well-organized phase III randomized trial, the median overall survival and relapse-free survival of patients treated with CC-486 were greatly improved.149 Importantly, fewer grades 3 or 4 adverse events were observed during CC-486 treatment, allowing the preservation of overall health-related quality of life.149 The doses of CC-486 used in clinical settings are approximately four times higher than the standard doses administered by intravenous or subcutaneous routes due to their reduced bioavailability.150 Meanwhile, the poor bioavailability of oral decitabine has led to the development of ASTX727, an oral combination of decitabine with cedazuridine. This cytidine deaminase enzyme inhibitor enhances decitabine exposure after oral administration. This combination has been approved for marketing in treating MDS and AML in some countries.151,152 However, in China, the therapeutic potential of CC-486 and ASTX727 for AML and MDS is still under evaluation in clinical trials (NCT05413018, NCT04102020, NCT06091267, NCT02649790). Furthermore, there has been an increased focus on the synergistic effects of HMA and traditional antitumor treatments to enhance therapeutic outcomes and prevent resistance in hematologic malignancies refractory to monotherapy. This topic is further summarized in the subsequent section on drug combination applications.
Inspired by the successful application of HMAs in hematologic malignancies, their therapeutic potential in treating solid tumors is also being explored. However, as of now, HMAs are not approved for treating solid tumors. In 2017, Linnekamp et al.153 conducted a systematic review to illustrate the clinical and biological effects of HMAs on solid tumors based on previously completed clinical trials. The efficacy of azacitidine and decitabine in solid tumors was less pronounced than in hematological malignancies, primarily because most study participants had advanced-stage tumors with short life expectancies.153 Moreover, many early-stage studies were small-sized cohorts, lacking sufficient evidence to generalize therapeutic effects across different tumor types. With significant advances in optimizing HMA formulations and dosages, as well as the increasing number of combination therapies showing promising effects on solid tumors in vitro and in vivo, clinical trials of HMAs, particularly the oral formulations CC-486 and ASTX727, among patients with solid tumors, are being extensively conducted, and their results are eagerly anticipated.154,155,156
HDAC inhibitors
Over the past two decades, substantial progress has been made in developing HDAC inhibitors, with six approved for clinical use. These include vorinostat (SAHA), romidepsin (FK228), belinostat (PXD101), and panobinostat (LBH589, although the FDA canceled it in 2022). These drugs have been approved by various regulatory bodies, such as the US FDA, the European Medicines Agency, and the Pharmaceuticals and Medical Devices Agency (PMDA). They are used to treat conditions such as multiple myeloma (MM), cutaneous T-cell lymphoma (CTCL), and peripheral T-cell lymphoma (PTCL).157,158 Additionally, chidamide (tucidinostat) was approved by PMDA in Japan and National Medical Products Administration (NMPA) in China for the treatment of PTCL and advanced breast cancer,159,160 and more recently, givinostat (ITF2357) was approved by the FDA in March 2024 as the first nonsteroidal treatment for Duchenne muscular dystrophy (DMD) for patients aged six years and older.161
Vorinostat was the first pan-inhibitor of HDACs approved by the FDA in October 2006 for CTCL.162 Soon after, in July 2011, it was also approved for clinical therapy by PMDA. In addition to CTCL, the application of vorinostat to AML, MM, malignant pleural mesothelioma, newly diagnosed high-grade glioma (NCT01236560), and advanced non-small cell lung cancer (NCT00473889) therapy has entered phase III clinical trials.163,164,165,166 Disappointingly, though vorinostat exerts effective therapeutic effects in diverse hematological malignancies, limited efficacy has been observed in solid tumors.166 Another thing to note when using vorinostat as a clinical medication is the potential adverse events that may occur. While generally mild, adverse events related to vorinostat can include thrombosis, QT interval prolongation, and potentially fatal ventricular tachycardia or torsional tachycardia.167,168,169 These findings have driven the development of other HDAC inhibitors, with the expectation of elevated safety and efficacy in vivo.
Romidepsin, another pan-HDAC inhibitor, was approved by the FDA in November 2009 for CTCL and later for PTCL. It has shown a higher affinity to class I HDAC proteins.170 Subsequently, it was approved for treating patients with PTCL by the FDA and PMDA. Phase III randomized controlled trials are performed to evaluate the therapeutic value of the first-line treatment of PTCL, referring to the combination of cyclophosphamide, doxorubicin, vincristine, prednisone (CHOP), and romidepsin plus CHOP in patients with PTCL, while the addition of romidepsin failed to increase efficacy as expected.171,172 However, after a six-year follow-up, the application of romidepsin shows beneficial effects in prolonging median progression-free survival.171 In addition, the combination of romidepsin with other therapeutics, such as oral 5-azacytidine, tenalisib (an inhibitor of phosphoinositide-3-kinase and salt-inducible-kinase-3), and lenalidomide (a new generation of immune modulator) shows promising therapeutic potential in various types of T-cell lymphoma in the initial stages of clinical practice, supporting further exploration.173,174,175,176 Meanwhile, investigations on the therapeutic effects of romidepsin against other diseases are ongoing, particularly in antiretroviral treatment in human immunodeficiency virus-1 (HIV-1) infection.177,178
Belinostat was FDA-approved in July 2014 for relapsed or refractory (R/R) PTCL, showing pan-inhibitory effects on HDAC proteins.179 Common adverse effects of belinostat include nausea, vomiting, diarrhea, dysgeusia, fatigue, and severe hematologic treatment-related adverse events.180,181 Further, dosing considerations are needed for patients with hepatic impairment due to liver metabolism.182 Belinostat is being explored for other myeloid malignancies and solid tumors, including glioblastoma and small-cell lung cancer.183,184,185,186
Panobinostat, an oral broad-spectrum HDAC inhibitor approved in January 2015 for R/R MM in combination with dexamethasone and bortezomib, showed a slight overall survival benefit in phase II and III trials.187 However, many participants experienced adverse events like thrombocytopenia, lymphopenia, asthenia, and fatigue, which raises concerns about its tolerability.187,188,189 In a recent randomized phase II clinical trial, it was proposed that administering bortezomib via subcutaneous application rather than intravenous injection could improve the safety and tolerability of the triplet regimen, including panobinostat.190 Beyond its primary indications, panobinostat is being explored for its efficacy in various other tumor diseases such as lymphoma, primary myelofibrosis, glioma, clear cell renal cell carcinoma, and prostate cancer, both as monotherapy and in combination with other tumor therapeutics, showing promising efficacy across multiple malignancies.191,192,193,194,195 However, the safety of panobinostat continues to be a primary concern and requires further evaluation in advanced clinical studies.
Chidamide, the first orally administered selective HDAC inhibitor targeting HDAC1, HDAC2, HDAC3, and HDAC10,160 is currently under investigation for a variety solid and hematological malignancies, autoimmune diseases, and neurodegenerative diseases.196,197,198,199,200,201 It offers advantages over pan-inhibitors in the treatment of tumor diseases and in minimizing severe adverse effects.160 Recent therapeutic strategies using chidamide in combination with a second antitumor intervention have shown promising prospects. For instance, a combination of chidamide and exemestane has proven effective as a neoadjuvant treatment for patients with stage II-III breast cancer that is hormone receptor-positive and human epidermal growth factor receptor 2-negative.199,202 A recent phase III clinical trial reported an increased occurrence of grades 3–4 hematological adverse events in the tucidinostat plus exemestane group, while the median progression-free survival of these patients was notably improved.202 Furthermore, synergistic effects have been observed when chidamide is used in conjunction with immunotherapy, endocrine therapy, or chemoradiation, offering novel adjuvant approaches for tumor therapy.203,204,205,206
Givinostat, developed by Italfarmaco SpA, is a potent inhibitor of HDAC1 and HDAC3 recently approved for clinical use. In a pivotal, multicenter, randomized phase III clinical trial involving 179 patients with DMD aged at least six years, givinostat effectively delayed disease progression.207 The most common adverse events reported were diarrhea and vomiting.207 Additionally, givinostat shows promise as a treatment for polycythemia vera, particularly in patients unresponsive to hydroxycarbamide monotherapy.208 In phase I/II clinical trials, givinostat demonstrated promising efficacy and tolerability in patients with polycythemia vera.209,210 Subsequently, long-term follow-up over four years has further substantiated the therapeutic benefits and safety profile of givinostat.211 Throughout the follow-up period, the overall response rate consistently exceeded 80% among patients with PV, while only 10% of these patients experienced grade 3 treatment-related adverse events, suggesting its potential for prolonged clinical use.211
IDH inhibitors
IDH is a key enzyme in the TCA cycle that normally catalyzes the conversion of isocitrate to α-KG and carbon dioxide. In cells with mutated IDH, this enzyme instead produces 2-hydroxyglutarate (2-HG), a metabolite that inhibits α-KG-dependent epigenetic enzymes and contributes to the aberrant epigenetic landscape seen in various diseases, particularly tumors.212,213 Currently, three IDH inhibitors are approved for clinical use: enasidenib (AG-221), ivosidenib (AG-120), and olutasidenib (FT-2102), targeting different forms of the enzyme mutation.214,215,216,217 Additional IDH1/2 inhibitors that are allowed to be investigated in clinical practice include the dual inhibitor of mutant IDH1/2 vorasidenib (AG-881),218 the irreversible mutant IDH1 inhibitor LY3410738 (NCT06181045, NCT06181084), and the pan-mutant-IDH1 inhibitor BAY1436032.219,220
Enasidenib is an allosteric inhibitor targeting mutated IDH2, approved by the FDA in August 2017 for the treatment of R/R AML with IDH2 mutations.221 Based on preliminary animal experiments and preclinical evidence, enasidinib effectively reduces the 2-HG levels derived from IDH2 mutations, reversing excessive histone and DNA methylation landscapes.222,223,224 Subsequently, enasidinib has entered clinical trials and demonstrated good efficacy in treating AML and MDS patients, which is further considered a promising option for elderly AML patients over 60 years old, especially those who are not suitable for intensified chemotherapy.225,226,227,228,229,230 Combination therapy with enasidenib and azacitidine has shown acceptable tolerability and potential to improve outcomes for patients with IDH-mutated AML.229,231,232 However, potential severe adverse effects include hyperbilirubinemia, thrombocytopenia, pneumonia, and IDH differentiation syndrome, the latter of which can be life-threatening and requires careful management.233,234,235 Noticeably, IDH differentiation syndrome is one of the potentially lethal entities that require prompt recognition and more appropriate management.236 Enasidenib is also being explored for other conditions caused by IDH2 mutations, such as D-2-hydroxyglutaric aciduria type II,237 chondrosarcoma,238 angioimmunoblastic T-cell lymphoma (NCT02273739), and malignant sinonasal or skull base tumors (NCT06176989).
Ivosidenib, targeting mutated IDH1, was first approved by the FDA in July 2018 for R/R AML.239 In April 2022, with the data from a completed phase III clinical trial being made public, the therapeutic potentials and good safety of the combination of azacitidine and ivosidinib among patients with AML received broader attention.240 Subsequently, the FDA approved this regimen for elderly patients with newly diagnosed IDH1-mutated AML in May 2022.241 Besides AML, ivosidenib is approved for MDS and cholangiocarcinoma, with ongoing phase III studies in unresectable or metastatic cholangiocarcinoma with IDH1 mutations.216,242 The most significant adverse events include ascites and other severe conditions, necessitating vigilant monitoring.242 Ivosidenib has also shown promising results in phase I clinical trials for IDH-mutated advanced glioma, with a daily dose of 500 mg proving effective in reducing 2-HG levels and controlling the disease.243,244,245,246
Olutasidenib, an oral IDH1 inhibitor, was approved by the FDA in December 2022 for treating R/R AML with specific IDH1 mutations. It has also shown promise as a therapeutic option for patients with IDH1-mutated AML who are insensitive to venetoclax, offering a new avenue for treatment where previous therapies may have failed.217 Clinical trials have demonstrated that olutasidenib, combined with azacitidine, provides comparable efficacy and tolerability in AML and MDS patients harboring mutant IDH1.247 Treatment-emergent side effects of grade 3-4, such as febrile neutropenia, anemia, thrombocytopenia, and neutropenia, occur at a low frequency with olutasidenib monotherapy or in combination therapies, suggesting a manageable safety profile.247,248 Beyond hematological malignancies, the therapeutic potential of olutasidenib is also being explored in other diseases, such as IDH-mutated R/R gliomas. In a multicenter, open-label, phase Ib/II clinical trial involving 26 patients, olutasidenib achieved a disease control rate of 48%. Notable grade 3-4 adverse events included increases in alanine aminotransferase and aspartate aminotransferase, indicating the need for careful liver function monitoring during treatment.249
EZH2 inhibitors
Currently, two EZH2 inhibitors, tazemetostat (EPZ-6438) and valemetostat tosilate (DS-3201, DS-3201B), targeting EZH1/2 or EZH2 have been approved and are being utilized in various therapeutic strategies.
Tazemetostat, the first oral EZH2 inhibitor, was approved by the FDA in January 2020 for patients over 16 years of age with advanced epithelioid sarcoma that is ineligible for complete resection.250 It is also the first targeted drug for epithelioid sarcoma treatment.251 In a phase II clinical trial (NCT02601950), tazemetostat demonstrated good tolerability and clinical activity, with a low incidence of severe treatment-related adverse events such as anemia and weight loss.252 Tazemetostat has also been studied as a monotherapy in R/R follicular lymphoma (FL), showing promising, durable responses and an acceptable safety profile.253,254 Common severe adverse events included thrombocytopenia, neutropenia, and anemia.254 In Japan, a phase I/II trial evaluated tazemetostat 800 mg twice daily in R/R EZH2 mutation-positive FL, showing encouraging response rates and tolerability, which helped to accelerate its approval by the FDA and PMDA for this indication.255,256 Furthermore, tazemetostat is being investigated as a single agent for malignant mesothelioma, with ongoing efforts to refine biomarkers for its activity in malignant pleural mesothelioma.257 Research is also underway to assess the efficacy and tolerability of tazemetostat in combination with other therapeutic agents, including programmed cell death 1 (PD-1)/programmed cell death 1 ligand 1 (PD-L1) inhibitors, chemotherapy, and targeted therapeutics across different tumor types.258,259,260 Notably, phase III clinical trials are exploring tazemetostat in combination with lenalidomide and rituximab (NCT04224493) or doxorubicin (NCT04204941) focusing on R/R FL and epithelioid sarcoma, which are highly anticipated for their potential to redefine treatment paradigms.
Valemetostat tosilate is an innovative dual inhibitor of EZH1/2 that received approval from the PMDA in Japan for the treatment of R/R adult T-cell leukemia/lymphoma (ATL) in September 2022.261 It is administered orally and should be taken on an empty stomach to avoid adverse food effects.262 Dosage adjustments are necessary when valemetostat is used concurrently with strong inhibitors of cytochrome P450 3 A and P-glycoprotein, which can affect its metabolism and excretion.263 In a multicenter phase 2 trial involving patients with R/R aggressive ATL, valemetostat demonstrated promising efficacy, even in heavily pretreated patients. The common treatment-associated adverse effects reported were manageable, including thrombocytopenia, anemia, alopecia, dysgeusia, neutropenia, lymphopenia, leukopenia, decreased appetite, and pyrexia.264 However, resistance to valemetostat has been observed in some patients with ATL, potentially due to acquired mutations in the polycomb repressive complex 2 (PRC2) within tumor cells, highlighting a significant challenge in long-term treatment scenarios.59 Currently, valemetostat and its analogs are also being investigated in various preclinical studies and animal models for conditions such as tumor protein p63 gene-rearranged lymphoma, sinonasal teratocarcinosarcoma, ibrutinib-resistant mantle cell lymphoma, and human T-cell leukemia virus type 1-associated myelopathy. These studies further expand the potential therapeutic applications of valemetostat and warrant continued exploration of valemetostat-based treatment strategies.265,266,267,268
Other epigenetics-targeted drugs under research as clinical candidates
Although there have been many advances in the research of marketed drugs, they still belong to the tip of the iceberg relative to the entire field of epigenetics-targeting drug development. Many small molecule inhibitors and agonists targeting epigenetic-modifying enzymes are being identified and progressively advancing into the early stages of clinical trials. This section categorizes and summarizes these drugs based on their mechanisms, highlighting agents that show potential therapeutic value in clinical settings (Fig. 4).

The development direction and major categories of epigenetics-targeted drugs. a Epigenetics-targeted drugs are developed through the virtual screening of compound libraries, drug design based on molecular structure, and the exploration of potential mechanisms of known agents. Subsequently, applying PROTAC, CRISPR/Cas, and other technologies or mechanisms to optimize the physical properties, and inhibitory or agonistic effects of compounds. Finally, the druggability of possible agents should be improved in experimental, preclinical and clinical studies. b The classification of epigenetics-targeted drugs and their corresponding marketed representative agents are depicted in this section. Among them, epigenetics-targeted drugs that have already been approved and applied in clinical treatment are highlighted in corresponding colors
Epigenetics-targeted drugs and DNA methylation
This critical regulatory mechanism of gene expression involves adding methyl groups to DNA, primarily at cytosine bases in CpG dinucleotides, which generally leads to gene silencing. The process is dynamically regulated by DNMT and TET enzymes. The aberrant activity of these enzymes is closely linked with the pathogenesis of a wide range of diseases, not only cancers but also metabolic, inflammatory, and neurological disorders, underscoring the therapeutic relevance of targeting these pathways.269,270,271,272 Therefore, DNA methylation provides a promising platform for developing epigenetics-targeted drugs in clinical practice (Table 3).
Targeting the writer of DNA methylation: DNMT
Current therapeutic strategies primarily involve DNMT inhibitors, which suppress the expression or enzymatic activities of DNMTs, thereby counteracting improper DNA methylation patterns. These inhibitors are crucial in correcting the abnormal addition of methyl groups to DNA, a common feature in many pathologies.
DNMT inhibitors
Most DNMT inhibitors under investigation are employed in treating hematological or solid tumors, with a smaller portion used for inflammatory or proliferative benign diseases.153,273 DNMT inhibitors fall into two principal categories based on their mechanisms of action: nucleoside DNA methylation inhibitors and non-nucleoside DNA methylation inhibitors.
Among the nucleoside analogs, the FDA-approved drugs azacitidine and decitabine are noteworthy. These compounds integrate into the DNA structure and are recognized by DNMTs during DNA replication, thereby obstructing normal DNA methylation processes.274 Another significant compound, guadecitabine (SGI110, an antimetabolite of decitabine), represents a second-generation DNA methylation inhibitor. It is an antinucleotide molecule that resists degradation by cytidine deaminase.275 Research primarily focuses on its application against various malignant tumors. However, although guadecitabine has demonstrated good tolerance and favorable outcomes in many clinical trials, there is still clinical evidence indicating that its application may cause serious treatment-related adverse effects, such as pneumonia, sepsis, aspiration pneumonia, metabolic disorders, neutropenia, leukopenia, and pruritis.276,277 Furthermore, a recent phase II clinical trial conducted among patients with succinate dehydrogenase-deficient tumors was terminated due to low objective response rates.278 Additionally, when combined with traditional antitumor agents such as chemotherapeutic drugs and immune checkpoint blockade agents, guadecitabine demonstrates a potent synergistic effect, enhancing long-term clinical benefits.279,280,281,282 Promisingly, its use in treating patients with AML has advanced to phase III clinical trials, indicating high response rates and comparable safety, positioning it as a promising future alternative.279,283 Other nucleoside DNA methylation inhibitors include CP-4200, with cellular uptake less dependent on the nucleoside transporters involved in azacytidine uptake;284 F-aza-T-dCyd (NSC801845), optimized structurally from T-dCyd, F-T-dCyd, and Aza-T-dCyd;285 DHDAC, which is less cytotoxic and more stable;286 NPEOC-DAC, a decitabine derivative modified at the N4 position of the azacitidine ring, displaying significantly reduced potency at low doses in inhibiting DNA methylation;287 and zebularine, known for its high selectivity and better biocompatibility towards pathological cells,288 demonstrating significant therapeutic effects not only on tumors but also on non-tumor diseases like renal fibrosis,289 T2DM,290 and NAFLD.291 Additionally, clofarabine, an FDA-approved purine nucleoside analog for treating pediatric AML, primarily inhibits DNA biosynthesis and the ribonucleotide reductase enzyme and has shown potential in early-stage carcinogenesis through DNMT1 inhibition.292
Beyond these, various non-nucleoside DNMT inhibitors have been identified. These include specific artificially synthesized inhibitors and naturally occurring agents with a demethylation function.293,294 Enhancements to the physical properties of these natural compounds, such as solubility and stability, benefit the development of more effective DNMT-targeted inhibitors.295,296 Non-nucleoside DNMT inhibitors are categorized based on their diverse mechanisms of action into competitors of S-adenosylmethionine (SAM), competitive or non-competitive inhibitors of DNMT, regulators of DNMT expression, and binders of DNA substrates.53 MG98 and hydralazine are the only two drugs currently in clinical trials. MG98, an antisense oligodeoxynucleotide, reduces DNMT1 mRNA levels by targeting its 3’ untranslated region. However, several clinical trials are far from satisfactory.297 A two-stage phase II clinical trial evaluated the antitumor efficacy of MG98 in seventeen patients with metastatic renal carcinoma.298 However, it failed to detect a decrease in DNMT1 activity caused by MG98, urging caution against potential side effects like transaminase elevation and fatigue from excessive dosages during intravenous administration.298,299 Hydralazine, a low molecular weight molecule, interacts with DNMT through a network of hydrogen bonds with arginine and glutamic acid residues.300 Its combination with traditional chemotherapeutic agents has been found to mitigate the progression of both hematological malignancies and solid tumors.301,302,303,304 In a completed phase II clinical trial, 15 patients with solid tumors qualified for the assessment of the therapeutic response to hydralazine and magnesium valproate. The majority of patients (80%), benefited from treatment and exhibited satisfactory clinical efficacy and tolerability.304 These findings underpin the hypothesis that epigenetic aberrations induced by chemotherapeutic agents are a primary cause of chemoresistance, providing a theoretical basis for the combined use of epigenetics-targeted drugs and chemotherapy in tumor therapy.
Targeting the eraser of DNA methylation: TET
Due to the significant heterogeneity in the roles that TET enzymes play across various diseases, the effectiveness of targeting TET enzymes as a therapeutic strategy depends on the specific disease or even different stages within a disease.305 While there are currently no epigenetic-targeted drugs that modulate TET available on the market, experimental studies and clinical trials suggest that reshaping methylation landscapes through TET inhibitors and agonists may be a viable approach to treating diseases.
TET inhibitors
To date, numerous small molecules have been identified that inhibit TET enzymes. Research into these TET inhibitors primarily focuses on elucidating their underlying molecular mechanisms. We categorize these inhibitors into three groups based on their distinct mechanisms of action, which will be discussed in detail below.
Auranofin, C35, and eltrombopag specifically target and bind to the catalytic domains of TET proteins, directly inhibiting their enzymatic activities. C35 exhibits potent inhibitory effects on all members of the TET family.306,307 In contrast, the effects of auranofin and eltrombopag are specific to TET1 and TET2, respectively.308,309 Notably, eltrombopag, a nonpeptidyl thrombopoietin receptor agonist approved by the US FDA for use in patients with aplastic anemia as an iron chelator.310 Recently, Guan et al.309 reported the negative effects of eltrombopag on TET2. Intriguingly, this mechanism is independent of its iron chelation properties, presenting it as a potential TET2-targeted epigenetic agent and providing new insights for epigenetics-oriented therapy.309 Further, well-designed studies are essential to evaluate the clinical application potential of these molecular-level discoveries.
Itaconic acid, fumarate, and succinate are in vivo synthesized metabolites that indirectly impair TET catalytic activity by competitively binding to TET2 alongside α-KG, a crucial cofactor.311,312,313 These metabolites are promising precursors for developing TET-targeted epigenetic drugs, as they are well tolerated in vivo.314,315 However, considerable work is necessary before clinical application, such as designing appropriate carriers that can deliver these agents directly to pathological cells, given their potential impact on the vital activities of normal cells. Additionally, synthetic compounds like dimethyloxallylglycine and TETi76, which mimic the properties of α-KG, serve as competitive inhibitors of TET.316,317 These agents represent a novel approach to developing TET inhibitors.
Bobcat339,318,319 NSC-311068,320 NSC-370284,320 and UC-514321,320 inhibit DNA methylation by reducing intracellular TET levels. Bobcat339 induces the degradation of TET3 directly,318 and its inhibitory effects on TET1 and TET2 are observed only in the presence of coordinating copper(II).321 NSC-370284 and UC-514321 bind directly to the DNA-binding domain of signal transducer and activator of transcription 3 (STAT3) or STAT5, transcriptional activators of TET1, leading to suppressed expression of TET1 in vivo.320 This mechanism has been confirmed in mouse models of AML and medulloblastoma, showing synergistic effects with standard chemotherapy.320,322 The therapeutic potential of these compounds in additional diseases is an exciting area for future research.
TET agonists
As previously discussed, TET inhibitors are highly valued for treating diseases. Conversely, research into TET agonists is also anticipated to yield promising breakthroughs and pave the way for clinical applications. Most TET agonists currently under investigation are drugs that upregulate cofactors of TET, such as vitamin C and enzymes involved in α-KG metabolism; other small molecules, including 3-nitroflavanones,295 retinoic acid,323 ioperamide hydrochloride,324 and mitoxantrone,325 are reported to directly upregulate TET expression.
Vitamin C, or ascorbate, uniquely interacts with the C-terminal catalytic domain of TET, positioning it as a novel epigenetic-modifying agent.326,327 As an antioxidant, it also helps maintain the divalent state of iron ions, indirectly supporting TET activity.323 Characterized by TET repair and increased 5hmC levels, vitamin C administration can exert therapeutic roles in various tumors and non-tumor diseases.328,329,330 Furthermore, it has been used as an adjuvant, synergizing with other immunotherapeutic or chemotherapeutic agents.331,332,333,334 The synergistic treatment of vitamin C with azacitidine or decitabine in clinical trials has shown positive outcomes for patients with myeloid tumors.335,336 However, the optimal doses, frequency, and duration of vitamin C administration remain debated, with long-term treatment and follow-up required for further investigation.337 Therefore, the full exploration of the therapeutic role of vitamin C as an epigenetic-modifying drug is crucial for its future clinical applications.
Inhibitors of IDH and α-KG dehydrogenase elevate α-KG levels. In tumor cells, IDH1/2 mutations lead to the production of the oncometabolite 2-HG, which competes with α-KG for binding sites on TET, potentially leading to reversible inhibition of TET proteins and dysregulation of DNA methylation levels.338,339 The administration of enasidinib and siRNA against IDH2 has been shown to restore the low methylated state of the genome, consistent with the reactivation of TET enzymatic activities.340,341,342 However, observations suggest that other α-KG-dependent enzymes, such as histone demethylases and prolyl hydroxylases, might play a more dominant role in the progression of IDH-mutant diseases.343 These findings highlight the potential for developing TET agonists based on IDH inhibitors to reshape the epigenetic landscape, warranting further investigation. Additionally, inhibiting α-KG dehydrogenase enhances α-KG levels and TET activities, restoring DNA demethylation and ameliorating the progression of T2DM and breast cancer.344,345 Similarly, IOX1, an inhibitor of α-KG oxygenases and a potent inhibitor of lysine demethylase 3 A (KDM3A) and KDM4A, has been found to reduce TET enzymatic activities in helper T cells, emerging as a potential epigenetic drug for various autoimmune diseases.346 In conclusion, α-KG represents a promising target for TET-targeted drug development that should be further explored in clinical trials.
Targeting the reader of DNA methylation: MBD and UHRF1
With the discovery of proteins that read methylated DNA sites, burgeoning research has aimed to identify small molecules targeting these enzymes, sparking considerable enthusiasm for developing novel therapeutic targets.
MBD inhibitors
The MBD protein family, critical readers of DNA methylation, consists of six members: methyl-CpG-binding protein 1 (MeCP1), MeCP2, MBD1, MBD2, MBD3, and MBD4.347 The aberrant activities and expression of these proteins observed in various diseases have recently positioned them as potential targets for epigenetic drugs. Current research predominantly focuses on MBD2 inhibitors, which are rapidly progressing.
The development of MBD2 inhibitors hinges on two prerequisite factors. Firstly, the knockdown of MBD2 or the application of targeted siRNA has demonstrated positive effects in tumor treatment, underscoring the therapeutic potential of targeting MBD2.348 Secondly, successfully elucidating MBD2’s molecular structure and associated mechanisms lays the scientific groundwork for identifying and designing inhibitory molecules. MBD2 inhibitors can be categorized into three groups based on their mechanisms of action. The first group disrupts the binding of the N-terminal MBD to methylated DNA.349 Through docking analysis, molecules such as CID3100583 and 8,8-ethylenebistheophylline have been identified to target the interaction between MBD2 and DNA.350 The second category aims to block the interaction between the C-terminal coiled-coil domain and the GATA zinc finger domain containing 2A, which has shown potent inhibitory effects on MBD2-dependent DNA methylation.351 However, no drugs based on this mechanism are currently in use, highlighting a gap in research that demands further exploration. The third group prevents the interaction with HDAC and the formation of the nucleosome remodeling and deacetylase complex via an intrinsically disordered region.352,353,354 Utilizing this concept, Na et al.355 developed a novel technique that efficiently discriminates potential compounds interacting with intrinsically disordered proteins through expanded virtual screening. This approach led to identifying two MBD2 inhibitors, ABA and APC. These findings provide a sound basis for a therapeutic strategy targeting MBD2 and advocate for more comprehensive in vivo studies to assess their efficacy and safety.
UHRF1 inhibitors
Ubiquitin-like with plant homeodomain (PHD) and RING finger domains 1 (UHRF1) plays a pivotal role in recruiting DNMT1 during replication, primarily through the recognition of hemimethylated DNA and the subsequent flipping of hemimethylated CpG sites via the SET and RING associated domain (SRA).356,357,358 UHRF1 also interacts with HDAC1 and facilitates the di- and tri-methylation of H3, contributing to the ubiquitination of histones and the formation of heterochromatin.359 The upregulation of UHRF1 has been observed in various pathologies, particularly in tumors, making it a promising target for therapeutic intervention.360,361
The initial identification of small inhibitors targeting the SRA domain of UHRF1 was based on structure-based screening and computational analyses. Compounds such as NSC232003,362 UM63,363 UF146,364 chicoric acid,365 have been shown to block the interaction between UHRF1 and 5mC sites, effectively preventing the proliferation of diverse cancer cell lines. Advanced screening techniques, such as nonequilibrium capillary electrophoresis of the equilibrium mixture, have facilitated the identification of proanthocyanidins and baicalein as promising inhibitors.366 Furthermore, Ciaco et al.367 reported the development of novel UHRF1 inhibitors, AMSA2 and MPB7, based on the structure of UM63. These inhibitors suppress SRA-mediated base-flipping activities without DNA intercalation and demonstrate minimal effects on non-cancer cells, offering a basis for further optimization. In addition to the SRA domain, the tandem Tudor domain and PHD domain of UHRF1, which are involved in recognizing methylated lysine and arginine residues on H3, have also become targets for inhibitor design.368,369 While some inhibitors targeting these domains have been reported, their effects have only been validated in vitro, and more evidence is needed before proceeding to clinical trials.370,371,372 Current research suggests that inhibiting UHRF1 alone may not be sufficient to restore gene silencing affected by hypermethylation. However, combining UHRF1 inhibitors with other epigenetic inhibitors, such as HDAC inhibitors, can lead to synergistic effects and improved therapeutic outcomes.373,374 Consequently, multi-target inhibitors have been developed and are emerging as clinical candidates for tumor therapy.375,376,377,378 Additionally, the use of small molecules that act as UHRF1 degraders, such as diosgenin and MK2206, has been explored for prostate cancer treatment, representing a novel therapeutic approach.379,380 Natural substances like hinokitiol have shown therapeutic effects in mouse models in a UHRF1 depletion-dependent manner although the underlying mechanisms remain to be fully elucidated and represent a direction for future research.381
Epigenetics-targeted drugs and histone acetylation
The dynamic equilibrium and normal function of histone acetylation and deacetylation are regulated by the cooperative actions of the lysine acetyltransferase (KAT) and HDAC families, along with various reader proteins. Acetylation of lysine residues at the N-terminus of histones induces negative charges that trigger gene transcription, while decreased acetylation downregulates gene expression. Conversely, an imbalance in histone acetylation/deacetylation disrupts normal gene expression patterns, leading to the onset and progression of diseases. The development of related epigenetic drugs is ongoing, offering new therapeutic options for treating these conditions (Table 4).
Targeting the writer of histone acetylation: KAT
Histone acetylation serves various functions within cells. However, aberrant acetylation catalyzed by KAT can trigger the pathogenesis of various human diseases, including neurodegenerative diseases, metabolic diseases, and tumors.382,383,384,385 Developing epigenetic drugs that regulate KAT activity is a promising avenue for treating these diseases.
KAT inhibitors
Numerous inhibitors targeting KAT have been developed, primarily focusing on the P300/CBP, GNAT/PCAF, and MYST classes.386,387,388 These enzymes contain two accessible domains: the acetyl-lysine binding BD and the catalytic domain, utilizing acetyl CoA as a cofactor to transfer acetyl groups. Thus, designed inhibitors can target the enzymatic activity and the binding sites for acetyl CoA. Moreover, research into KAT degraders is advancing with the advent of proteolysis-targeting chimeras (PROTACs). These degraders are ternary complexes comprising ligands for targeted proteins and E3 ubiquitin ligase, along with a connecting linker, allowing targeted degradation via a ubiquitination-dependent method.389
Five KAT inhibitors have entered clinical practice, including CCS1477, FT-7051, NEO2734, PRI-724, and PF-07248144. CCS1477 targets the P300/CBP (KAT3A/KAT3B) via interaction with the BD fragment, exhibiting potent antitumor effects in cancer cell lines and animal models.390,391 This has led to its application in monotherapy and in combination with chemotherapeutic drugs in phase I and II clinical trials (NCT04068597, NCT03568656), with the potential to improve therapeutic strategies for both advanced solid tumors and hematological malignancies. FT-7051, another P300/CBP inhibitor targeting the BD domain, has been shown to reduce H3K27Ac at specific promoter sites and is currently under study in a phase I clinical trial for patients with hormone receptor-positive prostate cancer.392 NEO2734, a dual P300/CBP and BET inhibitor, demonstrates therapeutic potential comparable to the combination of a BET inhibitor and a P300/CBP inhibitor in treating certain cancers.393,394 It is currently being evaluated in a phase I clinical trial focusing on castration-resistant prostate cancer and other advanced solid tumors to assess its maximum tolerated oral dose (NCT05488548). PRI-724 effectively disrupts the interaction between β-catenin and CBP, ameliorating various diseases by inhibiting the Wnt/β-catenin signaling pathway.395 Its safety, tolerability, and antifibrotic effects have been further evaluated in two completed clinical trials among patients with hepatitis C virus (HCV)- and HBV-induced cirrhosis.396,397 However, PRI-724 fails to exhibit sufficient evidence of improvement in hepatic function according to existing data.396 Lastly, PF-07248144, a selective inhibitor of KAT6 (a member of the MOZ/MORF family), is currently under clinical investigation for the treatment of advanced breast cancer (NCT04606446). In summary, KATs represent compelling targets for therapeutic strategies, and developing novel and high-quality inhibitors with improved safety and efficacy is reaching an exciting phase.
Targeting the eraser of histone acetylation: HDAC
Zn2+-dependent classical HDACs and nicotinamide adenine dinucleotide (NAD)+-dependent HDACs (sirtuins) are crucial for dynamic deacetylation modifications on histones and non-histone proteins, playing significant roles in ontogeny and tumorigenesis. Despite the HDAC family’s broad substrate range in vitro, their specific subcellular localization restricts their biological functions and target proteins. Using inhibitors and agonists of HDACs and sirtuins to correct abnormal acetylation patterns is a promising therapeutic strategy.398,399 Notably, the therapeutic effectiveness of these interventions, in an epigenetic-dependent manner, hinges on the participation of target enzymes in histone deacetylation.
HDAC inhibitors
HDAC inhibitors are designed based on the spatial structure of their targets, characterized by highly conserved and homologous catalytic domains, including a catalytic channel, a zinc cation, and secondary pockets. Most HDAC inhibitors consist of a surface binding region, binding to the catalytic channel, and a zinc-binding group along with the linker, chelating the zinc ion.400 Four main categories of HDAC inhibitors are extensively studied: pan-inhibitors, selective inhibitors, multitarget agents, and PROTACs-based HDAC degraders.398 We will now discuss the recent applications of these HDAC inhibitors in clinical trials.
Four FDA-approved HDAC inhibitors—vorinostat, romidepsin, belinostat, and panobinostat—demonstrate a pan-inhibitory effect on almost all HDAC members and have made significant progress in treating some hematological malignancies. This success has fueled enthusiasm for developing additional pan-inhibitors to expand the clinical indications of these drugs. Currently, several pan-inhibitors, including ivaltinostat (CG200745), AR-42, abexinostat (PCI-24781), bisthianostat (CF-367), and sodium valproate, are under clinical trials for various tumors. The phase II study on ivaltinostat for advanced pancreatic ductal adenocarcinoma reports enhanced sensitivity of tumor cells to gemcitabine and erlotinib, presenting it as a potential treatment option.401 Another phase II study aims to determine the maximum tolerated dose and dose-limiting toxicity of ivaltinostat in combination with gemcitabine and erlotinib in patients with advanced pancreatic cancer, although clinical data have not been publicly disclosed, suggesting potential challenges (NCT02737228). In phase I trials, single-agent AR-42 has shown promise in treating type 2-associated meningiomas and schwannomas, with patients exhibiting good tolerance and therapeutic potential.402,403 However, a phase I trial focusing on advanced sarcoma and kidney cancer was terminated early due to observed dose-limiting toxicities in six patients (NCT02795819). Abexinostat, an oral small pan-inhibitor, whether used as monotherapy or in combination with chemotherapeutic agents, has shown promising therapeutic potential and acceptable safety profiles in solid tumors and hematological malignancies.404,405,406 Notably, a phase III study on abexinostat for locally advanced or metastatic renal cell carcinoma is ongoing in various regions, highlighting its potential as a clinical candidate (NCT03592472). Bisthianostat, a novel bisthiazole-derived pan-HDAC inhibitor, was studied in phase 1a clinical trial.407 Although preliminary data suggested modest efficacy and tolerability as a single agent in patients with R/R MM, this study has been terminated for undisclosed reasons (NCT03618602).
The non-selective inhibition characteristic of pan-HDAC inhibitors often leads to a broad spectrum of adverse effects and off-target toxicities, which restrict their widespread clinical application.408 Given the diverse roles of different HDAC classes, there is increasing interest in developing selective HDAC inhibitors, viewed as promising alternatives with better tolerance.409,410 However, due to a lack of evidence supporting the involvement of HDAC5/6/7/8/10 in histone deacetylation, selective inhibitors targeting these enzymes are not typically included in summaries of epigenetic-targeted drugs.411 Chidamide and givinostat, both FDA-approved selective inhibitors, have shown superior therapeutic efficacy and safety profiles. Givinostat, in particular, has promisingly expanded the clinical indications of HDAC inhibitors to include non-tumor diseases. Beyond these marketed drugs, several selective inhibitors have entered clinical practice. Notably, four such inhibitors are undergoing phase III clinical trials: pracinostat (NCT03151408), entinostat,412 magnesium valproate (NCT00533299), and tacedinaline (NCT00005093). Among these, only the phase III trial of entinostat combined with exemestane in treating hormone receptor-positive advanced breast cancer has shown satisfactory efficacy and manageable toxicities.412 However, among patients with other types of tumors such as colorectal carcinoma, lung cancer, endometrial endometrioid adenocarcinoma, and hematologic malignancies, entinostat fails to improve survival despite exhibiting good clinical efficacy.413,414,415,416 Importantly, according to an early-terminated, phase I clinical trial that evaluated the combination of entinostat, hydroxychloroquine, and regorafenib, the drug regimen among patients with metastatic colorectal carcinoma was poorly tolerated, with higher risks of weight loss, fatigue, and anorexia.413 Despite these advancements, selective inhibitors still face significant developmental challenges as they emerge as the next generation of HDAC inhibitors.
Recently, multitarget agents-based HDAC inhibitors have gained attention and have been posited to perform versatile roles in disease treatment.417,418,419 Various such agents, including those dual-targeting HDACs and kinases, receptors, DNA, transcriptional factors, and apoptosis-related proteins, are under preclinical investigation. Examples include curcumin (previously mentioned as a DNMT/METTL3 inhibitor), CUDC-101, tinostamustine (EDO-S101), fimepinostat (CUDC-907), domatinostat (4SC-202), and dacinostat (NVP-LAQ824, LAQ824), all of which are involved in various clinical trials.398 It has been widely reported that these multitarget agents enhance safety and reduce drug resistance in various diseases, both as monotherapy and in combination with radiotherapy or chemotherapy.420,421,422,423 However, emerging research offers a contrasting perspective. In a recent phase Ib clinical trial focusing on domatinostat (a dual inhibitor of LSD1/HDAC) in patients with advanced melanoma, the drug failed to enhance the efficacy of treatments targeting anti-PD-1 and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) while unexpected severe skin toxicity was observed.424 Furthermore, as current knowledge about these multitarget agents is still primarily derived from early-stage clinical trials, extensive investigations are necessary to validate their therapeutic value in a broader population.
HDAC agonists
While HDAC inhibitors have been extensively studied, research on HDAC agonists has been less prevalent. However, the therapeutic value of these agents in specific diseases has been demonstrated. Theophylline, used initially as an inhibitor of phosphodiesterase and adenosine receptors in treating asthma and chronic obstructive pulmonary disease, has recently shown activated effects on HDAC in low doses. These effects synergistically enhance the anti-inflammatory properties of cortisol in asthma and chronic obstructive pulmonary disease treatments.425 Nonetheless, a phase III clinical study revealed that additional administration of low-dose theophylline, along with inhaled long-acting β2-agonists and corticosteroids, failed to enhance HDAC activity in vivo. This left no significant difference from the anti-inflammatory properties of standard therapy.426
SIRT inhibitors and agonists
The sirtuin family’s role in developing various diseases, including inflammation, cardiovascular diseases, metabolic disorders, neurodegenerative diseases, and cancer, underscores the importance of exploring molecules that modulate their activity. Notably, SIRT2 is involved in the deacetylation of histone H4 during the G2/M transition and mitosis but is predominantly found in the cytosol, where it participates in non-histone deacetylation.427,428 SIRT3-5 are mainly located in mitochondria and possess a mitochondrial targeting sequence,429 while SIRT7 is primarily found in the nucleus, though fewer studies have addressed molecules that regulate its activity.430 Consequently, the inhibitors and agonists of SIRT1 and SIRT6 are highlighted as promising epigenetics-targeted drugs with significant potential.
From a mechanistic perspective, five classes of inhibitors of SIRT1 have been identified: First, competitive inhibitors that vie for acylated substrates at the binding sites, exemplified by natural products such as sirtinol, splitomicin, and cambinol analogs;431,432 Second, competitive inhibitors that challenge NAD+ for binding sites, including selisistat (EX-527) and Sosbo;433,434,435 Third, adenosine analogs such as Ro 31-8220;436,437 Further, binary inhibitors that compete with substrates or cofactors at separate binding sites, represented by ELT-31, a non-selective SIRT1-3 inhibitor;438 And the last, non-competitive inhibitors, including nicotinamide and its analogs, tenovins, thioacetyl-lysine peptides, and other small peptides.439,440,441 EX527, one of the few sirtuin inhibitors in clinical use, has demonstrated antitumor effects in vitro and potential as an adjunct in tumor therapy.442 Additionally, it has proven safe and well-tolerated within the therapeutic concentration range for treating neurodegenerative diseases.443 However, minimal therapeutic effects were observed in a phase I clinical trial focusing on early-stage HD, with further large-scale trials needed to explore its clinical potential.444 Ongoing research also investigates EX-527’s potential roles in improving other metabolic diseases, including endotoxemia,445 diabetic nephropathy,446 and infertility (NCT04184323) remains ongoing. Utilizing computational tools to predict potential allosteric sites has led to the identification of some allosteric SIRT6 inhibitors, including JYQ-42,447 compound 11e,448 and a pyrrole-pyridinimidazole derivative.449,450 Given that histone deacetylation catalyzed by SIRT6 promotes both tumor and non-tumor diseases, designing and in-depth study of these allosteric SIRT6 inhibitors represent a promising research field for human disease treatment.451,452
The agonists of the sirtuin family have been widely studied since the discovery of the first SIRT1 agonist, resveratrol, in 2003.453 The initially discovered sirtuin agonists mainly upregulate target enzyme activity through allosteric effects and are classified into two primary categories based on their origins. The first category comprises natural products extracted from plants, including resveratrol and other polyphenolic molecules.454,455,456,457,458,459 The second category consists of synthesized agonists that exhibit greater selectivity, focusing particularly on SIRT1, such as SRT1460, SRT1720, SRT2104, SRT2183, and SRT3025, as well as those targeting SIRT6, including UBCS039 and MDL-800.460 Additionally, compounds that are suggested to upregulate SIRT1 expression have been identified. These are predominantly involved in the activation of the mitogen-activated protein kinase pathway, including DDIT3,461 phloretin,462 puerarin,463 and atractylenolide III.464 Other reported agonists include include astragaloside intravenous,465 hesperidin,466 caffeic acid phenethyl ester,467 agomelatine,468 ligustilide,469 tanshinone IIA,470 and farnesol.471 These studies emphasize the role of SIRT1 activation in the deacetylation of various non-histones. However, the potential of these molecules as epigenetics-targeted drugs requires further exploration. Moreover, NAD+-enhancing molecules, which promote NAD+ generation or rescue their levels, represent a novel class of sirtuin agonists. These molecules may activate all sirtuin members with a single compound, attracting considerable attention.472 Given the diverse roles of NAD+ in multiple signaling pathways, additional discussion is needed to determine whether drugs that increase NAD+ levels have therapeutic effects in a sirtuin-dependent manner.
Targeting the reader of histone acetylation: BET, YEATS, and PHD
BET, YAF9, eleven-nineteen-leukemia protein (ENL), acute lymphocytic leukemia 1-fused gene from chromosome 9 protein (AF9), TAF14, and SAS5 (YEATS) domain, and PHD finger proteins are critical “readers” of acetylated residues and play essential roles as epigenetics-modifying enzymes in the transcription of downstream target genes. Drugs that target aberrant levels or activities of acetyl-recognition domain-containing proteins represent an emerging class of therapies for various diseases.
BET inhibitors
In recent years, a substantial number of BET inhibitors have been identified, encompassing pan-inhibitors, BD1/BD2 selective inhibitors, dual inhibitors of kinases and BET, and PROTACs-based inhibitors.60,473,474 From a therapeutic standpoint, BET inhibitors are primarily developed for treating tumors, with some also showing potential in non-tumor diseases, such as VYN-201 and VYN-202, among other BD2 selective inhibitors.475
More than twenty BET inhibitors have progressed to clinical trials, with several undergoing advanced phase evaluations. Notably, apabetalone (RVX-208) stands out as the sole BD2-selective inhibitor in phase III trials for addressing cardiovascular diseases and metabolic disorders such as T2DM.476,477,478 Apabetalone demonstrates significant anti-inflammatory properties, providing a robust scientific basis for its ongoing clinical evaluation.479,480,481 Another BET inhibitor, pelabresib (CPI-0610), has also reached phase III trials and shows promise as a treatment for myelofibrosis.482 In earlier phase II studies, pelabresib combined with ruxolitinib surpassed the efficacy of Janus kinase inhibitor monotherapy in treating symptomatic myelofibrosis while maintaining a manageable safety profile.483,484 ZEN-3694, a leading pan-BET inhibitor, has advanced to phase II trials, demonstrating efficacy when used with cyclin-dependent kinases (CDKs) inhibitors and conventional chemotherapy in cancer treatment.485 Preliminary phase Ib/IIa trials indicate that ZEN-3694, in combination with enzalutamide, is beneficial for patients with metastatic castration-resistant prostate cancer.486 An increasing number of trials focusing on ZEN-3694 are currently underway, which will provide further data to evaluate its therapeutic promise. Furthermore, recent reports highlight dinaciclib, a well-known CDK inhibitor, now recognized for its novel activity in BET suppression.487 The dual inhibitory capability of dinaciclib presents a potential strategy to counteract BET resistance in AML treatment.487 These developments underscore the potential of these drugs to achieve market approval for broad clinical use. However, some BET inhibitors as single-agent therapies have shown mixed outcomes in clinical trials for distinct cancer settings, despite their excellent results in preclinical models.488 For example, several phases 1 and 2 clinical trials investigating the therapeutic effect of birabresib on solid or hematological malignancies were terminated prematurely because of limited efficacy (NCT02698176, NCT02698189, NCT02698176, NCT02296476). Therefore, combining BET inhibitors with other traditional drugs may open new possibilities for the development of antitumor strategies.
Numerous novel BET inhibitors have been identified recently, enhancing the landscape of therapeutic options. These include OPN-51107, a pan-BET inhibitor that mitigates T cell dysfunction in chronic lymphocytic leukemia;489 XL-126, a BD1-selective inhibitor noted for its potent anti-inflammatory effects;490 and DW-71177, another BD1-selective inhibitor geared towards AML treatment.491 Additional developments involve brain-permeable BD1-selective inhibitors for multiple sclerosis treatment,492 compounds with dual HDAC/BET inhibitory action for challenging tumors,493 phenoxyaryl pyridone derivatives as BD2-selective inhibitors for AML,494 and SRX3177, a potent triple-action CDK4/6-phosphoinositide 3-kinase-BET inhibitor for respiratory diseases linked to β-coronavirus.495 These advancements significantly contribute to understanding BET-targeted drug development, designing small molecule inhibitors tailored to the diverse pathological characteristics of human diseases.
YEATS domain inhibitors
Identified in 2014, the YEATS domain-comprising YAF9, ENL, AF9, TAF14, and SAS5—serves as a novel reader for histone acetylation. This domain also recognizes histone crotonylation and benzoylation, which are critical in regulating gene expression.496,497,498 The human genome encodes four YEATS domain-containing proteins: ENL, YEATS domain-containing 2 (YEATS2), AF9, and glioma amplified sequence 41 (GAS41). These proteins are primarily implicated in the pathogenesis of tumors, particularly hematologic malignancies, and represent promising targets for epigenetic therapies.499,500,501,502 Research has shown that the YEATS domain binds to acylated lysine side chains through a common binding pocket and engages in π-π-π stacking interactions, providing a structural and theoretical foundation for developing targeted inhibitors.503 A significant milestone was the identification of the first small-molecule chemical probe, SGC-iMLLT, which targets ENL and its paralog AF9. This probe’s inhibitory effects were validated in biological assays.504 Furthermore, another approach involves blocking the protein-protein interaction (PPI) between YEATS domain proteins and disruptor of telomeric silencing 1-like (DOT1L), effectively suppressing the activity of YEATS domain proteins.505,506 Current research is focused on developing selective inhibitors for various YEATS domain proteins, with the deepest insights into ENL inhibitors. In 2022, Liu et al.507 highlighted the promising potential of the oral ENL inhibitor TDI-11055 in treating AML in mouse models, advancing the clinical application of ENL inhibitors for AML treatment. Additionally, combination therapies involving ENL inhibitors with KAT or BET inhibitors have been emphasized.508,509 In 2020, Jiang et al.510 introduced the first selective inhibitor targeting the AF9 YEATS domain, presenting a novel cyclopeptide for in-depth exploration of the functional similarities and differences between AF9 and ENL, thereby laying the groundwork for novel YEATS domain inhibitor development. An optimized method for the solid-phase synthesis of these inhibitory cyclopeptides has since been proposed, significantly reducing preparation time and enhancing yield.510 Moreover, the study of amide-π interactions between histone acyl-lysine and the AF9 YEATS domain has led to the development of chemical compounds that disrupt this noncovalent interaction, notably those incorporating urea or aromatic rings.511,512 In 2021, the first selective GAS41 inhibitor was reported; this synthesized molecule binds to dimerized GAS41 YEATS domains and blocks interaction with acetylated histone H3 in cancer cell lines.513
PHD finger domain inhibitors
BD and PHD finger-containing protein (BRPF) and BD and PHD finger transcription factor (BPTF) are crucial targets involved in tumor progression and the development of resistance to molecularly targeted therapy drugs, such as kinase inhibitors and poly ADP-ribose polymerase (PARP) inhibitors.514,515,516 To date, an array of BRPF inhibitors featuring distinctive scaffolds—such as 3-acetyl-indole, 1,3-dimethylquinolin-2-one, 1,3-dimethyl benzimidazole, 1-(indolin-1-yl)ethan-1-one, 1,3-dimethylquinolin-2-one, and 2,3-dioxo-quinoxaline—has been identified. These compounds represent novel avenues for therapeutic innovation.517,518,519,520,521,522 However, as the inhibitory effects of these agents have primarily been confirmed in vitro, extensive efforts are required to advance these drugs to clinical trials. BPTF inhibitor development has not kept pace with those targeting other proteins with BD motifs, primarily remaining within fragment-based drug discovery. Only a handful have been tested in vivo or in vitro to demonstrate their inhibitory actions and therapeutic potential. AU1, the first small molecule selective for BPTF, has shown effectiveness in mouse models of gastric cancer and neuroblastoma.516,523,524 Bromosporine has exhibited significant antitumor effects in breast cancer and melanoma, suggesting promising therapeutic strategies for solid tumors.525,526 The novel selective inhibitor C620-0696 has shown cytotoxic effects in non-small-cell lung cancer cells overexpressing BPTF.527 The continued exploration of these inhibitors in oncology is highly anticipated.
Epigenetics-targeted drugs and histone methylation
Histone methylation is a highly dynamic regulator crucial for activating or suppressing gene transcription. Histone methyltransferases, demethylases, and reader proteins modify and maintain epigenetic signals that influence chromatin structure and cellular functions. Their dysregulation is linked to a variety of diseases, particularly malignant tumors. Recent advances in biochemistry and understanding of pathogenesis have led to identifying and developing small-molecule inhibitors that target aberrant demethylation patterns (Table 5).
Targeting the writer of histone methylation: KMT and PRMT
Histone methyltransferases (HMTs), including KMTs and protein arginine methyltransferases (PRMTs), are central to regulating histone methylation and are implicated in numerous biological and pathological processes. Inhibitors of HMTs are extensively researched as potential therapeutic agents. Notably, innovative drug discovery strategies for HMT proteins—such as covalent inhibition independent of SAM-competitive or substrate-competitive mechanisms, dual-target inhibition, and targeted degradation strategies—have received considerable attention and have rapidly progressed.528,529,530 These inhibitors, in addition to marketed drugs, are being advanced to clinical practice for further evaluation and oversight.
EZH2 inhibitors
Since the identification of the suppressor of variegation 3-9 homolog 1 (SUV39H1), the inaugural histone KMT8 discovered in 2000, numerous proteins mediating histone methylation have been reported. These include EZH1/2, euchromatic histone-lysine N-methyltransferase 2 (G9a/EHMT2), G9a-like protein (GLP/EHMT1), DOT1L, and various SET domain-containing histone lysine methyltransferase (SETD) and nuclear receptor binding SET domain protein (NSD) families.531,532,533 Over recent decades, considerable efforts have focused on developing efficient and selective inhibitors targeting various histone KMT subfamilies with potential therapeutic applications in disease treatment.534,535,536,537
In addition to the two marketed drugs summarized in the previous section, tazemetostat (EPZ-6438) and valemetostat (DS-3201b), numerous novel EZH2 inhibitors are under investigation, with several advances in clinical studies, particularly compounds featuring the 2-pyridone moiety which encompass both bicyclic heteroaromatic and monocyclic aromatic rings.538 CPI-1205 (lirametostat) has undergone evaluation in three clinical trials (NCT03480646, NCT03525795, NCT02395601) to assess its tolerance and therapeutic potential. Although CPI-1205 has shown good tolerability in phase I stages, phase II trials have yet to provide sufficient data to confirm its antitumor efficacy. CPI-0209 (tulmimetostat), an oral, next-generation dual EZH2/EZH1 inhibitor developed by the same company, is currently under clinical trial for treating both solid tumors and hematological malignancies (NCT05944562, NCT05942300, NCT04104776). SHR2554, a highly selective EZH2 inhibitor, has demonstrated potent efficacy both in vitro and in vivo.539 Its first-in-human, dose-escalation, and dose-expansion phase 1 trial conducted at 13 hospitals in China in 2018 indicated good tolerance and promising antitumor activity in patients with R/R lymphomas.540,541 A pharmacokinetic study revealed that combining itraconazole, an inhibitor of CYP3A4-metabolizing enzymes, with SHR2554 improves its plasma concentration while maintaining a favorable safety profile, suggesting a new therapeutic strategy.542 PF-06821497, a lactam-derived EZH2 inhibitor, was optimized from a series of similar compounds using ligand-based and physicochemical-property-based strategies, showing optimal inhibitory and therapeutic effects in mouse models.543 It also has demonstrated synergistic effects in combination with HDAC inhibitors, inhibiting proliferation and inducing apoptosis in cancer cell lines.544 Currently, two clinical studies are exploring appropriate administration methods for PF-06821497, such as intravenous injection or oral intake, and whether it can be consumed with food (NCT06392230, NCT05767905). In addition to demonstrating a strong therapeutic effect in tumor treatment in animal models, GSK126 has also achieved significant breakthroughs in enhancing β-like cell regeneration among patients with T1DM.111,545 However, a terminated phase I clinical trial revealed insufficient evidence of clinical activity for GSK126 at tolerable doses.546 XNW5004 (NCT06022757) and AXT-1003 (NCT05965505) are innovative EZH2 inhibitors currently in clinical trials, reflecting ongoing advancements in this therapeutic area. Moreover, astemizole, originally an antiallergy medication inhibiting histamine receptor H1, has recently been shown to disrupt the EZH2-embryonic ectoderm development (EED) PPI within the PRC2, offering new perspectives in developing EZH2/PRC2 inhibitors.547
DOT1L inhibitors
EPZ-5676 (pinometostat), EPZ004777, and SGC0946 are three selective inhibitors of DOT1L that are currently under extensive research. EPZ-004777 was the first SAM-competitive inhibitor of DOT1L to demonstrate in vivo efficacy.548 Despite showing promising therapeutic effects in various subtypes of AML through cell experiments and animal models, EPZ-004777’s preclinical application has been largely constrained by its pharmacokinetic characteristics.549,550 EPZ-5676 has been developed to improve selectivity and inhibition effects, showing potential as a therapeutic agent for mixed lineage leukemia (MLL).551 Early investigations using patient-derived xenografts and mouse models have indicated that EPZ-5676 exhibits potent antileukemic activities, facilitating further evaluation.552,553 In three completed clinical trials (NCT01684150, NCT02141828, NCT03701295), EPZ-5676 has been assessed for safety, tolerability, and preliminary antitumor activity in pediatric patients with MLL, with the combination of EPZ-5676 and azacytidine in a phase Ib/II study expected to show synergistic antiproliferative activities (NCT03701295). SGC0946, a brominated analog, serves as another selective inhibitor of DOT1L. Its therapeutic potential, either as monotherapy or in combination with other inhibitors such as HDACs and the mitogen-activated protein kinase pathway, has been observed in various solid tumors, setting the groundwork for clinical trials of SGC0946.554,555,556
Beyond EZH2 and DOT1L, several inhibitors targeting other subfamilies are being investigated in clinical studies, including EZM0414 and KTX-1001. EZM0414, a novel inhibitor of SETD2 derived from the optimization of EPZ-719, exhibits improved pharmacokinetic properties and potent pharmacodynamic activity in mouse xenograft models.557 A phase I/Ib clinical trial is currently underway to explore the safety, tolerability, and therapeutic efficacy of EZM0414 in patients with R/R MM and R/R diffuse large B-cell lymphoma (NCT05121103). KTX-1001, a selective NSD2 inhibitor, has been FDA-approved for clinical trials since 2022 and is being studied in a phase I trial to treat patients with R/R MM (NCT05651932). These meticulously organized clinical trials focusing on KMTs are drawing increasing attention, leading to significant breakthroughs in understanding the relationship between human diseases and aberrant histone methylation.
PRMT inhibitors
Significant progress has been made in developing inhibitors for type I PRMTs (PRMT1-4, 6, and 8) and a selective inhibitor targeting PRMT5, with several agents entering the early phases of clinical trials.
Two type I PRMTs inhibitors are already in clinical stages, including GSK3368715 and CTS-2190. GSK3368715 (EPZ019997), an oral, reversible inhibitor of PRMT1/6/8 developed for treating tumors and pulmonary disorders.558,559,560 GSK3368715 underwent a phase 1 clinical trial for treating solid tumors and diffuse large B-cell lymphoma in 2018. However, the first clinical application of a PRMT1 inhibitor did not meet expectations and was terminated early in 2022 due to its ineffectivenes.561 Given the adverse events potentially caused by high and sustained concentrations of the inhibitor in vivo, research into PROTAC-based degraders of GSK3368715 has intensified, potentially offering therapeutic benefits at lower doses and reducing adverse effects.562 CTS-2190, another inhibitor targeting PRMT1/3/4/6, received clinical trial approvals from the US FDA and China NMPA in February and April 2023, respectively. A phase I/II clinical trial is being conducted to evaluate its tolerability and preliminary antitumor activity in healthy participants and patients with solid tumors (NCT06224387).
Thirteen PRMT5 inhibitors have advanced to phase I and II clinical trials. Among these, GSK3326595, JNJ64619178, and PF06939999 were the earliest selective PRMT5 inhibitors to receive clinical trial approvals. GSK3326595 is a substrate-competitive inhibitor, while JNJ64619178 and PF-06939999 function as SAM-competitive agents.563,564,565 The efficacy and understanding of GSK3326595 primarily rely on animal model data due to a lack of published results from completed clinical trials. This inhibitor has been shown to induce DNA damage in cancer cells and enhance the antiproliferative effects of poly ADP-ribose polymerase inhibitors, such as niraparib;566 however, long-term or chronic use of GSK3326595 is associated with potential liver-related adverse effects.567 A completed phase I clinical trial of JNJ-64619178 determined that a daily dose of 0.5 mg was better tolerated by participants with R/R B cell non-Hodgkin lymphoma, though it demonstrated limited therapeutic effects.568 Conversely, PF-06939999 has shown an acceptable safety profile and clinical efficacy in its phase I trial.569 TNG908 and MRTX1719, both brain-penetrant PRMT5 inhibitors, have shown promise in selectively targeting cancer cells deficient in methylthioadenosine phosphorylase in both preclinical models and clinical trials.570,571 Phase I/II clinical trials for these drugs recruit participants to assess their therapeutic effects on various solid tumors (NCT05245500). Prelude Therapeutics has developed PRT543 and PRT811, leading oral PRMT5 inhibitors whose safety profiles and preliminary therapeutic potential have been evaluated in phase I clinical trials (NCT04089449, NCT03886831). PRT543 has demonstrated good tolerance and efficacy among patients with adenoid cystic carcinoma, warranting further advanced clinical testing.572 A phase I/II clinical trial of SKL27969 began in 2022 to evaluate its safety, tolerability, pharmacokinetics, pharmacodynamics, and preliminary efficacy in patients with advanced solid tumors. However, this study was terminated in 2024 due to portfolio prioritization, with no significant safety trends or issues identified during its execution (NCT05388435). Other PRMT5 inhibitors, such as AMG193, SH3765, TNG462, SCR6920, and SYHX-2001, are currently under investigation in clinical trials and are in the “recruiting” phase. Given that most of the small molecules or core scaffolds of PRMT5 inhibitors have been examined only in cellular experiments, there remains a significant gap in knowledge regarding their efficacy and therapeutic effects in vivo. Therefore, it is imperative to bridge the crucial divide between fundamental research and clinical application.
Targeting the eraser of histone methylation: KDM
KDMs are enzymes that remove histone and nonhistone methylation. They can be divided into two categories based on their molecular structures: flavin adenine dinucleotide-dependent KDM (KDM1) and Fe(II)- and α-KG-dependent KDM (KDM2-7), also called Jumonji C (JmjC)-KDMs.573 Both upregulation and downregulation of KDMs can affect the expression of pathological genes in cancers or other disorders. Currently, representative inhibitors of diverse KDM proteins are being investigated. Based on catalytic mechanisms, lysine specific demethylase 1 (LSD1/KDM1A) inhibitors can be divided into irreversible and reversible inhibitors; KDM2-7 inhibitors are classified into four types: α-KG cofactor mimics or inhibitors of α-KG oxygenases (such as N-oxalylglycine), metal cofactor disruptors, histone substrate competitive inhibitors, and other substrate- and cofactor-independent inhibitors.574
KDM2/7 inhibitors
KDM2 and KDM7 proteins, which belong to the JmjC-KDM subfamilies, share high similarity in their Fe(II)- and α-KG-binding residues.575 The development of inhibitors for KDM7 and KDM2 has typically co-occurred. In 2013, a series of hydroxamate analogs featuring an alkyl chain were identified. These compounds demonstrated antiproliferative activity in cancer cells by inhibiting KDM2A, KDM7A, and KDM7B.576 Similarly, Gerken et al.577 developed a series of novel KDM2A/7A inhibitors characterized by saturated indoline ring systems. These indoline-containing compounds exhibited potent and selective effects on KDM2A/7A at low micromolar concentrations, with notable cellular activity. Nonetheless, addressing limitations such as cytotoxicity and off-target effects remains challenging for future research. Other selective inhibitors have also been identified, including a cyclic peptide inhibitor, OC9, designed to target the PHD finger domain of KDM7. This inhibitor results in the inhibition of KDM7B and the activation of KDM7A.578 Through virtual screening of α-KG oxygenases, daminozide, a plant growth regulator, was found to selectively inhibit KDM2A. The therapeutic potential of daminozide was observed in mouse models of osteoarthritis, pointing to new directions for developing 2KG-competed inhibitors with enhanced selectivity, although its use in humans is unlikely.579,580
KDM3 inhibitors
In addition to IOX1, various inhibitors of the KDM3 family have been identified, most exhibiting a pan-inhibitory effect across all family members. Through virtual screening of natural products and traditional Chinese medicine components, compounds JDI-4, JDI-12, and JDI-16 selectively bind to the JmjC domains of KDM3B and KDM3C.581 Subsequent in vitro and in vivo studies confirmed the inhibitory effect and antitumor potential of JDI-16 in a KDM3-dependent manner.581 Another compound, JDM-7, also identified from this screening, inhibits KDM3A and KDM3B in AML cell lines, although initial observations indicated limited effects on the KDM3 family.582 Additionally, through high-throughput screening of benzhydryl amine derivatives, CBA-1 was found to be a potent inhibitor of KDM3A, exhibiting antiproliferative effects on colorectal cancer cell lines.583 The use of CBA-1 in zebrafish models also showed minimal toxicity, suggesting its potential as a promising drug for clinical application.583
KDM4 inhibitors
Given the critical roles of KDM4s in cancers and the inherent complexity of the KDM4 subfamily, significant efforts have been dedicated to developing KDM4 inhibitors. These inhibitors are categorized into four previously reviewed classes: α-KG cofactor mimics, Metal cofactor disruptors, histone substrate competitive inhibitors, and inhibitors targeted reader domains, having been summarized extensively in previous work.584,585 In addition to these established categories, we emphasize the progress in novel inhibitors that have not yet been summarized, further expanding the scope of therapeutic options against KDM4-related cancers.
TACH101 is a novel pan-inhibitor of KDM4A-D, competitively inhibiting α-KG without affecting other KDM subfamilies. The therapeutic effects of TACH101 have been demonstrated in organoids and xenograft models, suggesting its potential as an anticancer agent worthy of further investigation in animal studies.586 SD49-7, a derivative of SD70, is another novel KDM4 inhibitor. It has shown a stronger effect than SD70 in suppressing the proliferation of AML cell lines and enhancing the progression of resistant tumors in mouse models.587 Based on virtual screening, 2-(methylcarbamoyl)isonicotinic acid has been identified as an initial active fragment specifically inhibiting KDM4A by preventing its binding to H3K9me3 in a substrate-competitive manner.588 Molecular docking and dynamics approaches have recently revealed that a series of natural products containing sugars, aromatic rings, and OH or O− groups can interact with KDM4 and inhibit its activities.589 However, the mechanisms of these interactions remain unclear, underscoring the need for further development of these potential drugs.
KDM5 inhibitors
KDM5 inhibitors have shown significant therapeutic potential, though many compounds still lack sufficient evidence to confirm their efficacy and safety in vivo.
A prevailing approach in KDM5 inhibition involves designing small molecules that compete with α-KG for binding sites.590 Among these, KDOAM-25 is a potent and selective inhibitor affecting MM and triple-negative breast cancer cells, with minimal adverse effects observed in vivo applications. Nevertheless, its poor cell membrane permeability hinders its efficacy.591 RS3195 exhibits inhibitory effects on KDM5B and KDM5D in vitro. Due to potential toxicity, RS5033 was developed as an alternative, featuring a phenyl ring instead of a pyrrole nucleus to improve tolerance.592 KDM5-C49, an analog of 2,4-PDCA, binds to KDM5B in vitro and inhibits its enzymatic activities.593 To enhance cell membrane permeability and selectivity, derivatives KDM5-C48 and KDM5-C70 have been developed.593,594,595 Through high-throughput virtual screening, a series of cyclopenta[c]chromen derivatives targeting KDM5A have been identified as promising drugs due to their potent inhibitory effects and low toxicity.596 N70, a thienopyridine-based selective KDM5A inhibitor, displays α-KG-competitive inhibition, while its analog, N71, binds irreversibly to KDM5A through covalent modifications.597
Numerous compounds, identified through virtual screening and optimization of reported inhibitory molecules, employ different mechanisms of action.598,599 Among these, KDM5-inh1 and CPI-455 are broadly studied pan-inhibitors of KDM5. Using either KDM5-inh1 or CPI-455 has demonstrated therapeutic effects on cancer cell lines and has facilitated synergistic interactions with conventional antitumor agents.600,601 Further research should explore the potential for this synergy in animal models. GS-5801, designed from GS-080—one of the most potent KDM5 inhibitors—shows significant anti-HBV activity. Despite its promise, the in vivo effects of GS-5801 have not met expectations, underscoring the need for additional studies to enhance its efficacy.602 Utilizing the AlphaScreen method, ryuvidine was identified as an inhibitor of KMD5A/B/C, exhibiting substantial therapeutic impact on drug-tolerant cells.603 Dexmedetomidine, recently identified as a KDM5 inhibitor, is utilized to manage acute kidney injury in a KDM5-dependent manner.604 A novel approach was introduced by Yang et al.,605 who reported the first selective metal-based KDM5A inhibitor, rhodium(III) complex1. This compound disrupts the interaction between KDM5A and H3K4me2/3, offering a new scaffold for optimizing KDM5A-targeted drugs. The screening of imidazopyridine-analogs of zolpidem led to the discovery of O4I3, a novel chemical inhibitor of KDM5A. O4I3 generates and sustains patient-specific induced pluripotent stem cells in vitro.606 Additionally, TK-129, a pyrazole-based KDM5B inhibitor, is applied in treating cardiovascular diseases.607 High-throughput screening technology has facilitated the identification of PBIT, another novel KDM5B inhibitor. Despite its promising attributes, PBIT exhibits unstable therapeutic effects across different cell lines, necessitating careful consideration of its application in treatment.608 Similarly, several pyrazole derivatives that inhibit KDM5B have been recognized, with several demonstrating potent activity in cells, suggesting new therapeutic strategies.609 Furthermore, Iida et al.610 designed a selective KDM5C inhibitor with a triazole scaffold and subsequently synthesized a KDM5C degrader using PROTAC techniques. This selective degrader shows enhanced inhibitory effects on prostate cancer cell lines compared to its prodrug, thus expanding the possibilities for anticancer agent design.
KDM6 inhibitors
The KDM6 subfamily has gained attention as a therapeutic target for various diseases. GSK-J1 and GSK-J4 are two well-studied classical inhibitors of KDM6B, showing significant potential in treating autoimmune diseases, metabolic disorders, and tumors, and enhancing the effectiveness of traditional antitumor agents.611,612,613,614 Using optimized delivery systems for GSK-J1 has further advanced the development of effective in vivo strategies.615 Beyond these compounds, novel inhibitors have been introduced. For instance, KDOBA67, a hydroxyl derivative of GSK-J4, demonstrates favorable cell permeability in chordoma cell lines and inhibits the progression of chordoma.616 Employing a virtual fragment screening approach, Giordano et al.617 identified a series of benzoxazole scaffold compounds that bind to the KDM6B subfamily with high affinity, showing therapeutic promise in melanoma cell lines. Zhang et al.618 developed a simple capillary electrophoresis method for screening KDM6B inhibitors, leading to the identification of salvianic acid A and puerarin 6”-O-xyloside as effective agents. Additionally, Jones et al.619 used computational methods to develop an optimized peptide derived from the H3 C-terminus, which may enhance selectivity when linked with known inhibitors.
LSD1 inhibitors
Extensive research has been conducted on the biological and pathological functions of LSD1 and its inhibitors. Compared to other KDM subfamilies, LSD1 inhibitors have seen significant advances.620 Currently, several LSD1 inhibitors such as tranylcypromine (TCP), ORY‐1001 (ladademstat), ORY‐2001, GSK‐2879552, IMG-7289 (bomedemstat), INCB059872, SP‐2577 (seclidemstat), CC-90011 (pulrodemstat), 4SC‐202 (domatinostat), JBI‐802, TAK-418, LH‐1802, and SYHA1807, are undergoing clinical trials.
TCP, an irreversible inhibitor, is being used in clinical practice among patients with AML and MDS, showing promising effects either alone or in combination with all-trans-retinoic acid in phase I/II clinical trials, with overall response rates exceeding 20%.621,622 Building on TCP’s structure, novel inhibitors like ORY-1001, ORY-2001, GSK-2879552, INCB059872, and IMG-7289 have been developed, which also bind irreversibly to LSD1.623 These advancements have broadened the spectrum of treatable diseases with LSD1 inhibitors. Notably, ORY-1001 and ORY-2001, both orally administered, have been evaluated for their effectiveness in R/R hematologic malignancies and neurological disorders such as borderline personality disorder and AD.624,625,626,627,628 In completed phase I clinical trials, ORY-1001 exhibited a good safety profile without significant extrahematologic toxicity among healthy volunteers and patients with AML, indicating good therapeutic potential.624,627 GSK2879552 has shown antitumor efficacy in animal models,629 yet several clinical trials have been terminated due to a high incidence of adverse events.630,631 Similarly, clinical trials for INCB059872 were halted due to business decisions, among other reasons (NCT02959437, NCT03514407, NCT03132324, NCT02712905). Greater attention must be dedicated to evaluating the tolerability and efficacy of novel treatments. According to completed clinical trials, IMG-7289 demonstrates potential in ameliorating several blood disorders (NCT04254978, NCT03136185, NCT02842827), with numerous recent registrations for novel clinical trials concerning this drug. SP-2577 and CC-9001 are reversible LSD1 inhibitors,632 which, compared to their irreversible counterparts, exhibit enhanced safety profiles and have been extensively studied in both solid tumors and hematological malignancies.633,634 Domatinostat and JBI-802, dual inhibitors targeting LSD1 and HDAC, selectively interact with class I HDAC isoenzymes and HDAC6, respectively.620 Although promising antitumor effects have been observed in cancer cell lines, the therapeutic potential of domatinostat requires further exploration due to its unfavorable toxicity.424,635 TAK-418, a novel LSD1 inhibitor noted for its effective brain penetration, is considered a potential treatment for central nervous system disorders.636,637 The administration of TAK-418 was well tolerated by healthy volunteers in a phase I clinical trial, laying a solid foundation for further investigation.638 LH-1802 and SYHA1807, novel inhibitors, are currently under clinical trial evaluation for metastatic prostate cancer and small-cell lung cancer, respectively (NCT03678025, NCT04404543). The encouraging outcomes from these clinical-stage applications have spurred greater interest in the development of LSD1 inhibitors, with ongoing efforts to discover effective and tolerable agents.
Targeting the reader of histone methylation: reader domains
The identification of histone lysine and arginine methylation is attributed to proteins possessing malignant brain tumor (MBT) domains, chromodomains, Tudor domains, proline-tryptophan-tryptophan-proline (PWWP) domains, PHD fingers, and WD40 repeat (WDR) domains.639 Notably, enzymes that serve as writers or erasers for histone methylation may also contain these reader modules, such as PHD fingers and Tudor domains, aiding in recognizing residues they catalyze.640 Although numerous inhibitors targeting reader domains have been discovered, nearly half of these originate from structure-based virtual screenings and lack in vivo evaluation of their inhibitory effects and therapeutic activity.641 Encouragingly, MAK683, an inhibitor targeting EED—a representative histone methylation reader containing the WDR domain—has entered clinical trials.642 Currently, MAK683 is in a phase I clinical trial for treating diffuse large B-cell lymphoma (NCT02900651). Inspired by this milestone, many potent and selective inhibitors of EDD and other molecules targeting reader domain proteins are expected to advance into clinical trials as promising therapeutic strategies.
Epigenetics-targeted drugs and m6A
RNA m6A methylation, a prevalent and conserved modification in eukaryotic RNAs, is crucial in determining transcript fate at the post-transcriptional level through RNA processing, export, degradation, and translation. Dysregulated m6A regulators contribute to various pathological conditions, particularly in the pathogenesis of diverse tumors.643 With the identification of various enzymes involved in m6A modification—including writers, erasers, and readers—the reversibility of m6A modification has been increasingly recognized, providing a foundation for developing epigenetics-targeted drugs that regulate RNA m6A as a core mechanism.
Targeting the writer of m6A: METTL3
METTL3 plays a critical role in the m6A modification process by transferring methyl groups from SAM to target RNA, catalyzing the conversion of adenosine to methyladenosine. This function of METTL3, the most extensively studied m6A writer, has been linked to the development of various pathologies, notably various tumors.644 Recent research has highlighted a range of inhibitors and agonists targeting METTL3, with several epigenetic drugs demonstrating promising efficacy both in vitro and in vivo, thus reinforcing the significance of METTL3 regulation in disease pathology and its potential as a therapeutic target.
METTL3 inhibitors
The study of METTL3 inhibitors has attracted increasing attention due to their diverse roles in regulating gene expression across different diseases. These inhibitors are categorized into competitive and allosteric inhibitors and gene expression suppressors, each leveraging distinct mechanisms of action.
SAM analog is the dominant part of the competitive inhibitors, initially developed based on a fulfilled screening of compounds containing the adenosine moiety (the fragment responsible for the combination with METTL3 at the binding sites for SAM). In 2020, Bedi et al.645 performed a series of docking studies on over 4000 adenosine-moiety compounds, identifying seven molecules with potential binding affinity to METTL3; however, their inhibitory effects in vivo were minimal. Similarly, Moroz-Omori et al.646 and Dolbois et al.647 reported on adenine-based libraries, identifying UZH1a and UZH2 as compounds that occupy the catalytic pocket of METTL3, suggesting their role as potential competitive inhibitors in vitro. Cpd-564, an METTL3 inhibitor identified from ChemDiv and MCE screening libraries, has shown significant reno-protective effects in mouse models of acute kidney injury induced by cisplatin and ischemia-reperfusion.648 Coptisine chloride, identified via molecular docking-based virtual screening from the Vitas-M chemical library, displayed high affinity to METTL3, exerting competitive inhibitory effects by occupying the SAM binding pocket.649 STM2457 and STM3006 are novel small molecules that bind non-covalently to the catalytic center of METTL3, reducing its enzymatic activity.650,651 Specifically, STM2457 has demonstrated promising antitumor effects and tolerability in mouse models, improving drug resistance to chemotherapy.650,652,653,654 In comparison, although STM3006 exhibits enhanced cellular potency, its in vivo efficacy is constrained by its shorter half-life.651 In 2023, STC-15, an oral inhibitor optimized from STM2457, became the first and the only RNA m6A target drug to be applied in phase I clinical trials (NCT05584111). Through detailed studies on the spatial structure of the catalytic domain of METTL3, a series of branched, Y-shaped molecules are designed. These were synthesized by integrating chemical fragments from the most effective inhibitors, resulting in molecules with selectivity and binding affinities surpassing those of STM2457, the only commercially available METTL3 inhibitor.655 This advancement not only underscores the potential of METTL3 as a therapeutic target but also guides future drug design. Additionally, several natural products with METTL3-inhibitory capabilities have been identified. Quercetin, known as a DNMT inhibitor, has been found to interact with the adenosine moiety pocket in METTL3, forming a stable complex that reduces its catalytic activity.656 This interaction decreases METTL3 hyperactivation and lowers m6A levels in protein kinase D2 mRNA, improving insulin sensitivity under palmitic acid stimulation—a benefit in hyperinsulinemia conditions.657 Other natural compounds like berberine and curcumin, also noted for DNMT/HDAC inhibition, have shown METTL3 inhibitory activity, though their mechanisms require further clarification.658,659 Moreover, molecules F039-0002 and 7460-0250 have been designed to target METTL3’s catalytic pocket, showing potential in treating inflammatory bowel disease.660 Several candidates identified through silico analysis of South African natural products—SANCDB0370, SANCDB0867, and SANCDB1033—also exhibit METTL3 inhibitory properties, with further validation needed.661 More recently, Li et al.662 designed a stapled peptide inhibitor, RSM3, targeting the PPI at the METTL3-METTL14 interface. This inhibitor offers a unique approach compared to other small-molecule competitive inhibitors, providing a novel avenue for therapeutic intervention.
Allosteric inhibitors prevent METTL3/14-dependent m6A methylation in a non-competitive manner. To date, three allosteric inhibitors have been identified. The first two allosteric inhibitors are CDIBA and CDIBA-43n, which initially function as cytosolic phospholipase A2 inhibitors preventing inflammation. They show an inhibitory effect in the presence of METTL3/14 complex, instead of separate METTL3 and METTL14 subunits.663 The third compound, eltrombopag (previously mentioned as a TET agonist), is recently reported to bind with the METTL3 subunit at an allosteric site and has shown potential in treating AML.663,664
Metformin, traditionally used as a first-line treatment for T2DM, has recently been found to inhibit METTL3 expression, possibly contributing to its beneficial effects in patients with malignant tumors.665 The role of metformin in inhibiting METTL3 expression, at the post-transcriptional level, in breast tumors, is first reported in breast cancer.666 Subsequently, the application of metformin is also found to inhibit METTL3 expression at the transcriptional level, mediated by the recruitment of DNMT.667 This dual action of metformin, combined with chemotherapy, offers potential benefits for patients resistant to traditional chemotherapy, potentially mitigating poor prognoses.667,668 Given its safety profile, metformin is a promising candidate as an epigenetic drug targeting METTL3.
METTL3 agonists
While research has predominantly focused on METTL3 inhibitors, there is also interest in agonists, given their potential benefits in DNA damage repair, tumor therapies, and regenerative medicine.669,670,671,672 In 2019, Selberg et al.673 predicted interactions between four small-molecule ligands with METTL3 involving piperidine and piperazine rings, similar to SAM’s binding. These interactions enhanced cell viability and promoted proliferation, although differing onset times among the compounds suggest the need for further development of more effective METTL3 complex activators.673 Melatonin seems to act as an agonist of METTL3. Lv et al.674 proposed that melatonin pretreatment can upregulate the expression level of METTL3, restore m6A levels in spermatogonial stem cells, and help them resist the destructive effect of Cr(VI) on reproductive function. However, this viewpoint has recently been questioned. In mouse models with colon inflammation, melatonin inhibits METTL3 expression through melatonin receptor 1B.675 Further research is necessary to clarify melatonin’s role in METTL3 regulation.
Targeting the eraser of m6A: FTO and ALKBH5
FTO and alkB homolog 5 (ALKBH5) are established m6A erasers, each playing significant roles in epigenetic regulation. FTO is primarily involved in energy homeostasis, demethylating m6A in various RNA species, including cellular mRNA, which impacts multiple biological processes.676 ALKBH5 not only demethylates m6A-marked mRNA but also m6A-marked single-stranded DNA (ssDNA), influencing oncogenic or tumor-suppressive activities.677 Over the years, numerous small molecules targeting these m6A writers have been identified and designed, showing promising therapeutic efficacy in vitro and in vivo and advancing the development of epigenetic drugs.
FTO inhibitors
Current strategies for developing FTO inhibitors are multifaceted. Based on the spatial structure of FTO, competitive or non-competitive inhibitors that bind to FTO covalently or non-covalently have been developed. With a deeper understanding of FTO functionality, metabolites in vivo possibly related to FTO have been identified, represented by D-2-HG, a metabolite produced by mutant IDH.678 Furthermore, exploring the mechanisms underlying medical agents that treat FTO-related diseases provides a theoretical foundation for drug discovery.679 Subsequently, optimizing the molecular structures of these initially detected compounds will contribute to the development of FTO inhibitors with high selectivity and inhibitory effects, providing promise for the clinical application of FTO inhibitors in the future. Here, we summarize the typical drugs that inhibit FTO, which are the foundation for developing novel inhibitors through constant iterations.
In 2012, the first FTO inhibitor, rhein, was identified. Rhein impairs FTO activity by disrupting its interaction with ssDNA at the catalytic domain.680 Although rhein increases m6A levels in vitro, its weak selectivity and low inhibitory efficacy have limited its clinical potential, highlighting the need for more effective FTO inhibitors.680 New FTO inhibitors need to be designed to overcome these drawbacks. Meclofenamic acid (MA), an FDA-approved nonsteroidal anti-inflammatory drug, binds selectively to similar sites on FTO.681 MA and its prodrug, MA2, have shown promising results in reversing tumor progression and enhancing the efficacy of chemotherapeutic drugs, significantly prolonging survival.681,682 Inspired by MA, various compounds have been developed, such as the fluorescein derivative FL1, which retains the benzyl carboxylic acid structure critical for interaction with FTO. The complex formed between FTO and FL1 inhibits the enzyme’s activity and facilitates the study of FTO-related signaling pathways through fluorescein labeling.683 Other optimization molecules include GNPIPP12-MA,684 13a,685 FB23/FB23-2,686 Dac-51/Dac-85,687 ZLD115,688 and FTO-02/FTO-04/FTO-43.689,690 These compounds significantly improve MA in inhibitory activity, cell permeability, and biosafety while reducing off-target effects and potential resistance. Another similar mechanism inhibitor is diacerein, a structural analog of rhein, which has shown antitumor effects in breast cell lines.691
In addition to interfering with the interaction between ssDNA and FTO, inhibitors of this enzyme also compete with cofactors such as α-KG and iron(II). For instance, fumarate hydrazide 2 and compounds with the aminohydroxyfuranone core exemplify this approach.692,693 Furthermore, the discovery of N-CDPCB, a competitive inhibitor that binds to non-conserved fragments of FTO, provides novel insights into the development of inhibitory agents. Mechanistically, compounds like benzene-1,3-diol and 4-Cl-1,3-diol are crucial in mediating and enhancing the specific interaction between FTO and N-CDPCB.694 Additional potential inhibitors, such as CHTB and radicicol, have been identified through virtual screening; these compounds have similar structures.695,696 However, related evidence is lacking to exhibit their efficacy. Moreover, Su et al.697 reported on CS1 and CS2, which tightly bind to the catalytic pocket of FTO, activating immune checkpoint genes and reversing immune evasion in tumor diseases. Clausine E, another FTO inhibitor, targets the enzyme’s hydrophobic cavity, exhibiting antitumor activity.698
These ongoing discoveries provide a deeper understanding of the diverse structures of molecules interacting with FTO and their mechanisms of action, promoting large-scale virtual screenings to identify more potential inhibitors. For example, mupirocin, entacapone, compounds “18,077” and “18,079”, several quinolone derivatives, and a series of 1,2,3-triazole analogs have been identified as potential FTO inhibitors.699,700,701,702,703 Notably, quinolone derivatives and their antitumor properties have shown the potential to improve symptoms in neurodegenerative diseases by inhibiting FTO activity.702 These findings broaden the potential clinical applications of FTO inhibitors.
FTO agonists
Recent studies have identified that certain tricyclic antidepressants, such as imipramine and amitriptyline, exert their antidepressant effects by activating FTO in N2a cells.704 This emerging area of research highlights the potential therapeutic benefits of FTO activators and calls for more attention to their development and evaluation.
ALKBH5 inhibitors
The RNA demethylase ALKBH5 is recognized as a pro-oncogene, playing a vital role in the post-transcriptional regulation of various targets in cancer biology.677 Interest in targeting ALKBH5 for therapeutic purposes has significantly increased. We classify the identified ALKBH5 inhibitors into three main categories based on their mechanisms of action. The first category comprises typical competitive inhibitors that compete with cofactors for binding sites. These agents consist of IOX1 (also known as a TET/KDM inhibitor), MV1035, and Ena21, which exhibit the therapeutic potential of targeting ALKBH5 in antitumor therapies.705,706,707 Compounds that non-covalently interact with the active pocket of ALKBH5 are classified as the second group. Through structure-based virtual screening and optimization, the current compounds include DDO-2728, 2-((1-hydroxy-2-oxo-2-phenylethyl)thio)acetic acid, and 4-((furan-2-ylmethyl)amino)tetrahydropyridazine-3,6-dione.708,709 The third category includes molecules that bind to the m6A-binding pocket of ALKBH5, directly disrupting the interaction between the enzyme and its substrates. For instance, compounds 20 m and TD19 are representative of this type.710,711 Some compounds still exist whose potential mechanisms for inhibiting ALKBH5 have not been elucidated, such as ALK-04, Ena15, ZINC78774792, and ZINC00546946, although their antiproliferative effects have been revealed in vitro and in vivo.707,712,713 In brief, the continued development and in-depth research into ALKBH5 inhibitors hold significant potential for disease treatment, necessitating further efforts.
Targeting the reader of m6A: IGF2BP and YTH domain family
The discovery of m6A readers with specific motifs has spurred significant interest in developing drugs targeting these proteins, expanding the possibilities for therapeutic interventions.
IGF2BP inhibitors
Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) are newly identified m6A readers that enhance the stability and maintenance of their target mRNAs.714 IGF2BP plays an oncogenic role in various cancers, making its inhibition a promising strategy for antitumor therapy.715
Six IGF2BP inhibitors have been developed, demonstrating antitumor effects by disrupting IGF2BP-RNA interactions. BTYNB, the first identified IGF2BP1 inhibitor, suppresses melanoma and ovarian cancer cell proliferation by blocking IGF2BP1’s interaction with c-Myc mRNA.716 BTYNB’s therapeutic effects are being studied across various tumor models, including esophageal squamous carcinoma,717 neuroblastoma,718 and cholangiocarcinoma.719 CWI1-2 and JX5 are novel IGF2BP2 inhibitors with antileukemic activities that inactivate the Notch1 signaling pathway. CWI1-2 forms a hydrophobic interaction with IGF2BP2’s RNA-binding core, while JX5 binds tightly to the protein. Further research is needed to enhance their safety and reduce cytotoxicity.720,721 Cucurbitacin B, a natural product, exerts a pharmacological allosteric effect on IGF2BP1. In hepatocellular carcinoma mouse models, it modifies IGF2BP1’s configuration, reducing its efficacy.722 Another compound, “7773,” specifically disrupts the IGF2BP1-Kras mRNA interaction, effectively inhibiting IGF2BP1’s pro-oncogenic activity.723 Isoliquiritigenin is the only small molecule identified targeting IGF2BP3.724 Derived from the Chinese herb licorice, it downregulates IGF2BP3 expression, showing promise in treating non-small cell lung cancer.724
YTH domain family inhibitors
YTH domain family (YTHDF) comprises a group of readers featuring a YTH domain at the C-terminus. This domain forms a hydrophobic pocket essential for recognizing m6A modifications.725 Elevated levels of YTHDF proteins have been associated with the progression of various cancers.726 Conversely, reducing these proteins can synergistically enhance the effectiveness of ionizing radiation and anti-PD-L1 therapies in reducing cancer burdens.727,728 This underscores the potential of YTHDF inhibitors as a promising direction for improving antitumor treatments.
The binding sites between YTHDF proteins and m6A modifications are primary targets for most YTHDF inhibitors.729 The successful elucidation of the crystallographic structures of the YTH domains in YTHDF proteins has provided critical opportunities for drug design.730 High-throughput screening technology has identified three small molecules—ebselen, DC-Y13, and DC-Y13-27—as effective YTHDF inhibitors.727,731 Ebselen targets YTHDF1 and YTHDF2, either covalently or non-covalently binding to the YTH domain.731 DC-Y13 and DC-Y13-27, particularly the latter, act as selective inhibitors of YTHDF2, offering therapeutic benefits.727 Additionally, studies have shown that disrupting O-GlcNAcylation of YTHDF proteins can also decrease their stability and enzymatic activities, providing new avenues for identifying YTH-inhibiting small molecules.732,733
Epigenetics-targeted drugs and chromatin remodeling
SWI/SNF complexes are intricate multimeric structures composed of diverse, variable subunits that play distinct roles, emphasizing the importance of personalized characteristics and frequent mutations in these subunits in various human diseases. Recently, the design of small molecules targeting different components of the SWI/SNF complex has expanded, yielding numerous potential therapeutic interventions. Those that have progressed to clinical trials are detailed in Table 6.
The active DNA-dependent ATPase A domain inhibitor (ADAADi) was the first discovered inhibitor targeting the SWI/SNF complex. It was identified during studies on mammalian cell resistance to certain antibiotics in vitro.734 ADAADi binds to specific motifs in the enzyme complex, inducing conformational changes that inhibit SWI2/SNF2’s catalytic activities.734 Currently, ADAADi shows promising therapeutic effects in prostate cancer in preclinical studies, laying the groundwork for further development of SWI/SNF-targeted epigenetic drugs.735
Research has also focused on specific inhibitors targeting the SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin A4 (SMARCA4) and its paralog SMARCA2, which are DNA-stimulated ATPases within the SWI/SNF complexes. SMARCA4, commonly mutated in various tumors, is associated with reduced sensitivity to traditional cancer treatments.736,737 Inhibiting SMARCA4/2 is an effective strategy for curbing tumor growth and improving patient outcomes. Papillon et al.738 reported the earliest selective allosteric inhibitors of the SMARCA4/SMARCA2 subunits, with confirmed effects on pediatric H3K27M diffuse midline glioma and AML in both in vivo and in vitro settings.739,740 FHD-286, a novel orally bioavailable SMARCA4/SMARCA2 allosteric inhibitor, has shown preclinical efficacy. Combined treatment with FHD-286 and other epigenetic drugs, such as decitabine, BET inhibitors, and menin inhibitors, has demonstrated synergistic effects in reducing AML burden without significant toxicity.741 Notably, FHD-286 has entered clinical development for treating various malignant tumors, including metastatic uveal melanoma (NCT04879017) and several malignant hematological disorders (NCT04891757).
An alternative approach to inhibiting SMARCA4/SMARCA2 involves using specific inhibitors that target their BDs. This strategy extends to other BD-containing proteins within the SWI/SNF complexes, such as polybromo-1 (PBRM1), BD containing 7 (BRD7), and BRD9, also considered promising targets for epigenetic drug development. Notably, SMARCA4/SMARCA2 and BRD9/BRD7 each contain one BD, whereas PBRM1 contains six tandem BDs, providing numerous potential interaction points for inhibitors.742 Current research primarily focuses on inhibitors for family VIII BD in SMARCA4/SMARCA2 and PBRM1, with four major classes of inhibitors reported: salicylic acid fragment hits such as PFI-3;743,744,745 aminopyridazines represented by GNE-064;746 quinazolinones represented by LM146,747,748 and dihydroquinazolinones represented by compound16 and GNE-235.749,750 These inhibitors are categorized as either pan-inhibitors, affecting multiple proteins, or selective inhibitors, targeting specific proteins. PFI-3, its analogs, and GNE-064 are pan-inhibitors, whereas LM146 shows a higher affinity for SMARCA2, and compound16 and GNE-235 are selective for PBRM1. The therapeutic applications of these inhibitors, particularly the pan-inhibitors, have been extensively studied in various diseases.751,752,753,754,755 However, the efficacy of PFI-3 as a standalone treatment for malignancies has been less satisfactory. The application of compound16, on the other hand, demonstrates promising therapeutic effects in PBRM1-dependent prostate cancer, suggesting its potential as a foundational treatment for PBRM1-driven cancers.749 As for the other molecules, though their binding ability and inhibitory effects have been validated at the molecular level, sufficient evidence is still lacking in vivo or in vitro to demonstrate their clinical value. Furthermore, targeting family IV BD of BRD9 and BRD7 has led to the development of many selective inhibitors. Current research includes inhibitors like BI-7271,756 BI-7273,756 BI-9564,756 I-BRD9,757 iBRD9,758 GNE-375,759 and newly identified inhibitors developed through integrated computational approaches.760 Selective inhibitors for BRD7, such as 1-78 and 2-77,761 and molecules like LP99, TP-472, 4-acylpyrroles, and GSK6776, which inhibit both BRD9 and BRD7, are being evaluated for their therapeutic effects in various pathologies.762,763,764,765
PROTAC technology plays a significant role in developing SWI/SNF inhibitors, with novel agents such as AU-24118 and AU-15330 being tested in preclinical studies and clinical trials. AU-24118 and AU-15330 are degraders targeting family VIII BD in SMARCA4/SMARCA2 and PBRM1, which are valuable tools in castration-resistant prostate cancer treatment.766,767 AU-24118 has shown promise in inducing tumor regression at therapeutic doses.766 However, long-term treatment at high doses can lead to mutations in the BD and overexpression of ATP-binding cassette subfamily B member 1 (ABCB1), which contributes to drug resistance development.766 Combining these treatments with ABCB1 inhibitors could potentially mitigate resistance to SMARCA4/SMARCA2 inhibitors in vivo. Additionally, applying PROTACs to previously reported inhibitors can enhance their selectivity and reduce off-target effects. For instance, the linkage of BI-7273 with an E3 ubiquitin ligase has led to the design of dBRD9-A, the first BRD9-directed degrader, which is undergoing optimization.768,769 The current focus on dBRD9-A, which is being tested for its efficacy in AML, MM, and interferon-induced inflammation in animal models, highlights its potential as a promising therapeutic for both tumor and non-tumor conditions.768,770,771 Additional PROTACs-based SWI/SNF inhibitors, such as those derived from dihydropyrrolo-quinazolin scaffolds (targeting SMARCA4, SMARCA2, and PBRM1),747 A947 (targeting SMARCA2),772 VZ-185 (targeting BRD9 and BRD7),773 CFT8634 as well as FHD-609 (targeting BRD9), further illustrate the breadth of ongoing research.774 Notably, FHD-609 have recently advanced to clinical trials for synovial sarcoma (NCT04965753), underscoring the critical role of drug development targeting mutations in SWI/SNF complexes. Moreover, CFT8634 was originally planned for investigation in a phase 1/2 clinical trial of synovial sarcoma and other SMARCB1-perturbed soft-tissue sarcomas (NCT05355753). However, the clinical trial was terminated because of the less significant clinical activity of CFT8634 as a single agent. Considering the prevalence of mutations in SWI/SNF complexes in cancers, continued research into the in vivo therapeutic effects, potential applications, and long-term risks of these drugs is essential for assessing their clinical utility.
Epigenetics-targeted drugs and non-coding RNA
A deep understanding of ncRNA’s role in disease progression, particularly in various cancers, has led to innovative epigenetic strategies for disease management.775 RNA interference (RNAi) technologies, which utilize small double-stranded RNA to selectively interact and degrade specific intracellular RNAs, mimic gene deletion phenotypes.776,777 RNAi-based therapies targeting ncRNA are categorized based on their action mechanisms and intended outcomes: silencing overexpressed ncRNAs to curb disease-related expressions, restoring downregulated ncRNAs to regain lost functions, and blocking ncRNA localization to prevent ncRNA from functioning by interfering with its subcellular localization. RNAi-oriented drugs altering ncRNA patterns have been widely studied and applied in clinical practice (Table 7). Herein, we summarize the emerging technologies for ncRNA-targeted agent development, aiming at supplementing the current understanding of drug design.
Significant advancements in molecular editing and delivery systems have bolstered the clinical viability of these innovative ncRNA-targeted therapies.778,779 One pivotal development has been the chemical modification of synthetic nucleic acids to enhance their stability and delivery efficiency. The initial focus on replacing phosphodiester bonds with phosphorothioate has been a cornerstone in numerous FDA-approved oligonucleotide therapies,780 although concerns about inflammation and toxicity in vivo have prompted research into alternative modifications for RNA-targeted based on RNAi.781 Over the years, various modification strategies have emerged, including those based on 2’-O-methyl, 2’-O-methoxyethyl, 2’-fluoro, and n-acetylgalactosaminyl, aiming to preserve the therapeutic attributes of nucleic acids while enhancing their stability.782 Locked nucleic acids (LNAs) represent a notable innovation, linking the 2’ and 4’ carbons of ribose rings with methylene bridges, thus improving hybridization affinity and resistance to nucleases.783 Cobomarsen, an LNA-based inhibitor targeting miR-155, exemplifies this technology’s potential, having shown promising results in preclinical studies for hematological malignancies and solid tumors and exhibiting positive effects in mycosis fungoides patients.784,785,786,787,788 In addition, the therapeutic efficacy of cobomarsen has been further validated in patients with mycosis fungoides, indicating well-tolerated and positive clinical potentials (NCT03713320).
Furthermore, integrating nanomedicine-based delivery systems, such as lipid-based, polymeric, inorganic, and biomimetic nanoparticles, has significantly advanced the development of RNAi drugs. These delivery techniques enhance the stability and bioavailability of oligonucleotide drugs and improve their efficacy in modulating target ncRNA expression and functionality.789 The recent emphasis on nanoparticles designed for targeted delivery of therapeutic nucleic acids to specific subcellular organelles marks a significant advancement in ncRNA therapy. As ncRNAs are present not only in the cytoplasm but also the nucleus and various organelles, targeting these subcellular locations can enhance the efficacy of treatments.790,791 Researchers are exploring opportunities to integrate subcellular organelle-targeting signals into nanoparticle delivery systems. Current studies have reported RNAi nanoparticle systems designed to target the nucleus, mitochondria, endoplasmic reticulum, and Golgi apparatus.792,793,794,795 Though nucleus-targeted nanoparticles have been studied in regulating ncRNAs, only a few of them have proposed the incorporation of nucleus-targeting TAT peptide and nucleus-targeting peptide amphiphile into nanoparticle delivery systems to achieve active transportation.792 Many researchers have only reported the high concentrations of therapeutic oligonucleotides in the nucleus without elucidating the mechanisms involved in nucleus-targeting. This approach leaves a gap in our understanding of the complex and precise intracellular delivery processes that involve biomembrane systems and cytoskeletal interactions.796 The variability in cell types, delivery materials, and therapeutic nucleic acids means that successful results in specific contexts may not universally apply, highlighting the challenges in translating these strategies from experimental to clinical settings.
Additionally, the integration of clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated system (CRISPR/Cas) technologies, particularly CRISPR/Cas9, into ncRNA research offers novel avenues for manipulating ncRNA expression, including lncRNAs and microRNAs (miRNAs).797,798,799,800 CRISPR/Cas9’s flexibility and high specificity make it an advantageous tool for gene-targeted cancer therapies, such as CRISPR interference, activation, and knockout strategies, now moving into preclinical trials. This gene-editing technology promises to enhance the efficacy of traditional cancer treatments and aims to minimize off-target effects, thus fostering the development of personalized medicine.801,802
However, several challenges persist despite the growing number of ncRNAs identified as potential therapeutic targets. Technological advancements are still needed, and safety concerns must be rigorously addressed.803 The premature termination of the Phase I clinical trial for MRX34, a liposomal mimic of miR-34a, due to severe immune reactions and fatalities, underscores the critical need for comprehensive preclinical data and cautious progression to clinical trials.804
Practical challenges in the epigenetic drug development and application
Massive efforts have been made in epigenetics-targeting drug development, whether approved in different countries for the treatment of specific indications or currently identified and further evaluated in fundamental experiments or clinical trials, exhibiting notable potential. However, there are still practical issues that should be dealt with when applying them to a large scale in clinical practice.
First of all, a thorough understanding of epigenetic mechanisms is crucial for successfully applying epigenetic-targeted drugs. Alterations in epigenetic enzymes can change DNA, histones, or chromatin structures, impacting cellular processes like transcription, replication, and DNA repair. Even minor modifications in epigenetic enzyme activity can significantly affect cellular functions. Therefore, deepening our understanding of epigenetic biology is essential. However, gaps remain in our knowledge, particularly regarding the roles of epigenetic regulation and its proteins in mammals, such as new DNA modifications like m6A, RNA modifications beyond m6A, novel reader domains, and PPI networks related to epigenetic regulation. These areas represent potential targets for novel epigenetic drugs but require further exploration.805,806,807,808,809,810 Second, many molecules identified through virtual screening and molecular docking techniques are primarily used as molecular probes to study enzyme localization and function. However, transitioning from a potent molecular probe to a viable therapeutic agent involves rigorous in vivo evaluation to confirm their inhibitory effects and therapeutic potential. This step is crucial as it determines whether these compounds can be safely and effectively used in clinical settings.811,812 These small molecules are potent probes to detect the localization and function of targeted enzymes, and their potential for medical use needs confirmation. Notably, another challenge that needs to be overcome is associated with the precise localization and targeted activity of epigenetics-targeted drugs is critical for enhancing their clinical efficacy and reducing potential side effects. The need for precise delivery of epigenetic drugs to subcellular structures is underscored by the diverse localizations and functions of their target enzymes and ncRNAs. For instance, enzymes responsible for histone modifications are found both in the nucleus, where they modify histones, and in the cytoplasm, where they regulate non-histones and related signaling pathways at the post-translational level.813,814 Similarly, the function of ncRNAs depends significantly on their localization and distribution within the cell.790 To address these challenges, a deeper understanding of the intracellular trafficking mechanisms is required. Additionally, developing sophisticated drug delivery systems, such as lipid-based nanoparticles or targeted delivery vehicles, can enhance the specificity and efficiency of these therapies by directing them to their precise intracellular sites of action. Furthermore, the challenge of achieving selective inhibition within the HDAC family exemplifies the broader issue of specificity in drug design. Many drugs targeting epigenetic enzymes exhibit pan-inhibitory effects, leading to significant off-target effects and adverse reactions, particularly when the drugs indiscriminately affect multiple members of an enzyme family.408 This necessitates the development of more selective inhibitors that specifically target single proteins or subfamilies of proteins. By focusing on selective inhibition, researchers can potentially improve the safety and efficacy of these treatments, minimizing unwanted interactions and enhancing their therapeutic impact.
Importantly, the possibility of developing resistance to epigenetic-targeted drugs, which is another factor limiting their further application, cannot be ignored. Several studies have investigated the mechanisms involved in the development of resistance to epigenetic agents in different cancers.815,816,817 The activation of certain signaling pathways and gene mutations in tumor cells play an indispensable role in inducing the development of drug resistance to epigenetic agents. Research has shown that the activation of enhanced Wingless/Integrated (Wnt)/β-catenin signaling contributes to developing resistance to BET inhibitors in leukemia cells. However, inhibition of this pathway helps rescue drug sensitivity in vitro and in vivo.818,819 Furthermore, enhanced activity of the protein kinase B (AKT)/mTOR complex 1 (mTORC1) signaling pathway is also responsible for drug resistance to BET inhibitors in prostate cancer.820 In another study on HDAC resistance in solid tumors, the potential role of the activation of some kinases and downstream pathways was also reported.821 Additionally, altered TME may be one of the culprits in promoting drug resistance. Tumor-associated macrophage (TAM), a pivotal mediator in inducing tumor cells to develop resistance to traditional antitumor therapies, has recently been proposed to be involved in the occurrence of epigenetic drug resistance.822,823 A 2020 study on triple-negative breast cancer reported that interleukin-6 (IL-6) and IL-10 derived from TAM activated STAT3 signaling in tumor cells, conferring them with drug resistance to BET inhibitors.823 Additionally, these findings pave a theoretical foundation for the combination of epigenetics-targeted drugs and other pharmaceutical molecules to optimize their long-term efficacy.824 The relationship between drug resistance to epigenetic agents and mutations in certain genes is observed in tumor cells, especially in the case of applying EZH2 and IDH inhibitors.59,825,826 Based on this idea, the loss-of-function mutation of specific genes by CRISPR/Cas techniques may provide better platforms for coping with drug resistance.
In conclusion, for epigenetics-targeted drugs, it is crucial to balance pharmacokinetics (how the drug is processed in the body), tolerability (how well the drug is tolerated), and therapeutic efficacy (how to avoid off-target off-target toxicities and resistance). Developing optimal dosing regimens that maximize efficacy while minimizing side effects and resistance requires a thorough understanding of the drug’s behavior in the body, including its absorption, distribution, metabolism, and excretion. Innovative dosing strategies, possibly involving controlled release formulations or real-time monitoring of drug levels, could play a vital role in achieving this balance. Continued research into the biological and pathological roles of targets for epigenetic drugs is essential.
Developing trends and forthcoming prospects in epigenetics-targeted drugs
Research on epigenetics-targeted drugs has progressed rapidly, with a growing focus on their potential as next-generation clinical candidates. Current trends, illustrated in Fig. 5, emphasize the synergy between these agents and other therapeutic modalities such as chemotherapy, radiotherapy, kinase inhibitors, and immunotherapy. This integration promotes precision medicine and personalized treatment strategies and enhances the overall effectiveness of cancer therapies.827,828 Furthermore, developing epigenetic degraders, which can hydrolyze targeted proteins, complements the inhibitory functions of traditional epigenetic drugs.829 Notably, the swift advancement of sequencing technology has empowered the detection of epigenetic irregularities with growing efficacy, substantially enhancing the integration of epigenetics into personalized medicine.

The promising trends and practical challenges in the clinical application of epigenetics-targeted drugs. From the perspective of clinical practice, epigenetic agents are expected to become promising adjuvants in combination with traditional antitumor therapeutics, contributing to superior efficacy and decreased resistance. Based on this idea, multitarget anticancer agents inhibiting both HDAC and other pathological molecules have gained much attention as a new strategy. Further, epigenetic degraders based on PROTAC or other techniques responsible for TPD help supplement the catalytic function of epigenetic inhibitors. Notably, the successful transport of epigenetic regulators to specific tissues or cells, or even the finite subcellular structures, is the prerequisite for exerting therapeutic effects. The further optimization of different types of nanoparticles makes them inspiring tools for the delivery system
Epigenetics-based combination therapy in cancer cells
The integration of epigenetics-targeted drugs with conventional cancer therapies—such as chemotherapy, targeted therapy, immunotherapy, and hormone therapy—is emerging as a promising strategy for cancer treatment. Increasing experimental studies and clinical trials are assessing the safety and efficacy of various combination regimens. The benefits of these combinations can be categorized into two primary aspects:
Epigenetic drugs can synergize with other cancer therapies by modulating the metabolic and pathological characteristics of cancer cells, immune cells, and stromal cells within the TME.830 Although immune checkpoint inhibitors (ICIs), which target immune checkpoint proteins (ICPs), such as PD-L1, PD-1, and CTLA-4, show significant potential, their effectiveness may be limited by factors such as insufficient antigen presentation and suboptimal T cell responses in the TME.831,832 Epigenetic modifications can enhance the expression of tumor antigens and ICPs, overcoming these limitations. The use of epigenetic therapies not only disrupts immunosuppressive pathways but also enhances the recruitment of tumor-reactive immune cells, resulting in synergistic effects with ICIs.136,833,834,835
Interfering with aberrant epigenetic features is crucial for combating drug resistance, a major challenge in oncology. Chemoresistance often correlates with changes in DNA methylation and histone acetylation, among other epigenetic characteristics.139,836,837 Combining chemotherapeutic agents with epigenetic drugs has become an important strategy to address resistance,838,839 also helping to mitigate chemotherapy-related side effects.840,841 Additionally, reversing epigenetic alterations in chemoresistant tumor cells can restore their sensitivity to conventional therapies, offering a renewed opportunity for treatment.842 In the context of targeted therapy, inhibitors of mutant kinases initially provide rapid benefits but often lead to the development of resistance over time.843 The potential of epigenetic treatments, particularly HDAC and DNMT inhibitors, to reverse such resistance is currently being explored.844,845 The development of dual inhibitors, like CUDC907, CUDC101, and 4SC-202, shows promising results in overcoming resistance in kinase-driven cancers and warrants further investigation.846 For hormone-dependent cancers, such as estrogen receptor-positive breast cancer and androgen receptor-positive prostate cancer, endocrine therapy remains a crucial treatment option.847 However, epigenetic alterations can lead to resistance during endocrine therapy.848,849 Targeting these epigenetic changes can help sustain the effectiveness of endocrine therapies and reduce the proliferation of cancer cells.848,850
At present, various combination regimens based on HMA and traditional anticancer drugs have entered clinical trials, gaining the potential to become an alternative for patients with certain diseases. For example, the combination of the oral B-cell leukemia/lymphoma 2 inhibitor venetoclax with HMAs has become a standard regimen among patients with AML or MDS who are ineligible for intensive chemotherapy. In November 2018, the FDA approved this combination for AML therapy.851,852,853 Further, triplet regimens that include HMAs, venetoclax, and other targeted agents are being developed for AML with specific gene mutations.229,854,855,856 Early results from these clinical trials have demonstrated a good safety profile and promising effects, with ongoing studies needed to confirm their efficacy and potential adverse events. In May 2022, the combination of ivosidenib, an IDH1 inhibitor, and azacitidine was approved by the FDA for older patients with newly diagnosed IDH1-mutated AML.240 Other drugs being combined with HMAs include HDAC inhibitors, polo-like kinase 1 inhibitors, T-cell immunoglobulin domain and mucin domain-3 antibodies, and PD-L1 antibodies.857,858,859,860 These combinations are currently under evaluation in ongoing registrational clinical trials across different stages, with promising results anticipated for updating clinical strategies. Additionally, the combination of HMAs with targeted therapy and immunotherapy, as well as chemotherapy, is showing promising application prospects, especially in hematologic malignancies with acquired chemoresistance caused by aberrant DNA methylation.861,862 Currently, azacitidine is approved in multiple countries and is widely used in patients with myeloproliferative disorders, such as MDS, AML, chronic myelomonocytic leukemia (CMML), and juvenile myelomonocytic leukemia, whereas decitabine is approved for the treatment of MDS, AML, and CMML.863,864,865
Small molecules serving as epigenetics-targeted degraders
As an emerging therapeutic strategy, epigenetics-targeted degraders for targeted protein degradation (TPD), respected by molecules based on PROTAC, autophagy-targeting chimera (AUTAC), hydrophobic tagging (HyT), molecular glue (MG), and other novel techniques for drug discovery are worth trying as a remarkable alternative presenting pioneering approaches.866
PROTACs, first proposed in 2001, are considered revolutionary technologies in drug discovery. They consist of a ligand for targeted proteins, a ligand for E3 ubiquitin ligase, such as Von Hippel-Lindau and Cereblon (CRBN), and a linker connecting the two ligands.867 Many degraders targeting diverse epigenetic enzymes have been developed, including the newly designed or derived from the optimization of known selective inhibitors.868,869 This technology has led to the creation of various degraders targeting a wide range of epigenetic enzymes, enhancing their potency, duration of action, safety profile, and ability to counteract resistance mechanisms compared to conventional inhibitors.867,868,869 For instance, dBET1, derived from the BRD inhibitor JQ-1, was the first PROTAC-based degrader targeting BET proteins. It has demonstrated superior anticancer effects in both AML cell lines and mouse xenograft models compared to JQ-1 alone.836,870 Further optimization led to dBET6, which increased cell permeability significantly improved survival rates in solid tumor models, and reduced the emergence of resistance.836 Innovative approaches to enhance the selectivity and safety of PROTACs include antibody-PROTAC technology, which combines monoclonal antibodies targeting specific pathological cells with degraders. This strategy facilitates targeted delivery, minimizing side effects while maximizing efficacy. Antibody-PROTACs have been developed for breast cancer cells overexpressing human epidermal growth factor receptor 2 and prostate cancer cells expressing six transmembrane epithelial antigens of the prostate 1, showing enhanced degradation specificity in these cell types.871,872 Compared with general degradation agents, these small molecules present the preferential degradation of target proteins in specific cell lines. Additionally, integrating control elements into PROTAC molecules allows for activation in specific physiological or pathological conditions, reducing potential off-target effects.873 Techniques such as photocaged PROTACs, photo-switchable PROTACs, and radiotherapy-triggered PROTAC prodrugs represent cutting-edge strategies in this area. These methods ensure that the degradation activity of PROTACs can be spatially and temporally controlled, enhancing their clinical applicability and safety.874,875,876 Overall, the evolution of PROTACs and other PROTACs-oriented TPD strategies is shaping a promising future for epigenetics-targeted therapies, offering more precise and effective treatment modalities for various diseases, particularly cancer.
Other types of epigenetics-targeted degraders, such as AUTAC-based agents, HyT-based degraders, and MG, offer innovative alternatives to PROTACs for TPD. Each technology employs distinct mechanisms and offers unique advantages for therapeutic applications. Unlike PROTACs, which utilize the proteasomal degradation pathway, AUTAC agents promote lysosome-dependent degradation of target proteins.877 One of the key benefits of AUTAC degraders is their enhanced membrane permeability due to their typically low molecular weights, making them potent therapeutic candidates.878 For instance, AUTAC-based degraders targeting BRD4, developed from the covalent interaction between autophagy key proteins and JQ-1, have shown significant antiproliferative activity across multiple tumor cell lines. This demonstrates their potential as effective medical tools for treating various diseases.879 Introduced in 2011, HyT technology uses small molecules composed of a targeted protein ligand, a hydrophobic tag, and a linker. Unlike PROTACs that often target the ubiquitin-proteasome system, HyT-based degraders work by increasing the hydrophobicity of the target protein, facilitating its degradation.880 MS1943, an EZH2 HyT degrader, illustrates this technology’s effectiveness.881 It has shown superior inhibitory effects on tumor cell lines and greater selectivity towards cancer cells over normal cells, demonstrating significant tumor suppression and good tolerance in mouse models.881 Other HyT-based degraders are also developed, including those targeting HDAC and YEATS domain readers, providing therapeutic strategies for diseases caused by mutations or dysfunction in specific proteins.882,883,884 Further development of HyT-based degraders is ongoing, with efforts to improve their bioavailability and therapeutic effects in vivo by exploring new hydrophobic labels.885 MG differs fundamentally from PROTACs and other degraders by inducing degradation through promoting tight binding between the target protein and proteasome components, leading to the protein’s subsequent degradation.886,887 A notable example is the MG-based degrader DD-1-073, targeting HDAC1/3, derived from SAHA (a known HDAC inhibitor). This was among the first applications of MG in HDAC degrader development.888 Similarly, another MG-based agent targeting BRD4, termed JP-2-197, is further established as an optimal derivative of JQ-1.888 Due to their low molecular weights, DD-1-073 and JP-2-197 have favorable pharmacokinetic properties, enhancing cell permeability and drug-ability.888 Despite their potential, the development of MG-based agents faces challenges due to the lack of systematic strategies for their design and identification, making large-scale screening and optimization difficult.
Developing epigenetic-targeting degraders, especially those targeting HDACs and epigenetic readers, has made significant progress, opening new avenues for clinical practice. These novel small molecules offer promising therapeutic alternatives, but several challenges and limitations must be addressed to enhance their clinical applicability and effectiveness.
Combining epigenetics-targeted drugs and sequencing technology
Owing to the substantial relationship among epigenetic signatures, lifestyle choices, and environmental influences, drugs that target epigenetic mechanisms are highly promising for advancing personalized medicine.889 However, leveraging these drugs in this field is challenging. As disease research enters a new phase owing to advancements in sequencing technologies, the potential for epigenetic therapies to enhance personalized healthcare is being progressively realized.890
The advent of these cutting-edge technologies, ranging from whole-genome sequencing to single-cell analysis, has facilitated the detection of gene mutations and expression alterations associated with epigenetic changes throughout disease progression.891 This advancement significantly enhances our understanding of the heterogeneity in epigenetic modifications across various cell types, thereby revealing new therapeutic targets for clinical application. In recent years, advancements in single-cell methodologies have allowed researchers to further explore the multiple dimensions of the epigenome, including chromatin accessibility, DNA methylation patterns, histone modification profiles, and chromatin interaction networks.892,893,894 Collectively known as “single-cell epigenomics”, this burgeoning field offers an enhanced comprehension of epigenomic regulation in physiological and pathological settings at the level of individual cells from a more intuitive perspective.895,896,897 These comprehensive “omic” profilings support the distinct biological identity of individuals, thus providing a solid theoretical foundation for refining therapeutic strategies to achieve individual targeting.898 Furthermore, by integrating CRISPR/cas9 gene-editing technology with various sequencing, researchers can conduct high-throughput functional genomic screens and identify pathological genes that are responsive to epigenetic therapies. This step not only aids in identifying novel targets for epigenetic agents but also promotes the assessment of therapeutic responses of target tissues or cells.899,900 For example, these methods may help detect the occurrence of drug resistance and optimize the efficacy of epigenetic interventions.901
The significant potential of epigenetics in customizing personalized medicine has generated high enthusiasm, and advancements in this field have been consistently focused in medical research. Notably, the introduction of sequencing technologies has enabled us to investigate the correlation between epigenetic markers and disease pathology more comprehensively, thus facilitating the development of targeted therapeutic strategies. Nonetheless, the current epigenetic technologies used in the laboratory present several technical challenges that require refinement, such as the necessity for high-demand algorithms,902 sufficient amounts of training data,903 limited genome coverage per cell,904,905 and uncertain reproducibility.906 These techniques require significant improvement before they can be effectively used in clinical practice.
Conclusions and perspectives
Since the term “epigenetics” was first introduced in 1942, there has been a significant focus on elucidating the mechanisms and pivotal roles of epigenetic modifications and their associated enzymes in human physiology and pathology. Drugs targeting epigenetic enzymes have shown promising potential for treating diseases, particularly cancers. This review comprehensively examines the major epigenetic mechanisms involved in the pathogenesis and progression of various diseases. It also highlights recent advances in epigenetics-targeted drugs, underscoring their potential in clinical settings. Additionally, we explore the integration of novel technologies in drug development and the synergistic value of these drugs in conjunction with other cancer therapies, pointing to the future direction of epigenetics-oriented therapeutic strategies. Despite these advancements, significant challenges remain. For instance, certain enzymes that regulate the epigenetic landscape still lack effective targeted drugs, and those identified through virtual screening require further in vivo and in vitro investigation to validate their efficacy and safety profiles. Addressing these gaps will be crucial for integrating epigenetics-targeted drugs into clinical practice.
Advances in in-depth understanding of epigenetics have largely enhanced possibilities of curing disease. To date, the application of epigenetics-modifying drugs in preclinical and clinical setting has provided promise for the beginning of the era of epigenetic-orientated therapeutic strategies. Given the future needs in this field, great attention should be focused on exploring the heterogeneity of epigenetic hallmarks in different diseases to design and develop epigenetics-targeted drugs with high selectivity and improved targeting efficiency, based on the elucidation for the biological and pathological roles of epigenetics. These results are imperative for designing and developing agonists and inhibitors of epigenetic enzymes with enhanced selectivity and bioactivity. Furthermore, apart from the knowledge based on experimental research and preclinical studies, assessing the therapeutic potential of epigenetic drugs for specific diseases, particularly in advanced clinical trials, is also vital for advancing this field. Concurrently, emphasis should be placed on the clinical potential of integrating innovative drug discovery technologies into developing epigenetic-based drugs. Moreover, while capitalizing on unique strengths of epigenetics, efforts should be made to combine these novel agents with traditional therapeutic modalities, with a view to achieving synergic effects in treating disease, especially in the case of tumors with genomic complexity. In conclusion, we believe that deepened research in this field will catalyze innovation in treatment approaches for diseases involving epigenetic mechanisms, offering new hope to patients.
References
Allis, C. D. & Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500 (2016).
Parry, A., Rulands, S. & Reik, W. Active turnover of DNA methylation during cell fate decisions. Nat. Rev. Genet. 22, 59–66 (2021).
Millán-Zambrano, G., Burton, A., Bannister, A. J. & Schneider, R. Histone post-translational modifications - cause and consequence of genome function. Nat. Rev. Genet. 23, 563–580 (2022).
Delaunay, S., Helm, M. & Frye, M. RNA modifications in physiology and disease: towards clinical applications. Nat. Rev. Genet. 25, 104–122 (2024).
Eustermann, S. et al. Energy-driven genome regulation by ATP-dependent chromatin remodellers. Nat. Rev. Mol. Cell. Biol. 25, 309–332 (2024).
Tian, Y. et al. Exploring non-coding RNA mechanisms in hepatocellular carcinoma: implications for therapy and prognosis. Front. Immunol. 15, 1400744 (2024).
Wang, Y. et al. Epigenetic modification of m(6)A regulator proteins in cancer. Mol. Cancer 22, 102 (2023).
Liu, R. et al. Methylation across the central dogma in health and diseases: new therapeutic strategies. Signal. Transduct. Target. Ther. 8, 310 (2023).
Gourisankar, S., Krokhotin, A., Wenderski, W. & Crabtree, G. R. Context-specific functions of chromatin remodellers in development and disease. Nat. Rev. Genet. 25, 340–361 (2024).
Zhou, S. et al. Targeting tumor endothelial cells with methyltransferase inhibitors: mechanisms of action and the potential of combination therapy. Pharmacol. Ther. 247, 108434 (2023).
Zhang, X., Zhang, Y., Wang, C. & Wang, X. TET (Ten-eleven translocation) family proteins: structure, biological functions and applications. Signal. Transduct. Target. Ther. 8, 297 (2023).
Shi, J. et al. The concurrence of DNA methylation and demethylation is associated with transcription regulation. Nat. Commun. 12, 5285 (2021).
Lin, T. C. et al. Light-activatable MBD-readers of 5-methylcytosine reveal domain-dependent chromatin association kinetics in vivo. Adv. Sci. 11, e2307930 (2024).
Zhang, P. et al. Methyl-CpG binding domain protein 1 regulates localization and activity of Tet1 in a CXXC3 domain-dependent manner. Nucl. Acids Res. 45, 7118–7136 (2017).
Martin, B. J. E. et al. Global identification of SWI/SNF targets reveals compensation by EP400. Cell 186, 5290–5307.e5226 (2023).
Nemeth, K., Bayraktar, R., Ferracin, M. & Calin, G. A. Non-coding RNAs in disease: from mechanisms to therapeutics. Nat. Rev. Genet. 25, 211–232 (2024).
Fabbri, M., Girnita, L., Varani, G. & Calin, G. A. Decrypting noncoding RNA interactions, structures, and functional networks. Genom. Res. 29, 1377–1388 (2019).
Wilkinson, A. L., Zorzan, I. & Rugg-Gunn, P. J. Epigenetic regulation of early human embryo development. Cell. Stem Cell. 30, 1569–1584 (2023).
van Voorden, A. J. et al. EP300 facilitates human trophoblast stem cell differentiation. Proc. Natl Acad. Sci. USA 120, e2217405120 (2023).
Gao, L. et al. Structure of DNMT3B homo-oligomer reveals vulnerability to impairment by ICF mutations. Nat. Commun. 13, 4249 (2022).
Tsang, E. et al. Ketogenic diet modifies ribosomal protein dysregulation in KMT2D Kabuki syndrome. EBioMedicine 104, 105156 (2024).
Duan, J. E. et al. Methylome dynamics of bovine gametes and in vivo early embryos. Front. Genet. 10, 512 (2019).
Miura, F., Enomoto, Y., Dairiki, R. & Ito, T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 40, e136 (2012).
Greenberg, M. V. C. & Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell. Biol. 20, 590–607 (2019).
Lee, H. J., Hore, T. A. & Reik, W. Reprogramming the methylome: erasing memory and creating diversity. Cell. Stem Cell. 14, 710–719, (2014).
Wu, K. et al. Dynamics of histone acetylation during human early embryogenesis. Cell. Discov. 9, 29 (2023).
Regadas, I. et al. A unique histone 3 lysine 14 chromatin signature underlies tissue-specific gene regulation. Mol. Cell. 81, 1766–1780.e1710 (2021).
Yu, H. et al. Dynamic reprogramming of H3K9me3 at hominoid-specific retrotransposons during human preimplantation development. Cell. Stem Cell. 29, 1031–1050.e1012 (2022).
Ovadia, S. et al. SWI/SNF complexes are required for retinal pigmented epithelium differentiation and for the inhibition of cell proliferation and neural differentiation programs. Development 150, dev201488 (2023).
Saha, D., Animireddy, S. & Bartholomew, B. The SWI/SNF ATP-dependent chromatin remodeling complex in cell lineage priming and early development. Biochem. Soc. Trans. 52, 603–616 (2024).
Yao, Y. et al. Regulatory role of m(6)A epitranscriptomic modifications in normal development and congenital malformations during embryogenesis. Biomed. Pharmacother. 173, 116171 (2024).
Wang, M. K., Gao, C. C. & Yang, Y. G. Emerging roles of RNA methylation in development. Acc. Chem. Res. 56, 3417–3427 (2023).
Han, Y. et al. A Mettl16/m(6)A/mybl2b/Igf2bp1 axis ensures cell cycle progression of embryonic hematopoietic stem and progenitor cells. EMBO J. 43, 1990–2014 (2024).
Guo, J. et al. Zebrafish Mbd5 binds to RNA m5C and regulates histone deubiquitylation and gene expression in development metabolism and behavior. Nucleic Acids Res. 52, 4257–4275 (2024).
Zhang, M. et al. DNA methylation regulates RNA m(6)A modification through transcription factor SP1 during the development of porcine somatic cell nuclear transfer embryos. Cell. Prolif. 57, e13581 (2024).
Hupalowska, A. et al. CARM1 and paraspeckles regulate pre-implantation mouse embryo development. Cell 175, 1902–1916.e1913 (2018).
Wang, J. et al. Asymmetric expression of lincGET biases cell fate in two-cell mouse embryos. Cell 175, 1887–1901.e1818 (2018).
Wang, J. et al. Alternative splicing of CARM1 regulated by LincGET-guided paraspeckles biases the first cell fate in mammalian early embryos. Nat. Struct. Mol. Biol. 31, 1341–1354 (2024).
Reza, A. et al. Roles of microRNAs in mammalian reproduction: from the commitment of germ cells to peri-implantation embryos. Biol. Rev. Camb. Philos. Soc. 94, 415–438 (2019).
Kong, X. et al. LncRNA-Smad7 mediates cross-talk between Nodal/TGF-β and BMP signaling to regulate cell fate determination of pluripotent and multipotent cells. Nucleic Acids Res. 50, 10526–10543 (2022).
Hazra, R. et al. Platr4 is an early embryonic lncRNA that exerts its function downstream on cardiogenic mesodermal lineage commitment. Dev. Cell. 57, 2450–2468.e2457 (2022).
Movahed, E. et al. Aberrant expression of miR-29a/29b and methylation level of mouse embryos after in vitro fertilization and vitrification at two-cell stage. J. Cell. Physiol. 234, 18942–18950 (2019).
Wilson, V. L. & Jones, P. A. DNA methylation decreases in aging but not in immortal cells. Science 220, 1055–1057 (1983).
Yang, J. H. et al. Loss of epigenetic information as a cause of mammalian aging. Cell 186, 305–326.e327 (2023).
Johnstone, S. E., Gladyshev, V. N., Aryee, M. J. & Bernstein, B. E. Epigenetic clocks, aging, and cancer. Science 378, 1276–1277 (2022).
Tao, Y. et al. Aging-like spontaneous epigenetic silencing facilitates wnt activation, stemness, and braf(V600E)-induced tumorigenesis. Cancer Cell. 35, 315–328.e316 (2019).
Amatori, S., Bagaloni, I., Viti, D. & Fanelli, M. Premature senescence induced by DNA demethylating agent (Decitabine) as therapeutic option for malignant pleural mesothelioma. Lung Cancer 71, 113–115 (2011).
Bi, S. et al. SIRT7 antagonizes human stem cell aging as a heterochromatin stabilizer. Protein Cell. 11, 483–504 (2020).
Zhang, N. et al. Dual Inhibition of H3K9me2 and H3K27me3 Promotes Tumor Cell Senescence without Triggering the Secretion of SASP. Int. J. Mol. Sci. 23, 3911 (2022).
Rossi, M. et al. Increased PTCHD4 expression via m6A modification of PTCHD4 mRNA promotes senescent cell survival. Nucleic Acids Res. 52, 7261–7278 (2024).
Heissenberger, C. et al. The ribosomal RNA m(5)C methyltransferase NSUN-1 modulates healthspan and oogenesis in Caenorhabditis elegans. Elife. 9, (2020).
Krarup, J. et al. A brain anti-senescence transcriptional program triggered by hypothalamic-derived exosomal microRNAs. Int J Mol Sci. 25, e56205 (2024).
Lee, A. V., Nestler, K. A. & Chiappinelli, K. B. Therapeutic targeting of DNA methylation alterations in cancer. Pharmacol. Ther. 258, 108640 (2024).
Yamaguchi, K. et al. Non-canonical functions of UHRF1 maintain DNA methylation homeostasis in cancer cells. Nat. Commun. 15, 2960 (2024).
Janic, A., Abad, E. & Amelio, I. Decoding p53 tumor suppression: a crosstalk between genomic stability and epigenetic control? Cell Death Differ. 2024, 1–8 (2024).
Xue, F., Liu, L., Tao, X. & Zhu, W. TET3-mediated DNA demethylation modification activates SHP2 expression to promote endometrial cancer progression through the EGFR/ERK pathway. J. Gynecol. Oncol. 35, e64 (2024).
Brückmann, N. H., Pedersen, C. B., Ditzel, H. J. & Gjerstorff, M. F. Epigenetic reprogramming of pericentromeric satellite DNA in premalignant and malignant lesions. Mol. Cancer Res. 16, 417–427 (2018).
Ma, S. et al. Recent advances in targeting histone H3 lysine 36 methyltransferases for cancer therapy. Eur. J. Med. Chem. 274, 116532 (2024).
Yamagishi, M. et al. Mechanisms of action and resistance in histone methylation-targeted therapy. Nature 627, 221–228 (2024).
Wang, Z. Q. et al. Bromodomain and extraterminal (BET) proteins: biological functions, diseases, and targeted therapy. Signal. Transduct. Target. Ther. 8, 420 (2023).
Guo, J., Zheng, Q. & Peng, Y. BET proteins: biological functions and therapeutic interventions. Pharmacol. Ther. 243, 108354 (2023).
Wang, X. et al. Bromodomain protein BRDT directs ΔNp63 function and super-enhancer activity in a subset of esophageal squamous cell carcinomas. Cell. Death Differ. 28, 2207–2220 (2021).
Lee, J. K. et al. Complex chromosomal rearrangements by single catastrophic pathogenesis in NUT midline carcinoma. Ann. Oncol. 28, 890–897 (2017).
Hu, Y. et al. Lactylation: the novel histone modification influence on gene expression, protein function, and disease. Clin. Epigenetics 16, 72 (2024).
Zhang, Y., Song, H., Li, M. & Lu, P. Histone lactylation bridges metabolic reprogramming and epigenetic rewiring in driving carcinogenesis: oncometabolite fuels oncogenic transcription. Clin. Transl. Med. 14, e1614 (2024).
Wei, S. et al. Histone lactylation promotes malignant progression by facilitating USP39 expression to target PI3K/AKT/HIF-1α signal pathway in endometrial carcinoma. Cell. Death Discov. 10, 121 (2024).
Li, F. et al. Positive feedback regulation between glycolysis and histone lactylation drives oncogenesis in pancreatic ductal adenocarcinoma. Mol. Cancer 23, 90 (2024).
De Leo, A. et al. Glucose-driven histone lactylation promotes the immunosuppressive activity of monocyte-derived macrophages in glioblastoma. Immunity 57, 1105–1123.e1108 (2024).
Chen, X. Y. et al. CircUGGT2 downregulation by METTL14-dependent m(6)A modification suppresses gastric cancer progression and cisplatin resistance through interaction with miR-186-3p/MAP3K9 axis. Pharmacol. Res. 204, 107206 (2024).
Jin, H. et al. YTHDF2 favors protumoral macrophage polarization and implies poor survival outcomes in triple negative breast cancer. iScience 27, 109902 (2024).
Wang, S. et al. ALKBH5-mediated m6A modification of circFOXP1 promotes gastric cancer progression by regulating SOX4 expression and sponging miR-338-3p. Commun. Biol. 7, 565 (2024).
Wang, Q. et al. The demethylase ALKBH5 mediates ZKSCAN3 expression through the m(6)A modification to activate VEGFA transcription and thus participates in MNNG-induced gastric cancer progression. J. Hazard. Mater. 473, 134690 (2024).
Zhao, L. et al. Abnormal changes in metabolites caused by m(6)A methylation modification: the leading factors that induce the formation of immunosuppressive tumor microenvironment and their promising potential for clinical application. J. Adv. Res. S2090-1232 (2024).
Feng, G. et al. Small molecule inhibitors targeting m(6)A regulators. J. Hematol. Oncol. 17, 30 (2024).
Calin, G. A. et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl Acad. Sci. USA 99, 15524–15529 (2002).
Wells, A. C. et al. Let-7 enhances murine anti-tumor CD8 T cell responses by promoting memory and antagonizing terminal differentiation. Nat. Commun. 14, 5585 (2023).
Chen, H. et al. LINC00355 promotes gastric carcinogenesis by scaffolding p300 to activate CDC42 transcription and enhancing HNRNPA2B1 to stabilize CDC42 mRNA dependent on m6A. Mol. Carcinog. 63, 430–447 (2024).
Luongo, M. et al. The molecular conversations of sarcomas: exosomal non-coding RNAs in tumor’s biology and their translational prospects. Mol. Cancer 23, 172 (2024).
Li, Y., Sui, S. & Goel, A. Extracellular vesicles associated microRNAs: their biology and clinical significance as biomarkers in gastrointestinal cancers. Semin. Cancer Biol. 99, 5–23 (2024).
Kumar, M. A. et al. Extracellular vesicles as tools and targets in therapy for diseases. Signal. Transduct. Target. Ther. 9, 27 (2024).
Peng, X. X. et al. Correlation of plasma exosomal microRNAs with the efficacy of immunotherapy in EGFR/ALK wild-type advanced non-small cell lung cancer. J Immunother Cancer. 8, e000376 (2020).
Mjelle, R. et al. Serum small RNAs in metastatic colorectal cancer predict response to chemotherapy and characterize high-risk patients. Mol. Cancer 23, 133 (2024).
Yang, C. K. et al. EV-miRome-wide profiling uncovers miR-320c for detecting metastatic colorectal cancer and monitoring the therapeutic response. Cell. Oncol. 45, 621–638 (2022).
Su, Y. et al. Plasma extracellular vesicle long RNA profiles in the diagnosis and prediction of treatment response for breast cancer. NPJ Breast Cancer 7, 154 (2021).
Alberti, K. G., Zimmet, P. & Shaw, J. The metabolic syndrome–a new worldwide definition. Lancet 366, 1059–1062, (2005).
Han, Y. et al. New advances of adiponectin in regulating obesity and related metabolic syndromes. J. Pharmacol. Anal. 14, 100913 (2024).
Wu, Y. L. et al. Epigenetic regulation in metabolic diseases: mechanisms and advances in clinical study. Signal. Transduct. Target. Ther. 8, 98 (2023).
Alka, K., Mohammad, G. & Kowluru, R. A. Regulation of serine palmitoyl-transferase and Rac1-Nox2 signaling in diabetic retinopathy. Sci. Rep. 12, 16740 (2022).
Zhang, Q. et al. A maternal high-fat diet induces dna methylation changes that contribute to glucose intolerance in offspring. Front. Endocrinol. 10, 871 (2019).
Georgel, P. T. & Georgel, P. Where epigenetics meets food intake: their interaction in the development/severity of gout and therapeutic perspectives. Front. Immunol. 12, 752359 (2021).
Chen, H. C., Chen, Y. Z., Wang, C. H. & Lin, F. J. The nonalcoholic fatty liver disease-like phenotype and lowered serum VLDL are associated with decreased expression and DNA hypermethylation of hepatic ApoB in male offspring of ApoE deficient mothers fed a with Western diet. J. Nutr. Biochem. 77, 108319 (2020).
Davison, G. W., Irwin, R. E. & Walsh, C. P. The metabolic-epigenetic nexus in type 2 diabetes mellitus. Free Radic. Biol. Med. 170, 194–206 (2021).
Domínguez-Barragán, J. et al. Blood DNA methylation signature of diet quality and association with cardiometabolic traits. Eur. J. Prev. Cardiol. 31, 191–202 (2024).
Nadiger, N., Veed, J. K., Chinya Nataraj, P. & Mukhopadhyay, A. DNA methylation and type 2 diabetes: a systematic review. Clin. Epigenetics 16, 67 (2024).
Xiao, F. et al. Epigenetic regulation of B cells and its role in autoimmune pathogenesis. Cell. Mol. Immunol. 19, 1215–1234 (2022).
Liu, H. et al. Regulation of T cell differentiation and function by epigenetic modification enzymes. Semin. Immunopathol. 41, 315–326 (2019).
Han, X. et al. RNA m(6)A methylation modulates airway inflammation in allergic asthma via PTX3-dependent macrophage homeostasis. Nat. Commun. 14, 7328 (2023).
Ahmadi, M. et al. Epigenetic modifications and epigenetic based medication implementations of autoimmune diseases. Biomed. Pharmacother. 87, 596–608 (2017).
Cardenas, A., Fadadu, R. P. & Koppelman, G. H. Epigenome-wide association studies of allergic disease and the environment. J. Allergy Clin. Immunol. 152, 582–590 (2023).
Yue, X. et al. Control of Foxp3 stability through modulation of TET activity. J. Exp. Med. 213, 377–397 (2016).
Souza, N. H. et al. Low-level laser therapy suppresses the oxidative stress-induced glucocorticoids resistance in U937 cells: relevance to cytokine secretion and histone deacetylase in alveolar macrophages. J. Photochem. Photobiol. B 130, 327–336 (2014).
Kim, R. Y. et al. MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase-mediated suppression of histone deacetylase 2. J. Allergy Clin. Immunol. 139, 519–532 (2017).
Wu, H. et al. The IL-21-TET2-AIM2-c-MAF pathway drives the T follicular helper cell response in lupus-like disease. Clin. Transl. Med. 12, e781 (2022).
Chen, F. et al. Circulating exosomal microRNAs as biomarkers of lupus nephritis. Front. Immunol. 14, 1326836 (2023).
Jog, N. R. et al. Neutrophils isolated from systemic lupus erythematosus patients exhibit a distinct functional phenotype. Front. Immunol. 15, 1339250 (2024).
Regna, N. L. et al. Class I and II histone deacetylase inhibition by ITF2357 reduces SLE pathogenesis in vivo. Clin. Immunol. 151, 29–42 (2014).
Banham, G. D. et al. Bromodomain inhibitors modulate FcγR-mediated mononuclear phagocyte activation and chemotaxis. Front. Immunol. 13, 885101 (2022).
Fang, Y. et al. Epigenetic regulatory axis MIR22-TET3-MTRNR2L2 represses fibroblast-like synoviocyte-mediated inflammation in rheumatoid arthritis. Arthritis Rheumatol. 76, 845–856 (2024).
Lee, H. J., Stefan-Lifshitz, M., Li, C. W. & Tomer, Y. Genetics and epigenetics of autoimmune thyroid diseases: translational implications. Best. Pr. Res. Clin. Endocrinol. Metab. 37, 101661 (2023).
Lee, H. G. et al. Disease-associated astrocyte epigenetic memory promotes CNS pathology. Nature 627, 865–872 (2024).
Al-Hasani, K. et al. EZH2 inhibitors promote β-like cell regeneration in young and adult type 1 diabetes donors. Signal. Transduct. Target. Ther. 9, 2 (2024).
Qi, W. et al. Abnormal histone acetylation of CD8(+) T cells in patients with severe aplastic anemia. Int. J. Hematol. 104, 540–547 (2016).
Lavertu-Jolin, M. et al. Acan downregulation in parvalbumin GABAergic cells reduces spontaneous recovery of fear memories. Mol. Psychiatry 28, 2946–2963 (2023).
Gomez-Pinilla, F. & Thapak, P. Exercise epigenetics is fueled by cell bioenergetics: supporting role on brain plasticity and cognition. Free Radic. Biol. Med. 220, 43–55 (2024).
Prasanth, M. I. et al. Role of epigenetic modulation in neurodegenerative diseases: implications of phytochemical interventions. Antioxidants 13, 606.(2024).
Xu, D. C. et al. Histone acetylation in an Alzheimer’s disease cell model promotes homeostatic amyloid-reducing pathways. Acta Neuropathol. Commun. 12, 3 (2024).
Pan, R. Y. et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell. Metab. 34, 634–648.e636 (2022).
Wei, L. et al. H3K18 lactylation of senescent microglia potentiates brain aging and Alzheimer’s disease through the NFκB signaling pathway. J. Neuroinflammation 20, 208 (2023).
Park, H. et al. CRISPR/dCas9-Dnmt3a-mediated targeted DNA methylation of APP rescues brain pathology in a mouse model of Alzheimer’s disease. Transl. Neurodegener. 11, 41 (2022).
Bie, B. et al. Epigenetic suppression of neuroligin 1 underlies amyloid-induced memory deficiency. Nat. Neurosci. 17, 223–231 (2014).
Jörg, M. et al. N1-methylation of adenosine (m(1)A) in ND5 mRNA leads to complex I dysfunction in Alzheimer’s disease. Mol. Psychiatry 29, 1427–1439 (2024).
Lahiri, D. K. et al. The seeds of its regulation: natural antisense transcripts as single-gene control switches in neurodegenerative disorders. Ageing Res. Rev. 99, 102336 (2024).
Gonzalez-Latapi, P. et al. Alterations in blood methylome as potential epigenetic biomarker in sporadic Parkinson’s disease. Ann. Neurol. 95, 1162–1172 (2024).
Navarro-Sánchez, L., Águeda-Gómez, B., Aparicio, S. & Pérez-Tur, J. Epigenetic study in Parkinson’s disease: a pilot analysis of DNA methylation in candidate genes in brain. Cells. 7, 150 (2018).
Marshall, L. L. et al. Epigenomic analysis of Parkinson’s disease neurons identifies Tet2 loss as neuroprotective. Nat. Neurosci. 23, 1203–1214 (2020).
Guhathakurta, S. et al. Targeted attenuation of elevated histone marks at SNCA alleviates α-synuclein in Parkinson’s disease. EMBO Mol. Med. 13, e12188 (2021).
Huang, M. et al. Mitochondrial dysfunction-induced H3K27 hyperacetylation perturbs enhancers in Parkinson’s disease. JCI Insight. 6, e138088 (2021).
Thapa, R. et al. ncRNAs and their impact on dopaminergic neurons: autophagy pathways in Parkinson’s disease. Ageing Res. Rev. 98, 102327 (2024).
Zsindely, N., Siági, F. & Bodai, L. DNA methylation in Huntington’s disease. Int. J. Mol. Sci. 22, 4529 (2021).
Hecklau, K. et al. The effects of selective inhibition of histone deacetylase 1 and 3 in Huntington’s disease mice. Front. Mol. Neurosci. 14, 616886 (2021).
Beatriz, M. et al. Extracellular vesicles improve GABAergic transmission in Huntington’s disease iPSC-derived neurons. Theranostics 13, 3707–3724 (2023).
Pupak, A. et al. Altered m6A RNA methylation contributes to hippocampal memory deficits in Huntington’s disease mice. Cell. Mol. Life Sci. 79, 416 (2022).
Pang, L. et al. Epigenetic regulation of tumor immunity. J. Clin. Investig. 134, e178540 (2024).
Mabe, N. W., Perry, J. A., Malone, C. F. & Stegmaier, K. Pharmacological targeting of the cancer epigenome. Nat. Cancer 5, 844–865 (2024).
Watanabe, J. et al. BET bromodomain inhibition potentiates radiosensitivity in models of H3K27-altered diffuse midline glioma. J. Clin. Investig. 134, e174794 (2024).
Qin, S. et al. New insights into immune cells in cancer immunotherapy: from epigenetic modification, metabolic modulation to cell communication. MedComm 5, e551 (2024).
Tang, F. et al. Therapeutic applications of histone deacetylase inhibitors in sarcoma. Cancer Treat. Rev. 59, 33–45 (2017).
Zhuang, H. et al. The role of m6A methylation in therapy resistance in cancer. Mol. Cancer 22, 91 (2023).
Bharti, R. et al. Cell surface CD55 traffics to the nucleus leading to cisplatin resistance and stemness by inducing PRC2 and H3K27 trimethylation on chromatin in ovarian cancer. Mol. Cancer 23, 121 (2024).
Xu, Y. et al. ZNF397 deficiency triggers TET2-driven lineage plasticity and AR-targeted therapy resistance in prostate cancer. Cancer Discov. 14, 1496–1521 (2024).
Wang, F. et al. Combined anti-PD-1, HDAC inhibitor and anti-VEGF for MSS/pMMR colorectal cancer: a randomized phase 2 trial. Nat. Med. 30, 1035–1043 (2024).
Issa, J. P. & Kantarjian, H. Azacitidine. Nat Rev Drug Discov. Suppl, S6-S7, (2005).
Gore, S. D., Jones, C. & Kirkpatrick, P. Decitabine. Nat. Rev. Drug. Discov. 5, 891–892 (2006).
Silverman, L. R. et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J. Clin. Oncol. 20, 2429–2440 (2002).
Fenaux, P. et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 10, 223–232 (2009).
Kantarjian, H. et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 106, 1794–1803 (2006).
Jabbour, E. et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood 130, 1514–1522 (2017).
Kantarjian, H. et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood 109, 52–57 (2007).
Wei, A. H. et al. Oral azacitidine maintenance therapy for acute myeloid leukemia in first remission. N. Engl. J. Med. 383, 2526–2537 (2020).
Savona, M. R. et al. Extended dosing with CC-486 (oral azacitidine) in patients with myeloid malignancies. Am. J. Hematol. 93, 1199–1206 (2018).
Dhillon, S. Decitabine/cedazuridine: first approval. Drugs 80, 1373–1378 (2020).
Candoni, A. Fully oral regimen with decitabine and cedazuridine plus venetoclax: a new step forward for older or unfit patients with acute myeloid leukaemia. Lancet Haematol. 11, e245–e246 (2024).
Linnekamp, J. F. et al. Clinical and biological effects of demethylating agents on solid tumours—a systematic review. Cancer Treat. Rev. 54, 10–23 (2017).
Von Hoff, D. D. et al. Phase I study of CC-486 alone and in combination with carboplatin or nab-paclitaxel in patients with relapsed or refractory solid tumors. Clin. Cancer Res. 24, 4072–4080 (2018).
Luke, J. J. et al. Phase I/II sequencing study of azacitidine, epacadostat, and pembrolizumab in advanced solid tumors. Br. J. Cancer 128, 2227–2235 (2023).
Ye, C. et al. Epigenetic therapy: research progress of decitabine in the treatment of solid tumors. Biochim. Biophys. Acta Rev. Cancer 1879, 189066 (2024).
Pu, J. et al. Exploring the role of histone deacetylase and histone deacetylase inhibitors in the context of multiple myeloma: mechanisms, therapeutic implications, and future perspectives. Exp. Hematol. Oncol. 13, 45 (2024).
West, A. C. & Johnstone, R. W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 124, 30–39 (2014).
Shi, Y. et al. Chidamide in relapsed or refractory peripheral T cell lymphoma: a multicenter real-world study in China. J. Hematol. Oncol. 10, 69 (2017).
Sun, Y. et al. Therapeutic potential of tucidinostat, a subtype-selective HDAC inhibitor, in cancer treatment. Front. Pharmacol. 13, 932914 (2022).
Mozzetta, C., Sartorelli, V., Steinkuhler, C. & Puri, P. L. HDAC inhibitors as pharmacological treatment for Duchenne muscular dystrophy: a discovery journey from bench to patients. Trends Mol. Med. 30, 278–294 (2024).
Grant, S., Easley, C. & Kirkpatrick, P. Vorinostat. Nat. Rev. Drug. Discov. 6, 21–22 (2007).
Garcia-Manero, G. et al. A randomized phase III study of standard versus high-dose cytarabine with or without vorinostat for AML. Leukemia 38, 58–66 (2024).
Dimopoulos, M. et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol. 14, 1129–1140 (2013).
Jenner, M. W. et al. The addition of vorinostat to lenalidomide maintenance for patients with newly diagnosed multiple myeloma of all ages: results from ‘Myeloma XI’, a multicentre, open-label, randomised, phase III trial. Br. J. Haematol. 201, 267–279 (2023).
Krug, L. M. et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol. 16, 447–456 (2015).
Duvic, M. & Vu, J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin. Investig. Drugs 16, 1111–1120 (2007).
Mann, B. S. et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin. Cancer Res. 13, 2318–2322 (2007).
Lynch, D. R. Jr., Washam, J. B. & Newby, L. K. QT interval prolongation and torsades de pointes in a patient undergoing treatment with vorinostat: a case report and review of the literature. Cardiol. J. 19, 434–438 (2012).
Bertino, E. M. & Otterson, G. A. Romidepsin: a novel histone deacetylase inhibitor for cancer. Expert Opin. Investig. Drugs 20, 1151–1158 (2011).
Camus, V. et al. Romidepsin plus cyclophosphamide, doxorubicin, vincristine, and prednisone versus cyclophosphamide, doxorubicin, vincristine, and prednisone in patients with previously untreated peripheral T-cell lymphoma: final analysis of the Ro-CHOP trial. J. Clin. Oncol. 42, 1612–1618 (2024).
Bachy, E. et al. Romidepsin plus CHOP versus CHOP in patients with previously untreated peripheral T-cell lymphoma: results of the Ro-CHOP phase III study (Conducted by LYSA). J. Clin. Oncol. 40, 242–251 (2022).
Falchi, L. et al. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: a multicenter phase 2 study. Blood 137, 2161–2170 (2021).
Iyer, S. P. et al. Safety and efficacy of tenalisib in combination with romidepsin in patients with relapsed/refractory T-cell lymphoma: results from a phase I/II open-label multicenter study. Haematologica 109, 209–219 (2024).
Mehta-Shah, N. et al. Romidepsin and lenalidomide-based regimens have efficacy in relapsed/refractory lymphoma: combined analysis of two phase I studies with expansion cohorts. Am. J. Hematol. 96, 1211–1222 (2021).
Ruan, J. et al. Multicenter phase 2 study of romidepsin plus lenalidomide for previously untreated peripheral T-cell lymphoma. Blood Adv. 7, 5771–5779 (2023).
Gruell, H. et al. Effect of 3BNC117 and romidepsin on the HIV-1 reservoir in people taking suppressive antiretroviral therapy (ROADMAP): a randomised, open-label, phase 2A trial. Lancet Microbe 3, e203–e214 (2022).
Gunst, J. D. et al. Early intervention with 3BNC117 and romidepsin at antiretroviral treatment initiation in people with HIV-1: a phase 1b/2a, randomized trial. Nat. Med. 28, 2424–2435 (2022).
Poole, R. M. Belinostat: first global approval. Drugs 74, 1543–1554 (2014).
Shafer, D. et al. Phase 1 study of belinostat and adavosertib in patients with relapsed or refractory myeloid malignancies. Cancer Chemother. Pharmacol. 91, 281–290 (2023).
O’Connor, O. A. et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J. Clin. Oncol. 33, 2492–2499 (2015).
Takebe, N. et al. A phase I pharmacokinetic study of belinostat in patients with advanced cancers and varying degrees of liver dysfunction. Br. J. Clin. Pharmacol. 85, 2499–2511 (2019).
Holkova, B. et al. Phase 1 study of belinostat (PXD-101) and bortezomib (Velcade, PS-341) in patients with relapsed or refractory acute leukemia and myelodysplastic syndrome. Leuk. Lymphoma 62, 1187–1194 (2021).
Xu, K. et al. Final report on clinical outcomes and tumor recurrence patterns of a pilot study assessing efficacy of belinostat (PXD-101) with chemoradiation for newly diagnosed glioblastoma. Tomography 8, 688–700 (2022).
Balasubramaniam, S. et al. Phase I trial of belinostat with cisplatin and etoposide in advanced solid tumors, with a focus on neuroendocrine and small cell cancers of the lung. Anticancer Drugs 29, 457–465 (2018).
Thomas, A. et al. A phase I/II trial of belinostat in combination with cisplatin, doxorubicin, and cyclophosphamide in thymic epithelial tumors: a clinical and translational study. Clin. Cancer Res. 20, 5392–5402 (2014).
San-Miguel, J. F. et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 15, 1195–1206 (2014).
Rathkopf, D. E. et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 72, 537–544 (2013).
San-Miguel, J. F. et al. Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): a randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 3, e506–e515 (2016).
Laubach, J. P. et al. Efficacy and safety of oral panobinostat plus subcutaneous bortezomib and oral dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma (PANORAMA 3): an open-label, randomised, phase 2 study. Lancet Oncol. 22, 142–154 (2021).
Wieduwilt, M. J. et al. Histone deacetylase inhibition with panobinostat combined with intensive induction chemotherapy in older patients with acute myeloid leukemia: phase I study results. Clin. Cancer Res. 25, 4917–4923 (2019).
Mascarenhas, J. et al. A phase I study of panobinostat and ruxolitinib in patients with primary myelofibrosis (PMF) and post–polycythemia vera/essential thrombocythemia myelofibrosis (post–PV/ET MF). Leuk. Res. 88, 106272 (2020).
Monje, M. et al. Phase I trial of panobinostat in children with diffuse intrinsic pontine glioma: a report from the pediatric brain tumor consortium (PBTC-047). Neuro Oncol. 25, 2262–2272 (2023).
Wood, A. et al. Phase I study of the mTOR inhibitor everolimus in combination with the histone deacetylase inhibitor panobinostat in patients with advanced clear cell renal cell carcinoma. Investig. New. Drugs 38, 1108–1116 (2020).
Ferrari, A. C. et al. Epigenetic therapy with panobinostat combined with bicalutamide rechallenge in castration-resistant prostate cancer. Clin. Cancer Res. 25, 52–63 (2019).
Wang, T. et al. Second-line endocrine therapy of hormone receptor-positive/her2- negative advanced breast cancer: a systematic review and network meta-analysis. Curr. Cancer Drug. Targets 23, 718–730 (2023).
Rai, S. et al. Oral HDAC inhibitor tucidinostat in patients with relapsed or refractory peripheral T-cell lymphoma: phase IIb results. Haematologica 108, 811–821 (2023).
Li, L. et al. Chidamide enhances T-cell-mediated anti-tumor immune function by inhibiting NOTCH1/NFATC1 signaling pathway in ABC-type diffuse large B-cell lymphoma. Leuk. Lymphoma 65, 895–910 (2024).
Zhao, H. et al. Tucidinostat plus exemestane as a neoadjuvant in early-stage, hormone receptor-positive, human epidermal growth factor receptor 2-negative breast cancer. Oncologist 29, e763–e770 (2024).
Zhao, H. Y. et al. Low-dose chidamide restores immune tolerance in ITP in mice and humans. Blood 133, 730–742 (2019).
Dennison, J. et al. Low-dose chidamide treatment displays sex-specific differences in the 3xTg-AD mouse. Biomolecules. 13, 1324 (2023).
Jiang, Z. et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 20, 806–815 (2019).
Tonozuka, Y. et al. The combination of brentuximab vedotin and chidamide synergistically suppresses the proliferation of T-cell lymphoma cells through the enhancement of apoptosis. Cancer Chemother. Pharmacol. 93, 137–149 (2024).
Zhang, P. et al. Optimized dose selective HDAC inhibitor tucidinostat overcomes anti-PD-L1 antibody resistance in experimental solid tumors. BMC Med. 20, 435 (2022).
Zhou, J. et al. Clinical outcomes of tucidinostat-based therapy after prior CDK4/6 inhibitor progression in hormone receptor-positive heavily pretreated metastatic breast cancer. Breast 66, 255–261 (2022).
Chai, Y. et al. First-line chemoradiation with or without chidamide (tucidinostat) in patients with intermediate- and high-risk early-stage extranodal nasal-type natural killer/T-cell lymphoma: a randomized phase 2 study in China. Int. J. Radiat. Oncol. Biol. Phys. 113, 833–844 (2022).
Mercuri, E. et al. Safety and efficacy of givinostat in boys with Duchenne muscular dystrophy (EPIDYS): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 23, 393–403 (2024).
Chifotides, H. T., Bose, P. & Verstovsek, S. Givinostat: an emerging treatment for polycythemia vera. Expert Opin. Investig. Drugs 29, 525–536 (2020).
Rambaldi, A. et al. Safety and efficacy of the maximum tolerated dose of givinostat in polycythemia vera: a two-part Phase Ib/II study. Leukemia 34, 2234–2237 (2020).
Finazzi, G. et al. A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br. J. Haematol. 161, 688–694 (2013).
Rambaldi, A. et al. Long-term safety and efficacy of givinostat in polycythemia vera: 4-year mean follow up of three phase 1/2 studies and a compassionate use program. Blood Cancer J. 11, 53 (2021).
Xu, W. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 19, 17–30 (2011).
Tommasini-Ghelfi, S. et al. Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease. Sci. Adv. 5, eaaw4543 (2019).
Enasidenib Approved for AML, but Best Uses Unclear. Cancer Discov. 7, Of4, (2017).
Ivosidenib Gets Go-Ahead for AML. Cancer Discov. 8, Of3, (2018).
Lavacchi, D. et al. Ivosidenib in IDH1-mutated cholangiocarcinoma: clinical evaluation and future directions. Pharmacol. Ther. 237, 108170 (2022).
Kang, C. Olutasidenib: first approval. Drugs 83, 341–346 (2023).
Mellinghoff, I. K. et al. Vorasidenib, a dual inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first-in-human phase I trial. Clin. Cancer Res. 27, 4491–4499 (2021).
Wick, A. et al. Phase I assessment of safety and therapeutic activity of BAY1436032 in patients with IDH1-mutant solid tumors. Clin. Cancer Res. 27, 2723–2733 (2021).
Heuser, M. et al. Safety and efficacy of BAY1436032 in IDH1-mutant AML: phase I study results. Leukemia 34, 2903–2913 (2020).
Kim, E. S. Enasidenib: first global approval. Drugs 77, 1705–1711 (2017).
Quek, L. et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat. Med. 24, 1167–1177 (2018).
Thomas, D. & Majeti, R. Optimizing next-generation AML therapy: activity of mutant IDH2 inhibitor AG-221 in preclinical models. Cancer Discov. 7, 459–461 (2017).
Amatangelo, M. D. et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 130, 732–741 (2017).
Positive First Trial of Enasidenib for AML. Cancer Discov. 7, Of1, (2017).
DiNardo, C. D. & Wei, A. H. How I treat acute myeloid leukemia in the era of new drugs. Blood 135, 85–96 (2020).
Santini, V. Enasidenib: a magic bullet for myelodysplastic syndromes? Lancet Haematol. 7, e275–e276 (2020).
de Botton, S. et al. Enasidenib vs conventional care in older patients with late-stage mutant-IDH2 relapsed/refractory AML: a randomized phase 3 trial. Blood 141, 156–167 (2023).
Venugopal, S. et al. Efficacy and safety of enasidenib and azacitidine combination in patients with IDH2 mutated acute myeloid leukemia and not eligible for intensive chemotherapy. Blood Cancer J. 12, 10 (2022).
Pollyea, D. A. et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia 33, 2575–2584 (2019).
DiNardo, C. D. et al. Enasidenib plus azacitidine versus azacitidine alone in patients with newly diagnosed, mutant-IDH2 acute myeloid leukaemia (AG221-AML-005): a single-arm, phase 1b and randomised, phase 2 trial. Lancet Oncol. 22, 1597–1608 (2021).
Cai, S. F. et al. A study to assess the efficacy of enasidenib and risk-adapted addition of azacitidine in newly diagnosed IDH2-mutant AML. Blood Adv. 8, 429–440 (2024).
Stein, E. M. et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 133, 676–687 (2019).
Stein, E. M. et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130, 722–731 (2017).
Stein, E. M. et al. Enasidenib in patients with mutant IDH2 myelodysplastic syndromes: a phase 1 subgroup analysis of the multicentre, AG221-C-001 trial. Lancet Haematol. 7, e309–e319 (2020).
Fathi, A. T. et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol. 4, 1106–1110 (2018).
Geoerger, B. et al. Enasidenib treatment in two individuals with D-2-hydroxyglutaric aciduria carrying a germline IDH2 mutation. Nat. Med. 29, 1358–1363 (2023).
Rey, V. et al. A personalized medicine approach identifies enasidenib as an efficient treatment for IDH2 mutant chondrosarcoma. EBioMedicine 102, 105090 (2024).
Norsworthy, K. J. et al. FDA approval summary: ivosidenib for relapsed or refractory acute myeloid leukemia with an isocitrate dehydrogenase-1 mutation. Clin. Cancer Res. 25, 3205–3209 (2019).
Montesinos, P. et al. Ivosidenib and Azacitidine in IDH1-mutated acute myeloid leukemia. N. Engl. J. Med. 386, 1519–1531 (2022).
Woods, A. et al. FDA approval summary: ivosidenib in combination with azacitidine for treatment of patients with newly diagnosed acute myeloid leukemia with an IDH1 mutation. Clin. Cancer Res. 30, 1226–1231 (2024).
Zhu, A. X. et al. Final overall survival efficacy results of ivosidenib for patients with advanced cholangiocarcinoma with IDH1 mutation: the phase 3 randomized clinical clarIDHy trial. JAMA Oncol. 7, 1669–1677 (2021).
Rudà, R. et al. IDH inhibition in gliomas: from preclinical models to clinical trials. Nat. Rev. Neurol. 20, 395–407 (2024).
Mellinghoff, I. K. et al. Ivosidenib in isocitrate dehydrogenase 1-mutated advanced glioma. J. Clin. Oncol. 38, 3398–3406 (2020).
Tap, W. D. et al. Phase I study of the mutant IDH1 inhibitor ivosidenib: safety and clinical activity in patients with advanced chondrosarcoma. J. Clin. Oncol. 38, 1693–1701 (2020).
Mellinghoff, I. K. et al. Vorasidenib and ivosidenib in IDH1-mutant low-grade glioma: a randomized, perioperative phase 1 trial. Nat. Med. 29, 615–622 (2023).
Watts, J. M. et al. Olutasidenib alone or with azacitidine in IDH1-mutated acute myeloid leukaemia and myelodysplastic syndrome: phase 1 results of a phase 1/2 trial. Lancet Haematol. 10, e46–e58 (2023).
de Botton, S. et al. Olutasidenib (FT-2102) induces durable complete remissions in patients with relapsed or refractory IDH1-mutated AML. Blood Adv. 7, 3117–3127 (2023).
de la Fuente, M. I. et al. Olutasidenib (FT-2102) in patients with relapsed or refractory IDH1-mutant glioma: a multicenter, open-label, phase Ib/II trial. Neuro Oncol. 25, 146–156 (2023).
Hoy, S. M. Tazemetostat: first approval. Drugs 80, 513–521 (2020).
First EZH2 Inhibitor Approved-for Rare Sarcoma. Cancer Discov. 10, 333-334, (2020).
Gounder, M. et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol. 21, 1423–1432 (2020).
Italiano, A. et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 19, 649–659 (2018).
Morschhauser, F. et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 21, 1433–1442 (2020).
Munakata, W. et al. Phase 1 study of tazemetostat in Japanese patients with relapsed or refractory B-cell lymphoma. Cancer Sci. 112, 1123–1131 (2021).
Izutsu, K. et al. Phase II study of tazemetostat for relapsed or refractory B-cell non-Hodgkin lymphoma with EZH2 mutation in Japan. Cancer Sci. 112, 3627–3635 (2021).
Zauderer, M. G. et al. EZH2 inhibitor tazemetostat in patients with relapsed or refractory, BAP1-inactivated malignant pleural mesothelioma: a multicentre, open-label, phase 2 study. Lancet Oncol. 23, 758–767 (2022).
Liu, J. et al. Sunitinib attenuates reactive MDSCs enhancing anti-tumor immunity in HNSCC. Int. Immunopharmacol. 119, 110243 (2023).
Zhou, L. et al. Targeting EZH2 enhances antigen presentation, antitumor immunity, and circumvents anti-PD-1 resistance in head and neck cancer. Clin. Cancer Res. 26, 290–300 (2020).
Nastoupil, L. J. When to use targeted therapy for the treatment of follicular lymphoma. Curr. Hematol. Malig. Rep. 16, 45–51 (2021).
Keam, S. J. Valemetostat tosilate: first approval. Drugs 82, 1621–1627 (2022).
Tachibana, M. et al. Safety, tolerability, and pharmacokinetics of valemetostat tablets and the effect of food on valemetostat pharmacokinetics in healthy subjects: two phase 1 studies. Clin. Pharmacol. Drug. Dev. 13, 77–86 (2024).
Tachibana, M. et al. Effect of itraconazole and fluconazole on the pharmacokinetics of valemetostat: an open-label, phase I study in healthy subjects. Clin. Transl. Sci. 16, 2153–2162 (2023).
Izutsu, K. et al. An open-label, single-arm phase 2 trial of valemetostat for relapsed or refractory adult T-cell leukemia/lymphoma. Blood 141, 1159–1168 (2023).
Wu, G. et al. TP63 fusions drive multicomplex enhancer rewiring, lymphomagenesis, and EZH2 dependence. Sci. Transl. Med. 15, eadi7244 (2023).
Lorenzo-Guerra, S. L. et al. Characterization of a preclinical in vitro model derived from a SMARCA4-mutated sinonasal teratocarcinosarcoma. Cells. 13, 81 (2023).
Kagiyama, Y. et al. CDKN1C-mediated growth inhibition by an EZH1/2 dual inhibitor overcomes resistance of mantle cell lymphoma to ibrutinib. Cancer Sci. 112, 2314–2324 (2021).
Koseki, A. et al. EZH1/2 dual inhibitors suppress HTLV-1-infected cell proliferation and hyperimmune response in HTLV-1-associated myelopathy. Front. Microbiol. 14, 1175762 (2023).
Chavali, V., Tyagi, S. C. & Mishra, P. K. MicroRNA-133a regulates DNA methylation in diabetic cardiomyocytes. Biochem. Biophys. Res. Commun. 425, 668–672 (2012).
Hedrich, C. M., Mäbert, K., Rauen, T. & Tsokos, G. C. DNA methylation in systemic lupus erythematosus. Epigenomics 9, 505–525 (2017).
Nakano, K., Boyle, D. L. & Firestein, G. S. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J. Immunol. 190, 1297–1303 (2013).
Yang, L. et al. Functional characterization of age-dependent p16 epimutation reveals biological drivers and therapeutic targets for colorectal cancer. J. Exp. Clin. Cancer Res. 42, 113 (2023).
Hoang, N. M. & Rui, L. DNA methyltransferases in hematological malignancies. J. Genet Genom. 47, 361–372 (2020).
Mahfouz, R. Z. et al. Increased CDA expression/activity in males contributes to decreased cytidine analog half-life and likely contributes to worse outcomes with 5-azacytidine or decitabine therapy. Clin. Cancer Res. 19, 938–948 (2013).
Tellez, C. S. et al. SGI-110 and entinostat therapy reduces lung tumor burden and reprograms the epigenome. Int. J. Cancer 135, 2223–2231 (2014).
Prebet, T. et al. A phase 1b study of atezolizumab in combination with guadecitabine for the treatment of acute myeloid leukemia. Leuk. Lymphoma 63, 2180–2188 (2022).
Bever, K. M. et al. A feasibility study of combined epigenetic and vaccine therapy in advanced colorectal cancer with pharmacodynamic endpoint. Clin. Epigenetics 13, 25 (2021).
Ligon, J. A. et al. A phase II trial of guadecitabine in children and adults with SDH-deficient GIST, pheochromocytoma, paraganglioma, and HLRCC-associated renal cell carcinoma. Clin. Cancer Res. 29, 341–348 (2023).
Roboz, G. J. et al. Guadecitabine vs TC in relapsed/refractory AML after intensive chemotherapy: a randomized phase 3 ASTRAL-2 trial. Blood Adv. 8, 2020–2029 (2024).
Lee, V. et al. A phase II study of guadecitabine combined with irinotecan vs regorafenib or TAS-102 in irinotecan-refractory metastatic colorectal cancer patients. Int. J. Cancer 154, 1794–1801 (2024).
Noviello, T. M. R. et al. Guadecitabine plus ipilimumab in unresectable melanoma: five-year follow-up and integrated multi-omic analysis in the phase 1b NIBIT-M4 trial. Nat. Commun. 14, 5914 (2023).
Amaro, A. et al. Guadecitabine increases response to combined anti-CTLA-4 and anti-PD-1 treatment in mouse melanoma in vivo by controlling T-cells, myeloid derived suppressor and NK cells. J. Exp. Clin. Cancer Res. 42, 67 (2023).
Fenaux, P. et al. Guadecitabine vs treatment choice in newly diagnosed acute myeloid leukemia: a global phase 3 randomized study. Blood Adv. 7, 5027–5037 (2023).
Brueckner, B. et al. Delivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacy. Mol. Cancer Ther. 9, 1256–1264 (2010).
Morris, J. et al. F-aza-T-dCyd (NSC801845), a novel cytidine analog, in comparative cell culture and xenograft studies with the clinical candidates T-dCyd, F-T-dCyd, and Aza-T-dCyd. Mol. Cancer Ther. 20, 625–631 (2021).
Matoušová, M. et al. 2'-deoxy-5,6-dihydro-5-azacytidine—a less toxic alternative of 2'-deoxy-5-azacytidine: a comparative study of hypomethylating potential. Epigenetics 6, 769–776 (2011).
Byun, H. M. et al. 2'-Deoxy-N4-[2-(4-nitrophenyl)ethoxycarbonyl]-5-azacytidine: a novel inhibitor of DNA methyltransferase that requires activation by human carboxylesterase 1. Cancer Lett. 266, 238–248 (2008).
Takemura, Y., Satoh, M., Hatanaka, K. & Kubota, S. Zebularine exerts its antiproliferative activity through S phase delay and cell death in human malignant mesothelioma cells. Biosci. Biotechnol. Biochem. 82, 1159–1164 (2018).
Koh, E. S. et al. The protective effect of zebularine, an inhibitor of DNA methyltransferase, on renal tubulointerstitial inflammation and fibrosis. Int. J. Mol. Sci. 23, 14045 (2022).
Shah, R. et al. Reversal of dual epigenetic repression of non-canonical Wnt-5a normalises diabetic corneal epithelial wound healing and stem cells. Diabetologia 66, 1943–1958 (2023).
Pant, R. et al. Pharmacological inhibition of DNMT1 restores macrophage autophagy and M2 polarization in Western diet-induced nonalcoholic fatty liver disease. J. Biol. Chem. 299, 104779 (2023).
Lubecka-Pietruszewska, K. et al. Clofarabine, a novel adenosine analogue, reactivates DNA methylation-silenced tumour suppressor genes and inhibits cell growth in breast cancer cells. Eur. J. Pharmacol. 723, 276–287 (2014).
Vrânceanu, M. et al. The anticancer potential of plant-derived nutraceuticals via the modulation of gene expression. Plants 11, 2524 (2022).
Qadir Nanakali, N. M. et al. The role of dietary polyphenols in alternating DNA methylation in cancer. Crit. Rev. Food Sci. Nutr. 63, 12256–12269 (2023).
Marques-Magalhães, Â. et al. Anti-neoplastic and demethylating activity of a newly synthetized flavanone-derived compound in renal cell carcinoma cell lines. Biomed. Pharmacother. 141, 111681 (2021).
Wang, L. et al. Design and synthesis of water-soluble grifolin prodrugs for DNA methyltransferase 1 (DNMT1) down-regulation. RSC Adv. 11, 38907–38914 (2021).
Stewart, D. J. et al. A phase I pharmacokinetic and pharmacodynamic study of the DNA methyltransferase 1 inhibitor MG98 administered twice weekly. Ann. Oncol. 14, 766–774 (2003).
Winquist, E. et al. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: a National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Investig. New. Drugs 24, 159–167 (2006).
Davis, A. J. et al. Phase I and pharmacologic study of the human DNA methyltransferase antisense oligodeoxynucleotide MG98 given as a 21-day continuous infusion every 4 weeks. Investig. New. Drugs 21, 85–97 (2003).
Singh, N., Dueñas-González, A., Lyko, F. & Medina-Franco, J. L. Molecular modeling and molecular dynamics studies of hydralazine with human DNA methyltransferase 1. ChemMedChem 4, 792–799 (2009).
Arce, C. et al. A proof-of-principle study of epigenetic therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS ONE 1, e98 (2006).
Coronel, J. et al. A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results. Med. Oncol. 28(Suppl 1), S540–S546 (2011).
Candelaria, M. et al. Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial. Ann. Hematol. 90, 379–387 (2011).
Candelaria, M. et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann. Oncol. 18, 1529–1538 (2007).
Wu, X. & Zhang, Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 18, 517–534 (2017).
Ghosh, K. et al. DNA demethylation in the hypothalamus promotes transcription of Agtr1a and Slc12a2 and hypertension development. J. Biol. Chem. 300, 105597 (2024).
Singh, A. K. et al. Selective targeting of TET catalytic domain promotes somatic cell reprogramming. Proc. Natl. Acad. Sci. USA 117, 3621–3626 (2020).
Chen, L. et al. Direct inhibition of dioxygenases TET1 by the rheumatoid arthritis drug auranofin selectively induces cancer cell death in T-ALL. J. Hematol. Oncol. 16, 113 (2023).
Guan, Y. et al. Eltrombopag inhibits TET dioxygenase to contribute to hematopoietic stem cell expansion in aplastic anemia. J. Clin. Investig. 132, e149856 (2022).
Gilreath, J., Lo, M. & Bubalo, J. Thrombopoietin receptor agonists (TPO-RAs): drug class considerations for pharmacists. Drugs 81, 1285–1305 (2021).
Chen, L. L. et al. Itaconate inhibits TET DNA dioxygenases to dampen inflammatory responses. Nat. Cell. Biol. 24, 353–363 (2022).
Sciacovelli, M. et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547 (2016).
Carey, B. W. et al. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416 (2015).
O’Neill, L. A. J. & Artyomov, M. N. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 19, 273–281 (2019).
Mills, E. L., Kelly, B. & O’Neill, L. A. J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 18, 488–498 (2017).
Zhao, B. et al. Redox-active quinones induces genome-wide DNA methylation changes by an iron-mediated and Tet-dependent mechanism. Nucl. Acids Res. 42, 1593–1605 (2014).
Guan, Y. et al. A therapeutic strategy for preferential targeting of TET2 mutant and TET-dioxygenase deficient cells in myeloid neoplasms. Blood Cancer Discov. 2, 146–161 (2021).
Lv, H. et al. A small-molecule degrader of TET3 as treatment for anorexia nervosa in an animal model. Proc. Natl Acad. Sci. USA 120, e2300015120 (2023).
Chen, F. et al. TET family members are integral to porcine oocyte maturation and parthenogenetic pre-implantation embryogenesis. Int. J. Mol. Sci. 24, 12455 (2023).
Jiang, X. et al. Targeted inhibition of STAT/TET1 axis as a therapeutic strategy for acute myeloid leukemia. Nat. Commun. 8, 2099 (2017).
Weirath, N. A. et al. Small molecule inhibitors of TET dioxygenases: Bobcat339 activity is mediated by contaminating copper(II). ACS Med. Chem. Lett. 13, 792–798 (2022).
Kim, H. et al. Ten-eleven translocation protein 1 modulates medulloblastoma progression. Genome. Biol. 22, 125 (2021).
Hore, T. A. et al. Retinol and ascorbate drive erasure of epigenetic memory and enhance reprogramming to naïve pluripotency by complementary mechanisms. Proc. Natl. Acad. Sci. USA 113, 12202–12207 (2016).
Zhao, H. et al. Opioid receptor signaling suppresses leukemia through both catalytic and non-catalytic functions of TET2. Cell. Rep. 38, 110253 (2022).
Kim, H. et al. Development of novel epigenetic anti-cancer therapy targeting TET proteins. Int. J. Mol. Sci. 24, 16375 (2023).
Yin, R. et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 135, 10396–10403 (2013).
Cimmino, L., Neel, B. G. & Aifantis, I. Vitamin C in stem cell reprogramming and cancer. Trends Cell. Biol. 28, 698–708 (2018).
Shenoy, N. et al. Ascorbic acid-induced TET activation mitigates adverse hydroxymethylcytosine loss in renal cell carcinoma. J. Clin. Investig. 129, 1612–1625 (2019).
Röhr, D. et al. Sodium-dependent Vitamin C transporter 2 deficiency impairs myelination and remyelination after injury: Roles of collagen and demethylation. Glia 65, 1186–1200 (2017).
Yuan, Y. et al. Vitamin C inhibits the metabolic changes induced by Tet1 insufficiency under high fat diet stress. Mol. Nutr. Food Res. 65, e2100417 (2021).
Bensberg, M. et al. TET2 as a tumor suppressor and therapeutic target in T-cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA. 118, e2110758118 (2021).
Giansanti, M. et al. Poly(ADP-Ribose) polymerase inhibitors for arsenic trioxide-resistant acute promyelocytic leukemia: synergistic in vitro antitumor effects with hypomethylating agents or high-dose vitamin C. J. Pharmacol. Exp. Ther. 377, 385–397 (2021).
Liu, M. et al. Vitamin C increases viral mimicry induced by 5-aza-2’-deoxycytidine. Proc. Natl. Acad. Sci. USA 113, 10238–10244 (2016).
Peng, D. et al. Ascorbic acid induced TET2 enzyme activation enhances cancer immunotherapy efficacy in renal cell carcinoma. Int. J. Biol. Sci. 18, 995–1007 (2022).
Gillberg, L. et al. Oral vitamin C supplementation to patients with myeloid cancer on azacitidine treatment: Normalization of plasma vitamin C induces epigenetic changes. Clin. Epigenet. 11, 143 (2019).
Welch, J. S. et al. Combination decitabine, arsenic trioxide, and ascorbic acid for the treatment of myelodysplastic syndrome and acute myeloid leukemia: a phase I study. Am. J. Hematol. 86, 796–800 (2011).
Carr, A. C. & Cook, J. Intravenous Vitamin C for cancer therapy—identifying the current gaps in our knowledge. Front. Physiol. 9, 1182 (2018).
McBrayer, S. K. et al. Transaminase Inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 175, 101–116.e125 (2018).
Li, T. et al. D-2-hydroxyglutarate is necessary and sufficient for isocitrate dehydrogenase 1 mutant-induced MIR148A Promoter Methylation. Mol. Cancer Res. 16, 947–960 (2018).
Yen, K. et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 7, 478–493 (2017).
MacBeth, K. J. et al. Combination of azacitidine and enasidenib enhances leukemic cell differentiation and cooperatively hypomethylates DNA. Exp. Hematol. 98, 47–52.e46 (2021).
Yeung, B. H. Y. et al. Role of isocitrate dehydrogenase 2 on DNA hydroxymethylation in human airway smooth muscle cells. Am. J. Respir. Cell. Mol. Biol. 63, 36–45 (2020).
Fortin, J. et al. Distinct and opposite effects of leukemogenic Idh and Tet2 mutations in hematopoietic stem and progenitor cells. Proc. Natl. Acad. Sci. USA 120, e2208176120 (2023).
Spallotta, F. et al. Stable oxidative cytosine modifications accumulate in cardiac mesenchymal cells from type2 diabetes patients: rescue by α-ketoglutarate and TET-TDG functional reactivation. Circ. Res. 122, 31–46 (2018).
Atlante, S. et al. α-ketoglutarate dehydrogenase inhibition counteracts breast cancer-associated lung metastasis. Cell. Death Dis. 9, 756 (2018).
Hu, X. et al. Epigenetic drug screen identified IOX1 as an inhibitor of Th17-mediated inflammation through targeting TET2. EBioMedicine 86, 104333 (2022).
Ginder, G. D. & Williams, D. C. Jr. Readers of DNA methylation, the MBD family as potential therapeutic targets. Pharmacol. Ther. 184, 98–111 (2018).
Mahmood, N., Arakelian, A., Szyf, M. & Rabbani, S. A. Methyl-CpG binding domain protein 2 (Mbd2) drives breast cancer progression through the modulation of epithelial-to-mesenchymal transition. Exp. Mol. Med. 56, 959–974 (2024).
Pan, H. et al. CpG and methylation-dependent DNA binding and dynamics of the methylcytosine binding domain 2 protein at the single-molecule level. Nucl. Acids Res. 45, 9164–9177 (2017).
Çalışkaner, Z. O. Computational discovery of novel inhibitory candidates targeting versatile transcriptional repressor MBD2. J. Mol. Model 28, 296 (2022).
Gnanapragasam, M. N. et al. p66Alpha-MBD2 coiled-coil interaction and recruitment of Mi-2 are critical for globin gene silencing by the MBD2-NuRD complex. Proc. Natl. Acad. Sci. USA 108, 7487–7492 (2011).
Desai, M. A. et al. An intrinsically disordered region of methyl-CpG binding domain protein 2 (MBD2) recruits the histone deacetylase core of the NuRD complex. Nucl. Acids Res. 43, 3100–3113 (2015).
Leighton, G. O. et al. Analysis of the complex between MBD2 and the histone deacetylase core of NuRD reveals key interactions critical for gene silencing. Proc. Natl. Acad. Sci. USA 120, e2307287120 (2023).
Kim, M. Y. et al. Rational discovery of antimetastatic agents targeting the intrinsically disordered region of MBD2. Sci. Adv. 5, eaav9810 (2019).
Na, I. et al. Drug discovery targeting the disorder-to-order transition regions through the conformational diversity mimicking and statistical analysis. Int. J. Mol. Sci. 21, 5248 (2020).
Sharif, J. et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450, 908–912 (2007).
Kilin, V. et al. Dynamics of methylated cytosine flipping by UHRF1. J. Am. Chem. Soc. 139, 2520–2528 (2017).
Bostick, M. et al. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317, 1760–1764 (2007).
Jiao, D. et al. UHRF1 promotes renal cell carcinoma progression through epigenetic regulation of TXNIP. Oncogene 38, 5686–5699 (2019).
Colyn, L. et al. Dual targeting of G9a and DNA methyltransferase-1 for the treatment of experimental cholangiocarcinoma. Hepatology 73, 2380–2396 (2021).
Verdikt, R. et al. Novel role of UHRF1 in the epigenetic repression of the latent HIV-1. EBioMedicine 79, 103985 (2022).
Myrianthopoulos, V. et al. Tandem virtual screening targeting the SRA domain of UHRF1 identifies a novel chemical tool modulating DNA methylation. Eur. J. Med. Chem. 114, 390–396 (2016).
Zaayter, L. et al. A molecular tool targeting the base-flipping activity of human UHRF1. Chemistry 25, 13363–13375 (2019).
Hu, C. L. et al. Targeting UHRF1-SAP30-MXD4 axis for leukemia initiating cell eradication in myeloid leukemia. Cell. Res. 32, 1105–1123 (2022).
Awal, M. A. et al. Structural-guided identification of small molecule inhibitor of UHRF1 methyltransferase activity. Front. Genet. 13, 928884 (2022).
Lou, C. et al. Screening inhibitors for blocking UHRF1-methylated DNA interaction with capillary electrophoresis. J. Chromatogr. A 1636, 461790 (2021).
Ciaco, S. et al. Inhibitors of UHRF1 base flipping activity showing cytotoxicity against cancer cells. Bioorg. Chem. 137, 106616 (2023).
Senisterra, G. et al. Discovery of small-molecule antagonists of the H3K9me3 binding to UHRF1 tandem tudor domain. SLAS Discov. 23, 930–940 (2018).
Rajakumara, E. et al. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol. Cell. 43, 275–284 (2011).
Kori, S. et al. Structure-based screening combined with computational and biochemical analyses identified the inhibitor targeting the binding of DNA Ligase 1 to UHRF1. Bioorg Med. Chem. 52, 116500 (2021).
Chang, L. et al. Discovery of small molecules targeting the tandem tudor domain of the epigenetic factor UHRF1 using fragment-based ligand discovery. Sci. Rep. 11, 1121 (2021).
Liu, W. H. et al. Discovery and mechanism of small molecule inhibitors selective for the chromatin-binding domains of oncogenic UHRF1. Biochemistry 61, 354–366 (2022).
Niinuma, T. et al. UHRF1 depletion and HDAC inhibition reactivate epigenetically silenced genes in colorectal cancer cells. Clin. Epigenet. 11, 70 (2019).
Kim, J. K. et al. UHRF1 downmodulation enhances antitumor effects of histone deacetylase inhibitors in retinoblastoma by augmenting oxidative stress-mediated apoptosis. Mol. Oncol. 14, 329–346 (2020).
Demir, S. et al. Targeting G9a/DNMT1 methyltransferase activity impedes IGF2-mediated survival in hepatoblastoma. Hepatol. Commun. 8, e0378 (2024).
Abdullah, O. et al. Thymoquinone is a multitarget single epidrug that inhibits the UHRF1 protein complex. Genes 12, 622 (2021).
Fang, T. et al. Lenvatinib inhibited HCC cell migration and invasion through regulating the transcription and ubiquitination of UHRF1 and DNMT1. Biochem. Pharmacol. 210, 115489 (2023).
Chow, M. et al. Maintenance and pharmacologic targeting of ROR1 protein levels via UHRF1 in t(1;19) pre-B-ALL. Oncogene 37, 5221–5232 (2018).
Peng, Y. et al. Diosgenin inhibits prostate cancer progression by inducing UHRF1 protein degradation. Eur. J. Pharmacol. 942, 175522 (2023).
Fu, Y. et al. AKT1 regulates UHRF1 protein stability and promotes the resistance to abiraterone in prostate cancer. Oncogenesis 12, 1 (2023).
Wang, Y. et al. UHRF1 inhibition epigenetically reprograms cancer stem cells to suppress the tumorigenic phenotype of hepatocellular carcinoma. Cell. Death Dis. 14, 381 (2023).
Bousiges, O. et al. Spatial memory consolidation is associated with induction of several lysine-acetyltransferase (histone acetyltransferase) expression levels and H2B/H4 acetylation-dependent transcriptional events in the rat hippocampus. Neuropsychopharmacology 35, 2521–2537 (2010).
Kawabe, Y. et al. ACE2 exerts anti-obesity effect via stimulating brown adipose tissue and induction of browning in white adipose tissue. Am. J. Physiol. Endocrinol. Metab. 317, E1140–e1149 (2019).
Malone, C. F. et al. The KAT module of the SAGA complex maintains the oncogenic gene expression program in MYCN-amplified neuroblastoma. Sci. Adv. 10, eadm9449 (2024).
Sheikh, B. N. & Akhtar, A. The many lives of KATs - detectors, integrators and modulators of the cellular environment. Nat. Rev. Genet. 20, 7–23 (2019).
Strachowska, M. & Robaszkiewicz, A. Characteristics of anticancer activity of CBP/p300 inhibitors - Features of their classes, intracellular targets and future perspectives of their application in cancer treatment. Pharmacol. Ther. 257, 108636 (2024).
Suryanarayanan, V., Rajavel, T., Devi, K. P. & Singh, S. K. Structure based identification and biological evaluation of novel and potent inhibitors of PCAF catalytic domain. Int. J. Biol. Macromol. 120, 823–834 (2018).
Baell, J. B. et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 560, 253–257 (2018).
Vannam, R. et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell. Chem. Biol. 28, 503–514.e512 (2021).
Nicosia, L. et al. Therapeutic targeting of EP300/CBP by bromodomain inhibition in hematologic malignancies. Cancer Cell. 41, 2136–2153.e2113 (2023).
Welti, J. et al. Targeting the p300/CBP axis in lethal prostate cancer. Cancer Discov. 11, 1118–1137 (2021).
Caligiuri, M. et al. FT-6876, a potent and selective inhibitor of CBP/p300, is active in preclinical models of androgen receptor-positive breast cancer. Target. Oncol. 18, 269–285 (2023).
Yan, Y. et al. The novel BET-CBP/p300 dual inhibitor NEO2734 is active in SPOP mutant and wild-type prostate cancer. EMBO Mol. Med. 11, e10659 (2019).
Morrison-Smith, C. D. et al. Combined targeting of the BRD4-NUT-p300 axis in NUT midline carcinoma by dual selective bromodomain inhibitor, NEO2734. Mol. Cancer Ther. 19, 1406–1414 (2020).
Yamaji, K. et al. Molecular insights of a CBP/β-catenin-signaling inhibitor on nonalcoholic steatohepatitis-induced liver fibrosis and disorder. Biomed. Pharmacother. 166, 115379 (2023).
Kimura, K. et al. Safety, tolerability, and anti-fibrotic efficacy of the CBP/β-catenin inhibitor PRI-724 in patients with hepatitis C and B virus-induced liver cirrhosis: an investigator-initiated, open-label, non-randomised, multicentre, phase 1/2a study. EBioMedicine 80, 104069 (2022).
Kimura, K. et al. Safety, tolerability, and preliminary efficacy of the anti-fibrotic small molecule PRI-724, a CBP/β-catenin inhibitor, in patients with hepatitis C virus-related cirrhosis: a single-center, open-label, dose escalation phase 1 trial. EBioMedicine 23, 79–87 (2017).
Liang, T. et al. Targeting histone deacetylases for cancer therapy: trends and challenges. Acta Pharmacol. Sin. B 13, 2425–2463 (2023).
Jiang, Y. et al. Sirtuin inhibition: strategies, inhibitors, and therapeutic potential. Trends Pharmacol. Sci. 38, 459–472 (2017).
Li, Y. et al. Zinc-dependent deacetylase (HDAC) inhibitors with different zinc binding groups. Curr. Top. Med. Chem. 19, 223–241 (2019).
Jo, J. H. et al. A phase I/II study of ivaltinostat combined with gemcitabine and erlotinib in patients with untreated locally advanced or metastatic pancreatic adenocarcinoma. Int. J. Cancer 151, 1565–1577 (2022).
Collier, K. A. et al. A phase 1 trial of the histone deacetylase inhibitor AR-42 in patients with neurofibromatosis type 2-associated tumors and advanced solid malignancies. Cancer Chemother. Pharmacol. 87, 599–611 (2021).
Welling, D. B. et al. Early phase clinical studies of AR-42, a histone deacetylase inhibitor, for neurofibromatosis type 2-associated vestibular schwannomas and meningiomas. Laryngosc. Investig. Otolaryngol. 6, 1008–1019 (2021).
Aggarwal, R. et al. Inhibiting histone deacetylase as a means to reverse resistance to angiogenesis inhibitors: phase I study of abexinostat plus pazopanib in advanced solid tumor malignancies. J. Clin. Oncol. 35, 1231–1239 (2017).
Ribrag, V. et al. Safety and efficacy of abexinostat, a pan-histone deacetylase inhibitor, in non-Hodgkin lymphoma and chronic lymphocytic leukemia: results of a phase II study. Haematologica 102, 903–909 (2017).
Evens, A. M. et al. A phase I/II multicenter, open-label study of the oral histone deacetylase inhibitor abexinostat in relapsed/refractory lymphoma. Clin. Cancer Res. 22, 1059–1066 (2016).
Zhou, Y. B. et al. Pharmacodynamic, pharmacokinetic, and phase 1a study of bisthianostat, a novel histone deacetylase inhibitor, for the treatment of relapsed or refractory multiple myeloma. Acta Pharmacol. Sin. 43, 1091–1099 (2022).
Shah, R. R. Safety and tolerability of histone deacetylase (HDAC) inhibitors in oncology. Drug. Saf. 42, 235–245 (2019).
Ho, T. C. S., Chan, A. H. Y. & Ganesan, A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J. Med. Chem. 63, 12460–12484 (2020).
Roche, J. & Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 121, 451–483 (2016).
Adhikari, N., Jha, T. & Ghosh, B. Dissecting histone deacetylase 3 in multiple disease conditions: selective inhibition as a promising therapeutic strategy. J. Med. Chem. 64, 8827–8869 (2021).
Xu, B. et al. Entinostat, a class I selective histone deacetylase inhibitor, plus exemestane for chinese patients with hormone receptor-positive advanced breast cancer: a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial. Acta Pharmacol. Sin. B 13, 2250–2258 (2023).
Karasic, T. B. et al. Phase I trial of regorafenib, hydroxychloroquine, and entinostat in metastatic colorectal cancer. Oncologist 27, 716–e689 (2022).
Gentzler, R. D. et al. Phase I study of entinostat, atezolizumab, carboplatin, and etoposide in previously untreated extensive-stage small cell lung cancer, ETCTN 10399. Oncologist 28, 1007–e1107 (2023).
Duska, L. R. et al. A surgical window trial evaluating medroxyprogesterone acetate with or without entinostat in patients with endometrial cancer and validation of biomarkers of cellular response. Clin. Cancer Res. 27, 2734–2741 (2021).
Carraway, H. E. et al. Phase 1 study of the histone deacetylase inhibitor entinostat plus clofarabine for poor-risk philadelphia chromosome-negative (newly diagnosed older adults or adults with relapsed refractory disease) acute lymphoblastic leukemia or biphenotypic leukemia. Leuk. Res. 110, 106707 (2021).
Lai, C. J. et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 70, 3647–3656 (2010).
Yu, C. C. et al. A novel small molecule hybrid of vorinostat and DACA displays anticancer activity against human hormone-refractory metastatic prostate cancer through dual inhibition of histone deacetylase and topoisomerase I. Biochem. Pharmacol. 90, 320–330 (2014).
Moertl, S. et al. Comparison of radiosensitization by HDAC inhibitors CUDC-101 and SAHA in pancreatic cancer cells. Int. J. Mol. Sci. 20, 3259 (2019).
Galloway, T. J. et al. A phase I study Of CUDC-101, a multitarget inhibitor of HDACs, EGFR, and HER2, in combination with chemoradiation in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 21, 1566–1573 (2015).
Shimizu, T. et al. Phase I first-in-human study of CUDC-101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin. Cancer Res. 20, 5032–5040 (2014).
Oki, Y. et al. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: results from an expanded phase I trial. Haematologica 102, 1923–1930 (2017).
Landsburg, D. J. et al. Fimepinostat (CUDC-907) in patients with relapsed/refractory diffuse large B cell and high-grade B-cell lymphoma: report of a phase 2 trial and exploratory biomarker analyses. Br. J. Haematol. 195, 201–209 (2021).
Reijers, I. L. M. et al. IFN-γ signature enables selection of neoadjuvant treatment in patients with stage III melanoma. J Exp Med. 220, e20221952 (2023).
Ito, K. et al. A molecular mechanism of action of theophylline: induction of histone deacetylase activity to decrease inflammatory gene expression. Proc. Natl. Acad. Sci. USA 99, 8921–8926 (2002).
Cosío, B. G. et al. Oral low-dose theophylline on top of inhaled fluticasone-salmeterol does not reduce exacerbations in patients with severe COPD: a pilot clinical trial. Chest 150, 123–130 (2016).
Vaquero, A. et al. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes. Dev. 20, 1256–1261 (2006).
Gomes, P., Fleming Outeiro, T. & Cavadas, C. Emerging role of sirtuin 2 in the regulation of mammalian metabolism. Trends Pharmacol. Sci. 36, 756–768 (2015).
Shen, H. et al. Mitochondrial sirtuins in cancer: a revisited review from molecular mechanisms to therapeutic strategies. Theranostics 14, 2993–3013 (2024).
Ianni, A. et al. SIRT7: a novel molecular target for personalized cancer treatment? Oncogene 43, 993–1006 (2024).
Bedalov, A. et al. Identification of a small molecule inhibitor of Sir2p. Proc. Natl. Acad. Sci. USA 98, 15113–15118 (2001).
Grozinger, C. M. et al. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 276, 38837–38843 (2001).
Broussy, S., Laaroussi, H. & Vidal, M. Biochemical mechanism and biological effects of the inhibition of silent information regulator 1 (SIRT1) by EX-527 (SEN0014196 or selisistat). J. Enzym. Inhib. Med. Chem. 35, 1124–1136 (2020).
Peck, B. et al. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer Ther. 9, 844–855 (2010).
Spinck, M. et al. Discovery of dihydro-1,4-benzoxazine carboxamides as potent and highly selective inhibitors of sirtuin-1. J. Med. Chem. 64, 5838–5849 (2021).
Trapp, J. et al. Adenosine mimetics as inhibitors of NAD+-dependent histone deacetylases, from kinase to sirtuin inhibition. J. Med. Chem. 49, 7307–7316 (2006).
Shim, K. H. et al. Small-molecule drug screening identifies drug Ro 31-8220 that reduces toxic phosphorylated tau in Drosophila melanogaster. Neurobiol. Dis. 130, 104519 (2019).
Disch, J. S. et al. Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. J. Med. Chem. 56, 3666–3679 (2013).
Lain, S. et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 13, 454–463 (2008).l
Moreno-Yruela, C. et al. Hydroxamic acid-modified peptide microarrays for profiling isozyme-selective interactions and inhibition of histone deacetylases. Nat. Commun. 12, 62 (2021).
Asaba, T. et al. Inhibition of human sirtuins by in situ generation of an acetylated lysine-ADP-ribose conjugate. J. Am. Chem. Soc. 131, 6989–6996 (2009).
Shin, D. H. et al. Oncogenic KRAS mutation confers chemoresistance by upregulating SIRT1 in non-small cell lung cancer. Exp. Mol. Med. 55, 2220–2237 (2023).
Smith, M. R. et al. A potent and selective sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease. Hum. Mol. Genet. 23, 2995–3007 (2014).
Süssmuth, S. D. et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br. J. Clin. Pharmacol. 79, 465–476 (2015).
Huang, J. et al. The SIRT1 inhibitor EX-527 suppresses mTOR activation and alleviates acute lung injury in mice with endotoxiemia. Innate Immun. 23, 678–686 (2017).
Kundu, A. et al. Protective effect of EX-527 against high-fat diet-induced diabetic nephropathy in Zucker rats. Toxicol. Appl. Pharmacol. 390, 114899 (2020).
Zhang, Q. et al. Targeting a cryptic allosteric site of SIRT6 with small-molecule inhibitors that inhibit the migration of pancreatic cancer cells. Acta Pharmacol. Sin. B 12, 876–889 (2022).
Xu, X. et al. Discovery of a potent and highly selective inhibitor of SIRT6 against pancreatic cancer metastasis in vivo. Acta Pharmacol. Sin. B 14, 1302–1316 (2024).
Song, N. et al. Discovery of a pyrrole-pyridinimidazole derivative as novel SIRT6 inhibitor for sensitizing pancreatic cancer to gemcitabine. Cell. Death Dis. 14, 499 (2023).
Song, N. et al. A SIRT6 inhibitor, marine-derived pyrrole-pyridinimidazole derivative 8a, suppresses angiogenesis. Mar. Drugs. 21, 517 (2023).
Wang, Z. A. et al. Structural basis of sirtuin 6-catalyzed nucleosome deacetylation. J. Am. Chem. Soc. 145, 6811–6822 (2023).
Deng, Q. et al. Smooth muscle liver kinase B1 inhibits foam cell formation and atherosclerosis via direct phosphorylation and activation of SIRT6. Cell. Death Dis. 14, 542 (2023).
Howitz, K. T. et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196 (2003).
Wang, Z. et al. Diosgenin protects against podocyte injury in early phase of diabetic nephropathy through regulating SIRT6. Phytomedicine 104, 154276 (2022).
Luo, D. et al. Capsaicin attenuates arterial calcification through promoting SIRT6-mediated deacetylation and degradation of Hif1α (hypoxic-inducible factor-1 alpha). Hypertension 79, 906–917 (2022).
Xu, K. et al. Apigenin alleviates oxidative stress-induced myocardial injury by regulating SIRT1 signaling pathway. Eur. J. Pharmacol. 944, 175584 (2023).
Xue, C. et al. Pachymic acid protects hepatic cells against oxygen-glucose deprivation/reperfusion injury by activating sirtuin 1 to inhibit HMGB1 acetylation and inflammatory signaling. Chin. J. Physiol. 66, 239–247 (2023).
Sun, Y. et al. GB1a activates SIRT6 to regulate lipid metabolism in mouse primary hepatocytes. Int. J. Mol. Sci. 24, 9540 (2023).
Tu, Q. et al. Andrographolide improves the dysfunction of endothelial progenitor cells from angiotensin II-induced hypertensive mice through SIRT1 signaling. Biochem. Biophys. Res. Commun. 642, 11–20 (2023).
Wu, Q. J. et al. The sirtuin family in health and disease. Signal. Transduct. Target. Ther. 7, 402 (2022).
Yu, X. et al. DDIT3/CHOP mediates the inhibitory effect of ER stress on chondrocyte differentiation by AMPKα-SIRT1 pathway. Biochim. Biophys. Acta Mol. Cell. Res. 1869, 119265 (2022).
Han, L. et al. Phloretin attenuation of hepatic steatosis via an improvement of mitochondrial dysfunction by activating AMPK-dependent signaling pathways in C57BL/6J mice and HepG2 cells. Food Funct. 12, 12421–12433 (2021).
Pham, T. H. et al. Puerarin attenuates hepatic steatosis via G-protein-coupled estrogen receptor-mediated calcium and SIRT1 signaling pathways. Phytother. Res. 36, 3601–3618 (2022).
Liu, X. et al. Atractylenolide III from atractylodes macrocephala Koidz promotes the activation of brown and white adipose tissue through SIRT1/PGC-1α signaling pathway. Phytomedicine 104, 154289 (2022).
Tang, X., Li, X., Zhang, D. & Han, W. Astragaloside-IV alleviates high glucose-induced ferroptosis in retinal pigment epithelial cells by disrupting the expression of miR-138-5p/Sirt1/Nrf2. Bioengineered 13, 8240–8254 (2022).
Chen, H. et al. Low-dose deoxynivalenol exposure inhibits hepatic mitophagy and hesperidin reverses this phenomenon by activating SIRT1. J. Hazard. Mater. 468, 133854 (2024).
Zhang, Y. et al. Caffeic acid phenethyl ester inhibits neuro-inflammation and oxidative stress following spinal cord injury by mitigating mitochondrial dysfunction via the SIRT1/PGC1α/DRP1 signaling pathway. J. Transl. Med. 22, 304 (2024).
Hafez, H. M., Waz, S., El-Tahawy, N. F. G. & Mohamed, M. Z. Agomelatine ameliorates cadmium-induced toxicity through the modification of HMGB-1/TLR-4/NFκB pathway. Toxicol. Appl. Pharmacol. 457, 116313 (2022).
Li, G., Hu, C., Liu, Y. & Lin, H. Ligustilide, a novel SIRT1 agonist, alleviates lipopolysaccharide-induced acute lung injury through deacetylation of NICD. Int. Immunopharmacol. 121, 110486 (2023).
Wu, S. et al. Tanshinone IIA ameliorates experimental diabetic cardiomyopathy by inhibiting endoplasmic reticulum stress in cardiomyocytes via SIRT1. Phytother. Res. 37, 3543–3558 (2023).
Li, Y. et al. Farnesol exerts protective effects against chronic sleep deprivation-induced cognitive impairment via activation SIRT1/Nrf2 pathway in the hippocampi of adult mice. Mol. Nutr. Food Res. 67, e2200735 (2023).
Peng, F. et al. 2-APQC, a small-molecule activator of Sirtuin-3 (SIRT3), alleviates myocardial hypertrophy and fibrosis by regulating mitochondrial homeostasis. Signal. Transduct. Target. Ther. 9, 133 (2024).
Feng, L. et al. Dual-target inhibitors of bromodomain and extra-terminal proteins in cancer: a review from medicinal chemistry perspectives. Med. Res. Rev. 42, 710–743 (2022).
Viviano, M. et al. Successes and challenges in the development of BD1-selective BET inhibitors: a patent review. Expert Opin. Ther. Pat. 34, 529–545 (2024).
Wang, Z. et al. Discovery of a bromodomain and extra terminal domain (BET) inhibitor with the selectivity for the second bromodomain (BD2) and the capacity for the treatment of inflammatory diseases. J. Med. Chem. 66, 10824–10848 (2023).
Dhulkifle, H. et al. Apabetalone (RVX-208): a potential epigenetic therapy for the treatment of cardiovascular, renal, neurological, viral, and cancer disorders. ACS Pharmacol. Transl. Sci. 7, 546–559 (2024).
Ray, K. K. et al. Effect of apabetalone added to standard therapy on major adverse cardiovascular events in patients with recent acute coronary syndrome and type 2 diabetes: a randomized clinical trial. Jama 323, 1565–1573 (2020).
Picaud, S. et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 110, 19754–19759 (2013).
Wasiak, S. et al. The BET inhibitor apabetalone decreases neuroendothelial proinflammatory activation in vitro and in a mouse model of systemic inflammation. Transl. Neurosci. 14, 20220332 (2023).
Sun, M. et al. Selective BET inhibitor RVX-208 ameliorates periodontal inflammation and bone loss. J. Clin. Periodontol. 50, 1658–1669 (2023).
Fu, L. et al. Inhibition of epigenetic reader proteins by apabetalone counters inflammation in activated innate immune cells from Fabry disease patients receiving enzyme replacement therapy. Pharmacol. Res. Perspect. 10, e00949 (2022).
Gangat, N. & Tefferi, A. New drugs in myelofibrosis: critical assessment of additional value to monotherapy with JAK inhibitors. Am. J. Hematol. 99, 978–981 (2024).
Gupta, V. et al. Matching-adjusted indirect comparison of the pelabresib-ruxolitinib combination vs JAKi monotherapy in myelofibrosis. Blood Adv. 7, 5421–5432 (2023).
Mascarenhas, J. et al. MANIFEST: pelabresib in combination with ruxolitinib for janus kinase inhibitor treatment-naïve myelofibrosis. J. Clin. Oncol. 41, 4993–5004 (2023).
Kharenko, O. A., Patel, R. G., Calosing, C. & van der Horst, E. H. Combination of ZEN-3694 with CDK4/6 inhibitors reverses acquired resistance to CDK4/6 inhibitors in ER-positive breast cancer. Cancer Gene Ther. 29, 859–869 (2022).
Aggarwal, R. R. et al. A phase Ib/IIa study of the pan-BET inhibitor ZEN-3694 in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer. Clin. Cancer Res. 26, 5338–5347 (2020).
Marr, A. R. et al. The multi-CDK inhibitor dinaciclib reverses bromo- and extra-terminal domain (BET) inhibitor resistance in acute myeloid leukemia via inhibition of Wnt/β-catenin signaling. Exp. Hematol. Oncol. 13, 27 (2024).
Stathis, A. & Bertoni, F. BET proteins as targets for anticancer treatment. Cancer Discov. 8, 24–36 (2018).
Smith, A. L. et al. BET inhibition reforms the immune microenvironment and alleviates T cell dysfunction in chronic lymphocytic leukemia. JCI Insight 9, e177054 (2024).
Li, Y. et al. Structure-guided design and synthesis of pyridinone-based selective bromodomain and extra-terminal domain (BET)-first bromodomain (BD1) inhibitors. J. Med. Chem. 67, 2712–2731 (2024).
Ali, I. et al. DW71177: A novel [1,2,4]triazolo[4,3-a]quinoxaline-based potent and BD1-Selective BET inhibitor for the treatment of acute myeloid leukemia. Eur. J. Med. Chem. 265, 116052 (2024).
Chen, X. et al. Discovery of a brain-permeable bromodomain and extra terminal domain (BET) inhibitor with selectivity for BD1 for the treatment of multiple sclerosis. Eur. J. Med. Chem. 265, 116080 (2024).
Bauer, N. et al. Development of potent dual BET/HDAC inhibitors via pharmacophore merging and structure-guided optimization. ACS Chem. Biol. 19, 266–279 (2024).
Jiang, W. et al. Discovery of novel phenoxyaryl pyridones as bromodomain and extra-terminal domain (BET) inhibitors with high selectivity for the second bromodomain (BD2) to potentially treat acute myeloid leukemia. J. Med. Chem. 67, 1513–1532 (2024).
Pandey, K. et al. SRX3177, a CDK4/6-PI3K-BET inhibitor, in combination with an RdRp inhibitor, molnupiravir, or an entry inhibitor MU-UNMC-2, has potent antiviral activity against the omicron variant of SARS-CoV-2. Antivir. Res. 227, 105904 (2024).
Li, Y. et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 159, 558–571 (2014).
Li, Y. et al. Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Mol. Cell. 62, 181–193 (2016).
Ren, X. et al. Histone benzoylation serves as an epigenetic mark for DPF and YEATS family proteins. Nucl. Acids Res. 49, 114–126 (2021).
Wan, L. et al. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269 (2017).
Mi, W. et al. YEATS2 links histone acetylation to tumorigenesis of non-small cell lung cancer. Nat. Commun. 8, 1088 (2017).
Ji, K. et al. Unveiling the role of GAS41 in cancer progression. Cancer Cell. Int. 23, 245 (2023).
Bilgin, N. et al. Reading and erasing of histone crotonyllysine mimics by the AF9 YEATS domain and SIRT2 deacylase. Bioorg. Med. Chem. 95, 117500 (2023).
Li, X. et al. Structure-guided development of YEATS domain inhibitors by targeting π-π-π stacking. Nat. Chem. Biol. 14, 1140–1149 (2018).
Moustakim, M. et al. Discovery of an MLLT1/3 YEATS domain chemical probe. Angew. Chem. Int. Ed. Engl. 57, 16302–16307 (2018).
Yang, Y. et al. Structural studies of intrinsically disordered MLL-fusion protein AF9 in complex with peptidomimetic inhibitors. Protein Sci. 33, e5019 (2024).
Yuan, Y. et al. Design, synthesis, and biological evaluations of DOT1L peptide mimetics targeting the protein-protein interactions between DOT1L and MLL-AF9/MLL-ENL. J. Med. Chem. 65, 7770–7785 (2022).
Liu, Y. et al. Small-molecule inhibition of the acyl-lysine reader ENL as a strategy against acute myeloid leukemia. Cancer Discov. 12, 2684–2709 (2022).
Yan, F. et al. KAT6A and ENL form an epigenetic transcriptional control module to drive critical leukemogenic gene-expression programs. Cancer Discov. 12, 792–811 (2022).
Chen, Y. et al. Targeting the epigenetic reader ENL inhibits super-enhancer-driven oncogenic transcription and synergizes with BET inhibition to suppress tumor progression. Cancer Res. 84, 1237–1251 (2024).
Jiang, Y. et al. Selective targeting of AF9 YEATS domain by cyclopeptide inhibitors with preorganized conformation. J. Am. Chem. Soc. 142, 21450–21459 (2020).
Travis, C. R., Francis, D. Y., Williams, D. C. Jr. & Waters, M. L. Evaluation of acyllysine isostere interactions with the aromatic pocket of the AF9 YEATS domain. Protein Sci. 32, e4533 (2023).
Liu, Y. et al. Fragment-based discovery of AF9 YEATS domain inhibitors. Int. J. Mol. Sci. 23, 3893 (2022).
Listunov, D. et al. Development of potent dimeric inhibitors of GAS41 YEATS domain. Cell. Chem. Biol. 28, 1716–1727.e1716 (2021).
Cheng, C. L. et al. Bromodomain-containing protein BRPF1 is a therapeutic target for liver cancer. Commun. Biol. 4, 888 (2021).
Bitler, B. G. et al. Targeting BRPF3 moderately reverses olaparib resistance in high grade serous ovarian carcinoma. Mol. Carcinog. 62, 1717–1730 (2023).
Li, F. et al. BPTF drives gastric cancer resistance to EGFR inhibitor by epigenetically regulating the C-MYC/PLCG1/Perk Axis. Adv. Sci. 10, e2303091 (2023).
Zhang, W. et al. Development of 3-acetylindole derivatives that selectively target BRPF1 as new inhibitors of receptor activator of NF-κB ligand (RANKL)-induced osteoclastogenesis. Bioorg. Med. Chem. 96, 117440 (2023).
Ghazy, E. et al. Design, synthesis, and biological evaluation of dual targeting inhibitors of histone deacetylase 6/8 and bromodomain BRPF1. Eur. J. Med. Chem. 200, 112338 (2020).
Palmer, W. S. et al. Structure-guided design of IACS-9571, a selective high-affinity dual TRIM24-BRPF1 bromodomain inhibitor. J. Med. Chem. 59, 1440–1454 (2016).
Xiang, Q. et al. Discovery, optimization and evaluation of 1-(indolin-1-yl)ethan-1-ones as novel selective TRIM24/BRPF1 bromodomain inhibitors. Eur. J. Med. Chem. 236, 114311 (2022).
Igoe, N. et al. Design of a biased potent small molecule inhibitor of the bromodomain and PHD finger-containing (BRPF) proteins suitable for cellular and in vivo studies. J. Med. Chem. 60, 668–680 (2017).
Zhu, J., Zhou, C. & Caflisch, A. Structure-based discovery of selective BRPF1 bromodomain inhibitors. Eur. J. Med. Chem. 155, 337–352 (2018).
Urick, A. K. et al. Dual screening of BPTF and Brd4 using protein-observed fluorine NMR uncovers new bromodomain probe molecules. ACS Chem. Biol. 10, 2246–2256 (2015).
Jiang, C. et al. BPTF in bone marrow provides a potential progression biomarker regulated by TFAP4 through the PI3K/AKT pathway in neuroblastoma. Biol. Proced. Online 25, 11 (2023).
Bezrookove, V. et al. BPTF promotes the progression of distinct subtypes of breast cancer and is a therapeutic target. Front Oncol. 12, 1011173 (2022).
Khan, I. & Kashani-Sabet, M. Bromodomain inhibition targeting BPTF in the treatment of melanoma and other solid tumors. Clin. Exp. Metastasis 509—515 (2024).
Xu, J. et al. Compound C620-0696, a new potent inhibitor targeting BPTF, the chromatin-remodeling factor in non-small-cell lung cancer. Front Med. 14, 60–67 (2020).
Zhu, Y. et al. Promising role of protein arginine methyltransferases in overcoming anti-cancer drug resistance. Drug Resist Updat. 72, 101016 (2024).
Tong, C. et al. Overview of the development of protein arginine methyltransferase modulators: achievements and future directions. Eur. J. Med. Chem. 267, 116212 (2024).
Li, D. et al. Small molecules targeting selected histone methyltransferases (HMTs) for cancer treatment: current progress and novel strategies. Eur. J. Med. Chem. 264, 115982 (2024).
Rea, S. et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599 (2000).
Haebe, J. R., Bergin, C. J., Sandouka, T. & Benoit, Y. D. Emerging role of G9a in cancer stemness and promises as a therapeutic target. Oncogenesis 10, 76 (2021).
Husmann, D. & Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 26, 880–889 (2019).
Yang, C. et al. Role of NSD1 as potential therapeutic target in tumor. Pharmacol. Res. 173, 105888 (2021).
Saha, N. & Muntean, A. G. Insight into the multi-faceted role of the SUV family of H3K9 methyltransferases in carcinogenesis and cancer progression. Biochim. Biophys. Acta Rev. Cancer 1875, 188498 (2021).
Ma, T. et al. SETDB1: progress and prospects in cancer treatment potential and inhibitor research. Bioorg. Chem. 145, 107219 (2024).
Ma, Z., Bolinger, A. A., Chen, H. & Zhou, J. Drug discovery targeting nuclear receptor binding SET domain protein 2 (NSD2). J. Med. Chem. 66, 10991–11026 (2023).
Fioravanti, R. et al. Six years (2012-2018) of researches on catalytic EZH2 inhibitors: the boom of the 2-pyridone compounds. Chem. Rec. 18, 1818–1832 (2018).
Wang, X. et al. The synergistic anti-tumor activity of EZH2 inhibitor SHR2554 and HDAC inhibitor chidamide through ORC1 reduction of DNA replication process in diffuse large B cell lymphoma. Cancers 13, 4249 (2021).
Song, Y. et al. SHR2554, an EZH2 inhibitor, in relapsed or refractory mature lymphoid neoplasms: a first-in-human, dose-escalation, dose-expansion, and clinical expansion phase 1 trial. Lancet Haematol. 9, e493–e503 (2022).
Song, Y. et al. Enhancer of zeste homolog 2 inhibitor SHR2554 in relapsed or refractory peripheral T-cell lymphoma: data from the first-in-human phase I study. Clin. Cancer Res. 30, 1248–1255 (2024).
Deng, K. et al. Study on pharmacokinetic interactions between SHR2554 and itraconazole in healthy subjects: a single-center, open-label phase I trial. Cancer Med. 12, 1431–1440 (2023).
Kung, P. P. et al. Optimization of orally bioavailable enhancer of zeste homolog 2 (EZH2) inhibitors using ligand and property-based design strategies: identification of development candidate (R)-5,8-dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497). J. Med. Chem. 61, 650–665 (2018).
Zhang, Y. et al. EZH2i EPZ-6438 and HDACi vorinostat synergize with ONC201/TIC10 to activate integrated stress response, DR5, reduce H3K27 methylation, ClpX and promote apoptosis of multiple tumor types including DIPG. Neoplasia 23, 792–810 (2021).
Al-Hasani, K. et al. Inhibition of pancreatic EZH2 restores progenitor insulin in T1D donor. Signal. Transduct. Target. Ther. 7, 248 (2022).
Yap, T. A. et al. Phase I study of the novel enhancer of zeste homolog 2 (EZH2) inhibitor GSK2816126 in patients with advanced hematologic and solid tumors. Clin. Cancer Res. 25, 7331–7339 (2019).
Du, D. et al. Structure-guided development of small-molecule PRC2 inhibitors targeting EZH2-EED interaction. J. Med. Chem. 64, 8194–8207 (2021).
Yu, W. et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 3, 1288 (2012).
Daigle, S. R. et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 20, 53–65 (2011).
Rau, R. E. et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 128, 971–981 (2016).
Godfrey, L. et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nat. Commun. 10, 2803 (2019).
Perner, F. et al. Novel inhibitors of the histone methyltransferase DOT1L show potent antileukemic activity in patient-derived xenografts. Blood 136, 1983–1988 (2020).
Klaus, C. R. et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J. Pharmacol. Exp. Ther. 350, 646–656 (2014).
Wong, M. et al. The histone methyltransferase DOT1L promotes neuroblastoma by regulating gene transcription. Cancer Res. 77, 2522–2533 (2017).
Evanno, E. et al. Tri-methylation of H3K79 is decreased in TGF-β1-induced epithelial-to-mesenchymal transition in lung cancer. Clin. Epigenet. 9, 80 (2017).
Zhang, J. et al. Gain-of-function mutations in the catalytic domain of DOT1L promote lung cancer malignant phenotypes via the MAPK/ERK signaling pathway. Sci. Adv. 9, eadc9273 (2023).
Alford, J. S. et al. Conformational-design-driven discovery of EZM0414: a Selective, potent SETD2 inhibitor for clinical studies. ACS Med. Chem. Lett. 13, 1137–1143 (2022).
Fedoriw, A. et al. Anti-tumor activity of the type I PRMT inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP loss. Cancer Cell. 36, 100–114.e125 (2019).
Fedoriw, A. et al. Inhibiting type I arginine methyltransferase activity promotes T cell-mediated antitumor immune responses. Cancer Immunol. Res. 10, 420–436 (2022).
Zhou, S. et al. Targeting type I PRMTs as promising targets for the treatment of pulmonary disorders: asthma, COPD, lung cancer, PF, and PH. Life Sci. 342, 122538 (2024).
El-Khoueiry, A. B. et al. Phase 1 study of GSK3368715, a type I PRMT inhibitor, in patients with advanced solid tumors. Br. J. Cancer 129, 309–317 (2023).
Martin, P. L. et al. towards the targeted protein degradation of PRMT1. ChemMedChem, e202400269 (2024).
Li, X., Wang, C., Jiang, H. & Luo, C. A patent review of arginine methyltransferase inhibitors (2010-2018). Expert Opin. Ther. Pat. 29, 97–114 (2019).
Brehmer, D. et al. Discovery and pharmacological characterization of JNJ-64619178, a novel small-molecule inhibitor of PRMT5 with potent antitumor activity. Mol. Cancer Ther. 20, 2317–2328 (2021).
Jensen-Pergakes, K. et al. SAM-competitive PRMT5 inhibitor PF-06939999 demonstrates antitumor activity in splicing dysregulated NSCLC with decreased liability of drug resistance. Mol. Cancer Ther. 21, 3–15 (2022).
O’Brien, S. et al. Inhibiting PRMT5 induces DNA damage and increases anti-proliferative activity of Niraparib, a PARP inhibitor, in models of breast and ovarian cancer. BMC Cancer 23, 775 (2023).
Zhang, Y., Verwilligen, R. A. F., Van Eck, M. & Hoekstra, M. PRMT5 inhibition induces pro-inflammatory macrophage polarization and increased hepatic triglyceride levels without affecting atherosclerosis in mice. J. Cell. Mol. Med. 27, 1056–1068 (2023).
Haque, T. et al. Phase 1 study of JNJ-64619178, a protein arginine methyltransferase 5 inhibitor, in patients with lower-risk myelodysplastic syndromes. Leuk. Res. 134, 107390 (2023).
Rodon, J. et al. A phase I study to evaluate the safety, pharmacokinetics, and pharmacodynamics of PF-06939999 (PRMT5 inhibitor) in patients with selected advanced or metastatic tumors with high incidence of splicing factor gene mutations. ESMO Open 9, 102961 (2024).
Cottrell, K. M. et al. Discovery of TNG908: a selective, brain penetrant, MTA-cooperative PRMT5 inhibitor that is synthetically lethal with MTAP-deleted cancers. J. Med. Chem. 67, 6064–6080 (2024).
Engstrom, L. D. et al. MRTX1719 Is an MTA-cooperative PRMT5 inhibitor that exhibits synthetic lethality in preclinical models and patients with MTAP-deleted cancer. Cancer Discov. 13, 2412–2431 (2023).
Ferrarotto, R. et al. PRT543, a protein arginine methyltransferase 5 inhibitor, in patients with advanced adenoid cystic carcinoma: an open-label, phase I dose-expansion study. Oral. Oncol. 149, 106634 (2024).
Tong, D., Tang, Y. & Zhong, P. The emerging roles of histone demethylases in cancers. Cancer Metastasis Rev. 43, 795–821 (2024).
Arifuzzaman, S., Khatun, M. R. & Khatun, R. Emerging of lysine demethylases (KDMs): from pathophysiological insights to novel therapeutic opportunities. Biomed. Pharmacother. 129, 110392 (2020).
Onuora, S. Targeting KDM2/7 histone demethylases could protect against OA. Nat. Rev. Rheumatol. 19, 326 (2023).
Suzuki, T. et al. Identification of the KDM2/7 histone lysine demethylase subfamily inhibitor and its antiproliferative activity. J. Med. Chem. 56, 7222–7231 (2013).
Gerken, P. A. et al. Discovery of a highly selective cell-active inhibitor of the histone lysine demethylases KDM2/7. Angew. Chem. Int. Ed. Engl. 56, 15555–15559 (2017).
Coleman, O. D. et al. Cyclic peptides target the aromatic cage of a PHD-finger reader domain to modulate epigenetic protein function. Chem. Sci. 14, 7136–7146 (2023).
Rose, N. R. et al. Plant growth regulator daminozide is a selective inhibitor of human KDM2/7 histone demethylases. J. Med. Chem. 55, 6639–6643 (2012).
Assi, R. et al. Inhibition of KDM7A/B histone demethylases restores H3K79 methylation and protects against osteoarthritis. Ann. Rheum. Dis. 82, 963–973 (2023).
Xu, X. et al. Small molecular modulators of JMJD1C preferentially inhibit growth of leukemia cells. Int. J. Cancer 146, 400–412 (2020).
Yang, Y. et al. Modulators of histone demethylase JMJD1C selectively target leukemic stem cells. FEBS Open. Bio. 11, 265–277 (2021).
Zhang, W. et al. Epigenetic regulation of wnt signaling by carboxamide-substituted benzhydryl amines that function as histone demethylase inhibitors. iScience 23, 101795 (2020).
Lee, D. H. et al. Advances in histone demethylase KDM4 as cancer therapeutic targets. FASEB J. 34, 3461–3484 (2020).
Wu, Q. et al. Recent advances with KDM4 inhibitors and potential applications. J. Med. Chem. 65, 9564–9579 (2022).
Chandhasin, C. et al. TACH101, a first-in-class pan-inhibitor of KDM4 histone demethylase. Anticancer Drugs 34, 1122–1131 (2023).
Li, Y. et al. KDM4 inhibitor SD49-7 attenuates leukemia stem cell via KDM4A/MDM2/p21(CIP1) axis. Theranostics 12, 4922–4934 (2022).
Lombino, J. et al. In-silico guided chemical exploration of KDM4A fragments hits. Clin. Epigenet. 15, 197 (2023).
Del Moral-Morales, A. et al. Transcriptomic and drug discovery analyses reveal natural compounds targeting the KDM4 subfamily as promising adjuvant treatments in cancer. Front Genet. 13, 860924 (2022).
Tarhonskaya, H. et al. Studies on the interaction of the histone demethylase KDM5B with tricarboxylic acid cycle intermediates. J. Mol. Biol. 429, 2895–2906 (2017).
Tumber, A. et al. Potent and selective KDM5 inhibitor stops cellular demethylation of H3K4me3 at transcription start sites and proliferation of MM1S myeloma cells. Cell. Chem. Biol. 24, 371–380 (2017).
Pippa, S. et al. Small molecule inhibitors of KDM5 histone demethylases increase the radiosensitivity of breast cancer cells overexpressing JARID1B. Molecules 24, 1739 (2019).
Johansson, C. et al. Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat. Chem. Biol. 12, 539–545 (2016).
Liang, J. et al. Lead optimization of a pyrazolo[1,5-a]pyrimidin-7(4H)-one scaffold to identify potent, selective and orally bioavailable KDM5 inhibitors suitable for in vivo biological studies. Bioorg. Med. Chem. Lett. 26, 4036–4041 (2016).
Terao, M. et al. Structural optimization of a lysine demethylase 5 inhibitor for improvement of its cellular activity. Bioorg. Med. Chem. 98, 117579 (2024).
Yang, G. J. et al. Structure-based discovery of a selective KDM5A inhibitor that exhibits anti-cancer activity via inducing cell cycle arrest and senescence in breast cancer cell lines. Cancers 11, 92 (2019).
Horton, J. R. et al. Structure-based engineering of irreversible inhibitors against histone lysine demethylase KDM5A. J. Med. Chem. 61, 10588–10601 (2018).
Miyake, Y. et al. Identification of novel lysine demethylase 5-selective inhibitors by inhibitor-based fragment merging strategy. Bioorg. Med. Chem. 27, 1119–1129 (2019).
Liang, J. et al. From a novel HTS hit to potent, selective, and orally bioavailable KDM5 inhibitors. Bioorg. Med. Chem. Lett. 27, 2974–2981 (2017).
Paroni, G. et al. HER2-positive breast-cancer cell lines are sensitive to KDM5 inhibition: definition of a gene-expression model for the selection of sensitive cases. Oncogene 38, 2675–2689 (2019).
Leadem, B. R. et al. A KDM5 inhibitor increases global H3K4 trimethylation occupancy and enhances the biological efficacy of 5-aza-2'-deoxycytidine. Cancer Res. 78, 1127–1139 (2018).
Gilmore, S. A. et al. Characterization of a KDM5 small molecule inhibitor with antiviral activity against hepatitis B virus. PLoS One 17, e0271145 (2022).
Mitsui, E. et al. Identification of ryuvidine as a KDM5A inhibitor. Sci. Rep. 9, 9952 (2019).
Liu, Y., Yu, Y., Zhang, J. & Wang, C. The therapeutic effect of dexmedetomidine on protection from renal failure via inhibiting KDM5A in lipopolysaccharide-induced sepsis of mice. Life Sci. 239, 116868 (2019).
Yang, G. J. et al. Selective inhibition of lysine-specific demethylase 5A (KDM5A) using a rhodium(III) complex for triple-negative breast cancer therapy. Angew. Chem. Int. Ed. Engl. 57, 13091–13095 (2018).
Dabiri, Y. et al. Imidazopyridines as potent KDM5 demethylase inhibitors promoting reprogramming efficiency of human iPSCs. iScience 12, 168–181 (2019).
Tang, K. et al. Discovery of novel pyrazole-based KDM5B inhibitor TK-129 and its protective effects on myocardial remodeling and fibrosis. J. Med. Chem. 65, 12979–13000 (2022).
Sayegh, J. et al. Identification of small molecule inhibitors of Jumonji AT-rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J. Biol. Chem. 288, 9408–9417 (2013).
Zhao, B. et al. Discovery of pyrazole derivatives as cellular active inhibitors of histone lysine-specific demethylase 5B (KDM5B/JARID1B). Eur. J. Med. Chem. 192, 112161 (2020).
Iida, T. et al. Design, synthesis, and biological evaluation of lysine demethylase 5 C degraders. ChemMedChem 16, 1609–1618 (2021).
Kruidenier, L. et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488, 404–408 (2012).
Montano, E. N. et al. α-Ketoglutarate-dependent KDM6 histone demethylases and interferon-stimulated gene expression in lupus. Arthritis Rheumatol. 76, 396–410 (2024).
Hung, P. H. et al. The histone demethylase inhibitor GSK-J4 is a therapeutic target for the kidney fibrosis of diabetic kidney disease via DKK1 modulation. Int. J. Mol. Sci. 23, 9407 (2022).
Zhang, W. et al. Therapeutically targeting head and neck squamous cell carcinoma through synergistic inhibition of LSD1 and JMJD3 by TCP and GSK-J1. Br. J. Cancer 122, 528–538 (2020).
Wang, J. J. et al. The JMJD3 histone demethylase inhibitor GSK-J1 ameliorates lipopolysaccharide-induced inflammation in a mastitis model. J. Biol. Chem. 298, 102017 (2022).
Cottone, L. et al. Inhibition of histone H3K27 demethylases inactivates brachyury (TBXT) and promotes chordoma cell death. Cancer Res. 80, 4540–4551 (2020).
Giordano, A. et al. Identification of the 2-benzoxazol-2-yl-phenol scaffold as new hit for JMJD3 inhibition. ACS Med. Chem. Lett. 10, 601–605 (2019).
Zhang, Y. et al. Screening of inhibitors against histone demethylation jumonji domain-containing protein 3 by capillary electrophoresis. J. Chromatogr. A 1613, 460625 (2020).
Jones, S. E. et al. Peptides derived from histone 3 and modified at position 18 inhibit histone demethylase KDM6 enzymes. Chembiochem 19, 1817–1822 (2018).
Shen, L. et al. Combination therapy and dual-target inhibitors based on LSD1: new emerging tools in cancer therapy. J. Med. Chem. 67, 922–951 (2024).
Wass, M. et al. A proof of concept phase I/II pilot trial of LSD1 inhibition by tranylcypromine combined with ATRA in refractory/relapsed AML patients not eligible for intensive therapy. Leukemia 35, 701–711 (2021).
Tayari, M. M. et al. Clinical responsiveness to all-trans retinoic acid is potentiated by LSD1 inhibition and associated with a quiescent transcriptome in myeloid malignancies. Clin. Cancer Res. 27, 1893–1903 (2021).
Dai, X. J. et al. Tranylcypromine based lysine-specific demethylase 1 inhibitor: summary and perspective. J. Med. Chem. 63, 14197–14215 (2020).
Salamero, O. et al. First-in-human phase I study of Iadademstat (ORY-1001): a first-in-class lysine-specific histone demethylase 1A inhibitor, in relapsed or refractory acute myeloid leukemia. J. Clin. Oncol. 38, 4260–4273 (2020).
Maes, T. et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell. 33, 495–511.e412 (2018).
Cuyàs, E. et al. The LSD1 inhibitor iadademstat (ORY-1001) targets SOX2-driven breast cancer stem cells: a potential epigenetic therapy in luminal-B and HER2-positive breast cancer subtypes. Aging 12, 4794–4814 (2020).
Antonijoan, R. M. et al. First-in-human randomized trial to assess safety, tolerability, pharmacokinetics and pharmacodynamics of the KDM1A inhibitor vafidemstat. CNS Drugs 35, 331–344 (2021).
Maes, T. et al. Modulation of KDM1A with vafidemstat rescues memory deficit and behavioral alterations. PLoS One 15, e0233468 (2020).
Benyoucef, A., Haigh, K., Cuddihy, A. & Haigh, J. J. JAK/BCL2 inhibition acts synergistically with LSD1 inhibitors to selectively target ETP-ALL. Leukemia 36, 2802–2816 (2022).
Bauer, T. M. et al. Phase I, open-label, dose-escalation study of the safety, pharmacokinetics, pharmacodynamics, and efficacy of GSK2879552 in relapsed/refractory SCLC. J. Thorac. Oncol. 14, 1828–1838 (2019).
Roboz, G. J. et al. Phase I trials of the lysine-specific demethylase 1 inhibitor, GSK2879552, as mono- and combination-therapy in relapsed/refractory acute myeloid leukemia or high-risk myelodysplastic syndromes. Leuk. Lymphoma 63, 463–467 (2022).
Dai, X. J. et al. Reversible lysine specific demethylase 1 (LSD1) inhibitors: a promising wrench to impair LSD1. J. Med. Chem. 64, 2466–2488 (2021).
Kanouni, T. et al. Discovery of CC-90011: a potent and selective reversible inhibitor of lysine specific demethylase 1 (LSD1). J. Med. Chem. 63, 14522–14529 (2020).
Soldi, R. et al. The novel reversible LSD1 inhibitor SP-2577 promotes anti-tumor immunity in SWItch/Sucrose-NonFermentable (SWI/SNF) complex mutated ovarian cancer. PLoS One 15, e0235705 (2020).
Granieri, L. et al. Targeting the USP7/RRM2 axis drives senescence and sensitizes melanoma cells to HDAC/LSD1 inhibitors. Cell. Rep. 40, 111396 (2022).
Baba, R. et al. LSD1 enzyme inhibitor TAK-418 unlocks aberrant epigenetic machinery and improves autism symptoms in neurodevelopmental disorder models. Sci Adv. 7, eaba1187 (2021).
Baba, R. et al. Investigating the therapeutic potential of LSD1 enzyme activity-specific inhibition by TAK-418 for social and memory deficits in Rodent Disease Models. ACS Chem. Neurosci. 13, 313–321 (2022).
Yin, W. et al. Safety, pharmacokinetics and pharmacodynamics of TAK-418, a novel inhibitor of the epigenetic modulator lysine-specific demethylase 1A. Br. J. Clin. Pharmacol. 87, 4756–4768 (2021).
Huang, X. et al. Chemical inhibitors targeting histone methylation readers. Pharmacol. Ther. 256, 108614 (2024).
Musselman, C. A., Lalonde, M. E., Côté, J. & Kutateladze, T. G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227 (2012).
Liu, S., Li, X., Li, X. & Li, X. D. Recent advances in the development of peptide-based inhibitors targeting epigenetic readers of histone lysine acetylation and methylation marks. Curr. Opin. Chem. Biol. 75, 102334 (2023).
Huang, Y. et al. Discovery of the clinical candidate MAK683: an EED-directed, allosteric, and selective PRC2 inhibitor for the treatment of advanced malignancies. J. Med. Chem. 65, 5317–5333 (2022).
Jiang, X. et al. The role of m6A modification in the biological functions and diseases. Signal. Transduct. Target. Ther. 6, 74 (2021).
Qi, Y. N. et al. Methyltransferase-like proteins in cancer biology and potential therapeutic targeting. J. Hematol. Oncol. 16, 89 (2023).
Bedi, R. K. et al. Small-molecule inhibitors of METTL3, the major human epitranscriptomic writer. ChemMedChem 15, 744–748 (2020).
Moroz-Omori, E. V. et al. METTL3 inhibitors for epitranscriptomic modulation of cellular processes. ChemMedChem 16, 3035–3043 (2021).
Dolbois, A. et al. 1,4,9-Triazaspiro[5.5]undecan-2-one derivatives as potent and selective METTL3 inhibitors. J. Med. Chem. 64, 12738–12760 (2021).
Wang, J. N. et al. Inhibition of METTL3 attenuates renal injury and inflammation by alleviating TAB3 m6A modifications via IGF2BP2-dependent mechanisms. Sci. Transl. Med. 14, eabk2709 (2022).
Zhou, X. et al. Inhibition of METTL3 alleviates NLRP3 inflammasome activation via increasing ubiquitination of NEK7. Adv. Sci. 11, e2308786 (2024).
Yankova, E. et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601 (2021).
Guirguis, A. A. et al. Inhibition of METTL3 results in a cell-intrinsic interferon response that enhances antitumor immunity. Cancer Discov. 13, 2228–2247 (2023).
Pomaville, M. et al. Small-molecule inhibition of the METTL3/METTL14 complex suppresses neuroblastoma tumor growth and promotes differentiation. Cell. Rep. 43, 114165 (2024).
Sun, Y. et al. METTL3 promotes chemoresistance in small cell lung cancer by inducing mitophagy. J. Exp. Clin. Cancer Res. 42, 65 (2023).
Xuan, Y. F. et al. The combination of methionine adenosyltransferase 2A inhibitor and methyltransferase like 3 inhibitor promotes apoptosis of non-small cell lung cancer cells and produces synergistic anti-tumor activity. Biochem. Biophys. Res. Commun. 716, 150011 (2024).
Ganguly, M., Gupta, R., Roychowdhury, A. & Hazra, D. De novo drug designing coupled with brute force screening and structure guided lead optimization gives highly specific inhibitor of METTL3: a potential cure for Acute Myeloid Leukaemia. J. Biomol. Struct. Dyn., 1–14 (2023).
Du, Y. et al. Discovery of METTL3 small molecule inhibitors by virtual screening of natural products. Front Pharmacol. 13, 878135 (2022).
Jiao, Y., Williams, A. & Wei, N. Quercetin ameliorated insulin resistance via regulating METTL3-mediated N6-methyladenosine modification of PRKD2 mRNA in skeletal muscle and C2C12 myocyte cell line. Nutr. Metab. Cardiovasc Dis. 32, 2655–2668 (2022).
Hu, J. et al. METTL3-dependent N6-methyladenosine modification is involved in berberine-mediated neuroprotection in ischemic stroke by enhancing the stability of NEAT1 in astrocytes. Aging 16, 299–321 (2024).
Cui, J., Wang, X., Dong, L. & Wang, Q. Curcumin reduces myocardial ischemia-reperfusion injury, by increasing endogenous H(2)S levels and further modulating m(6)A. Mol. Biol. Rep. 51, 558 (2024).
Yin, H. et al. Inhibition of METTL3 in macrophages provides protection against intestinal inflammation. Cell. Mol. Immunol. 21, 589–603 (2024).
Issahaku, A. R. et al. Multi-dimensional structural footprint identification for the design of potential scaffolds targeting METTL3 in cancer treatment from natural compounds. J. Mol. Model 29, 122 (2023).
Li, Z. et al. A stapled peptide inhibitor targeting the binding interface of N6-adenosine-methyltransferase subunits METTL3 and METTL14 for cancer therapy. Angew. Chem. Int. Ed. Engl. 63, e202402611 (2024).
Lee, J. H., Kim, S., Jin, M. S. & Kim, Y. C. Discovery of substituted indole derivatives as allosteric inhibitors of m(6) A-RNA methyltransferase, METTL3-14 complex. Drug. Dev. Res. 83, 783–799 (2022).
Lee, J. H. et al. Eltrombopag as an allosteric inhibitor of the METTL3-14 complex affecting the m(6)A methylation of RNA in acute myeloid leukemia cells. Pharmaceuticals. 15, 440 (2022).
Chen, C. J. et al. Metformin attenuates multiple myeloma cell proliferation and encourages apoptosis by suppressing METTL3-mediated m6A methylation of THRAP3, RBM25, and USP4. Cell. Cycle 22, 986–1004 (2023).
Cheng, L. et al. Metformin exhibits antiproliferation activity in breast cancer via miR-483-3p/METTL3/m(6)A/p21 pathway. Oncogenesis 10, 7 (2021).
Li, K. et al. Stimulation of let-7 maturation by metformin improved the response to tyrosine kinase inhibitor therapy in an m6A dependent manner. Front. Oncol. 11, 731561 (2021).
Zhang, Q. et al. Hypoxia-responsive PPARGC1A/BAMBI/ACSL5 axis promotes progression and resistance to lenvatinib in hepatocellular carcinoma. Oncogene 42, 1509–1523 (2023).
Xiang, Y. et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576 (2017).
Meng, M. et al. mTOR signaling promotes rapid m6A mRNA methylation to regulate NK-cell activation and effector functions. Immunol. Res. 12, 1039–1057 (2024).
Yang, X. et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol. Cancer 19, 46 (2020).
Gao, L., Lee, H., Goodman, J. H. & Ding, L. Hematopoietic stem cell niche generation and maintenance are distinguishable by an epitranscriptomic program. Cell 187, 2801–2816.e2817 (2024).
Selberg, S. et al. Discovery of small molecules that activate RNA methylation through cooperative binding to the METTL3-14-WTAP complex active site. Cell. Rep. 26, 3762–3771.e3765 (2019).
Lv, Y. et al. Melatonin attenuates chromium (VI)-induced spermatogonial stem cell/progenitor mitophagy by restoration of METTL3-mediated RNA N(6)-methyladenosine modification. Front Cell. Dev. Biol. 9, 684398 (2021).
Li, Y. et al. The effect mechanism of N6-adenosine methylation (m6A) in melatonin regulated LPS-induced colon inflammation. Int. J. Biol. Sci. 20, 2491–2506 (2024).
Li, Y. et al. FTO in cancer: functions, molecular mechanisms, and therapeutic implications. Trends Cancer 8, 598–614 (2022).
Qu, J. et al. RNA demethylase ALKBH5 in cancer: from mechanisms to therapeutic potential. J. Hematol. Oncol. 15, 8 (2022).
Chou, F. J., Liu, Y., Lang, F. & Yang, C. D-2-hydroxyglutarate in glioma biology. Cells. 10, 2345 (2021).
Sun, K. et al. Saikosaponin D exhibits anti-leukemic activity by targeting FTO/m(6)A signaling. Theranostics 11, 5831–5846 (2021).
Chen, B. et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J. Am. Chem. Soc. 134, 17963–17971 (2012).
Huang, Y. et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucl. Acids Res. 43, 373–384 (2015).
Chen, H., Jia, B., Zhang, Q. & Zhang, Y. Meclofenamic acid restores gefinitib sensitivity by downregulating breast cancer resistance protein and multidrug resistance protein 7 via FTO/m6A-demethylation/c-myc in non-small cell lung cancer. Front. Oncol. 12, 870636 (2022).
Wang, T. et al. Fluorescein derivatives as bifunctional molecules for the simultaneous inhibiting and labeling of FTO protein. J. Am. Chem. Soc. 137, 13736–13739 (2015).
Cao, K. et al. Glutathione-bioimprinted nanoparticles targeting of n6-methyladenosine FTO demethylase as a strategy against leukemic stem cells. Small 18, e2106558 (2022).
Liu, Z. et al. Structure-activity relationships and antileukemia effects of the tricyclic benzoic acid FTO inhibitors. J. Med. Chem. 65, 10638–10654 (2022).
Huang, Y. et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. 35, 677–691.e610 (2019).
Liu, Y. et al. Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance. Cell. Metab. 33, 1221–1233.e1211 (2021).
Xiao, P. et al. Rational design of RNA demethylase FTO inhibitors with enhanced antileukemia drug-like properties. J. Med. Chem. 66, 9731–9752 (2023).
Huff, S. et al. m(6)A-RNA demethylase FTO inhibitors impair self-renewal in glioblastoma stem cells. ACS Chem. Biol. 16, 324–333 (2021).
Huff, S. et al. Rational design and optimization of m(6)A-RNA demethylase FTO inhibitors as anticancer agents. J. Med. Chem. 65, 10920–10937 (2022).
Zhang, Y. et al. Identification of specific N(6)-methyladenosine RNA demethylase FTO inhibitors by single-quantum-dot-based FRET nanosensors. Anal. Chem. 92, 13936–13944 (2020).
Toh, J. D. W. et al. A strategy based on nucleotide specificity leads to a subfamily-selective and cell-active inhibitor of N(6)-methyladenosine demethylase FTO. Chem. Sci. 6, 112–122 (2015).
Zheng, G. et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem. Neurosci. 5, 658–665 (2014).
He, W. et al. Identification of a novel small-molecule binding site of the fat mass and obesity-associated protein (FTO). J. Med. Chem. 58, 7341–7348 (2015).
Qiao, Y. et al. A novel inhibitor of the obesity-related protein FTO. Biochemistry 55, 1516–1522 (2016).
Wang, R. et al. Identification of natural compound radicicol as a potent FTO inhibitor. Mol. Pharm. 15, 4092–4098 (2018).
Su, R. et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 38, 79–96.e11 (2020).
Wang, Y. et al. Identification of clausine E as an inhibitor of fat mass and obesity-associated protein (FTO) demethylase activity. J. Mol. Recognit. 32, e2800 (2019).
Qiao, Y. et al. Targeting FTO induces colorectal cancer ferroptotic cell death by decreasing SLC7A11/GPX4 expression. J. Exp. Clin. Cancer Res. 43, 108 (2024).
Peng, S. et al. Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Sci. Transl. Med. 11, eaau7116 (2019).
Xie, G. et al. A novel inhibitor of N (6)-methyladenosine demethylase FTO induces mRNA methylation and shows anti-cancer activities. Acta Pharmacol. Sin. B 12, 853–866 (2022).
Selberg, S. et al. Small-molecule inhibitors of the RNA M6A demethylases FTO potently support the survival of dopamine neurons. Int. J. Mol. Sci. 22, 4537 (2021).
Qin, B. et al. Discovery of novel mRNA demethylase FTO inhibitors against esophageal cancer. J. Enzym. Inhib. Med. Chem. 37, 1995–2003 (2022).
Wu, P. F. et al. Erasing m(6)A-dependent transcription signature of stress-sensitive genes triggers antidepressant actions. Neurobiol. Stress 15, 100390 (2021).
Chen, J. et al. Inhibition of ALKBH5 attenuates I/R-induced renal injury in male mice by promoting Ccl28 m6A modification and increasing Treg recruitment. Nat. Commun. 14, 1161 (2023).
Malacrida, A. et al. 3D proteome-wide scale screening and activity evaluation of a new ALKBH5 inhibitor in U87 glioblastoma cell line. Bioorg. Med. Chem. 28, 115300 (2020).
Takahashi, H. et al. Discovery of two novel ALKBH5 selective inhibitors that exhibit uncompetitive or competitive type and suppress the growth activity of glioblastoma multiforme. Chem. Biol. Drug. Des. 100, 1–12 (2022).
Wang, Y. Z. et al. Discovery of pyrazolo[1,5-a]pyrimidine derivative as a novel and selective ALKBH5 inhibitor for the treatment of AML. J. Med. Chem. 66, 15944–15959 (2023).
Selberg, S., Seli, N., Kankuri, E. & Karelson, M. Rational design of novel anticancer small-molecule RNA m6A demethylase ALKBH5 inhibitors. ACS Omega 6, 13310–13320 (2021).
Fang, Z. et al. Discovery of a potent, selective and cell active inhibitor of m(6)A demethylase ALKBH5. Eur. J. Med. Chem. 238, 114446 (2022).
Lai, G. Q. et al. A covalent compound selectively inhibits RNA demethylase ALKBH5 rather than FTO. RSC Chem. Biol. 5, 335–343 (2024).
Li, N. et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc. Natl. Acad. Sci. USA 117, 20159–20170 (2020).
Komal, S. et al. ALKBH5 inhibitors as a potential treatment strategy in heart failure-inferences from gene expression profiling. Front Cardiovasc. Med. 10, 1194311 (2023).
Huang, H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell. Biol. 20, 285–295 (2018).
Ramesh-Kumar, D. & Guil, S. The IGF2BP family of RNA binding proteins links epitranscriptomics to cancer. Semin Cancer Biol. 86, 18–31 (2022).
Mahapatra, L. et al. A Novel IMP1 inhibitor, BTYNB, targets c-Myc and inhibits melanoma and ovarian cancer cell proliferation. Transl. Oncol. 10, 818–827 (2017).
Wang, J. J. et al. Elevated expression of the RNA-binding protein IGF2BP1 enhances the mRNA stability of INHBA to promote the invasion and migration of esophageal squamous cancer cells. Exp. Hematol. Oncol. 12, 75 (2023).
Hagemann, S. et al. IGF2BP1 induces neuroblastoma via a druggable feedforward loop with MYCN promoting 17q oncogene expression. Mol. Cancer 22, 88 (2023).
Xiao, P. et al. IGF2BP1-mediated N6-methyladenosine modification promotes intrahepatic cholangiocarcinoma progression. Cancer Lett. 557, 216075 (2023).
Weng, H. et al. The m(6)A reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell. 40, 1566–1582.e1510 (2022).
Feng, P. et al. Inhibition of the m(6)A reader IGF2BP2 as a strategy against T-cell acute lymphoblastic leukemia. Leukemia 36, 2180–2188 (2022).
Liu, Y. et al. Allosteric Regulation of IGF2BP1 as a novel strategy for the activation of tumor immune microenvironment. ACS Cent. Sci. 8, 1102–1115 (2022).
Wallis, N. et al. Small molecule inhibitor of Igf2bp1 represses Kras and a pro-oncogenic phenotype in cancer cells. RNA Biol. 19, 26–43 (2022).
Cui, Y. et al. Isoliquiritigenin inhibits non-small cell lung cancer progression via m(6)A/IGF2BP3-dependent TWIST1 mRNA stabilization. Phytomedicine 104, 154299 (2022).
Wang, X. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014).
Liao, J. et al. Insight into the structure, physiological function, and role in cancer of m6A readers-YTH domain-containing proteins. Cell. Death Discov. 8, 137 (2022).
Wang, L. et al. YTHDF2 inhibition potentiates radiotherapy antitumor efficacy. Cancer Cell. 41, 1294–1308.e1298 (2023).
Bao, Y. et al. Targeting m(6)A reader YTHDF1 augments antitumour immunity and boosts anti-PD-1 efficacy in colorectal cancer. Gut 72, 1497–1509 (2023).
Zhou, L. et al. Hypoxia-induced lncRNA STEAP3-AS1 activates Wnt/β-catenin signaling to promote colorectal cancer progression by preventing m(6)A-mediated degradation of STEAP3 mRNA. Mol. Cancer 21, 168 (2022).
Cazzanelli, G. et al. Pliability in the m(6)A-binding region extends druggability of YTH domains. J. Chem. Inf. Model 64, 1682–1690 (2024).
Micaelli, M. et al. Small-molecule ebselen binds to YTHDF proteins interfering with the recognition of N (6)-methyladenosine-modified RNAs. ACS Pharmacol. Transl. Sci. 5, 872–891 (2022).
Chen, Y. et al. O-GlcNAcylation determines the translational regulation and phase separation of YTHDF proteins. Nat. Cell. Biol. 25, 1676–1690 (2023).
Yang, Y. et al. O-GlcNAcylation of YTHDF2 promotes HBV-related hepatocellular carcinoma progression in an N(6)-methyladenosine-dependent manner. Signal. Transduct. Target. Ther. 8, 63 (2023).
Dutta, P. et al. Global epigenetic changes induced by SWI2/SNF2 inhibitors characterize neomycin-resistant mammalian cells. PLoS One 7, e49822 (2012).
Dreier, M. R., Walia, J. & de la Serna, I. L. Targeting SWI/SNF complexes in cancer: pharmacological approaches and implications. Epigenome 8, 7 (2024).
Xue, Y. et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat. Commun. 10, 557 (2019).
Xue, Y. et al. SMARCA4/2 loss inhibits chemotherapy-induced apoptosis by restricting IP3R3-mediated Ca(2+) flux to mitochondria. Nat. Commun. 12, 5404 (2021).
Papillon, J. P. N. et al. Discovery of orally active inhibitors of brahma homolog (BRM)/SMARCA2 ATPase activity for the treatment of brahma related gene 1 (BRG1)/SMARCA4-mutant cancers. J. Med. Chem. 61, 10155–10172 (2018).
Rago, F. et al. Exquisite sensitivity to dual BRG1/BRM ATPase inhibitors reveals broad SWI/SNF dependencies in acute myeloid leukemia. Mol. Cancer Res. 20, 361–372 (2022).
Panditharatna, E. et al. BAF complex maintains glioma stem cells in pediatric H3K27M glioma. Cancer Discov. 12, 2880–2905 (2022).
Fiskus, W. et al. BRG1/BRM inhibitor targets AML stem cells and exerts superior preclinical efficacy combined with BET or menin inhibitor. Blood 143, 2059–2072 (2024).
Charlop-Powers, Z., Zeng, L., Zhang, Q. & Zhou, M. M. Structural insights into selective histone H3 recognition by the human polybromo bromodomain 2. Cell. Res. 20, 529–538 (2010).
Gerstenberger, B. S. et al. Identification of a chemical probe for family VIII bromodomains through optimization of a fragment hit. J. Med. Chem. 59, 4800–4811 (2016).
Fedorov, O. et al. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci. Adv. 1, e1500723 (2015).
He, Y. et al. Novel structural-related analogs of PFI-3 (SRAPs) that target the BRG1 catalytic subunit of the SWI/SNF complex increase the activity of temozolomide in glioblastoma cells. Bioorg Med. Chem. 53, 116533 (2022).
Taylor, A. M. et al. GNE-064: a potent, selective, and orally bioavailable chemical probe for the bromodomains of SMARCA2 and SMARCA4 and the fifth bromodomain of PBRM1. J. Med. Chem. 65, 11177–11186 (2022).
Sutherell, C. L. et al. Identification and development of 2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one inhibitors targeting bromodomains within the switch/sucrose nonfermenting complex. J. Med. Chem. 59, 5095–5101 (2016).
Mélin, L. et al. Design and synthesis of LM146, a potent inhibitor of PB1 with an improved selectivity profile over SMARCA2. ACS Omega 6, 21327–21338 (2021).
Shishodia, S. et al. Selective and cell-active PBRM1 bromodomain inhibitors discovered through NMR fragment screening. J. Med. Chem. 65, 13714–13735 (2022).
Cochran, A. G. & Flynn, M. GNE-235: a lead compound selective for the second bromodomain of PBRM1. J. Med. Chem. 66, 13116–13134 (2023).
Wu, X. et al. Ablation of Brg1 in fibroblast/myofibroblast lineages attenuates renal fibrosis in mice with diabetic nephropathy. Life Sci. 344, 122578 (2024).
Hong, W. et al. The chromatin remodeling protein BRG1 mediates Ang II induced pro-fibrogenic response in renal fibroblasts. Life Sci. 340, 122320 (2024).
Li, N. et al. The chromatin remodeling protein BRG1 contributes to liver ischemia-reperfusion injury by regulating NOXA expression. Life Sci. 334, 122235 (2023).
Jolly, A. J. et al. Redistribution of the chromatin remodeler Brg1 directs smooth muscle-derived adventitial progenitor-to-myofibroblast differentiation and vascular fibrosis. JCI Insight 8, e164862 (2023).
Li, N. et al. Targetable Brg1-CXCL14 axis contributes to alcoholic liver injury by driving neutrophil trafficking. EMBO Mol. Med. 15, e16592 (2023).
Martin, L. J. et al. Structure-based design of an in vivo active selective BRD9 inhibitor. J. Med. Chem. 59, 4462–4475 (2016).
Theodoulou, N. H. et al. Discovery of I-BRD9, a selective cell active chemical probe for bromodomain containing protein 9 inhibition. J. Med. Chem. 59, 1425–1439 (2016).
Basuroy, T. et al. Epigenetic and pharmacological control of pigmentation via bromodomain protein 9 (BRD9). Pigment Cell. Melanoma Res. 36, 19–32 (2023).
Crawford, T. D. et al. Inhibition of bromodomain-containing protein 9 for the prevention of epigenetically-defined drug resistance. Bioorg. Med. Chem. Lett. 27, 3534–3541 (2017).
Ali, M. M. et al. Identification of selective BRD9 inhibitor via integrated computational approach. Int. J. Mol. Sci. 23, 13513 (2022).
Ordonez-Rubiano, S. C. et al. Rational design and development of selective BRD7 bromodomain inhibitors and their activity in prostate cancer. J. Med. Chem. 66, 11250–11270 (2023).
Clark, P. G. et al. LP99: discovery and synthesis of the first selective BRD7/9 bromodomain inhibitor. Angew. Chem. Int. Ed. Engl. 54, 6217–6221 (2015).
Mason, L. D., Chava, S., Reddi, K. K. & Gupta, R. The BRD9/7 inhibitor TP-472 blocks melanoma tumor growth by suppressing ECM-mediated oncogenic signaling and inducing apoptosis. Cancers 13, 5516 (2021).
Hügle, M. et al. 4-acyl pyrroles as dual BET-BRD7/9 bromodomain inhibitors address BETi insensitive human cancer cell lines. J. Med. Chem. 63, 15603–15620 (2020).
Clegg, M. A. et al. Application of atypical acetyl-lysine methyl mimetics in the development of selective inhibitors of the bromodomain-containing protein 7 (BRD7)/bromodomain-containing protein 9 (BRD9) bromodomains. J. Med. Chem. 63, 5816–5840 (2020).
He, T. et al. Development of an orally bioavailable mSWI/SNF ATPase degrader and acquired mechanisms of resistance in prostate cancer. Proc. Natl. Acad. Sci. USA 121, e2322563121 (2024).
Xiao, L. et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature 601, 434–439 (2022).
Remillard, D. et al. Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew. Chem. Int. Ed. Engl. 56, 5738–5743 (2017).
Bouguenina, H. et al. A degron blocking strategy towards improved CRL4(CRBN) recruiting PROTAC selectivity. Chembiochem 24, e202300351 (2023).
Kurata, K. et al. BRD9 degradation disrupts ribosome biogenesis in multiple myeloma. Clin. Cancer Res. 29, 1807–1821 (2023).
Ahmed, N. S. et al. BRD9 regulates interferon-stimulated genes during macrophage activation via cooperation with BET protein BRD4. Proc. Natl. Acad. Sci. USA 119, e2110812119 (2022).
Cantley, J. et al. Selective PROTAC-mediated degradation of SMARCA2 is efficacious in SMARCA4 mutant cancers. Nat. Commun. 13, 6814 (2022).
Zoppi, V. et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel-lindau (VHL) based dual degrader probe of BRD9 and BRD7. J. Med. Chem. 62, 699–726 (2019).
Hescheler, D. A. et al. Targeted therapy for adrenocortical carcinoma: a genomic-based search for available and emerging options. Cancers 14, 2721 (2022).
Alahdal, M. & Elkord, E. Non-coding RNAs in cancer immunotherapy: predictive biomarkers and targets. Clin. Transl. Med. 13, e1425 (2023).
Jadhav, V., Vaishnaw, A., Fitzgerald, K. & Maier, M. A. RNA interference in the era of nucleic acid therapeutics. Nat. Biotechnol. 42, 394–405 (2024).
Adams, B. D. et al. Targeting noncoding RNAs in disease. J. Clin. Investig. 127, 761–771 (2017).
Mahato, R. K. et al. Targeting long non-coding RNAs in cancer therapy using CRISPR-Cas9 technology: a novel paradigm for precision oncology. J. Biotechnol. 379, 98–119 (2024).
Wei, C. et al. Role of long non-coding RNAs in cancer: from subcellular localization to nanoparticle-mediated targeted regulation. Mol. Ther. Nucl. Acids 33, 774–793 (2023).
Crooke, S. T., Vickers, T. A. & Liang, X. H. Phosphorothioate modified oligonucleotide-protein interactions. Nucl. Acids Res. 48, 5235–5253 (2020).
Sergeeva, O. et al. Structure-activity relationship study of mesyl and busyl phosphoramidate antisense oligonucleotides for unaided and PSMA-mediated uptake into prostate cancer cells. Front. Chem. 12, 1342178 (2024).
Egli, M. & Manoharan, M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 51, 2529–2573 (2023).
Kamali, M. J. et al. Locked nucleic acid (LNA): a modern approach to cancer diagnosis and treatment. Exp. Cell. Res. 423, 113442 (2023).
Anastasiadou, E. et al. Cobomarsen, an oligonucleotide inhibitor of miR-155, slows DLBCL tumor cell growth in vitro and in vivo. Clin. Cancer Res. 27, 1139–1149 (2021).
Cheng, M., Zain, J., Rosen, S. T. & Querfeld, C. Emerging drugs for the treatment of cutaneous T-cell lymphoma. Expert Opin. Emerg. Drugs 27, 45–54 (2022).
Seto, A. G. et al. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 183, 428–444 (2018).
Witten, L. & Slack, F. J. miR-155 as a novel clinical target for hematological malignancies. Carcinogenesis 41, 2–7 (2020).
Kim, S. et al. Targeted eicosanoids profiling reveals a prostaglandin reprogramming in breast Cancer by microRNA-155. J. Exp. Clin. Cancer Res. 40, 43 (2021).
Ozpolat, B., Sood, A. K. & Lopez-Berestein, G. Nanomedicine based approaches for the delivery of siRNA in cancer. J. Intern. Med. 267, 44–53 (2010).
Chen, L. L. Linking long noncoding RNA localization and function. Trends Biochem. Sci. 41, 761–772 (2016).
Guo, C. J., Xu, G. & Chen, L. L. Mechanisms of long noncoding RNA nuclear retention. Trends Biochem. Sci. 45, 947–960 (2020).
Gong, N., Teng, X., Li, J. & Liang, X. J. Antisense oligonucleotide-conjugated nanostructure-targeting lncRNA MALAT1 inhibits cancer metastasis. ACS Appl. Mater. Interfaces 11, 37–42 (2019).
Xu, R. et al. Remodeling of mitochondrial metabolism by a mitochondria-targeted RNAi nanoplatform for effective cancer therapy. Small 20, e2305923 (2024).
Shi, Y. et al. Pharmaceutical strategies for endoplasmic reticulum-targeting and their prospects of application. J. Control Release 329, 337–352 (2021).
Zhang, M. et al. Potential therapies and diagnosis based on Golgi-targeted nano drug delivery systems. Pharmacol. Res. 175, 105861 (2022).
Van de Vyver, T., De Smedt, S. C. & Raemdonck, K. Modulating intracellular pathways to improve non-viral delivery of RNA therapeutics. Adv. Drug. Deliv. Rev. 181, 114041 (2022).
Xun, G. et al. Harnessing noncanonical crRNA for highly efficient genome editing. Nat. Commun. 15, 3823 (2024).
Wang, L. et al. CRISPR-Cas13d screens identify KILR, a breast cancer risk-associated lncRNA that regulates DNA replication and repair. Mol. Cancer 23, 101 (2024).
Li, S., Wu, H. & Chen, L. L. Screening circular RNAs with functional potential using the RfxCas13d/BSJ-gRNA system. Nat. Protoc. 17, 2085–2107 (2022).
Hussen, B. M. et al. Targeting miRNA by CRISPR/Cas in cancer: advantages and challenges. Mil. Med. Res. 10, 32 (2023).
Capelletti, S., García Soto, S. C. & Gonçalves, M. On RNA-programmable gene modulation as a versatile set of principles targeting muscular dystrophies. Mol. Ther. S1525–0016 (2024).
Narayanan, A. et al. In vivo mutagenesis of miRNA gene families using a scalable multiplexed CRISPR/Cas9 nuclease system. Sci. Rep. 6, 32386 (2016).
Prinz, F. et al. MicroRNA mimics can distort physiological microRNA effects on immune checkpoints by triggering an antiviral interferon response. RNA Biol. 19, 1305–1315 (2022).
Hong, D. S. et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 122, 1630–1637 (2020).
Feng, X. & He, C. Mammalian DNA N(6)-methyladenosine: challenges and new insights. Mol. Cell. 83, 343–351 (2023).
Xie, L. et al. Emerging roles for DNA 6mA and RNA m6A methylation in mammalian genome. Int. J. Mol. Sci. 24, 13897 (2023).
Wiener, D. & Schwartz, S. The epitranscriptome beyond m(6)A. Nat. Rev. Genet. 22, 119–131 (2021).
Chen, P. C. et al. Leveraging a phage-encoded noncanonical amino acid: a novel pathway to potent and selective epigenetic reader protein inhibitors. ACS Cent. Sci. 10, 782–792 (2024).
Tomassi, S. et al. Polycomb repressive complex 2 modulation through the development of EZH2-EED interaction inhibitors and EED binders. J. Med. Chem. 64, 11774–11797 (2021).
Li, X. & Song, Y. Structure, function and inhibition of critical protein-protein interactions involving mixed lineage leukemia 1 and its fusion oncoproteins. J. Hematol. Oncol. 14, 56 (2021).
Dong, G. et al. Structure-based design of the indole-substituted triazolopyrimidines as new EED-H3K27me3 inhibitors for the treatment of lymphoma. J. Med. Chem. 66, 1063–1081 (2023).
Perfetti, M. T. et al. Identification of a fragment-like small molecule ligand for the methyl-lysine binding protein, 53BP1. ACS Chem. Biol. 10, 1072–1081 (2015).
Dang, F. & Wei, W. Targeting the acetylation signaling pathway in cancer therapy. Semin Cancer Biol. 85, 209–218 (2022).
Hanquier, J. N. et al. Identification of nonhistone substrates of the lysine methyltransferase PRDM9. J. Biol. Chem. 299, 104651 (2023).
Shu, S. et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529, 413–417 (2016).
Zhang, Y. et al. Elevating PLK1 overcomes BETi resistance in prostate cancer via triggering BRD4 phosphorylation-dependent degradation in mitosis. Cell. Rep. 43, 114431 (2024).
Paolini, R. L. & Souroullas, G. P. The cell cycle: a key to unlock EZH2-targeted therapy resistance. Cancer Discov. 14, 903–905 (2024).
Fong, C. Y. et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 525, 538–542 (2015).
Rathert, P. et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547 (2015).
Zhang, P. et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat. Med. 23, 1055–1062 (2017).
Hao, B. B. et al. Proteomics analysis of histone deacetylase inhibitor-resistant solid tumors reveals resistant signatures and potential drug combinations. Acta Pharmacol. Sin. 45, 1305–1315 (2024).
Mills, G. B. & Labrie, M. Enhancing anticancer activity of macrophages through rational drug combinations. J. Clin. Investig. 134, e180512 (2024).
Qiao, J. et al. Macrophages confer resistance to BET inhibition in triple-negative breast cancer by upregulating IKBKE. Biochem. Pharmacol. 180, 114126 (2020).
Kazansky, Y. et al. Overcoming clinical resistance to EZH2 inhibition using rational epigenetic combination therapy. Cancer Discov. 14, 965–981 (2024).
Intlekofer, A. M. et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 559, 125–129 (2018).
Wang, F. et al. Leukemia stemness and co-occurring mutations drive resistance to IDH inhibitors in acute myeloid leukemia. Nat. Commun. 12, 2607 (2021).
Yuan, Z. et al. Design, synthesis and anticancer potential of NSC-319745 hydroxamic acid derivatives as DNMT and HDAC inhibitors. Eur. J. Med. Chem. 134, 281–292 (2017).
Ocaña-Paredes, B. et al. The pharmacoepigenetic paradigm in cancer treatment. Front. Pharmacol. 15, 1381168 (2024).
Peng, X. et al. Overview of epigenetic degraders based on PROTAC, molecular glue, and hydrophobic tagging technologies. Acta Pharmacol. Sin. B 14, 533–578 (2024).
Hogg, S. J., Beavis, P. A., Dawson, M. A. & Johnstone, R. W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug. Discov. 19, 776–800 (2020).
Bonaventura, P. et al. Cold tumors: a therapeutic challenge for immunotherapy. Front. Immunol. 10, 168 (2019).
Qian, X., Hu, W. & Yan, J. Nano-chemotherapy synergize with immune checkpoint inhibitor- a better option? Front. Immunol. 13, 963533 (2022).
Kong, Y., Yu, J., Ge, S. & Fan, X. Novel insight into RNA modifications in tumor immunity: promising targets to prevent tumor immune escape. Innovation 4, 100452 (2023).
Wang, D. X. et al. Mutation status of the KMT2 family associated with immune checkpoint inhibitors (ICIs) therapy and implicating diverse tumor microenvironments. Mol. Cancer 23, 15 (2024).
Jiang, Z. et al. A combination of a TLR7/8 agonist and an epigenetic inhibitor suppresses triple-negative breast cancer through triggering anti-tumor immune. J. Nanobiotechnol. 22, 296 (2024).
Bauer, K. et al. BRD4 degraders may effectively counteract therapeutic resistance of leukemic stem cells in AML and ALL. Am. J. Hematol. 99, 1721-1731(2024).
Zhang, J. et al. Research progress of long non-coding RNA in tumor drug resistance: a new paradigm. Drug. Des. Dev. Ther. 18, 1385–1398 (2024).
Solta, A. et al. Entinostat enhances the efficacy of chemotherapy in small cell lung cancer through s-phase arrest and decreased base excision repair. Clin. Cancer Res. 29, 4644–4659 (2023).
Cervena, K. et al. Methylation-based therapies for colorectal cancer. Cells 9, 1540 (2020).
Manengu, C. et al. HDAC inhibitors as a potential therapy for chemotherapy-induced neuropathic pain. Inflammopharmacology 32, 2153–2175(2024).
He, Y. et al. Inhibiting DNA methylation alleviates cisplatin-induced hearing loss by decreasing oxidative stress-induced mitochondria-dependent apoptosis via the LRP1-PI3K/AKT pathway. Acta Pharmacol. Sin. B 12, 1305–1321 (2022).
Clozel, T. et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov. 3, 1002–1019 (2013).
Zhong, L. et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal. Transduct. Target. Ther. 6, 201 (2021).
Parag-Sharma, K. et al. Synergistic efficacy of combined EGFR and HDAC inhibitors overcomes tolerance to EGFR monotherapy in salivary mucoepidermoid carcinoma. Oral. Oncol. 115, 105166 (2021).
Wang, J. H. et al. A novel small-molecule antagonist enhances the sensitivity of osteosarcoma to cabozantinib in vitro and in vivo by targeting DNMT-1 correlated with disease severity in human patients. Pharmacol. Res. 173, 105869 (2021).
Bass, A. K. A. et al. Comprehensive review for anticancer hybridized multitargeting HDAC inhibitors. Eur. J. Med. Chem. 209, 112904 (2021).
Heath, H. et al. Targeting systemic and gut microbial metabolism in ER(+) breast cancer. Trends Endocrinol. Metab. 35, 321–330 (2024).
Llinas-Bertran, A., Bellet-Ezquerra, M. & Seoane, J. A. Epigenetic control of cancer cell dormancy and awakening in endocrine therapy resistance. Cancer Discov. 14, 704–706 (2024).
Culig, Z. & Puhr, M. Androgen receptor-interacting proteins in prostate cancer development and therapy resistance. Am. J. Pathol. 194, 324–334 (2024).
Fischetti, I. et al. Combined therapy targeting AR and EZH2 curbs castration-resistant prostate cancer enhancing anti-tumor T-cell response. Epigenomics 16, 653–670 (2024).
Manda, S. et al. A phase 3b study of venetoclax and azacitidine or decitabine in an outpatient setting in patients with acute myeloid leukemia. Hematol. Oncol. 42, e3274 (2024).
Bazinet, A. et al. Oral decitabine and cedazuridine plus venetoclax for older or unfit patients with acute myeloid leukaemia: a phase 2 study. Lancet Haematol. 11, e276–e286 (2024).
Bataller, A. et al. Oral decitabine plus cedazuridine and venetoclax in patients with higher-risk myelodysplastic syndromes or chronic myelomonocytic leukaemia: a single-centre, phase 1/2 study. Lancet Haematol. 11, e186–e195 (2024).
Lachowiez, C. A. et al. A phase Ib/II study of ivosidenib with venetoclax ± azacitidine in IDH1-mutated myeloid malignancies. Blood Cancer Discov. 4, 276–293 (2023).
Pereira, M. P., Herrity, E. & Kim, D. D. H. TP53-mutated acute myeloid leukemia and myelodysplastic syndrome: biology, treatment challenges, and upcoming approaches. Ann. Hematol. 103, 1049–1067 (2024).
DiNardo, K. W., LeBlanc, T. W. & Chen, H. Novel agents and regimens in acute myeloid leukemia: latest updates from 2022 ASH annual meeting. J. Hematol. Oncol. 16, 17 (2023).
Pommert, L. et al. Decitabine and vorinostat with FLAG chemotherapy in pediatric relapsed/refractory AML: Report from the therapeutic advances in childhood leukemia and lymphoma (TACL) consortium. Am. J. Hematol. 97, 613–622 (2022).
Croucher, P. J. P. et al. Spliceosome mutations are associated with clinical response in a phase 1b/2 study of the PLK1 inhibitor onvansertib in combination with decitabine in relapsed or refractory acute myeloid leukemia. Ann. Hematol. 102, 3049–3059 (2023).
Zeidan, A. M. et al. Sabatolimab plus hypomethylating agents in previously untreated patients with higher-risk myelodysplastic syndromes (STIMULUS-MDS1): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Haematol. 11, e38–e50 (2024).
Saxena, K. et al. A phase 1b/2 study of azacitidine with PD-L1 antibody avelumab in relapsed/refractory acute myeloid leukemia. Cancer 127, 3761–3771 (2021).
Ruan, J. et al. Multicenter phase 2 study of oral azacitidine (CC-486) plus CHOP as initial treatment for PTCL. Blood 141, 2194–2205 (2023).
Martin, P. et al. Phase 1 study of oral azacitidine (CC-486) plus R-CHOP in previously untreated intermediate- to high-risk DLBCL. Blood 139, 1147–1159 (2022).
Short, N. J. & Kantarjian, H. Hypomethylating agents for the treatment of myelodysplastic syndromes and acute myeloid leukemia: past discoveries and future directions. Am. J. Hematol. 97, 1616–1626 (2022).
Patnaik, M. M. & Tefferi, A. Chronic myelomonocytic leukemia: 2024 update on diagnosis, risk stratification and management. Am. J. Hematol. 99, 1142–1165 (2024).
Mayerhofer, C., Niemeyer, C. M. & Flotho, C. Current treatment of juvenile myelomonocytic leukemia. J. Clin. Med. 10, 3084 (2021).
Dai, X. J. et al. Degraders in epigenetic therapy: PROTACs and beyond. Theranostics 14, 1464–1499 (2024).
Sakamoto, K. M. et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 98, 8554–8559 (2001).
Sinatra, L. et al. Solid-phase synthesis of cereblon-recruiting selective histone deacetylase 6 degraders (HDAC6 PROTACs) with antileukemic activity. J. Med. Chem. 65, 16860–16878 (2022).
Min, J. et al. Phenyl-glutarimides: alternative cereblon binders for the design of PROTACs. Angew. Chem. Int. Ed. Engl. 60, 26663–26670 (2021).
Winter, G. E. et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015).
Maneiro, M. A. et al. Antibody-PROTAC conjugates enable HER2-dependent targeted protein degradation of BRD4. ACS Chem. Biol. 15, 1306–1312 (2020).
Dragovich, P. S. et al. Antibody-mediated delivery of chimeric brd4 degraders. part 1: exploration of antibody linker, payload loading, and payload molecular properties. J. Med. Chem. 64, 2534–2575 (2021).
Pfaff, P., Samarasinghe, K. T. G., Crews, C. M. & Carreira, E. M. Reversible spatiotemporal control of induced protein degradation by bistable photoPROTACs. ACS Cent. Sci. 5, 1682–1690 (2019).
Xue, G. et al. Light-induced protein degradation with photocaged PROTACs. J. Am. Chem. Soc. 141, 18370–18374 (2019).
Li, X. et al. Chemical proteomic profiling of bromodomains enables the wide-spectrum evaluation of bromodomain inhibitors in living cells. J. Am. Chem. Soc. 141, 11497–11505 (2019).
Yang, C. et al. Radiotherapy-triggered proteolysis targeting chimera prodrug activation in tumors. J. Am. Chem. Soc. 145, 385–391 (2023).
Takahashi, D. et al. AUTACs: cargo-specific degraders using selective autophagy. Mol. Cell. 76, 797–810.e710 (2019).
Ding, Y., Xing, D., Fei, Y. & Lu, B. Emerging degrader technologies engaging lysosomal pathways. Chem. Soc. Rev. 51, 8832–8876 (2022).
Pei, J. et al. Developing potent LC3-targeting AUTAC tools for protein degradation with selective autophagy. Chem. Commun. 57, 13194–13197 (2021).
Xie, S. et al. Small-molecule hydrophobic tagging: a promising strategy of druglike technology for targeted protein degradation. J. Med. Chem. 66, 10917–10933 (2023).
Ma, A. et al. Discovery of a first-in-class EZH2 selective degrader. Nat. Chem. Biol. 16, 214–222 (2020).
Schiedel, M. et al. Halotag-targeted sirtuin-rearranging ligand (sirreal) for the development of proteolysis-targeting chimeras (PROTACs) against the lysine deacetylase sirtuin 2 (Sirt2)*. Chembiochem 21, 3371–3376 (2020).
Huang, M. et al. A 18β-glycyrrhetinic acid conjugate with Vorinostat degrades HDAC3 and HDAC6 with improved antitumor effects. Eur. J. Med. Chem. 188, 111991 (2020).
Li, X., Yao, Y., Wu, F. & Song, Y. A proteolysis-targeting chimera molecule selectively degrades ENL and inhibits malignant gene expression and tumor growth. J. Hematol. Oncol. 15, 41 (2022).
Xie, S. et al. Discovery of norbornene as a novel hydrophobic tag applied in protein degradation. Angew. Chem. Int. Ed. Engl. 62, e202217246 (2023).
Domostegui, A., Nieto-Barrado, L., Perez-Lopez, C. & Mayor-Ruiz, C. Chasing molecular glue degraders: screening approaches. Chem. Soc. Rev. 51, 5498–5517 (2022).
Kozicka, Z. & Thomä, N. H. Haven’t got a glue: protein surface variation for the design of molecular glue degraders. Cell. Chem. Biol. 28, 1032–1047 (2021).
Toriki, E. S. et al. Rational chemical design of molecular glue degraders. ACS Cent. Sci. 9, 915–926 (2023).
Cavalli, G. & Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 571, 489–499 (2019).
Schwartzman, O. & Tanay, A. Single-cell epigenomics: techniques and emerging applications. Nat. Rev. Genet. 16, 716–726, (2015).
Hawkins, R. D., Hon, G. C. & Ren, B. Next-generation genomics: an integrative approach. Nat. Rev. Genet. 11, 476–486, (2010).
Cusanovich, D. A. et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 174, 1309–1324.e1318 (2018).
Preissl, S., Gaulton, K. J. & Ren, B. Characterizing cis-regulatory elements using single-cell epigenomics. Nat. Rev. Genet 24, 21–43 (2023).
Kelsey, G., Stegle, O. & Reik, W. Single-cell epigenomics: recording the past and predicting the future. Science 358, 69–75 (2017).
Gaulton, K. J., Preissl, S. & Ren, B. Interpreting non-coding disease-associated human variants using single-cell epigenomics. Nat. Rev. Genet. 24, 516–534 (2023).
Shapiro, E., Biezuner, T. & Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 14, 618–630 (2013).
Liu, H. et al. Epigenomic and transcriptomic analyses define core cell types, genes and targetable mechanisms for kidney disease. Nat. Genet. 54, 950–962 (2022).
Ren, B. et al. High-resolution Hi-C maps highlight multiscale 3D epigenome reprogramming during pancreatic cancer metastasis. J. Hematol. Oncol. 14, 120 (2021).
Stricker, S. H., Köferle, A. & Beck, S. From profiles to function in epigenomics. Nat. Rev. Genet. 18, 51–66 (2017).
Thakore, P. I., Black, J. B., Hilton, I. B. & Gersbach, C. A. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat. Methods 13, 127–137 (2016).
Wright, S. et al. Interrogating bromodomain inhibitor resistance in KMT2A-rearranged leukemia through combinatorial CRISPR screens. Proc. Natl. Acad. Sci. USA 120, e2220134120 (2023).
Ruan, P. et al. NEpiC: a network-assisted algorithm for epigenetic studies using mean and variance combined signals. Nucl. Acids Res. 44, e134 (2016).
Liu, Q. et al. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun. 10, 2449 (2019).
Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015).
Nagano, T. et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013).
Shao, D. et al. PEGR: a flexible management platform for reproducible epigenomic and genomic research. Genome Biol. 23, 99 (2022).
Du, Q., Wang, Z. & Schramm, V. L. Human DNMT1 transition state structure. Proc. Natl. Acad. Sci. USA 113, 2916–2921 (2016).
Huang, Z. X. et al. Position 34 of tRNA is a discriminative element for m5C38 modification by human DNMT2. Nucl. Acids Res. 49, 13045–13061 (2021).
Yang, L., Rau, R. & Goodell, M. A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 15, 152–165 (2015).
Yu, J. et al. DNA methyltransferases: emerging targets for the discovery of inhibitors as potent anticancer drugs. Drug. Discov. Today 24, 2323–2331 (2019).
Xu, T. H. et al. Structure of nucleosome-bound DNA methyltransferases DNMT3A and DNMT3B. Nature 586, 151–155 (2020).
Suetake, I. et al. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J. Biol. Chem. 279, 27816–27823 (2004).
Liu, W., Wu, G., Xiong, F. & Chen, Y. Advances in the DNA methylation hydroxylase TET1. Biomark. Res. 9, 76 (2021).
Ko, M. et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 497, 122–126 (2013).
Gu, T. P. et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610 (2011).
Feng, Q. & Zhang, Y. The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes. Dev. 15, 827–832 (2001).
Adkins, N. L. & Georgel, P. T. MeCP2: structure and function. Biochem. Cell. Biol. 89, 1–11 (2011).
Ichimura, T. et al. Transcriptional repression and heterochromatin formation by MBD1 and MCAF/AM family proteins. J. Biol. Chem. 280, 13928–13935 (2005).
Klose, R. J. & Bird, A. P. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97 (2006).
Ng, H. H. et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet. 23, 58–61 (1999).
Yildirim, O. et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147, 1498–1510 (2011).
Le Guezennec, X. et al. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol. Cell. Biol. 26, 843–851 (2006).
Busque, L. & Godley, L. A. MBD4: guardian of the epigenetic galaxy. Blood 132, 1468–1469 (2018).
Zhang, J. et al. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell. Res. 21, 1723–1739 (2011).
Zhou, T. et al. Structural basis for hydroxymethylcytosine recognition by the SRA domain of UHRF2. Mol. Cell. 54, 879–886 (2014).
Liu, X. et al. UHRF2 commissions the completion of DNA demethylation through allosteric activation by 5hmC and K33-linked ubiquitination of XRCC1. Mol. Cell. 81, 2960–2974.e2967 (2021).
Sterner, D. E. & Berger, S. L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64, 435–459 (2000).
Ortega, M. A. et al. Understanding HAT1: a comprehensive review of noncanonical roles and connection with disease. Genes. 14, 915 (2023).
Poziello, A. et al. Recent insights into Histone Acetyltransferase-1: biological function and involvement in pathogenesis. Epigenetics 16, 838–850 (2021).
González-Medina, A., Hidalgo, E. & Ayté, J. Gcn5-mediated acetylation at MBF-regulated promoters induces the G1/S transcriptional wave. Nucl. Acids Res. 47, 8439–8451 (2019).
Ogryzko, V. V. et al. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87, 953–959 (1996).
Jin, Q. et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262 (2011).
Hess, L. et al. A toolbox for class I HDACs reveals isoform specific roles in gene regulation and protein acetylation. PLoS Genet. 18, e1010376 (2022).
Li, J. et al. Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat. Med. 18, 783–790 (2012).
Yang, C., Croteau, S. & Hardy, P. Histone deacetylase (HDAC) 9: versatile biological functions and emerging roles in human cancer. Cell. Oncol. 44, 997–1017 (2021).
Vaquero, A. et al. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell. 16, 93–105 (2004).
Bosch-Presegué, L. et al. Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensures genome protection. Mol. Cell. 42, 210–223 (2011).
Serrano, L. et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes. Dev. 27, 639–653 (2013).
Vempati, R. K. et al. p300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J. Biol. Chem. 285, 28553–28564 (2010).
Gil, R., Barth, S., Kanfi, Y. & Cohen, H. Y. SIRT6 exhibits nucleosome-dependent deacetylase activity. Nucl. Acids Res. 41, 8537–8545 (2013).
Tasselli, L. et al. SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat. Struct. Mol. Biol. 23, 434–440 (2016).
Barber, M. F. et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 487, 114–118 (2012).
Umehara, T. et al. Structural basis for acetylated histone H4 recognition by the human BRD2 bromodomain. J. Biol. Chem. 285, 7610–7618 (2010).
Lee, J. E. et al. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 8, 2217 (2017).
Liu, Y. et al. Chromatin looping shapes KLF5-dependent transcriptional programs in human epithelial cancers. Cancer Res. 80, 5464–5477 (2020).
Yokoyama, A. Leukemogenesis via aberrant self-renewal by the MLL/AEP-mediated transcriptional activation system. Cancer Sci. 112, 3935–3944 (2021).
Andrews, F. H., Shanle, E. K., Strahl, B. D. & Kutateladze, T. G. The essential role of acetyllysine binding by the YEATS domain in transcriptional regulation. Transcription 7, 14–20 (2016).
Zhao, D. et al. YEATS2 is a selective histone crotonylation reader. Cell. Res. 26, 629–632 (2016).
Cho, H. J. et al. GAS41 recognizes diacetylated histone h3 through a bivalent binding mode. ACS Chem. Biol. 13, 2739–2746 (2018).
Kikuchi, M. et al. GAS41 promotes H2A.Z deposition through recognition of the N terminus of histone H3 by the YEATS domain. Proc. Natl. Acad. Sci. USA 120, e2304103120 (2023).
Margueron, R. et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell. 32, 503–518 (2008).
Stulemeijer, I. J. et al. Dot1 histone methyltransferases share a distributive mechanism but have highly diverged catalytic properties. Sci. Rep. 5, 9824 (2015).
Strepkos, D. et al. Histone methyltransferase SETDB1: a common denominator of tumorigenesis with therapeutic potential. Cancer Res. 81, 525–534 (2021).
Shinkai, Y. & Tachibana, M. H3K9 methyltransferase G9a and the related molecule GLP. Genes. Dev. 25, 781–788, (2011).
Huang, J. et al. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 285, 9636–9641 (2010).
Brown, M. A., Sims, R. J. 3rd, Gottlieb, P. D. & Tucker, P. W. Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol. Cancer 5, 26 (2006).
Huang, J. et al. Repression of p53 activity by Smyd2-mediated methylation. Nature 444, 629–632 (2006).
Li, W. et al. Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature 590, 498–503 (2021).
Thiebaut, C., Eve, L., Poulard, C. & Le Romancer, M. Structure, activity, and function of PRMT1. Life 11, 1147 (2021).
Liu, F. et al. PRMT5-mediated histone arginine methylation antagonizes transcriptional repression by polycomb complex PRC2. Nucl. Acids Res. 48, 2956–2968 (2020).
Liu, R. et al. PHD finger protein 1 (PHF1) is a novel reader for histone H4R3 symmetric dimethylation and coordinates with PRMT5-WDR77/CRL4B complex to promote tumorigenesis. Nucl. Acids Res. 46, 6608–6626 (2018).
Whyte, W. A. et al. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482, 221–225 (2012).
Reischl, S. & Kramer, A. Fbxl11 is a novel negative element of the mammalian circadian clock. J. Biol. Rhythm. 30, 291–301 (2015).
Tsukada, Y., Ishitani, T. & Nakayama, K. I. KDM7 is a dual demethylase for histone H3 Lys 9 and Lys 27 and functions in brain development. Genes. Dev. 24, 432–437 (2010).
Hoyle, R. G. et al. IOX1 suppresses wnt target gene transcription and colorectal cancer tumorigenesis through inhibition of KDM3 histone demethylases. Mol. Cancer Ther. 20, 191–202 (2021).
Agger, K. et al. The KDM4/JMJD2 histone demethylases are required for hematopoietic stem cell maintenance. Blood 134, 1154–1158 (2019).
Zhang, Y. et al. The PHD1 finger of KDM5B recognizes unmodified H3K4 during the demethylation of histone H3K4me2/3 by KDM5B. Protein Cell. 5, 837–850 (2014).
Tran, N., Broun, A. & Ge, K. Lysine demethylase KDM6A in differentiation, development, and cancer. Mol. Cell Biol. 40, e00341–e00320 (2020).
Lagunas-Rangel, F. A. KDM6B (JMJD3) and its dual role in cancer. Biochimie 184, 63–71 (2021).
Li, H. et al. Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Mol. Cell. 28, 677–691 (2007).
Gonzalez-Sandoval, A. et al. Perinuclear anchoring of H3K9-methylated chromatin stabilizes induced cell fate in C. elegans embryos. Cell 163, 1333–1347 (2015).
Tavares, L. et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 148, 664–678 (2012).
Lu, R. & Wang, G. G. Tudor: a versatile family of histone methylation ‘readers’. Trends Biochem Sci. 38, 546–555 (2013).
Vezzoli, A. et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat. Struct. Mol. Biol. 17, 617–619 (2010).
Qiu, Y. et al. Solution structure of the Pdp1 PWWP domain reveals its unique binding sites for methylated H4K20 and DNA. Biochem. J. 442, 527–538 (2012).
Black, J. C. & Kutateladze, T. G. Atypical histone targets of PHD fingers. J. Biol. Chem. 299, 104601 (2023).
Wysocka, J. et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121, 859–872 (2005).
Huang, J. & Yin, P. Structural Insights into N(6)-methyladenosine (m(6)A) Modification in the transcriptome. Genom. Proteom. Bioinforma. 16, 85–98 (2018).
Wang, X. et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534, 575–578 (2016).
Jia, G. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011).
Bartosovic, M. et al. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3’-end processing. Nucl. Acids Res. 45, 11356–11370 (2017).
Zheng, G. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 49, 18–29 (2013).
Esteve-Puig, R. et al. Epigenetic loss of m1A RNA demethylase ALKBH3 in Hodgkin lymphoma targets collagen, conferring poor clinical outcome. Blood 137, 994–999 (2021).
Wang, X. et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015).
Einstein, J. M. et al. Inhibition of YTHDF2 triggers proteotoxic cell death in MYC-driven breast cancer. Mol. Cell. 81, 3048–3064.e3049 (2021).
Buscarlet, M. et al. Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood 123, 1720–1728 (2014).
Singh, M., Popowicz, G. M., Krajewski, M. & Holak, T. A. Structural ramification for acetyl-lysine recognition by the bromodomain of human BRG1 protein, a central ATPase of the SWI/SNF remodeling complex. Chembiochem 8, 1308–1316, (2007).
Sanchez, O. F., Williamson, D., Cai, L. & Yuan, C. A sensitive protein-based sensor for quantifying histone acetylation levels. Talanta 140, 212–218 (2015).
Peng, C. et al. The transcriptional regulation role of BRD7 by binding to acetylated histone through bromodomain. J. Cell. Biochem. 97, 882–892 (2006).
Li, S. et al. LncRNA LENGA acts as a tumor suppressor in gastric cancer through BRD7/TP53 signaling. Cell. Mol. Life Sci. 80, 5 (2022).
Flynn, E. M. et al. A subset of human bromodomains recognizes butyryllysine and crotonyllysine histone peptide modifications. Structure 23, 1801–1814 (2015).
Niemeyer, C. M. et al. Response to upfront azacitidine in juvenile myelomonocytic leukemia in the AZA-JMML-001 trial. Blood Adv. 5, 2901–2908 (2021).
Adès, L. et al. Pevonedistat plus azacitidine vs azacitidine alone in higher-risk MDS/chronic myelomonocytic leukemia or low-blast-percentage AML. Blood Adv. 6, 5132–5145 (2022).
Sekeres, M. A. et al. Randomized phase 2 trial of pevonedistat plus azacitidine versus azacitidine for higher-risk MDS/CMML or low-blast AML. Leukemia 35, 2119–2124 (2021).
Ravandi, F. et al. Management of adverse events in patients with acute myeloid leukemia in remission receiving oral azacitidine: experience from the phase 3 randomized QUAZAR AML-001 trial. J. Hematol. Oncol. 14, 133 (2021).
Santini, V. et al. A phase II, multicentre trial of decitabine in higher-risk chronic myelomonocytic leukemia. Leukemia 32, 413–418 (2018).
Garcia-Manero, G. et al. Oral decitabine-cedazuridine versus intravenous decitabine for myelodysplastic syndromes and chronic myelomonocytic leukaemia (ASCERTAIN): a registrational, randomised, crossover, pharmacokinetics, phase 3 study. Lancet Haematol. 11, e15–e26 (2024).
Itzykson, R. et al. Decitabine versus hydroxyurea for advanced proliferative chronic myelomonocytic leukemia: results of a randomized phase III trial within the EMSCO network. J. Clin. Oncol. 41, 1888–1897 (2023).
Garcia-Manero, G. et al. Oral cedazuridine/decitabine for MDS and CMML: a phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood 136, 674–683 (2020).
Kim, Y. H. et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 19, 1192–1204 (2018).
Duvic, M. et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 109, 31–39 (2007).
Bates, S. E. et al. Romidepsin in peripheral and cutaneous T-cell lymphoma: mechanistic implications from clinical and correlative data. Br. J. Haematol. 170, 96–109 (2015).
Foss, F. et al. Romidepsin for the treatment of relapsed/refractory peripheral T cell lymphoma: prolonged stable disease provides clinical benefits for patients in the pivotal trial. J. Hematol. Oncol. 9, 22 (2016).
Brunvand, M. W. & Carson, J. Complete remission with romidepsin in a patient with T-cell acute lymphoblastic leukemia refractory to induction hyper-CVAD. Hematol. Oncol. 36, 340–343 (2018).
Gimsing, P. et al. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur. J. Haematol. 81, 170–176 (2008).
Foss, F. et al. A phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 168, 811–819 (2015).
Dimicoli, S. et al. Phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in patients with low or intermediate-1 risk myelodysplastic syndrome. Am. J. Hematol. 87, 127–129 (2012).
Garcia-Manero, G. et al. A phase 1b/2b multicenter study of oral panobinostat plus azacitidine in adults with MDS, CMML or AML with ⩽30% blasts. Leukemia 31, 2799–2806 (2017).
Zhang, M. C. et al. Clinical efficacy and molecular biomarkers in a phase II study of tucidinostat plus R-CHOP in elderly patients with newly diagnosed diffuse large B-cell lymphoma. Clin. Epigenet. 12, 160 (2020).
Wang, Y. et al. Chidamide plus prednisone, etoposide, and thalidomide for untreated angioimmunoblastic T-cell lymphoma in a Chinese population: a multicenter phase II trial. Am. J. Hematol. 97, 623–629 (2022).
DiNardo, C. D. et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 378, 2386–2398 (2018).
Abou-Alfa, G. K. et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 21, 796–807 (2020).
Albany, C. et al. A phase 1 study of combined guadecitabine and cisplatin in platinum refractory germ cell cancer. Cancer Med. 10, 156–163 (2021).
Papadatos-Pastos, D. et al. Phase 1, dose-escalation study of guadecitabine (SGI-110) in combination with pembrolizumab in patients with solid tumors. J. Immunother Cancer. 10, e004495. (2022).
Wei, C. X. et al. A brief report of a phase II trial evaluating efficacy and safety of hypomethylating agent guadecitabine in combination with carboplatin in extensive stage small cell lung cancer. Clin. Lung Cancer 24, 347–352 (2023).
Jang, H. J. et al. A phase II trial of guadecitabine plus atezolizumab in metastatic urothelial carcinoma progressing after initial immune checkpoint inhibitor therapy. Clin. Cancer Res. 29, 2052–2065 (2023).
Chen, S. et al. Epigenetic priming enhances antitumor immunity in platinum-resistant ovarian cancer. J. Clin. Investig. 132, e158800 (2022).
Sébert, M. et al. A phase II study of guadecitabine in higher-risk myelodysplastic syndrome and low blast count acute myeloid leukemia after azacitidine failure. Haematologica 104, 1565–1571 (2019).
Sheikh, T. N. et al. Growth inhibition and induction of innate immune signaling of chondrosarcomas with epigenetic inhibitors. Mol. Cancer Ther. 20, 2362–2371 (2021).
Oza, A. M. et al. A randomized phase II trial of epigenetic priming with guadecitabine and carboplatin in platinum-resistant, recurrent ovarian cancer. Clin. Cancer Res. 26, 1009–1016 (2020).
Zakharia, Y. et al. Durvalumab and guadecitabine in advanced clear cell renal cell carcinoma: results from the phase Ib/II study BTCRC-GU16-043. Nat. Commun. 15, 972 (2024).
O’Connell, C. L. et al. Safety, outcomes, and T-cell characteristics in patients with relapsed or refractory MDS or CMML treated with atezolizumab in combination with guadecitabine. Clin. Cancer Res. 28, 5306–5316 (2022).
Garcia-Manero, G. et al. Guadecitabine (SGI-110) in patients with intermediate or high-risk myelodysplastic syndromes: phase 2 results from a multicentre, open-label, randomised, phase 1/2 trial. Lancet Haematol. 6, e317–e327 (2019).
Chung, W. et al. Genomic and epigenomic predictors of response to guadecitabine in relapsed/refractory acute myelogenous leukemia. Clin. Epigenet. 11, 106 (2019).
Kantarjian, H. M. et al. Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol. 18, 1317–1326 (2017).
Issa, J. J. et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 16, 1099–1110 (2015).
Sharma, A. et al. Hypomethylating agents synergize with irinotecan to improve response to chemotherapy in colorectal cancer cells. PLoS ONE 12, e0176139 (2017).
Liu, Y. C. et al. A clinical trial with valproic acid and hydralazine in combination with gemcitabine and cisplatin followed by doxorubicin and dacarbazine for advanced hepatocellular carcinoma. Asia Pac. J. Clin. Oncol. 18, 19–27 (2022).
Kim, B., Huh, K. Y., Yu, K. S. & Lee, S. Pharmacokinetics, pharmacodynamics and safety of oral formulation (CG-750) of ivaltinostat, a histone deacetylase inhibitor, compared to IV formulation (CG-745). Br. J. Clin. Pharmacol. 90, 1103–1114 (2024).
Vey, N. et al. Phase 1 dose-escalation study of oral abexinostat for the treatment of patients with relapsed/refractory higher-risk myelodysplastic syndromes, acute myeloid leukemia, or acute lymphoblastic leukemia. Leuk. Lymphoma 58, 1880–1886 (2017).
Choy, E. et al. Phase 1 study of oral abexinostat, a histone deacetylase inhibitor, in combination with doxorubicin in patients with metastatic sarcoma. Cancer 121, 1223–1230 (2015).
Morschhauser, F. et al. Phase 1 study of the oral histone deacetylase inhibitor abexinostat in patients with Hodgkin lymphoma, non-Hodgkin lymphoma, or chronic lymphocytic leukaemia. Investig. New. Drugs 33, 423–431 (2015).
Liva, S. G. et al. Phase I study of AR-42 and decitabine in acute myeloid leukemia. Leuk. Lymphoma 61, 1484–1492 (2020).
Sborov, D. W. et al. A phase 1 trial of the HDAC inhibitor AR-42 in patients with multiple myeloma and T- and B-cell lymphomas. Leuk. Lymphoma 58, 2310–2318 (2017).
Abaza, Y. M. et al. Phase 1 dose escalation multicenter trial of pracinostat alone and in combination with azacitidine in patients with advanced hematologic malignancies. Cancer 123, 4851–4859 (2017).
Yong, W. P. et al. Phase I and pharmacodynamic study of an orally administered novel inhibitor of histone deacetylases, SB939, in patients with refractory solid malignancies. Ann. Oncol. 22, 2516–2522 (2011).
Razak, A. R. et al. Phase I clinical, pharmacokinetic and pharmacodynamic study of SB939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br. J. Cancer 104, 756–762 (2011).
Garcia-Manero, G. et al. Pracinostat plus azacitidine in older patients with newly diagnosed acute myeloid leukemia: results of a phase 2 study. Blood Adv. 3, 508–518 (2019).
Bose, P. et al. A phase 2 study of pracinostat combined with ruxolitinib in patients with myelofibrosis. Leuk. Lymphoma 60, 1767–1774 (2019).
Yalniz, F. F. et al. A phase II study of addition of pracinostat to a hypomethylating agent in patients with myelodysplastic syndromes who have not responded to previous hypomethylating agent therapy. Br. J. Haematol. 188, 404–412 (2020).
Eigl, B. J. et al. A phase II study of the HDAC inhibitor SB939 in patients with castration resistant prostate cancer: NCIC clinical trials group study IND195. Investig. New. Drugs 33, 969–976 (2015).
Garcia-Manero, G. et al. Phase 2, randomized, double-blind study of pracinostat in combination with azacitidine in patients with untreated, higher-risk myelodysplastic syndromes. Cancer 123, 994–1002 (2017).
Quintás-Cardama, A. et al. Therapy with the histone deacetylase inhibitor pracinostat for patients with myelofibrosis. Leuk. Res. 36, 1124–1127 (2012).
Chu, Q. S. et al. A phase II study of SB939, a novel pan-histone deacetylase inhibitor, in patients with translocation-associated recurrent/metastatic sarcomas-NCIC-CTG IND 200. †. Ann. Oncol. 26, 973–981 (2015).
Garcia-Manero, G. et al. Pracinostat combined with azacitidine in newly diagnosed adult acute myeloid leukemia (AML) patients unfit for standard induction chemotherapy: PRIMULA phase III study. Leuk. Res. 140, 107480 (2024).
Ikeda, M. et al. Phase I study of resminostat, an HDAC inhibitor, combined with S-1 in patients with pre-treated biliary tract or pancreatic cancer. Investig. New. Drugs 37, 109–117 (2019).
Brunetto, A. T. et al. First-in-human, pharmacokinetic and pharmacodynamic phase I study of Resminostat, an oral histone deacetylase inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 19, 5494–5504 (2013).
Walewski, J. et al. Resminostat in patients with relapsed or refractory Hodgkin lymphoma: results of the phase II SAPHIRE study. Leuk. Lymphoma 60, 675–684 (2019).
Bitzer, M. et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma - The SHELTER study. J. Hepatol. 65, 280–288 (2016).
Ueno, M. et al. A randomized, double-blind, phase II study of oral histone deacetylase inhibitor resminostat plus S-1 versus placebo plus S-1 in biliary tract cancers previously treated with gemcitabine plus platinum-based chemotherapy. Cancer Med. 10, 2088–2099 (2021).
Tak, W. Y. et al. Phase I/II study of first-line combination therapy with sorafenib plus resminostat, an oral HDAC inhibitor, versus sorafenib monotherapy for advanced hepatocellular carcinoma in east Asian patients. Investig. New. Drugs 36, 1072–1084 (2018).
Tambo, Y. et al. Phase I/II study of docetaxel combined with resminostat, an oral hydroxamic acid HDAC inhibitor, for advanced non-small cell lung cancer in patients previously treated with platinum-based chemotherapy. Investig. New. Drugs 35, 217–226 (2017).
Pauer, L. R. et al. Phase I study of oral CI-994 in combination with carboplatin and paclitaxel in the treatment of patients with advanced solid tumors. Cancer Investig. 22, 886–896 (2004).
Undevia, S. D. et al. A phase I study of the oral combination of CI-994, a putative histone deacetylase inhibitor, and capecitabine. Ann. Oncol. 15, 1705–1711 (2004).
Nemunaitis, J. J. et al. Phase I study of oral CI-994 in combination with gemcitabine in treatment of patients with advanced cancer. Cancer J. 9, 58–66 (2003).
Prakash, S. et al. Chronic oral administration of CI-994: a phase 1 study. Investig. New. Drugs 19, 1–11 (2001).
Richards, D. A. et al. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann. Oncol. 17, 1096–1102 (2006).
Ljubenkov, P. A. et al. Effect of the histone deacetylase inhibitor FRM-0334 on progranulin levels in patients with progranulin gene haploinsufficiency: a randomized clinical trial. JAMA Netw. Open. 4, e2125584 (2021).
Venugopal, B. et al. A phase I study of quisinostat (JNJ-26481585), an oral hydroxamate histone deacetylase inhibitor with evidence of target modulation and antitumor activity, in patients with advanced solid tumors. Clin. Cancer Res. 19, 4262–4272 (2013).
Moreau, P. et al. Quisinostat, bortezomib, and dexamethasone combination therapy for relapsed multiple myeloma. Leuk. Lymphoma 57, 1546–1559 (2016).
Child, F. et al. Phase II multicentre trial of oral quisinostat, a histone deacetylase inhibitor, in patients with previously treated stage IB-IVA mycosis fungoides/Sézary syndrome. Br. J. Derm. 175, 80–88 (2016).
Booth, S. W. et al. A Phase 2a cohort expansion study to assess the safety, tolerability, and preliminary efficacy of CXD101 in patients with advanced solid-organ cancer expressing HR23B or lymphoma. BMC Cancer 21, 851 (2021).
Saunders, M. P. et al. CXD101 and nivolumab in patients with metastatic microsatellite-stable colorectal cancer (CAROSELL): a multicentre, open-label, single-arm, phase II trial. ESMO Open. 7, 100594 (2022).
Heath, E. I. et al. Phase Ia dose escalation study of OBP-801, a cyclic depsipeptide class I histone deacetylase inhibitor, in patients with advanced solid tumors. Investig. New. Drugs 40, 300–307 (2022).
Haverkos, B. et al. Targeted therapy with nanatinostat and valganciclovir in recurrent EBV-positive lymphoid malignancies: a phase 1b/2 study. Blood Adv. 7, 6339–6350 (2023).
Lin, J. et al. Phase I study of entinostat in combination with enzalutamide for treatment of patients with metastatic castration-resistant prostate cancer. Oncologist 26, e2136–e2142 (2021).
Wang, J. et al. Phase I study and pilot efficacy analysis of entinostat, a novel histone deacetylase inhibitor, in chinese postmenopausal women with hormone receptor-positive metastatic breast cancer. Target. Oncol. 16, 591–599 (2021).
Gojo, I. et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood 109, 2781–2790 (2007).
Kummar, S. et al. Phase I trial of MS-275, a histone deacetylase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clin. Cancer Res. 13, 5411–5417 (2007).
Bewersdorf, J. P. et al. A multicenter phase Ib trial of the histone deacetylase inhibitor entinostat in combination with pembrolizumab in patients with myelodysplastic syndromes/neoplasms or acute myeloid leukemia refractory to hypomethylating agents. Ann. Hematol. 103, 105–116 (2024).
Pili, R. et al. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br. J. Cancer 106, 77–84 (2012).
Prebet, T. et al. Azacitidine with or without Entinostat for the treatment of therapy-related myeloid neoplasm: further results of the E1905 North American Leukemia Intergroup study. Br. J. Haematol. 172, 384–391, (2016).
Figueroa, M. E. et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 114, 3448–3458 (2009).
Fandy, T. E. et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood 114, 2764–2773 (2009).
Masuda, N. et al. Phase 1 trial of entinostat as monotherapy and combined with exemestane in Japanese patients with hormone receptor-positive advanced breast cancer. BMC Cancer 21, 1269 (2021).
Bukowinski, A. et al. A phase 1 study of entinostat in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: Trial ADVL1513, Pediatric Early Phase-Clinical Trial Network (PEP-CTN). Pediatr. Blood Cancer 68, e28892 (2021).
Lim, B. et al. A phase Ib study of entinostat plus lapatinib with or without trastuzumab in patients with HER2-positive metastatic breast cancer that progressed during trastuzumab treatment. Br. J. Cancer 120, 1105–1112 (2019).
Ngamphaiboon, N. et al. A phase I study of the histone deacetylase (HDAC) inhibitor entinostat, in combination with sorafenib in patients with advanced solid tumors. Investig. New. Drugs 33, 225–232 (2015).
Torres, Roussos et al. Entinostat, nivolumab and ipilimumab for women with advanced HER2-negative breast cancer: a phase Ib trial. Nat. Cancer 5, 866–879 (2024).
Iwata, H. et al. Efficacy and exploratory biomarker analysis of entinostat plus exemestane in advanced or recurrent breast cancer: phase II randomized controlled trial. Jpn J. Clin. Oncol. 53, 4–15 (2023).
Jespersen, H. et al. Concomitant use of pembrolizumab and entinostat in adult patients with metastatic uveal melanoma (PEMDAC study): protocol for a multicenter phase II open label study. BMC Cancer 19, 415 (2019).
Ny, L. et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat. Commun. 12, 5155 (2021).
Batlevi, C. L. et al. ENGAGE- 501: phase II study of entinostat (SNDX-275) in relapsed and refractory Hodgkin lymphoma. Haematologica 101, 968–975 (2016).
Prebet, T. et al. Prolonged administration of azacitidine with or without entinostat for myelodysplastic syndrome and acute myeloid leukemia with myelodysplasia-related changes: results of the US Leukemia Intergroup trial E1905. J. Clin. Oncol. 32, 1242–1248 (2014).
Yardley, D. A. et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 31, 2128–2135 (2013).
Hellmann, M. D. et al. Entinostat plus pembrolizumab in patients with metastatic NSCLC previously treated with anti-PD-(L)1 therapy. Clin. Cancer Res. 27, 1019–1028 (2021).
van Tilburg, C. M. et al. INFORM2 NivEnt: the first trial of the INFORM2 biomarker driven phase I/II trial series: the combination of nivolumab and entinostat in children and adolescents with refractory high-risk malignancies. BMC Cancer 20, 523 (2020).
Pili, R. et al. Immunomodulation by Entinostat in renal cell carcinoma patients receiving high-dose interleukin 2: a multicenter, single-arm, phase I/II trial (NCI-CTEP#7870). Clin. Cancer Res. 23, 7199–7208 (2017).
Juergens, R. A. et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 1, 598–607 (2011).
Witta, S. E. et al. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J. Clin. Oncol. 30, 2248–2255 (2012).
Connolly, R. M. et al. E2112: randomized phase III trial of endocrine therapy plus entinostat or placebo in hormone receptor-positive advanced breast cancer. a trial of the ecog-acrin cancer research group. J. Clin. Oncol. 39, 3171–3181 (2021).
Ballinger, T. J. et al. Impact of muscle measures on outcome in patients receiving endocrine therapy for metastatic breast cancer: analysis of ECOG-ACRIN E2112. J. Natl Compr. Cancer Netw. 21, 915–923.e911 (2023).
Garcia-Manero, G. et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood 112, 981–989 (2008).
Weber, J. S. et al. Clinical and immune correlate results from a phase 1b study of the histone deacetylase inhibitor mocetinostat with ipilimumab and nivolumab in unresectable stage III/IV melanoma. Melanoma Res. 32, 324–333 (2022).
Grivas, P. et al. Mocetinostat for patients with previously treated, locally advanced/metastatic urothelial carcinoma and inactivating alterations of acetyltransferase genes. Cancer 125, 533–540 (2019).
Younes, A. et al. Mocetinostat for relapsed classical Hodgkin’s lymphoma: an open-label, single-arm, phase 2 trial. Lancet Oncol. 12, 1222–1228 (2011).
Batlevi, C. L. et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br. J. Haematol. 178, 434–441 (2017).
Blum, K. A. et al. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br. J. Haematol. 147, 507–514 (2009).
Chan, E. et al. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother. Pharmacol. 81, 355–364 (2018).
Qualls, D. et al. Molecularly targeted epigenetic therapy with mocetinostat in relapsed and refractory non-Hodgkin lymphoma with CREBBP or EP300 mutations: an open label phase II study. Leuk. Lymphoma 64, 738–741 (2023).
Johnson, M. L. et al. Mocetinostat in combination with durvalumab for patients with advanced NSCLC: results from a phase I/II Study. Clin. Lung Cancer 24, 218–227 (2023).
von Tresckow, B. et al. Phase I study of domatinostat (4SC-202), a class I histone deacetylase inhibitor in patients with advanced hematological malignancies. Eur. J. Haematol. 102, 163–173 (2019).
Cartwright, E. et al. Phase II trial of domatinostat (4SC-202) in combination with avelumab in patients with previously treated advanced mismatch repair proficient oesophagogastric and colorectal adenocarcinoma: EMERGE. ESMO Open. 9, 102971 (2024).
Younes, A. et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. Lancet Oncol. 17, 622–631 (2016).
Ossenkoppele, G. J. et al. A phase I first-in-human study with tefinostat—a monocyte/macrophage targeted histone deacetylase inhibitor—in patients with advanced haematological malignancies. Br. J. Haematol. 162, 191–201 (2013).
Ford, P. A. et al. Treatment effects of low-dose theophylline combined with an inhaled corticosteroid in COPD. Chest 137, 1338–1344 (2010).
Cosio, B. G. et al. Low-dose theophylline enhances the anti-inflammatory effects of steroids during exacerbations of COPD. Thorax 64, 424–429 (2009).
Bo, S. et al. Impact of sirtuin-1 expression on H3K56 acetylation and oxidative stress: a double-blind randomized controlled trial with resveratrol supplementation. Acta Diabetol. 55, 331–340 (2018).
Moreno, V. et al. Trotabresib, an oral potent bromodomain and extraterminal inhibitor, in patients with high-grade gliomas: a phase I, “window-of-opportunity” study. Neuro Oncol. 25, 1113–1122 (2023).
Moreno, V. et al. BET inhibitor trotabresib in heavily pretreated patients with solid tumors and diffuse large B-cell lymphomas. Nat. Commun. 14, 1359 (2023).
Moreno, V. et al. Phase I study of CC-90010, a reversible, oral BET inhibitor in patients with advanced solid tumors and relapsed/refractory non-Hodgkin’s lymphoma. Ann. Oncol. 31, 780–788 (2020).
Brown, J. A. et al. A randomized study of the safety and pharmacokinetics of GSK3358699, a mononuclear myeloid-targeted bromodomain and extra-terminal domain inhibitor. Br. J. Clin. Pharmacol. 88, 2140–2155 (2022).
Roboz, G. J. et al. A dose escalation study of RO6870810/TEN-10 in patients with acute myeloid leukemia and myelodysplastic syndrome. Leuk. Lymphoma 62, 1740–1748 (2021).
Shapiro, G. I. et al. A Phase 1 study of RO6870810, a novel bromodomain and extra-terminal protein inhibitor, in patients with NUT carcinoma, other solid tumours, or diffuse large B-cell lymphoma. Br. J. Cancer 124, 744–753 (2021).
Ramasamy, K. et al. A phase 1b dose-escalation/expansion study of BET inhibitor RO6870810 in patients with advanced multiple myeloma. Blood Cancer J. 11, 149 (2021).
Dickinson, M. et al. Phase 1b study of the BET protein inhibitor RO6870810 with venetoclax and rituximab in patients with diffuse large B-cell lymphoma. Blood Adv. 5, 4762–4770 (2021).
Ameratunga, M. et al. First-in-human Phase 1 open label study of the BET inhibitor ODM-207 in patients with selected solid tumours. Br. J. Cancer 123, 1730–1736 (2020).
Lewin, J. et al. Phase Ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J. Clin. Oncol. 36, 3007–3014 (2018).
Amorim, S. et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 3, e196–e204 (2016).
Berthon, C. et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 3, e186–e195 (2016).
Cousin, S. et al. Safety, pharmacokinetic, pharmacodynamic and clinical activity of molibresib for the treatment of nuclear protein in testis carcinoma and other cancers: results of a Phase I/II open-label, dose escalation study. Int. J. Cancer 150, 993–1006 (2022).
Krishnatry, A. S. et al. Population pharmacokinetic modeling of molibresib and its active metabolites in patients with solid tumors: a semimechanistic autoinduction model. CPT Pharmacomet. Syst. Pharmacol. 10, 709–722 (2021).
Cescon, D. W. et al. A phase I/II Study of GSK525762 combined with fulvestrant in patients with hormone receptor-positive/HER2-negative advanced or metastatic breast cancer. Clin. Cancer Res. 30, 334–343 (2024).
Riddell, K. et al. An adaptive physiologically based pharmacokinetic-driven design to investigate the effect of itraconazole and rifampicin on the pharmacokinetics of molibresib (GSK525762) in healthy female volunteers. J. Clin. Pharmacol. 61, 125–137 (2021).
Dawson, M. A. et al. A phase I/II open-label study of molibresib for the treatment of relapsed/refractory hematologic malignancies. Clin. Cancer Res. 29, 711–722 (2023).
Borthakur, G. et al. A phase 1 study of the pan-bromodomain and extraterminal inhibitor mivebresib (ABBV-075) alone or in combination with venetoclax in patients with relapsed/refractory acute myeloid leukemia. Cancer 127, 2943–2953 (2021).
Piha-Paul, S. A. et al. First-in-human study of mivebresib (ABBV-075), an oral pan-inhibitor of bromodomain and extra terminal proteins, in patients with relapsed/refractory solid tumors. Clin. Cancer Res. 25, 6309–6319 (2019).
Falchook, G. et al. Development of 2 bromodomain and extraterminal inhibitors with distinct pharmacokinetic and pharmacodynamic profiles for the treatment of advanced malignancies. Clin. Cancer Res. 26, 1247–1257 (2020).
Hamilton, E. P. et al. First-in-human Study of AZD5153, a small-molecule inhibitor of bromodomain protein 4, in patients with relapsed/refractory malignant solid tumors and lymphoma. Mol. Cancer Ther. 22, 1154–1165 (2023).
Schöffski, P. et al. Phase Ia dose-escalation trial with the BET protein inhibitor BI 894999 in patients with advanced or metastatic solid tumours. Eur. J. Cancer 191, 112987 (2023).
Provencher, S. et al. BET protein inhibition for pulmonary arterial hypertension: a pilot clinical trial. Am. J. Respir. Crit. Care Med. 205, 1357–1360 (2022).
Nicholls, S. J. et al. Efficacy and safety of a novel oral inducer of apolipoprotein a-I synthesis in statin-treated patients with stable coronary artery disease a randomized controlled trial. J. Am. Coll. Cardiol. 57, 1111–1119 (2011).
Siebel, A. L. et al. Effects of the BET-inhibitor, RVX-208 on the HDL lipidome and glucose metabolism in individuals with prediabetes: a randomized controlled trial. Metabolism 65, 904–914 (2016).
Nicholls, S. J. et al. ApoA-I induction as a potential cardioprotective strategy: rationale for the SUSTAIN and ASSURE studies. Cardiovasc. Drugs Ther. 26, 181–187 (2012).
Shishikura, D. et al. The effect of bromodomain and extra-terminal inhibitor apabetalone on attenuated coronary atherosclerotic plaque: insights from the ASSURE trial. Am. J. Cardiovasc. Drugs 19, 49–57 (2019).
Nicholls, S. J. et al. Effect of the BET protein inhibitor, RVX-208, on progression of coronary atherosclerosis: results of the phase 2b, randomized, double-blind, multicenter, ASSURE trial. Am. J. Cardiovasc. Drugs 16, 55–65 (2016).
Kulikowski, E. et al. Apabetalone mediated epigenetic modulation is associated with favorable kidney function and alkaline phosphatase profile in patients with chronic kidney disease. Kidney Blood Press Res. 43, 449–457 (2018).
Schwartz, G. G. et al. Relation of insulin treatment for type 2 diabetes to the risk of major adverse cardiovascular events after acute coronary syndrome: an analysis of the BETonMACE randomized clinical trial. Cardiovasc. Diabetol. 20, 125 (2021).
Ray, K. K. et al. Effect of selective BET protein inhibitor apabetalone on cardiovascular outcomes in patients with acute coronary syndrome and diabetes: rationale, design, and baseline characteristics of the BETonMACE trial. Am. Hear. J. 217, 72–83 (2019).
Cummings, J. et al. Cognitive effects of the BET protein inhibitor apabetalone: a prespecified montreal cognitive assessment analysis nested in the BETonMACE randomized controlled trial. J. Alzheimers Dis. 83, 1703–1715 (2021).
Kalantar-Zadeh, K. et al. Effect of apabetalone on cardiovascular events in diabetes, CKD, and recent acute coronary syndrome: results from the BETonMACE randomized controlled trial. Clin. J. Am. Soc. Nephrol. 16, 705–716 (2021).
Nicholls, S. J. et al. Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: a prespecified analysis of the BETonMACE study. Cardiovasc. Diabetol. 20, 13 (2021).
Blum, K. A. et al. A phase I study of pelabresib (CPI-0610), a small-molecule inhibitor of BET proteins, in patients with relapsed or refractory lymphoma. Cancer Res. Commun. 2, 795–805 (2022).
Stein, E. M. et al. Results from phase 1 of the MANIFEST clinical trial to evaluate the safety and tolerability of pelabresib in patients with myeloid malignancies. Leuk. Lymphoma 65, 503–510 (2024).
Eickhoff, N., Bergman, A. M. & Zwart, W. Homing in on a moving target: androgen receptor cistromic plasticity in prostate cancer. Endocrinology 163, bqac153 (2022).
Stein, E. M. et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 131, 2661–2669 (2018).
Vieito, M. et al. Phase 1 study of JNJ-64619178, a protein arginine methyltransferase 5 inhibitor, in advanced solid tumors. Clin. Cancer Res. 29, 3592–3602 (2023).
Guo, C. et al. PK/PD model-informed dose selection for oncology phase I expansion: case study based on PF-06939999, a PRMT5 inhibitor. CPT Pharmacomet. Syst. Pharmacol. 12, 1619–1625 (2023).
Hollebecque, A. et al. Clinical activity of CC-90011, an oral, potent, and reversible LSD1 inhibitor, in advanced malignancies. Cancer 128, 3185–3195 (2022).
Hollebecque, A. et al. Phase I study of lysine-specific demethylase 1 inhibitor, CC-90011, in patients with advanced solid tumors and relapsed/refractory non-hodgkin lymphoma. Clin. Cancer Res. 27, 438–446 (2021).
Abplanalp, W. T. et al. Efficiency and target derepression of anti-mir-92a: results of a first in human study. Nucl. Acid. Ther. 30, 335–345 (2020).
Beg, M. S. et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New. Drugs 35, 180–188 (2017).
van Zandwijk, N. et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: a first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 18, 1386–1396 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation grant of China (No. 82371647, 82071607, 82071651, 82403489); National Key Research and Development Program (2022YFC3600304, 2022YFC2704700); Science and Technology Department of Sichuan Province (23ZDYF2489); Cadre Health Care Committee of Sichuan Province, China (2023-1701); Chengdu Science and Technology Bureau (2021-YF05-02106-SN, 2017-GH02-00030-HZ); Outstanding Scientific Fund of Shengjing Hospital (No. 202003); Sichuan University 0-1 Innovation Research project (2023SCUH0019); The Frontiers Medical Center, Tianfu Jincheng Laboratory Foundation (T FJC2023010001); China Postdoctoral Science Foundation (No. 2023M733902, 2023M743909); PHD Research Initiated Fund Project in Liaoning Province (No. 2023-BSBA-331, 2023-BSBA-352); Science and Technology Plan of Liaoning Province (2022JH2/20200066); 345 Talent Project of Shengjing Hospital of China Medical University (No. M1344).
Author information
Authors and Affiliations
Contributions
D.L., X.X., and Z.N. proposed the topic and main idea. W.D., X.Q., and Y.F. wrote the original manuscript and drew the figures. W.D., R.G., and P.B. were responsible for the revision of the manuscript. W.D., X.Q., Y.F., and S.L. were responsible for collecting data and making tables. W.D., T.L., Y.J., and S.W. were responsible for the literature search. D.L., X.X., and Z.N. commented on and revised the manuscript. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dai, W., Qiao, X., Fang, Y. et al. Epigenetics-targeted drugs: current paradigms and future challenges. Sig Transduct Target Ther 9, 332 (2024). https://doi.org/10.1038/s41392-024-02039-0
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41392-024-02039-0
This article is cited by
-
The promise of epigenetic editing strategies in functionally curing chronic hepatitis B virus infections
Epigenetics Communications (2026)
-
Recent perspectives on macrophage memory: types, mechanisms, and characteristics in pulmonary diseases
Cell Communication and Signaling (2026)
-
Bromodomain and extra-terminal protein inhibitors modulate natural killer cell function and differentiation
Scientific Reports (2026)
-
Characterization and therapy of fertilization failure in murine and human models with HNRNPR mutations
EMBO Molecular Medicine (2026)
-
Targeting epigenetic readers
Nature Chemical Biology (2026)
