
Abstract
Macrophages are immune cells belonging to the mononuclear phagocyte system. They play crucial roles in immune defense, surveillance, and homeostasis. This review systematically discusses the types of hematopoietic progenitors that give rise to macrophages, including primitive hematopoietic progenitors, erythro-myeloid progenitors, and hematopoietic stem cells. These progenitors have distinct genetic backgrounds and developmental processes. Accordingly, macrophages exhibit complex and diverse functions in the body, including phagocytosis and clearance of cellular debris, antigen presentation, and immune response, regulation of inflammation and cytokine production, tissue remodeling and repair, and multi-level regulatory signaling pathways/crosstalk involved in homeostasis and physiology. Besides, tumor-associated macrophages are a key component of the TME, exhibiting both anti-tumor and pro-tumor properties. Furthermore, the functional status of macrophages is closely linked to the development of various diseases, including cancer, autoimmune disorders, cardiovascular disease, neurodegenerative diseases, metabolic conditions, and trauma. Targeting macrophages has emerged as a promising therapeutic strategy in these contexts. Clinical trials of macrophage-based targeted drugs, macrophage-based immunotherapies, and nanoparticle-based therapy were comprehensively summarized. Potential challenges and future directions in targeting macrophages have also been discussed. Overall, our review highlights the significance of this versatile immune cell in human health and disease, which is expected to inform future research and clinical practice.
Similar content being viewed by others
Introduction
Macrophages are immune cells widely distributed in the blood and tissue of the body. They, along with peripheral blood mononuclear cells and dendritic cells (DCs), belong to the mononuclear phagocyte system (MPS), playing important roles in immune defense, surveillance, and homeostasis.1,2 At least three types of hematopoietic progenitors: primitive hematopoietic progenitors, erythro-myeloid progenitors (EMPs), and hematopoietic stem cells (HSCs) exist in vertebrates, which have distinct genetic backgrounds and developmental process.3,4 Primitive hematopoietic progenitors are of RUNX1-independent origin, and have been reported to produce macrophages in zebrafish and mice; however, macrophages fail to develop at the stage of RUNX1 absence in humans and mice.5,6,7,8,9,10 Thus, whether macrophages could originate from primitive hematopoietic progenitors is uncertain.3 Existing research suggests that tissue-resident cells mainly originate from the yolk sac EMPs and EMP-derived macrophage precursors (PreMacs) in the embryonic stage, and they migrate and colonize in specific tissue sites for further differentiation and maturation. They could self-renew locally and persist in the tissue with or without the complement of HSCs-derived monocytes.11,12 Noticeably, the identity of macrophage populations is imprinted by their resident tissue, and tissue-specific transcriptional programs are essential for the maintenance, phenotype, and function features of tissue macrophages.12,13,14,15,16,17
Macrophages play complex and diverse roles in almost all aspects of biological processes in the body.18 As an important component of innate immunity, macrophages could migrate into damaged sites induced by chemokines, such as CCL2 and CX3CL1, during infection, inflammation, and tissue damage. They ingest diverse pathogens, apoptotic and dead cells, and tissue fragments by phagocytosis and further digest them in phagolysosomes.3,19,20,21 In addition, they could capture and endocytose antigens and further present them to other immune cells, such as T and B cells, thus involved in initiating the adaptive immune responses.1,22,23 In addition, macrophages could secrete various active substances (cytokines, chemokines, complements, enzymes, etc.) to regulate inflammation and immune responses.1,24,25,26 Besides, macrophages could sense various physiological and pathological signals and then participate in tissue repair and remodeling as well as metabolic regulation by removing necrotic cells and cell debris, regulating inflammation, and providing nutrition support for the proliferation and repair of cells, etc.27,28,29,30
The tumor microenvironment (TME) is a complex environment composed of cellular components such as tumor cells, multiple immune cells, endothelial cells, and non-cellular components such as extracellular matrix.31,32 Tumor-associated macrophages (TAMs) is a key component of TME, accounting for about 50% of hematopoietic cells.33 TAMs have both anti-tumor and pro-tumor properties. TAMs could promote tumor progression by promoting tumor cell proliferation and invasion, increasing the activity of tumor stem cells, inducing angiogenesis, and inhibiting the activity of cytotoxic T cells and natural killer (NK) cells,34 while they could also play an anti-tumor role by the phagocytosis and secretion of pro-inflammatory cytokines for the activation of adaptive immune cells.35 Furthermore, the functional status of macrophages is closely related to the development of various diseases, such as rheumatoid arthritis (RA), atherosclerosis (AS), Alzheimer’s disease (AD), diabetes, obesity, trauma, etc.36,37,38,39,40,41 With the development of diverse technologies (gene editing, nano-drug delivery system, etc.) and immunotherapies, targeting macrophages to treat diseases has become a promising therapeutic strategy.42,43,44
In this review, we summarize the research history, origin heterogeneity, and polarization of tissue macrophages. We also discuss their biological functions and roles in cancerous and non-cancerous diseases. Correspondingly, we illustrate multiple therapeutic strategies for targeting tissue macrophages.
Origins and heterogeneity of tissue macrophages
Research history and milestone events of study on macrophages
In 1882, experimental pathologist Elie Metchnikoff found that a group of mobile cells around the rose thorn-pierced starfish larvae could quickly clear foreign material. Under the suggestion of zoologist Claus, he named them “phagocytes”, which was considered the discovery of macrophages, and eventually published his first paper on phagocytosis in 188345 (Fig. 1). In 1887, Metchnikoff classified phagocytes into two populations: macrophages and microphages (later known as neutrophils).46,47 Since then, a series of concepts about the mononuclear phagocytic system (MPS), such as the “reticulo-endothelial system” of Aschoff and the “reticulo-histiocyte system” proposed and reintroduced by Volterra and Thomas, have been published. However, both of them are proven limited and incorrect several years later.47,48

A timeline of research history and milestones of macrophages. NO Nitric Oxide, TAMs tumor-associated macrophages, CAR-M CAR-macrophage, CAR-iMac induced pluripotent stem cells (iPSCs)-derived CAR-expressing macrophage cells
In 1968, based on the similarities in origin, morphology, function, and kinetics of the phagocytes, Ralph van Furth et al. redefined the concept of MPS. They excluded the reticular cells, endothelial cells, dendritic cells, and fibroblasts from the MPS and proposed that macrophages mainly arise from monocytes derived from myeloid progenitor cells.48 Over the past few decades after the proposal and amendment, macrophages were thought to have lost their differential and proliferative potential, only to be continuously replenished by circulating monocytes in the blood. However, from the 1990s to 2010s, scientists realized that there is an embryo-derived macrophage lineage with completely different characteristics in heredity, development, and function compared to those derived from HSCs and circulating monocytes.3,49,50 These macrophages are termed “tissue-resident macrophages” (TRMs) due to their persistence in the body and close association with specialized tissue cells.3
Colony stimulating factor-1 (CSF-1), also known as macrophage colony-stimulating factor (M-CSF), was discovered by Bradley et al. and purified by Stanley et al. It is the first cytokine identified to stimulate hematopoietic cells to differentiate into macrophage colonies.51,52,53 In 1987–1988, Hibbs et al. found that macrophages could kill tumor cells by producing nitric oxide (NO), a product of arginine metabolism.54,55 In 2000, Oldenborg et al. demonstrated that CD47, an important ‘self’ marker on the cell surface, could bind to SIRPɑ on the surface of macrophages, generating a series of signaling cascades that inhibit the phagocytosis of macrophages.56,57 In 2000, Mills et al. classified macrophages into two subtypes, M1 and M2, based on their differences in metabolism, secretion, and function,58,59 laying the foundation for a series of follow-up studies on macrophage polarization. Between the 1990s and 2010s, Mantovani and Pollard et al. detailed the role of macrophages in tumor growth, invasion and metastasis, angiogenesis, and immunosuppression.60,61,62,63,64
In 1990, Andreesen’s team used monocyte-derived macrophages (MDMs) on 15 advanced cancer patients who had failed in conventional treatments. It is the first clinical trial of cancer immunotherapy involving macrophages. The results showed that while the primary tumor tissue did not completely disappear, some patients remained stable within six months after treatment. Importantly, no serious adverse events were found except for low fever and discomfort at the intraperitoneal injection site. However, challenges such as the failure to transport macrophages to the tumor site or the lack of plasticity of macrophages leads to a rapid loss of their anti-tumor phenotype.65,66 In the following decades, researchers explored numerous methods to enhance the efficacy of macrophage-associated therapies, including combining them with other treatments and applying new techniques such as gene editing. In 2020, Gill et al. engineered human macrophages using chimeric antigen receptor (CAR) to direct their phagocytic activity against tumors. CAR macrophages (CAR-M) exhibited the antigen-specific phagocytosis and ability to clear tumor in vitro, and it was further shown to induce a proinflammatory TME and enhance anti-tumor T cell activity in humanized mouse models.67 Besides, in 2020, Zhang et al. developed induced pluripotent stem cells (iPSCs)-derived, CAR-expressing macrophage cells(CAR-iMac) by introducing CAR into hiPSCs and making it differentiate into macrophages. This study showed that CAR-mediated signaling could significantly improve the ability of CAR-iMac to engulf tumor cells and lead to the transformation of CAR-iMac from M2 to M1 type in the presence of specific antigens such as CD19.68 In 2020, the FDA approved a Phase I clinical trial, NCT04660929, aimed at treating tumor patients with relapsed/refractory HER2 over-expression with anti-HER2 CAR macrophages (CT-0508, CARISMA Therapeutics). It is the first trial to study the effects of adenovirus transduction CAR-M in humans.69
Developmental origins of macrophages
Macrophages’ origin, migration, and development is a complex process with high similarity between humans and mice.11,70 As described earlier in the MPS section, from the beginning, it was thought that macrophages in the human body’s tissues were entirely derived from the HSCs. However, a series of studies showed that macrophages in adult tissues were mainly derived from the yolk sac or fetal liver during the embryonic development.3,11 Mouse fate mapping studies have shown that yolk sac-derived EMPs have at least two distinct waves at the origin: at embryonic day 7.5 (E7.5), the yolk sac produces the first wave of EMPs, and they differentiate into macrophages in situ. Then macrophages migrate into the brain rudiment and become the major source of microglia.8,71,72,73 The yolk sac at E8.25 produced the second wave of EMPs. Firstly, this wave of EMPs migrates into the fetal liver. In the fetal liver, EMPs could differentiate into EMP-derived macrophage precursors (PreMacs), which could further develop into heterogeneous TRMs, such as epidermal Langerhans cells and liver Kupffer cells, after transmitting with blood and colonizing in various tissues(except brain tissue).3,8,72,73,74,75,76 EMPs disappear during fetal life, but TRMs and some mast cells persist and self-renew in the adulthood.3
The definition of TRMs and whether they could originate from HSCs remains controversial. Except for one study, the existing fate-mapping studies have shown that fetal HSCs derived from the Aorta-gonad-mesonephros region could not produce macrophages.3,50,77,78,79 Besides, multiple experimental evidence indicates that TRMs are locally self-renewing long-lived cells in tissues. In contrast, short-lived HSC-derived macrophages rely on circulating monocytes for renewal and could expand massively when receiving stimuli.3 Several studies about experimental brain inflammation (autoimmune encephalitis (EAE) and stroke) have shown that monocytes and bone marrow monocyte-derived macrophages (BMDMs) recruited in the brain mediate inflammation and gradually disappear during inflammatory remission. In contrast, microglia do not mediate inflammation and persist consistently.77,80 However, the following two examples may support TRMs’ HSC origin. Osteoclasts in the embryonic period contain EMP-derived nuclei and self-maintain in adult bones, and they could integrate HSC-derived nuclei by fusion, resulting in individual adult mouse osteoclasts being a chimera containing nuclei of EMPs and HSCs.3,81 Besides, gut lamina propria is replenished by HSCs and BMDMs, which have the property of self-maintaining.3,82,83,84
Classically activated and polarized macrophages
Macrophage polarization is a process in which macrophages produce specific phenotypes and functional responses to microenvironmental stimuli and signals.85,86 In 2000, Mills et al. classified macrophages into two types, M1 and M2, according to their differences in activation patterns and functions, etc.59 (Fig. 2). CD68 is a marker expressed by all monocytes and tissue macrophages.87 Firstly, monocytes are stimulated by CSF-1 to form M0 macrophages. Then, M0 macrophages differentiate into M1 macrophages upon activating lipopolysaccharide (LPS) and Th1-type cytokines such as IFN-γ and TNF-ɑ. In contrast, M2 macrophages are formed by activating Th2-type cytokines such as IL-4, IL-10, and TGF-β.88 In addition to the activation patterns, there are other differences in receptor expression, cytokine production, and functions between these two types of macrophages.

CSF-1 could induce monocytes to differentiate into M0 macrophages. Then, M0 macrophages could further evolve into M1 or M2 macrophages stimulated by Th1-type or Th2-type cytokines. Due to their differences in activation patterns and other aspects, M2 macrophages could be further divided into four subtypes: M2a, M2b, M2c, and M2d. Various macrophage types express different molecular markers and secrete different materials, which play important roles in various physiological and pathological processes. CSF-1 colony stimulating factor-1, MHC-II major histocompatibility complex class II, iNOS inducible nitric oxide synthase, LPS Lipopolysaccharides IFN-γ interferon-gamma, TNF-ɑ tumor necrosis factor-alpha, IL-1β interleukin-1β, IL-8 interleukin-8, IL-12 interleukin-12
From the perspective of signaling pathways, the polarization of macrophages is primarily governed by several key pathways. The TLR4/NF-κB89 and IFN-γ/JAK/STAT190 pathways predominantly drive M1 polarization, activating pro-inflammatory profiles in macrophages. Conversely, the PI3K/Akt/mTOR,91 TGF-β/Smad,92,93 NOTCH,94 Wnt/β-catenin,95,96,97,98 IL-4/IL-6/JAK/STAT399,100/STAT6,101 TRAF3/STAT6,102 and Hedgehog103 pathways mainly dictate M2 polarization, promoting anti-inflammatory and pro-tumoral activities. Research has shown that PI3Kγ, a critical isoform of PI3K,104 promotes M2 macrophage polarization through MTORC1-dependent activation of CCAAT enhancer binding protein β (CEBPB) and integrin subunit α 4 (ITGA4).105,106 This action inhibits the key pro-inflammatory transcription factor NF-κB signaling pathway,107 thus contributing to an immunosuppressive TME. In colorectal cancer, tumor cells release IL-4 to promote the expression of CD155 on TAMs,108 thus facilitating their M2 polarization and increasing the expression of IL-10 and TGF-β, thereby supporting tumor progression. In glioblastoma, CXCL8 supports the M2-like phenotype in TAMs through the CXCR2-JAK2/STAT3 axis,109 further highlighting the role of these signaling pathways in influencing macrophage behavior and impacting disease outcomes in different types of cancers.
M1 macrophages, also known as classically activated macrophages, mediate ROS-induced tissue damage and exhibit strong anti-microbial and anti-tumoral activity.86 These macrophages express or secrete markers such as MHC-II, iNOS, CD80, TGF-α, IL-1β, IL-8, and IL-12, contributing to their pro-inflammatory and immune-stimulating roles.86,110 On the other hand, M2 macrophages, often referred to as alternatively activated or immunosuppressive macrophages, are further classified into four subtypes based on different stimulatory factors: M2a, M2b, M2c, and M2d. Each subtype exhibits distinct surface markers and secretes specific cytokines and chemokines, playing crucial roles in inflammation resolution, tissue repair, and tumor progression.86,88
M2a macrophages, primarily induced by the cytokines IL-4 and IL-13, are one of the most widely studied subsets of M2 macrophages. First characterized in 1992, these macrophages are distinguished by the high surface expression of markers such as CD206, CD209, and Dectin-1. These are critical for recognizing and eliminating invading pathogens like bacteria, fungi, and parasites. Alongside these markers, M2a macrophages exhibit variable expression of CD14, CD163, and CD80/86, ranging from low to medium levels. Functionally, M2a macrophages secrete a variety of anti-inflammatory and tissue remodeling molecules, including IL-10, CCL17, CCL18, CCL22, CCL24, and the enzyme arginase 1 (Arg1), which plays a key role in amino acid metabolism and matrix reorganization.111,112,113 A key function of M2a macrophages is promoting tissue repair and remodeling. Upon tissue injury, IL-4 activates M2a macrophages, producing L-ornithine, a precursor for collagen and polyamines, essential for extracellular matrix (ECM) formation. M2a macrophages also secrete fibronectin and chitinase-like proteins, which aid in ECM reorganization and wound healing. However, fibronectin supports tissue repair and promotes tumor cell proliferation, invasion, and migration, contributing to tumor progression. Additionally, arginase-1 (Arg1) expressed by M2a macrophages depletes L-arginine, inhibiting T-cell proliferation and suppressing immune responses, while IL-10 further reduces pro-inflammatory cytokine production and T-cell activation. This immunosuppressive environment facilitates tumor growth, angiogenesis, and metastasis, making M2a macrophages and their secreted factors potential targets for cancer therapy.114,115
M2b macrophages, primarily activated by TLR agonists, immune complexes (ICs), and IL-1β, are known for their dual role in immune regulation and tumor progression.86,116 Unlike other M2 subsets, M2b macrophages produce high levels of anti-inflammatory cytokines, such as IL-10, while suppressing pro-inflammatory IL-12, facilitating a shift from Th1 to Th2 responses. They express CD86 and secrete IL-6, TGF-α, and CCL1, contributing to immune escape mechanisms and promoting tumor growth. The polarization of M2b macrophages requires two stimuli, typically ICs and LPS or IL-1β, which activate signaling pathways like NF-κB and PI3K/Akt. Their ability to suppress immune responses while promoting tissue repair and tumor progression highlights their unique regulatory function within the TME.117,118
M2c macrophages, stimulated by immunosuppressive molecules such as IL-10, TGF-β, and glucocorticoids, are characterized by CD163, MerTK, and CD206 expression. These macrophages secrete anti-inflammatory cytokines like IL-10 and TGF-β and chemokines such as CCL16, CCL18, and CXCL13,119,120 contributing to immune suppression and tissue remodeling. They play a key role in promoting tumor immune evasion and efficiently clearing apoptotic cells via MerTK-mediated phagocytosis.116,121,122 Additionally, M2c macrophages degrade the extracellular matrix through the secretion of matrix metalloproteinases (MMP7, MMP8, MMP9) and TIMP1, further aiding tissue repair and sustaining anti-inflammatory responses. Their ability to capture and sequester inflammatory chemokines through decoy receptors like CCR2 and CCR5 also highlights their immunoregulatory functions.119,120
M2d macrophages, first identified in the ascites of ovarian cancer patients, are polarized by IL-6 and leukemia inhibitory factor (LIF), with additional activation by adenosine receptor agonists and TLR agonists.86,123,124,125 They exhibit a typical M2 cytokine profile with high IL-10 and low IL-12, expressing markers like CD14, CD163, and TGF-β while showing low levels of CD86. M2d macrophages secrete VEGF, IL-10, and CCL18, contributing to tumor progression by promoting angiogenesis and suppressing T cell proliferation, thereby facilitating immune evasion and tumor growth.126,127
The plasticity of macrophages, often called ‘repolarization’ or ‘reprogramming,’ allows them to shift between these phenotypes in response to environmental cues.128,129 While this flexibility is crucial for mitigating inflammation and facilitating tissue repair in chronic infections, it can also promote tumor malignancy. The presence of M2 macrophages, with their immunosuppressive and pro-tumoral activities, is often associated with poor prognosis in various cancers. Thus, understanding each macrophage subtype’s specific markers and secreted factors provides key insights into their roles in both physiological processes and disease progression.
The M1/M2 polarization classification of macrophages is currently one of the dominant perspectives in the field and is widely applied to describe the functional roles of macrophages in various pathological conditions. The advantage of this classification is that it provides researchers with a clear framework to understand the dual roles of macrophages in immune responses. M1 macrophages are generally associated with pro-inflammatory and anti-tumor activities, capable of eliminating pathogens and inhibiting tumor growth by secreting inflammatory factors such as IL-12, tumor necrosis factor-alpha (TNF-α), and NO. On the other hand, M2 macrophages are involved in anti-inflammatory responses, tissue repair, and immunosuppression, secreting factors like IL-10 and TGF-β, which aid in tissue regeneration and immune regulation. Therefore, the M1/M2 classification offers an important directional guide for understanding the biological characteristics of macrophages in various diseases. This classification allows researchers to quickly identify the general functional tendencies of macrophages in a particular disease, especially in areas like inflammatory response, tumor immunity, and tissue repair, making it a foundational framework for macrophage research.
However, the limitations of this binary classification have become increasingly apparent, particularly in complex diseases such as cancer, autoimmune disorders, and cardiovascular diseases. While M1 and M2 polarization help researchers outline macrophage functions, the situation is far more complex. With the advancement of single-cell sequencing technology, we now recognize that macrophages exhibit significant heterogeneity across different disease microenvironments, and the simplistic M1/M2 classification fails to capture the full scope of macrophage functionality. Various macrophage subpopulations display distinct biological characteristics in different disease contexts, and the functional differences between these subtypes play a crucial role in disease progression and treatment response. Furthermore, the general M1/M2 classification may hinder the development of precision therapies in specific disease settings. For example, in the TME, some M2-type macrophages may both promote tumor progression and participate in tissue repair, while M1 macrophages, under certain conditions, may exacerbate inflammation or tissue damage. Therefore, although the M1/M2 classification provides a simple framework, its limitations in explaining and treating complex diseases have led researchers to adopt more refined subpopulation analyses. This more nuanced approach helps uncover the specific roles of different macrophage subtypes in various diseases, providing a critical foundation for personalized precision therapies.
Heterogeneity, molecular markers, and phenotypic characteristics of macrophages
With the development of single-cell sequencing, lineage tracing, and other technologies, the heterogeneity of macrophages has been gradually understood and deciphered.71,130 Its heterogeneity is manifested in differences in many aspects, such as origin, distribution, stimuli, antigen expression, secretory factors and functions, etc.130,131 By using single-cell sequencing techniques, researchers could track and map the pathways of macrophage maturation from precursor cells, providing key insights into their origin and developmental processes.11,50,132,133 Furthermore, macrophages display high heterogeneity across different tissues and microenvironments, which is difficult to capture by traditional bulk transcriptome sequencing methods. Single-cell sequencing analysis allows for high-resolution analysis of gene and protein expression in macrophages, revealing unique subpopulations and functional characteristics in various locations, such as the liver, lungs, kidneys, brain, etc.134,135,136,137,138 Additionally, researchers could use a range of softwares or websites(such as CellChat, CellPhoneDB, etc.) to analyze single-cell sequencing data and construct interaction networks between different cell populations in the immune microenvironment, which could be applied for the identification of interactions between macrophages and other microenvironmental cells, as well as related secreted factors, ligands/receptors, and signaling pathways. These insights are crucial for understanding the roles and mechanisms of macrophages within the immune microenvironment.139,140,141
Human and mouse macrophages share a high degree of similarity in important functions such as phagocytosis, antigen presentation, inflammatory responses, and tissue repair, which makes the mouse an effective animal model for macrophage-related research.142,143 However, these two types of macrophages differ from one another in many aspects, such as the regulation of transcription factors, expression of surface markers, molecules involved in antigen presentation pathways, and drug responses, etc.144,145 For example, human macrophages express high levels of CD14 and human homolog of MHC-II(HLA-DR). In contrast, mouse macrophages could specifically express F4/80, Arginase-1(Arg-1) and Chitinase-like protein 3(YM1).146,147,148,149,150,151 Furthermore, the grouping of macrophage subtypes differs between humans and mice in different tissue and organs.152 Therefore, when designing experiments and analyzing data involving macrophages, it is essential to carefully consider the species-specific differences to ensure accurate and relevant results.
Macrophages in the bone
Bone macrophages could be divided into the following types: bone marrow macrophage (such as erythroblastic island macrophage, HSC niche macrophage, etc.), osteoclasts, and osteal macrophages (osteomacs).153,154
The erythroblastic island (EBI), the first hematopoietic niche to be discovered, primarily consists of a central erythroblastic island macrophage(EIM) surrounded by a cluster of immature erythroid precursor cells.155,156 HSCs-derived myeloid progenitors could differentiate into monocytes, and when monocytes enter the EBI microenvironment in the bone marrow, they further differentiate into EIMs.155,157 Research has shown that over 90% of mouse native EBIs are F4/80+ EPOR+ macrophages and EBI in the human fetal liver was also found to contain macrophages that express the EPOR.157 Besides, F4/80+ EPOR+ EIMs also express molecules such as CD169, VCAM1, Mertk, and DNase2α, etc., which are closely related to their functions.157The highly expressed adhesion molecules VCAM1 and CD169 (Siglec-1) on EIMs could bind to integrin ɑ4β1(VLA-4) and CD43, the ligands on the surface of erythroid precursor cells, respectively. This intermolecular interaction enables the EIMs to support the proliferation and differentiation of erythroid precursor cells.158,159 During the erythropoiesis, erythroid precursor cells undergo enucleation, where they shed their nuclei to become fully functional red blood cells.160,161 Mertk mediates the engulfment of pyrenocytes by EIMs, and DNase2ɑ is the key enzyme responsible for removing DNA left over from the enucleation of erythroid precursor cells, which enables EIMs to maintain a clean erythropoiesis environment for supporting the differentiation and maturation of erythroid precursor cells.162,163
Osteoclasts are present in the bone marrow, spleen, and blood. In bone, they are located in the resorption bays within the bone endosteum and specialize in bone resorption and remodeling by releasing proteolytic enzymes and acids.164,165 Of all the enzymes secreted by osteoclasts, tartrate-resistant acidic phosphatase (TRAP) is unique to osteoclasts and often serves as an important marker for identifying osteoclasts.12 Besides, the receptor activator for the Nuclear Factor-κb ligand (RANKL) is considered one of the most important factors in promoting the maturation and maintenance of the activity of osteoclasts. M-CSF could induce RANK expression on the cell membrane of the osteoclast precursor, which binds RANKL from osteoblasts, stromal cells, or T cells and produces an effect inducing osteoclast maturation and differentiation.12,13 As mentioned above, they are embryonically derived but could fuse with nuclei from HSC-derived macrophages, finally becoming multinucleated cells in adults. Defective osteoclast activity could contribute to osteopetrosis and bone marrow failure, while excess activity could result in bone loss and osteoporosis.164 Besides, an iterative fusion of circulating blood monocytic cells with long-lived osteoclast syncytia is crucial for the postnatal maintenance of osteoclasts, bone mass, and the bone marrow cavity.164
Osteal macrophages(OsteoMacs) originate from a resident population of macrophages and express typical macrophage markers such as CD68, F4/80(mouse), and CD169, but lack TRAP expression, which distinguishes them from osteoclasts.166,167,168,169 They are located adjacent to osteoblasts, osteoclasts, and dormant cells. They exhibit a stellate morphology that allows them to extend across bone surfaces, suggesting they may form an extensive communication network.167 OsteoMacs play an important role in bone formation and repair. They could regulate the activity of osteoblasts and the mineralization process of the bone matrix, but more experimental evidence is needed to support the mechanisms involved.170 Besides, it has been reported that they could also serve as immune surveillance cells in the bone microenvironment. This subset of macrophages is capable of clearing necrotic cells and debris through phagocytosis as well as responding to antigens.168,171,172,173 Notably, OsteoMacs support the maintenance of murine hematopoiesis by the megakaryocyte-induced up-regulation of Embigin and CD166.174,175
Macrophages in the peripheral blood
Monocytes, one of the precursors of macrophages, circulate in peripheral blood for approximately 1–3 days after producing and releasing by the bone marrow. Then, they migrate into tissues and subsequently differentiate into various types of macrophages or DCs.176,177 Human monocytes could be divided into three subsets: the classical (CD14++ CD16-), intermediate (CD14++ CD16+), and nonclassical (CD14+ CD16++).177 When incubated with GM-CSF or CSF-1, all three subsets acquired macrophage morphology, secreted cytokines associated with macrophages, and showed increased phagocytic activity.178 In mice, monocytes are categorized into two subsets: the Ly6Chigh and Ly6Cmiddle subsets based on their Ly6C expression. The Ly6Chigh subset, which perform pro-inflammatory functions and express high levels of CCR2, are more likely to differentiate into inflammatory M1 macrophages.Conversely, the Ly6Clow monocytes patrol along the vascular endothelium, participate in tissue repair, and tend to mature into M2 macrophages.176
Macrophages in the lung
In the homeostatic lung, there are several major macrophage populations separated by their anatomical location: alveolar macrophages (AMs) in the air-exposed space of the alveolus and two or three interstitial macrophage populations in the interstitial region of lung.131,179
Sialic acid-binding immunoglobulin-like lectin (Siglec)-F (SiglecF), a mouse cell surface glycoprotein, could be used to distinguish AMs from other types of lung macrophages, playing an important role in inflammation regulation, pathogen clearance, and the inhibition of autoimmune responses.180,181,182,183,184,185 AMs derived from Yolk sac EMPs and characterized with CD11c+ SiglecF+ CXC3R1−. They fill the alveolar spaces of the lungs after birth and are self-maintained in homeostasis.12,186 The production and maturation of AMs depend on GM-CSF and TGF-β-mediated induction of peroxisome proliferator-activated receptor γ (PPARγ), a transcription factor crucial for regulating lipid metabolism.12,187,188,189 AMs play an important role in the clearance of pulmonary surfactant, phagocytosis of inhaled particles, and immunosurveillance.12,130
The level of CX3CR1 expressed on monocytes is increased with maturation in bone marrow, which is inversely correlated with the Ly6C marker and CCR2 in the blood. Notably, CX3CR1 was reported to be associated with maturation, recruitment into specific sites, and immunomodulatory effects of monocytes and macrophages.190,191,192,193 Lung interstitial macrophages (CXC3R1+ CD11b+ SiglecF-) are located primarily between the epithelium and the capillary, which are crucial for immune surveillance in the lungs.17,194 They originate from monocytes in the embryonic stage, whose development is largely dependent on homeostatic CSF-1R signaling and could be supplemented by circulating monocytes in adulthood.131 They are rare in homeostasis but could increase significantly when faced with immune challenges.194 Besides, they could be further divided into two subsets (LYVE1hi MHC-IIlow and LYVE1low MHC-IIhi) or three subsets (based on the expression levels of FOLR2, CCR2, TIM4, LYVE1, and MHC-II).131,136,195,196,197 In addition, with the development and application of single-cell sequencing techniques, multiple studies have been conducted to investigate the heterogeneity and functions of macrophages in lung homeostasis and disease states, enhancing our understanding of lung macrophages and diseases associated with them.136,137,198,199
Macrophages in the liver
The macrophages in the liver include many populations: Kupffer cells (KCs), liver capsular macrophages (LCMs), central vein macrophages, lipid-associated macrophages, etc.200 KCs constitute the largest group of resident macrophages in the human body, accounting for approximately 80% to 90% of the resident macrophage population.201,202 They are derived from Yolk sac EMPs and line in the sinusoids of the liver in adulthood. KCs have various functions, with F4/80+, CLEC4F+, and TIM4+ recognized as their markers.12,186 C-type lectin domain family 4 member F (CLEC4F), an inducible C-type lectin, could be considered a characteristic marker of KCs.1,203 It could recognize and bind to specific carbohydrate structures, thus participating in the recognition and clearance of pathogens.203,204
KCs could remove cellular debris, senescent cells, and pathogens from the blood. Besides, They promote immune tolerance and are involved in the systemic metabolism of iron, cholesterol, and other lipids.205 Two distinct KC populations were reported in mice: KC1 (CD206Low ESAM- CD36Low) and KC2 (CD206hi ESAM+ CD36hi). KC2 populations have higher metabolic activity, express high lipid and carbohydrate metabolism-related genes, and could regulate liver metabolism via the fatty acid transporter CD36.206 LCMs are monocyte-derived F4/80+ CX3CR1+ MHC-II+ cells and have been reported to join in neutrophil recruitment and immune surveillance.201,207 In addition, a single-cell RNA sequencing (scRNA-seq) study based on human liver tissue attempted to explore the grouping of intrahepatic macrophages, showing that there are two distinct populations of intrahepatic macrophages expressing CD68 across all liver samples analyzed.CD68+ macrophage population 1, characterized by high LYZ, CSTA, and CD74 expression, could represent inflammatory macrophages. As for CD68+ macrophage population 2, it is considered to have a tolerogenic function due to their high expression of MARCO and VSIG4, etc, which are associated with immune tolerance.136
Macrophages in the heart
Cardiac and pericardial macrophages are crucial in maintaining heart homeostasis and responding to pathological conditions.208 In recent research based on the scRNA-seq and other experimental technologies, human cardiac macrophages could be divided into three distinct groups according to the expression levels of CCR2 and HLA-DR: CCR2+ HLA‐DRlow, CCR2+ HLA‐DRhi and CCR2‐ HLA‐DRhi cells.209 Similarly, in a recent study utilizing a combination of cell tracking and scRNA-seq, researchers identified four transcriptionally distinct macrophage populations in mice: TIMD4 cluster (TIMD4+ LYVE1+ MHC-IIlow CCR2-), MHC-II cluster (TIMD4- LYVE1- MHC-IIhi CCR2-), CCR2 cluster (TIMD4- LYVE1- MHC-IIhi CCR2+), and ISG cluster (TIMD4- LYVE1- MHC-IIhi CCR2+) were observed.210 TIMD4 cluster operates independently of blood monocytes while maintaining the ability for self-renewal. In contrast, the MHC-II cluster shows partial replacement by monocytes, and the two CCR2+ subpopulations, the CCR2 and ISG clusters, are fully derived from monocytes.210 The population of TIMD4+ macrophages plays a key role in reducing inflammation and facilitating tissue repair through the phagocytosis of apoptotic cardiomyocytes.211,212 Moreover, they are crucial in regulating fibrosis and aiding in the repair process after cardiac injury, contributing to the maintenance of the structural and functional integrity of the heart.209 Notably, LYVE1 on macrophages plays a vital role in preserving vascular homeostasis by interacting with hyaluronic acid, which is expressed on the surface of smooth muscle cells.213
Beyond the heart tissue, the mouse pericardial cavity surrounding the heart is populated by two main macrophage populations: Gata6+ pericardial and MHCII+ pericardial macrophages.208,214 Gata6+ pericardial macrophages displayed transcriptional profiles similar to those of Gata6+ macrophages found in the peritoneal and pleural cavities, but differed from the profiles of resident cardiac macrophages.These cells express the transcription factor GATA6, which is crucial in regulating cardiac damage and preventing fibrosis after myocardial infarction.208,214 In contrast, MHCII+ pericardial macrophages, characterized by high levels of MHCII molecules, are primarily responsible for antigen presentation, which promotes adaptive immune responses. These macrophages assist in the recognition and clearance of damaged tissues during cardiac injury or inflammation.208,214
Macrophages in the spleen
In the spleen, at least four distinct subsets of macrophages are present in discrete anatomic regions: the red pulp, the white pulp, and the marginal zone separating the two, which have significant spatial, phenotypic, and functional diversities.130,215 Red pulp macrophages (RPMs) are thought to originate from the Yolk sac and fetal liver progenitors. In contrast, the other three subsets(marginal zone metallophilic macrophages, marginal zone macrophages, and white pulp macrophages) derive from adult bone marrow/blood monocytes.12 RPMs(F4/80+ VCAM1+ CD11bLow) could engulf and clear the senescent red blood cells, platelets, and other cells from the blood, which is vital for avoiding the development of autoimmune responses as well as the recovery of iron and heme.1,27,216 Noticeably, they rely on a series of transcription factors (Spi-C, NRF2, PPARγ, LXRɑ, and SREBP1) to regulate the active metabolism of iron and lipids, further regulate the homeostasis of splenic cells and red blood cells.3,186 Furthermore, VCAM1, a downstream molecule of Spi-C, has also been reported to be associated with immune regulation and iron metabolism.217,218,219 However, the effect of its expression on RPMs, particularly in relation to macrophage function, still requires further research.
A distinct CD169+ metallophilic macrophage subpopulation in the marginal zone of the mouse spleen could interact with the antibody-producing B lymphocytes and DCs, playing important roles in sinusoidal immunity.220,221 Besides, a MARCO+ SIGNR-1+ macrophage subpopulation in the outer section of the marginal zone is reported to participate in antigen capture.222 White pulp macrophages (WPMs) (CD68+, F4/80-) are located in B cell follicles of the white pulp, which are similar to tingible body macrophages in the germinal centers of lymph nodes,222 and they participate in the phagocytosis and clearance of apoptotic B cells.130
Macrophages in the lymph nodes
A group of sinusoidal CD169+ macrophages that resemble the metallophilic cells in the marginal zone of the spleen are located in the subcapsular sinuses of the lymph nodes, and they could deliver captured antigens to DCs for the activation of B and T lymphocytes.130,223 Medullary macrophages express CD68 and F4/80, the expression of which could be strongly enhanced by the phagocytosis of apoptotic lymphocytes. Notably, lymph nodes are often referred to as a graveyard for macrophages, as they undergo local turnover within this site.130
Macrophages in the intestine
In anatomy, the walls of both the large and small intestines could be subdivided into four layers: mucosa, submucosa, muscularis propria, and serosa (or adventitia in certain regions). The mucosa is further subdivided into three sections: the epithelium, lamina propria, and muscularis mucosae.131,224 Different macrophage subsets have been identified in different intestinal layers, with the lamina propria being the most abundant.225
Lamina propria macrophages (CD64+ MHC-IIhi CD206+) are in the mucosa lamina propria, located beneath the intestinal epithelial layer.226,227 They are initially fetal-derived and rapidly replaced by short-lived MDMs in a CCR2-dependent manner after birth and require live microbiota to thrive.228,229 Notably, Lamina propria macrophages are essential for gut barrier homeostasis. First, small intestinal lamina propria macrophages engulf surrounding material (such as apoptotic cells (ACs)) within the lumen and lamina propria, collect antigens, and support epithelial stem cell proliferation within intestinal crypts by providing Wnt ligands.230,231,232,233 Furthermore, macrophages in the lamina propria of the small intestine secrete large amounts of IL-10, which are critical for the induction of microbiota-specific regulatory T Cells.229,234
Besides, a group of long-lived macrophages could be found at the sub-tissular niches of the submucosa and the muscular external layers.231 The macrophages in the external layers express markers such as TIM4 and MHC-II and could be self-renewal in niches.1,228,235 Besides, they are close to blood vessels, as well as the submucosal plexus and muscular plexus, playing important roles in the maintenance of intestinal movement, as well as support for the growth and function of neuronal bodies in the enteric nervous system as well as blood vessels.227,236,237,238,239
Macrophages in the central nervous system
In the central nervous system (CNS), different populations of TRMs are found in defined anatomical locations: microglia in the CNS parenchyma and other macrophage subgroups located in the CNS interfaces, including ventricles, meninges, and perivascular space.
The mouse microglia have been reported to express F4/80, CX3CR1, and CD11b, which originate from Yolk sac EMPs without any contribution from HSCs in homeostasis.71,236,240 Specifically, a research indicated that CD45- c-kit+ erythromyeloid progenitors in the yolk sac could be identified as the source of immigrating macrophages in the developing brain and represent the direct precursor of the definitive microglia population in the CNS.241 Besides, other two researches using mouse fate mapping deemed that microglia may derive from Runx1+ or CD206+ macrophages.71,242 Notably, In the brain, only microglia express marker CX3CR1.241,243 In addition, microglia could be distinguished from other macrophage subtypes in the CNS by a group of markers, including SALL1, P2RY12, TMEM119, and HEXB.244 These molecules have been reported to participate in the transcriptional regulation, development, and differentiation, inflammation regulation, as well as neuroprotection function of microglia.245,246,247,248,249,250 After their settle-down in the CNS, the embryonic microglia could be self-maintained through a cell-autonomous proliferation.251,252 Once planted in its tissue niche, microglia depend on CSF-1 receptor (CSF-1R) ligands locally released by histiocytes, primarily CSF-1 released by neurons and IL-34 released by astrocytes, to mature, thus performing specific functions during the CNS development and homeostasis.253,254,255 Notably, adult microglia are long-lived cells that maintain a stable network throughout their life cycle only with rare proliferation.256,257 They act as immune sentinels to protect the brain from pathogens. Besides, they could also maintain brain homeostasis through various mechanisms, such as scavenging ACs as well as regulating neurogenesis and synaptic activity.258,259,260,261,262
Brain perivascular macrophages (PVMs) are a group of macrophages localized in the perivascular spaces of CNS, including the leptomeningeal macrophages, stromal choroid plexus macrophages, etc., with distinct zonation and phenotype compared to microglia.1,263 Besides, compared to the single origin of microglia, the origin of other macrophages in the CNS is more diverse. Some researches support that PVMs, such as leptomeningeal macrophages, seem to be only derived from Yolk sac EMPs without the contribution of HSC-derived progenitors and circulating monocytes during adulthood.242,264,265,266 However, another research shows that stromal choroid plexus macrophages originate initially from embryonic EMPs, but could be postnatally replaced by circulating monocytes. In addition, intraventricular macrophages, including the Kolmer epiplexus cells, are of embryonic origin, while dural macrophages could be partialliy replenish by monocytes.264,266 Brain PVMs were reported to regulate cerebrospinal fluid (CSF) flow dynamics via the control of arterial motion, and the TIM4+ subgroup in them could promote proper dynamics of the ECM.267
It is worth noting that once the blood-brain barrier (BBB) is compromised, monocytes and macrophages from peripheral blood could also enter the brain.268,269,270
Macrophages in the skeletal muscle
The lineage tracing and bone marrow transplant experiment results demonstrate that mouse skeletal muscle-resident macrophages are CD11b+ F4/80+ CD64+. They originate from embryonic hematopoietic progenitors in the yolk sac and fetal liver and definitive HSC in the bone marrow of adult mice.271 By using single-cell sequencing technology, researchers have identified three different macrophage subpopulations in skeletal muscle: a population of locally self-renewing F4/80+ LYVE1+ TIM4+ macrophages (also named self-renewing resident macrophages) and two other populations F4/80 + TIM4- macrophages and F4/80Low CD11C+ MHCII+ cells that are monocyte-derived.272 It has been reported that local CSF-1 from fibro-adipogenic progenitors (FAPs) is essential for the survival of both TIM4- monocyte-derived and TIM4+ self-renewing resident macrophages in adult skeletal muscle.273 Regardless of the muscle type, three transcription factor genes, Maf, Mef2c, and Tcf4, are differentially expressed by skeletal muscle macrophages.271 Notably, these macrophages could be important in maintaining tissue homeostasis and promoting muscle growth and regeneration.271
Macrophages in the kidney
There are two different kinds of macrophages in the kidney: kidney-resident macrophages (KRMs) and bone marrow-derived kidney macrophages (BMKMs).274 KRMs originate from Yolk sac EMPs, and the predominant markers in mice are CD64, F4/80, and CD11c.275,276 Besides, scRNAseq analysis and experimental results indicate that CD74 and CD81 may be potential cell surface markers for kidney resident macrophages in multiple species.138 Compared to those in other developing organs (brain, lung, and liver, etc.), kidney macrophages show increased expression of the transcriptional regulators Ahr, Irf9, Nfatc1, and Nfatc2, which are closely associated with the cytokine expression, secretion of NO and arginine, as well as activation of macrophages.50,277,278,279,280 KRMs could monitor and clear macromolecules, especially circulating immune complexes, which are transported through the capillary around the renal tubules. In addition, they may be involved in promoting renal vascular and ureteric bud branching development.275,276 Mouse KRMs showed metabolic quiescence in the homeostasis. In the lupus nephritis mouse model, the expression of OXPHOS and glycolysis genes was up-regulated, while the expression of fatty acid metabolism genes was down-regulated, suggesting that inhibition of this glycolytic switch by KRMs may be a therapeutic approach to control renal inflammation.281
Macrophages in white adipose tissue
Macrophages have been reported to play important roles in lipid metabolism, inflammatory responses, and energy expenditure in adipose tissue. Lean white adipose tissue (WAT) macrophages are predominantly derived from Yolk sac EMPs, and express markers like F4/80, CD11b, and CD206. Compared to macrophages in obese WAT, those located in lean WAT are generally metabolically quiescent, showing lower dependency on oxidative phosphorylation (OXPHOS).3,12 In obesity, mouse macrophages in WAT undergo dramatic remodeling in their state of cellular metabolism and function, leading to a pro-inflammatory state.12,282 Specifically, many factors induced by excessive lipid accumulation and hypertrophy of WAT, such as mechanical stress, hypoxia, etc., could stimulate macrophages in adipose tissue to secrete pro-inflammatory mediators, such as TNF-ɑ or IL-1β, which could further activate inflammatory pathways, such as JNK or IKKβ pathway in adipocytes. This mechanism is involved in many metabolic pathological processes or diseases, such as non-alcoholic fatty liver disease (NAFLD), insulin resistance, etc.12,283,284 Furthermore, inflammatory cytokines secreted by senescent cells could induce macrophages to proliferate and express the nicotinamide adenine dinucleotide (NAD)-consuming enzyme CD38, thus promoting a decrease in tissue NAD+ level during senescence.285
Macrophages in skin
Skin resident macrophages have two main cell types: Langerhans cells (LCs) and dermal macrophages (DMs). Fate-mapping studies revealed that skin LCs originate from embryonic fetal liver monocytes and yolk sac-derived macrophages.80,240,286 Adult epidermal LCs are live-long cells and could self-renew under homeostatic conditions. In contrast, endogenous LCs are replaced by monocyte-derived progenitors within their niche287,288 under severe inflammation, infection, or injury. Depending on transcription factors RUNX3, AHR, and ID2, LCs could further differentiate.7,289 By single-cell sequencing and mass-cytometry analysis of CD34+ HSCs derived human LCs obtained from cord blood, researchers successfully identified four distinct subgroups of human LCs:two steady-state subgroups LC1 (CD207hi, CD1a, EpCAM) and LC2 (CD207low, CD1b, CD1c, HLA-DR), as well as two activated-state subgroups activated LCs (aLC) (CCR7low, CD83, CD40), and migratory LCs (migLC) (CCR7hi, CXCR4).290 The LC1 and LC2 subgroups could be distinguished due to their distinct expression levels of Langerin (CD207), the CD1 family members (CD1a, CD1b, CD1c), and EpCAM.291 CD207 could be considered as a specific marker for LCs, which are involved in the capture, internalization, and presentation of antigens.292,293,294 CD83, a well-characterized marker of DC activation, could express on mature LCs and might be involved in T-cell activation.295,296,297 CCR7 could promote the migration of LCs to the lymph nodes.298 LCs play an essential role in skin immune surveillance and the maintenance of homeostasis. They are the only antigen-presenting cells in the epidermis that could migrate into draining lymph nodes after exposure to antigens and present antigens to T cells to initiate immune responses.293 In addition, LCs could reorganize the epidermal layer of the Keratinocytes by continuously isolating external antigens, keeping regulatory T cells in a steady state, thus controlling epidermal tolerance towards autoantigens and commensal microbiota, as well as the pattern of undifferentiated Keratinocytes in the suprabasal layers.131,299,300,301,302
DMs mainly derived from EMPs and could self-sustain in a CSF-1-dependent manner in a steady state. Besides, they could also be minimally supplemented by monocyte-derived macrophages after birth. It has been reported that several macrophage subsets with different anatomical locations and tissue functions have been identified in the adult dermis, such as sensory nerve-associated macrophages(CX3CR1hi, LYVE1low, and MHC-IIhi) as well as vascular-associated macrophages (CX3CR1low, LYVE1hi, and MHCIIlow).196,303,304 Sensory nerve-associated macrophages could promote nerve regeneration after injury by degrading myelin in damaged fibers. Newly grown axons at lesion sites appear to recruit macrophages from other dermal sources, and these cells could acquire a sensory nerve-associated macrophage phenotype over time.131,196,303 In addition, vessel-associated macrophages are crucial for dermal blood vessel integrity, and the regulation of antifibrotic activity and immune cell recruitment.196,304
Biological functions of tissue macrophages
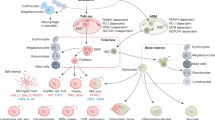
TRMs play a pivotal role in many physiological processes, including clearance of cellular debris, inflammation and resolution, tissue remodeling, defense, and metabolic function. There are diverse channels or receptors on the cell membrane of TRMs that can sense microenvironment changes, making them respond to maintain tissue homeostasis (Fig. 3).

TRMs in inflammation and homeostasis. a The canonical pro-inflammatory response is initiated by either PRRs or opsonin receptors. PRRs can directly recognize DAMPs (usually cell-derived molecules, e.g., Biglycan, versican, F-actin) and PAMPs (usually microorganism-derived molecules, e.g., Foreign DNA, flagellin, mannose). The opsonin receptor-mediated recognition process involves binding foreign particles labeled by opsonins and opsonic receptors, including Fcγ receptors. The recognition activates actin polymerization, pro-inflammatory cytokines, and other responses. The phagosome fuses with the lysosome. In late endosomal MIICs, most newly synthesized MHC-II molecules are likely loaded in an HLA-DM-dependent mechanism. The antigens and MHC-II will form MHC-II peptide complexes and then be delivered to the plasma membrane to be available to stimulate antigen-specific CD4+ T lymphocytes. Pro-inflammatory cytokines (e.g., TNF-α, IL-1β, LPS). b Resolution of inflammation. Apoptotic cells can release “find me” signals (EG. ATP, Lys phosphatidylcholine, CX3CL1) to recruit TAMs. Phagocytosis is facilitated by receptors (e.g., BAI1, Mer, Axl, αvβ3-integrin) directly binding with eat-me signals (e.g., PtdSer, Calreticulin, LPC) on apoptotic cells or indirectly recognizing bridging molecules, such as MFGE8, C1q, protein S, etc., which bind to eat-me signals. The recognition activates actin polymerization, immune-resolution cytokines, and other responses. Pro-inflammation cytokines (TNF-α, IL-6, etc.) decrease, and immune-resolution cytokines (IL-10, TGF-β) increase. Some viruses display PtdSer on their surface and mimic apoptotic cells, which allows them to evade the immune system and facilitate entry into host cells. Tolerant responses have also been attributed to Dectin-1/2 and SIRPα. Altered glycosylation is a universal feature of cancer cells; some abnormal glycans (e.g., galectin 9) promote cancer growth and immune tolerance through Dectin 1/2 activation. Some tumor cells express CD47 that interacts with SIRPα, expressed on the surface of macrophages and dendritic cells, inhibiting phagocytosis and maintaining self-tolerance. c TRMs sense physical factors and cytokines in the microenvironment. SIRPα signal regulatory protein alpha, LPC lysophosphatidylcholine, PRRs pattern recognition receptors, PAMPs pathogen-associated molecular patterns, DAMPs damage-associated molecular patterns, MIIC MHC class II compartment, LPS lipopolysaccharides, LPC lysophosphatidylcholines, HLA human leukocyte antigen
Phagocytosis and clearance of cellular debris
Phagocytosis is the engulfment and clearance process of granule cell or cell debris, including microorganisms, foreign matter, senescent cells, damaged cells, and mutated cells. This function is predominantly in charge of professional phagocytes with high phagocytosis efficiency, including macrophages, neutrophils, dendritic cells, monocytes, etc.305 As the principal phagocytes, macrophage has the powerful function of phagocytosis and clearance of cellular debris. Accurate and specific recognition of the phagocytic target is macrophage’s first and foremost step.
Pathogen-associated molecular patterns (PAMPs) are molecular structures found on the surface or inside of microorganisms, such as bacteria, viruses, fungi, and parasites, such as LPS, peptidoglycan, viral RNA, unmethylated CpG DNA. Damage-associated molecular patterns (DAMPs) are the other critical part in the activation of the immune system. DAMPs originate from internal sources, which are usually tissue damage, trauma, ischemia, cancer, and autoimmune diseases. The examples of DAMPs include HMGB1, ATP, uric acid, heat shock proteins, DNA from necrotic cells. Macrophages can directly recognize PAMPs and DAMPs through surface pattern recognition receptors (PRRs) and opsonic receptors and introduce them into the cell through receptor-mediated endocytosis. The effect of recognition can be divided into two types. Some PRRs induce phagocytosis by recognition of PAMPs/DAMPs. Other PRRs can activate macrophage secreting proinflammatory cytokine but cannot trigger phagocytosis.
The PRRs with phagocytosis-inducing function mainly include C-type lectins (e.g., mannose receptor and dectins), scavenger receptors, and partial toll-like receptors (TLRs). Mannose receptor and scavenger receptor (SR) can effectively mediate macrophage phagocytosis, kill and eliminate pathogenic bacteria or apoptotic tissue cells through the recognition and binding of mannose/fucose residues on the surface of bacteria or fungi and LPS/lipoteichoic acid or phosphatidylserine on the surface of ACs.306 Controversially, whether the role of SR in inducing macrophage phagocytosis is direct or indirect is unclear.307 Scavenger receptor A (SR-A), MARCO, CD36, and CD14 are also in this question.305 In humans and other mammals, TLRs are divided into 10 main types (TLR1 to TLR10), each with a different extracellular domain that recognizes specific molecular patterns of PAMPs/DAMPs. As for the cytosolic domain, there are two main transduction modes, one is myeloid differentiation factor 88 (MyD88) dependent, and the other is MyD88 independent. The MyD88-dependent pathway is common to TLR signal transduction except TLR3. MyD88 is the main adaptor protein in the TLR signal transduction pathway. MyD88 then recruits IL-1R-associated protein kinase (IRAK) through DD, and then initiates downstream signal transduction through signal molecules tumor necrosis factor receptor-associated factor 6 (TRAF6), β-transforming growth factor-activated protein kinase (TAK1), TAK1 binding protein 1, 2 (TAB1, 2), activating nuclear factor (NF-κB) or activator protein-1 (AP-1), and finally inducing the expression of inflammatory cytokines such as IL-1, IL-6, IL-8, IL-12, TNF-α and other genes.308 Other PRRs without the capability of triggering phagocytosis can induce the production of inflammatory cytokines. After the sensation of lipoproteins of pathogens, including broad bacteria, fungi, and viruses, TLR2 can bind with TLR1 or TLR6 to form heterodimer for recognition of its ligands, namely triacyl and diacyl lipoproteins, respectively.309 Sequentially, the heterodimers induce macrophages and DCs to secret various inflammatory cytokines. Besides TLR1/2/6, the relatively functional specificity of TLRs is well identified (such as TLR4 & LP, MD2, DAMPs310; TLR3, TLR7, TLR8, TLR9 & nucleic acids of bacteria and viruses311). However, the mechanistic bridge of TLR recognition and macrophage phagocytosis is not perspicuously established, but phagocytic gene programs307 and a series of pathways are demonstrated in this process.306 TLR3 plays a key role in macrophage- hematopoietic stem and progenitor cells (HSPCs) interactions. High ROS levels within the HSPCs are associated with increased surface calreticulin (Calr), leading to macrophage phagocytosis, whereas low ROS levels allow HSPCs to continue dividing after interacting with macrophages. TLR3 signaling induces surface Calr in a manner that promotes “grooming” rather than cell death, indicating a protective effect on HSPCs.312
Another type of recognition pattern is mediated by opsonic receptors, among which antibody molecules (IgG) and complement components are well-researched. The recognition process involves the binding of foreign particles labeled by opsonins and opsonic receptors, including Fcγ receptors313 and complement receptors (CRs).314 Meanwhile, the effect of these receptors is not isolated, and there are interdependent interactions and cooperation between phagocytic receptors. For example, many receptors need to interact with several lgG.315 These scattered receptors are recruited and aggregated, resulting from the alteration of a well-flowing phospholipid bilayer, the effect of transmembrane glycoproteins and cytoskeleton316,317 (Fig. 3a).
Besides receptor-mediated endocytosis, pinocytosis is a type of non-phagocytic endocytosis that allowing cells to engulf and digest large particles or cells. Based on the vesicle size, there are two types of pinocytosis—macropinocytosis and micropinocytosis. Macrophages can intake larger target cells and cell debris by micropinocytosis, which usually helps in nutrient uptake, immune responses, and cell signaling. Under the trigger of specific stimuli, membrane folds are extended to form giant macropinocytosomes containing large amounts of extracellular fluid, nutrients, pathogens, soluble antigens, and liquid macromolecules into macrophage.318 Micropinocytosis is to uptake smaller vesicles that helps in the general maintenance of the cellular environment. Micropinocytosis occurs in most cells by three recognized mechanisms – clathrin-mediated endocytosis, caveolin-mediated endocytosis, non-coated vesicle endocytosis that is clathrin- and caveolin-independent.319
After the recognition, a series of signal pathways are activated to form a phagocytic cup, pseudopod, and phagosome. Emerging techniques are applied to measure phagosome dynamics, especially imaging and fluorescence-based methods in observing phagocyte formation.320 The forming of phagosome can be summarized in 4 steps (namely dynamic probing, receptor clustering induced by particle binding and receptor recognition, phagocytic cup formation, and phagosome sealing).321 Driven by Arp2/3-dependent construction of branched actin networks, membrane protrusions are extended. The hydrolysis of PI (4,5) P2322 and Rho-family GTPase323 seems to be determinate in actin remodeling during the phagocyte formation by activating WASP (Wiskott-Aldrich syndrome protein) and N-WASP for Arp2/3 complex activation.324 BAR (Bin-Amphiphysin-Rvs) domain-containing proteins can polarize actin and recruit scissor-associated proteins such as dynamin to facilitate phagosome sealing.325,326 Immunofluorescence localization shows that contractility of myosin, actin, and actin-binding proteins participate in phagocyte formation in macrophages. In the formation of macrophage phagosome, Myosins II and IXb, myosin IC, and myosin V were concentrated in the early stage, later stage, and phagosome sealing, respectively.327After forming new and early phagosome, phagosome maturation was performed to construct a hostile and degradative environment to facilitate phagocytic prey destruction.328 Phagosome maturation is a process where newly formed phagosomes undergo fusion and fission with endocytic organelles via a “kiss-and-run” mechanism.329 This interaction allows the phagosome to acquire necessary molecules for each stage of maturation. Eventually, phagosomes fuse with lysosomes to form phagolysosomes, which degrade the phagosomal contents.330 Subsequently, lysosomes form from complicated fission and fusion of phagolysosomes, early endosomes, and late lysosomes.331
Apoptosis is a form of programmed cell death crucial for organ development, tissue remodeling, regeneration, lesion healing, and homeostasis.332 The process of AC engulfment by responding phagocytes, predominantly macrophages, is called efferocytosis.333 Efferocytosis is a multi-stage apoptotic cell clearance mechanism, usually considered the final step of apoptosis. Efferocytosis is performed by phagocytic cells but distinct from phagocytosis and is crucial for the resolution of inflammation and tissue homeostasis. During apoptosis, dying cells release ‘find me’ signals to attract circulating phagocytes to their location for clearance. These ‘find me’ signals are diverse, which include nucleotides (ATP, UTP), lipids (LPC), chemokines (CX3CL1, S1P).334,335,336 To ensure that apoptotic cells are efficiently recognized and removed, ACs expose ‘eat-me’ singals on the outer leaflet of their plasma membrane. The most well-known ‘eat me’ signal molecule is phosphatidylserine (PtdSer), and others include calreticulin, oxidized lipids and altered/abnormal glycosylation.337,338,339,340 For preventing unwarranted phagocytosis of healthy cells, ‘do not eat me’ signals are essential to build up self-tolerance. Key ‘do not eat me’ signals include CD47, MHC I, and protective glycans57,341,342,343 (Fig. 3b).
As people age, they experience a gradual decline in overall physical function and accumulate more senescent cells. Senescent cells are cells that have stopped dividing and have entered a state of irreversible growth arrest, and untimely phagocytosis of them can lead to the breakdown of homeostasis, tissue deterioration, and tumorigenesis. Like ACs, these senescent cells also express ‘eat-me’ signals. Additionally, they secrete various bioactive molecules known as the senescence-associated secretory phenotype (SASP). These signals help macrophages recognize senescent cells and further clearance. However, it was found that senescent cells can upregulate the “do not eat me” CD47-QPCT/L axis to evade efferocytosis and inhibit macrophage-mediated clearance.344 Moreover, both senescent and aged macrophages exhibit impaired efferocytosis, contributing to STING signaling mediated inflammation.345
Apart from canonical roles of macrophages, TRMs in different tissue have additional roles. Splenic macrophages are specialized in facilitating blood filtration. For example, splenic red pulp macrophages (RPMs) are a specialized population which is derived from fetal monocyte. RPMs play a role in maintaining blood homeostasis and immune function, for example, taking up splenic red pulp by direct contact and phagocytosing blood-borne pathogens. In the clearance of eryptotic red blood cells, RPMs can recycle heme and iron from broken down senescent red blood cells.346 During this process, A transcription factor, Bach1, in red-pulp macrophages senses heme, an iron-containing product from erythrocyte degradation and ensure effective heme degradation by controlling HO-1 levels, a heme enzyme.216 The skin outmost layer is host to Langerhans cells, the primary immune cells in epidermis. Langerhans cells are marked with high CD207 (Langerin), EpCAM, MHC-II, and CD11c expression levels. Langerhans cells is important for skin immunity because they are known to migrate to the skin-draining lymph nodes after capturing antigens and undergo a maturation process, and then present antigens to T cells to initiate immune responses.293 They contribute to skin homeostasis because of their phagocytosis in cleaning up debris such as apoptotic keratinocytes347 and control regulatory T cells at steady state.299
Antigen presentation and immune response
Antigen cross-presentation is vital for initiating adaptive immune responses against cancer, infections, and immune tolerance. TRMs, such as those in the liver (Kupffer cells), lungs (alveolar macrophages), and spleen (splenic macrophages), can capture extracellular antigens through phagocytosis and receptor-mediated endocytosis. They are professional antigen-presenting cells (APCs) that display peptides from internalized intracellular and extracellular antigens on major histocompatibility complex class I (MHC-I) proteins for presentation to T cells.348 The exact phenotype and antigen access of TRMs vary depending on the tissue. However, macrophages resident in the spleen, lymph nodes, liver, and peritoneum regularly encounter antigens carried by blood or lymph. This makes them optimal for antigen uptake and well-suited for cross-presenting to CD8+ T lymphocytes.349
In the spleen, macrophages are distinguished by their localization in the red and white pulp regions, which are demarcated by the marginal zone. Marginal Zone Macrophages (MZMs) and Marginal Metallophilic Macrophages (MMMs) are situated in the marginal zone. These macrophages are defined by their expression of sialic-acid binding immunoglobulin-like lectin 1 (CD169), macrophage receptor with collagenous structure (MARCO), and DC-Sign-related protein 1 (SIGNR1, CD209b). They are instrumental in capturing antigens from the bloodstream. Targeting antigens to metallophilic macrophages has been shown to facilitate the generation of cytotoxic T lymphocytes (CTLs) following the transfer of blood-borne antigens or adenovirus to CD8+ dendritic cells.222,350
In lymph nodes, macrophages can be categorized into several distinct subpopulations based on their anatomical locations. TAMs in the subcapsular sinus are identified as F4/80− CD169+, while those in the medullary sinus are identified as F4/80+ CD169+, and medullary cord macrophages are dentified as F4/80+ CD169−.351 These macrophages are directly exposed to lymph fluid, enabling them to effectively capture lymph-borne antigens for presentation to T cells. In vivo studies have demonstrated that when a nanogel containing a tumor-specific synthetic long peptide antigen (LPA) and a TLR9 agonist is administered, F4/80+ CD169+ MSMs and F4/80+ CD169− MCMs exhibit cross-presentation capabilities. This is evidenced by their ability to induce antitumor responses through the activation of tumor-specific CD8+ T lymphocytes.352 Further research indicates that CD169+ macrophages residing in lymph nodes can phagocytose apoptotic tumor cells and are crucial for the initial activation of tumor antigen-specific CD8+ T cells. Moreover, CD169+ CD11c+ macrophages demonstrate superior cross-presentation compared to CD169+ CD11c− macrophages and CD169− CD11c+ CD8+ dendritic cells.353 Tonsils harbor CD11c HLA-DR CD14+ cells, which have been identified as macrophages rather than DCs. These tonsillar macrophages efficiently phagocytose fluorescently labeled necrotic cells, as confirmed by flow cytometry.354 However, in vitro studies involving MelanA and NS3 antigen cross-presentation reveal that these macrophages are less effective in cross-presentation compared to major dendritic cell subsets.355
FOLR2+ TAMs found in both healthy and malignant breast tissues. The density of FOLR2+ macrophages within tumors has been positively correlated with improved patient survival outcomes. A robust correlation exists between FOLR2 expression in tumors and various immune pathways, including T cell receptor (TCR) signaling, PD-1 signaling, and antigen processing.356 Additionally, macrophages engaged in these processes exhibit high TIM4 expression, a receptor known to modulate cholesterol metabolism in macrophages by suppressing type I interferon signaling and enhancing SREBP2 activation.357
TIM4 also directs the slow progression of phagosomes, preserving antigens for cross-presentation. The peritoneal cavity, where gut and ovarian tumors commonly metastasize, is primarily populated by two distinct classes of macrophages.358 Small peritoneal macrophages (SPMs) originate from bone marrow myeloid precursors, are sparse under normal conditions, but undergo significant expansion during inflammation and tumor advancement. In contrast, large peritoneal macrophages (LPMs) constitute the predominant population under steady-state conditions, originating from embryonic precursors.84,359 Function displays high TIM4 expression and is associated with better prognosis in patients. During initiation of primary tumors or early colonization of metastatic sites, TIM4-mediated uptake of tumor cells can induce specific transcriptional remodeling of LPMs, further prolonging the integrity of ingested antigens, facilitating cross-presentation, and finally inducing anti-tumoral CD8 responses.360
Kupffer cells show antigen cross-presentation and efficient CD8+ T-cell proliferation, similar to classical DCs from the spleen. Antigen cross-presentation by Tie2 CD11blow liver endothelial cells and CD11b F4/80 Kupffer cells depend on intercellular adhesion molecule-1 rather than intracellular interferon-gamma.361 However, the function of Kupffer cell cross-presentation may be immunosuppressive, as they contribute to the induction of tolerance of orthotopic liver transplantation in rats.362
Kidney-resident macrophages hold their homeostatic ability to monitor and clear macromolecules transported across peritubular capillaries, particularly of small antigen-antibody immune complexes in a FcγRIV- mediated recognition manner.274 Furthermore, they are potentially involved in kidney organogenesis by promoting proper vascular network assembly.363
Regulation of inflammation and cytokine production
Tissue-specific macrophages are pivotal in promoting and resolving inflammation through the production and regulation of cytokines. When exposed to an inflammatory stimulus, such as an infection, macrophages can undertake several critical responses. Macrophages promote the recruitment of leukocytes to the infection site by secreting chemokines and various cytokines. Activating Vascular Endothelium: Through the release of TNF-α, macrophages enhance the activation of the vascular endothelium, thereby facilitating the ingress of leukocytes. Macrophages activate a range of immune cells, including natural killer (NK) cells, T cells, and B cells, by producing cytokines such as TNF-α, interleukin-6 (IL-6), interleukin-12 (IL-12), and interleukin-1β (IL-1β). Engaging in Adaptive Immunity: Macrophages also contribute to the activation of the adaptive immune system through antigen presentation and the production of additional cytokines. These mechanisms collectively enhance the body’s ability to respond to and manage inflammatory challenges.25
Granulocyte-macrophage colony-stimulating factor (GM-CSF, also known as colony stimulating factor 2, CSF2) is a cytokine that stimulates the production of various myeloid cell subsets in response to stress, infections, and cancers. For example, GM-CSF stimulates the functional activity of mature granulocytes and macrophages, enhancing their capacity under immune stress. The differentiation of megakaryocytic progenitors and erythroid progenitor cells can also be activated by GM-CSF, thereby influencing the production of platelets and red blood cells. Myelopoiesis refers to differentiating cells into the myeloid, non-lymphoid cell lineage. This process is initiated by binding GM-CSF to GM-CSFR on myeloid cell precursors, which triggers a cascade of signaling events downstream of the GM-CSFR. This cascade ultimately produces myeloid-specific transcription factors, including PU.1 and interferon regulatory factor 4 (IRF4).364,365 The critical role of GM-CSF in myeloid cells is demonstrated in GM-CSF-transgenic (Tg) mice, which exhibit significantly increased counts of myeloid subsets such as macrophages, neutrophils, and eosinophils compared to control mice.366,367 Furthermore, studies on GM-CSF-deficient (GMCSF−/−) mice reveal that while GM-CSF is essential for emergency myelopoiesis in response to infection, cancer, and stress, it is not required for basal myelopoiesis.368,369
Macrophages exhibit significant heterogeneity, with their functions and activation states influenced by their tissue-specific microenvironments. For example, intestinal macrophages help maintain gut homeostasis by sampling luminal contents and secreting anti-inflammatory cytokines like IL-10 and IL-1β to regulate the activity and function of regulatory T cells (Tregs)370 and Th17 cells,371 respectively. Macrophages expressing CX3CR1, a receptor crucial for tissue-specific migration and adhesion, are important in counteracting inflammatory responses and maintaining barrier integrity in the gut lamina propria and the mucosal layers. They present antigens to T cells in the gut-associated lymphoid tissues (GALT), such as Peyer’s patches and mesenteric lymph nodes, and release microbial products and cytokines such as IL-22 released.25 They uptake ACs and induce an anti-inflammatory phenotype through TGF-β and IL-10 production by macrophages, supplemented by cytokines produced by local fibroblasts. Inflammatory macrophages, recruited during tissue damage or infection, release pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-6, crucial for initiating and sustaining immune responses.
In the typical pro-inflammatory response, broad metabolic adaptations are essential for TRMs to effectively perform their roles. Pro-inflammatory macrophages often rely on glycolysis, while anti-inflammatory macrophages increase their TCA cycle activity and fatty acid oxidation. These metabolic pathways are regulated by various signals, including cytokines like IL-4, which promote the anti-inflammatory phenotype.372,373,374 Additionally, heightened TCA cycle activity leads to the accumulation of α-ketoglutarate (α-KG). α-KG has been shown to attenuate canonical pro-inflammatory cytokine production by inhibiting the inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ) and to stimulate the production of canonical anti-inflammatory cytokines through the activation of the histone demethylase Jumonji domain-containing protein D3 (JMJD3).375
MicroRNAs (miRNAs) are small, noncoding RNAs that play crucial roles in gene regulation in animals and plants by binding to the mRNAs of protein-coding genes, thereby guiding their post-transcriptional repression. MiRNAs are important regulators of macrophage polarization. Certain miRNAs, such as miR-155 and miR-125a/b, are upregulated in pro-inflammatory M1 macrophages, enhancing the production of inflammatory cytokines. Conversely, miRNAs like miR-187 and miR-378-3p support the M2 phenotype, aiding in anti-inflammatory responses and tissue repair.376,377
Recent in vitro studies indicate that efferocytosis also stimulates the proliferation of pro-resolution macrophages, partially depending on pathways downstream of the TAM receptor Mer (also known as MerTK) and phagolysosomal degradation.378 Co-culturing ACs with either LysMCreMertkfl/fl BMDMs or wild-type BMDMs treated with a phagolysosomal acidification inhibitor diminished BMDM proliferation.378 After phagolysosomal degradation of ACs, macrophages activate metabolite sensing pathways, including the nuclear receptor family of transcriptional regulators, which can suppress inflammation.379,380,381 For instance, macrophages from LXRα/β-deficient mice (Nhr1h3−/− Nhr1h2−/−), which lack downstream activation of the LXR metabolite sensing pathway, exhibit increased expression of pro-inflammatory genes such as IL-1β in response to ACs.382
In conclusion, to restore homeostasis after inflammatory responses, pro-resolution macrophages are essential. The macrophages clear ACs,380 produce anti-inflammatory factors such as IL-10 and transforming growth factor beta (TGF-β).383 They inhibit leukocyte recruitment through several mechanisms matrix metalloproteinase (MMP).384 Further facilitation of tissue repair by producing growth factors and remodeling the ECM385 (Fig. 3b).
Tissue remodeling and repair
Efferocytosis, mediated by TRMs, is critical for maintaining tissue homeostasis and enabling tissue remodeling and repair after injury. This process, which involves the clearance of apoptotic cells, is intricately linked to tissue renewal and the prevention of pathological conditions. Here, we summarize the roles of macrophage-mediated efferocytosis across various systems.
In the CNS, microglia, the resident macrophages, regulate tissue homeostasis and repair. Within the hippocampal dentate gyrus, microglia balance neurogenesis and apoptosis through efferocytosis.386 During retinal development, Morales et al. recently clarified the involvement of microglia in efferocytosis during retinal development and that microglia collaborate with Müller glia to facilitate this process.387 Microglias also prevent the degeneration of myelin, which covers neuronal axons and maintains their structural integrity. The myelin disorders could lead to neurodegenerative diseases such as dementia, Parkinson’s disease (PD) and Alzheimer’s disease (AD). In acute demyelination, microglia synthesize desmosterol to resolve inflammation, accelerate remyelination, and maintain axonal integrity.388 Dysregulation in myelin maintenance, often tied to signaling pathways like TGFβ1-TGFβR1, contributes to neurodegenerative diseases such as PD and AD through abnormal lipid metabolism. SRI-011381 hydrochloride, a small-molecule activator targeting these pathways has shown promise in addressing myelin-related disorders.389 In the visual system, certain scavenger receptors directly participate in regulating the circadian rhythm by clearing outer segments of photoreceptors via retinal pigment epithelial cells.390 While not strictly efferocytosis, this process involves scavenger receptors, suggesting a potential role for efferocytosis in circadian rhythm maintenance.
Macrophage-mediated efferocytosis is fundamental to immune regulation. In lymphoid follicles, apoptotic B cells activate follicular macrophages, transforming them into tingible body macrophages, which prevent autoimmune diseases.391 Similarly, large peritoneal macrophages execute efferocytosis to maintain peritoneal equilibrium and self-tolerance.392 During pregnancy, decidual macrophages perform efferocytosis to uphold homeostasis at the maternal-fetal interface.393
In the heart, macrophages play essential roles in development, repair, and remodeling. Resident cardiac macrophages expressing MHC-II facilitate arachidonic acid metabolism and efferocytosis, while their dysfunction can disrupt cardiac growth.394 Following ischemic injury, resident macrophages orchestrate inflammation to improve remodeling, while recruited macrophages influence infarct size.395 Overexpression of EphrinB2 enhances these reparative processes, while its deletion exacerbates cardiac dysfunction by impairing lymphangiogenesis.396 These findings highlight the dual roles of macrophages in both injury repair and pathological progression.
Kupffer cells, the liver-resident macrophages, are indispensable for liver repair. They secrete WNT ligands and hepatocyte growth factors, promoting the differentiation of hepatic progenitor cells into functional hepatocytes.397 The absence of Kupffer cells delays liver recovery significantly, highlighting their role in tissue regeneration.397,398
In bone tissue, efferocytosis is central to in the digestion and recycling of ECM. Osteoclasts, specialized macrophages located in bone dndosteum,164 mediate bone resorption by secreting enzymes like cathepsin K, while macrophages release IGF1 to stimulate osteoblasts for bone formation.82,165,399 Osteoclasts are recruited to resorption sites by TGF-β1 and secrete IGF1, thereby enhancing osteoblast activity and promoting bone formation in vivo.400,401 Bone marrow stromal cells also perform efferocytosis to regulate bone marrow homeostasis and mitigate bone loss.402 This coordinated activity ensures skeletal integrity and adaptation to physiological demands.
Macrophages also secrete soluble mediators that stimulate local stromal and progenitor cell proliferation, crucial for the repair process.403 Dermal macrophages are vital for skin wound healing.404,405 Subsets such as TIM4+ and MHC-II+ perivascular macrophages interact with sensory neurons to promote proper dermal innervation.303,406 During injury recovery, macrophages produce IGF1 and PDGF-CC to stimulate fibroblast activity and facilitate wound closure.406,407 Early-stage depletion of macrophages during skin wound repair in mice impairs normal re-epithelialization and vascularization. Depletion of macrophages in the later stages of tissue repair leads to fibrosis in both the skin and liver398. In muscle tissue, macrophages secrete paracrine factors like IGF1 and glutamine, enhancing satellite cell proliferation and myogenesis. Macrophages associated with muscle tissue secrete paracrine molecules such as IGF1,408 the metalloprotease ADAMTS1,407 and glutamine in response to injury. These molecules stimulate the proliferation and differentiation of muscle-resident stem cells (satellite cells) and promote myogenesis.409
Within the male genitourinary system, Sertoli cells function as specialized phagocytes responsible for preventing the accumulation of apoptotic germ cells in the seminiferous tubules through efferocytosis. Smoothelin-like 2 has been identified as a regulator of efferocytosis and lactate metabolism in mouse Sertoli cells to achieve homeostasis.410 Additionally, “find me” signals attract neutrophils, and neutrophil-mediated efferocytosis has been implicated in inflammation411,412 and colorectal cancer.413 Macrophage-mediated efferocytosis supports regeneration in various contexts, including neonatal heart repair,414 kidney recovery,415 peripheral nerve restoration,416,417 and etc.418,419,420 Evolutionary conservation of these functions is evident in limb regeneration in salamanders421 and tail fin regrowth in zebrafish,288 as well as for repair after injury in zebrafish422 and Drosophila.423,424 This underscores the universal importance of macrophages in tissue regeneration across species.
Macrophage-mediated efferocytosis is a cornerstone of tissue remodeling and repair, playing versatile roles across diverse systems. Its broad regulatory and regenerative capacities offer critical insights into both physiological processes and therapeutic potential.
Multi-level regulatory signaling pathways/crosstalk involved in homeostasis and physiology
Endocrine organs maintain systemic balance by producing hormones that regulate target tissues, stabilizing deviations from set points. Similarly, tissue homeostasis ensures a steady state free of inflammation or damage. Examples of homeostatically maintained variables include cell number and composition within tissue compartments, ECM density, composition, and stiffness, as well as the volume, oxygen concentration, pH, temperature, and osmolarity of interstitial fluids.292 TRMs, alongside autonomic nervous system afferents, C-fiber nociceptors, and mast cells, function as key homeostatic controllers, sensing changes in the tissue microenvironment and modulating these variables. TRMs achieve this by detecting ECM signals, producing growth factors, and releasing cytokines to either promote ECM synthesis or facilitate its degradation.275,293,294 For instance, macrophages release proteases to degrade ECM components and clear cellular debris upon sensing damage signals, such as extracellular ATP, low pH, or ECM fragments.271 They also recruit immune cells like monocytes and neutrophils, critical for tissue disinfection and repair during early injury stages.160,271
Macrophages contribute to vascular homeostasis and repair by supporting angiogenesis and vascular remodeling.295 In hypoxic conditions, macrophages secrete proangiogenic factors like VEGF-A, regulated by HIF1 and HIF2, to promote endothelial cell formation.293,296 Conversely, retinal macrophages counteract excessive vascular growth by producing WNT ligands, maintaining the retinal vascular plexus.297
In the pulmonary system, alveolar macrophages maintain surfactant homeostasis by recycling surfactant lipids and proteins produced by alveolar type 2 epithelial cells.298 The homeostasis of pulmonary surfactant in the lungs is maintained by a delicate balance: alveolar epithelial cells secrete surfactant lipids and proteins, while alveolar macrophages remove these substances. Specifically, macrophages in the alveoli eliminate excess surfactant phospholipids and proteins, with PPAR-γ in alveolar macrophages sensing surfactant lipids.102 Innate immune cells in the lungs also respond to mechanical forces via mechanosensory ion channels (MSICs) (e.g., PIEZO1). Activation via PIEZO1 leads to the secretion of EDN1 and promotes HIF1-alpha stabilization and CXCL2 expression, mediating immune responses like neutrophilia and bacterial clearance during abnormal cyclical hydrostatic pressure299 (Fig. 3c).
Fever is a beneficial response in host defense, yet its underlying mechanism remains unclear. Elevated body temperature sensitizes transient receptor potential melastatin 2 (TRPM2), a Ca2+-permeable cation channel found in various immune cells, including macrophages. Depletion of TRPM2 in macrophages has been shown to reduce cytokine release and fever-induced phagocytic activity, possibly mediated through redox signals. However, the precise mechanism requires further investigation.300
TRMs sense osmolarity to regulate the growth of lymphatic vessels via VEGF-C.301,302 They also influence the proliferation and differentiation of various local parenchymal, stromal, and progenitor cells.303 The increased density and proliferation of the lymphatic capillary network are monitored by tonicity-responsive enhancer binding protein (TonEBP) within MPS cells that infiltrate the skin’s interstitial spaces. TonEBP interacts with the promoter region of the vascular endothelial growth factor-C (VEGF-C) gene (Vegfc), leading to the secretion of VEGF-C by macrophages.300 Additionally, macrophages sense hypoxia and produce VEGF-A to promote angiogenesis425.
Macrophages detect pH variations in their microenvironments through pH-sensing G protein-coupled receptor 65 (GPR65) and two other proton-sensing receptors, GPR4 and GPR68 (also known as ovarian cancer G protein-coupled receptor 1, OGR1). When exposed to acidic extracellular pH, GPR65 triggers an anti-inflammatory response, whereas GPR4 and GPR68 promote pro-inflammatory responses.304,305,306 A murine model study showed that deficiency in GPR65 enhances the recruitment of macrophages and neutrophils to the colon, accompanied by increased expression of pro-inflammatory mediators.307 Moreover, tumors in patients with obesity and multiple cancers like CRC and HCC exhibited increased GPR65 expression, which drives intensified macrophage signaling and tumor cell-derived acid production, and finally promotes accelerated tumor growth.424 These ion-receptors detect changes in the ionic environment, particularly during inflammation, tissue damage, or infection. Epithelial sodium channel (ENaC), expressed on macrophages, plays a role in detecting changes in sodium levels, particularly in high-salt environments such as inflamed tissues. Under cardiac oxidative stress fibrosis and maladaptive remodeling, ENaC can activate macrophage recruitment and M1 polarization.426 ENaC may be a critical molecule in promoting macrophage migration and polarization.427 K2P2.1, KCa3.1 and Kv1.3 in macrophages can sense environmental potassium. K2P2.1, a two-pore potassium channel regulates NLRP3 inflammasome activation by controlling potassium efflux and maintaining plasma membrane potential in macrophages. This channel promotes the secretion of pro-inflammatory cytokines like IL-1β and the activation of caspase-1 during inflammatory responses.428 KCa3.1, an intermediate conductance calcium-activated potassium channel, reduces plaque instability in advanced atherosclerosis by limiting macrophage polarization toward the pro-inflammatory M1 phenotype.429 Inhibiting KCa3.1 may also offer therapeutic potential in macrophage-related disorders such as asthma,430 multiple sclerosis,431 and stroke,432 where controlling inflammation is critical. Kv1.3 influences macrophages in acute liver injury (ALI) by regulating their migration and infiltration into damaged liver tissues.433 Kv1.3 regulates macrophage inflammatory responses in atherosclerosis by modulating the ERK/NF-κB signaling pathway.434 In microglia, Kv1.3 promotes a pro-inflammatory state in disease-associated microglia by interacting with immune signaling proteins like STAT1 and TLR2.435 ZnR/GPR39 responds to extracellular zinc and plays an anti-inflammatory role by enhancing IL-10 production.436 ZnR/GPR39 helps in controlling hepatic insulin receptor signaling and mitigating liver fibrosis and inflammation.437
After acute depletion of Kupffer cells, NOTCH, TGF-β family receptors, and LXR signaling pathways play crucial roles in repopulating liver macrophages to maintain cell population homeostasis. DLL4 regulates the NOTCH transcriptional effector RBPJ, activating poised enhancers that rapidly induce LXRα and other factors determining the Kupffer cell lineage 4.
Splenic red pulp macrophages (RPM) and bone marrow macrophages (BMM) play roles in degrading senescent erythrocytes and recycling heme-associated iron. The transcription factor SPI-C is normally inhibited by the transcriptional repressor BACH1 in both RPM and F4/80+ VCAM1+ BMM. During pathologic hemolysis that leads to the loss of RPM and BMM, excessive heme triggers BACH1 degradation and derepression of Spic in monocytes. This process generates new RPM and BMM to facilitate iron recycling.116
The tissue-specific roles of macrophages are integral to normal physiology. They aid in vascularization, pulmonary function, fever responses, osmolarity and pH regulation, and iron recycling, underscoring their centrality in maintaining homeostasis across diverse systems. These specialized functions highlight macrophages as indispensable players in both systemic and tissue-specific regulatory networks.
Role of tissue macrophages in diseases
As pivotal cell types within the innate immune system, Macrophages perform multifaceted biological functions across various diseases. They serve as the primary line of defense and play crucial roles in maintaining tissue homeostasis, modulating inflammatory responses, and promoting wound healing. Recent studies have unveiled the dual roles of macrophages in disease progression, particularly in conditions such as cancer, cardiovascular diseases, autoimmune disorders, metabolic diseases, and neurodegenerative diseases. They influence disease development through various mechanisms, including regulating the immune milieu, promoting or inhibiting inflammation, participating in tissue repair, and affecting cell death pathways. In this section, we will focus on elucidating the specific mechanisms by which macrophages operate within these diseases and their diversity and plasticity throughout disease progression. This exploration aims to provide a foundational understanding of their complex roles in disease regulation.
Macrophage and cancer
Macrophages in the tumor microenvironment
The TME is a habitat for tumor cells and typically features extensive cellular infiltration. Tumor cells engage in crosstalk with surrounding stromal and immune cells, reshaping the microenvironment.438,439 This interaction facilitates tumor immune evasion, angiogenesis, increased drug resistance, phenotypic plasticity, and co-adaptive evolution under environmental pressures.440,441,442 As a crucial component of the TME, Macrophages play a dual role in the progression of almost all types of cancers.443
Cancer, characterized as a distinct chronic inflammatory condition, chemotactically attracts a significant infiltration of macrophages known as TAMs444 during its progression. Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) is the principal cytokine driving TAM recruitment to the tumor tissue. In the early stages of tumor development, low levels of GM-CSF induce effective chemotaxis and antigen presentation by DCs, exerting an anti-tumor effect. However, as cancer progresses into its later stages, GM-CSF levels gradually increase, promoting TAM recruitment and thus facilitating tumor progression.445,446 Moreover, GM-CSF also enhances TAMs’ production of IL-8, which generates lymphotoxicity and induces tumor cells to produce more GM-CSF, thereby exacerbating TAMs’ impact on shaping the microenvironment. Beyond GM-CSF, cytokines such as IL-17, IL-34, CXCL4, CXCL12, CCL2, CCL5, CCL20, and CSF2 also play significant roles in TAM recruitment, with other tumor-associated stromal cells participating in this process.21,447,448,449,450,451,452,453,454,455 Pan-cancer analysis has revealed that macrophages are significantly expanded in the blood of most cancer patients, and the number of macrophages is notably higher in patients who are non-responsive to immune checkpoint inhibitors compared to those who are responsive. More importantly, antigen processing and presentation ability is significantly downregulated in myeloid cells in the peripheral blood of cancer patients.456 Recent research has shown that tumor nucleotide metabolism can regulate TAM recruitment. Researchers have demonstrated that cytidine deaminase (CDA) in tumor cells aids in producing and releasing uridine diphosphate (UDP) and other uracil nucleotides. These nucleotides act as signaling molecules that bind to the P2Y6 receptor, predominantly expressed on TAMs. This interaction promotes TAM recruitment and an immunosuppressive phenotype, thereby protecting the tumor from cytotoxic T-cell infiltration and rendering it resistant to immune checkpoint blockade (ICB) therapies, such as anti-PD-1.457 Interferon-Induced Protein 35 (IFI35) recruits TAMs through the proteasomal regulation of non-classical NF-κB signaling via p105458 in glioblastoma.
M1-type macrophages in the tumor microenvironment
In the early stages of tumor development, when tumor cells have not fully “evolved”, and their ability to shape the microenvironment is not yet perfected, their surface ligands/receptors facilitate recognition and phagocytosis by macrophages, enabling effective antigen presentation. Furthermore, the early stages of a tumor are akin to damage to native tissues, and the induced inflammatory response facilitates the polarization of macrophages to M1, thereby secreting pro-inflammatory and immune-activating cytokines. In collaboration with Th1 cells, they effectively inhibit tumor progression by producing nitric oxide (NO) and reactive oxygen species (ROS).459 Chemokines produced by M1 macrophages, such as CCL5, CXCL9, and CXCL10, also recruit activated T cells and NK cells to exert anti-tumor immunity.460 Therefore, M1 polarization of macrophages is critically important in anti-tumor immunity. For instance, in ovarian cancer, overexpression of guanylate binding protein 5 (GBP5) not only promotes classic cell pyroptosis in ovarian cancer cells via the JAK2/STAT1 pathway but also induces the secretion of CXCL9/10/11, which promotes M1 polarization of macrophages to reverse the immunosuppressive TME.461 In tumors, knocking down the YTHDF2 protein (an m6A modification reader protein) can encourage macrophage and M1 polarization recruitment, thereby enhancing CD8+ T cell anti-tumor immunity.462 A novel platinum drug, naphplatin, can activate endoplasmic reticulum calcium release in macrophages, thus activating the MAPK p38 and NF-κB signaling pathways, which promote the reprogramming of M2-type TAMs to M1-type macrophages with anti-tumor effects.463 In hematological malignancies, polarizing TAMs to M1 is crucial for curbing tumor progression.464
M2-type macrophages in the tumor microenvironment
Cancer, characterized as a unique form of chronic, uncontrollable inflammation, predominantly involves M2-type macrophages in the composition of TAMs, with a smaller proportion of M1-type macrophages. However, TAMs exhibit high heterogeneity, meaning they often represent an intermediate phenotype between M1 and M2 types. Moreover, the presence of TAMs is closely linked to cancer staging and prognosis. Early-stage tumors predominantly harbor anti-tumor M1-type TAMs, while pro-tumor M2-type TAMs characterize later stages. Consequently, the ratio of M2 to M1 TAMs, known as the TAMs polarization index, has been recognized as a prognostic marker for cancer.465 Increasing evidence suggests that some TAM subgroups can express M1 and M2 macrophage genes,466 and the functional heterogeneity of TAMs is closely associated with their spatial distribution. Therefore, the spatial distribution of M1 and M2 TAMs provides a more accurate reflection of disease prognosis than merely assessing the presence of different TAM phenotypes in the TME.467 Furthermore, TAMs secrete various cytokines that promote the development of corresponding tumor phenotypes,468 such as direct secretion of growth factors that stimulate tumor growth.469 For instance, M1 macrophages upregulate inducible nitric oxide synthase (iNOS), which metabolizes L-arginine to L-citrulline and nitric oxide, creating a microenvironment unfavorable for tumor progression. In contrast, M2 macrophages exhibit an opposite metabolic pattern, where upregulated arginase (Arg) 1 metabolizes L-arginine into L-ornithine and polyamines, tumor-supporting factors.470 Additionally, TAMs secrete vascular endothelial growth factor (VEGF) and matrix metalloproteinases (MMPs) to remodel the extracellular matrix, thereby facilitating tumor invasion and metastasis.471 They also release TGF-β, Arg1, Indoleamine 2,3-dioxygenase (IDO), IL-4, and IL-10 to shape the overall tumor immunosuppressive microenvironment, thereby limiting the maturation of dendritic cells and the normal function of CD8+ T cells.472 TAMs also recruit Treg and Th2 cells by releasing Aryl hydrocarbon receptor (AhR), CCL17, CCL18, CCL22, and CCL24.473 At the same time, they decrease the expression of anti-tumor effector T cell (CTL, Th1) surface markers such as MHC-I, MHC-II, CD80, and CD86 while upregulating surface PD-L1 to facilitate immune escape by tumor cells.474
The transformation and regulation mechanisms of tumor-associated macrophage phenotypes
In the TME, as tumors progress and evolve under conditions such as hypoxia and through the reshaping of the immune and metabolic landscape, M1 macrophages gradually transition into M2 macrophages, thus fostering tumor development. This polarization is influenced by cytokines such as IL-4 and IL-13, secreted by Th2 cells, eosinophils,475 and basophils.476,477,478 Additionally, tumor cells further drive M2 polarization by secreting macrophage colony-stimulating factors (CSFs) and transforming growth factor (TGF)-β. After interacting with tumor cells, macrophages can undergo a phenotypic shift from expressing M1 markers to increasing expression of M2 markers such as Arg1 and CD163.479 Interestingly, even the death of tumor cells can promote M2 polarization within the microenvironment.480,481 In pancreatic ductal adenocarcinoma (PDAC), extracellular vesicles release the KRASG12D protein into the microenvironment during autophagy-dependent ferroptosis. Macrophages that ingest these vesicles undergo M2 polarization via the STAT3 signaling pathway due to the presence of this protein.482 Surprisingly, the β2-microglobulin (B2M) subunit of Class I Major Histocompatibility Complex (MHC-I), which is crucial for the function of CD8+ T cells, can interact with PIP5K1A in gliomas and promote the secretion of MYC-induced TGF-β1, leading to M2 polarization of TAMs.483 OAS1 (2’-5’-oligoadenylate synthetase 1) is a member of the interferon-stimulated gene family and plays a crucial role in antiviral processes. Interestingly, pan-cancer analysis has revealed significant differential expression of OAS1 across various tumors. Overexpression of OAS1 can result in CTL (cytotoxic T lymphocyte) dysfunction and promote M2 polarization of macrophages.484 Interestingly, TRIM56, an interferon-induced E3 ubiquitin ligase that is overexpressed upon double-stranded DNA stimulation and regulates the production of type I interferon via the stimulator of interferon genes (STING) pathway, has been found through pan-cancer analysis to promote M2 polarization of macrophages in gliomas. Additionally, it can serve as an immunological biomarker for glioma prognosis.485 The stiffness of the extracellular matrix also affects TAM polarization. In stiff matrices, tumor cells secrete more CSF-1, which promotes the accumulation of M2 macrophages.486,487 Viral infections can also influence TAM polarization; Epstein-Barr Virus (EBV) induces M2 polarization in nasopharyngeal carcinoma by producing Ataxia Telangiectasia and Rad3-related protein (ATR), which supports tumor progression.488 Other cells within the TME also contribute to the M2 phenotype transition of TAMs. Platelets, for example, can bind to TAMs through CD62P interacting with P-selectin glycoprotein ligand-1 (PSGL-1) expressed on TAMs, activating the JNK/STAT1 pathway, which promotes the transcription of C5 and the release of C5a, leading to a pro-tumoral phenotype of TAMs.489
The distinct microenvironmental localization of M1 and M2 macrophages,490 also significantly reflects their biological functions. Generally, macrophages are primarily located at the tumor core, at the interface between tumor cells and the stroma, and within the tumor stroma itself.491 M2-type TAMs tend to be positioned near blood vessels and necrotic areas,492 demonstrating high adaptability to the hypoxic tumor environment. They frequently interact with endothelial cells,493 promoting angiogenesis and assisting in migrating tumor cells towards invasive areas.494 TAMs also play varying roles in different types of tumors. In gliomas, TAMs predominantly foster tumor angiogenesis492,495 and promote glioma metastasis. Microglia exhibit strong pro-inflammatory effects.496,497 In lung cancer, TAMs mainly facilitate tumor growth and metastasis.498 In triple-negative breast cancer (TNBC), depletion of TAMs significantly reduces tumor growth, recurrence, and invasion.478,499 In pancreatic ductal adenocarcinoma (PDAC), TAMs primarily contribute to tissue fibrosis, creating an environment conducive to tumor cell invasion, metastasis, and immune escape.500 Reprogramming M2-type TAMs into M1-type has been proven to be an extremely effective tumor immunotherapy approach,461,463,501 offering new avenues for cancer treatment by altering the TME to stimulate an anti-tumor immune response.
Due to differences in ontogeny and/or local stimuli, the simple M1/M2 dichotomy cannot fully characterize macrophages for advancing research and precision clinical treatment. With the rise of pan-cancer analysis, single-cell sequencing, and spatial transcriptomics, several key molecules have been identified that play critical roles in defining functional subgroups of TAMs. These molecules help distinguish macrophage subtypes and uncover their diverse functions within the TME. SPP1 is closely associated with M2 polarization, particularly in lung adenocarcinoma, where it promotes tumor angiogenesis and supports TAM infiltration. TREM2 is linked to immunotherapy resistance in multiple cancers, including melanoma, where TAMs with high TREM2 expression exhibit immunosuppressive properties. Inhibiting TREM2 has been shown to enhance immunotherapy response and inhibit tumor growth. APOE and APOC1 are widely expressed in breast and lung cancers, playing important roles in lipid metabolism and highlighting TAMs’ significance in metabolic regulation. Inhibiting APOC1 can reprogram M2-type TAMs into M1-type, thereby enhancing the efficacy of anti-PD1 immunotherapy. VEGFA, a key factor in angiogenesis, is highly expressed in TAM subgroups that promote tumor growth by enhancing endothelial cell proliferation and migration. CXCL9 and CXCL10, chemokines highly expressed in M1-type TAMs, especially those associated with inflammation, promote T cell recruitment, boosting anti-tumor immune responses, and are often observed in patients who respond well to immune checkpoint inhibitors. COL1A1, COL1A2, and COL3A1 are associated with collagen production in TAMs, particularly in renal cell carcinoma and lung cancer, influencing matrix remodeling and invasive tumor growth and potentially affecting responses to immune checkpoint therapies. HMOX1, a molecule involved in heme clearance, is highly expressed in specific TAM subgroups, indicating its role in maintaining antioxidant balance within the TME. These molecules help differentiate M1 and M2 macrophages and reveal the functional diversity of TAMs across various cancers. Understanding these pathways may provide novel therapeutic targets for improving cancer immunotherapy.502
Metabolic differences between M1 and M2 macrophages
From a metabolic perspective, M1 macrophages engage in anaerobic glycolysis and the pentose phosphate pathway to synthesize large quantities of proteins and fatty acids,503 whereas M2 macrophages prefer oxidative phosphorylation and fatty acid oxidation.504 Compared to normal macrophages, TAMs exhibit lower activities of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and succinate dehydrogenase (SDH),505 indicating that they require fewer nutrients to function within the TME, reducing their competition with tumor cells for glucose and thereby diminishing macrophage-tumor cell nutrient competition.506 High levels of glycolysis conducted by tumor cells can extensively deplete glucose in the microenvironment, inhibiting the anti-tumor activity of M1 macrophages. Furthermore, in gliomas, the reduced level of glycolysis in TAMs is often associated with poor patient prognosis.497 Although M2 TAMs show an increased tendency for glycolysis in breast and pancreatic cancers, even if they compete with tumor cells for nutrients like glucose, TAMs still favor tumor invasion and growth.507,508 Interestingly, tumor cells and TAMs form a compartmentalized metabolic unit where tumor cells secrete lactate and CSF-1. High lactate levels promote histone lactylation in macrophages, facilitating their transition to the M2 phenotype,509,510,511,512,513 while CSF-1R enhances the recruitment of circulating monocytes and their polarization towards M2 macrophages. Pan-cancer analysis revealed that lactate dehydrogenase LDHA is significantly overexpressed in various cancers and is closely associated with poor prognosis. Moreover, high LDHA expression is often positively correlated with macrophage infiltration and reduced antitumor activity of CD8+ T cells.514,515 Interestingly, research has unexpectedly found that the microbiome metabolite D-lactate (DL) can actually inhibit the PI3K/Akt signaling pathway and enhance the NF-κB signaling pathway, thereby reprogramming M2-type TAMs into M1-type, contrary to previous conclusions.516 Macrophages reciprocate by supplying tumor cells with growth factors such as epidermal growth factor (EGF), VEGF, interleukin-10 (IL-10), and matrix metalloproteinases (MMPs), thereby remodeling the tumor immune-suppressive microenvironment and promoting invasion and metastasis.517 Conversely, the metabolic relationship between tumor cells and cancer-associated fibroblasts (CAFs) is somewhat opposite.518 CAFs release large amounts of lactate into the matrix, which tumor cells take up and shift their metabolic mode from glycolysis to mitochondrial metabolism. CAFs even transfer healthy mitochondria to tumor cells to boost their mitochondrial function and invasion.519 The interaction between CAFs and TAMs also promotes the M2 polarization of TAMs,520 evidencing the cooperative adaptation and evolution of macrophages with surrounding stromal and tumor cells. Furthermore, studies show that TAMs upregulate the mTOR inhibitory regulator REDD1 under hypoxic conditions, reducing glycolysis and promoting tumor angiogenesis.521 However, overexpression of HIF-1α can stimulate glycolysis and PPP intermediates in macrophages, paradoxically inducing their polarization towards the M1 phenotype522 (Fig. 4).

Metabolic differences between M1 and M2 macrophages. In the tumor microenvironment, the metabolic differences between M1 and M2 macrophages are closely linked to the onset and progression of cancer. Typically, both M1 macrophages and cancer cells primarily undergo glycolysis, leading to metabolic competition. As the malignancy of the tumor progresses, M1 macrophages in the tumor microenvironment are gradually reprogrammed into M2 macrophages, shifting their metabolism to oxidative phosphorylation. Furthermore, the exchange of metabolic products between M2 macrophages and cancer cells further promotes the manifestation of the tumor’s biological characteristics. This process highlights the complexity of the tumor microenvironment and underscores the crucial role of macrophages in tumor development. ppp pentose phosphate pathway, NADPH nicotinamide adenine dinucleotide phosphate (reduced form), HIF-1α hypoxia-inducible factor-1 alpha, OXPHOS oxidative phosphorylation, GLUT1 glucose transporter type 1, MCT4 monocarboxylate transporter 4, MCT1 monocarboxylate transporter 1, SLC1A5 solute carrier family 1 member 5, IL-4 interleukin-4, IL-13 interleukin-13, TGF-β transforming growth factor-beta, FAO fatty acid oxidation, 25HC 25-hydroxycholesterol, AMPKα AMP-activated protein kinase alpha, MAO-A monoamine oxidase A, ROS reactive oxygen species, MAT2A methionine adenosyltransferase 2A
M2-type TAMs exhibit high glutamine metabolic enzymes and transport proteins expression, indicating a high glutamine metabolism.523,524 Glutamine synthetase (GLUL), which converts glutamate to glutamine, facilitates the polarization of TAMs towards the M2 phenotype.525 Experimentally, depleting glutamine or inhibiting GLUL activity can effectively reduce the production of α-ketoglutarate, thus diminishing epigenetic modifications associated with macrophage polarization and ultimately inhibiting M2 polarization.375,526 This significant reduction in glutamine metabolism can even reprogram M2-type TAMs into M1-type. Furthermore, glutamine produced by TAMs can be absorbed by cancer cells through SLC1A5 and metabolized by GLS1 to counteract the damage induced by DNA-damaging agents.527 Inhibiting the fatty acid oxidation (FAO) of TAMs can also promote reprogramming from M2 to M1, which aligns with the metabolic characteristics of M2-type TAMs.528,529 Additionally, high-density lipoprotein can polarize M2-like macrophages to M1-like phenotypes by activating the MAPK p38 and NF-κB p65 pathways, mitigating immune suppression in the TME.530 Cholesterol metabolism also impacts the immunoregulatory capabilities of TAMs. Studies have found that 25-hydroxycholesterol (25HC) accumulation in TAMs alters the typical regulatory pathways of IL-6 and IL-13. 25HC promotes the phosphorylation of STAT6 by activating AMP kinase α (AMPKa), activating the STAT6-dependent signaling pathway, and promoting the expression of the anti-inflammatory gene Arg1.531 Interestingly, arachidonic acid can inhibit M2 polarization of macrophages, but its inflammatory metabolite PGE2, by inhibiting PPARγ, can indirectly enhance macrophage OXPHOS and thus promote M2 polarization.532 Recent research indicates that methionine metabolism also plays a significant role in TAM phenotypic transformations. Methionine adenosyl transferase II α (MAT2A) promotes the expression of RIP1 by facilitating the trimethylation of histone H3K4 (H3K4me3), which leads to the transformation of TAMs into the M2 subtype and contributes to the malignant progression of gastric cancer.533 Furthermore, the metabolic confrontation of nicotinamide between macrophages and cancer-associated fibroblasts (CAFs) can regulate the tumor immune microenvironment by modulating the activity of CD8+ T cells.534 In hepatocellular carcinoma, the absence of SIRT5, in conjunction with oncogenes, synergistically promotes the production of bile acids by liver cancer cells, thereby promoting M2 polarization of TAMs.535 Interestingly, monoamine oxidase A (MAO-A), typically active only in the brain, also promotes the immunosuppressive phenotype polarization of TAMs through oxidative stress responses.536
The relationship between macrophages and tumor development
Role in promoting tumor growth and metastasis
Cancer stem cells (CSCs) are often characterized by high expression of the anti-inflammatory gene NRF2 and by shaping a TME rich in TGF-β, thereby promoting chemotherapy resistance, metastasis, and recurrence. In glioblastomas, TAMs secreted TGF-β1 promote the growth of glioblastoma stem cells driven by the integrin αvβ5-Src-STAT3 signaling pathway.537 CD11b+ /CD163+ TAMs can also stimulate glioblastoma stem cell development by secreting pleiotrophin (PTN), which activates the protein tyrosine phosphatase receptor type Z1 (PTPRZ1) receptor on the surface of these cells.538 Recent studies have shown that cancer stem cells (CSCs) release large oncosomes (LOs) to mimic the presence of necrotic cells in the microenvironment, thereby “deceiving” macrophage precursors and inducing them to adopt an immunosuppressive phenotype. The activation of NRF2 signaling in CSCs promotes the translocation of IL-33 from the nucleus to the cytoplasm, where NRF2 also acts as a transcriptional activator to increase the expression of the membrane protein ATG9B. ATG9B encapsulates IL-33, forming large oncosomes that are released into the microenvironment. Interestingly, ATG9B, as a lipid transferase, creates a docking site for ANXA1 on the surface of the oncosome. ANXA1, a glucocorticoid-regulated anti-inflammatory protein, can be released extracellularly and mediate the resolution of inflammation. Additionally, ANXA1 binds to the membrane phospholipid phosphatidylserine (PtdSer), a well-known “eat-me” signal. The ANXA1 protein on the large oncosomes binds to FPR+ macrophage precursors in the microenvironment, driving their differentiation into immunosuppressive Arg1hi CD206+ TAMs, further assisting CSCs in establishing a TGF-β-rich microenvironment.539 Pan-cancer analysis has revealed that Family with sequence similarity 109, member B (FAM109B) is significantly elevated across various tumor types and is associated with poor prognosis. Its expression is linked to aggressive progression and poor prognosis in low-grade glioma (LGG) patients, serving as an independent prognostic marker for LGG. Glioma grading is negatively correlated with FAM109B DNA promoter methylation. Additionally, immune infiltration and single-cell analysis show that FAM109B is significantly expressed in TAMs.540 Recent studies have shown that TAMs in the glioblastoma microenvironment, after engulfing cholesterol-rich myelin debris, transform into lipid-laden macrophages (LLMs), undergoing significant metabolic rewiring, accumulating large amounts of lipids, and altering their inflammatory response. The uptake of myelin debris not only triggers epigenetic changes in TAMs but also leads to the suppression of chromatin accessibility and immune activation-related genes, thus driving TAMs toward an immunosuppressive phenotype and weakening their anti-tumor immune function. LLMs transfer myelin-derived lipids to mesenchymal-like glioblastoma cells, supporting the high metabolic demands of the tumor, promoting its growth and proliferation, and managing lipotoxicity through the esterification of cholesterol. The LXR signaling pathway plays a key role in regulating cholesterol accumulation and export in LLMs, with lipid transporters ATP Binding Cassette Subfamily A Member 1 (ABCA1) and ABCG1 facilitating lipid efflux, which protects tumor cells from lipotoxicity and provides essential components for membrane construction. A symbiotic relationship forms between TAMs and mesenchymal-like glioblastoma cells, with tumor cells instructing TAMs to enhance their lipid scavenging abilities. TAMs provide the lipids necessary for tumor growth. This crosstalk is critical in the lipid-poor microenvironment of glioblastoma.541 In breast cancer, LSECtin on TAMs enhances the stemness of breast cancer cells by interacting with its receptor BTN3A3.542 TAMs also release the soluble glycoprotein NMB (GPNMB), which binds to the CD44 receptor on tumor cells, activating the expression of IL-33 and its receptor IL-1R1L, ultimately enhancing the stemness of the tumor cells.543 TAMs release exosomes containing lncMMPA, which interacts with miR-548s to increase mRNA levels of ALDH1A3, promoting tumor cell glucose metabolism and proliferation. Moreover, lncMMPA promotes M2 polarization of TAMs within the microenvironment.544 Under hypoxic conditions, TAMs increase the synthesis and secretion of galectin 3. The elevated galectin-3 promotes tumor growth through ROS generation and NF-κB activation.545 SnRNA/snoRNA-derived nuclear RNA 3, a major dicer-independent RNA, selectively inhibits transcription of Nos2 in macrophages, reducing iNOS expression by lowering chromatin accessibility at the Nos2 promoter and enriching Mi-2β and H3K27me3, thus promoting tumor growth.546 TAMs’ RON signaling activation promotes the secretion of IL-35, enhancing the growth of breast cancer,547 while their secretion of IL-6 also promotes breast cancer stem cell enrichment through STAT3 activation.548 Through integrated multi-omics analysis, JUN and its regulatory network positively correlate with TAMs and fibroblasts. Macrophage- and fibroblast-derived fibronectin 1 (FN1) can activate the Hippo pathway via JUN, promoting tumor metastasis. This mechanism may be universally present across various types of tumors.549 Moreover, TAMs facilitate tumor invasion and distant metastasis, which are closely linked with poor prognosis across various cancers. The collaborative interaction between cancer-associated fibroblasts (CAFs) and TAMs also promotes TME matrix remodeling. CAFs secrete CXCL14 and chitinase 3-like 1 (Chi3L1) to recruit and promote M2 polarization of TAMs. In return, M2 TAMs secrete MMPs and tissue proteases, degrading the ECM.550 TAMs also promote the growth of bladder cancer by stimulating the PI3K/AKT pathway through collagen.551 ECM degradation fragments further recruit TAMs, reshaping the ECM to optimize conditions for tumor cell metastasis and angiogenesis.552 In melanoma, the absence of TRIM59 in M2 TAMs, through upregulation of MMP9 and Madcam1, promotes tumor migration and invasion.553 In breast cancer, under the stimulation of TGF-β, TAMs promote the expression of collagen cross-linking enzymes such as lysyl hydroxylase 2 (LH2) and lysyl oxidase (LOX), which directly participate in collagen cross-linking, hardening the tumor ECM and facilitating invasion.554 Interestingly, TAMs at potential metastatic sites and those extravasating through endothelium re-arrange collagen in the ECM, creating micro-tracks that facilitate tumor cell invasion.555 Conversely, TAMs can spontaneously secrete collagen molecules and tightly cross-link them within the ECM, forming a physical barrier that impedes the infiltration and killing by external immune cells.556 Beyond reshaping the microenvironment, TAMs can further induce epithelial-mesenchymal transition (EMT) in tumor cells, promoting metastasis by releasing IL-1, IL-6, TNF-α, and TGF-β, which upregulate EMT markers in tumor cells such as vimentin and β-catenin.557,558,559
Role in inhibiting tumor growth and spread
While many TAMs exhibit an M2 phenotype that promotes tumor growth, the M1 subtype of TAMs serves as a critical mechanism for tumor elimination. Repolarizing M2 TAMs to the M1 phenotype may also represent a novel and effective anti-cancer strategy.560 In the context of ependymomas, a subset of inflammatory CCL2+ TAMs has been identified, characterized by high pro-apoptotic gene expression and immune response-related factors such as IL-1β, CCL3, and CCL4.561 This suggests that CCL2+ TAMs may be active in initiating immune responses by promoting inflammatory reactions and regulating tumor cell apoptosis, thereby influencing the TME. Interestingly, tumor cells release small amounts of the tumor suppressor factor PTEN into the microenvironment. PTEN binds to PLXCD2 on the surface of M2-type TAMs and activates the downstream JAK2/STAT1 signaling pathway, inducing the reprogramming of M2-type TAMs into M1-type. This reprogramming enhances the anti-tumor abilities of CD8+ T cells and NK cells.562 Additionally, studies have shown that macrophages can regulate nicotinamide metabolism in fibroblasts by secreting exosomes (EVs) containing NAMPT, which inhibit NNMT expression in fibroblasts through the SIRT1/NICD axis, thereby enhancing the cytotoxicity of CD8+ T cells.534 This cholinergic metabolic communication mediated by EVs enhances the efficacy of anti-PD-1 therapy. Furthermore, exosomes from M1-like macrophages expressing OX40L (CD134) (OX40L M1-exos) can effectively inhibit the progression of nasopharyngeal carcinoma by engaging the OX40/OX40L pathway and reprogramming M2-like TAMs into M1-like macrophages.563 TAMs with high enolase (PCB) expression exhibit enhanced phagocytic capacity, and PCB downregulates PD-L1 transcription via transcription factor IRF, promoting the infiltration of CD8+ T cells.564 Moreover, TAMs lacking the YTH N6-methyladenosine RNA binding protein F2 (YTHDF2) can be effectively reprogrammed into an M1 phenotype by IFN-γ-STAT1, exerting antitumor effects.565 Ferroptosis, a recently discovered form of cell death, has also been utilized in cancer treatment. Recent studies have shown that 1-stearoyl-2-15-HpETE-sn-glycero-3-phosphatidylethanolamine (SAPE-OOH) is an important surface marker of ferroptotic tumor cells and binds to TLR-2 on the surface of macrophages, promoting the phagocytosis of ferroptotic tumor cells by macrophages. However, ferroptosis inducers are not cell-specific and can lead to phospholipid peroxidation in both tumor and non-tumor cells. The accumulation of lipid peroxides in the endoplasmic reticulum (ER) of macrophages caused by ferroptosis inducers inhibits the transport of TLR2 to the plasma membrane. It leads to its retention in the ER by disrupting the interaction between TLR2 and its chaperone, CNPY3. Subsequently, the ER-retained TLR2 recruits the E3 ligase MARCH6 and initiates proteasome-dependent degradation.566,567 ScRNA-seq and pan-cancer analysis have revealed that CLDN18 is a key gene regulating antibody-dependent cellular phagocytosis (ADCP) immunotherapy in hepatocellular carcinoma. Its expression is closely associated with the infiltration of M1 macrophages in the TME of liver cancer.568 Additionally, based on extensive single-cell and spatial transcriptomic data, Proteasome Activator Complex Subunit 2 (PSME2) has been identified as a pan-cancer biomarker for M1 macrophage infiltration. In osteosarcoma cells, overexpression of PSME2 can significantly inhibit tumor proliferation, migration, and invasion activities.569 Interestingly, pan-cancer analysis has revealed that macrophage migration inhibitory factor (MIF) and CD74 are both significantly overexpressed in various cancers, with MIF serving as a marker for M0 macrophage infiltration and CD74 being a marker for M1 macrophage infiltration in the TME. Studies have shown that blocking the MIF-CD74 signaling pathway on macrophages and dendritic cells can effectively activate anti-tumor immune responses in “cold” tumors, such as melanoma. Additionally, CD74 expression significantly increases in certain cancers after receiving immune checkpoint blockade (ICB) therapy, strongly indicating that CD74 activation triggers immune responses in most cancers.570,571
Impact on tumor angiogenesis
Cancer fundamentally represents a chronic, uncontrolled inflammatory injury. Correspondingly, most TAMs predominantly exhibit M2-like functions, playing a role in counterbalancing and repairing the tissue damage caused by cancer. A critical step in tissue repair is angiogenesis,572 which ensures adequate nutrient supply by restoring blood flow to the damaged area. Similarly, the uncontrollable proliferation of tumor cells compresses the surrounding normal tissue, creating necrotic zones, and results in numerous avascular areas unless new blood vessels form promptly.573 Under the pressures of hypoxia and nutrient deprivation, tumor tissues can also undergo necrosis. Therefore, angiogenesis is a critical target in cancer therapy.573,574 Long-standing research has identified TAMs as accomplices in promoting tumor angiogenesis.575,576 For instance, TAMs associated with matrix remodeling in the TME are highly correlated with angiogenesis. In contrast, M1 pro-inflammatory macrophages tend to induce vascular quiescence. Consequently, reversing M2-like TAMs to an M1 phenotype can be highly beneficial for targeting tumor angiogenesis.577,578,579
TAMs secrete a plethora of angiogenic factors such as EGF,580 PDGF,581 PAF, VEGF,582 FGF2,115 and MYDGF,583 as well as inflammatory cytokines like TNF-α,584 IL-1,585 chemokines (CXCL8,586,587 CCL18588), and other pro-angiogenic activators including adrenomedullin (ADM),589 PGE2,590 thymidine phosphorylase (TP),591 and urokinase-type plasminogen activator (uPA). These factors induce endothelial cell proliferation and chemotaxis, thereby driving angiogenesis. Particularly, a subset of monocytes expressing Tie2, termed Tie2-expressing monocytes (TEMs), are a crucial source of angiogenic TAMs within tumor tissues.592,593,594,595 These cells are chemotactically attracted by angiopoietin-2 (ANG2) derived from tumor endothelial cells (EC),594,596 and their interaction with ECs stimulates further expression of Tie2 in TAMs. On the other hand, tumor ECs also secrete IL-6, which further promotes the M2 polarization of macrophages, thus facilitating tumor progression.597 Moreover, tumor cells can deliver Tie2 proteins to TAMs via exosomes.598 However, recent studies have found that Tie2-expressing TAMs may be dispensable for tumor angiogenesis and post-chemotherapy tumor recurrence.599,600 Under hypoxic conditions, HIFs601,602,603 promote transcription of vast amounts of VEGF, further regulated by transcription factors induced by IL-1β, such as STAT3 and NF-κB,604,605,606,607 and CCL18.588 Additionally, TAMs enhance pericyte coverage and increase vascular density by secreting PDGF-B and adenosine deaminase 2 (CECR1),608 further promoting vascular maturation. TAMs expressing lymphatic endothelial hyaluronan receptor 1 (Lyve-1) coordinate with αSMA+ cancer-associated fibroblasts (CAFs) in the tumor vascular microenvironment to create a conducive environment for angiogenesis.581 Neurotransmitters can also enhance tumor angiogenesis, with catecholamines promoting the M2 polarization of TAMs, thereby significantly increasing their expression of factors such as VEGFA.609 Blocking β-adrenergic receptors can effectively inhibit tumor growth and angiogenesis.610 Abnormalities in cholinergic metabolism also promote the M2 subtype polarization of TAMs and the extensive proliferation of endothelial cells, thus enhancing tumor angiogenesis.611 In glioblastoma, single-cell sequencing has identified a group of hypoxia-TAMs that respond strongly to hypoxic environments. These TAMs are activated by adrenomedullin-mediated paracrine signals, leading to unstable tumor vascular structures. This instability results in high vascular permeability, impacting drug delivery efficiency.612 Single-cell sequencing and SCENIC (Single-cell regulatory network inference and clustering) analysis of ependymoma cells have identified a group of CD44+ TAMs that highly express VEGFA driven by the transcription factor TEAD1, thereby promoting tumor angiogenesis.561 In head and neck squamous cell carcinoma, TAM-derived exosomes containing miR21-5p enhance tumor angiogenesis through the YAP1/HIF-1α axis.613 Tumor cells can also release exosomes containing miR-301a-3p, which, through the PTEN/PI3K/AKT signaling pathway, induces M2 polarization of macrophages and promotes angiogenesis.614 Annotation of single-cell data from various tumors has identified a new type of TAM, SPP1+ TAMs, highly associated with tumor angiogenesis.615,616 SPP1+ TAMs are identified by the specific expression of the core gene SPP1 and often express genes such as FN1, IL1RN,617 MARCO, and VEGFA.618 They were first discovered in colorectal cancer and later found in lung and breast cancers.619,620
Traditional therapies targeting tumor angiogenesis typically suppress vascular formation, hoping to “starve” tumor cells. However, this approach can exacerbate hypoxia in the tumor core, further promoting proliferation and metastasis. Recent studies have demonstrated that protocatechuic acid nanoliposomes (PCN) can inhibit tumor growth and metastasis by stabilizing vascular tight junctions and increasing pericyte coverage. This is achieved through the occupation of EPCR and the activation of PAR-1, which induces heterodimerization of PAR-1 and PAR-3. Consequently, this leads to Gα13-RhoA-mediated activation of Tie2 and stabilization of vascular structures via the AKT-FOXO3a signaling pathway.621
Formation of immune evasion
In the metabolic immunological microenvironment of tumors, M1 macrophages, which originally possess anti-tumor functions, are ‘tamed’ and transformed into M2-type TAMs that promote tumor development, becoming significant ‘accomplices’ in tumor initiation and progression. The primary function of M2-type TAMs is immune suppression, mainly through inhibiting the cytotoxic functions of T cells or inducing their exhaustion,622 thereby facilitating tumor immune evasion. TAMs release immunosuppressive cytokines such as TGF-β623 and IL-10, which not only weaken the killing ability of NK cells and induce their exhaustion624 but also promote the further differentiation of Th cells into Treg cells,625,626 suppressing anti-tumor immunity while also inhibiting the maturation and chemotaxis of dendritic cells.627 TAMs release a variety of chemokines, including CCL5, CCL17, CCL20, and CCL22, which alter the infiltration spectrum of T cells within the TME, resulting in reduced infiltration of CD8+ T cells and increased infiltration of Tregs.628 TAMs also release arginase Arg1, which consumes significant amounts of L-arginine in the microenvironment. L-arginine is crucial for T cell proliferation, TCR formation, and the shaping of immune memory.629 Some M2 TAMs also present antigens to T cells, but disruptions occur during the immunological synapse formation, leading to T cell non-responsiveness to external antigens.630 In the early stages of cancer development, tumor cells upregulate phagocytic signals on their surface, leading to early macrophage phagocytosis. However, fragments of apoptotic tumor cells or certain components can form DAMPs that induce inflammatory responses when phagocytosed by macrophages. Macrophages that ingest apoptotic bodies containing tumor cell DNA activate the interferon-inducible protein AIM2, thus upregulating their expression of PD-L1 and IDO481 and promoting the synthesis of TGF-β and IL-10 through activation of LXR and RXR by the lipid components of tumor apoptotic fragments.480 Although apoptotic tumor cells can induce TAM immune tolerance to some extent, avoiding inflammatory damage also promotes tumor immune evasion. However, recent studies have found that thymosin alpha-1 can reverse the M2 polarization of macrophages induced by exocytosis by activating the TLR7/SHIP1 axis, thereby reversing the immunosuppressive process.631
In breast cancer, TAMs synthesize large amounts of collagen by activating TGF-β signaling, promoting fibrosis. This process consumes significant amounts of arginine in the microenvironment (CD8+ T cell proliferation and cytotoxic activity are highly dependent on arginine metabolism). It drives the synthesis of proline and ornithine (tumor-supporting factors), severely inhibiting the killing power of CD8+ T cells.554,632,633 Pan-cancer analyses have found that apolipoprotein E (APOE+) TAMs in triple-negative breast cancer promote the exhaustion of CD8+ T cells.634,635 Moreover, co-culturing triple-negative breast cancer-derived cancer-associated fibroblasts (CAFs) leads to the reprogramming of blood monocytes into an immunosuppressive subset of STAB1+ TREM2+ high-lipid-associated macrophages (LAM), reducing anti-tumor immune effects.636 The absence of neutral ceramidase can also lead to the emergence of an immunosuppressive TREM2-related macrophage subset.637 On the other hand, although PARP inhibitors (PARPi) can promote TAMs to revert to an anti-tumor phenotype,638 the response to PARPi in BRCA1-deficient breast tumors is severely limited by pro-tumorigenic TAMs, which not only inhibit CD8+ T cells but also suppress tumor cell DNA damage induced by PARPi, thus inhibiting the activation of the DNA-sensing STING pathway and reducing inherent anti-tumor immune responses.639 In colorectal cancer, high expression of CXCL1 promotes the infiltration of TAMs, suppressing the killing power of both CD4+ and CD8+ T cells, thereby facilitating immune escape.640 Inhibition of CXCL1 expression can also suppress M2 polarization of TAMs.641 Colorectal cancer cells regulate the PTEN/AKT and SCOS1/STAT1 pathways by releasing extracellular vesicles containing miR-21-5p and miR-200a, inducing PD-L1 expression and M2 polarization in TAMs.642 TAMs’ released extracellular vesicles can also promote upregulation of PD-L1 in tumor cells.643 Pan-cancer analysis has identified that Krüppel-like factor 3 (KLF3), a key transcriptional repressor, is abnormally expressed across various tumor types, particularly in pancreatic cancer, and is closely linked to immune pathways. It is also highly expressed in TAMs.644 Interestingly, after cancer cells are attacked in liver cancer, they secrete prostaglandin E2 (PGE2). In addition to its inherent ability to inhibit the growth of CD8+ T cells, PGE2 can also induce CX3CR1+ TAMs to shift into a pro-tumor phenotype. These TAMs then secrete interleukin-27 (IL-27), which induces CD8+ T cells to upregulate the immune checkpoint molecule TIM-3 and decrease the expression of TNF-α and IFN-γ, leading to CD8+ T cell exhaustion and facilitating immune evasion.645 Furthermore, liver cancer cells activate the Smad signaling pathway through TGF-β in the microenvironment, which in turn activates the expression of the downstream molecule SOX18. SOX18 acts as a transcriptional activator to upregulate the expression of CXCL12 and PD-L1 in liver cancer cells, thereby recruiting TAMs and Tregs and suppressing the anti-tumor immune response.646 In hepatocellular carcinoma, integrated multi-omics analyses have revealed that three key transcription factors—ILF2, YBX1, and HMGA1—are regulated by the long non-coding RNA HCG18. ScRNA-seq shows that HCG18 co-localizes with macrophages and stem cells and is positively correlated with M0-type macrophages but negatively correlated with M1-type and M2-type macrophages. High expression levels of HCG18, ILF2, YBX1, and HMGA1 are strongly positively associated with cancer stem cells. Moreover, pan-cancer analysis indicates that high expression of HCG18 implies high sensitivity to immune checkpoint therapy.647 Moreover, the role of TAMs in the TME is closely related to the overall metabolic state of the body. For example, in obesity, specific inflammatory factors such as interferon-γ, tumor necrosis factor (TNF), leptin, insulin, and palmitate activate the mTORC1 pathway, enhancing macrophage glycolysis activity, thereby increasing PD-1 surface expression and suppressing T cell activity. However, high PD-1 expression on TAMs inhibits their glycolysis (a typical feature of M2 macrophages). In obese models, anti-PD-1 treatment significantly enhances the glycolysis activity of TAMs (a feature of M1 macrophages). It boosts their antigen-presenting ability and the expression of co-stimulatory signals. It suggests that while obesity promotes immune suppression in the TME, it may respond more positively to tumor immune therapy.648,649 TAMs’ ketone metabolism also suppresses anti-tumor immunity. In hepatocellular carcinoma, TAMs highly express 3-oxoacid CoA-transferase 1 (OXCT1), promoting the formation of ketone metabolism by-product succinate, which promotes H3K4me3 at the upstream promoter of Arg1. Transcriptionally activated Arg1 leads to CD8+ T cell exhaustion.650 Interestingly, single-cell sequencing revealed a novel subset of interstitial CD68+ SOX2+ double-positive TAMs in glioblastoma under conditions of cytomegalovirus infection, which is closely associated with poor clinical prognosis and resistance to ICB therapy. Additionally, this subset promotes the generation of immunosuppressive FXYD6+ T cells.651 Pan-cancer analysis has revealed that elevated CD93 expression is closely associated with poor prognosis and immune evasion in the majority of cancers. CD93 expression correlates strongly with the presence of monocyte/macrophage lineages and neutrophils while showing a negative correlation with Th1, MDSCs, NK cells, and T follicular helper cells in nearly all cancers. Therefore, CD93 could serve as a prognostic marker for malignant cancers.652 CD44 and CD147 have also been found to be abnormally expressed in various cancers and are often closely associated with resistance to immune checkpoint therapy. The specific knockout of CD44 can inhibit M2 polarization of macrophages in the TME.653,654 Tumor cells can also induce TAMs to highly express IL-15Rα, thereby reducing tumor cell production of CX3CL1 and lowering the recruitment of CD8+ T cells, which is a significant reason for the inefficacy of anti-PD-1 agents.655 Additionally, tumor cell oxidative stress-generated ROS suppresses the release of their miR-155-5p exosomes, thereby increasing PD-L1 expression in TAMs.656 TAMs also release IL-23 to promote Foxp3 expression, thereby stabilizing the immunosuppressive function of Tregs in the microenvironment.657 Type I IFNs can effectively reprogram TAMs into an anti-tumor phenotype, but when interferon signals coexist with M-CSF signals, they promote Arg1 expression through the STAT3 signaling pathway.658 Through single-cell sequencing analysis of numerous patients across multiple cancer types who did not respond to immune checkpoint blockade (ICB) therapy, a unique niche of TIM3+ VISTA+ TAMs was identified. This genetic signature is associated with negative prognostic and predictive impacts across various cancers. The interaction between HMGB1/VISTA exposed by immunogenic cell death (ICD) of cancer cells and TIM3/VISTA on TAMs inhibits the paracrine IFN response, thereby hindering the production of pro-inflammatory TAMs. This further reduces the neoantigen load, ultimately causing PD-1/PD-L1-targeted ICB therapy to function inadequately. Instead, it increases the ecological proportion of this unique TAM subset within the overall macrophage population.659
Current macrophage-targeted cancer immunotherapies primarily include inhibiting TAM recruitment, inducing TAM repolarization, depleting TAMs within the TME, and enhancing TAM phagocytic activity. Targeting chemokine ligands and receptors can reduce TAM recruitment. In contrast, antibody-mediated therapies targeting TAM-specific markers, selective chemotherapeutic agents that eliminate TAMs, nanoparticles (NP), and other nanotechnologies for receptor-mediated depletion can effectively mitigate TAM’s negative impact on antitumor therapies. Furthermore, TAMs can be reprogrammed to adopt an antitumor phenotype, or the CD47-SIRPα axis can be blocked to enhance TAM phagocytic activity, promoting their role in antitumor immunity.
However, despite these advancements, clinical trial outcomes may not always meet expectations, with challenges in translating preclinical success into sustained clinical efficacy. For instance, inducing TAM depletion can lead to off-target effects, such as the non-specific depletion of monocytes, which may compromise immune homeostasis beyond the TME. Therefore, selective targeting of TAMs while preserving TRMs is critical to reducing side effects and ensuring therapeutic safety and efficacy. Additionally, approaches like targeting inflammatory cytokines, blocking inhibitory receptors, and enhancing antigen presentation can provoke immune-related adverse effects, as the immune system may start attacking normal tissues. Another promising strategy is to inhibit TAM recruitment to the TME, which could enhance the effectiveness of existing immunotherapies. This method involves disrupting chemokine signaling and using monoclonal antibodies or small molecule inhibitors. However, future clinical studies must carefully consider potential resistance mechanisms and side effects, such as accelerated metastasis. Stimulating TAM phagocytic activity is another promising approach, as it can potentially destroy large populations of tumor cells, enhancing the efficacy of chemotherapy and immunotherapy. Though research on this technology is still limited, focusing on the CD47-SIRPα signaling pathway and leveraging macrophages as key players in the fight against tumor cells could yield encouraging results in future studies.
Lastly, the repolarization of TAMs towards an M1-like phenotype has garnered significant interest. While reducing M2 TAM abundance or promoting the switch from M2 to M1 could be crucial in cancer therapy, this approach faces challenges due to the poorly understood polarization mechanisms and the unpredictable nature of patients’ immune responses. Additionally, it remains difficult to fully attribute TAM’s tumor-promoting role solely to M2 macrophages, as M1 macrophages may contribute to inflammation and tumor development. The subtle distinctions between these phenotypes and the numerous factors influencing their plasticity—differing across cancer types and patients—complicate the definition of therapeutic strategies.
In conclusion, while macrophage-targeted therapies hold great promise in precision medicine, developing effective immunotherapies requires a multidisciplinary approach. This involves devising comprehensive treatments that address the complex mechanisms regulating macrophage behavior within the TME. As research advances, future strategies must integrate multiple approaches to precisely modulate TAM functions, considering both the pro- and anti-tumor effects of macrophages in cancer progression.
Inflammation and autoimmune disease
As previously mentioned, macrophages play a critical coordinating role in the body’s physiological and pathophysiological states. These cells are not only broadly active in inflammation response, immune modulation, and tissue repair but are also increasingly recognized for their role in autoimmune diseases. Although the pathogenesis of autoimmune diseases primarily involves T cells and B cells, these cells may not fully explain all the etiologies and pathological processes of autoimmune diseases, making the role of macrophages particularly significant.660 In conditions such as RA, systemic lupus erythematosus (SLE), and systemic sclerosis (SSc), macrophages display high plasticity, exhibiting pro-inflammatory or anti-inflammatory properties depending on the microenvironmental conditions. For example, macrophages promote tissue healing and fibrosis in SSc by producing TGF-β and other anti-inflammatory factors. Conversely, in rheumatoid arthritis, macrophages exacerbate inflammation and tissue damage by releasing pro-inflammatory cytokines such as IL-6 and TNF-α. Additionally, the failure of macrophages to clear apoptotic cells and process self-antigens may be a contributing factor in triggering and sustaining autoimmune responses.
Despite the increasing recognition of the role of macrophages in many autoimmune diseases, their precise roles in disease progression and their specific functions and subtypes in different types of autoimmune diseases are still not fully understood. A deeper understanding of macrophages’ functions and regulatory mechanisms is crucial for developing new therapeutic strategies.
Rheumatoid arthritis (RA)
RA is characterized by extensive infiltration of macrophages in the synovium, with the severity of the disease positively correlated with the degree of macrophage infiltration.661 This phenomenon serves as a reliable biomarker for the disease.662 Increasing research indicates that the activation of macrophages plays a key role in the pathogenesis of RA, where excessive activation of pro-inflammatory M1 macrophages and incomplete polarization of anti-inflammatory M2 macrophages significantly exacerbate the condition. Recent studies have identified a specific type of macrophage, characterized by high expression of CX3CR1 and low expression of Ly6C, F4/80, and IA/IE, termed arthritis-associated osteoclastogenic macrophages (AtoM). These are precursors to pathogenic osteoclasts in arthritis.37 Local disease conditions are typically directly related to synovial macrophage production of IL-6 and TNF-α.663,664,665,666,667 IL-6 activates STAT3, which promotes the upregulation of nuclear factor κB receptor activator ligand (RANKL) in osteoblasts and enhances osteoclast differentiation, leading to joint destruction in RA.668 Studies show that the histone demethylase inhibitor GSK-J4, by reducing H3K27me3, decreases macrophage IL-6 expression, effectively delaying disease progression.669 The NOTCH signaling pathway is another crucial factor contributing to RA’s imbalance between M1 and M2 macrophages. Thus, using NOTCH inhibitors in arthritis models can effectively alleviate TNF-induced M1 polarization of macrophages,670 and NOTCH signaling is critical in regulating osteoclast differentiation and bone resorptive activity.671 ERK, a vital member of the MAPK family, also plays an important role in macrophage polarization. The adipocytokine nesfatin-1 promotes high expression of CCL2 in RA synovial fibroblasts via the ERK/MAPK pathway, enhancing M1 polarization and chemotaxis, thereby exacerbating RA conditions.672 Nesfatin-1 has been identified as a potential risk factor for RA.673 As previously mentioned, M1 macrophages primarily rely on aerobic glycolysis,674 while M2 macrophages depend on oxidative phosphorylation. Hypoxic conditions in the synovial microenvironment promote the transcriptional enhancement of glycolytic enzymes through HIF-α, the expression of the key pro-inflammatory cytokine IL-1β, and M1 polarization of macrophages.503,675 Additionally, lysine acetyltransferase 2 A (KAT2A) supports glycolytic reprogramming of macrophages by inhibiting NRF2 activity and its downstream antioxidant molecules, promoting M1 polarization and synthesis of NLRP3 and IL-1β. Specific KAT2A chemical inhibitor MB-3 significantly improves synovitis and bone destruction in a collagen-induced arthritis model.676 Therefore, inhibiting M1 polarization and enhancing M2 polarization can effectively alleviate inflammatory responses in RA.677,678 For instance, activation of SIRT1 promotes phosphorylation of AMPKα, increases M2 gene expression, and inhibits M1 gene expression, and SIRT1 transgenic mice exhibit milder arthritis symptoms.679 Proteins c-Fos and c-Jun, whose expression levels are elevated in RA synovial tissues, act by reducing the synthesis of Arg-1, diminishing the anti-inflammatory properties of macrophages.680,681 Similarly, IL-10 effectively reduces the release of pro-inflammatory cytokines by macrophages by inhibiting the NF-κB signaling pathway.682,683 GRK2 inhibits the migration and pro-angiogenic characteristics of Flt-1+ macrophages through the PPARγ signaling pathway, reducing synovitis and M1 polarization in a mouse model of RA.684 Recent research has found that exosomes derived from anti-inflammatory (M2) macrophages, modified on their surface with oligo-lysine and matrix metalloproteinase (MMP)-cleavable polyethylene glycol (PEG), can clear cfDNA after MMP cleavage and induce M2 polarization, significantly inhibiting RA and providing robust cartilage and bone protection.685 Additionally, single-cell sequencing has identified a large number of IL-1B highly expressed macrophages with enhanced NLRP3 inflammasome activity in the peripheral blood and synovial fluid of inflammatory arthritis induced by PD-1 inhibitors (PD-1-IA). Yet, this macrophage subtype is not observed in RA.686 Significant research potential remains concerning the heterogeneity of macrophage subtypes in RA.
In the synovial lining, synovial lining cells (SLCs), key players in RA, can be categorized into type A (macrophage-like synoviocytes, MLS) and type B (fibroblast-like synoviocytes, FLS).687,688 Both cell types coexist in the synovial layer, typically contributing to the production of synovial fluid and the maintenance of joint function. However, in RA, MLS secrete a plethora of pro-inflammatory cytokines, chemokines, and growth factors that not only activate FLS but also cause them to release IL-6, prostaglandin E2 (PGE2), and matrix metalloproteinases (MMPs). This cascade of reactions leads to persistent extracellular matrix degradation and exacerbates synovial inflammation.689 Furthermore, FLS promotes osteoclastogenesis in RA by producing fibroblast growth factor 2 (FGF2), further aggravating the disease.690 A newly discovered gene, Merlot, regulates the NFATc1-GSK3β axis to inhibit osteoclast differentiation and promote apoptosis, effectively alleviating the progression of RA.691 Studies have shown that citrullinated and malondialdehyde-acetaldehyde-modified fibrinogen, by activating macrophages to release soluble mediators like PDGF-BB, can induce FLS to transform into an invasive phenotype, worsening RA.692 Macrophage extracellular traps (METs) can also induce severe synovial inflammation. METs stimulate the DNA sensor cGAS in FLS, promoting proliferation, invasion, migration, and tumor-like biological behaviors and releasing many inflammatory cytokines such as TNF-α, IL-1β, MMP9, and MMP13.693 Moreover, FLS can also influence the phenotype of macrophages in RA. For instance, exosomes produced by FLS that contain PTX3 not only promote the expression of IL-6 and TNF-α in macrophages and induce an invasive phenotype in these cells. Interestingly, FLS can also induce macrophages to develop a non-M1/M2 polarized phenotype; inflammatory mediators like PGE2 produced by FLS can polarize macrophages toward heparin-binding EGF-like growth factor (HBEGF) polarization. HBEGF-type inflammatory macrophages, besides releasing a vast array of inflammatory mediators, can conversely prompt FLS to convert into a destructive invasive phenotype.694 This complex interplay highlights the critical role of cell-cell interactions within the RA synovial microenvironment, influencing disease progression and offering potential targets for therapeutic intervention.
Systemic lupus erythematosus (SLE)
In SLE, macrophages play a critical role in disease progression by regulating adaptive immunity. Macrophages in SLE patients exhibit an abnormally high activity level, with the degree of activation correlating directly with disease activity and potentially life-threatening conditions.695,696 Macrophages activate humoral immunity through the CD40/CD40L co-stimulatory molecule, promoting plasma cell differentiation, antibody secretion, and class switching, thus influencing the disease process.697 Compared to healthy controls, SLE patients have increased expression of CD40L on peripheral blood macrophages.698 Mouse experiments have shown that overexpression of CD40L can induce lupus-like autoimmune diseases, whereas neutralizing CD40L can significantly reduce autoreactive B cell activation and antibody production.699,700 Single-cell sequencing of SLE samples revealed that macrophages and DCs in SLE patients express higher levels of interferon-stimulated genes (ISGs) and that M1 macrophages dominate in SLE.701 High levels of IFN-γ and TNF-α in the serum of SLE patients promote the polarization of macrophages towards the M1 phenotype.702 M1 macrophages further exacerbate SLE inflammation by secreting IL-1β, IFN-γ, CXCL10, CCL2, IL-6, and TNF-α.703,704,705,706,707,708 IL-6, in particular, appears to enhance the activity of B cells in SLE, and inhibiting IL-6R reduces the number of circulating plasma cells and the levels of autoantibodies.708 Moreover, the impaired phagocytic function of macrophages709 and the serum of SLE patients can accelerate macrophage apoptosis, another significant factor contributing to SLE.710,711 This primarily manifests in the inability to clear apoptotic cells and immune complexes in a timely manner. Recent studies show that macrophages lacking Late Endosomal/Lysosomal Adaptor, MAPK And MTOR Activator 5 (LAMTOR5) suffer from lysosomal dysfunction and abnormal mTORC1 activation, leading to ineffective phagocytosis and digestion of external apoptotic cell debris.712 Macrophages usually bind immune complexes through Fc receptors, and the genetic diversity of Fc receptors typically determines the efficiency of immune complex clearance, which is closely associated with regional SLE prevalence and directly affects disease progression.713 SLE patients’ macrophages highly express pyruvate kinase M2 (PKM2), and overexpression of PKM2 enhances the activity of TLR4, TLR7, and TLR9 pathways.714 In SLE, impaired gut barriers lead to the release of serum LPS, which induces macrophage pyroptosis through the Caspase 11-GSDMD pathway. Notably, pyroptotic macrophages promote the differentiation of mature B cells independently of T cells.715 Interestingly, during differentiation, the red blood cells of SLE patients fail to switch from glycolysis to oxidative phosphorylation, thereby retaining mitochondria. These mitochondria-containing red blood cells become a new source of interferons upon being phagocytosed by macrophages.716
Research indicates that disulfiram can effectively alleviate inflammation caused by SLE by inhibiting gasdermin D (GSDMD) and IL-1β-mediated pyroptosis.717 An increase in oxysterols, especially 7α, 25-dihydroxycholesterol (7α, 25-OHC), suppresses STAT activation and the production of IFN-β, chemokines, and cytokines in macrophages when 7α, 25-OHC binds to its receptor EBI2.718 Additionally, miR-4512 can inhibit innate immune activation and the formation of neutrophil extracellular traps (NETs) in SLE by targeting TLR4 and CXCL2.719 Recent studies show that spermine, by binding to the FERM and SH2 domains of JAK1, inhibits the phosphorylation of JAK1 induced by IFN-I, IFN-II, IL-2, and IL-6, thus suppressing cytokine signaling. Treatment with spermine alleviates autoimmune lesions in SLE mice and reduces IFN-I signaling in monocytes from SLE patients.720 IL-4-induced M2 macrophages can effectively inhibit the development of SLE.721,722 Meanwhile, knockout of scavenger receptors and low levels of TGF-β are associated with poor prognosis in SLE.723,724 Interestingly, despite the detection of significant levels of the anti-inflammatory cytokine IL-10 in SLE patients, the levels of IL-10 unexpectedly correlate positively with disease activity.725,726 This may reflect a feedback mechanism where the body controls excessive inflammation by increasing IL-10 production. However, in the presence of IFN-α, the anti-inflammatory effects of IL-10 can be reversed to pro-inflammatory actions. IL-10 activates STAT1, promoting the expression of CXCL9, CXCL10, IFN-γ, and platelet-activating factor, further enhancing inflammation and M1 macrophage polarization.727 Therefore, inhibiting IFN-α can effectively improve the inflammatory damage caused by SLE.728 Overexpression of AKT2 in macrophages can interact with IRF3 and phosphorylate IRF3 at Thr207, weakening its nuclear translocation and thereby reducing the production of IFN-α, which in turn slows the progression of SLE.729 Research has shown that the 21-mer phosphopeptide P140 can effectively halt the progression of SLE by inhibiting the formation of NETs. However, in a localized Imiquimod (IMQ)-induced lupus model, following the administration of P140, a significant accumulation of CX3CR1-positive macrophages was observed in the lungs of lupus-prone mice, along with the development of pulmonary fibrosis. This macrophage response was associated with increased citrullinated histone H3 (H3cit) in the cytosol, the expression of interleukin-1 receptor type 1 (IL-1R1) on the cell membrane, and the production of intracellular ROS.730
Systemic sclerosis (SSc)
SSc is a complex chronic autoimmune disease characterized by a significant gender disparity, with a much higher incidence in women than in men.731 The progression of the disease primarily involves three core processes: extensive vascular abnormalities, immune system dysregulation, and fibrosis of the skin and internal organs.732,733 Decades ago, researchers first discovered high levels of CD163+ macrophages infiltrating SSc.734 Macrophages are situated at a crucial intersection of inflammation and fibrosis in SSc, playing an indispensable role. Increasing evidence demonstrates that macrophage activation, particularly M2 polarization, is crucial in the pathogenesis of SSc. M2 polarized macrophages can promote the activation of fibroblasts,735 releasing a large amount of factors such as TGF-β, PDGF, and CCL18, which further promote tissue fibrosis.736 In SSc, immune complexes in patients can induce monocytes to secrete secreted phosphoprotein 1 (SPP1),737 macrophage colony-stimulating factor (M-CSF), and IL-6.738 These factors collectively influence the disease process: SPP1 can promote the activation and migration of lung fibroblasts. At the same time, the autocrine effects of M-CSF and IL-6 drive further polarization of macrophages toward the M2 phenotype. Single-cell sequencing studies have revealed a subgroup of CD163+ macrophages in SSc, where SPP1+ macrophages accumulate in areas with higher degrees of fibrosis739,740 and show enhanced proliferative capabilities. Additionally, in patients with diffuse cutaneous systemic sclerosis (dcSSc), a group of macrophages expressing FCGR3A has been identified that express pro-fibrotic cytokines and chemokines such as IL-6 or CCL18, further driving the progression of the disease.741 Research has found that novel nanoparticle polymers of lactic acid and glycolic acid (PLG) can mitigate TGF-β induced fibrotic reactions by inhibiting the activation of MARCO+ macrophages.742 These findings highlight the critical roles of macrophages in SSc and suggest potential targets for therapeutic interventions to modulate macrophage behavior and mitigate disease symptoms.
Although initial analyses of the lungs, skin, and blood of patients with SSc suggested that macrophages primarily polarize into an M2 pro-fibrotic phenotype, recent studies indicate that macrophages in SSc exhibit a mixed M1/M2 phenotype.743,744,745 These cells express both M2 markers, such as CD204, CD163, and CD206, and M1 markers, including CD80, CD86, and TLR4.746 SSc predominantly shows a shift from a pro-inflammatory state in the early stages to a balanced pro-inflammatory and anti-inflammatory state in the later stages.747,748 ADAR1 is extensively expressed in macrophages during the early stages of bleomycin-induced SSc and plays a crucial role in developing skin and lung fibrosis. The absence of ADAR1 significantly alleviates skin and lung sclerosis by inhibiting the expression of inducible nitric oxide synthase (iNOS) and IL-1β in macrophages through weakening the NF-κB signaling pathway.749 Recent studies have found that downregulating the transcription factor Fli1 in macrophages might be the primary reason for mixed polarization. This leads to concurrent upregulation of the M2 characteristic CD163 and the M1 characteristic CXCL10 when Fli1 is downregulated.750 As previously mentioned, the M1 polarization of macrophages is driven by JAK/STAT-dependent type II IFN signaling, while M2 polarization is associated with JAK/STAT-dependent IL-4/IL-13 signaling. Therefore, using broad inhibitors of the JAK signaling pathway is effective for treating SSc. For example, ruxolitinib, which blocks both IFN and IL-4/IL-13 pathways, can reduce M1 and M2 markers and successfully prevent skin and lung fibrosis.747 Additionally, studies have found that depleting B cells in SSc can promote the differentiation of pro-fibrotic macrophages, thereby inhibiting tissue fibrosis.751
In autoimmune diseases, pro-inflammatory macrophages are often key drivers of disease progression or primary contributors to inflammatory tissue damage. Anti-macrophage therapies in these diseases focus primarily on reducing the production of abnormal pro-inflammatory cytokines derived from macrophages, inhibiting monocyte recruitment and differentiation in inflamed areas, and upregulating anti-inflammatory cytokines. For example, in treating RA, using CD64-targeted immunotoxins to selectively eliminate synovial inflammatory macrophages and inhibit M1 macrophage polarization while inducing M2 polarization has been identified as a promising drug development strategy. Additionally, macrophage-derived extracellular vesicles are considered optimal drug carriers due to their minimal toxicity and strong targeting capabilities. Research has shown that macrophage-derived microvesicle-coated poly (lactic-co-glycolic acid) (PLGA) nanoparticles encapsulating tacrolimus can significantly inhibit the progression of RA in mice, demonstrating potential as an effective biomimetic targeted therapy for RA.752
With the rapid advancement of single-cell sequencing technologies, macrophage classification in inflammatory diseases has evolved beyond the simple M1/M2 dichotomy, revealing a more diverse set of macrophage subtypes that better align with different disease states. This offers more potential targets for precision and personalized therapies, paving the way for future treatment strategies.
Cardiovascular diseases
Macrophages play a crucial role in the development of cardiovascular diseases, especially in conditions such as myocardial infarction and atherosclerosis. In the heart, 8% of non-myocardial cells are macrophages. Under disease conditions, both the number of these cells and their phenotypes undergo significant changes. As key components of the innate immune system, macrophages are massively recruited to the damaged area via CC chemokine receptor type 2 (CCR2) following cardiovascular injury, becoming the predominant immune cells in the region.753,754,755 They participate in crosstalk with other cells by releasing various mediators, influencing the chemotaxis and function of other immune cells to modulate immune responses, promoting or inhibiting the formation of endothelial cells (ECs), and directly driving fibroblast activation and proliferation. Their differentiation into myofibroblasts, thus regulating fibrosis.756,757 During cardiac pathology, macrophages, through their diverse phenotypes and functions, significantly influence the course of the disease. For instance, macrophages contribute to myocardial repair after a myocardial infarction by clearing dead myocardial cells. In atherosclerosis, they exacerbate vascular inflammation and plaque formation by ingesting oxidized low-density lipoproteins and transforming into foam cells. Additionally, macrophages play a key role in cardiac fibrosis, promoting extracellular matrix production and cell proliferation. Cardiac fibrosis is a common pathological outcome of various cardiovascular diseases. Excessive deposition and abnormal distribution of collagen can lead to dysfunction in cardiac contraction and relaxation.758,759
Typing of macrophages in the cardiovascular system
The cardiovascular system’s macrophages primarily originate from MDM and TRM that develop from the embryonic yolk sac during early gestation.760 As mentioned earlier, MDM can be subdivided into pro-inflammatory M1 and anti-inflammatory M2 types.761 TRMs establish stable spatial and functional connections with specific tissue cells762,763 and proliferate in fixed tissues in adult organisms to maintain their function. Additionally, these cells have significant anti-inflammatory effects, including clearing apoptotic cells and mitochondria and inhibiting myocardial fibrosis and hypertrophy.756 Regarding vascular macrophages, 60% of the macrophages in the vessels of newborn mice originate from the yolk sac, but this decreases to only 20% in adults. As age increases, embryo-derived macrophages are gradually replaced.762 Arterial resident macrophages typically express the Lyve-1 positive marker; some also express CCR2,764 and they have weaker phagocytic abilities than MDMs. In newborn mice, these macrophages exhibit low MHC-II expression, but as they mature, high MHC-II expressing resident macrophages gradually dominate.765 Normally, arterial resident macrophages maintain their numbers mainly through local self-renewal, a process dependent on M-CSF and CX3CL1 produced by endothelial and mesenchymal cells,753 with only a small portion deriving from infiltrating monocytes.766 Regarding cardiac macrophages, heart macrophages are spindle-shaped cells in the interstitial spaces between myocytes, fibroblasts, and endothelial cells.767 They are closely associated with blood vessels and are significant in signaling processes.768 Unlike vascular resident macrophages, heart resident macrophages almost do not express CCR2; macrophages that do express CCR2 are primarily derived from monocytes.70 Similarly, in cardiac tissue, macrophages that originally do not express CCR2 are gradually replaced by those that do express CCR2, while the expression of MHC-II molecules also continuously increases.195,209,769,770 Based on MHC-II, Ly-6C, CCR2, and CD11c, cardiac macrophages can be categorized into four major types: 1. Ly-6C− MHC-IIhi CX3CR1hi CD206int MerTK+ CD11clow CCR2− CD64+ Macrophages; 2. Ly-6C− MHC-IIlow CX3CR1int CD206hi MerTK+ CD11clow CCR2− CD64+ Macrophages; 3. Ly-6C+ MHC-IIhi/low CX3CR1hi CD206hi/int MerTK+ CD11clow CCR2− CD64+ Macrophages; 4. Ly-6C− MHC-IIhi CX3CR1hi CD206int MerTK+ CD11hi CCR2+ CD103− CD64+ Macrophages. Subgroups 1 and 2 are most common under steady-state conditions.770
In vascular diseases, TRMs primarily perform phagocytic and immune surveillance roles, while MDMs primarily engage in inflammation and vascular remodeling. During the early stages of vascular diseases, macrophage proliferation mainly relies on the recruitment and differentiation of circulating monocytes. Among these, monocytes expressing high levels of Ly-6C predominantly differentiate into M1-type MDMs and promote vascular inflammation by releasing IL-6 and TNF-α.763 In cardiac diseases, circulating monocytes are chemotaxed to lesion sites via CCL2/CX3CL1 and differentiate into macrophages expressing high levels of MHC-II and CCR2. CCR2+ macrophages primarily exhibit pro-inflammatory characteristics similar to M1 macrophages, while CCR2- macrophages that appear in the later stages of injury possess anti-inflammatory and reparative functions similar to M2 macrophages.771 The extent of myocardial fibrosis induced by macrophages varies across different diseases, and macrophages play a dual role in regulating fibrosis.757,772,773 As previously mentioned, macrophages can promote fibrosis by releasing pro-fibrotic mediators such as TGF-β, PDGF, IL-10, VEGF, and amphiregulin (AREG), which induce activation of fibroblasts via receptors like TGF-βR, EGFR, and PDGFR on their surfaces, leading to excessive collagen production. Additionally, they can secrete inhibitors of matrix-degrading enzymes, such as tissue inhibitors of metalloproteinases (TIMPs), resulting in myocardial scarring. Conversely, fibroblasts may interact with macrophages by expressing CSF-1 and secreting macrophage chemoattractants like CCL2. Fibroblasts can also support macrophage activation by providing IL-6. In the TME, macrophages can undergo partial EMT, differentiating into fibroblast-like macrophages capable of secreting collagen and remodeling the TME. A similar subtype is expected in the heart, although it has not yet been identified.774,775,776,777 Moreover, macrophages can secrete MMPs to degrade the ECM and promote other cells in the microenvironment to release the same, collectively balancing myocardial fibrosis. While the activation of fibroblasts by macrophages is closely associated with M2 polarization, it is generally believed that polarization towards the M2 phenotype over time can suppress inflammation, thereby reducing fibrosis.778 Additionally, macrophages help resolve chronic inflammation in heart diseases through phagocytic actions and secretion of anti-inflammatory mediators such as TGF-β and IL-10, thus promoting cardiac repair.779,780 However, the pro-inflammatory mediators released by macrophages during the phagocytosis of necrotic tissue, such as IL-1β, IL-6, and IL-23, bind to corresponding receptors on fibroblasts, inducing an increase in pro-fibrotic factor release, thereby inducing fibrosis and adverse cardiac remodeling.776,781
Atherosclerosis (AS)
Atherosclerosis (AS) is the most extensively studied arterial disease. It is a chronic arterial condition marked by inflammation and lipid accumulation within the vessel walls, leading to plaque formation. Areas of disturbed blood flow within vessels are prone to atherosclerosis. Low shear stress from varying directions and angles of blood flow can disrupt endothelial function, which promotes morphological changes in endothelial cells and allows large molecules such as low-density lipoprotein (LDL) to more easily penetrate the endothelial layer of the vessel wall. Simultaneously, LDL binds to proteoglycans in the intima, retaining it within the endothelial layer.782,783 The transformation of LDL into its oxidized state, oxLDL, is a critical step in the pathogenesis of atherosclerosis. oxLDL further activates endothelial cells, causing them to release adhesion molecules and chemokines (M-CSF, CCL2, and VCAM-1).784,785,786 During the formation of atherosclerosis, monocytes from the bone marrow and liver are recruited to the plaque via CCL2/CCL7-CCR2 and CX3CR1 pathways.787 Early in the formation process, the recruitment predominantly involves monocytes expressing high levels of Ly-6C. Once recruited to the plaque, these monocytes differentiate into macrophages.
M1 macrophages dominate in progressive atherosclerotic plaques, primarily releasing a plethora of pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) that influence the progression of atherosclerosis (AS), including plaque instability, thrombus formation, and the chronic inflammation caused by plaques.38,788,789 Similar to rheumatoid arthritis, M1 macrophages in AS can also promote thrombosis and plaque formation through mechanisms such as macrophage recruitment, angiogenesis, and endothelial activation mediated by the IL-1β/NLRP3 axis.790,791,792 Interestingly, the absence of macrophage mTORC2, through signaling pathways involving FOXO1 and IL-1β, can lead to atherosclerosis.793 Additionally, lysosomal damage induced by cholesterol crystals can promote NLRP3-mediated inflammatory responses, with cholesterol content closely related to the number of circulating monocytes.794,795 In THP-1-derived macrophages, TRIM64 can activate NF-κB through the ubiquitination of IκBα, creating a positive feedback loop that exacerbates inflammation and atherosclerosis in these cells.796 IL-6, another significant pro-inflammatory cytokine, can exacerbate chronic inflammation in AS through trans-signaling, leading to the proliferation of vascular smooth muscle cells, thrombus formation, and increased lipid accumulation in macrophages.797 Studies have found that the inflammasome AIM2 promotes macrophage foam cell formation by inhibiting the ABCA1 protein. TNF can induce the production of ROS and further endothelial dysfunction, promoting the formation of oxLDL.798 Moreover, inhibiting the Hedgehog signaling pathway can improve early atherosclerosis by promoting autophagy and reducing foam cell formation.799 Most M2-polarized macrophages release anti-inflammatory cytokines such as IL-10 and TGF-β, which significantly inhibit the progression of AS. IL-10 and TGF-β can also reduce or stabilize plaque through cholesterol efflux and promote collagen formation.800,801,802 However, M2 macrophages expressing CD163 can enhance angiogenesis through the HIF-α/VEGFA pathway, promoting thrombus formation and macrophage infiltration.803,804
In atherosclerotic plaques, macrophages ingest apolipoprotein B-containing lipoproteins (apoB-LPs) to form lipid-laden cells called foam cells.757,786 Foam cells express fewer inflammatory genes, primarily expressing genes involved in the uptake, processing, and efflux of lipids.805 Macrophages primarily use scavenger receptors such as SR-A, CD36, and lectin-like oxidized LDL receptor-1 (LOX-1) to intake circulating lipids.806,807 LOX-1 can promote the development of atherosclerosis (AS) inflammation by activating NF-κB and MAPK signaling pathways.808 Oxidized LDL (oxLDL) exacerbates AS by inhibiting the expression of KLF2 in M2 macrophages, which induces these cells to produce pro-inflammatory cytokines.809 Additionally, oxLDL restricts the autophagic degradation of the NLRP3 inflammasome, thereby promoting the formation of the inflammatory environment in AS.810 After foam cell formation, endoplasmic reticulum stress and apoptosis-related molecular pathways cause them to release MMPs extensively, expanding the necrotic core of the plaque. In the early stages of AS, macrophages react to other apoptotic cells or components, which can inhibit the formation of the necrotic core within the plaque. However, in the mid to late stages, the phagocytic capacity of macrophages significantly decreases, and their rate of apoptosis increases, leading to uncontrollable chronic inflammation and the formation and expansion of the necrotic core within the arterial intima.811,812 In late-stage plaques, reduced expression of Baf60a, a component of the SWI/SNF chromatin remodeling complex, leads to decreased mitochondrial integrity and increases adhesion, apoptosis, and plaque formation.813 The rupture of atherosclerotic plaques, leading to acute myocardial infarction and stroke, is one of the direst consequences of atherosclerosis. Plaques prone to rupture contain a large necrotic core and a thin fibrous cap. They are characterized by high MMP activity, hydrolysis of ECM proteins, dedifferentiation of VSMCs, impaired exocytosis, and chronic inflammation.757,766 Macrophages contribute to plaque rupture by secreting different MMPs; M1 macrophages primarily release MMP1, MMP3, and MMP10, while M2 macrophages mainly release MMP11, MMP12, and MMP15 814,815。
Research has discovered that asprosin upregulates the expression of ABCA1 and ABCG1 via the p38/Elk-1 pathway, inhibiting lipid accumulation in macrophages and reducing atherosclerosis burden in apoE-/- mice.816 Additionally, inhibiting the MCT4 on the surface of macrophages can decrease lactate efflux, thereby enhancing p300-mediated histone lactylation at H3K18la, initiating macrophage repair, and promoting the resolution of inflammation.817 Increasing studies suggest that activation of PPARs can effectively prevent atherosclerosis.818,819 PPARα can reduce the uptake of glycated LDL (glyLDL) by inhibiting lipoprotein lipase (LPL) and decrease the levels of triglyceride-rich lipoproteins (Tg-Lp) by inhibiting the apolipoprotein B48 receptor (apoB48R) pathway.820 PPARα also increases the expression of ABCA1, ABCG1, and SR-BI in macrophages to promote cholesterol efflux.786 Furthermore, PPARγ promotes the polarization of M2 macrophages and inhibits the polarization of macrophages towards the M1 phenotype.819
Myocardial Infarction (MI)
Myocardial infarction (MI) refers to the process where myocardial ischemia leads to extensive death of cardiac muscle cells. MI primarily progresses through three phases: the inflammatory phase, the anti-inflammatory phase, and the repair phase, all of which involve extensive participation of macrophages.775 At the onset of injury, many necrotic cardiac cells release damage-associated molecular patterns (DAMPs), which release numerous chemotactic signals via pattern recognition receptors such as TLRs. Ly6Chi monocytes are recruited to the ischemic infarct area via the CCR2/CCL2 signal and differentiate into CCR2+ MHC-IIhi macrophages,821 playing a primary role in the inflammatory phase. Compared to CCR2+ TRMs, monocyte-derived CCR2+ macrophages possess stronger pro-inflammatory capabilities. Interestingly, tissue-resident CCR2- macrophages can inhibit the recruitment of monocytes, playing a crucial role in preventing myocardial fibrosis after heart damage.210 CCR2+ macrophages have a strong phagocytic function, working alongside neutrophils to clear debris. Still, they also secrete proteases, ROS, and inflammatory cytokines such as TNF-α, IL-1, IL-6, monocyte chemoattractant protein-1 (MCP-1), and MMPs. NETs can promote the pro-inflammatory polarization of macrophages, leading to myocardial remodeling.822 The cytokines synthesized by macrophages during the inflammatory phase are not conducive to benign repair outcomes following myocardial infarction; they promote fibrosis. For instance, IL-1, NLRP3, IL-6, IL-34, and angiotensin II757,823,824,825,826,827,828 contribute to this process. IL-1α promotes myocardial tissue fibrosis and remodeling by inducing the release of IL-6, MCP-1, and connective tissue growth factor (CTGF).829 Compared to IL-1α, IL-1β has a dual role in myocardial tissue fibrosis. Studies have found that IL-1R1-dependent IL-1β inhibits TGF-β-induced fibroblast contractile activity and α-smooth muscle actin expression, promoting matrix metalloproteinase synthesis, thereby reducing fibrosis. On the other hand, IL-1β induces upregulation of the AT1 receptor faster than TNF-α, and their combined effects further promote extracellular matrix remodeling and fibrosis.830,831 LncRNA MIAT, by downregulating IL-1β and TNF-α, inhibits macrophage inflammation, but its activation by ATP-induced NLRP3 inflammasomes is inhibited.832 IL-34 activates the NF-κB signaling pathway to promote recruitment and polarization of macrophages after myocardial ischemia in mice and humans, and knocking out IL-34 reduces cardiac remodeling, dysfunction, and fibrosis.824 Additionally, the adipocyte-derived adiponectin (ADPN) via the ADPN/AdipoR2/HMGB1 axis promotes macrophage M2 polarization, reduces IL-6 release, and stabilizes mitochondrial metabolism.833 IL-7, by regulating macrophage infiltration and polarization, promotes apoptosis of myocardial cells, thereby exacerbating myocardial I/R injury. Anti-IL-7 antibodies affect the production of cytokines by T helper cells (Th)1 and Th2 and promote macrophage polarization towards M2, thus reducing myocardial damage.834 Endothelial cells produce small extracellular vesicles (EVs) containing KLF2 that alleviate mouse myocardial ischemic injury by inhibiting the recruitment of Ly6Chi monocytes.835 The caspase recruitment domain family member 9 (CARD9) can upregulate macrophage expression of lipocalin 2 (LCN2) and MMP9, leading to myocardial cell apoptosis and adverse remodeling after MI.836
During the anti-inflammatory and repair phases (about three days after myocardial infarction), inflammation gradually subsides, and myofibroblasts proliferate to form scar tissue. Ly-6Chi monocytes differentiate into reparative macrophages. This differentiation depends on Nr4a1, and the lack of Nr4a1 can lead to abnormal inflammation in macrophages, poor healing, exacerbated fibrosis, and heart failure.837 Reparative macrophages express high levels of CD206 and MerTK, low levels of Ly-6C, MHC-II, and CCR2, and secrete a range of anti-inflammatory and tissue fibrogenic cytokines, such as IL-10, TGF-β, HIF-α, VEGFA, and SPP1.757,838,839 Among these, TGF-β/Smad3 signaling is crucial as it targets fibroblasts to stimulate their migration, transdifferentiation, and the synthesis of collagen and fibronectin.840 IL-10, as a multifunctional anti-inflammatory cytokine, has a dual role in whether myocardial tissue undergoes fibrosis. Hypoxia-induced VSIG4 promotes the expression of IL-10 in M2 macrophages, facilitating the transformation of cardiac fibroblasts into myofibroblasts.841 On the other hand, IL-10 can inhibit the human antigen R (HuR)/MMP9 signaling pathway and activate STAT3 to suppress collagen deposition.842 Basophils can promote the conversion of Ly6Chi macrophages into CD206+ macrophages by secreting IL-4 and IL-13, aiding in the healing of necrotic cardiac tissue.843 Research has shown that targeting NPM1 can reprogram the metabolism of reparative macrophages from inflammatory glycolysis to oxygen-driven mitochondrial energy production, thereby enhancing the reparative function of cardiac macrophages and promoting cardiac repair after myocardial infarction.844 However, other studies have found that mitochondrial metabolism influences the exocytosis of macrophages. Deficiency in mitochondrial complex I in macrophages can promote glycolysis, increase mitochondrial ROS production, exacerbate early inflammatory responses, and weaken exocytosis, thereby hindering the proliferation activation of fibroblasts and scar formation after myocardial infarction.845 This indicates that the metabolic mode of macrophages directly impacts the prognosis of MI. Studies have identified a group of Bhlhe41+ macrophages on day seven after MI. These cells can inhibit the activation of myofibroblasts, prevent excessive fibrosis, and limit infarct expansion.846 Additionally, sEVs derived from M2 macrophages can reduce the pro-inflammatory CCR2+ macrophage subgroup, which is beneficial for cardiac repair post-AMI847 (Fig. 5).

Role of Tissue Macrophages in Diseases. Macrophages play a pivotal role in developing a wide range of diseases, with distinct functions attributed to tissue-resident and monocyte-derived macrophages, as well as their M1/M2 polarization, which varies across different diseases. In autoimmune diseases such as rheumatoid arthritis and systemic sclerosis, macrophages damage tissue by releasing pro-inflammatory cytokines (e.g., IL-6 and TNF-α) and promote multi-tissue fibrosis through anti-inflammatory and fibrogenic factors. In cardiovascular diseases, including atherosclerosis and myocardial infarction, macrophage polarization facilitates phagocytosis of necrotic tissue, fibrotic repair, myocardial remodeling, and the formation and rupture of atherosclerotic plaques. AtoM arthritis-associated osteoclastogenic macrophages, IL-6 interleukin-6, TNF-α tumor necrosis factor-alpha, PANKL parathyroid hormone-related protein, CCL2 chemokine (C-C motif) ligand 2, ERK extracellular signal-regulated kinase, MAPK mitogen-activated protein kinase, FGF2 fibroblast growth factor 2, PGE2 prostaglandin E2, SPP1 secreted phosphoprotein 1, TGF-β transforming growth factor-beta, PDGF platelet-derived growth factor, CCL-18 chemokine (C-C motif) ligand 18, IL-34 interleukin-34, IL-7 interleukin-7, IL-1 interleukin-1, MCP-1 monocyte chemoattractant protein-1, NLRP3 NOD-like receptor pyrin domain-containing 3, KLF2 Krüppel-like factor 2, ADPN adiponectin, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells, HMGB1 high-mobility group box 1, IL-4 interleukin-4, IL-13 interleukin-13, OPN osteopontin, HIF-α hypoxia-inducible factor alpha
In the future, macrophage-targeted therapies hold great promise for treating cardiovascular diseases (CVD). Macrophages are crucial in heart injury, fibrosis, and diseases such as atherosclerosis (AS). By regulating macrophage recruitment, polarization, and function, we can effectively influence inflammation, tissue repair, and remodeling of the heart and blood vessels. Current research shows that different types of macrophages have distinct roles in various stages of cardiovascular diseases. For example, pro-inflammatory M1 macrophages are typically associated with acute inflammation and tissue damage, while anti-inflammatory M2 macrophages aid tissue repair and suppress inflammation. Future therapeutic strategies should focus on regulating these functions and account for the dynamic changes in macrophage behavior during different pathological stages.
In atherosclerosis, macrophages ingest oxidized low-density lipoproteins (oxLDL), transforming into foam cells, which promote plaque formation and inflammation. Dysfunctional foam cells exacerbate arterial hardening and increase the risk of plaque rupture, leading to acute events such as myocardial infarction (MI). Therefore, blocking the release of inflammatory mediators from macrophages or inducing the clearance of foam cells could be a potential strategy for preventing AS. In MI, macrophages play a role in the inflammatory response following injury and the repair process. CCR2+ macrophages are recruited during the acute inflammatory phase and exhibit pro-inflammatory effects, while CCR2- macrophages help tissue remodeling during the repair phase by releasing anti-inflammatory factors and promoting fibrosis. However, excessive fibrosis can lead to further deterioration of heart function. Thus, future therapies should aim to balance macrophages’ pro-inflammatory and anti-inflammatory functions, avoiding excessive inflammation or fibrosis. Utilizing small molecules, nanoparticles, or gene therapies to regulate macrophage polarization and function precisely will be key in future cardiovascular disease treatments.757
Furthermore, with advances in single-cell sequencing technology, the complexity of macrophage subtypes has become increasingly apparent. Different diseases and their stages may correspond to different macrophage subtypes, offering more targets for personalized therapies. Future research should also focus on exploring emerging macrophage functions, such as macrophage extracellular traps (MET), which have shown potential therapeutic value in infections and other diseases but remain understudied in the cardiovascular field. By gaining a deeper understanding of the diverse roles of macrophages and their interactions with other immune cells, endothelial cells, and fibroblasts, future therapeutic strategies will be able to intervene in the onset and progression of cardiovascular diseases more precisely, improving treatment outcomes and minimizing adverse effects.
Neurodegeneration disease
Over the past 15 years, numerous risk factors for neurodegenerative diseases have been identified. Among these, factors closely related to brain macrophages, such as TREM2 or APOE-positive macrophages, have been highlighted.848,849 These macrophage-related risk factors underscore the critical role of brain-associated macrophages/microglia in the pathogenesis of neurodegenerative diseases.850 This is particularly evident in Alzheimer’s Disease (AD) and Parkinson’s Disease (PD), the two most common neurodegenerative disorders. As the global population increasingly ages, the number of individuals suffering from neurodegenerative diseases such as AD and PD continues to rise. Although AD and PD represent distinct neurodegenerative pathologies, inflammatory responses play a crucial role in the progression of the pathophysiology of these diseases. These inflammatory responses can trigger toxic neuronal activation, leading to neuronal death and the formation of abnormal protein aggregates. The macrophages in the brain consist of various types, including microglia, the brain’s resident tissue macrophages originating from embryonic yolk sacs,71 and those located in the meninges, choroid plexus, and perivascular areas. Additionally, macrophages derived from circulating monocytes are recruited during disease or inflammatory processes. Historically, microglia have been primarily associated with neurodegenerative diseases. Recent research also finds that perivascular macrophages can participate in the pathology of Alzheimer’s disease by SPP1, which induces phagocytic states and synaptic phagocytosis in microglia, preventing synaptic loss when SPP1 expression is inhibited.851 Brain boundary-associated macrophages (BAMs) produce free radicals by binding to Aβ through the innate immune receptor CD36, leading to neurovascular dysfunction, cerebral amyloid angiopathy (CAA), and cognitive impairments852 Furthermore, anti-Aβ antibody (3D6) forms immune complexes with vascular amyloid deposits, which activate CD169+ perivascular macrophages, enhancing the expression of inflammatory signals and extracellular matrix remodeling genes (e.g., TIMP1 and MMP9), thereby increasing vascular permeability and recruitment of inflammatory monocytes.853 In PD, microglia are typically categorized into M1 and M2 phenotypes. Unlike many other diseases, in the early stages of Parkinson’s disease, M2 microglia primarily produce anti-inflammatory cytokines, alleviating neuroinflammation and promoting tissue repair. However, as the disease progresses, microglia, continually activated by stimuli such as α-synuclein, environmental toxins, or pathogens, transition to the M1 phenotype. This shift to M1 microglia exacerbates the inflammatory cascade, ultimately resulting in progressive neuronal loss.854,855,856
Alzheimer’s disease
Alzheimer’s disease (AD) is characterized by progressive cognitive decline and memory loss, closely linked to abnormal deposition of amyloid-β (Aβ) and pathological phosphorylation of tau protein in the brain.857,858 During this process, macrophages attempt to clear these pathological proteins through phagocytosis. However, their capacity is often limited as the disease progresses, potentially exacerbating inflammatory responses and neuronal damage. Moreover, studies have shown that anti-TNF-α antibodies may protect AD.859 The pathogenic macrophages in AD include disease-associated inflammatory (DIM) and disease-associated microglia (DAM). DIMs predominantly express various pro-inflammatory genes, such as IL-6, IL-1β, and TNF-α, which are crucial in promoting the pathological state.860 Conversely, DAMs primarily express genes like Spp1, Igf1, Gpnmb, and Dkk2, which are involved not only in the phagocytosis of amyloid-β aggregates but also in activating immune regulatory pathways, demonstrating their complex functionality.39,266 Research has found that 25-hydroxycholesterol (25-HC) can induce DAMs to produce IL-1β, leading to neuroinflammation.861 Dimethyl malonate (DMM), by inhibiting succinate dehydrogenase (SDH), reduces mitochondrial biogenesis in DIMs, converting them to an anti-neuroinflammatory phenotype.862 Despite commonalities in gene expression between DAM and DIM, particularly in Apoe and Trem2863 expression, their origins and functions differ significantly. DAMs primarily originate from embryonic sources and depend on the expression of the triggering receptor expressed on myeloid cells 2 (TREM2) to exert protective effects in the brain. In contrast, DIMs arise from circulating monocytes and, although they also express TREM2, do not depend on it and are typically associated with inflammatory states in the brain.850,860 This distinction reflects their differing pathological roles and potential therapeutic targets in AD. Peripheral MDMs can effectively reduce amyloid plaques in AD brain tissue,864 but the infiltration of aged macrophages can exacerbate the condition.865 The erythropoietin-related signaling pathway plays a significant role in halting AD progression. It enhances phagocytic activity, levels of Aβ-degrading enzymes, and the clearance rate of Aβ in peripheral macrophages. A deficiency in the EPO receptor in peripheral macrophages lowers the clearance rate of Aβ, leading to increased peripheral and cerebral levels of Aβ and accelerated progression of AD.866 IL-34 inhibits the differentiation of bone marrow-derived monocytes into macrophages, reducing the uptake of Aβ42 fibrils and oligomers and decreasing the expression of proteins such as CD36, TREM2, and MMP9, thereby contributing to the progression of AD.867 Furthermore, enhanced glycolysis-induced histone lactylation in macrophages can induce the production of β-amyloid (Aβ).868 Inhibiting the smad3 signaling pathway in macrophages can effectively promote the efflux of Aβ from the brain.869 The knockout of METTL3 in macrophages weakens m6A modification in the mRNA of DNA methyltransferase 3 A (DNMT3A), impairing the YTHDF1-mediated translation of DNMT3A. Depletion of METTL3 leads to a downregulation of ATAT1 and reduced acetylation of α-tubulin, thus enhancing macrophage migration and clearance of Aβ.870
Parkinson’s disease
Parkinson’s disease (PD) is a well-recognized neurodegenerative disorder, primarily manifesting as motor dysfunction and autonomic nervous system impairments. The neuropathology of PD is characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), and the formation of intraneuronal protein aggregates known as Lewy bodies and Lewy neurites, predominantly composed of insoluble α-synuclein.871 Microglia-mediated neuroinflammation plays a crucial role in the pathogenesis and progression of PD and is inversely correlated with the survival of dopaminergic neurons.872,873 Under normal conditions, α-synuclein exists as a soluble monomer. However, under cellular stress, misfolded α-synuclein directly contributes to neurotoxicity and activates immune cells, triggering neuroinflammatory lesions. Pathological α-synuclein activates the expression and kinase activity of LRRK2 in monocytes, inducing their recruitment to the brain. Inhibition of LRRK2 kinase may alleviate the detrimental pro-inflammatory monocyte responses in the brain.874 Moreover, macrophages or microglia overexpressing LRRK2 exhibit significantly reduced lysosomal degradation activity, exacerbating PD pathology.875 Furthermore, α-synuclein promotes the polarization of microglia to the M1 phenotype while activating NADPH oxidase, expressing pro-inflammatory cytokines and ROS, leading to sustained and progressive neurotoxicity, forming a vicious cycle.876 Additionally, overexpression of α-synuclein in microglia leads to phagocytic exhaustion and produces a large amount of oxidative toxicity, ultimately causing severe degeneration of DA neurons.877 α-Synuclein also enhances microglial glycolysis through PKM2, stabilizing the M1 phenotype and blocking mitochondrial metabolism.878,879 Disruptions in microglial mitochondrial fission, autophagy interruptions, and other disturbances lead to the activation of NLRP3, resulting in a more severe neuroinflammatory response.880,881,882 Inhibition of NLRP3 effectively promotes microglial polarization to the M2 phenotype and reduces the accumulation of α-synuclein.883,884 Research also reveals that microglia-derived exosomes can transfer misfolded α-synuclein between cells, exacerbating neuronal damage.885,886,887 Perivascular macrophages play a significant role in PD, predominantly expressing genes like CD68 and MHC-II for antigen presentation and T-cell activation.888,889 PAAN/MIF nuclease inhibition can prevent neurodegenerative changes in Parkinson’s disease, protecting against neurodegeneration induced by α-syn PFF, AAV-α-syn overexpression, or MPTP toxicity.890
The heterogeneity of macrophages in Alzheimer’s and Parkinson’s disease presents new directions for future treatments. In both neurodegenerative diseases, inflammatory responses are closely linked to neuronal damage, with macrophages playing a critical role. Studies have shown that different macrophage subtypes exhibit distinct functions at various stages of these diseases, such as disease-associated microglia (DAM) and disease inflammatory macrophages (DIM) in Alzheimer’s disease and BAM (border-associated macrophages) in Parkinson’s disease. These cells contribute to regulating inflammation, clearance of pathological proteins, and either protecting or damaging neurons through different pathways. Future research should further investigate the specific roles of these macrophage subtypes in disease progression, especially how they influence the development of the disease through molecular pathways.
Additionally, the integrity of the BBB is becoming increasingly important in Alzheimer’s and Parkinson’s diseases. It has been found that damage to the BBB may affect the function of macrophages and microglia, exacerbating inflammatory responses. Therefore, future therapeutic strategies need to focus on repairing the BBB and modulating macrophage functions when the BBB is compromised to reduce neuroinflammation and protect neurons. With the rapid advancement of single-cell sequencing technologies, new macrophage subtypes are being discovered, offering more possibilities for personalized therapies. For example, targeting specific pro-inflammatory or anti-inflammatory macrophage subtypes, along with molecular-targeted drugs, antibody treatments, or gene editing technologies, may reduce disease-related inflammation and improve patient outcomes. Research into genes like TREM2 has revealed the potential protective role of macrophages in clearing pathological protein aggregates and regulating inflammation, which could lead to innovative therapies based on these targets.
Metabolic disorders
In current medical research, obesity and insulin resistance are recognized as key precursor states for metabolic diseases such as type 2 diabetes and non-alcoholic fatty liver disease (NAFLD). Obesity is not merely an independent health issue but also a catalyst for various metabolic disorders, particularly when it leads to insulin resistance, significantly increasing the risk of type 2 diabetes. NAFLD, which directly reflects the liver’s response to obesity and insulin resistance, is one of the most common chronic liver diseases worldwide,891 and is closely linked to type 2 diabetes. These metabolic conditions and diseases are often associated with chronic low-grade inflammation,892,893 where cytokine signaling can interfere with insulin pathways, impairing glucose uptake and uncontrolled fat breakdown, ultimately resulting in ectopic lipid storage and propagating insulin resistance in a vicious cycle.894
Macrophages are the most common type of immune cells in adipose tissue, both in genetic and diet-induced obesity. Studies have shown that obesity significantly increases the number of macrophages in the adipose tissue of mice; for instance, while macrophages may constitute about 5% of all adipose tissue cells in mice of normal weight, this proportion can surge to as much as 50% in obese mice.895,896,897,898 The classification and polarization of adipose tissue macrophages (ATMs) remain a focal point of research in metabolic immunology. Beyond the traditional M1/M2 polarization applicable across various tissues and disease models, macrophages’ unique metabolic activation (MMe) is distinguished as a distinct subtype due to the adipose tissue (AT) environment rich in lipids, glucose, and insulin. The expression of metabolic-related genes and membrane receptors such as CD36, PLIN2, and ABCA1899,900 characterizes this subtype. MMe polarization features an enriched expression of lipid-handling genes,900 and adipocytes in the AT environment also release lipid-rich exosomes, inducing MMe polarization in MDMs.901 Additionally, CD9+ adipose tissue macrophages (ATMs) and TREM2 macrophages within AT display robust lipid uptake and processing capabilities902,903; TREM2 macrophages belong to a subset of lipid-associated macrophages (LAM).
Insulin resistance and Type 2 diabetes
Insulin resistance is a key precursor to Type 2 diabetes, with most patients experiencing obesity, a primary human cause of insulin resistance.904 Beyond insulin resistance, Type 2 diabetes also involves impaired insulin secretion, glucose intolerance, and hyperglycemia. In healthy/lean adipose tissue (AT), alternatively, activated M2 macrophages secrete anti-inflammatory factors to maintain tissue homeostasis. Glucocorticoids can activate anti-inflammatory macrophages to prevent insulin resistance.905 However, in obese individuals, AT induces M1 polarization of macrophages, as previously mentioned. M1 macrophages release a series of pro-inflammatory factors (such as TNF-α, IL-6, and IL-1β) and promote further M1 macrophage infiltration into AT. Pro-inflammatory cytokines activate inflammatory signaling pathways like JNK, ERK, p38, and NF-κB, promoting insulin resistance.41,896,906,907 Research has found that eosinophils can prevent this obesity-induced insulin resistance.908 Running and calorie expenditure can promote the production of M2-like macrophages through the TRIB3-AKT pathway, alleviating insulin resistance.909 Additionally, THP-1 human macrophages conditioned with IL-4 (THP1-IL-4-exo) enhance insulin-dependent glucose uptake by modulating the energy metabolism of macrophages and adipocytes and improving inflammatory responses.40 The absence of G protein-signaling modulator 1 (GPSM1) in macrophages can inhibit the pro-inflammatory state of adipose tissue, thus preventing the occurrence of insulin resistance. Mechanistically, the lack of GPSM1 primarily promotes the transcription of TNFAIP3 via the Gαi3/cAMP/PKA/CREB axis, thus inhibiting TLR4-induced NF-κB activation in macrophages.910 CITED2 can limit inflammatory responses and metabolic diseases by inhibiting STAT5 activation and promoting BCL6 expression, while macrophage CITED2 promotes obesity and insulin resistance.911 Silencing macrophage TXNIP improves hyperuricemia-induced insulin resistance through the IRS2/AKT and NRF2/HO-1 pathways. The rapamycin target complex 1 (mTORC1), and P53 have also been shown to play key roles in obesity-induced insulin resistance. High-nutrient conditions of obesity can shift ATMs from M2 to M1 through mTORC1, exacerbating insulin resistance.912,913,914 Intermittent fasting enhances visceral adipose tissue LPS-associated macrophage inflammatory phenotype through p53-driven adipocyte apoptosis, and inhibiting p53 prevents the accumulation of lipid-associated macrophages, enhancing systemic metabolic flexibility and insulin sensitivity.915 The NOTCH signaling pathway is a protective factor against insulin resistance.916 Recent studies have shown that IgG, an aging factor, can promote insulin resistance. IgG activates macrophages through the Ras signaling pathway and causes adipose tissue fibrosis, inflammation, and insulin resistance through the TGF-β/SMAD pathway.917 In addition, peptidase D (PEPD) can promote a fibro-inflammatory response in macrophages and adipocytes through the EGFR signal, exacerbating insulin resistance.918 Interestingly, embryonic vitamin D deficiency suppresses JARID2 expression through epigenetic mechanisms and activates the MEF2/PGC-1α pathway, leading to macrophage infiltration in adipose tissue, secretion of miR106-5p, inhibition of PIK3, and downregulation of AKT signaling, causing insulin resistance.919
Non-Alcoholic Fatty Liver Disease (NAFLD)
Non-alcoholic fatty liver disease (NAFLD) encompasses a spectrum of liver conditions, including non-alcoholic fatty liver (NAFL), non-alcoholic steatohepatitis (NASH), and cirrhosis. NAFL represents a basic form of fatty liver, characterized by significant lipid accumulation in hepatocytes without substantial inflammation or damage that would impair liver cell function. It is generally considered less severe and, with proper management, may not progress to more serious liver conditions. NASH, on the other hand, not only involves lipid accumulation but also significant steatosis accompanied by inflammation and some degree of fibrosis, which can progress to cirrhosis—a critical precursor to hepatocellular carcinoma.920,921,922 KCs, lipid-associated macrophages (LAMs), and MDMs are recognized as key players in the core pathophysiological development of NAFLD.923 Traditionally, it has been challenging to distinguish KCs from other liver macrophages, as they share many common macrophage surface markers. However, advancements in single-cell sequencing technologies have recently identified core genes unique to KCs, including CD5L, VSIG4, CD163, FOLR2, MARCO, and SLC40A1, with specific surface markers such as VSIG4, Clec4F, and FOLR2.77 Liver-associated LAMs, often called biliary-LAMs, predominantly express GPNMB, SPP1, TREM2, and CD9.77,902,924 MDMs in the liver are typically characterized by high expression of CD11b, intermediate expression of F4/80, and receptors CCR2 and CX3CR1.925
In the early stages of non-alcoholic fatty liver disease (NAFLD), excessive fat accumulation in liver cells leads to steatosis. These steatotic hepatocytes can activate KCs through CCL2/CXCL10 signaling and extracellular vesicles. Additionally, free cholesterol ingested by KCs can act as a pro-inflammatory agent, further stimulating their activity.926,927,928 Activated KCs release pro-inflammatory cytokines, such as IL-1β and TNF-α, which inhibit the peroxisome proliferator-activated receptor (PPAR)-α pathway in hepatocytes, thereby impairing lipid metabolism and exacerbating hepatic steatosis.929 Moreover, the IL-6/STAT3 pathway can significantly increase susceptibility to NAFLD.930 As inflammation progresses, the presence of TIM4+ mature Kupffer cells diminishes, replaced by TIM4- Ly6Chi CCR2+ MDMs, also known as monocyte-derived Kupffer cells (MoKCs). MoKCs are less effective in promoting triglyceride storage in the liver, thus exacerbating liver injury.931 Knocking out these Ly-6Chi macrophages markedly reduces liver inflammation and fibrosis.931,932,933,934 Research has found that macrophages deficient in STING enhance nuclear YAP activity, reduce lipid accumulation, and increase the expression of autophagy-related proteins. However, dual deficiency of STING and YAP exacerbates lipid deposition.543 Inhibiting the dopamine receptor D2 by selectively blocking the YAP signaling pathway in macrophages reduces steatohepatitis.935 The Hippo/YAP pathway also plays a significant role in hepatocytes, with YAP-expressing hepatocytes rapidly and effectively activating the expression of proteins that promote fibrosis (COL1A1, TIMP1, PDGFc, TGFβ2) and inflammation (TNF, IL-1β).936 Lipid-associated macrophages (LAMs) also play a significant role in promoting steatohepatitis. MS4A7 enhances TREM2+ macrophage-induced NLRP3 inflammasome activation through lipid droplet mediation, exacerbating steatohepatitis.937 Zbtb18 transcription activates FXR-mediated FAO and CLTC expression, inhibiting the activity of the NLRP3 inflammasome and thus alleviating hepatic steatosis.938 Research has shown that the transcription factor XBP1 can also regulate the progression of NASH; macrophages deficient in XBP1 alleviate steatohepatitis by reducing NLRP3 expression and pro-inflammatory cytokine secretion and promote their M2 polarization.939 Persistent obesity induces TNF and IL-1β-mediated proteolytic cleavage, leading to the shedding of TREM2 from LAMs, resulting in the loss of efferocytosis, thereby triggering chronic liver inflammation and NASH.940
Liver fibrosis is a critical hallmark of late-stage non-alcoholic steatohepatitis (NASH), where progressive, long-term fibrosis may lead to cirrhosis, portal hypertension, and even hepatocellular carcinoma.941 The complexity of liver fibrosis arises from the interplay among immune cells like macrophages, hepatic stellate cells, and myofibroblasts.942 The fibrotic microenvironment fosters the generation of scar-forming macrophages, which express high levels of IL-1β, SPP1, LGALS3, CCR2, and TNFSF12, contributing further to the fibrotic process.942 These macrophages release factors such as TGF-β, TNF, IL-1β, and galectin-3, which activate hepatic stellate cells to produce collagen and exacerbate fibrosis.943,944 Hepatic CYR61 polarizes infiltrating monocytes via the IRAK4/SYK/NF-κB signaling pathway, enhancing their inflammatory and fibrogenic transcriptional profiles.945 Additionally, XBP1 can promote fibrosis by impairing macrophage mitophagy, releasing mtDNA that activates the STING pathway in macrophages to drive fibrogenesis.946 Downregulation of METTL14 affects the translation efficiency mediated by YTHDF1, reducing GLS2 levels, creating an oxidative stress environment, and recruiting CX3CR1+ CCR2+ MDMs, which in turn activate hepatic stellate cells to promote fibrosis.947 Activation of MerTK in macrophages induces phosphorylation of AKT, STAT3, ERK1/2, p38, and upregulation of VEGF-A expression, enhancing the pro-fibrotic phenotype of human hepatic stellate cells.948 Mucosal-associated invariant T (MAIT) cells can promote macrophages to adopt a pro-fibrotic phenotype, thus facilitating the progression to cirrhosis.949
Hepatocellular carcinoma (HCC), a severe complication arising from inflammation and fibrosis in the context of NAFLD, exhibits higher levels of pro-inflammatory cytokines such as IL-8, IL-13, CCL3, CCL4, and CCL5 compared to patients with NASH, accompanied by an increase in activated monocytes in the bloodstream.950,951 Single-cell sequencing reveals that in HCC driven by liver fibrosis, TAMs predominantly express genes such as TREM2, GPNMB, SLC40A1, APOE, C1QA, and C1QB. Fibroblast growth factor (FGF) plays a significant role in fatty liver inflammation and fibrosis. Studies indicate that macrophage-specific knockout of FGFR1 alleviates liver inflammation induced by a high-fat diet (HFD) by inhibiting the activation of MAPKs and the TNF signaling pathway, reducing lipid deposition in hepatocytes, and preventing the activation of hepatic stellate cells.952 FGF21 also inhibits the transition from NASH to hepatocellular carcinoma via the hepatocyte-TLR4-IL-17A signaling pathway.953,954 Neuregulin 4 has been found to inhibit the development of NASH-associated HCC by limiting the proliferation of TAMs and the exhaustion of cytotoxic CD8+ T cells.955 Conversely, leptin exacerbates inflammation in NAFLD by promoting CD8+ T cell infiltration and mediating apoptosis of hepatocytes and macrophages.956 Recent research shows that fatty liver disease upregulates the expression of Rab27a in the liver, promoting the production of extracellular vesicles (EVs). These EVs enhance the activity of YAP protein in liver metastases of colorectal cancer, which, through the production of CYR61, promotes the infiltration of M2 macrophages, thereby fostering cancer cell growth and an immunosuppressive microenvironment957 (Fig. 6).

Role of Tissue Macrophages in Diseases. In neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases, macrophages are implicated in neuronal damage and neurotoxicity, involving the processing of amyloid-beta and alpha-synuclein. In metabolic disorders like obesity, which represents a state of systemic chronic inflammation, macrophages accelerate the development of insulin resistance and affect the efficacy of cell-based immunotherapies through tumor-associated macrophage interactions. Additionally, the diverse functions of macrophages are also evident in the progression of hepatic diseases, including hepatitis, liver cirrhosis, and hepatocellular carcinoma. p-Tau phosphorylated tau, APP amyloid precursor protein, SAPPβ soluble amyloid precursor protein beta, TREM2 triggering receptor expressed on myeloid cells 2, LRRK2 leucine-rich repeat kinase 2, NADPH nicotinamide adenine dinucleotide phosphate (reduced form), NOTCH NOTCH signaling pathway, mTOR1 mechanistic target of rapamycin complex 1, P53 tumor protein p53, CXCL10 C-X-C motif chemokine ligand 10, LGALS3 galectin-3 TNFsf12 tumor necrosis factor superfamily member 12
In the future treatment of Non-Alcoholic Fatty Liver Disease (NAFLD) and Non-Alcoholic Steatohepatitis (NASH), macrophage-targeted therapies are becoming a research focus. In recent years, as the critical role of macrophages in the progression of NAFLD and NASH has been further elucidated, therapeutic strategies such as CCR2/CCR5 dual antagonists and TREM2 inhibitors have shown promising results in preclinical and clinical trials. The heterogeneity of macrophages, especially the roles of different macrophage subtypes at various stages of disease, offers the potential for more precise targeted therapies.
Macrophage-related molecular targets, such as CD163 and TREM2, have been identified as potential biomarkers for early detection and monitoring of NAFLD progression. Moreover, adoptive cell transfer therapies targeting macrophages also show great potential in preclinical studies. With advancements in bioinformatics and single-cell sequencing technologies, researchers can more clearly analyze the transcriptomic characteristics of macrophage subtypes and their dynamic behavior in metabolic diseases. This will help better understand the interactions between macrophages and the liver microenvironment, gut, and adipose tissue, providing new targets for personalized treatment of NAFLD and NASH.
In terms of treatment strategies, in addition to traditional lifestyle interventions, drug development is shifting toward multi-targeted combination therapies. Drugs such as PPAR agonists and GLP-1 receptor agonists have shown potential in clinical trials for treating NAFLD and NASH. These drugs can reduce liver fibrosis and inflammation by targeting M2-like macrophage polarization and regulating inflammatory responses. Additionally, macrophage phagocytic activity is being explored as a potential strategy for drug delivery systems, with novel drug carriers like hard-shell microbubbles, liposomes, and polymers utilizing macrophage characteristics for more precise targeted treatments.
Trauma
Trauma typically refers to mechanical damage inflicted on the human body, resulting in compromised tissue integrity or functional impairment. Whether due to accidental injury, surgical intervention, or other forms of physical harm, trauma initiates a complex cascade of biological responses, with macrophages playing a pivotal role. Macrophages are primarily responsible for the inflammatory response and promoting tissue repair and remodeling following trauma. Immediately post-trauma, a local inflammatory response is typically elicited. During this phase, monocyte-macrophages migrate to the damaged tissue and polarize into the M1 phenotype to clear damaged or necrotic tissue. In the later stages of tissue repair, M2 macrophages dominate at the wound site, curbing local inflammation and facilitating tissue repair. M2 macrophages activate fibroblasts by secreting cytokines such as TGF-β, which significantly enhances collagen deposition and angiogenesis, collectively fostering granulation tissue formation. As previously mentioned, M2 macrophages can be further subdivided into four subtypes: the M2a phenotype, known as alternative macrophages, which promote angiogenesis and scar formation.36 The M2c phenotype, called Mreg-like macrophages, plays a crucial role in tissue remodeling by phagocytosing the matrix and preventing excessive fibrosis during repair processes.958
The role of macrophages in trauma inflammation and repair
During the early stages of trauma-induced inflammation, monocytes are recruited to the wound site under the influence of chemokines. These monocytes polarize into M1 macrophages in response to PAMPs or DAMPs, triggering a robust immune-inflammatory response that helps clear damaged tissue and dead cells. However, the inflammatory response mediated by M1 macrophages can further damage the initially affected tissue, potentially leading to severe sequelae. Reprogramming M1 macrophages into M2 macrophages can effectively mitigate local inflammation and enhance tissue repair by producing anti-inflammatory cytokines, fibrogenic factors, and angiogenic factors.36,959 Studies have shown that aspirin-triggered resolvin-D1 (AT-RvD1) and recombinant human interleukin-10 (IL-10), released from polyethylene glycol (PEG)-based hydrogels, promote the recruitment of regenerative immune cells, including CD206+ macrophages (M2a/c).960 Recent studies have shown that following trauma/inflammatory responses, the activation of GSDMD in macrophages leads to the release of 11,12-EET, which activates the MAPK cascade pathway in muscle stem cells, thereby promoting tissue repair.961
In the case of traumatic spinal cord injury, microglia within the lesion core progressively diminish and are replaced by an influx of blood-derived macrophages, exhibiting both anti-inflammatory and pro-inflammatory phenotypes. Peripherally, TMEM119+ microglia maintain their numbers through local proliferation and predominantly display a pro-inflammatory phenotype. As time progresses post-injury, inflammation in the lesion core gradually subsides, yet significant APP+/e06+ neuronal axonal and dendritic damage is detectable in the peripheral tissue, persisting for months or years post-injury.962 Studies have indicated that both central and peripheral microglia/macrophages display increased mitochondria and phagosomes upon spinal injury, suggesting these cells are in stress.963 However, a subset of MRC1+ spinal macrophages can upregulate the expression of the anti-inflammatory mediator CD163, promoting IL-10 release, which mitigates microgliosis and astrogliosis, and alleviates mechanical and thermal hypersensitivity in animal models of neural injury.964 Additionally, the lncGBP9 sponge absorbs miR-34a to rescue SOCS3 expression, thereby exacerbating spinal injury through p-STAT1 mediated enhancement of macrophage M1 polarization.965 However, biocompatible hyaluronic acid and methylcellulose (HAMC) hydrogels loaded with fatty extracts (FE) can promote macrophage polarization from the inflammatory M1 to the anti-inflammatory M2 phenotype via the STAT6/Arg1 pathway, thus reducing inflammation in spinal injury models.966 In intervertebral disc injury models, biomimetic nanomaterials encapsulating MnO2 nanoparticles and TrkA overexpressing macrophage membranes effectively bind various inflammatory and neurotrophic factors, inhibiting inflammation-induced NPC apoptosis, matrix degradation, and neurogenesis. This also significantly reduces ROS production and M1 polarization by macrophages, effectively alleviating disc inflammation.967 In skin injuries, LPS increases macrophage cholesterol accumulation in a DNMT1-dependent manner, and the cholesterol content determines cellular stiffness and motility, thus affecting the effective chemotaxis of macrophages at the trauma site.968 Studies have shown that in lung injuries caused by mechanical overventilation, besides macrophage WISP1 and TLR4 exacerbating the damage,969,970 the angiotensin II type receptor AT2R can inhibit macrophage M1 polarization and alveolar macrophage apoptosis, thereby curbing further injury progression.971 Silencing ROCK1 reduces lung injury by inhibiting NLRP3 signaling in M1 macrophages.972 Additionally, the absence of YAP lowers pro-inflammatory cytokine levels (IL-1β, IL-6, and TNF-α) and enhances M2 polarization, thus mitigating lung injury.973 In a model of long-segment urethral trauma, researchers found that a long-lasting anti-inflammatory and antioxidant hydrogel composed of lipoic acid, small molecule glycine, and γ-Fe2O3 nanoparticles, through mechanical stimulation, achieves rapid re-epithelialization and guides macrophages towards M2 polarization, effectively reducing inflammation and repairing trauma.974 Radiation trauma disrupts the vascular microenvironment of HSCs, with studies revealing that post-irradiation residual CD206+ macrophages exhibit activated M2 polarization, and the expression of the mechanosensitive ion channel Piezo1 is upregulated in response to mechanical environmental changes induced by bone marrow ablation. Piezo1 activation can promote VEGF-A expression, thereby facilitating the recovery of sinusoidal vessels.975
Diabetic trauma
Diabetes, characterized as a chronic low-grade inflammatory condition, often results in non-healing wounds in affected individuals.976 Unlike the orderly progression of normal wound healing, chronic wounds fail to transition from the inflammatory to the proliferative phase, resulting in persistent, unresolved inflammation.977 The pathophysiology of chronic wounds is complex, influenced by multiple factors, including hyperglycemia, venous insufficiency, arterial hyperperfusion, and sustained pressure, which affect the normal function of macrophages.36,978 In diabetic wound tissue, the lack of transcription factors FOXM1 and STAT3 significantly reduces chemotaxis of macrophages and neutrophils,979 and the phagocytic ability of M1 macrophages in chronic wounds is diminished. This leads to the prolonged presence of damaged tissue and necrotic cells at the wound site, serving as harmful stimuli. The ensuing persistent inflammation promotes the transition of immune cells to an exhausted state and impedes the transition from M1 to M2 macrophages, thus hindering wound healing.980 Only after these dysfunctional macrophages have completely cleared pathogens and damaged tissue can the transition from M1 to M2 occur; hence, diabetic wound macrophages exhibit a “slow in, slow out” characteristic.981 In diabetic wounds, excessive ROS activate the NF-κB pathway, leading to transcriptional silencing of the nuclear factor erythroid 2-related factor 2 (Nrf2), thereby perpetuating a vicious cycle of oxidative stress and inflammation (Table 1).
In diabetic wounds, keratinocytes induce the expression of the NLRP3 gene in wound macrophages through IL-1 receptor-mediated signaling, enhancing inflammasome activation in the presence of PAMPs and DAMPs, thereby inhibiting wound healing.982 However, suppose the content of Nrf2 is normal in the wound. In that case, it can promote keratinocytes to secrete CCL2, which drives macrophage chemotaxis and induces their epidermal growth factor (EGF) expression, thereby stimulating keratinocyte proliferation.983 Recent research has developed a nanoenzyme hydrogel spray composed of oxidized alginate and methacrylic acid gelatin, which can restore macrophage Nrf2 transcriptional activity in vitro, inhibiting the production of ROS, particularly hydroxyl radicals.984 Interestingly, the traditional Chinese medicine formula ‘San Huang Anti-inflammatory Prescription’ activates the AMPK/NRF2 signaling pathway, downregulating the HMGB1-mediated abnormal inflammatory microenvironment and improving diabetic foot conditions.985 The expression of CD64 also plays a crucial role in the healing of diabetic wounds; in CD64 knockout mice, wound healing is significantly delayed, particularly on the seventh day, with a marked reduction in CD163+ M2 macrophages infiltrating the wounds of diabetic mice.986 The lncH19 expressed by fibroblasts, by inhibiting p53 activity and GDF15 release, alleviates fibroblast cell cycle arrest and increases macrophage infiltration in damaged tissue, thus promoting the healing of diabetic wounds.987 Interestingly, exosomes secreted by adipose tissue macrophages from healthy lean mice containing miR-222-3p promote M2 polarization of macrophages, thereby enhancing wound healing in diabetic mice.988 Studies show that a flexible liposome containing phosphatidylserine (PS), mimicking apoptotic cells (D-PSLs), can persistently bind to macrophage membranes, effectively promoting M2 macrophage polarization, increasing the expression of the endothelial marker CD31 and accelerating anti-inflammatory and diabetic wound healing processes.989 Additionally, a ROS-scavenging hydrogel formed by cross-linking polyvinyl alcohol (PVA) with a ROS-responsive linker, by reducing ROS levels and upregulating the M2 macrophage phenotype, promotes the healing of bacterial-infected diabetic wounds.990 Research has found that a hydrogel composite of ginseng-derived sEVs (G-sEV) and Mg2+ not only promotes the chemotaxis and neurogenic differentiation of mesenchymal stem cells but also encourages M1 macrophages to reprogram into an M2 phenotype that promotes angiogenesis while creating a suitable immune microenvironment for wound healing.991 Studies indicate that macrophages cultured to mimic the reparative functions of TAMs, termed TAMEMs, outperform the primary macrophage phenotypes (M0, M1, M2) in vitro in terms of inhibiting inflammation, promoting angiogenesis, and activating fibroblasts, effectively enhancing the healing of diabetic mouse skin wounds992 (Table 2).
In trauma healing and the treatment of diabetic wounds, macrophages play a crucial role, with their dynamic changes during different stages of the wound determining the progression of inflammation and tissue repair. Studies have shown that the phenotypic plasticity of macrophages is essential for normal wound healing. The prolonged presence of M1 macrophages delays wound closure, while M2 macrophages help suppress inflammation and promote tissue repair. With the development of technologies such as single-cell sequencing, our understanding of the role of macrophages in chronic wounds has deepened, particularly in complex conditions like diabetic foot ulcers, where dysregulation of macrophage phenotypes may exacerbate wound healing delays.
Currently, macrophage-based therapeutic strategies can be categorized into two main approaches: pharmacological interventions and ex vivo macrophage transplantation. Pharmacological interventions aimed at modulating macrophage polarization to reduce the inflammatory effects of M1 macrophages or promote their transition to M2 have shown potential in treating chronic wounds. Additionally, the technology of gene manipulation to control macrophage phenotypes, especially through siRNA delivery, is advancing, but its clinical application requires further research. Furthermore, ex vivo activation of macrophages for transplantation has also been explored in wound treatment. Although this approach can accelerate wound healing under normal conditions, its application in diabetic wounds still faces challenges, such as phenotypic instability and interference with the wound microenvironment.
Therapeutic targets and strategies
Macrophages play an active role in inflammation and resolution, which makes them promising targets of autoimmune diseases and tumor immunotherapy. It is important to decide the direction of macrophage intervention when it comes to cancer or non-cancer disease. For example, in cancer treatment, inhibiting pro-tumoral M2 macrophages or reprogramming them toward an pro-inflammatory M1 phenotype can be beneficial to enhance anti-tumor immune responses. Or we can choose target immune checkpoints (e.g., PD-1, CTLA-4) that may involve macrophages to enhance anti-tumor immunity. For inflammatory diseases, modulating macrophage activity is the key to reduce inflammation and promote tissue repair. In chronic inflammatory diseases (e.g., rheumatoid arthritis, inflammatory bowel disease), M1 macrophages are often overactivated, contributing to tissue damage and sustained inflammation. In such cases, inhibiting M1 macrophages or promoting an M2-like anti-inflammatory response is beneficial for reducing inflammation and promoting tissue repair. However, as M2-like macrophages are pro-fibrotic and M1-like phenotype area nti-fibrotic. In fibrotic diseases, strategies include inhibiting pro-fibrotic signals like TGF-β, reprogramming macrophages to an anti-fibrotic M1 phenotype, promoting ECM degradation, and blocking macrophage recruitment. In conclusion, current targeting strategies towards macrophages in cancer include depleting, reprograming, activating/inhibiting the recruitment, and modulation of metabolism and function. (Table 3).
Besides medicine intervention, clinical trials of adoptive cell therapy are under rapid development. Using macrophages to bring cytokines (e.g., IFNα, IL-12) to the tumor site and consequently activate an immune response were investigated.993,994 Compared to traditional drug-delivery ways, the various delivery systems (cell, viral, and nanoparticle-based) provide more possibility for those with drug resistance or low permeability of BBB. Through such delivery systems, peptide, DNA, miRNA, circRNA, and etc. can be administrated as key modulators of macrophage differentiation and polarization, including TAMs.995,996
Macrophages-based targeted drugs (FDA-approved drugs and clinical trials)
Csf-1/Csf-1r axis is a signaling pathway crucial for the development and maturation of macrophages. After deletion of CSF-1R, the number of macrophages is significantly reduced, which has been studied in the clinic based on preclinical data showing a delay in tumor outgrowth in different tumor models.997,998,999 CSF-1R is primarily targeted for inhibition in therapeutic settings because CSF-1R signaling promotes the survival, proliferation, and differentiation of TAMs. CSF-1R blockade reduced the ability to unleash the immune-stimulatory capacity of TAMs with a skewing of MHC IIlow to MHC IIhi macrophages. Therefore, multiple clinical trials have been conducted with different antibodies and small molecules targeting CSF-1R, alone or in combination with standard treatment or immunotherapy. The small molecule drugs under clinical development and research mainly include Pexidartinib (PLX3397), JNJ-40346527, and BLZ945.
Preclinical studies have demonstrated that Pexidartinib significantly inhibits the growth and metastasis of osteosarcoma in mouse xenograft models.1000 Phase I and II clinical trials indicate that Pexidartinib has anti-tumor effects in patients with advanced tenosynovial giant cell tumor, which has been approved by FDA997,1001,1002; however, in patients with recurrent glioblastoma, the drug showed tolerability but did not significantly improve 6-month progression-free survival.1003 JNJ-40346527 has shown significant efficacy in treating relapsed or refractory Hodgkin lymphoma.1004 BLZ945, when combined with insulin-like growth factor receptor (IGF1R) inhibitors and phosphoinositide 3-kinase (PI3K) inhibitors, can reprogram TAMs from a pro-tumoral to an anti-tumoral phenotype and inhibit glioma progression,1005 with clinical trials targeting advanced solid tumors underway.
Emactuzumab (RG7155), a monoclonal antibody (mAb) targeting CSF-1R, has also entered clinical trials. Emactuzumab is a humanized mAb that binds to CSF-1R and prevents its dimerization. Preclinical data show that emactuzumab can significantly reduce the number of CSF-1R+ CD163+ macrophages and increase T cell infiltration in the TME.997 Results from a phase I clinical trial for tenosynovial giant cell tumor revealed that emactuzumab treatment had no dose-limiting toxicity, with common side effects being facial edema, weakness, and pruritus. During the dose-expansion phase of the trial, 24 out of 28 patients had objective responses, and 2 achieved complete responses.1006
However, attempts to target TAMs with Csf-1r-inhibiting therapies have met with disappointing clinical results. On the one hand, there is a restriction in the dosage that long-term loss of macrophages can cause an imbalance in body homeostasis. On the other hand, a small but discernible macrophage with high CD163 expression was found to impede the responses to T cell-based immunotherapy, which could be a potential therapeutic target.1007 The CD163hi M2 macrophages highly express several tumor-promoting macrophage markers and have a functional anti-inflammatory transcriptome profile, but the Csf-1r expression is low.
Another treatment strategy involves using bisphosphonates to selectively eliminate TAMs, with clodronate being the most widely used.1008 Clodronate is encapsulated in liposomes. When macrophages phagocytose these liposomes, lysosomal phosphatases dissolve them, gradually releasing the clodronate. Once accumulated in the macrophages sufficiently, clodronate induces macrophage apoptosis, leading to their clearance. Studies have shown that this therapy can significantly reduce macrophage infiltration and limit metastasis growth in models of lung cancer bone metastasis,1009 breast cancer metastasis,1010 and mouse melanoma.1011 In an RCT involving 1069 primary operable breast cancer patients, clodronate significantly reduced the occurrence of bone metastases.473 Moreover, clodronate combined with chemotherapy drugs such as cisplatin and sorafenib exhibits synergistic anti-tumor effects in treating various tumor types.1012,1013 However, at the clinical level, the results of clodronate treatment in different cancers have been inconsistent, indicating that optimization of combination treatment regimens or extended clinical trials may be necessary.473
Immunotherapies usually require a more activated immune environment. GM-CSF treatment can regulate the proliferation and/or activation of myeloid cells to enhance immune responses, which has diverse strategies. For example, direct administration of GM-CSF is often used in combination with other therapies, especially the monoclonal antibodies, nivolumab (Opdivo) and pembrolizumab (Keytruda) in the clinic and clinical development.1014,1015
Sargramostim (tradename Leukine) is a GM-CSF that functions as an immunostimulatory, mainly used in combination with other cancer immunotherapy or as vaccine adjuvant. Up to now, chemotherapy + sargramostim in the treatment of Acute Myelogenous Leukemia and sargramostim after autologous and allogenic BMT in Non-Hodgkin’s lymphoma (NHL), acute lymphoblastic leukemia (ALL) and Hodgkin’s disease are approved by FDA.1016 Similar to Sargramostim, Molgramostim is a recombinant GM-CSF that acts as an immunostimulatory. The addition of molgramostim to antibiotic therapy can also reverse sepsis-associated immunosuppression decreases the rate of infectious complications in sepsis.1017,1018,1019,1020
GM-CSF as a vaccine adjuvant can be beneficial. Still, it depends on the specific context and goals of the vaccine, as adding it may induce side effects and overstimulation of the immune system. In a phase 1 trial of a DNA vaccine (pTVG-AR, MVI-118) in prostate cancer, persistent IFNγ immune responses were observed irrespective of GM-CSF adjuvant.1021 In a phase 2 trial of a DNA vaccine (pTVG-HP) and nivolumab in prostate cancer, among 14 patients to whom GM-CSF was added, three (21%) developed any subsequent PSA decline.1014 From this, we conclude that while GM-CSF may have provided a modest improvement for a few patients, it was not required as an adjuvant.
However, GM-CSF also plays a more pronounced role in autoimmune diseases.GM-CSF inhibitors (e.g., mavrilimumab) are being explored in clinical trials for conditions like rheumatoid arthritis and COVID-19-induced cytokine storms.1022,1023,1024 Until now, other GM-CSF inhibitors, like Gimsilumab (KIN-1901) and Namilumab (MT203) have not shown significant suppression of key inflammatory pathways and therapeutic effects compared to placebo.1025,1026,1027
The increase of TAMs in tumor tissues is primarily driven by the recruitment of monocytes via the CCL2-CCR2 signaling axis. CCL2 is a potent chemoattractant for monocytes, T cells, and NK cells. Studies in mouse models have demonstrated the crucial role of CCL2 and other chemokines in TAM recruitment.1028 Tumor cells release CCL2, which attracts cells expressing the CCR2 receptor to the tumor site. Inhibition of CCL2 signaling has been shown to suppress tumor growth and metastasis in various experimental models.1029,1030,1031,1032,1033 However, concerns have arisen regarding the long-term efficacy of this approach. In mouse breast cancer models, discontinuation of anti-CCL2 treatment led to accelerated lung metastasis and increased monocyte recruitment, resulting in the death of the mice.1034,1035 Moreover, as several neutralizing antibodies targeting CCL2 have entered clinical trials, none of these studies have reported a sufficient therapeutic effect by inhibiting the CCL2/CCR2 axis as monotherapy.1036,1037,1038
Among the leading candidates are carlumab (CNTO888), a monoclonal antibody against CCL2, and PF-4136309 (INCB8761), a small molecule inhibitor targeting CCR2. Carlumab, a human immunoglobulin G1κ (IgG1κ) antibody, has been shown to inhibit TAM recruitment and angiogenesis in prostate and ovarian cancer mouse models, thereby suppressing tumor growth and enhancing the efficacy of chemotherapy.1029,1039,1040 In a phase Ib clinical trial for advanced pancreatic cancer, the combination of PF-04136309 with FOLFIRINOX (a regimen including oxaliplatin, irinotecan, folinic acid, and fluorouracil) showed improved therapeutic outcomes compared to FOLFIRINOX alone.1041 However, when combined with nab-paclitaxel and gemcitabine in a phase 1b trial of previously untreated metastatic pancreatic ductal adenocarcinoma (mPDAC), PF-04136309 showed synergistic pulmonary toxicity and no significantly better clinical responses above nab-paclitaxel and gemcitabine.1042
CD47-SIRPα axis inhibits the accumulation of myosin IIA at phagocytic synapses and inhibits the phagocytic function of macrophages, which prevents autoimmunity. However, tumor cells also highly express CD47, achieving immune escape and promoting tumor invasion and metastasis.1043 Experiments in mouse transplant tumor models have shown that inhibiting CD47 can promote macrophage phagocytosis of tumor cells and is an effective tumor treatment strategy.1044,1045
Several antibodies and small molecule inhibitors targeting CD47 or SIRPα have been tested in clinical trials, for example, Hu5F9, CC-90002, and TTI-621. Magrolimab (Hu5F9-G4) showed promising safety and therapeutic effects in preclinical studies of human AML and pediatric brain tumors. In a recent phase 1b clinical trial including 87 previously untreated AML patients, the median OS was 9.8 months for TP53-mutant patients and 18.9 months for wild-type patients.1046 TTI-621, a fully human recombinant protein that blocks the CD47-SIRPα axis, has enhanced macrophage-mediated phagocytosis of tumor cells in aggressive AML and B-cell lymphoma. In a first-in-human phase I study involving 164 patients with relapsed or refractory hematologic malignancies, an overall response rate (ORR) of 13% was observed.1047
Some apoptosis-resistant mechanisms in cancer cells along with the hypoxia of TME can limit the effectiveness of photodynamic therapy (PDT). To address this challenge, He et al. developed an innovative biomimetic nanoplatform that integrates oxygen-enhanced PDT, ferroptosis induction, and CD47-SIRPα blockade. This system ensures precise delivery of chlorin e6 (Ce6) as a photosensitizer, hemin, and PEP20, a CD47 inhibitory peptide. In preclinical trials, mice bearing primary breast tumors exhibited significantly reduced tumor growth and lung metastasis as a result of this approach.1048 Moreover, hybrid nanovesicles (hEL-RS17), which are functionalized with the RS17 peptide—an antitumor agent that blocks CD47-SIRPα signaling—were shown to actively target tumor cells and modulate TAM phenotypes to enhance tumor infiltration. When used in combination with the chemotherapeutic agent shikonin, photosensitizer IR820, and immunomodulator polymetformin, hEL-RS17 exhibited superior antitumor efficacy in both 4T1 breast cancer and B16F10 melanoma models.1049 Another research shows that genetically engineered cell-membrane-coated magnetic nanoparticles (gCM-MNs) can disable both CD47-SIRPα and CSF-1 mechanism of tumorigenic M2 phenotype, which significantly prolonged overall mouse survival.1050
However, the expression of CD47 in a wide range of cells poses a challenge, as CD47 monoclonal antibodies can bind to red blood cells, causing severe anemia. QPCTL (isoQC) offers a targeted approach by modifying CD47 on tumor cells. QPCTL is a pyroglutaminase that facilitates CD47-SIRPα interaction, thus inhibiting macrophage-mediated phagocytosis of tumor cells. In tumor cells not expressing QPCTL, there is reduced pyroglutamination in CD47- SIRPα binding and enhanced macrophage phagocytosis of tumor cells.1051,1052 Phase 1 or 2a studies evaluating QPCT or QPCTL inhibitors for neurodegenerative diseases reported no significant toxicity. The development of QPCTL inhibitors, whether used alone or combined with CD47-blocking antibodies, is a promising area for future research.
The CD40 pathway plays a critical role in macrophage-targeting therapy, particularly in immunotherapy and cancer treatment. CD40 is a costimulatory receptor expressed on the surface of macrophages, DCs, and B cells. Its activation, through binding with its ligand CD40L (CD154) expressed on T cells, leads to a series of immune responses that are crucial for activating both innate and adaptive immunity. Engagement of the CD40 pathway significantly increases the production of pro-inflammatory cytokines like TNF-α, IL-12, and IL-6, which are crucial in driving the anti-tumor immune response and promoting inflammation. In TAMs, it promotes the transition from an immunosuppressive M2-like phenotype to a more inflammatory and anti-tumor M1-like phenotype.473 Trials of CD40 agonists on macrophages, such as Sotigalimab (APX005M) have reported good tolerance with chemotherapy and immunotherapy.1053,1054 And another CD40 agonist, Selicrelumab, In solid tumors, Selicrelumab showed promising effect, especially when combined with immune checkpoint inhibitors.1055 However, CD40 activation can lead to a heightened immune response, which could result in cytokine release syndrome. Managing such side effects is a key area of focus.
These molecular targets (Csf-1/Csf-1r, CCL2-CCR2, CD47-SIRPα, CD40-CD40L, etc.) are at the forefront of macrophage-targeting therapy development. They play critical roles in modulating macrophage behavior, either by reprogramming macrophages in the TME, controlling inflammation, or reducing fibrosis. Therapeutics focusing on these hotspots are actively being investigated in clinical trials for a wide range of diseases, including cancer, autoimmune conditions, and chronic inflammatory diseases(Table 3).
Macrophages-based immunotherapies (FDA-approved drugs and clinical trials)
Due to the critical role macrophages play in tissue repair and inflammation, macrophage-based cell therapies have been utilized in the treatment of various diseases. Current strategies are mainly divided into three: (1) Ex vivo educated or generated macrophages utilizing their innate properties; (2) macrophages as delivery vehicles for small molecules, plasmid DNA, and other therapies; (3) genetically engineered macrophages enhanced for therapeutic benefits, like chimeric antigen receptor-macrophage (CAR-M). Ex vivo educated cells have the most extensive history in macrophage-based therapies. This approach is grounded in the observation of elevated monocyte and macrophage recruitment to tumors in vivo, as well as animal studies that highlight the cytotoxic capabilities of primary macrophages treated with IFNγ.1056,1057,1058 Among current completed and ongoing trials of macrophage cell therapies, most are regarding the adoptive transfer of macrophages and ex vivo polarization with different strategies, while some are CAR-M trials.
Ixmyelocel-T is a multicellular therapy designed to treat ischemic conditions. It is composed of three key cell types derived from a patient’s bone marrow: mesenchymal stem cells (MSCs), M2-like macrophages, and hematopoietic cells. These cells work together to promote tissue repair, reduce inflammation, and stimulate angiogenesis. Ixmyelocel-T has been tested in several clinical trials for its potential to treat ischemic cardiovascular conditions. Ixmyelocel-T has shown positive outcomes, improving amputation-free survival and wound healing in critical limb ischemia patients. Clinical studies have demonstrated that Ixmyelocel-T can improve heart function and reduce the occurrence of major adverse cardiovascular events (MACE), slowing the progression of heart failure. In a phase IIA clinical trial (open label), heart failure patients receiving Ixmyelocel-T cells via intramyocardial delivery exhibited improved functional, clinical and quality-of-life outcomes.1059 Ixmyelocel-T has been explored as a treatment to acute myocardial infarction. After a heart attack, the heart tissue is damaged by a lack of blood flow. Ixmyelocel-T has been tested to aid in the repair of this damaged tissue. The ixCELL-DCM trial, a Phase IIB study, has completed recruiting patients with end-stage heart failure due to ischemic dilated cardiomyopathy, with 66 patients receiving Ixmyelocel-T and 60 receiving a placebo. This trial aims to assess the efficacy, tolerability, and safety of Ixmyelocel-T compared to placebo, with the cells administered via catheter-based, transendocardial injections. Initial 12-month results indicate that Ixmyelocel-T significantly reduces clinical cardiac events and improves patient outcomes, even though there were no notable changes in left ventricular ejection fraction (LVEF), left ventricular volumes, or six-minute walk distance (6MWD). These early findings suggest that while Ixmyelocel-T may not drastically change certain heart function measurements, it offers meaningful clinical benefits for patients with ischemic heart failure.1060 By modifying macrophages to express CARs that target specific tumor antigens, tumor cells expressing CAR molecular ligands can be specifically targeted and directly phagocytosed. Such macrophages are called CAR-macrophages (CAR-M). CAR-M therapies are primarily in preclinical and clinical development, with ongoing trials assessing their safety, feasibility, and efficacy in cancer patients.
A study in mouse tumor models embedded CAR molecules targeting HER2 in macrophages and evaluated the tumor-killing effect of CAR-M.67 The results demonstrated that in a human ovarian cancer xenograft mouse model, tail vein injection of CAR-M significantly inhibited tumor growth and prolonged the overall survival of tumor-bearing mice. Additionally, CAR-M reduced the rate of lung metastasis from ovarian cancer cells. The study also found that CAR-M, besides its inherent anti-tumor M1-like phenotype, could convert M2-like TAMs in the TME into an M1-like phenotype and promote T-cell infiltration in tumor tissues. HER2 CAR-M and CD47 CAR-M were generated for target antigen-positive ovarian cancer. Their antigen-specific phagocytosis of ovarian cancer cells was preliminarily verified in vitro and in vivo.1061 Preclinical studies of CAR-M have reported promising results in anti-PSCA against pancreatic cancer.1062 The phase 1, first-in-human (FIH) study of the anti-HER2 CAR-M, CT-0508 (NCT04660929), and CT-0525 (NCT06254807) in subjects with HER2 overexpressing solid tumors is ongoing. Preliminary results of CT-0508 showed safety, tolerability, CT-0508 infiltration, and immune activation in 7 participants with breast, esophageal, cholangiocarcinoma, ovarian, and parotid gland cancers.
iPSCs-derived, CAR-expressing macrophage cells (CAR-iMac) were generated and tested for tumor treatment.68 CAR-iMacs have high yield and purity and exhibit macrophage gene expression profiles, phagocytosis, polarization, and other functions. When co-cultured with CD19-positive lymphoma cells and ovarian cancer cells expressing mesothelin antigen, CAR-iMacs exhibited antigen-dependent phagocytosis and killing functions and displayed an M1-like macrophage phenotype. CAR-iMacs also demonstrated the ability to inhibit tumor growth in mouse models of hematological and solid tumors. However, there is a major hurdle of unstable CAR gene expression in hiPSCs. To solve it, another team used an elongation factor short promoter and the ubiquitous chromatin opening CBX3 element to reduce epigenetic methylation and set up a scalable differentiation system to generate CAR-iMacs that efficiently eradicate CD19-positive leukemia.1063 In mouse models, the CD19-CAR-iMacs effectively eradicated CD19-positive leukemia cells, significantly inhibiting tumor growth and improving survival rates of the treated mice. Preclinical studies of CAR-iMacs reported potent anticancer activity against GBM cells.1064
CAR-M can be combined with other therapies, such as checkpoint inhibitors, to enhance overall treatment efficacy.
Nanoparticle-based therapy
Nano-drug delivery system (NDDS) is a drug delivery method developed by using nanotechnology, which encapsulates drugs in nano-carriers and delivers them to specific cells or tissues in a targeted and controlled manner.1065,1066 NDDS could deliver drugs to target sites and simultaneously improve the stability and bioavailability of drugs, thereby improving drug treatment’s efficacy and reducing side effects.1065,1067 Notably, specifically edited or modified NDDS could penetrate biological barriers, including the BBB, making it difficult to treat certain areas for traditional drugs to reach.1068,1069 In clinical and preclinical studies, it has been found that multi-drug combination therapy could reduce the incidence of adverse events and achieve more therapeutic benefits compared to single-drug therapy.1070,1071,1072 NDDS may provide a new way or chance for multi-drug therapy of diseases.
Considering the different fractions in which macrophages are involved, macrophage-associated NDDS could be developed based on two main strategies.1073,1074,1075 One is macrophage-targeted NDDS. It could specifically target macrophages by nanoparticles modified with specific molecules capable of recognizing receptors/ligands on macrophage surfaces, thereby affecting the pathological processes of macrophages and blocking disease progression.1073,1074 The other is NDDS based on macrophages or their secreted extracellular vesicles (EVs). It utilizes one of the following components: living macrophages, macrophage membranes, EVs secreted by macrophages or membranes of EVs to phagocytize or coat nanoparticles, then deliver them to specific tissue sites and help with their functions based on the properties of macrophages or their secreted EVs.1050,1065,1074,1076,1077
CSF-1 could affect the proliferation of CSF-1R-expressing cells and promote M2 polarization of macrophages.761,1078 pIL-12 + PLX@cR-PssPD could disintegrate at the tumor site and release CSF-1R inhibitor PLX3397 (PLX) and pIL-12. The synchronous inhibition of CSF-1R and the expression of IL-12 ameliorated the TME by stimulating the activation and proliferation of T lymphocytes, reducing myeloid-derived suppressor cells (MDSCs), as well as promoting the repolarization of TAMs and the maturation of DCs, thereby efficiently suppressing tumor growth and metastasis of melanoma and colon cancer.1079 Sun et al. developed a nanoparticle that could achieve specific delivery to M2 macrophages by the sialic acid-CD169 axis, and it has been reported to reprogram TAM due to the downregulation of CSF-1R via delivering CSF-1R siRNA in the prostate cancer model.1080
CCR2-MM@PLGA/Cur, a CCL2/CCR2 axis targeted multifunctional biomimetic nanoplatform, features an engineered macrophage membrane (MM) coating with increased CCR2 expression and a therapeutic drug-loaded PLGA nanoparticle core. Overexpression of CCR2 on the MM enhanced the delivery of targeted drug to the injury site and reduced macrophage infiltration, pro-inflammatory polarization of microglia, and neuronal apoptosis by sequestering CCL2. It promoted neural regeneration and motor function recovery in mice with spinal cord injury (SCI), providing a comprehensive treatment strategy for SCI.1081 Two-dimensional Mg2Si nanosheet(MSN) is developed as a high biocompatibility nanomaterial with super-persistent hydrogen release, and the incorporation of MSN into the chitosan/hyaluronic acid hydrogel (MSN@CS/HA) is designed as a dressing for the repair of deeply burned skin.1082 This hydrogel dressing could continuously generate hydrogen molecules for the support of long-term and abundant hydrogen supply, which assists with the wound repair through diverse mechanisms.Specifically, hydrogen molecules promote anti-inflammatory M2 macrophage polarization via increasing CCL2 expression to facilitate angiogenesis and reduce fibrosis.Besides, it locally scavenges overexpressed ROS, leading to the enhanced proliferation and migration capacity of skin cells.1082
A nanogel BBLZ-945@PAC-PTX offers a “dominolike” barrier elimination strategy in tumors, which promotes immunocyte infiltration for potential immune responses. Deeper spatial penetration of shrunk nanogel (PAC-PTX) could block cell membrane CXCR4,thus decreasing the recruitment of immunosuppressive cell,as well as internalize into tumor cells for tumor-killing and T-cell priming. Once it reaches the tumor site, BLZ-945 conjugated albumin (BBLZ-945) could be released by BBLZ-945@PAC-PTX under the trigger of adenosine triphosphate (ATP) and then deplete TAMs.1083 An engineering CXCL12 biomimetic decoy-integrated versatile immunosuppressive nanoparticle (VIN) for treating ischemic stroke has been reported. The shell of VIN (mesenchymal stem cell membrane overexpressing CXCR4) could facilitate the delivery of nanoparticles to the cerebral ischemic lesions as well as adsorb and neutralize CXCL12 for the inhibition of infiltration of peripheral neutrophils and mononuclear macrophages. Besides, the cGAS inhibitor A151 released from the VIN could inhibit the cGAS-STING pathway in microglia, leading to an M2-type polarization of microglia.1084
Kühne et al. designed an efficient NDDS carrying the class I/IIa selective histone deacetylase (HDAC) inhibitor valproic acid (VPA), denoted as the cellulose-based sulfated VPA-coupled (CV-S) NPs. CV-S NPs could release VPA to suppress the TLR4-MyD88-NF-κB signaling axis, attenuating the LPS-mediated inflammatory response in human primary macrophages via reducing the expression and secretion of TNF-α.1085 A biomimetic polymer magnetic nanocarrier, PLGA-ION-R837 @ M (PIR @ M), was developed to target TAMs by coating LPS-treated macrophage membranes on the surface. It encapsulated Fe3O4 NPs and TLR7 agonist imiquimod (R837), which could synergistically repolarize TAMs from M2 to antitumor M1 phenotype.1086 Resiquimod, also known as R848, is a TLR7/8 agonist shown to enhance cancer immunotherapy by promoting the transformation of M2 macrophages into M1.1087,1088 In vitro studies have shown that cathepsin B (CTSB)-responsive programmed brain-targeted delivery system (D&R-HM-MCA) could go through the BBB of glioblastoma (GBM) with high endocytosis efficiency and promote the repolarization of M2 macrophages into M1 type by the delivery of resiquimod.1089
CD47 is a glycoprotein highly expressed in many types of tumor cells, and the Inhibition of CD47 binding to SIRPɑ on macrophages could promote the repolarization of TAMs from M2 to M1.1090 Zhou et al. developed a BBB-penetrating nano-capsule named NAcp@CD47 to deliver anti-CD47 antibodies and STING agonists. Both in vivo and in vitro studies have shown that NACP@CD47 could improve tumor immunosuppression by promoting phagocytosis of macrophages/microglia and increasing M1-type polarization of TAMs.1091 Guo et al. use a layered double hydroxide nanosheet carrier to deliver a CD47 inhibitor RRX-001 and a T-type calcium channel inhibitor TTA-Q6 into lung tumors. TTA-Q6 is released in the tumor’s acidic environment. It induces calreticulin transfer to the cell surface, which could activate macrophages and mature DC, leading to the activation of anti-tumor T cells by promoting antigen presentation. Furthermore, RRX-001 reduced CD47 protein levels to prevent the immune escape of calreticulin-rich cancer cells by enhancing phagocytosis of tumor cells by macrophages.1092 In osteosarcoma (OS), RRX-001 and a sonosensitizer IR780 were combined with PEG-PCL nano micelles and OS cell membranes to assemble a biomimetic nano-drug, MPIRx. This nano drug could direct the migration of macrophages into tumor cells, facilitate M1-type polarization, and enhance the phagocytosis of macrophages towards OS cells.1093
Gao et al. developed a DNA nanocarrier. When delivered locally, the RP-182 peptide in the nanocarrier could target macrophages and repolarize M2-type TAMs into M1-type.1094 Specifically, the RP-182 peptide could activate phagocytosis and autophagy in M2 macrophages by activating CD206, restoring these cells to M1-type and further enhancing their abilities to engulf tumor cells and assist with intratumoral CD8+ T cells in tumor antigen recognition.1095 Subsequently, the nanoparticles could introduce ErbB2-targeted CAR into macrophages, programmed to be CAR-macrophages (CAR-M). CAR-M could engulf tumor cells and recruit T cells, NK cells, and APCs, especially DCs, leading to a wide range of adaptive immune responses.1094
Wang et al. developed a biodegradable NDDS named siCD40/NPs to deliver CD40 siRNA (siCD40) into HSCs, myeloid progenitors, mature DCs, and macrophages. The inhibition of CD40 in HSCs and myeloid progenitors could prevent their differentiation towards DCs and macrophages. Besides, siCD40/NPs could reduce the amounts of peripheral DCs and macrophages as well as their abilities to activate alloreactive T cells, which suppresses alloimmune responses and prolongs allograft survival in mouse models of skin allotransplantation.1096 For improving the treatment of colon cancer, Ding et al. developed a liposome-based nano-drug named LIC, which encapsulates phosphoinositide 3-kinase gamma (PI3Kγ) inhibitor IPI-549 and photosensitizer chlorin e6 (Ce6). In tumor, LIC-mediated immunogenic photodynamic therapy(PDT) synergistically work with MDSCs-targeting immunotherapy, which could promote the DC maturation and CD8 + T cell infiltration, simultaneously reduce the infiltration of MDSCs, immunosuppressive Tregs, and M2-type TAMs, resulting in the inhibition of tumor growth.1097 Song et al. designed an albumin nanoparticle named Nano-PI. It contained the immunomodulators IPI-549 and paclitaxel (PTX), which could promote the repolarization of M2 macrophages to M1 type.1098
L-arginine (L-Arg) and an inhibitor of Arg-1 L-norvaline (L-Nor) were loaded in a multifunctional nanoplatform named HN-HFPA. This nanoplatform could deplete intracellular GSH, produce ROS under light irradiation and release L-Arg for the NO generation after the degradation of hyaluronic acid and GSH-mediated disintegration, leading to the tumor immunogenic cell death (ICD). Besides, the L-Nor could suppress the overexpression of Arg-1 in M2 macrophages, which interfered with the balance of arginine metabolism and reversed the immunosuppressive tumor microenvironment (ITM) via increasing the ratios of M1 macrophages and CD8+ T cells, finally resulting in the enhanced antitumor immune responses and tumor metastasis inhibition.1099
Gao et al. developed a mannosylated nanotrinity containing imatinib, which could effectively induce in situ macrophage remodeling through a dual approach: activating M1-type polarization while simultaneously inhibiting M2-type polarization. This two-pronged strategy successfully combated the infection of Candida albicans.1100 Guanabenz (GBZ)-loaded lipoaspirate nanoparticles (Lipo-NPs) could reduce TLR4-induced inflammatory cytokines secretion by macrophages, providing a promising approach for treating inflammatory diseases.1101 Shi et al. designed a prodrug-formulated liposome named lipoprodrug to safely and effectively deliver cabazitaxel, which could reduce its systemic toxicity in vivo. It could decrease tumor invasiveness and reprogram the TIM via promoting proinflammatory macrophage polarization.1102 Nanoparticles loaded with an antihelminthic drug, Niclosamide (Ncl) (Ncl-NPs), could prevent and reverse pulmonary fibrosis by inhibiting M2-type macrophage polarization and blocking TGF-β/Smad and STAT3 signaling pathways.1103
Although there is a lot of research on NDDS-based macrophage immunotherapy, the number of projects approved for clinical trials is very limited. OP-101, a nanotherapeutic compound composed of generation-4 hydroxyl-terminated poly(amidoamine) dendrimer and N-acetyl cysteine (NAC), could target activated macrophages and improve outcomes in many preclinical models of systemic inflammation and neuroinflammation.1104,1105,1106,1107,1108 Besides, in a multicenter 2a clinical trial, researchers evaluated the safety and preliminary efficacy of OP-101 in patients with severe COVID-19(NCT04458298). The findings demonstrated that OP-101 was well tolerated in patients with severe COVID-19 and showed promising preliminary efficacy. There was an improvement in the composite clinical outcome of mechanical ventilation or death up to day 60, along with a reduction in inflammation and neurological injury markers. No drug-related adverse events (AEs) were reported in the primary endpoint1109 (Table 4).
Potential challenges and future directions in targeting macrophages
The current mainstream classification of macrophages is based on their activation mode, molecular expression, and function, which divides them into pro-inflammatory and anti-tumor M1 type as well as pro-tumor and anti-inflammatory M2 type.58,59 However, this classification still has some limitations. First, the state of macrophages is dynamic, and they may vary dynamically between types M1 and M2, influenced by environmental and other factors. In addition, macrophages often present in various states and subsets in different tissues, not only characteristic M1 and M2 types. Today, although many studies are focusing on M1 and M2 markers, the specificity of these markers remains a problem, and the criteria used by different groups, as well as their studies, vary. Therefore, the diversity and plasticity of macrophages make the development and application of therapeutic methods targeting macrophage polarization face challenges.348,761,1110,1111 Research for identifying specific macrophage polarization markers and the correct and refined distinguishment of macrophage subpopulations are imperative for developing macrophage-targeted therapies.1074 In addition, activation of macrophages may induce excessive immune responses, triggering or exacerbating inflammation or several autoimmune diseases.1112,1113 Therefore, macrophage therapies need to be designed to strike a balance between safety and efficacy.
Although the research and development of NDDS have led to the optimization of macrophage-specific drugs and immunotherapy, their current application still poses certain challenges. First, the immunogenicity, potential toxicity, and long-term safety of many newly developed human nanomaterials have not been recognized. In addition, the production process of NDDS is complex and highly costly. Besides, the accurate release of drugs from NDDS in vivo still has some technical difficulties and needs further optimization.1074,1114,1115,1116
Conclusions and perspectives
Tissue macrophages play critical roles in maintaining tissue homeostasis, regulating immune responses, and mediating pathological processes in various diseases. The origins, heterogeneity, and functional polarization of tissue macrophages have been extensively investigated in recent decades. Primitive hematopoietic progenitors, EMPs, and HSCs have all been identified as sources for tissue macrophages, with EMPs and EMP-derived macrophage precursors (PreMacs) being the major contributors to TRMs during embryonic development. The tissue microenvironment significantly shapes the identity and function of tissue macrophages, as evidenced by the tissue-specific transcriptional profiles and phenotypic features. Further research is needed to fully elucidate these different progenitor populations’ ontogeny and relative contributions to TRMs.
Macrophages exhibit diverse biological functions, including pathogen clearance, antigen presentation, tissue repair, and iron metabolism regulation. The dysregulation of macrophage activities has been closely associated with the pathogenesis of various diseases, such as cancer, autoimmune disorders, neurodegenerative diseases, and metabolic disorders. In the TME, TAMs can promote or suppress tumor progression through complex crosstalk with other cellular components. In autoimmune disorders, macrophages release pro-inflammatory cytokines that can perpetuate inflammation and immunity. Likewise, macrophages infiltrate the adipose tissue and release pro-inflammatory cytokines, contributing to systemic insulin resistance. In neurodegenerative diseases, macrophages may interact with neurons and release neurotoxic substances that promote neuronal death. It should be noted that the precise molecular mechanisms underlying this tissue-specific programming still need to be fully understood and warrant further investigation. While we know more about how macrophages respond in their particular niches, we still need to learn how different macrophages (produced from HSCs versus EMPs) inside a tissue integrate and react to the same signals. Besides, it is still unknown how long-term this intercellular interaction impacts tissue integrity and function or if these different macrophage populations might affect the tissue niche. The specific mechanisms and factors regulating tissue macrophages’ self-renewal and long-term maintenance also require deeper exploration. Luckily, with advanced technologies, such as single-cell genomics, spatial transcriptomics, genetic manipulation, advanced imaging, high-dimensional flow cytometry and mass cytometry, and bioinformatics approaches, in combination with traditional cell and molecular biology techniques, we will gain unprecedented insights into the complex biology of tissue macrophages, their origins, interactions, and roles in maintaining tissue homeostasis and function.
Additionally, the increasing understanding of the heterogeneity and plasticity of tissue macrophages has provided promising opportunities for developing novel therapeutic strategies. Macrophages can exhibit pro-inflammatory (M1) or anti-inflammatory (M2) phenotypes in response to environmental cues. Therapies aim to shift the balance towards a desired macrophage phenotype, such as promoting M2 macrophages to enhance tissue repair or dampen excessive inflammation. Further elucidation of the molecular mechanisms governing macrophage polarization and function will help refine these therapeutic approaches and unlock the full clinical potential of macrophage-targeted interventions. Pathogenic macrophages can accumulate and contribute to disease progression in some diseases, such as cancer or fibrosis. Therapies focus on selectively depleting or reprogramming these harmful macrophage populations to restore tissue homeostasis. Strategies include using antibody-drug conjugates, liposomal drug delivery, or genetic approaches to target and eliminate specific macrophage subsets. Besides, Macrophages can be utilized as “Trojan horses” to deliver therapeutic agents to disease sites. Macrophages can internalize and transport drugs, nanoparticles, or gene therapy vectors to target tissues, facilitating localized drug delivery. This approach can improve the therapeutic index by increasing drug accumulation at the disease site and reducing off-target effects. Specially, autologous or allogeneic macrophages can be engineered or polarized ex vivo and then administered to patients. These macrophage-based cell therapies can supplement or replace dysfunctional macrophages, modulate the immune response, or promote tissue regeneration. Approaches include ex vivo polarization of macrophages to enhance their regenerative properties before reintroduction into the body. This has implications for treating conditions like myocardial infarction and chronic wounds, where promoting tissue repair is crucial. Challenges include the scalability of macrophage production and ensuring the long-term survival and functionality of the transferred cells. Specific signaling pathways that regulate macrophage function, such as those involved in inflammation, phagocytosis, or metabolism, can be targeted by small-molecule inhibitors or biologics. This approach aims to fine-tune macrophage behavior without depleting the entire macrophage population. Identifying the appropriate therapeutic targets and balancing macrophage modulation’s beneficial and deleterious effects remain key challenges. Lastly, a new field of study is the interaction between macrophages and the microbiota. According to recent research, the gut microbiota might affect the polarization and activity of macrophages, affecting immunological responses and systemic inflammation. Gaining insight into these relationships may help develop new treatment approaches that use the microbiota to control macrophage activity in conditions like metabolic disorders and inflammatory bowel disease. Some problems need to be solved: 1. How to better regulate the TME and realize the transformation of macrophages from tumor-promoting to inhibitory? 2. How to design a more effective macrophage-based drug delivery system? 3. How to enhance the cytotoxic and anti-tumor functions of CAR-M cells? 4. How to develop more effective strategies for eliminating or depleting specific harmful macrophage subsets? 5. More accurate macrophage typing and prediction models are needed to guide individualized therapy. 6. Ensuring macrophage-based therapies are safe and do not cause off-target effects. 7. Ongoing research is needed to understand and counteract resistance mechanisms involved in macrophage-targeted therapies. 8. Identifying which specific strains or metabolites of the microbiome are beneficial or harmful to macrophage function.
In conclusion, tissue macrophages are indispensable in maintaining homeostasis and mediating pathological processes. Continued research on the origins, heterogeneity, and functional regulation of tissue macrophages will advance our understanding of their roles in health and disease and guide the development of innovative macrophage-targeted therapies.
References
Zhao, J., Andreev, I. & Silva, H. M. Resident tissue macrophages: Key coordinators of tissue homeostasis beyond immunity. Sci. Immunol. 9, eadd1967 (2024).
Hume, D. A., Millard, S. M. & Pettit, A. R. Macrophage heterogeneity in the single-cell era: facts and artifacts. Blood 142, 1339–1347 (2023).
Lazarov, T., Juarez-Carreño, S., Cox, N. & Geissmann, F. Physiology and diseases of tissue-resident macrophages. Nature 618, 698–707 (2023).
Sakai, M. et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain kupffer cell identity. Immunity 51, 655–670.e8 (2019).
Tober, J., McGrath, K. E. & Palis, J. Primitive erythropoiesis and megakaryopoiesis in the yolk sac are independent of c-myb. Blood 111, 2636–2639 (2008).
Herbomel, P., Thisse, B. & Thisse, C. Ontogeny and behaviour of early macrophages in the zebrafish embryo. Dev. Camb. Engl. 126, 3735–3745 (1999).
Cox, N., Pokrovskii, M., Vicario, R. & Geissmann, F. Origins, biology, and diseases of tissue macrophages. Annu. Rev. Immunol. 39, 313–344 (2021).
Bertrand, J. Y. et al. Definitive hematopoiesis initiates through a committed erythromyeloid progenitor in the zebrafish embryo. Dev. Camb. Engl. 134, 4147–4156 (2007).
Okuda, T., van Deursen, J., Hiebert, S. W., Grosveld, G. & Downing, J. R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 84, 321–330 (1996).
Le Guyader, D. et al. Origins and unconventional behavior of neutrophils in developing zebrafish. Blood 111, 132–141 (2008).
Bian, Z. et al. Deciphering human macrophage development at single-cell resolution. Nature 582, 571–576 (2020).
Wculek, S. K., Dunphy, G., Heras-Murillo, I., Mastrangelo, A. & Sancho, D. Metabolism of tissue macrophages in homeostasis and pathology. Cell. Mol. Immunol. 19, 384–408 (2022).
T’Jonck, W., Guilliams, M. & Bonnardel, J. Niche signals and transcription factors involved in tissue-resident macrophage development. Cell. Immunol. 330, 43–53 (2018).
Okabe, Y. & Medzhitov, R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 157, 832–844 (2014).
Lavin, Y. et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326 (2014).
Gautier, E. L. et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 13, 1118–1128 (2012).
Mass, E. Delineating the origins, developmental programs and homeostatic functions of tissue-resident macrophages. Int. Immunol. 30, 493–501 (2018).
Park, M. D., Silvin, A., Ginhoux, F. & Merad, M. Macrophages in health and disease. Cell 185, 4259–4279 (2022).
Truman, L. A. et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112, 5026–5036 (2008).
Wynn, T. A., Chawla, A. & Pollard, J. W. Macrophage biology in development, homeostasis and disease. Nature 496, 445–455 (2013).
Chen, C. et al. LNMAT1 promotes lymphatic metastasis of bladder cancer via CCL2 dependent macrophage recruitment. Nat. Commun. 9, 3826 (2018).
Martinez-Pomares, L. & Gordon, S. Antigen presentation the macrophage way. Cell 131, 641–643 (2007).
Underhill, D. M., Bassetti, M., Rudensky, A. & Aderem, A. Dynamic interactions of macrophages with T cells during antigen presentation. J. Exp. Med. 190, 1909–1914 (1999).
DeNardo, D. G. & Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 19, 369–382 (2019).
Rodríguez-Morales, P. & Franklin, R. A. Macrophage phenotypes and functions: resolving inflammation and restoring homeostasis. Trends Immunol 44, 986–998 (2023).
Chavakis, T., Alexaki, V. I. & Ferrante, A. W. Macrophage function in adipose tissue homeostasis and metabolic inflammation. Nat. Immunol. 24, 757–766 (2023).
Soares, M. P. & Hamza, I. Macrophages and iron metabolism. Immunity 44, 492–504 (2016).
Bohaud, C. et al. The role of macrophages during mammalian tissue remodeling and regeneration under infectious and non-infectious conditions. Front. Immunol. 12, 707856 (2021).
Zhao, C., Yang, Z., Li, Y. & Wen, Z. Macrophages in tissue repair and regeneration: insights from zebrafish. Cell Regen. Lond. Engl. 13, 12 (2024).
Korolnek, T. & Hamza, I. Macrophages and iron trafficking at the birth and death of red cells. Blood 125, 2893–2897 (2015).
Bikfalvi, A. et al. Challenges in glioblastoma research: focus on the tumor microenvironment. Trends Cancer 9, 9–27 (2023).
de Visser, K. E. & Joyce, J. A. The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell 41, 374–403 (2023).
Robinson, A., Han, C. Z., Glass, C. K. & Pollard, J. W. Monocyte regulation in homeostasis and malignancy. Trends Immunol 42, 104–119 (2021).
Cassetta, L. & Pollard, J. W. A timeline of tumour-associated macrophage biology. Nat. Rev. Cancer 23, 238–257 (2023).
Mantovani, A., Marchesi, F., Jaillon, S., Garlanda, C. & Allavena, P. Tumor-associated myeloid cells: diversity and therapeutic targeting. Cell. Mol. Immunol. 18, 566–578 (2021).
Chen, C. et al. Epigenetic regulation of macrophage polarization in wound healing. Burns Trauma 11, tkac057 (2023).
Hasegawa, T. et al. Identification of a novel arthritis-associated osteoclast precursor macrophage regulated by FoxM1. Nat. Immunol. 20, 1631–1643 (2019).
Lin, P., Ji, H.-H., Li, Y.-J. & Guo, S.-D. Macrophage plasticity and atherosclerosis therapy. Front. Mol. Biosci. 8, 679797 (2021).
Keren-Shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017).
Phu, T. A., Ng, M., Vu, N. K., Bouchareychas, L. & Raffai, R. L. IL-4 polarized human macrophage exosomes control cardiometabolic inflammation and diabetes in obesity. Mol. Ther. J. Am. Soc. Gene Ther. 30, 2274–2297 (2022).
Lumeng, C. N., Deyoung, S. M., Bodzin, J. L. & Saltiel, A. R. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 56, 16–23 (2007).
Chen, Y. et al. Targeting tumor-associated macrophages: a potential treatment for solid tumors. J. Cell. Physiol. 236, 3445–3465 (2021).
Lin, D.-W. et al. Targeting macrophages: therapeutic approaches in diabetic kidney disease. Int. J. Mol. Sci. 25, 4350 (2024).
Li, C. et al. Tumor-associated macrophages: potential therapeutic strategies and future prospects in cancer. J. Immunother. Cancer 9, e001341 (2021).
Teti, G., Biondo, C. & Beninati, C. The phagocyte, Metchnikoff, and the foundation of immunology. Microbiol. Spectr. 4, 17–29 (2016).
Silva, M. T. & Correia-Neves, M. Neutrophils and macrophages: the main partners of phagocyte cell systems. Front. Immunol. 3, 174 (2012).
Yona, S. & Gordon, S. From the reticuloendothelial to mononuclear phagocyte system - the unaccounted years. Front. Immunol. 6, 328 (2015).
van Furth, R. et al. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ 46, 845–852 (1972).
Perdiguero, E. G. & Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 17, 2–8 (2016).
Mass, E. et al. Specification of tissue-resident macrophages during organogenesis. Science 353, aaf4238 (2016).
Stanley, E. R. & Heard, P. M. Factors regulating macrophage production and growth. Purification and some properties of the colony stimulating factor from medium conditioned by mouse L cells. J. Biol. Chem. 252, 4305–4312 (1977).
Bradley, T. R., Stanley, E. R. & Sumner, M. A. Factors from mouse tissues stimulating colony growth of mouse bone marrow cells in vitro. Aust. J. Exp. Biol. Med. Sci. 49, 595–603 (1971).
Bradley, T. R. & Metcalf, D. The growth of mouse bone marrow cells in vitro. Aust. J. Exp. Biol. Med. Sci. 44, 287–299 (1966).
Hibbs, J. B., Taintor, R. R., Vavrin, Z. & Rachlin, E. M. Nitric oxide: a cytotoxic activated macrophage effector molecule. Biochem. Biophys. Res. Commun. 157, 87–94 (1988).
Hibbs, J. B., Vavrin, Z. & Taintor, R. R. L-arginine is required for expression of the activated macrophage effector mechanism causing selective metabolic inhibition in target cells. J. Immunol. 138, 550–565 (1987).
Oldenborg, P. A., Gresham, H. D. & Lindberg, F. P. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcgamma and complement receptor-mediated phagocytosis. J. Exp. Med. 193, 855–862 (2001).
Oldenborg, P. A. et al. Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054 (2000).
Mills, C. D. Anatomy of a discovery: m1 and m2 macrophages. Front. Immunol. 6, 212 (2015).
Mills, C. D., Kincaid, K., Alt, J. M., Heilman, M. J. & Hill, A. M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 164, 6166–6173 (2000).
Qian, B.-Z. & Pollard, J. W. Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 (2010).
Mantovani, A., Ming, W. J., Balotta, C., Abdeljalil, B. & Bottazzi, B. Origin and regulation of tumor-associated macrophages: the role of tumor-derived chemotactic factor. Biochim. Biophys. Acta 865, 59–67 (1986).
Mantovani, A., Sozzani, S., Locati, M., Allavena, P. & Sica, A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23, 549–555 (2002).
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444 (2008).
Pollard, J. W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 4, 71–78 (2004).
Anderson, N. R., Minutolo, N. G., Gill, S. & Klichinsky, M. Macrophage-based approaches for cancer immunotherapy. Cancer Res. 81, 1201–1208 (2021).
Andreesen, R. et al. Adoptive transfer of tumor cytotoxic macrophages generated in vitro from circulating blood monocytes: a new approach to cancer immunotherapy. Cancer Res 50, 7450–7456 (1990).
Klichinsky, M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 38, 947–953 (2020).
Zhang, L. et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 13, 153 (2020).
Chen, Y. et al. CAR-macrophage: a new immunotherapy candidate against solid tumors. Biomed. Pharmacother. 139, 111605 (2021).
Ginhoux, F. & Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 44, 439–449 (2016).
Ginhoux, F. et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845 (2010).
Hoeffel, G. & Ginhoux, F. Ontogeny of tissue-resident macrophages. Front. Immunol. 6, 486 (2015).
Frame, J. M., McGrath, K. E. & Palis, J. Erythro-myeloid progenitors: ‘definitive’ hematopoiesis in the conceptus prior to the emergence of hematopoietic stem cells. Blood Cells Mol. Dis 51, 220–225 (2013).
Ivanovs, A. et al. Human haematopoietic stem cell development: from the embryo to the dish. Dev. Camb. Engl. 144, 2323–2337 (2017).
McGrath, K. E. et al. Distinct sources of hematopoietic progenitors emerge before HSCs and provide functional blood cells in the mammalian embryo. Cell Rep 11, 1892–1904 (2015).
Hoeffel, G. et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42, 665–678 (2015).
Guilliams, M. et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 185, 379–396.e38 (2022).
Chen, M. J., Yokomizo, T., Zeigler, B. M., Dzierzak, E. & Speck, N. A. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 457, 887–891 (2009).
Pei, W. et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 548, 456–460 (2017).
Gomez Perdiguero, E. et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551 (2015).
Boyle, W. J., Simonet, W. S. & Lacey, D. L. Osteoclast differentiation and activation. Nature 423, 337–342 (2003).
Sheng, J., Ruedl, C. & Karjalainen, K. Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity 43, 382–393 (2015).
Petraki, S., Alexander, B. & Brückner, K. Assaying blood cell populations of the Drosophila melanogaster Larva. J. Vis. Exp. 52733, https://doi.org/10.3791/52733 (2015).
Yona, S. et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 (2013).
Biswas, S. K. & Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab 15, 432–437 (2012).
Shapouri-Moghaddam, A. et al. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 233, 6425–6440 (2018).
Holness, C. L. & Simmons, D. L. Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood 81, 1607–1613 (1993).
Bosco, M. C. Macrophage polarization: reaching across the aisle? J. Allergy Clin. Immunol. 143, 1348–1350 (2019).
Hoover, A. A. et al. Increased canonical NF-kappaB signaling specifically in macrophages is sufficient to limit tumor progression in syngeneic murine models of ovarian cancer. BMC Cancer 20, 970 (2020).
Lu, H. et al. Quercetin ameliorates kidney injury and fibrosis by modulating M1/M2 macrophage polarization. Biochem. Pharmacol. 154, 203–212 (2018).
Zhou, D. et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell. Signal. 26, 192–197 (2014).
Yu, M. Y. et al. Exosomal miRNAs-mediated macrophage polarization and its potential clinical application. Int. Immunopharmacol. 117, 109905 (2023).
Kerneur, C., Cano, C. E. & Olive, D. Major pathways involved in macrophage polarization in cancer. Front. Immunol. 13, 1026954 (2022).
Mandula, J. K. et al. Jagged2 targeting in lung cancer activates anti-tumor immunity via Notch-induced functional reprogramming of tumor-associated macrophages. Immunity 57, 1124–1140.e9 (2024).
Sarode, P. et al. Reprogramming of tumor-associated macrophages by targeting β-catenin/FOSL2/ARID5A signaling: A potential treatment of lung cancer. Sci. Adv. 6, eaaz6105 (2020).
Song, J. et al. Hsa_circ_0009092/miR-665/NLK signaling axis suppresses colorectal cancer progression via recruiting TAMs in the tumor microenvironment. J. Exp. Clin. Cancer Res. CR 42, 319 (2023).
Tigue, M. L. et al. Wnt signaling in the phenotype and function of tumor-associated macrophages. Cancer Res 83, 3–11 (2023).
Tian, X. et al. Long noncoding RNA LINC00662 promotes M2 macrophage polarization and hepatocellular carcinoma progression via activating Wnt/β-catenin signaling. Mol. Oncol. 14, 462–483 (2020).
Shen, T. et al. Exosomal AP000439.2 from clear cell renal cell carcinoma induces M2 macrophage polarization to promote tumor progression through activation of STAT3. Cell Commun. Signal. 20, 152 (2022).
Sun, L. et al. IGFBP2 promotes tumor progression by inducing alternative polarization of macrophages in pancreatic ductal adenocarcinoma through the STAT3 pathway. Cancer Lett 500, 132–146 (2021).
Tang, B. et al. Macrophage xCT deficiency drives immune activation and boosts responses to immune checkpoint blockade in lung cancer. Cancer Lett 554, 216021 (2023).
Shi, J.-H. et al. TRAF3/STAT6 axis regulates macrophage polarization and tumor progression. Cell Death Differ 30, 2005–2016 (2023).
Petty, A. J. et al. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J. Clin. Investig. 129, 5151–5162 (2019).
De Henau, O. et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 539, 443–447 (2016).
Kaneda, M. M. et al. Macrophage PI3Kγ drives pancreatic ductal adenocarcinoma progression. Cancer Discov 6, 870–885 (2016).
Foubert, P., Kaneda, M. M. & Varner, J. A. PI3Kγ activates integrin α4 and promotes immune suppressive myeloid cell polarization during tumor progression. Cancer Immunol. Res. 5, 957–968 (2017).
Kaneda, M. M. et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 539, 437–442 (2016).
Zhu, X. et al. Tumor-associated macrophage-specific CD155 contributes to M2-phenotype transition, immunosuppression, and tumor progression in colorectal cancer. J. Immunother. Cancer 10, e004219 (2022).
Yuan, W. et al. Dual role of CXCL8 in maintaining the mesenchymal state of glioblastoma stem cells and M2-Like tumor-associated macrophages. Clin. Cancer Res. 29, 3779–3792 (2023).
Li, M. et al. Metabolism, metabolites, and macrophages in cancer. J. Hematol. Oncol. 16, 80 (2023).
Scodeller, P. et al. Precision targeting of tumor macrophages with a CD206 binding peptide. Sci. Rep. 7, 14655 (2017).
Chanmee, T., Ontong, P., Konno, K. & Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 6, 1670–1690 (2014).
Zhang, Q. & Sioud, M. Tumor-associated macrophage subsets: shaping polarization and targeting. Int. J. Mol. Sci. 24, 7493 (2023).
Gu, H. et al. NLRP3 activation in tumor-associated macrophages enhances lung metastasis of pancreatic ductal adenocarcinoma. Transl. Lung Cancer Res 11, 858–868 (2022).
Jetten, N. et al. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 17, 109–118 (2014).
Martinez, F. O., Sica, A., Mantovani, A. & Locati, M. Macrophage activation and polarization. Front. Biosci. J. Virtual Libr. 13, 453–461 (2008).
Jordakieva, G. et al. IgG4 induces tolerogenic M2-like macrophages and correlates with disease progression in colon cancer. Oncoimmunology 10, 1880687 (2021).
Liu, Y. et al. Tumor necrosis factor α inhibition overcomes immunosuppressive M2b macrophage-induced bevacizumab resistance in triple-negative breast cancer. Cell Death Dis 11, 993 (2020).
Kim, D. et al. Ubiquitin E3 Ligase Pellino-1 Inhibits IL-10-mediated M2c polarization of macrophages, thereby suppressing tumor growth. Immune Netw 19, e32 (2019).
Vidyarthi, A. et al. Predominance of M2 macrophages in gliomas leads to the suppression of local and systemic immunity. Cancer Immunol. Immunother. 68, 1995–2004 (2019).
Zizzo, G., Hilliard, B. A., Monestier, M. & Cohen, P. L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 189, 3508–3520 (2012).
Strizova, Z. et al. M1/M2 macrophages and their overlaps - myth or reality? Clin. Sci. 137, 1067–1093 (2023).
Ferrante, C. J. et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation 36, 921–931 (2013).
Haskó, G., Pacher, P., Deitch, E. A. & Vizi, E. S. Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol. Ther. 113, 264–275 (2007).
Pinhal-Enfield, G. et al. An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7, and 9 and adenosine A(2A) receptors. Am. J. Pathol. 163, 711–721 (2003).
Duluc, D. et al. Interferon-gamma reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int. J. Cancer 125, 367–373 (2009).
Li, L. et al. NF-κB RelA renders tumor-associated macrophages resistant to and capable of directly suppressing CD8+ T cells for tumor promotion. Oncoimmunology 7, e1435250 (2018).
Tonnesen, M. G., Feng, X. & Clark, R. A. F. Angiogenesis in Wound Healing. J. Investig. Dermatol. Symp. Proc. 5, 40–46 (2000).
Bosurgi, L. et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 356, 1072–1076 (2017).
Gordon, S. & Plüddemann, A. Tissue macrophages: heterogeneity and functions. BMC Biol 15, 53 (2017).
Mass, E., Nimmerjahn, F., Kierdorf, K. & Schlitzer, A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat. Rev. Immunol. 23, 563–579 (2023).
Wang, Z. et al. An immune cell atlas reveals the dynamics of human macrophage specification during prenatal development. Cell 186, 4454–4471.e19 (2023).
Evren, E. et al. Distinct developmental pathways from blood monocytes generate human lung macrophage diversity. Immunity 54, 259–275.e7 (2021).
Ochocka, N. et al. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 12, 1151 (2021).
MacParland, S. A. et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 9, 4383 (2018).
Ural, B. B. et al. Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci. Immunol. 5, eaax8756 (2020).
Schyns, J. et al. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat. Commun. 10, 3964 (2019).
Zimmerman, K. A. et al. Single-cell RNA sequencing identifies candidate renal resident macrophage gene expression signatures across species. J. Am. Soc. Nephrol. 30, 767–781 (2019).
Liu, Y., Zhang, L., Ju, X., Wang, S. & Qie, J. Single-cell transcriptomic analysis reveals macrophage-tumor crosstalk in hepatocellular carcinoma. Front. Immunol. 13, 955390 (2022).
Xiao, G. et al. Machine learning-based identification of SOX10 as an immune regulator of macrophage in gliomas. Front. Immunol. 13, 1007461 (2022).
Huang, L. et al. Mrc1+ macrophage-derived IGF1 mitigates crystal nephropathy by promoting renal tubule cell proliferation via the AKT/Rb signaling pathway. Theranostics 14, 1764–1780 (2024).
Bjornson-Hooper, Z. B. et al. A comprehensive Atlas of immunological differences between humans, mice, and non-human primates. Front. Immunol. 13, 867015 (2022).
Li, P. et al. Comparative proteomic analysis of polarized human THP-1 and mouse RAW264.7 macrophages. Front. Immunol. 12, 700009 (2021).
Schneemann, M. & Schoeden, G. Macrophage biology and immunology: man is not a mouse. J. Leukoc. Biol. 81, 579 (2007).
Mestas, J. & Hughes, C. C. W. Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738 (2004).
Nio, J. et al. Cellular expression of murine Ym1 and Ym2, chitinase family proteins, as revealed by in situ hybridization and immunohistochemistry. Histochem. Cell Biol. 121, 473–482 (2004).
Wright, S. D., Ramos, R. A., Tobias, P. S., Ulevitch, R. J. & Mathison, J. C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249, 1431–1433 (1990).
Chang, Y.-C. et al. Epigenetic control of MHC class II expression in tumor-associated macrophages by decoy receptor 3. Blood 111, 5054–5063 (2008).
Austyn, J. M. & Gordon, S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur. J. Immunol. 11, 805–815 (1981).
Gordon, S. & Taylor, P. R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964 (2005).
Pesce, J. T. et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog 5, e1000371 (2009).
Masuda, T. et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392 (2019).
Chen, K. et al. Communications between bone marrow macrophages and bone cells in bone remodeling. Front. Cell Dev. Biol. 8, 598263 (2020).
Sinder, B. P., Pettit, A. R. & McCauley, L. K. Macrophages: their emerging roles in bone. J. Bone Miner. Res. 30, 2140–2149 (2015).
Li, W., Guo, R., Song, Y. & Jiang, Z. Erythroblastic Island macrophages shape normal erythropoiesis and drive associated disorders in erythroid hematopoietic diseases. Front. Cell Dev. Biol. 8, 613885 (2020).
Bessis, M. Erythroblastic island, functional unity of bone marrow. Rev. Hematol. 13, 8–11 (1958).
Li, W. et al. Identification and transcriptome analysis of erythroblastic island macrophages. Blood 134, 480–491 (2019).
Chow, A. et al. CD169+ macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med. 19, 429–436 (2013).
Sadahira, Y., Yoshino, T. & Monobe, Y. Very late activation antigen 4-vascular cell adhesion molecule 1 interaction is involved in the formation of erythroblastic islands. J. Exp. Med. 181, 411–415 (1995).
Țichil, I. et al. A review of key regulators of steady-state and ineffective erythropoiesis. J. Clin. Med. 13, 2585 (2024).
Liang, R. et al. Mitochondrial localization and moderated activity are key to murine erythroid enucleation. Blood Adv 5, 2490–2504 (2021).
Kawane, K. et al. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science 292, 1546–1549 (2001).
Toda, S., Segawa, K. & Nagata, S. MerTK-mediated engulfment of pyrenocytes by central macrophages in erythroblastic islands. Blood 123, 3963–3971 (2014).
Jacome-Galarza, C. E. et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature 568, 541–545 (2019).
Saftig, P. et al. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc. Natl Acad. Sci. USA 95, 13453–13458 (1998).
Batoon, L. et al. CD169+ macrophages are critical for osteoblast maintenance and promote intramembranous and endochondral ossification during bone repair. Biomaterials 196, 51–66 (2019).
Chang, M. K. et al. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J. Immunol. 181, 1232–1244 (2008).
Raggatt, L. J. et al. Fracture healing via periosteal callus formation requires macrophages for both initiation and progression of early endochondral ossification. Am. J. Pathol. 184, 3192–3204 (2014).
Mohsenzadegan, M. et al. Direct immunomodulatory influence of IFN-β on human astrocytoma cells. Immunopharmacol. Immunotoxicol. 37, 214–219 (2015).
Miron, R. J. & Bosshardt, D. D. OsteoMacs: key players around bone biomaterials. Biomaterials 82, 1–19 (2016).
Heinemann, D. E. et al. Human osteoblast-like cells phagocytose metal particles and express the macrophage marker CD68 in vitro. J. Bone Jt. Surg. Br. 82, 283–289 (2000).
Ruiz, C., Pérez, E., Vallecillo-Capilla, M. & Reyes-Botella, C. Phagocytosis and allogeneic T cell stimulation by cultured human osteoblast-like cells. Cell. Physiol. Biochem. 13, 309–314 (2003).
Reyes-Botella, C., Montes, M. J., Vallecillo-Capilla, M. F., Olivares, E. G. & Ruiz, C. Expression of molecules involved in antigen presentation and T cell activation (HLA-DR, CD80, CD86, CD44 and CD54) by cultured human osteoblasts. J. Periodontol. 71, 614–617 (2000).
Mohamad, S. F. et al. Osteomacs promote maintenance of murine hematopoiesis through megakaryocyte-induced upregulation of Embigin and CD166. Stem Cell Rep. 19, 486–500 (2024).
Mohamad, S. F. et al. Osteomacs interact with megakaryocytes and osteoblasts to regulate murine hematopoietic stem cell function. Blood Adv 1, 2520–2528 (2017).
Yang, J., Zhang, L., Yu, C., Yang, X.-F. & Wang, H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2, 1 (2014).
Wong, K. L. et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 118, e16–e31 (2011).
Boyette, L. B. et al. Phenotype, function, and differentiation potential of human monocyte subsets. PloS ONE 12, e0176460 (2017).
Aegerter, H., Lambrecht, B. N. & Jakubzick, C. V. Biology of lung macrophages in health and disease. Immunity 55, 1564–1580 (2022).
Liao, C. et al. Siglec-F+ neutrophils in the spleen induce immunosuppression following acute infection. Theranostics 14, 2589–2604 (2024).
Tateyama, H. et al. Siglec-F is induced by granulocyte-macrophage colony-stimulating factor and enhances interleukin-4-induced expression of arginase-1 in mouse macrophages. Immunology 158, 340–352 (2019).
Feng, Y. & Mao, H. Expression and preliminary functional analysis of Siglec-F on mouse macrophages. J. Zhejiang Univ. Sci. B 13, 386–394 (2012).
Bolden, J. E. et al. Identification of a Siglec-F+ granulocyte-macrophage progenitor. J. Leukoc. Biol. 104, 123–133 (2018).
Borkner, L., Curham, L. M., Wilk, M. M., Moran, B. & Mills, K. H. G. IL-17 mediates protective immunity against nasal infection with Bordetella pertussis by mobilizing neutrophils, especially Siglec-F+ neutrophils. Mucosal Immunol 14, 1183–1202 (2021).
Sanfilippo, A. M., Furuya, Y., Roberts, S., Salmon, S. L. & Metzger, D. W. Allergic lung inflammation reduces tissue invasion and enhances survival from pulmonary pneumococcal infection in mice, which correlates with increased expression of transforming growth factor β1 and SiglecF(low) alveolar macrophages. Infect. Immun. 83, 2976–2983 (2015).
Wculek, S. K., Forisch, S., Miguel, V. & Sancho, D. Metabolic homeostasis of tissue macrophages across the lifespan. Trends Endocrinol. Metab. https://doi.org/10.1016/j.tem.2024.04.017 (2024). S1043-2760(24)00111–5.
Schneider, C. et al. Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat. Immunol. 15, 1026–1037 (2014).
Baker, A. D. et al. Targeted PPAR{gamma} deficiency in alveolar macrophages disrupts surfactant catabolism. J. Lipid Res. 51, 1325–1331 (2010).
Yu, X. et al. The Cytokine TGF-β promotes the development and homeostasis of alveolar macrophages. Immunity 47, 903–912.e4 (2017).
Xiong, Z., Leme, A. S., Ray, P., Shapiro, S. D. & Lee, J. S. CX3CR1+ lung mononuclear phagocytes spatially confined to the interstitium produce TNF-α and IL-6 and promote cigarette smoke-induced emphysema. J. Immunol. 186, 3206–3214 (2011).
Jacquelin, S. et al. CX3CR1 reduces Ly6Chigh-monocyte motility within and release from the bone marrow after chemotherapy in mice. Blood 122, 674–683 (2013).
Lee, M., Lee, Y., Song, J., Lee, J. & Chang, S.-Y. Tissue-specific Role of CX3CR1 expressing immune cells and their relationships with human disease. Immune Netw 18, e5 (2018).
Tighe, R. M. et al. Ozone inhalation promotes CX3CR1-dependent maturation of resident lung macrophages that limit oxidative stress and inflammation. J. Immunol. 187, 4800–4808 (2011).
Evren, E., Ringqvist, E. & Willinger, T. Origin and ontogeny of lung macrophages: from mice to humans. Immunology 160, 126–138 (2020).
Dick, S. A. et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci. Immunol. 7, eabf7777 (2022).
Chakarov, S. et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, eaau0964 (2019).
Gibbings, S. L. et al. Three unique interstitial macrophages in the murine lung at steady state. Am. J. Respir. Cell Mol. Biol. 57, 66–76 (2017).
Hu, Z. et al. Dissecting the single-cell transcriptome network of macrophage and identifies a signature to predict prognosis in lung adenocarcinoma. Cell. Oncol. 46, 1351–1368 (2023).
Kumar, S. et al. Single cell transcriptomic analyses reveal diverse and dynamic changes of distinct populations of lung interstitial macrophages in hypoxia-induced pulmonary hypertension. Front. Immunol. 15, 1372959 (2024).
Guilliams, M. & Scott, C. L. Liver macrophages in health and disease. Immunity 55, 1515–1529 (2022).
Blériot, C. & Ginhoux, F. Understanding the heterogeneity of resident liver macrophages. Front. Immunol. 10, 2694 (2019).
Elchaninov, A., Vishnyakova, P., Menyailo, E., Sukhikh, G. & Fatkhudinov, T. An eye on kupffer cells: development, phenotype and the macrophage niche. Int. J. Mol. Sci. 23, 9868 (2022).
Yang, C.-Y. et al. CLEC4F is an inducible C-type lectin in F4/80-positive cells and is involved in alpha-galactosylceramide presentation in liver. PloS ONE 8, e65070 (2013).
Barrios, A. A. et al. Mucins shed from the laminated layer in cystic Echinococcosis are captured by kupffer cells via the lectin receptor Clec4F. Infect. Immun. 91, e0003123 (2023).
Wen, Y., Lambrecht, J., Ju, C. & Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 18, 45–56 (2021).
Blériot, C. et al. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity 54, 2101–2116.e6 (2021).
Sierro, F. et al. A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity 47, 374–388.e6 (2017).
Isidoro, C. A. & Deniset, J. F. The role of macrophage subsets in and around the heart in modulating cardiac homeostasis and pathophysiology. Front. Immunol. 14, 1111819 (2023).
Bajpai, G. et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 24, 1234–1245 (2018).
Dick, S. A. et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 20, 29–39 (2019).
Wan, E. et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 113, 1004–1012 (2013).
Miyanishi, M. et al. Identification of Tim4 as a phosphatidylserine receptor. Nature 450, 435–439 (2007).
Lim, H. Y. et al. Hyaluronan Receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49, 326–341.e7 (2018).
Deniset, J. F. et al. Gata6+ pericardial cavity macrophages relocate to the injured heart and prevent cardiac fibrosis. Immunity 51, 131–140.e5 (2019).
Nobs, S. P. & Kopf, M. Tissue-resident macrophages: guardians of organ homeostasis. Trends Immunol 42, 495–507 (2021).
Haldar, M. et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell 156, 1223–1234 (2014).
Okreglicka, K. et al. PPARγ is essential for the development of bone marrow erythroblastic island macrophages and splenic red pulp macrophages. J. Exp. Med. 218, e20191314 (2021).
Kong, D.-H., Kim, Y. K., Kim, M. R., Jang, J. H. & Lee, S. Emerging roles of vascular cell adhesion molecule-1 (VCAM-1) in immunological disorders and cancer. Int. J. Mol. Sci. 19, 1057 (2018).
A-Gonzalez, N. & Castrillo, A. Origin and specialization of splenic macrophages. Cell. Immunol. 330, 151–158 (2018).
Martinez-Pomares, L. & Gordon, S. CD169+ macrophages at the crossroads of antigen presentation. Trends Immunol 33, 66–70 (2012).
den Haan, J. M. M. & Martinez-Pomares, L. Macrophage heterogeneity in lymphoid tissues. Semin. Immunopathol. 35, 541–552 (2013).
Lewis, S. M., Williams, A. & Eisenbarth, S. C. Structure and function of the immune system in the spleen. Sci. Immunol. 4, eaau6085 (2019).
Zhang, Y. et al. Migratory and adhesive cues controlling innate-like lymphocyte surveillance of the pathogen-exposed surface of the lymph node. eLife 5, e18156 (2016).
Kim, J., Ahn, J., Kang, G., Hwang, J. H. & Kim, C. High-resolution photoacoustic/ultrasound imaging of the porcine stomach wall: an ex vivo feasibility study. Biomed. Opt. Express 12, 6717–6729 (2021).
Hegarty, L. M., Jones, G.-R. & Bain, C. C. Macrophages in intestinal homeostasis and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 20, 538–553 (2023).
Bain, C. C. & Schridde, A. Origin, differentiation, and function of intestinal macrophages. Front. Immunol. 9, 2733 (2018).
De Schepper, S. et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell 175, 400–415.e13 (2018).
Shaw, T. N. et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J. Exp. Med. 215, 1507–1518 (2018).
Zigmond, E. et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity 37, 1076–1090 (2012).
Chikina, A. S. et al. Macrophages maintain epithelium integrity by limiting fungal product absorption. Cell 183, 411–428.e16 (2020).
Delfini, M., Stakenborg, N., Viola, M. F. & Boeckxstaens, G. Macrophages in the gut: masters in multitasking. Immunity 55, 1530–1548 (2022).
Cummings, R. J. et al. Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature 539, 565–569 (2016).
Niess, J. H. et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307, 254–258 (2005).
Murai, M. et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 10, 1178–1184 (2009).
Moura Silva, H. et al. c-MAF-dependent perivascular macrophages regulate diet-induced metabolic syndrome. Sci. Immunol. 6, eabg7506 (2021).
Gabanyi, I. et al. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell 164, 378–391 (2016).
Muller, P. A. et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell 158, 1210 (2014).
Matheis, F. et al. Adrenergic signaling in muscularis macrophages limits infection-induced neuronal loss. Cell 180, 64–78.e16 (2020).
Viola, M. F. et al. Dedicated macrophages organize and maintain the enteric nervous system. Nature 618, 818–826 (2023).
Schulz, C. et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90 (2012).
Kierdorf, K. et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 16, 273–280 (2013).
Masuda, T. et al. Specification of CNS macrophage subsets occurs postnatally in defined niches. Nature 604, 740–748 (2022).
Jung, S. et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20, 4106–4114 (2000).
Bennett, F. C. et al. A combination of ontogeny and CNS environment establishes microglial identity. Neuron 98, 1170–1183.e8 (2018).
Masuda, T. et al. Novel Hexb-based tools for studying microglia in the CNS. Nat. Immunol. 21, 802–815 (2020).
Scott, E. P., Breyak, E., Nishinakamura, R. & Nakagawa, Y. The zinc finger transcription factor Sall1 is required for the early developmental transition of microglia in mouse embryos. Glia 70, 1720–1733 (2022).
Gómez Morillas, A., Besson, V. C. & Lerouet, D. Microglia and neuroinflammation: what place for P2RY12? Int. J. Mol. Sci. 22, 1636 (2021).
Buttgereit, A. et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 17, 1397–1406 (2016).
Ruan, C. & Elyaman, W. A New Understanding of TMEM119 as a Marker of Microglia. Front. Cell. Neurosci. 16, 902372 (2022).
Shah, S. et al. Microglia-specific promoter activities of HEXB Gene. Front. Cell. Neurosci. 16, 808598 (2022).
Goldmann, T. et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 16, 1618–1626 (2013).
Hashimoto, D. et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804 (2013).
Erblich, B., Zhu, L., Etgen, A. M., Dobrenis, K. & Pollard, J. W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PloS ONE 6, e26317 (2011).
Wang, Y. et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 13, 753–760 (2012).
Greter, M. et al. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 37, 1050–1060 (2012).
Füger, P. et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 20, 1371–1376 (2017).
Tay, T. L. et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 20, 793–803 (2017).
Bernier, L.-P. et al. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat. Commun. 11, 1559 (2020).
Ginhoux, F. & Prinz, M. Origin of microglia: current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 7, a020537 (2015).
Aldana, B. I. Microglia-specific metabolic changes in neurodegeneration. J. Mol. Biol. 431, 1830–1842 (2019).
Lauro, C. & Limatola, C. Metabolic Reprograming of microglia in the regulation of the innate inflammatory response. Front. Immunol. 11, 493 (2020).
Montilla, A., Zabala, A., Matute, C. & Domercq, M. Functional and metabolic characterization of microglia culture in a defined medium. Front. Cell. Neurosci. 14, 22 (2020).
Prinz, M., Masuda, T., Wheeler, M. A. & Quintana, F. J. Microglia and central nervous system-associated macrophages-from origin to disease modulation. Annu. Rev. Immunol. 39, 251–277 (2021).
Goldmann, T. et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797–805 (2016).
Utz, S. G. et al. Early fate defines microglia and non-parenchymal brain macrophage development. Cell 181, 557–573.e18 (2020).
Van Hove, H. et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 22, 1021–1035 (2019).
Drieu, A. et al. Parenchymal border macrophages regulate the flow dynamics of the cerebrospinal fluid. Nature 611, 585–593 (2022).
Leibrand, C. R. et al. HIV-1 Tat disrupts blood-brain barrier integrity and increases phagocytic perivascular macrophages and microglia in the dorsal striatum of transgenic mice. Neurosci. Lett. 640, 136–143 (2017).
Winkler, B. et al. Brain inflammation triggers macrophage invasion across the blood-brain barrier in Drosophila during pupal stages. Sci. Adv. 7, eabh0050 (2021).
Sun, N. et al. Antibiotic-induced microbiome depletion in adult mice disrupts blood-brain barrier and facilitates brain infiltration of monocytes after bone-marrow transplantation. Brain. Behav. Immun. 92, 102–114 (2021).
Wang, X. et al. Heterogeneous origins and functions of mouse skeletal muscle-resident macrophages. Proc. Natl. Acad. Sci. USA 117, 20729–20740 (2020).
Babaeijandaghi, F. et al. Metabolic reprogramming of skeletal muscle by resident macrophages points to CSF1R inhibitors as muscular dystrophy therapeutics. Sci. Transl. Med. 14, eabg7504 (2022).
Babaeijandaghi, F. et al. DPPIV+ fibro-adipogenic progenitors form the niche of adult skeletal muscle self-renewing resident macrophages. Nat. Commun. 14, 8273 (2023).
Stamatiades, E. G. et al. Immune monitoring of trans-endothelial transport by kidney-resident macrophages. Cell 166, 991–1003 (2016).
Puranik, A. S. et al. Kidney-resident macrophages promote a proangiogenic environment in the normal and chronically ischemic mouse kidney. Sci. Rep. 8, 13948 (2018).
Munro, D. A. D. & Hughes, J. The origins and functions of tissue-resident macrophages in kidney development. Front. Physiol. 8, 837 (2017).
Elloumi, H. Z. et al. A cell permeable peptide inhibitor of NFAT inhibits macrophage cytokine expression and ameliorates experimental colitis. PloS ONE 7, e34172 (2012).
Climaco-Arvizu, S. et al. Aryl hydrocarbon receptor influences nitric oxide and arginine production and alters M1/M2 macrophage polarization. Life Sci 155, 76–84 (2016).
Ganta, V. C. et al. A MicroRNA93-Interferon Regulatory Factor-9-Immunoresponsive Gene-1-Itaconic Acid Pathway Modulates M2-Like Macrophage Polarization to Revascularize Ischemic Muscle. Circulation 135, 2403–2425 (2017).
Minematsu, H. et al. Nuclear presence of nuclear factor of activated T cells (NFAT) c3 and c4 is required for Toll-like receptor-activated innate inflammatory response of monocytes/macrophages. Cell Signal 23, 1785–1793 (2011).
Jing, C. et al. Macrophage metabolic reprogramming presents a therapeutic target in lupus nephritis. Proc. Natl. Acad. Sci. USA 117, 15160–15171 (2020).
De Maeyer, R. P. H. & Chambers, E. S. The impact of ageing on monocytes and macrophages. Immunol. Lett. 230, 1–10 (2021).
Lefere, S. & Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep 1, 30–43 (2019).
Orliaguet, L., Dalmas, E., Drareni, K., Venteclef, N. & Alzaid, F. Mechanisms of macrophage polarization in insulin signaling and sensitivity. Front. Endocrinol. 11, 62 (2020).
Covarrubias, A. J. et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat. Metab. 2, 1265–1283 (2020).
Hoeffel, G. et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J. Exp. Med. 209, 1167–1181 (2012).
Mielcarek, M. et al. Langerhans cell homeostasis and turnover after nonmyeloablative and myeloablative allogeneic hematopoietic cell transplantation. Transplantation 98, 563–568 (2014).
Merad, M. et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat. Immunol. 3, 1135–1141 (2002).
Chopin, M. et al. Langerhans cells are generated by two distinct PU.1-dependent transcriptional networks. J. Exp. Med. 210, 2967–2980 (2013).
Liu, X. et al. Distinct human Langerhans cell subsets orchestrate reciprocal functions and require different developmental regulation. Immunity 54, 2305–2320.e11 (2021).
Collin, M. & Bigley, V. Many Langerhans make light work of skin immunity. Immunity 54, 2188–2190 (2021).
Hunger, R. E. et al. Langerhans cells utilize CD1a and langerin to efficiently present nonpeptide antigens to T cells. J. Clin. Investig. 113, 701–708 (2004).
Merad, M., Ginhoux, F. & Collin, M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat. Rev. Immunol. 8, 935–947 (2008).
Valladeau, J. et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 12, 71–81 (2000).
Leitch, C. S. et al. Filaggrin-null mutations are associated with increased maturation markers on Langerhans cells. J. Allergy Clin. Immunol. 138, 482–490.e7 (2016).
Prechtel, A. T. & Steinkasserer, A. CD83: an update on functions and prospects of the maturation marker of dendritic cells. Arch. Dermatol. Res. 299, 59–69 (2007).
Di Gennaro, P. et al. IDO and CD83 expression in human epidermal Langerhans cells. J. Dermatol. Sci. 73, 172–174 (2014).
Yue, C. et al. IL-38 Aggravates Atopic Dermatitis Via Facilitating Migration Of Langerhans Cells. Int. J. Biol. Sci. 20, 3094–3112 (2024).
Seneschal, J., Clark, R. A., Gehad, A., Baecher-Allan, C. M. & Kupper, T. S. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity 36, 873–884 (2012).
Kitashima, D. Y. et al. Langerhans cells prevent autoimmunity via expansion of keratinocyte antigen-specific regulatory T cells. EBioMedicine 27, 293–303 (2018).
Kobayashi, T., Naik, S. & Nagao, K. Choreographing immunity in the skin epithelial barrier. Immunity 50, 552–565 (2019).
Park, S. et al. Skin-resident immune cells actively coordinate their distribution with epidermal cells during homeostasis. Nat. Cell Biol. 23, 476–484 (2021).
Kolter, J. et al. A subset of skin macrophages contributes to the surveillance and regeneration of local nerves. Immunity 50, 1482–1497.e7 (2019).
Abtin, A. et al. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat. Immunol. 15, 45–53 (2014).
Uribe-Querol, E. & Rosales, C. Phagocytosis: our current understanding of a universal biological process. Front. Immunol. 11, 1066 (2020).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Doyle, S. E. et al. Toll-like receptors induce a phagocytic gene program through p38. J. Exp. Med. 199, 81–90 (2004).
Ohnishi, H. et al. Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc. Natl Acad. Sci. USA 106, 10260–10265 (2009).
Jin, M. S. et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130, 1071–1082 (2007).
Park, B. S. et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 (2009).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Pessoa Rodrigues, C. et al. Transcripts of repetitive DNA elements signal to block phagocytosis of hematopoietic stem cells. Science 385, eadn1629 (2024).
Ravetch, J. V. & Bolland, S. IgG Fc receptors. Annu. Rev. Immunol. 19, 275–290 (2001).
Dustin, M. L. Complement receptors in myeloid cell adhesion and phagocytosis. Microbiol. Spectr. 4, 429–445 (2016).
Jaumouillé, V. & Grinstein, S. Receptor mobility, the cytoskeleton, and particle binding during phagocytosis. Curr. Opin. Cell Biol. 23, 22–29 (2011).
Ostrowski, P. P., Grinstein, S. & Freeman, S. A. Diffusion barriers, mechanical forces, and the biophysics of phagocytosis. Dev. Cell 38, 135–146 (2016).
Springer, T. A. Adhesion receptors of the immune system. Nature 346, 425–434 (1990).
Doodnauth, S. A., Grinstein, S. & Maxson, M. E. Constitutive and stimulated macropinocytosis in macrophages: roles in immunity and in the pathogenesis of atherosclerosis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 374, 20180147 (2019).
Stern, S. T., Adiseshaiah, P. P. & Crist, R. M. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part. Fibre Toxicol. 9, 20 (2012).
Levin-Konigsberg, R. et al. Phagolysosome resolution requires contacts with the endoplasmic reticulum and phosphatidylinositol-4-phosphate signalling. Nat. Cell Biol. 21, 1234–1247 (2019).
Flannagan, R. S., Jaumouillé, V. & Grinstein, S. The cell biology of phagocytosis. Annu. Rev. Pathol. 7, 61–98 (2012).
Mullins, R. D. & Pollard, T. D. Rho-family GTPases require the Arp2/3 complex to stimulate actin polymerization in Acanthamoeba extracts. Curr. Biol. CB 9, 405–415 (1999).
Mullins, R. D. How WASP-family proteins and the Arp2/3 complex convert intracellular signals into cytoskeletal structures. Curr. Opin. Cell Biol. 12, 91–96 (2000).
Prehoda, K. E., Scott, J. A., Mullins, R. D. & Lim, W. A. Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science 290, 801–806 (2000).
Pang, X. et al. A PH domain in ACAP1 possesses key features of the BAR domain in promoting membrane curvature. Dev. Cell 31, 73–86 (2014).
Daumke, O., Roux, A. & Haucke, V. BAR domain scaffolds in dynamin-mediated membrane fission. Cell 156, 882–892 (2014).
Diakonova, M., Bokoch, G. & Swanson, J. A. Dynamics of cytoskeletal proteins during Fcgamma receptor-mediated phagocytosis in macrophages. Mol. Biol. Cell 13, 402–411 (2002).
Fairn, G. D. & Grinstein, S. How nascent phagosomes mature to become phagolysosomes. Trends Immunol 33, 397–405 (2012).
Desjardins, M. Biogenesis of phagolysosomes: the ‘kiss and run’ hypothesis. Trends Cell Biol 5, 183–186 (1995).
Kinchen, J. M. & Ravichandran, K. S. Phagosome maturation: going through the acid test. Nat. Rev. Mol. Cell Biol. 9, 781–795 (2008).
Canton, J. Phagosome maturation in polarized macrophages. J. Leukoc. Biol. 96, 729–738 (2014).
Li, F. et al. Apoptotic cells activate the ‘phoenix rising’ pathway to promote wound healing and tissue regeneration. Sci. Signal. 3, ra13 (2010).
Erwig, L.-P. & Henson, P. M. Clearance of apoptotic cells by phagocytes. Cell Death Differ 15, 243–250 (2008).
Kono, H. & Rock, K. L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289 (2008).
Elliott, M. R. et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286 (2009).
Hume, D. A. Bring out your dead. Nat. Immunol. 9, 12–14 (2008).
Lichtenstein, R. G. & Rabinovich, G. A. Glycobiology of cell death: when glycans and lectins govern cell fate. Cell Death Differ 20, 976–986 (2013).
Kuypers, F. A. & de Jong, K. The role of phosphatidylserine in recognition and removal of erythrocytes. Cell. Mol. Biol. Noisy-Gd. Fr. 50, 147–158 (2004).
Takemura, Y. et al. Adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies. J. Clin. Investig. 117, 375–386 (2007).
Shaw, P. X. et al. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J. Clin. Investig. 105, 1731–1740 (2000).
Fadok, V. A., Xue, D. & Henson, P. If phosphatidylserine is the death knell, a new phosphatidylserine-specific receptor is the bellringer. Cell Death Differ 8, 582–587 (2001).
Rammensee, H. G., Falk, K. & Rötzschke, O. Peptides naturally presented by MHC class I molecules. Annu. Rev. Immunol. 11, 213–244 (1993).
van Kooyk, Y. & Rabinovich, G. A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 9, 593–601 (2008).
Schloesser, D. et al. Senescent cells suppress macrophage-mediated corpse removal via upregulation of the CD47-QPCT/L axis. J. Cell Biol. 222, e202207097 (2023).
Hu, H. et al. Defective efferocytosis by aged macrophages promotes STING signaling mediated inflammatory liver injury. Cell Death Discov 9, 236 (2023).
Kohyama, M. et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 457, 318–321 (2009).
Ramirez-Ortiz, Z. G. et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat. Immunol. 14, 917–926 (2013).
Murray, P. J. & Wynn, T. A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 (2011).
Backer, R. et al. Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc. Natl Acad. Sci. USA 107, 216–221 (2010).
A-Gonzalez, N. et al. The nuclear receptor LXRα controls the functional specialization of splenic macrophages. Nat. Immunol. 14, 831–839 (2013).
Gray, E. E. & Cyster, J. G. Lymph node macrophages. J. Innate Immun. 4, 424–436 (2012).
Muraoka, D. et al. Nanogel-based immunologically stealth vaccine targets macrophages in the medulla of lymph node and induces potent antitumor immunity. ACS Nano 8, 9209–9218 (2014).
Asano, K. et al. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity 34, 85–95 (2011).
Segura, E. et al. Characterization of resident and migratory dendritic cells in human lymph nodes. J. Exp. Med. 209, 653–660 (2012).
Segura, E., Durand, M. & Amigorena, S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J. Exp. Med. 210, 1035–1047 (2013).
Nalio Ramos, R. et al. Tissue-resident FOLR2+ macrophages associate with CD8+ T cell infiltration in human breast cancer. Cell 185, 1189–1207.e25 (2022).
Wang, Y. et al. Tim-4 reprograms cholesterol metabolism to suppress antiviral innate immunity by disturbing the Insig1-SCAP interaction in macrophages. Cell Rep 41, 111738 (2022).
Ghosn, E. E. B. et al. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc. Natl. Acad. Sci. USA 107, 2568–2573 (2010).
Bain, C. C. et al. Long-lived self-renewing bone marrow-derived macrophages displace embryo-derived cells to inhabit adult serous cavities. Nat. Commun. 7, ncomms11852 (2016).
Joshi, S. et al. Tim4 enables large peritoneal macrophages to cross-present tumor antigens at early stages of tumorigenesis. Cell Rep 43, 114096 (2024).
Ebrahimkhani, M. R., Mohar, I. & Crispe, I. N. Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatology 54, 1379–1387 (2011).
Chen, Y. et al. Role of Kupffer cells in the induction of tolerance of orthotopic liver transplantation in rats. Liver Transplant 14, 823–836 (2008).
Dad, M. et al. Macrophages restrict the nephrogenic field and promote endothelial connections during kidney development. eLife 8, e43271 (2019).
van de Laar, L., Coffer, P. J. & Woltman, A. M. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood 119, 3383–3393 (2012).
Zhan, Y., Lew, A. M. & Chopin, M. The pleiotropic effects of the GM-CSF Rheostat on myeloid cell differentiation and function: more than a numbers game. Front. Immunol. 10, 2679 (2019).
Lang, R. A. et al. Transgenic mice expressing a hemopoietic growth factor gene (GM-CSF) develop accumulations of macrophages, blindness, and a fatal syndrome of tissue damage. Cell 51, 675–686 (1987).
van Nieuwenhuijze, A. E. et al. Transgenic expression of GM-CSF in T cells causes disseminated histiocytosis. Am. J. Pathol. 184, 184–199 (2014).
Dougan, M., Dranoff, G. & Dougan, S. K. GM-CSF, IL-3, and IL-5 family of cytokines: regulators of inflammation. Immunity 50, 796–811 (2019).
Zhan, Y., Lieschke, G. J., Grail, D., Dunn, A. R. & Cheers, C. Essential roles for granulocyte-macrophage colony-stimulating factor (GM-CSF) and G-CSF in the sustained hematopoietic response of Listeria monocytogenes-infected mice. Blood 91, 863–869 (1998).
Hadis, U. et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34, 237–246 (2011).
Shaw, M. H., Kamada, N., Kim, Y.-G. & Núñez, G. Microbiota-induced IL-1β, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J. Exp. Med. 209, 251–258 (2012).
Huang, S. C.-C. et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity 45, 817–830 (2016).
Covarrubias, A. J. et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 5, e11612 (2016).
Wang, F. et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab 28, 463–475.e4 (2018).
Liu, P.-S. et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 18, 985–994 (2017).
Squadrito, M. L., Etzrodt, M., De Palma, M. & Pittet, M. J. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol 34, 350–359 (2013).
Rossato, M. et al. IL-10-induced microRNA-187 negatively regulates TNF-α, IL-6, and IL-12p40 production in TLR4-stimulated monocytes. Proc. Natl. Acad. Sci. USA 109, E3101–E3110 (2012).
Gerlach, B. D. et al. Efferocytosis induces macrophage proliferation to help resolve tissue injury. Cell Metab 33, 2445–2463.e8 (2021).
Elliott, M. R., Koster, K. M. & Murphy, P. S. Efferocytosis signaling in the regulation of macrophage inflammatory responses. J. Immunol. 198, 1387–1394 (2017).
Doran, A. C., Yurdagul, A. & Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 20, 254–267 (2020).
Kourtzelis, I. et al. DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat. Immunol. 20, 40–49 (2019).
A-Gonzalez, N. et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–258 (2009).
Watanabe, S., Alexander, M., Misharin, A. V. & Budinger, G. R. S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 129, 2619–2628 (2019).
Sugimoto, M. A., Sousa, L. P., Pinho, V., Perretti, M. & Teixeira, M. M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 7, 160 (2016).
Franklin, R. A. Fibroblasts and macrophages: collaborators in tissue homeostasis. Immunol. Rev. 302, 86–103 (2021).
Kamei, R. & Okabe, S. In vivo imaging of the phagocytic dynamics underlying efficient clearance of adult-born hippocampal granule cells by ramified microglia. Glia 71, 2005–2023 (2023).
Zuttion, M. S. S. R. et al. Interstitial macrophages mediate efferocytosis of alveolar epithelium during influenza infection. Am. J. Respir. Cell Mol. Biol. 70, 159–164 (2024).
Berghoff, S. A. et al. Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis. Nat. Neurosci. 24, 47–60 (2021).
Niu, X., Zhou, F. & Zhang, L. Microglia regulate the health of central nervous system through myelin. Mol. Biomed. 4, 14 (2023).
Rieu, Q. et al. Pleiotropic roles of scavenger receptors in circadian retinal phagocytosis: a new function for Lysosomal SR-B2/LIMP-2 at the RPE cell surface. Int. J. Mol. Sci. 23, 3445 (2022).
Grootveld, A. K. et al. Apoptotic cell fragments locally activate tingible body macrophages in the germinal center. Cell 186, 1144–1161.e18 (2023).
Ardavín, C., Alvarez-Ladrón, N., Ferriz, M., Gutiérrez-González, A. & Vega-Pérez, A. Mouse tissue-resident peritoneal macrophages in homeostasis, repair, infection, and tumor metastasis. Adv. Sci. 10, e2206617 (2023).
Abrahams, V. M., Kim, Y. M., Straszewski, S. L., Romero, R. & Mor, G. Macrophages and apoptotic cell clearance during pregnancy. Am. J. Reprod. Immunol. 51, 275–282 (2004).
Gu, Y. et al. Multi-omics profiling visualizes dynamics of cardiac development and functions. Cell Rep 41, 111891 (2022).
Weinberger, T. et al. Resident and recruited macrophages differentially contribute to cardiac healing after myocardial ischemia. eLife 12, RP89377 (2024).
Bai, Y. et al. EphrinB2-mediated CDK5/ISL1 pathway enhances cardiac lymphangiogenesis and alleviates ischemic injury by resolving post-MI inflammation. Signal Transduct. Target. Ther. 9, 326 (2024).
Meijer, C. et al. Kupffer cell depletion by CI2MDP-liposomes alters hepatic cytokine expression and delays liver regeneration after partial hepatectomy. Liver 20, 66–77 (2000).
Duffield, J. S. et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 115, 56–65 (2005).
Blanchard, L. & Girard, J.-P. High endothelial venules (HEVs) in immunity, inflammation and cancer. Angiogenesis 24, 719–753 (2021).
Miyagawa, K. et al. Osteoclast-derived IGF1 is required for pagetic lesion formation in vivo. JCI Insight 5, e133113 (2020).
Tang, Y. et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 15, 757–765 (2009).
Quarato, E. R. et al. Efferocytosis by bone marrow mesenchymal stromal cells disrupts osteoblastic differentiation via mitochondrial remodeling. Cell Death Dis 14, 428 (2023).
Wynn, T. A. & Vannella, K. M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462 (2016).
Hoeffel, G. et al. Sensory neuron-derived TAFA4 promotes macrophage tissue repair functions. Nature 594, 94–99 (2021).
Mirza, R., DiPietro, L. A. & Koh, T. J. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am. J. Pathol. 175, 2454–2462 (2009).
Shook, B. A. et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 362, eaar2971 (2018).
Du, H. et al. Macrophage-released ADAMTS1 promotes muscle stem cell activation. Nat. Commun. 8, 669 (2017).
Tonkin, J. et al. Monocyte/Macrophage-derived IGF-1 orchestrates murine skeletal muscle regeneration and modulates autocrine polarization. Mol. Ther. J. Am. Soc. Gene Ther. 23, 1189–1200 (2015).
Shang, M. et al. Macrophage-derived glutamine boosts satellite cells and muscle regeneration. Nature 587, 626–631 (2020).
Wu, D. et al. Smtnl2 regulates apoptotic germ cell clearance and lactate metabolism in mouse Sertoli cells. Mol. Cell. Endocrinol. 551, 111664 (2022).
Hellberg, L. et al. Proinflammatory stimuli enhance phagocytosis of apoptotic cells by neutrophil granulocytes. ScientificWorldJournal 11, 2230–2236 (2011).
Esmann, L. et al. Phagocytosis of apoptotic cells by neutrophil granulocytes: diminished proinflammatory neutrophil functions in the presence of apoptotic cells. J. Immunol. 184, 391–400 (2010).
Schimek, V. et al. Tumour cell apoptosis modulates the colorectal cancer immune microenvironment via interleukin-8-dependent neutrophil recruitment. Cell Death Dis 13, 113 (2022).
Aurora, A. B. et al. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 124, 1382–1392 (2014).
Lin, S.-L. et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc. Natl. Acad. Sci. USA 107, 4194–4199 (2010).
Schlundt, C. et al. Macrophages in bone fracture healing: their essential role in endochondral ossification. Bone 106, 78–89 (2018).
Vi, L. et al. Macrophage cells secrete factors including LRP1 that orchestrate the rejuvenation of bone repair in mice. Nat. Commun. 9, 5191 (2018).
Kigerl, K. A. et al. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 29, 13435–13444 (2009).
Cattin, A.-L. et al. Macrophage-induced blood vessels guide schwann cell-mediated regeneration of peripheral nerves. Cell 162, 1127–1139 (2015).
Simkin, J. et al. Macrophages are required to coordinate mouse digit tip regeneration. Dev. Camb. Engl. 144, 3907–3916 (2017).
Godwin, J. W., Pinto, A. R. & Rosenthal, N. A. Macrophages are required for adult salamander limb regeneration. Proc. Natl. Acad. Sci. USA 110, 9415–9420 (2013).
Mase, A., Augsburger, J. & Brückner, K. Macrophages and their organ locations shape each other in development and homeostasis - a drosophila perspective. Front. Cell Dev. Biol. 9, 630272 (2021).
Coates, J. A. et al. Identification of functionally distinct macrophage subpopulations in Drosophila. eLife 10, e58686 (2021).
Bagchi, S. et al. The acid-sensing receptor GPR65 on tumor macrophages drives tumor growth in obesity. Sci. Immunol. 9, eadg6453 (2024).
Lee, S. H. et al. Mannose receptor high, M2 dermal macrophages mediate nonhealing Leishmania major infection in a Th1 immune environment. J. Exp. Med. 215, 357–375 (2018).
Jia, G. et al. Enhanced endothelium epithelial sodium channel signaling prompts left ventricular diastolic dysfunction in obese female mice. Metabolism 78, 69–79 (2018).
Nemeth, Z. et al. Epithelial sodium channels in macrophage migration and polarization: role of proinflammatory cytokines TNFα and IFNγ. Am. J. Physiol. Regul. Integr. Comp. Physiol. 323, R763–R775 (2022).
Immanuel, C. N. et al. Two-pore potassium channel TREK-1 (K2P2.1) regulates NLRP3 inflammasome activity in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 326, L367–L376 (2024).
Xu, R. et al. Role of KCa3.1 channels in macrophage polarization and its relevance in atherosclerotic plaque instability. Arterioscler. Thromb. Vasc. Biol. 37, 226–236 (2017).
Hua, X. et al. The potassium channel KCa3.1 as new therapeutic target for the prevention of obliterative airway disease. Transplantation 95, 285–292 (2013).
Wulff, H. et al. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc. Natl. Acad. Sci. USA 97, 8151–8156 (2000).
Chen, Y.-J. et al. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J. Cereb. Blood Flow. Metab. 36, 2146–2161 (2016).
Wu, B. et al. Blockage of Kv1.3 regulates macrophage migration in acute liver injury by targeting δ-catenin through RhoA signaling. Int. J. Biol. Sci. 16, 671–681 (2020).
Zhang, Q. et al. Kv1.3 channel is involved In Ox-LDL-induced macrophage inflammation via ERK/NF-κB signaling pathway. Arch. Biochem. Biophys. 730, 109394 (2022).
Bowen, C. A. et al. Proximity labeling proteomics reveals Kv1.3 potassium channel immune interactors in microglia. Mol. Cell Proteom. 23, 100809 (2024).
Muneoka, S. et al. G protein-coupled receptor 39 plays an anti-inflammatory role by enhancing IL-10 production from macrophages under inflammatory conditions. Eur. J. Pharmacol. 834, 240–245 (2018).
Shendge, A. K., Sekler, I. & Hershfinkel, M. ZnR/GPR39 regulates hepatic insulin signaling, tunes liver bioenergetics and ROS production, and mitigates liver fibrosis and injury. Redox Biol 78, 103403 (2024).
Kloosterman, D. J. & Akkari, L. Macrophages at the interface of the co-evolving cancer ecosystem. Cell 186, 1627–1651 (2023).
Zhang, Y. & Zhang, Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 17, 807–821 (2020).
Polyak, K., Haviv, I. & Campbell, I. G. Co-evolution of tumor cells and their microenvironment. Trends Genet 25, 30–38 (2009).
Nam, A. S., Chaligne, R. & Landau, D. A. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat. Rev. Genet. 22, 3–18 (2021).
Anderson, A. R. A., Weaver, A. M., Cummings, P. T. & Quaranta, V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell 127, 905–915 (2006).
Mantovani, A. Macrophages as tools and targets in cancer therapy. Drug Discov 21, 799–820 (2022).
De Palma, M. & Lewis, C. E. Cancer: macrophages limit chemotherapy. Nature 472, 303–304 (2011).
Lu, C.-S. et al. Oct4 promotes M2 macrophage polarization through upregulation of macrophage colony-stimulating factor in lung cancer. J. Hematol. Oncol. 13, 62 (2020).
Wang, H. et al. Interactions between colon cancer cells and tumor-infiltrated macrophages depending on cancer cell-derived colony stimulating factor 1. Oncoimmunology 5, e1122157 (2016).
Zhang, J. et al. Tumoral NOX4 recruits M2 tumor-associated macrophages via ROS/PI3K signaling-dependent various cytokine production to promote NSCLC growth. Redox Biol 22, 101116 (2019).
Ren, G. et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 11, 812–824 (2012).
Su, S. et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 25, 605–620 (2014).
Liu, L. et al. Interleukin-17 and prostaglandin E2 are involved in formation of an M2 macrophage-dominant microenvironment in lung cancer. J. Thorac. Oncol. 7, 1091–1100 (2012).
Ségaliny, A. I. et al. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitment. Int. J. Cancer 137, 73–85 (2015).
Nandi, B. et al. Stromal CCR6 drives tumor growth in a murine transplantable colon cancer through recruitment of tumor-promoting macrophages. Oncoimmunology 5, e1189052 (2016).
Frankenberger, C. et al. Metastasis suppressors regulate the tumor microenvironment by blocking recruitment of prometastatic tumor-associated macrophages. Cancer Res 75, 4063–4073 (2015).
Zhang, R. et al. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis 10, 273 (2019).
Li, K. et al. Leptin promotes breast cancer cell migration and invasion via IL-18 expression and secretion. Int. J. Oncol. 48, 2479–2487 (2016).
Zhang, Y. et al. Pan-cancer scRNA-seq analysis reveals immunological and diagnostic significance of the peripheral blood mononuclear cells. Hum. Mol. Genet. 33, 342–354 (2024).
Scolaro, T. et al. Nucleotide metabolism in cancer cells fuels a UDP-driven macrophage cross-talk, promoting immunosuppression and immunotherapy resistance. Nat. Cancer https://doi.org/10.1038/s43018-024-00771-8 (2024).
Li, D. et al. IFI35 regulates non-canonical NF-κB signaling to maintain glioblastoma stem cells and recruit tumor-associated macrophages. Cell Death Differ 31, 738–752 (2024).
Ngambenjawong, C., Gustafson, H. H. & Pun, S. H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 114, 206–221 (2017).
Watkins, S. K., Egilmez, N. K., Suttles, J. & Stout, R. D. IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. J. Immunol. 178, 1357–1362 (2007).
Zou, C. et al. Immunoreactive microenvironment modulator GBP5 suppresses ovarian cancer progression by inducing canonical pyroptosis. J. Cancer 15, 3510 (2024).
Xiao, S. et al. The tumor-intrinsic role of the m 6 A reader YTHDF2 in regulating immune evasion. Sci. Immunol. 9, eadl2171 (2024).
Ma, J. et al. Lysosome blockade induces divergent metabolic programs in macrophages and tumours for cancer immunotherapy. J. Exp. Clin. Cancer Res. 42, 192 (2023).
Yang, X. et al. Repolarizing heterogeneous leukemia-associated macrophages with more M1 characteristics eliminates their pro-leukemic effects. OncoImmunology 7, e1412910 (2018).
Jayasingam, S. D. et al. Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: technicalities and challenges in routine clinical practice. Front. Oncol. 9, 1512 (2019).
Gubin, M. M. et al. High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell 175, 1014–1030.e19 (2018).
Mei, J. et al. Prognostic impact of tumor-associated macrophage infiltration in non-small cell lung cancer: a systemic review and meta-analysis. Oncotarget 7, 34217–34228 (2016).
Lewis, C. E. & Pollard, J. W. Distinct role of macrophages in different tumor microenvironments. Cancer Res 66, 605–612 (2006).
Yang, Q. et al. Single-Cell RNA Sequencing reveals the heterogeneity of tumor-associated macrophage in non-small cell lung cancer and differences between sexes. Front. Immunol. 12, 756722 (2021).
Orecchioni, M., Ghosheh, Y., Pramod, A. B. & Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. classically and M2(LPS–) vs. alternatively activated macrophages. Front. Immunol. 10, 1084 (2019).
De Palma, M., Biziato, D. & Petrova, T. V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 17, 457–474 (2017).
Ruffell, B. et al. Macrophage IL-10 Blocks CD8+ T Cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26, 623–637 (2014).
Cassetta, L. & Pollard, J. W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17, 887–904 (2018).
Pombo Antunes, A. R. et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 24, 595–610 (2021).
De Monte, L. et al. Basophil recruitment into tumor-draining lymph nodes correlates with Th2 inflammation and reduced survival in pancreatic cancer patients. Cancer Res 76, 1792–1803 (2016).
Mantovani, A. & Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 212, 435–445 (2015).
Shiao, S. L. et al. TH2-Polarized CD4+ T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol. Res. 3, 518–525 (2015).
DeNardo, D. G. et al. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102 (2009).
Feng, R. et al. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal. 16, 54 (2018).
Zhang, S. et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab 29, 443–456.e5 (2019).
Su, S. et al. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell 175, 442–457.e23 (2018).
Dai, E. et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 16, 2069–2083 (2020).
Li, D. et al. 2-Microglobulin maintains glioblastoma stem cells and induces M2-like polarization of tumor-associated macrophages. Cancer Res 82, 3321–3334 (2022).
Yang, R. et al. Multi-omics analysis reveals interferon-stimulated gene OAS1 as a prognostic and immunological biomarker in pan-cancer. Front. Immunol. 14, 1249731 (2023).
Wang, B. et al. TRIM56: a promising prognostic immune biomarker for glioma revealed by pan-cancer and single-cell analysis. Front. Immunol. 15, 1327898 (2024).
Taufalele, P. V. et al. Matrix stiffness enhances cancer-macrophage interactions and M2-like macrophage accumulation in the breast tumor microenvironment. Acta Biomater 163, 365–377 (2023).
Atcha, H. et al. Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat. Commun. 12, 3256 (2021).
Zhang, B. et al. EB virus-induced ATR activation accelerates nasopharyngeal carcinoma growth via M2-type macrophages polarization. Cell Death Dis 11, 742 (2020).
Li, X. et al. Platelets promote CRC by activating the C5a/C5aR1 axis via PSGL-1/JNK/STAT1 signaling in tumor-associated macrophages. Theranostics 13, 2040–2056 (2023).
Komohara, Y., Jinushi, M. & Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci 105, 1–8 (2014).
Llosa, N. J. et al. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer Is Balanced by Multiple Counter-Inhibitory Checkpoints. Cancer Discov 5, 43–51 (2015).
Chen, Z. et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res 77, 2266–2278 (2017).
Riabov, V. et al. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 5, 75 (2014).
Lim, S. Y., Yuzhalin, A. E., Gordon-Weeks, A. N. & Muschel, R. J. Tumor-infiltrating monocytes/macrophages promote tumor invasion and migration by upregulating S100A8 and S100A9 expression in cancer cells. Oncogene 35, 5735–5745 (2016).
Erbani, J., Boon, M. & Akkari, L. Therapy-induced shaping of the glioblastoma microenvironment: Macrophages at play. Semin. Cancer Biol. 86, 41–56 (2022).
Bowman, R. L. et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep 17, 2445–2459 (2016).
Müller, S. et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol 18, 234 (2017).
Loyher, P.-L. et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med. 215, 2536–2553 (2018).
Hirano, R. et al. Tissue-resident macrophages are major tumor-associated macrophage resources, contributing to early TNBC development, recurrence, and metastases. Commun. Biol. 6, 144 (2023).
Zhu, Y. et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47, 323–338.e6 (2017).
Li, Y. et al. Arenobufagin modulation of PCSK9-mediated cholesterol metabolism induces tumor-associated macrophages polarisation to inhibit hepatocellular carcinoma progression. Phytomedicine 128, 155532 (2024).
Coulton, A. et al. Using a pan-cancer atlas to investigate tumour associated macrophages as regulators of immunotherapy response. Nat. Commun. 15, 5665 (2024).
Tannahill, G. M. et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 (2013).
Vitale, I., Manic, G., Coussens, L. M., Kroemer, G. & Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab 30, 36–50 (2019).
Miller, A. et al. Exploring metabolic configurations of single cells within complex tissue microenvironments. Cell Metab 26, 788–800.e6 (2017).
Vats, D. et al. Oxidative metabolism and PGC-1β attenuate macrophage- mediated inflammation. Cell metab 4, 13–24 (2007).
Penny, H. L. et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. OncoImmunology 5, e1191731 (2016).
Liu, D. et al. Comprehensive proteomics analysis reveals metabolic reprogramming of tumor-associated macrophages stimulated by the tumor microenvironment. J. Proteome Res. 16, 288–297 (2017).
Cai, J. et al. Targeting SRSF10 might inhibit M2 macrophage polarization and potentiate anti-PD-1 therapy in hepatocellular carcinoma. Cancer Commun https://doi.org/10.1002/cac2.12607 (2024).
Liu, N. et al. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2α-mediated tumor progression. J. Clin. Investig. 129, 631–646 (2019).
Henze, A.-T. & Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 126, 3672–3679 (2016).
Chen, Y.-J. et al. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 12, 937–943 (2016).
Colegio, O. R. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014).
Chen, D. et al. Pan-cancer analysis implicates novel insights of lactate metabolism into immunotherapy response prediction and survival prognostication. J. Exp. Clin. Cancer Res. CR 43, 125 (2024).
Zhang, Q. et al. A systematic pan-cancer analysis identifies LDHA as a novel predictor for immunological, prognostic, and immunotherapy resistance. Aging 16, 8000–8018 (2024).
Han, S. et al. d-lactate modulates M2 tumor-associated macrophages and remodels immunosuppressive tumor microenvironment for hepatocellular carcinoma. Sci. Adv. 9, eadg2697 (2023).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Giannoni, E. et al. Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs OXPHOS and prostate cancer metastatic spread. Oncotarget 6, 24061–24074 (2015).
Guan, F. et al. Mitochondrial transfer in tunneling nanotubes—a new target for cancer therapy. J. Exp. Clin. Cancer Res. 43, 147 (2024).
Sheng, Y. et al. Cancer-associated fibroblasts exposed to high-dose ionizing radiation promote M2 polarization of macrophages, which induce radiosensitivity in cervical cancer. Cancers 15, 1620 (2023).
Wenes, M. et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab 24, 701–715 (2016).
Wang, T. et al. HIF1 α -Induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediators Inflamm 2017, 1–10 (2017).
Oh, M.-H. et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Investig. 130, 3865–3884 (2020).
Choi, J., Stradmann-Bellinghausen, B., Yakubov, E., Savaskan, N. E. & Régnier-Vigouroux, A. Glioblastoma cells induce differential glutamatergic gene expressions in human tumor-associated microglia/macrophages and monocyte-derived macrophages. Cancer Biol. Ther. 16, 1205–1213 (2015).
Palmieri, E. M. et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep 20, 1654–1666 (2017).
Jha, A. K. et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430 (2015).
Fan, H. et al. Osteoclast cancer cell metabolic cross-talk confers parp inhibitor resistance in bone metastatic breast cancer. Cancer Res 84, 449–467 (2024).
Wu, H. et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol. Med. 11, e10698 (2019).
Hossain, F. et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 3, 1236–1247 (2015).
Luo, F. et al. HDL-cholesterol confers sensitivity of immunotherapy in nasopharyngeal carcinoma via remodeling tumor-associated macrophages towards the M1 phenotype. J. Immunother. Cancer 12, e008146 (2024).
Xiao, J. et al. 25-Hydroxycholesterol regulates lysosome AMP kinase activation and metabolic reprogramming to educate immunosuppressive macrophages. Immunity 57, 1087–1104.e7 (2024).
Xu, M. et al. Arachidonic acid metabolism controls macrophage alternative activation through regulating oxidative phosphorylation in pparγ dependent manner. Front. Immunol. 12, 618501 (2021).
Zhang, Y. et al. Activation of MAT2A-RIP1 signaling axis reprograms monocytes in gastric cancer. J. Immunother. Cancer 9, e001364 (2021).
Jiang, Y. et al. Nicotinamide metabolism face-off between macrophages and fibroblasts manipulates the microenvironment in gastric cancer. Cell Metab. https://doi.org/10.1016/j.cmet.2024.05.013 (2024). S1550-4131(24)00189-X.
Sun, R. et al. Loss of SIRT5 promotes bile acid-induced immunosuppressive microenvironment and hepatocarcinogenesis. J. Hepatol. 77, 453–466 (2022).
Wang, Y.-C. et al. Targeting monoamine oxidase A-regulated tumor-associated macrophage polarization for cancer immunotherapy. Nat. Commun. 12, 3530 (2021).
Peng, P. et al. TGFBI secreted by tumor-associated macrophages promotes glioblastoma stem cell-driven tumor growth via integrin αvβ5-Src-Stat3 signaling. Theranostics 12, 4221–4236 (2022).
Shi, Y. et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 8, 15080 (2017).
Erickson, H. L. et al. Cancer stem cells release interleukin-33 within large oncosomes to promote immunosuppressive differentiation of macrophage precursors. Immunity 57, 1908–1922.e6 (2024).
Zhang, Z. et al. FAM109B plays a tumorigenic role in low-grade gliomas and is associated with tumor-associated macrophages (TAMs). J. Transl. Med. 22, 833 (2024).
Kloosterman, D. J. et al. Macrophage-mediated myelin recycling fuels brain cancer malignancy. Cell https://doi.org/10.1016/j.cell.2024.07.030 (2024). S0092-8674(24)00824–9.
Liu, D. et al. LSECtin on tumor-associated macrophages enhances breast cancer stemness via interaction with its receptor BTN3A3. Cell Res 29, 365–378 (2019).
Liguori, M. et al. The soluble glycoprotein NMB (GPNMB) produced by macrophages induces cancer stemness and metastasis via CD44 and IL-33. Cell. Mol. Immunol. 18, 711–722 (2021).
Xu, M. et al. Tumor associated macrophages-derived exosomes facilitate hepatocellular carcinoma malignance by transferring lncMMPA to tumor cells and activating glycolysis pathway. J. Exp. Clin. Cancer Res. CR 41, 253 (2022).
Wang, L. et al. Galectin-3 expression and secretion by tumor-associated macrophages in hypoxia promotes breast cancer progression. Biochem. Pharmacol. 178, 114113 (2020).
Shi, Y., Shi, Q., Shen, Q., Zhang, Q. & Cao, X. Dicer-independent snRNA/snoRNA-derived nuclear RNA 3 regulates tumor-associated macrophage function by epigenetically repressing inducible nitric oxide synthase transcription. Cancer Commun 41, 140–153 (2021).
Ruiz-Torres, S. J. et al. Macrophage-mediated RON signaling supports breast cancer growth and progression through modulation of IL-35. Oncogene 41, 321–333 (2022).
Radharani, N. N. V. et al. Tumor-associated macrophage derived IL-6 enriches cancer stem cell population and promotes breast tumor progression via Stat-3 pathway. Cancer Cell Int. 22, 122 (2022).
Zhang, L. et al. Fibronectin 1 derived from tumor-associated macrophages and fibroblasts promotes metastasis through the JUN pathway in hepatocellular carcinoma. Int. Immunopharmacol. 113, 109420 (2022).
Afik, R. et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J. Exp. Med. 213, 2315–2331 (2016).
Qiu, S. et al. Tumor-associated macrophages promote bladder tumor growth through PI3K/AKT signal induced by collagen. Cancer Sci 110, 2110–2118 (2019).
Bied, M., Ho, W. W., Ginhoux, F. & Blériot, C. Roles of macrophages in tumor development: a spatiotemporal perspective. Cell. Mol. Immunol. 20, 983–992 (2023).
Tian, Y. et al. TRIM59 loss in M2 macrophages promotes melanoma migration and invasion by upregulating MMP-9 and Madcam1. Aging 11, 8623–8641 (2019).
Maller, O. et al. Tumour-associated macrophages drive stromal cell-dependent collagen crosslinking and stiffening to promote breast cancer aggression. Nat. Mater. 20, 548–559 (2021).
Kim, H. et al. Macrophages-triggered sequential remodeling of endothelium-interstitial matrix to form pre-metastatic niche in microfluidic tumor microenvironment. Adv. Sci. 6, 1900195 (2019).
Winkler, J., Abisoye-Ogunniyan, A., Metcalf, K. J. & Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 11, 5120 (2020).
Suarez-Carmona, M., Lesage, J., Cataldo, D. & Gilles, C. EMT and inflammation: inseparable actors of cancer progression. Mol. Oncol. 11, 805–823 (2017).
Gao, S., Hu, J., Wu, X. & Liang, Z. PMA treated THP-1-derived-IL-6 promotes EMT of SW48 through STAT3/ERK-dependent activation of Wnt/β-catenin signaling pathway. Biomed. Pharmacother. 108, 618–624 (2018).
Cai, J. et al. Tumor-Associated Macrophages Derived TGF-β-Induced Epithelial to Mesenchymal Transition in Colorectal Cancer Cells through Smad2,3-4/Snail Signaling Pathway. Cancer Res. Treat. 51, 252–266 (2019).
Toledo, B. et al. Deciphering the performance of macrophages in tumour microenvironment: a call for precision immunotherapy. J. Hematol. Oncol. 17, 44 (2024).
Zhang, Q. et al. Interrogation of the microenvironmental landscape in spinal ependymomas reveals dual functions of tumor-associated macrophages. Nat. Commun. 12, 6867 (2021).
Zhang, C. et al. Secreted PTEN binds PLXDC2 on macrophages to drive antitumor immunity and tumor suppression. Dev. Cell https://doi.org/10.1016/j.devcel.2024.08.003 (2024). S1534-5807(24)00486–6.
Yu, Y. et al. OX40L-expressing M1-like macrophage exosomes for cancer immunotherapy. J. Control. Release 365, 469–479 (2024).
Shu, Y. et al. Intervening pyruvate carboxylase stunts tumor growth by strengthening anti-tumor actions of tumor-associated macrophages. Signal Transduct. Target. Ther. 7, 34 (2022).
Ma, S. et al. YTHDF2 orchestrates tumor-associated macrophage reprogramming and controls antitumor immunity through CD8+ T cells. Nat. Immunol. 24, 255–266 (2023).
Luo, X. et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ 28, 1971–1989 (2021).
Luo, X. et al. Phospholipid peroxidation in macrophage confers tumor resistance by suppressing phagocytic capability towards ferroptotic cells. Cell Death Differ 31, 1184–1201 (2024).
Zhang, Z. et al. Constructing immune and prognostic features associated with ADCP in hepatocellular carcinoma and pan-cancer based on scRNA-seq and bulk RNA-seq. Front. Immunol. 15, 1397541 (2024).
Li, R. et al. PSME2 offers value as a biomarker of M1 macrophage infiltration in pan-cancer and inhibits osteosarcoma malignant phenotypes. Int. J. Biol. Sci. 20, 1452–1470 (2024).
Pu, Y. et al. The Macrophage migration inhibitory factor is a vital player in Pan-Cancer by functioning as a M0 Macrophage biomarker. Int. Immunopharmacol. 134, 112198 (2024).
Li, R. Q., Yan, L., Zhang, L., Zhao, Y. & Lian, J. CD74 as a prognostic and M1 macrophage infiltration marker in a comprehensive pan-cancer analysis. Sci. Rep. 14, 8125 (2024).
Suarez-Lopez, L. et al. MAPKAP Kinase-2 drives expression of angiogenic factors by tumor-associated macrophages in a model of inflammation-induced colon cancer. Front. Immunol. 11, 607891 (2020).
Hashemi Goradel, N. et al. Nanoparticles as new tools for inhibition of cancer angiogenesis. J. Cell. Physiol. 233, 2902–2910 (2018).
Roviello, G. et al. The role of bevacizumab in solid tumours: a literature based meta-analysis of randomised trials. Eur. J. Cancer 75, 245–258 (2017).
Teuwen, L.-A. et al. Tumor vessel co-option probed by single-cell analysis. Cell Rep 35, 109253 (2021).
Yang, F., Lee, G. & Fan, Y. Navigating tumor angiogenesis: therapeutic perspectives and myeloid cell regulation mechanism. Angiogenesis https://doi.org/10.1007/s10456-024-09913-z (2024).
El Hafny-Rahbi, B. et al. Tumour angiogenesis normalized by myo-inositol trispyrophosphate alleviates hypoxia in the microenvironment and promotes antitumor immune response. J. Cell. Mol. Med. 25, 3284–3299 (2021).
Han, S. et al. Tumor microenvironment remodeling and tumor therapy based on M2-like tumor associated macrophage-targeting nano-complexes. Theranostics 11, 2892–2916 (2021).
Feng, K. et al. Encapsulation of LXR ligand by D-Nap-GFFY hydrogel enhances anti-tumorigenic actions of LXR and removes LXR-induced lipogenesis. Theranostics 11, 2634–2654 (2021).
Yin, M. et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Investig. 126, 4157–4173 (2016).
Opzoomer, J. W. et al. Macrophages orchestrate the expansion of a proangiogenic perivascular niche during cancer progression. Sci. Adv. 45, eabg9518 (2021).
Hughes, R. et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res 75, 3479–3491 (2015).
Wang, X. et al. Hypoxia-induced myeloid derived growth factor promotes hepatocellular carcinoma progression through remodeling tumor microenvironment. Theranostics 11, 209–221 (2021).
Leibovich, S. J. et al. Macrophage-induced angiogenesis is mediated by tumour necrosis factor-alpha. Nature 329, 630–632 (1987).
Voronov, E., Carmi, Y. & Apte, R. N. The role IL-1 in tumor-mediated angiogenesis. Front. Physiol. 5, 114 (2014).
Kimura, Y. N. et al. Inflammatory stimuli from macrophages and cancer cells synergistically promote tumor growth and angiogenesis. Cancer Sci 98, 2009–2018 (2007).
Koch, A. E. et al. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 258, 1798–1801 (1992).
Lin, L. et al. CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget 6, 34758–34773 (2015).
Zhao, Y. et al. PCR display identifies tamoxifen induction of the novel angiogenic factor adrenomedullin by a non estrogenic mechanism in the human endometrium. Oncogene 16, 409–415 (1998).
Kamiyama, M. et al. EP2, a receptor for PGE2, regulates tumor angiogenesis through direct effects on endothelial cell motility and survival. Oncogene 25, 7019–7028 (2006).
Akiyama, S. et al. The role of thymidine phosphorylase, an angiogenic enzyme, in tumor progression. Cancer Sci 95, 851–857 (2004).
Pucci, F. et al. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood ‘resident’ monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood 114, 901–914 (2009).
Chen, L. et al. Tie2 expression on macrophages is required for blood vessel reconstruction and tumor relapse after chemotherapy. Cancer Res 76, 6828–6838 (2016).
Coffelt, S. B. et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res 70, 5270–5280 (2010).
Stepanova, V. et al. Urokinase-type Plasminogen Activator (uPA) Promotes Angiogenesis by Attenuating Proline-rich Homeodomain Protein (PRH) Transcription Factor Activity and De-repressing Vascular Endothelial Growth Factor (VEGF) Receptor Expression. J. Biol. Chem. 291, 15029–15045 (2016).
De Palma, M. et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8, 211–226 (2005).
Yang, F. et al. Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat. Commun. 12, 3424 (2021).
Du, S. et al. Tumor cell-derived exosomes deliver TIE2 protein to macrophages to promote angiogenesis in cervical cancer. Cancer Lett 529, 168–179 (2022).
Jakab, M., Rostalski, T., Lee, K. H., Mogler, C. & Augustin, H. G. Tie2 receptor in tumor-infiltrating macrophages is dispensable for tumor angiogenesis and tumor relapse after chemotherapy. Cancer Res 82, 1353–1364 (2022).
Zhang, Y. & Brekken, R. A. Are TEMs Canceled? Questioning the Functional Relevance of Tie2-Expressing Macrophages. Cancer Res 82, 1172–1173 (2022).
Chen, P. et al. SiRNA-HIF-1α delivered by attenuated Salmonella enhances the efficacy of Lenvatinib against hepatocellular carcinoma. Int. Immunopharmacol. 130, 111728 (2024).
Fang, H.-Y. et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 114, 844–859 (2009).
Dong, Y. et al. HIF1α epigenetically repressed macrophages via CRISPR/Cas9-EZH2 system for enhanced cancer immunotherapy. Bioact. Mater. 6, 2870–2880 (2021).
Sui, A. et al. Inhibiting NF-κB signaling activation reduces retinal neovascularization by promoting a polarization shift in macrophages. Investig. Ophthalmol. Vis. Sci. 61, 4 (2020).
Tomolonis, J. A. et al. Interaction between tumor cell TNFR2 and monocyte membrane-bound TNF-α triggers tumorigenic inflammation in neuroblastoma. J. Immunother. Cancer 11, e005478 (2023).
Nam, E.-H., Park, S.-R. & Kim, P.-H. TGF-beta1 induces mouse dendritic cells to express VEGF and its receptor (Flt-1) under hypoxic conditions. Exp. Mol. Med. 42, 606–613 (2010).
Cs M. et al. Macrophage IL-1β promotes arteriogenesis by autocrine STAT3- and NF-κB-mediated transcription of pro-angiogenic VEGF-A. Cell Rep. 38, (2022).
Zhu, C. et al. CECR1-mediated cross talk between macrophages and vascular mural cells promotes neovascularization in malignant glioma. Oncogene 36, 5356–5368 (2017).
Xia, Y. et al. Catecholamines contribute to the neovascularization of lung cancer via tumor-associated macrophages. Brain. Behav. Immun. 81, 111–121 (2019).
Fjæstad, K. Y. et al. Blockade of beta-adrenergic receptors reduces cancer growth and enhances the response to anti-CTLA4 therapy by modulating the tumor microenvironment. Oncogene 41, 1364–1375 (2022).
Xiao, B. et al. Choline metabolism reprogramming mediates an immunosuppressive microenvironment in non-small cell lung cancer (NSCLC) by promoting tumor-associated macrophage functional polarization and endothelial cell proliferation. J. Transl. Med. 22, 442 (2024).
Wang, W. et al. Identification of hypoxic macrophages in glioblastoma with therapeutic potential for vasculature normalization. Cancer Cell 42, 815–832.e12 (2024).
Yan, Q. et al. Tumor-associated macrophage-derived exosomal miR21-5p promotes tumor angiogenesis by regulating YAP1/HIF-1α axis in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 81, 179 (2024).
Shou, Y. et al. Exosomal miR-301a-3p from esophageal squamous cell carcinoma cells promotes angiogenesis by inducing M2 polarization of macrophages via the PTEN/PI3K/AKT signaling pathway. Cancer Cell Int 22, 153 (2022).
Droho, S., Rajesh, A., Cuda, C. M., Perlman, H. & Lavine, J. A. CD11c+ macrophages are proangiogenic and necessary for experimental choroidal neovascularization. JCI Insight 8, e168142 (2023).
Chen, P. et al. Symbiotic macrophage-glioma cell interactions reveal synthetic lethality in PTEN-Null Glioma. Cancer Cell 35, 868–884.e6 (2019).
Wang, J., Zhu, N., Su, X., Gao, Y. & Yang, R. Novel tumor-associated macrophage populations and subpopulations by single cell RNA sequencing. Front. Immunol. 14, 1264774 (2023).
Zhang, L. et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 181, 442–459.e29 (2020).
Kim, N. et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 11, 2285 (2020).
Azizi, E. et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308.e36 (2018).
Choi, Y. S. et al. Tie2-mediated vascular remodeling by ferritin-based protein C nanoparticles confers antitumor and anti-metastatic activities. J. Hematol. Oncol. 13, 123 (2020).
Nixon, B. G. et al. Tumor-associated macrophages expressing the transcription factor IRF8 promote T cell exhaustion in cancer. Immunity 55, 2044–2058.e5 (2022).
Xia, Q. et al. Tumor-associated macrophages promote PD-L1 expression in tumor cells by regulating PKM2 nuclear translocation in pancreatic ductal adenocarcinoma. Oncogene 41, 865–877 (2022).
Krneta, T. et al. M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J. Leukoc. Biol. 101, 285–295 (2017).
Palazon, A., Goldrath, A. W., Nizet, V. & Johnson, R. S. HIF transcription factors, inflammation, and immunity. Immunity 41, 518–528 (2014).
Kanamori, M., Nakatsukasa, H., Ito, M., Chikuma, S. & Yoshimura, A. Reprogramming of Th1 cells into regulatory T cells through rewiring of the metabolic status. Int. Immunol. 30, 357–373 (2018).
Trebska-McGowan, K. et al. TGF-β alters the proportion of infiltrating immune cells in a pancreatic ductal adenocarcinoma. J. Gastrointest. Surg. 26, 113–121 (2022).
Sarkar, T. et al. FOXP3/HAT1 axis controls treg infiltration in the tumor microenvironment by inducing CCR4 expression in breast cancer. Front. Immunol. 13, 740588 (2022).
Czystowska-Kuzmicz, M. et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat. Commun. 10, 3000 (2019).
Alonso, R. et al. Induction of anergic or regulatory tumor-specific CD4+ T cells in the tumor-draining lymph node. Nat. Commun. 9, 2113 (2018).
Wei, Y.-T. et al. Thymosin α-1 Reverses M2 polarization of tumor-associated macrophages during efferocytosis. Cancer Res 82, 1991–2002 (2022).
Combes, A. J. et al. Discovering dominant tumor immune archetypes in a pan-cancer census. Cell 185, 184–203.e19 (2022).
Tharp, K. M. et al. Tumor-associated macrophages restrict CD8+ T cell function through collagen deposition and metabolic reprogramming of the breast cancer microenvironment. Nat. Cancer https://doi.org/10.1038/s43018-024-00775-4 (2024).
Masetti, M. et al. Lipid-loaded tumor-associated macrophages sustain tumor growth and invasiveness in prostate cancer. J. Exp. Med. 219, e20210564 (2022).
Liu, C. et al. Pan-Cancer Single-Cell and Spatial-Resolved Profiling Reveals the Immunosuppressive Role of APOE+ Macrophages in Immune Checkpoint Inhibitor Therapy. Adv. Sci. 2401061 https://doi.org/10.1002/advs.202401061 (2024).
Timperi, E. et al. Lipid-associated macrophages are induced by cancer-associated fibroblasts and mediate immune suppression in breast cancer. Cancer Res 82, 3291–3306 (2022).
Sun, R. et al. Neutral ceramidase regulates breast cancer progression by metabolic programming of TREM2-associated macrophages. Nat. Commun. 15, 966 (2024).
Wang, L. et al. PARP-inhibition reprograms macrophages toward an anti-tumor phenotype. Cell Rep 41, 111462 (2022).
Wang, Q. et al. STING agonism reprograms tumor-associated macrophages and overcomes resistance to PARP inhibition in BRCA1-deficient models of breast cancer. Nat. Commun. 13, 3022 (2022).
Zhuo, C., Ruan, Q., Zhao, X., Shen, Y. & Lin, R. CXCL1 promotes colon cancer progression through activation of NF-κB/P300 signaling pathway. Biol. Direct 17, 34 (2022).
Wu, H. et al. Tumor cell SPTBN1 inhibits M2 polarization of macrophages by suppressing CXCL1 expression. J. Cell. Physiol. 239, 97–111 (2024).
Yin, Y. et al. Colorectal cancer-derived small extracellular vesicles promote tumor immune evasion by upregulating PD-L1 expression in tumor-associated macrophages. Adv. Sci. 9, 2102620 (2022).
Yin, L. & Wang, Y. Extracellular vesicles derived from M2-polarized tumor-associated macrophages promote immune escape in ovarian cancer through NEAT1/miR-101-3p/ZEB1/PD-L1 axis. Cancer Immunol. Immunother. CII 72, 743–758 (2023).
Zhu, J. et al. Pan-cancer analysis of Krüppel-like factor 3 and its carcinogenesis in pancreatic cancer. Front. Immunol. 14, 1167018 (2023).
Xiang, X. et al. Blocking CX3CR1+ tumor-associated macrophages enhances the efficacy of Anti-PD-1 therapy in hepatocellular carcinoma. Cancer Immunol. Res. https://doi.org/10.1158/2326-6066.CIR-23-0627 (2024).
Chen, J. et al. TGF-β1-Induced SOX18 elevation promotes hepatocellular carcinoma progression and metastasis through transcriptionally upregulating PD-L1 and CXCL12. Gastroenterology 167, 264–280 (2024).
Zhang, L. et al. HCG18 participates in vascular invasion of hepatocellular carcinoma by regulating macrophages and tumor stem cells. Front. Cell Dev. Biol. 9, 707073 (2021).
Wang, Z. et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 25, 141–151 (2019).
Bader, J. E. et al. Obesity induces PD-1 on macrophages to suppress anti-tumour immunity. Nature https://doi.org/10.1038/s41586-024-07529-3 (2024).
Zhu, C.-X. et al. Targeting OXCT1-mediated ketone metabolism reprograms macrophages to promote antitumor immunity via CD8+ T cells in hepatocellular carcinoma. J. Hepatol. https://doi.org/10.1016/j.jhep.2024.05.007 (2024). S0168-8278(24)00342–8.
Long, X. et al. ScRNA-seq reveals novel immune-suppressive T cells and investigates CMV-TCR-T cells cytotoxicity against GBM. J. Immunother. Cancer 12, e008967 (2024).
Zhang, Z., Zheng, M., Ding, Q. & Liu, M. CD93 correlates with immune infiltration and impacts patient immunotherapy efficacy: a pan-cancer analysis. Front. Cell Dev. Biol. 10, 817965 (2022).
Zhang, J. et al. Large-scale single-cell and bulk sequencing analyses reveal the prognostic value and immune aspects of CD147 in Pan-Cancer. Front. Immunol. 13, 810471 (2022).
Zhang, Q., Wang, X., Liu, Y., Xu, H. & Ye, C. Pan-cancer and single-cell analyses identify CD44 as an immunotherapy response predictor and regulating macrophage polarization and tumor progression in colorectal cancer. Front. Oncol. 14, 1380821 (2024).
Zhang, W. et al. Crosstalk between IL-15Rα+ tumor-associated macrophages and breast cancer cells reduces CD8+ T cell recruitment. Cancer Commun 42, 536–557 (2022).
Li, X. et al. Reactive oxygen species reprogram macrophages to suppress antitumor immune response through the exosomal miR-155-5p/PD-L1 pathway. J. Exp. Clin. Cancer Res. CR 41, 41 (2022).
Wertheimer, T. et al. IL-23 stabilizes an effector Treg cell program in the tumor microenvironment. Nat. Immunol. 25, 512–524 (2024).
Tong, Y. et al. Concomitant type I IFN and M-CSF signaling reprograms monocyte differentiation and drives pro-tumoral arginase production. EBioMedicine 39, 132–144 (2019).
Vanmeerbeek, I. et al. Targeting conserved TIM3+VISTA+ tumor-associated macrophages overcomes resistance to cancer immunotherapy. Sci. Adv. 10, eadm8660 (2024).
Chen, S. et al. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 8, 207 (2023).
Weyand, C. M. & Goronzy, J. J. The immunology of rheumatoid arthritis. Nat. Immunol. 22, 10–18 (2021).
Janossy, G. et al. Rheumatoid arthritis: a disease of T-lymphocyte/macrophage immunoregulation. Lancet 2, 839–842 (1981).
Yokota, K. et al. Characterization and function of tumor necrosis factor and Interleukin-6-Induced osteoclasts in rheumatoid arthritis. Arthritis Rheumatol 73, 1145–1154 (2021).
Tak, P. P. et al. Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum 40, 217–225 (1997).
van Schouwenburg, P. A., Rispens, T. & Wolbink, G. J. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat. Rev. Rheumatol. 9, 164–172 (2013).
Yamanaka, H. TNF as a target of inflammation in rheumatoid arthritis. Endocr. Metab. Immune Disord. Drug Targets 15, 129–134 (2015).
Keffer, J. et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J 10, 4025–4031 (1991).
Yoshitake, F., Itoh, S., Narita, H., Ishihara, K. & Ebisu, S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways. J. Biol. Chem. 283, 11535–11540 (2008).
Zhao, Z. et al. Inhibition of Histone H3 Lysine-27 demethylase activity relieves rheumatoid arthritis symptoms via repression of IL6 transcription in macrophages. Front. Immunol. 13, 818070 (2022).
Sun, W. et al. Targeting Notch-Activated M1 macrophages attenuates joint tissue damage in a mouse model of inflammatory arthritis. J. Bone Miner. Res. 32, 1469–1480 (2017).
Yu, J. & Canalis, E. Notch and the regulation of osteoclast differentiation and function. Bone 138, 115474 (2020).
Chang, J.-W. et al. Nesfatin-1 stimulates CCL2-dependent Monocyte Migration And M1 Macrophage Polarization: implications for rheumatoid arthritis therapy. Int. J. Biol. Sci. 19, 281–293 (2023).
Robinson, C. et al. Nesfatin-1 and visfatin expression is associated with reduced atherosclerotic disease risk in patients with rheumatoid arthritis. Peptides 102, 31–37 (2018).
Xu, Y. et al. Glycolysis in innate immune cells contributes to autoimmunity. Front. Immunol. 13, 920029 (2022).
Hollander, A. P., Corke, K. P., Freemont, A. J. & Lewis, C. E. Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum. 44, 1540–1544 (2001).
Zhang, Y. et al. Targeting KAT2A inhibits inflammatory macrophage activation and rheumatoid arthritis through epigenetic and metabolic reprogramming. MedComm 4, e306 (2023).
Wen, Y. et al. Water extracts of Tibetan medicine Wuweiganlu attenuates experimental arthritis via inducing macrophage polarization towards the M2 type. J. Ethnopharmacol. 318, 116934 (2024).
Wan, L. et al. A novel pharmaceutical preparation of Tripterygium wilfordii Hook. f. regulates macrophage polarization to alleviate inflammation in rheumatoid arthritis. J. Pharm. Pharmacol. 75, 1442–1457 (2023).
Park, S. Y. et al. SIRT1/Adenosine monophosphate-activated protein kinase α signaling enhances macrophage polarization to an anti-inflammatory phenotype in rheumatoid arthritis. Front. Immunol. 8, 1135 (2017).
Hannemann, N. et al. The AP-1 Transcription Factor c-Jun promotes arthritis by regulating Cyclooxygenase-2 and Arginase-1 expression in macrophages. J. Immunol. 198, 3605–3614 (2017).
Hannemann, N. et al. Transcription factor Fra-1 targets arginase-1 to enhance macrophage-mediated inflammation in arthritis. J. Clin. Investig. 129, 2669–2684 (2019).
Yuba, E. et al. Suppression of rheumatoid arthritis by enhanced lymph node trafficking of engineered Interleukin-10 in Murine Models. Arthritis Rheumatol 73, 769–778 (2021).
Ye, L. et al. Interleukin-10 attenuation of collagen-induced arthritis is associated with suppression of interleukin-17 and retinoid-related orphan receptor γt production in macrophages and repression of classically activated macrophages. Arthritis Res. Ther. 16, R96 (2014).
Yang, X. et al. GRK2 inhibits Flt-1+ macrophage infiltration and its proangiogenic properties in rheumatoid arthritis. Acta Pharm. Sin. B 14, 241–255 (2024).
Wang, Z. et al. A targeted exosome therapeutic confers both CfDNA scavenging and macrophage polarization for ameliorating rheumatoid arthritis. Adv. Mater. 35, e2302503 (2023).
Zhou, Z. et al. Single-cell profiling identifies IL1Bhi macrophages associated with inflammation in PD-1 inhibitor-induced inflammatory arthritis. Nat. Commun. 15, 2107 (2024).
Cheng, L. et al. New insights from single-cell sequencing data: synovial fibroblasts and synovial macrophages in rheumatoid arthritis. Front. Immunol. 12, 709178 (2021).
You, S. et al. Identification of key regulators for the migration and invasion of rheumatoid synoviocytes through a systems approach. Proc. Natl Acad. Sci. USA 111, 550–555 (2014).
Matsuda, K., Shiba, N. & Hiraoka, K. New insights into the role of synovial fibroblasts leading to joint destruction in rheumatoid arthritis. Int. J. Mol. Sci. 24, 5173 (2023).
Zhao, S. et al. Pentraxin 3 inhibits fibroblast growth factor 2 induced osteoclastogenesis in rheumatoid arthritis. Biomed. Pharmacother. 131, 110628 (2020).
Yamakawa, T. et al. Novel gene Merlot inhibits differentiation and promotes apoptosis of osteoclasts. Bone 138, 115494 (2020).
Aripova, N. et al. Citrullinated and malondialdehyde-acetaldehyde modified fibrinogen activates macrophages and promotes an aggressive synovial fibroblast phenotype in patients with rheumatoid arthritis. Front. Immunol. 14, 1203548 (2023).
Weng, W. et al. Macrophage extracellular traps promote tumor-like biologic behaviors of fibroblast-like synoviocytes through cGAS-mediated PI3K/Akt signaling pathway in patients with rheumatoid arthritis. J. Leukoc. Biol. 115, 116–129 (2024).
Kuo, D. et al. HBEGF+ macrophages in rheumatoid arthritis induce fibroblast invasiveness. Sci. Transl. Med. 11, eaau8587 (2019).
Liu, A.-C. et al. Macrophage activation syndrome in systemic lupus erythematosus: a multicenter, case-control study in China. Clin. Rheumatol. 37, 93–100 (2018).
Labonte, A. C. et al. Identification of alterations in macrophage activation associated with disease activity in systemic lupus erythematosus. PloS ONE 13, e0208132 (2018).
Malkiel, S., Barlev, A. N., Atisha-Fregoso, Y., Suurmond, J. & Diamond, B. Plasma cell differentiation pathways in systemic lupus erythematosus. Front. Immunol. 9, 427 (2018).
Katsiari, C. G. et al. Aberrant expression of the costimulatory molecule CD40 ligand on monocytes from patients with systemic lupus erythematosus. Clin. Immunol. Orlando Fla 103, 54–62 (2002).
Higuchi, T. et al. Cutting edge: ectopic expression of CD40 ligand on B cells induces lupus-like autoimmune disease. J. Immunol. 168, 9–12 (2002).
Harigai, M. et al. Responsiveness of peripheral blood B cells to recombinant CD40 ligand in patients with systemic lupus erythematosus. Lupus 8, 227–233 (1999).
Zheng, M. et al. Single-cell sequencing shows cellular heterogeneity of cutaneous lesions in lupus erythematosus. Nat. Commun. 13, 7489 (2022).
Bennett, L. et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197, 711–723 (2003).
Zhang, F. et al. IFN-γ and TNF-α drive a CXCL10+ CCL2+ macrophage phenotype expanded in severe COVID-19 lungs and inflammatory diseases with tissue inflammation. Genome Med 13, 64 (2021).
Aringer, M. et al. Adverse events and efficacy of TNF-alpha blockade with infliximab in patients with systemic lupus erythematosus: long-term follow-up of 13 patients. Rheumatol. Oxf. Engl. 48, 1451–1454 (2009).
Umare, V. et al. Effect of proinflammatory cytokines (IL-6, TNF-α, and IL-1β) on clinical manifestations in Indian SLE patients. Mediators Inflamm 2014, 385297 (2014).
Santer, D. M., Yoshio, T., Minota, S., Möller, T. & Elkon, K. B. Potent induction of IFN-alpha and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus. J. Immunol. 182, 1192–1201 (2009).
Aringer, M. et al. Increased bioactive TNF in human systemic lupus erythematosus: associations with cell death. Lupus 11, 102–108 (2002).
Gröndal, G. et al. Cytokine production, serum levels and disease activity in systemic lupus erythematosus. Clin. Exp. Rheumatol. 18, 565–570 (2000).
Ren, Y. et al. Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus. Arthritis Rheum 48, 2888–2897 (2003).
Roberts, A. W. et al. Tissue-resident macrophages are locally programmed for silent clearance of apoptotic cells. Immunity 47, 913–927.e6 (2017).
Bengtsson, A. A., Sturfelt, G., Gullstrand, B. & Truedsson, L. Induction of apoptosis in monocytes and lymphocytes by serum from patients with systemic lupus erythematosus - an additional mechanism to increased autoantigen load? Clin. Exp. Immunol. 135, 535–543 (2004).
Zhang, W. et al. Defective Lamtor5 leads to autoimmunity by deregulating v-atpase and lysosomal acidification. Adv. Sci. 11, e2400446 (2024).
Dijstelbloem, H. M. et al. Fcgamma receptor polymorphisms in systemic lupus erythematosus: association with disease and in vivo clearance of immune complexes. Arthritis Rheum 43, 2793–2800 (2000).
Zhang, X. et al. Pyruvate Kinase M2 Contributes to TLR-Mediated inflammation and autoimmunity by promoting Pyk2 Activation. Front. Immunol. 12, 680068 (2021).
Xin, Y., Gao, C., Wang, L., Liu, Q. & Lu, Q. Lipopolysaccharide released from gut activates pyroptosis of macrophages via Caspase 11-Gasdermin D pathway in systemic lupus erythematosus. MedComm 5, e610 (2024).
Morel, L. Erythrocyte-derived mitochondria: an unexpected interferon inducer in lupus. Trends Immunol 42, 1054–1056 (2021).
Zhuang, L. et al. Disulfiram alleviates pristane-induced lupus via inhibiting GSDMD-mediated pyroptosis. Cell Death Discov 8, 379 (2022).
Zhang, F. et al. The oxysterol receptor EBI2 links innate and adaptive immunity to limit IFN response and systemic lupus erythematosus. Adv. Sci. 10, e2207108 (2023).
Yang, B. et al. Decreased miR-4512 levels in monocytes and macrophages of individuals with systemic lupus erythematosus contribute to innate immune activation and Neutrsophil NETosis by targeting TLR4 and CXCL2. Front. Immunol. 12, 756825 (2021).
Xu, H. et al. Cellular spermine targets JAK signaling to restrain cytokine-mediated autoimmunity. Immunity https://doi.org/10.1016/j.immuni.2024.05.025 (2024).
Mohammadi, S., Saghaeian-Jazi, M., Sedighi, S. & Memarian, A. Immunomodulation in systemic lupus erythematosus: induction of M2 population in monocyte-derived macrophages by pioglitazone. Lupus 26, 1318–1327 (2017).
Li, F., Yang, Y., Zhu, X., Huang, L. & Xu, J. Macrophage polarization modulates development of systemic lupus erythematosus. Cell. Physiol. Biochem. 37, 1279–1288 (2015).
Wermeling, F. et al. Class A scavenger receptors regulate tolerance against apoptotic cells, and autoantibodies against these receptors are predictive of systemic lupus. J. Exp. Med. 204, 2259–2265 (2007).
Becker-Merok, A., Eilertsen, G. Ø. & Nossent, J. C. Levels of transforming growth factor-beta are low in systemic lupus erythematosus patients with active disease. J. Rheumatol. 37, 2039–2045 (2010).
Mellor-Pita, S. et al. Monocytes and T lymphocytes contribute to a predominance of interleukin 6 and interleukin 10 in systemic lupus erythematosus. Cytom. B Clin. Cytom. 76, 261–270 (2009).
Godsell, J. et al. Clinical associations of IL-10 and IL-37 in systemic lupus erythematosus. Sci. Rep. 6, 34604 (2016).
Bussolati, B., Rollino, C., Mariano, F., Quarello, F. & Camussi, G. IL-10 stimulates production of platelet-activating factor by monocytes of patients with active systemic lupus erythematosus (SLE). Clin. Exp. Immunol. 122, 471–476 (2000).
Zhou, W. et al. Degradation of HDAC10 by autophagy promotes IRF3-mediated antiviral innate immune responses. Sci. Signal. 15, eabo4356 (2022).
Zheng, X. et al. AKT2 reduces IFNβ1 production to modulate antiviral responses and systemic lupus erythematosus. EMBO J 41, e108016 (2022).
Zhong, J. et al. Phosphopeptides P140 cause oxidative burst responses of pulmonary macrophages in an imiquimod-induced lupus model. Mol. Biomed. 4, 38 (2023).
Allanore, Y. et al. Systemic sclerosis. Nat. Rev. Dis. Prim. 1, 15002 (2015).
Lescoat, A., Varga, J., Matucci-Cerinic, M. & Khanna, D. New promising drugs for the treatment of systemic sclerosis: pathogenic considerations, enhanced classifications, and personalized medicine. Expert Opin. Investig. Drugs 30, 635–652 (2021).
Varga, J. & Abraham, D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J. Clin. Investig. 117, 557–567 (2007).
Ishikawa, O. & Ishikawa, H. Macrophage infiltration in the skin of patients with systemic sclerosis. J. Rheumatol. 19, 1202–1206 (1992).
Valenzi, E. et al. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann. Rheum. Dis. 78, 1379–1387 (2019).
Martinez, F. O. & Gordon, S. The evolution of our understanding of macrophages and translation of findings toward the clinic. Expert Rev. Clin. Immunol. 11, 5–13 (2015).
Gao, X. et al. Osteopontin links myeloid activation and disease progression in systemic sclerosis. Cell Rep. Med. 1, 100140 (2020).
Denton, C. P. et al. Therapeutic interleukin-6 blockade reverses transforming growth factor-beta pathway activation in dermal fibroblasts: insights from the faSScinate clinical trial in systemic sclerosis. Ann. Rheum. Dis. 77, 1362–1371 (2018).
Morse, C. et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 54, 1802441 (2019).
Papazoglou, A. et al. Epigenetic Regulation of Profibrotic Macrophages in Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheumatol 74, 2003–2014 (2022).
Xue, D. et al. Expansion of Fcγ Receptor IIIa-Positive Macrophages, Ficolin 1-Positive Monocyte-Derived dendritic cells, and plasmacytoid dendritic cells associated with severe skin disease in systemic sclerosis. Arthritis Rheumatol 74, 329–341 (2022).
Xu, D. et al. PLG nanoparticles target fibroblasts and MARCO+ monocytes to reverse multiorgan fibrosis. JCI Insight 7, e151037 (2022).
Lescoat, A., Lecureur, V. & Varga, J. Contribution of monocytes and macrophages to the pathogenesis of systemic sclerosis: recent insights and therapeutic implications. Curr. Opin. Rheumatol. 33, 463–470 (2021).
Higashi-Kuwata, N. et al. Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res. Ther. 12, R128 (2010).
Christmann, R. B. et al. Association of Interferon- and transforming growth factor β-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol 66, 714–725 (2014).
Soldano, S. et al. Increase in circulating cells coexpressing M1 and M2 macrophage surface markers in patients with systemic sclerosis. Ann. Rheum. Dis. 77, 1842–1845 (2018).
Lescoat, A. et al. Combined anti-fibrotic and anti-inflammatory properties of JAK-inhibitors on macrophages in vitro and in vivo: perspectives for scleroderma-associated interstitial lung disease. Biochem. Pharmacol. 178, 114103 (2020).
Skaug, B. et al. Global skin gene expression analysis of early diffuse cutaneous systemic sclerosis shows a prominent innate and adaptive inflammatory profile. Ann. Rheum. Dis. 79, 379–386 (2020).
Sun, C., Cai, D. & Chen, S.-Y. ADAR1 promotes systemic sclerosis via modulating classic macrophage activation. Front. Immunol. 13, 1051254 (2022).
Bujor, A. M., El Adili, F., Parvez, A., Marden, G. & Trojanowska, M. Fli1 downregulation in scleroderma myeloid cells has profibrotic and proinflammatory effects. Front. Immunol. 11, 800 (2020).
Numajiri, H. et al. B cell depletion inhibits fibrosis via suppression of profibrotic macrophage differentiation in a mouse model of systemic sclerosis. Arthritis Rheumatol 73, 2086–2095 (2021).
Li, R. et al. Route to rheumatoid arthritis by macrophage-derived microvesicle-coated nanoparticles. Nano Lett 19, 124–134 (2019).
Frodermann, V. & Nahrendorf, M. Macrophages and cardiovascular health. Physiol. Rev. 98, 2523–2569 (2018).
Kurihara, T., Warr, G., Loy, J. & Bravo, R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 186, 1757–1762 (1997).
Gerszten, R. E. et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 398, 718–723 (1999).
Frangogiannis, N. G. Cardiac fibrosis. Cardiovasc. Res. 117, 1450–1488 (2021).
Chen, R. et al. Macrophages in cardiovascular diseases: molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 9, 130 (2024).
Frangogiannis, N. G. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 65, 70–99 (2019).
López, B. et al. Diffuse myocardial fibrosis: mechanisms, diagnosis and therapeutic approaches. Nat. Rev. Cardiol. 18, 479–498 (2021).
Epelman, S., Lavine, K. J. & Randolph, G. J. Origin and functions of tissue macrophages. Immunity 41, 21–35 (2014).
Murray, P. J. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014).
Molawi, K. et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 211, 2151–2158 (2014).
Ensan, S. et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat. Immunol. 17, 159–168 (2016).
Hernandez, G. E. et al. Aortic intimal resident macrophages are essential for maintenance of the non-thrombogenic intravascular state. Nat. Cardiovasc. Res. 1, 67–84 (2022).
Weinberger, T. et al. Ontogeny of arterial macrophages defines their functions in homeostasis and inflammation. Nat. Commun. 11, 4549 (2020).
Williams, J. W. et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 21, 1194–1204 (2020).
Hulsmans, M. et al. Macrophages facilitate electrical conduction in the heart. Cell 169, 510–522.e20 (2017).
Pinto, A. R. et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PloS ONE 7, e36814 (2012).
Lavine, K. J. et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 111, 16029–16034 (2014).
Epelman, S. et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104 (2014).
Li, J., Chen, Q., Zhang, R., Liu, Z. & Cheng, Y. The phagocytic role of macrophage following myocardial infarction. Heart Fail. Rev. 28, 993–1007 (2023).
Lafuse, W. P., Wozniak, D. J. & Rajaram, M. V. S. Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells 10, 51 (2020).
DeBerge, M., Shah, S. J., Wilsbacher, L. & Thorp, E. B. Macrophages in heart failure with reduced versus preserved ejection fraction. Trends Mol. Med. 25, 328–340 (2019).
Maruyama, K. & Imanaka-Yoshida, K. The pathogenesis of cardiac fibrosis: a review of recent progress. Int. J. Mol. Sci. 23, 2617 (2022).
Yap, J. et al. Macrophages in cardiac remodelling after myocardial infarction. Nat. Rev. Cardiol. 20, 373–385 (2023).
Buechler, M. B., Fu, W. & Turley, S. J. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity 54, 903–915 (2021).
Kuppe, C. et al. Spatial multi-omic map of human myocardial infarction. Nature 608, 766–777 (2022).
Hu, S. et al. Different roles of resident and non-resident macrophages in cardiac fibrosis. Front. Cardiovasc. Med. 9, 818188 (2022).
Prabhu, S. D. & Frangogiannis, N. G. The Biological Basis For Cardiac Repair After Myocardial Infarction: From Inflammation To Fibrosis. Circ. Res. 119, 91–112 (2016).
Chen, S. et al. M2b macrophages protect against doxorubicin induced cardiotoxicity via alternating autophagy in cardiomyocytes. PloS ONE 18, e0288422 (2023).
Liu, Y., Wu, M., Zhong, C., Xu, B. & Kang, L. M2-like macrophages transplantation protects against the doxorubicin-induced heart failure via mitochondrial transfer. Biomater. Res. 26, 14 (2022).
Zhou, J., Li, Y.-S. & Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 34, 2191–2198 (2014).
Skålén, K. et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 417, 750–754 (2002).
Gerrity, R. G., Naito, H. K., Richardson, M. & Schwartz, C. J. Dietary induced atherogenesis in swine. Morphology of the intima in prelesion stages. Am. J. Pathol. 95, 775–792 (1979).
Li, A. C. & Glass, C. K. The macrophage foam cell as a target for therapeutic intervention. Nat. Med. 8, 1235–1242 (2002).
Moore, K. J. & Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355 (2011).
Tacke, F. et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 117, 185–194 (2007).
Poznyak, A. V. et al. Anti-inflammatory therapy for atherosclerosis: focusing on cytokines. Int. J. Mol. Sci. 22, 7061 (2021).
Weber, B. N., Giles, J. T. & Liao, K. P. Shared inflammatory pathways of rheumatoid arthritis and atherosclerotic cardiovascular disease. Nat. Rev. Rheumatol. 19, 417–428 (2023).
Carmi, Y. et al. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J. Immunol. 183, 4705–4714 (2009).
Abbate, A. et al. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 126, 1260–1280 (2020).
Hettwer, J. et al. Interleukin-1β suppression dampens inflammatory leucocyte production and uptake in atherosclerosis. Cardiovasc. Res. 118, 2778–2791 (2022).
Zhang, X. et al. Loss of macrophage mTORC2 drives atherosclerosis via FoxO1 and IL-1β signaling. Circ. Res. 133, 200–219 (2023).
Swirski, F. K. et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 117, 195–205 (2007).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010).
Zhu, C. et al. TRIM64 promotes ox-LDL-induced foam cell formation, pyroptosis, and inflammation in THP-1-derived macrophages by activating a feedback loop with NF-κB via IκBα ubiquitination. Cell Biol. Toxicol. 39, 607–620 (2023).
Tyrrell, D. J. & Goldstein, D. R. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat. Rev. Cardiol. 18, 58–68 (2021).
Ohta, H. et al. Disruption of tumor necrosis factor-alpha gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis 180, 11–17 (2005).
Zhang, Y. et al. Inhibition of Hedgehog signaling ameliorates foam cell formation by promoting autophagy in early atherosclerosis. Cell Death Dis 14, 740 (2023).
Low, E. L., Baker, A. H. & Bradshaw, A. C. TGFβ, smooth muscle cells and coronary artery disease: a review. Cell Signal 53, 90–101 (2019).
Fourman, L. T. et al. Anti-inflammatory interleukin 10 Inversely Relates To Coronary Atherosclerosis In Persons With Human Immunodeficiency Virus. J. Infect. Dis. 221, 510–515 (2020).
Han, X., Kitamoto, S., Lian, Q. & Boisvert, W. A. Interleukin-10 facilitates both cholesterol uptake and efflux in macrophages. J. Biol. Chem. 284, 32950–32958 (2009).
Maguire, E. M., Pearce, S. W. A. & Xiao, Q. Foam cell formation: a new target for fighting atherosclerosis and cardiovascular disease. Vasc. Pharmacol. 112, 54–71 (2019).
Guo, L. et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Investig. 128, 1106–1124 (2018).
Kim, K. et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ. Res. 123, 1127–1142 (2018).
Akhmedov, A. et al. Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1): a crucial driver of atherosclerotic cardiovascular disease. Eur. Heart J. 42, 1797–1807 (2021).
Mäkinen, P. I. et al. Silencing of either SR-A or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc. Res. 88, 530–538 (2010).
Mehta, J. L. et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ. Res. 100, 1634–1642 (2007).
van Tits, L. J. H. et al. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Krüppel-like factor 2. Atherosclerosis 214, 345–349 (2011).
Zhou, Z. et al. K63 ubiquitin chains target NLRP3 inflammasome for autophagic degradation in ox-LDL-stimulated THP-1 macrophages. Aging 12, 1747–1759 (2020).
Jinnouchi, H. et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell. Mol. Life Sci. CMLS 77, 1919–1932 (2020).
Gonzalez, L. & Trigatti, B. L. Macrophage apoptosis and necrotic core development in atherosclerosis: a rapidly advancing field with clinical relevance to imaging and therapy. Can. J. Cardiol. 33, 303–312 (2017).
Zhao, Y. et al. Myeloid BAF60a deficiency alters metabolic homeostasis and exacerbates atherosclerosis. Cell Rep 42, 113171 (2023).
Erbel, C. et al. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+ in vitro and in vivo. Innate Immun 21, 255–265 (2015).
Huang, W.-C., Sala-Newby, G. B., Susana, A., Johnson, J. L. & Newby, A. C. Classical macrophage activation up-regulates several matrix metalloproteinases through mitogen activated protein kinases and nuclear factor-κB. PloS ONE 7, e42507 (2012).
Zou, J., Xu, C., Zhao, Z.-W., Yin, S.-H. & Wang, G. Asprosin inhibits macrophage lipid accumulation and reduces atherosclerotic burden by up-regulating ABCA1 and ABCG1 expression via the p38/Elk-1 pathway. J. Transl. Med. 20, 337 (2022).
Zhang, Y. et al. Macrophage MCT4 inhibition activates reparative genes and protects from atherosclerosis by histone H3 lysine 18 lactylation. Cell Rep 43, 114180 (2024).
Zheng, Y. et al. PPARs in atherosclerosis: the spatial and temporal features from mechanism to druggable targets. J. Adv. Res. https://doi.org/10.1016/j.jare.2024.03.020 (2024). S2090-1232(24)00120–6.
Soskic, S. S. et al. Peroxisome proliferator-activated receptors and atherosclerosis. Angiology 62, 523–534 (2011).
Chinetti, G., Fruchart, J.-C. & Staels, B. Peroxisome proliferator-activated receptors: new targets for the pharmacological modulation of macrophage gene expression and function. Curr. Opin. Lipidol. 14, 459–468 (2003).
Heidt, T. et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 115, 284–295 (2014).
Wei, X. et al. EDIL3 deficiency ameliorates adverse cardiac remodelling by neutrophil extracellular traps (NET)-mediated macrophage polarization. Cardiovasc. Res. 118, 2179–2195 (2022).
Weber, K. T., Sun, Y., Bhattacharya, S. K., Ahokas, R. A. & Gerling, I. C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 10, 15–26 (2013).
Zhuang, L., Zong, X., Yang, Q., Fan, Q. & Tao, R. Interleukin-34-NF-κB signaling aggravates myocardial ischemic/reperfusion injury by facilitating macrophage recruitment and polarization. EBioMedicine 95, 104744 (2023).
Bujak, M. et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am. J. Pathol. 173, 57–67 (2008).
Bageghni, S. A. et al. Fibroblast-specific deletion of interleukin-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction. JCI Insight 5, e125074 (2019).
Liu, W. et al. Activation in M1 but not M2 macrophages contributes to cardiac remodeling after myocardial infarction in rats: a critical role of the calcium sensing Receptor/NRLP3 inflammasome. Cell. Physiol. Biochem. 35, 2483–2500 (2015).
Jing, R., Long, T.-Y., Pan, W., Li, F. & Xie, Q.-Y. IL-6 knockout ameliorates myocardial remodeling after myocardial infarction by regulating activation of M2 macrophages and fibroblast cells. Eur. Rev. Med. Pharmacol. Sci. 23, 6283–6291 (2019).
Lugrin, J. et al. The systemic deletion of interleukin-1α reduces myocardial inflammation and attenuates ventricular remodeling in murine myocardial infarction. Sci. Rep. 13, 4006 (2023).
Zhou, Y. et al. The novel vaccines targeting interleukin-1 receptor type I. Int. Immunopharmacol. 132, 111941 (2024).
Gurantz, D. et al. IL-1beta and TNF-alpha upregulate angiotensin II type 1 (AT1) receptors on cardiac fibroblasts and are associated with increased AT1 density in the post-MI heart. J. Mol. Cell. Cardiol. 38, 505–515 (2005).
Wang, Z. et al. LncRNA MIAT downregulates IL-1β, TNF-? to suppress macrophage inflammation but is suppressed by ATP-induced NLRP3 inflammasome activation. Cell Cycle Georget. Tex 20, 194–203 (2021).
Zheng, Y. et al. IL6/adiponectin/HMGB1 feedback loop mediates adipocyte and macrophage crosstalk and M2 polarization after myocardial infarction. Front. Immunol. 15, 1368516 (2024).
Yan, M. et al. Interleukin-7 aggravates myocardial ischaemia/reperfusion injury by regulating macrophage infiltration and polarization. J. Cell. Mol. Med. 25, 9939–9952 (2021).
Qiao, S. et al. Extracellular vesicles derived from Krüppel-Like Factor 2-overexpressing endothelial cells attenuate myocardial ischemia-reperfusion injury by preventing Ly6Chigh monocyte recruitment. Theranostics 10, 11562–11579 (2020).
Liu, Y. et al. Macrophage CARD9 mediates cardiac injury following myocardial infarction through regulation of lipocalin 2 expression. Signal Transduct. Target. Ther. 8, 394 (2023).
Hilgendorf, I. et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 114, 1611–1622 (2014).
Shiraishi, M. et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 126, 2151–2166 (2016).
DeBerge, M. et al. MerTK cleavage on resident cardiac macrophages compromises repair after myocardial ischemia reperfusion injury. Circ. Res. 121, 930–940 (2017).
Kong, P. et al. Opposing actions of fibroblast and cardiomyocyte Smad3 signaling in the infarcted myocardium. Circulation 137, 707–724 (2018).
Wang, Y. et al. Hypoxia induces M2 macrophages to express VSIG4 and mediate cardiac fibrosis after myocardial infarction. Theranostics 13, 2192–2209 (2023).
Krishnamurthy, P. et al. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ. Res. 104, e9–e18 (2009).
Sicklinger, F. et al. Basophils balance healing after myocardial infarction via IL-4/IL-13. J. Clin. Investig. 131, e136778 (2021).
Zhang, S., Zhang, Y., Duan, X., Wang, B. & Zhan, Z. Targeting NPM1 epigenetically promotes postinfarction cardiac repair by reprogramming reparative macrophage metabolism. Circulation 149, 1982–2001 (2024).
Cai, S. et al. Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction. J. Clin. Investig. 133, e159498 (2023).
Xu, Y. et al. A transient wave of Bhlhe41+ resident macrophages enables remodeling of the developing infarcted myocardium. Cell Rep 42, 113174 (2023).
Li, L. et al. M2 Macrophage-Derived sEV regulate pro-inflammatory CCR2+ Macrophage Subpopulations to Favor Post-AMI Cardiac Repair. Adv. Sci. 10, e2202964 (2023).
Winfree, R. L. et al. TREM2 gene expression associations with Alzheimer’s disease neuropathology are region-specific: implications for cortical versus subcortical microglia. Acta Neuropathol 145, 733–747 (2023).
Guerreiro, R. et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127 (2013).
Silvin, A., Qian, J. & Ginhoux, F. Brain macrophage development, diversity and dysregulation in health and disease. Cell. Mol. Immunol. 20, 1277–1289 (2023).
De Schepper, S. et al. Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer’s disease. Nat. Neurosci. 26, 406–415 (2023).
Uekawa, K. et al. Border-associated macrophages promote cerebral amyloid angiopathy and cognitive impairment through vascular oxidative stress. Mol. Neurodegener. 18, 73 (2023).
Taylor, X. et al. Amyloid-β (Aβ) immunotherapy induced microhemorrhages are associated with activated perivascular macrophages and peripheral monocyte recruitment in Alzheimer’s disease mice. Mol. Neurodegener. 18, 59 (2023).
Li, J. et al. Melatonin ameliorates Parkinson’s disease via regulating microglia polarization in a RORα-dependent pathway. NPJ Park. Dis. 8, 90 (2022).
Tang, Y. & Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181–1194 (2016).
Yu, H. et al. Metabolic reprogramming and polarization of microglia in Parkinson’s disease: role of inflammasome and iron. Ageing Res. Rev. 90, 102032 (2023).
Meraz-Ríos, M. A., Lira-De León, K. I., Campos-Peña, V., De Anda-Hernández, M. A. & Mena-López, R. Tau oligomers and aggregation in Alzheimer’s disease. J. Neurochem. 112, 1353–1367 (2010).
Hardy, J. A. & Higgins, G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185 (1992).
Butchart, J. et al. Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 84, 2161–2168 (2015).
Silvin, A. et al. Dual ontogeny of disease-associated microglia and disease inflammatory macrophages in aging and neurodegeneration. Immunity 55, 1448–1465.e6 (2022).
Wong, M. Y. et al. 25-Hydroxycholesterol amplifies microglial IL-1β production in an apoE isoform-dependent manner. J. Neuroinflamm. 17, 192 (2020).
Sangineto, M. et al. Metabolic reprogramming in inflammatory microglia indicates a potential way of targeting inflammation in Alzheimer’s disease. Redox Biol 66, 102846 (2023).
Nugent, A. A. et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105, 837–854.e9 (2020).
Yan, P. et al. Peripheral monocyte-derived cells counter amyloid plaque pathogenesis in a mouse model of Alzheimer’s disease. J. Clin. Investig. 132, e152565 (2022).
Wilk, C. M. et al. Circulating senescent myeloid cells infiltrate the brain and cause neurodegeneration in histiocytic disorders. Immunity 56, 2790–2802.e6 (2023).
Xu, L. et al. Erythropoietin signaling in peripheral macrophages is required for systemic β-amyloid clearance. EMBO J 41, e111038 (2022).
Zuroff, L. R. et al. Effects of IL-34 on macrophage immunological profile in response to Alzheimer’s-Related Aβ42 assemblies. Front. Immunol. 11, 1449 (2020).
Zhang, Y. et al. Porphyromonas gingivalis msRNA P.G_45033 induces amyloid-β production by enhancing glycolysis and histone lactylation in macrophages. Int. Immunopharmacol. 121, 110468 (2023).
Xu, L. et al. Inhibition of Smad3 in macrophages promotes Aβ efflux from the brain and thereby ameliorates Alzheimer’s pathology. Brain. Behav. Immun. 95, 154–167 (2021).
Yin, H. et al. Loss of the m6A methyltransferase METTL3 in monocyte-derived macrophages ameliorates Alzheimer’s disease pathology in mice. PLoS Biol 21, e3002017 (2023).
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M. & Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl Acad. Sci. USA 95, 6469–6473 (1998).
Ouchi, Y. et al. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 57, 168–175 (2005).
Poewe, W. et al. Parkinson disease. Nat. Rev. Dis. Prim. 3, 17013 (2017).
Xu, E. et al. Pathological α-synuclein recruits LRRK2 expressing pro-inflammatory monocytes to the brain. Mol. Neurodegener. 17, 7 (2022).
Yadavalli, N. & Ferguson, S. M. LRRK2 suppresses lysosome degradative activity in macrophages and microglia through MiT-TFE transcription factor inhibition. Proc. Natl. Acad. Sci. USA 120, e2303789120 (2023).
Zhang, W. et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J 19, 533–542 (2005).
Bido, S. et al. Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 12, 6237 (2021).
Lu, J., Zhou, W., Dou, F., Wang, C. & Yu, Z. TRPV1 sustains microglial metabolic reprogramming in Alzheimer’s disease. EMBO Rep 22, e52013 (2021).
Lu, J. et al. A breakdown in microglial metabolic reprogramming causes internalization dysfunction of α-synuclein in a mouse model of Parkinson’s disease. J. Neuroinflamm. 19, 113 (2022).
Yan, M., Jin, H., Pan, C. & Han, X. Chronic Microcystin-LR-Induced α-Synuclein promotes neuroinflammation through activation of the NLRP3 inflammasome in microglia. Mol. Neurobiol. 60, 884–900 (2023).
Pike, A. F. et al. Synuclein evokes NLRP3 inflammasome-mediated IL-1β secretion from primary human microglia. Glia 69, 1413–1428 (2021).
Lv, Q.-K. et al. Role of α-synuclein in microglia: autophagy and phagocytosis balance neuroinflammation in Parkinson’s disease. Inflamm. Res. 72, 443–462 (2023).
Yu, H.-Y. et al. Exendin-4 and linagliptin attenuate neuroinflammation in a mouse model of Parkinson’s disease. Neural Regen. Res. 18, 1818–1826 (2023).
Grotemeyer, A. et al. Inflammasome inhibition protects dopaminergic neurons from α-synuclein pathology in a model of progressive Parkinson’s disease. J. Neuroinflamm. 20, 79 (2023).
Sun, R. & Jiang, H. Border-associated macrophages in the central nervous system. J. Neuroinflamm. 21, 67 (2024).
Conway, K. A., Harper, J. D. & Lansbury, P. T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320 (1998).
Guo, M. et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain J. Neurol. 143, 1476–1497 (2020).
Frosch, M., Amann, L. & Prinz, M. CNS-associated macrophages shape the inflammatory response in a mouse model of Parkinson’s disease. Nat. Commun. 14, 3753 (2023).
Schonhoff, A. M. et al. Border-associated macrophages mediate the neuroinflammatory response in an alpha-synuclein model of Parkinson disease. Nat. Commun. 14, 3754 (2023).
Park, H. et al. PAAN/MIF nuclease inhibition prevents neurodegeneration in Parkinson’s disease. Cell 185, 1943–1959.e21 (2022).
Koyama, Y. & Brenner, D. A. Liver inflammation and fibrosis. J. Clin. Investig. 127, 55–64 (2017).
Jais, A. & Brüning, J. C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Investig. 127, 24–32 (2017).
Lumeng, C. N. & Saltiel, A. R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 121, 2111–2117 (2011).
Schleh, M. W. et al. Metaflammation in obesity and its therapeutic targeting. Sci. Transl. Med. 15, eadf9382 (2023).
Harman-Boehm, I. et al. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J. Clin. Endocrinol. Metab. 92, 2240–2247 (2007).
Lumeng, C. N., Bodzin, J. L. & Saltiel, A. R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 117, 175–184 (2007).
Weisberg, S. P. et al. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 112, 1796–1808 (2003).
Boutens, L. & Stienstra, R. Adipose tissue macrophages: going off track during obesity. Diabetologia 59, 879–894 (2016).
Coats, B. R. et al. Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet-induced obesity. Cell Rep 20, 3149–3161 (2017).
Kratz, M. et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab 20, 614–625 (2014).
Flaherty, S. E. et al. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science 363, 989–993 (2019).
Da, J. et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178, 686–696 (2019).
Hill, D. A. et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc. Natl. Acad. Sci. USA 115, E5096–E5105 (2018).
Ying, W., Fu, W., Sok Lee, Y. & Olefsky, J. M. Role of macrophages in obesity-associated islet inflammation and beta cell abnormalities. Nat. Rev. Endocrinol. 16, 81–90 (2020).
Caratti, G. et al. Glucocorticoid activation of anti-inflammatory macrophages protects against insulin resistance. Nat. Commun. 14, 2271 (2023).
Lu, X. et al. UBE2M-mediated neddylation of TRIM21 regulates obesity-induced inflammation and metabolic disorders. Cell Metab 35, 1390–1405.e8 (2023).
Castoldi, A., Naffah de Souza, C., Câmara, N. O. S. & Moraes-Vieira, P. M. The macrophage switch in obesity development. Front. Immunol. 6, 637 (2015).
de Oliveira, M. C. et al. Eosinophils protect from metabolic alterations triggered by obesity. Metabolism 146, 155613 (2023).
Luo, W. et al. Downhill running and caloric restriction attenuate insulin resistance associated skeletal muscle atrophy via the promotion of M2-like macrophages through TRIB3-AKT pathway. Free Radic. Biol. Med. 210, 271–285 (2024).
Yan, J. et al. GPSM1 impairs metabolic homeostasis by controlling a pro-inflammatory pathway in macrophages. Nat. Commun. 13, 7260 (2022).
Zafar A., Ng, H.P., Chan, E.R., Dunwoodie, S.L. & Mahabeleshwar, G.H. Myeloid-CITED2 deficiency exacerbates diet-induced obesity and pro-inflammatory macrophage response. Cells 12, (2023).
Chawla, A., Nguyen, K. D. & Goh, Y. P. S. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 11, 738–749 (2011).
Byles, V. et al. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 4, 2834 (2013).
Jiang, H., Westerterp, M., Wang, C., Zhu, Y. & Ai, D. Macrophage mTORC1 disruption reduces inflammation and insulin resistance in obese mice. Diabetologia 57, 2393–2404 (2014).
Reinisch, I. et al. Adipocyte p53 coordinates the response to intermittent fasting by regulating adipose tissue immune cell landscape. Nat. Commun. 15, 1391 (2024).
Siouti, E. et al. Notch signaling in adipose tissue macrophages prevents diet-induced inflammation and metabolic dysregulation. Eur. J. Immunol. 54, e2350669 (2024).
Yu, L. et al. IgG is an aging factor that drives adipose tissue fibrosis and metabolic decline. Cell Metab 36, 793–807.e5 (2024).
Pellegrinelli, V. et al. Dysregulation of macrophage PEPD in obesity determines adipose tissue fibro-inflammation and insulin resistance. Nat. Metab. 4, 476–494 (2022).
Oh, J. et al. Embryonic vitamin D deficiency programs hematopoietic stem cells to induce type 2 diabetes. Nat. Commun. 14, 3278 (2023).
Sheka, A. C. et al. Nonalcoholic steatohepatitis: a review. JAMA 323, 1175–1183 (2020).
Huby, T. & Gautier, E. L. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat. Rev. Immunol. 22, 429–443 (2022).
Wong, R. J. et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 148, 547–555 (2015).
Kazankov, K. et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 16, 145–159 (2019).
Remmerie, A. et al. Osteopontin expression identifies a subset of recruited macrophages distinct from kupffer cells in the fatty liver. Immunity 53, 641–657.e14 (2020).
Guillot, A. & Tacke, F. Liver macrophages: old dogmas and new insights. Hepatol. Commun. 3, 730–743 (2019).
Vonderlin, J., Chavakis, T., Sieweke, M. & Tacke, F. The multifaceted roles of macrophages in NAFLD pathogenesis. Cell. Mol. Gastroenterol. Hepatol. 15, 1311–1324 (2023).
Rada, P., González-Rodríguez, Á., García-Monzón, C. & Valverde, Á. M. Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver? Cell Death Dis 11, 802 (2020).
Horn, C. L., Morales, A. L., Savard, C., Farrell, G. C. & Ioannou, G. N. Role of cholesterol-associated steatohepatitis in the development of NASH. Hepatol. Commun. 6, 12–35 (2022).
Tosello-Trampont, A.-C., Landes, S. G., Nguyen, V., Novobrantseva, T. I. & Hahn, Y. S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 287, 40161–40172 (2012).
Park, J. et al. IL-6/STAT3 axis dictates the PNPLA3-mediated susceptibility to non-alcoholic fatty liver disease. J. Hepatol. 78, 45–56 (2023).
Tran, S. et al. Impaired Kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity 53, 627–640.e5 (2020).
Zigmond, E. & Varol, C. Two roads diverge in the sick liver, monocytes travel both. Immunity 53, 479–481 (2020).
Daemen, S. et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in NASH. Cell Rep 34, 108626 (2021).
Miura, K., Yang, L., van Rooijen, N., Ohnishi, H. & Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G1310–G1321 (2012).
Qing, J. et al. Dopamine receptor D2 antagonism normalizes profibrotic macrophage-endothelial crosstalk in non-alcoholic steatohepatitis. J. Hepatol. 76, 394–406 (2022).
Mooring, M. et al. Hepatocyte stress increases expression of yes-associated protein and transcriptional coactivator With PDZ-binding motif in hepatocytes to promote parenchymal inflammation and fibrosis. Hepatology 71, 1813–1830 (2020).
Zhou, L. et al. Hepatic danger signaling triggers TREM2+ macrophage induction and drives steatohepatitis via MS4A7-dependent inflammasome activation. Sci. Transl. Med. 16, eadk1866 (2024).
Zhang, L. et al. Hepatic Zbtb18 (Zinc Finger and BTB Domain Containing 18) alleviates hepatic steatohepatitis via FXR (Farnesoid X Receptor). Signal Transduct. Target. Ther. 9, 20 (2024).
Wang, Q. et al. Role of XBP1 in regulating the progression of non-alcoholic steatohepatitis. J. Hepatol. 77, 312–325 (2022).
Wang, X. et al. Prolonged hypernutrition impairs TREM2-dependent efferocytosis to license chronic liver inflammation and NASH development. Immunity 56, 58–77.e11 (2023).
Sanyal, A. J. et al. Prospective study of outcomes in adults with nonalcoholic fatty liver disease. N. Engl. J. Med. 385, 1559–1569 (2021).
Ramachandran, P. et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518 (2019).
Pradere, J.-P. et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58, 1461–1473 (2013).
Matsuda, M. & Seki, E. Hepatic stellate cell-macrophage crosstalk in liver fibrosis and carcinogenesis. Semin. Liver Dis. 40, 307–320 (2020).
Mooring, M. et al. Hepatocyte CYR61 polarizes profibrotic macrophages to orchestrate NASH fibrosis. Sci. Transl. Med. 15, eade3157 (2023).
Wang, Q. et al. XBP1-mediated activation of the STING signalling pathway in macrophages contributes to liver fibrosis progression. JHEP Rep. Innov. Hepatol. 4, 100555 (2022).
Wang, Y.-F. et al. METTL14 downregulation drives S100A4+ monocyte-derived macrophages via MyD88/NF-κB pathway to promote MAFLD progression. Signal Transduct. Target. Ther. 9, 91 (2024).
Pastore, M. et al. Macrophage MerTK promotes profibrogenic cross-talk with hepatic stellate cells via soluble mediators. JHEP Rep 4, 100444 (2022).
Mabire, M. et al. MAIT cell inhibition promotes liver fibrosis regression via macrophage phenotype reprogramming. Nat. Commun. 14, 1830 (2023).
Ponziani, F. R. et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 69, 107–120 (2019).
Zhang, Q. et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell 179, 829–845 (2019).
Zhao, Y.-N. et al. Macrophage-specific FGFR1 deletion alleviates high-fat-diet-induced liver inflammation by inhibiting the MAPKs/TNF pathways. Acta Pharmacol. Sin. 45, 988–1001 (2024).
Zheng, Q. et al. Lack of FGF21 promotes NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling. Theranostics 10, 9923–9936 (2020).
Liu, C. et al. FGF21 protects against hepatic lipotoxicity and macrophage activation to attenuate fibrogenesis in nonalcoholic steatohepatitis. eLife 12, e83075 (2023).
Zhang, P. et al. Neuregulin 4 suppresses NASH-HCC development by restraining tumor-prone liver microenvironment. Cell Metab 34, 1359–1376.e7 (2022).
Zhang, Q., Wang, J., Huang, F., Yao, Y. & Xu, L. Leptin induces NAFLD progression through infiltrated CD8+ T lymphocytes mediating pyroptotic-like cell death of hepatocytes and macrophages. Dig. Liver Dis. 53, 598–605 (2021).
Wang, Z. et al. Extracellular vesicles in fatty liver promote a metastatic tumor microenvironment. Cell Metab 35, 1209–1226.e13 (2023).
Miki, S. et al. S-1-Propenylcysteine promotes IL-10-induced M2c macrophage polarization through prolonged activation of IL-10R/STAT3 signaling. Sci. Rep. 11, 22469 (2021).
Xu, X. et al. The role of macrophages in the formation of hypertrophic scars and keloids. Burns Trauma 8, tkaa006 (2020).
Sok, M. C. P. et al. Dual delivery of IL-10 and AT-RvD1 from PEG hydrogels polarize immune cells towards pro-regenerative phenotypes. Biomaterials 268, 120475 (2021).
Chi, Z. et al. Gasdermin D-mediated metabolic crosstalk promotes tissue repair. Nature https://doi.org/10.1038/s41586-024-08022-7 (2024).
Zrzavy, T. et al. Acute and non-resolving inflammation associate with oxidative injury after human spinal cord injury. Brain J. Neurol. 144, 144–161 (2021).
St-Pierre, M.-K., González, Ibáñez, F., Kroner, A. & Tremblay, M.-È. Microglia/macrophages are ultrastructurally altered by their proximity to spinal cord injury in adult female mice. J. Neuroinflamm. 20, 273 (2023).
Niehaus, J. K., Taylor-Blake, B., Loo, L., Simon, J. M. & Zylka, M. J. Spinal macrophages resolve nociceptive hypersensitivity after peripheral injury. Neuron 109, 1274–1282 (2021).
Zhou, J. et al. LncGBP9/miR-34a axis drives macrophages toward a phenotype conducive for spinal cord injury repair via STAT1/STAT6 and SOCS3. J. Neuroinflamm. 17, 134 (2020).
Xu, G.-Y. et al. Cell-free extracts from human fat tissue with a hyaluronan-based hydrogel attenuate inflammation in a spinal cord injury model through M2 Microglia/Microphage polarization. Small 18, e2107838 (2022).
Yang, W. et al. An engineered bionic nanoparticle sponge as a cytokine trap and reactive oxygen species scavenger to relieve disc degeneration and discogenic pain. ACS Nano 18, 3053–3072 (2024).
Zhao, C. et al. DNA methyltransferase 1 deficiency improves macrophage motility and wound healing by ameliorating cholesterol accumulation. NPJ Regen. Med. 8, 29 (2023).
Yu, Z. et al. WISP1 and TLR4 on Macrophages Contribute to Ventilator-Induced Lung Injury. Inflammation 43, 425–432 (2020).
Su, K. et al. TLR4 is required for macrophage efferocytosis during resolution of ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 321, L787–L801 (2021).
Zheng, X. et al. Angiotensin II Type 2 receptor inhibits M1 polarization and apoptosis of alveolar macrophage and protects against mechanical ventilation-induced lung injury. Inflammation https://doi.org/10.1007/s10753-024-02037-y (2024).
Zhang, S., Zhu, L., Dai, H. & Pan, L. Silencing ROCK1 ameliorates ventilator-induced lung injury in mice by inhibiting macrophages’ NLRP3 signaling. Int. Immunopharmacol. 101, 108208 (2021).
Luo, Q., Luo, J. & Wang, Y. YAP deficiency attenuates pulmonary injury following mechanical ventilation through the regulation of M1/M2 macrophage polarization. J. Inflamm. Res. 13, 1279–1290 (2020).
Yang, W. et al. Mechanical stimulation of anti-inflammatory and antioxidant hydrogels for rapid re-epithelialization. Adv. Mater. 36, e2312740 (2024).
Zhang, X. et al. Piezo1-mediated mechanosensation in bone marrow macrophages promotes vascular niche regeneration after irradiation injury. Theranostics 12, 1621–1638 (2022).
Martin, P. & Nunan, R. Cellular and molecular mechanisms of repair in acute and chronic wound healing. Br. J. Dermatol. 173, 370–378 (2015).
Tottoli, E. M. et al. Skin wound healing process and new emerging technologies for skin wound care and regeneration. Pharmaceutics 12, 735 (2020).
Han, G. & Ceilley, R. Chronic wound healing: a review of current management and treatments. Adv. Ther. 34, 599–610 (2017).
Sawaya, A. P. et al. Deregulated immune cell recruitment orchestrated by FOXM1 impairs human diabetic wound healing. Nat. Commun. 11, 4678 (2020).
Krzyszczyk, P., Schloss, R., Palmer, A. & Berthiaume, F. The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front. Physiol. 9, 419 (2018).
Gu, X. et al. Effect of activated autologous monocytes/macrophages on wound healing in a rodent model of experimental diabetes. Diabetes Res. Clin. Pract. 102, 53–59 (2013).
Wolf, S. J. et al. Diabetic wound keratinocytes induce macrophage JMJD3-mediated Nlrp3 expression via IL-1R signaling. Diabetes https://doi.org/10.2337/db23-0968 (2024).
Villarreal-Ponce, A. et al. Keratinocyte-macrophage crosstalk by the Nrf2/Ccl2/EGF signaling axis orchestrates tissue repair. Cell Rep 33, 108417 (2020).
Zhu, Z. et al. Enhanced ·OH-Scavenging Activity of Cu-CeOx nanozyme via resurrecting macrophage nrf2 transcriptional activity facilitates diabetic wound healing. Adv. Healthcare Mater. 13, e2303229 (2024).
Zhang, Z. et al. San Huang Xiao Yan recipe modulates the HMGB1-mediated abnormal inflammatory microenvironment and ameliorates diabetic foot by activating the AMPK/Nrf2 signalling pathway. Phytomed. Int. J. Phytother. Phytopharm. 118, 154931 (2023).
Zhang, X. et al. CD64 plays a key role in diabetic wound healing. Front. Immunol. 15, 1322256 (2024).
Yu, P. et al. lncRNA-H19 in fibroblasts promotes wound healing in. Diabetes. Diabetes 71, 1562–1578 (2022).
Xia, W. et al. Lean adipose tissue macrophage derived exosome confers immunoregulation to improve wound healing in. diabetes. J. Nanobiotechnol. 21, 128 (2023).
Zhang, G. et al. Soft apoptotic-cell-inspired nanoparticles persistently bind to macrophage membranes and promote anti-inflammatory and pro-healing effects. Acta Biomater 131, 452–463 (2021).
Zhao, H. et al. ROS-scavenging hydrogel to promote healing of bacteria infected diabetic wounds. Biomaterials 258, 120286 (2020).
Xiong, Y. et al. A whole-course-repair system based on neurogenesis-angiogenesis crosstalk and macrophage reprogramming promotes diabetic wound healing. Adv. Mater. 35, e2212300 (2023).
Mu, R. et al. Tumor-associated macrophages-educated reparative macrophages promote diabetic wound healing. EMBO Mol. Med. 15, e16671 (2023).
De Palma, M. et al. Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell 14, 299–311 (2008).
Kaczanowska, S. et al. Genetically engineered myeloid cells rebalance the core immune suppression program in metastasis. Cell 184, 2033–2052 (2021).
Curtale, G., Rubino, M. & Locati, M. MicroRNAs as molecular switches in macrophage activation. Front. Immunol. 10, 799 (2019).
Locati, M., Curtale, G. & Mantovani, A. Diversity, mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 15, 123–147 (2020).
Ries, C. H. et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25, 846–859 (2014).
Strachan, D. C. et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8+ T cells. Oncoimmunology 2, e26968 (2013).
Sluijter, M. et al. Inhibition of CSF-1R supports T-cell mediated melanoma therapy. PloS ONE 9, e104230 (2014).
Smeester, B. A. et al. PLX3397 treatment inhibits constitutive CSF1R-induced oncogenic ERK signaling, reduces tumor growth, and metastatic burden in osteosarcoma. Bone 136, 115353 (2020).
Molena, B. et al. Synovial colony-stimulating factor-1 mRNA expression in diffuse pigmented villonodular synovitis. Clin. Exp. Rheumatol. 29, 547–550 (2011).
Tap, W. D. et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N. Engl. J. Med. 373, 428–437 (2015).
Butowski, N. et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy foundation early phase clinical trials consortium phase II study. Neuro-Oncol 18, 557–564 (2016).
von Tresckow, B. et al. An open-label, multicenter, Phase I/II Study of JNJ-40346527, a CSF-1R inhibitor, in patients with relapsed or refractory hodgkin lymphoma. Clin. Cancer Res. 21, 1843–1850 (2015).
Pyonteck, S. M. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–1272 (2013).
Cassier, P. A. et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol 16, 949–956 (2015).
van Elsas, M. J. et al. Invasive margin tissue-resident macrophages of high CD163 expression impede responses to T cell-based immunotherapy. J. Immunother. Cancer 11, e006433 (2023).
Piaggio, F. et al. A novel liposomal Clodronate depletes tumor-associated macrophages in primary and metastatic melanoma: Anti-angiogenic and anti-tumor effects. J. Control. Release. 223, 165–177 (2016).
Hiraoka, K. et al. Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci 99, 1595–1602 (2008).
Tacconi, C. et al. CD169+ lymph node macrophages have protective functions in mouse breast cancer metastasis. Cell Rep 35, 108993 (2021).
Gazzaniga, S. et al. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J. Investig. Dermatol. 127, 2031–2041 (2007).
Zhang, W. et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin. Cancer Res. 16, 3420–3430 (2010).
Zhou, Y., Chen, Y., Ma, C., Shao, B. & Zhang, F. Liposomal clodronate combined with Cisplatin or Sorafenib inhibits hepatocellular carcinoma cell proliferation, migration and invasion by suppressing FOXQ1 expression. Cell. Mol. Biol. 66, 49–54 (2020).
McNeel, D. G. et al. Phase 2 trial of a DNA vaccine (pTVG-HP) and nivolumab in patients with castration-sensitive non-metastatic (M0) prostate cancer. J. Immunother. Cancer 11, e008067 (2023).
Haakensen, V. D. et al. NIPU: a randomised, open-label, phase II study evaluating nivolumab and ipilimumab combined with UV1 vaccination as second line treatment in patients with malignant mesothelioma. J. Transl. Med. 19, 232 (2021).
Hm, L., Ce, R., Rp, G. & Gh, L. Sargramostim (rhu GM-CSF) as Cancer Therapy (Systematic Review) and An Immunomodulator. A Drug Before Its Time? Front. Immunol. 12, 706186 (2021).
Pei, F., Gu, B., Miao, S.-M., Guan, X.-D. & Wu, J.-F. Clinical practice of sepsis-induced immunosuppression: Current immunotherapy and future options. Chin. J. Traumatol. 27, 63–70 (2024).
H, O. et al. Molgramostim (GM-CSF) associated with antibiotic treatment in nontraumatic abdominal sepsis: a randomized, double-blind, placebo-controlled clinical trial. Arch. Surg. 141, 150–153 (2006).
C, M. et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am. J. Respir. Crit. Care Med. 180, 640–648 (2009).
Em, P. et al. Randomised controlled trial of GM-CSF in critically ill patients with impaired neutrophil phagocytosis. Thorax 73, 918–925 (2018).
Kyriakopoulos, C. E. et al. Multicenter Phase I Trial of a DNA vaccine encoding the androgen receptor ligand-binding domain (pTVG-AR, MVI-118) in patients with metastatic prostate cancer. Clin. Cancer Res. 26, 5162–5171 (2020).
Weinblatt, M. E. et al. A randomized phase IIb Study of Mavrilimumab and Golimumab in Rheumatoid Arthritis. Arthritis Rheumatol. 70, 49–59 (2018).
Burmester, G. R. et al. Mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor α monoclonal antibody: long-term safety and efficacy in patients with rheumatoid arthritis. Arthritis Rheumatol. 70, 679–689 (2018).
Cremer, P. C. et al. Mavrilimumab in patients with severe COVID-19 pneumonia and systemic hyperinflammation (MASH-COVID): an investigator initiated, multicentre, double-blind, randomised, placebo-controlled trial. Lancet Rheumatol. 3, e410–e418 (2021).
Papp, K. A. et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) as a therapeutic target in psoriasis: randomized, controlled investigation using namilumab, a specific human anti-GM-CSF monoclonal antibody. Br. J. Dermatol. 180, 1352–1360 (2019).
Criner, G. J. et al. Anti-granulocyte-macrophage colony-stimulating factor monoclonal antibody Gimsilumab for COVID-19 pneumonia: a randomized, double-blind, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 205, 1290–1299 (2022).
Worth, C. et al. Granulocyte-macrophage colony-stimulating factor neutralisation in patients with axial spondyloarthritis in the UK (NAMASTE): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Rheumatol. 6, e537–e545 (2024).
Fei, L., Ren, X., Yu, H. & Zhan, Y. Targeting the CCL2/CCR2 Axis in cancer immunotherapy: one stone, three birds? Front. Immunol 12, 771210 (2021).
Loberg, R. D. et al. CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia 9, 556–562 (2007).
Chen, X. et al. PIM1/NF-κB/CCL2 blockade enhances anti-PD-1 therapy response by modulating macrophage infiltration and polarization in tumor microenvironment of NSCLC. Oncogene https://doi.org/10.1038/s41388-024-03100-6 (2024).
De la Fuente López, M. et al. The relationship between chemokines CCL2, CCL3, and CCL4 with the tumor microenvironment and tumor-associated macrophage markers in colorectal cancer. Tumour Biol 40, 1010428318810059 (2018).
Zheng, Y., Wang, Z., Wei, S., Liu, Z. & Chen, G. Epigenetic silencing of chemokine CCL2 represses macrophage infiltration to potentiate tumor development in small cell lung cancer. Cancer Lett 499, 148–163 (2021).
Steinberger, K. J. et al. Stress-induced norepinephrine downregulates CCL2 in macrophages to suppress tumor growth in a model of malignant melanoma. Cancer Prev. Res. 13, 747–760 (2020).
Hitchcock, J. R. & Watson, C. J. Anti-CCL2: building a reservoir or opening the floodgates to metastasis? Breast Cancer Res 17, 68 (2015).
Bonapace, L. et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 515, 130–133 (2014).
Pienta, K. J. et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. N. Drugs 31, 760–768 (2013).
Tu, M. M. et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun. Biol. 3, 720 (2020).
Colombo, A. et al. A double-blind randomised study to evaluate the efficacy and safety of bindarit in preventing coronary stent restenosis. EuroIntervention 12, e1385–e1394 (2016).
Moisan, F. et al. Enhancement of paclitaxel and carboplatin therapies by CCL2 blockade in ovarian cancers. Mol. Oncol. 8, 1231–1239 (2014).
Loberg, R. D. et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res 67, 9417–9424 (2007).
Nywening, T. M. et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol 17, 651–662 (2016).
Noel, M. et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Investig. N. Drugs 38, 800–811 (2020).
Chen, J. et al. SLAMF7 is critical for phagocytosis of haematopoietic tumour cells via Mac-1 integrin. Nature 544, 493–497 (2017).
Edris, B. et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. USA 109, 6656–6661 (2012).
Xiao, Z. et al. Antibody mediated therapy targeting CD47 inhibits tumor progression of hepatocellular carcinoma. Cancer Lett 360, 302–309 (2015).
Daver, N. G. et al. Tolerability and efficacy of the anticluster of differentiation 47 antibody magrolimab combined with azacitidine in patients with previously untreated AML: phase Ib results. J. Clin. Oncol. 41, 4893–4904 (2023).
Ansell, S. M. et al. Phase I study of the CD47 Blocker TTI-621 in Patients with Relapsed or Refractory Hematologic Malignancies. Clin. Cancer Res. 27, 2190–2199 (2021).
He, Z. et al. Oxygen-boosted biomimetic nanoplatform for synergetic phototherapy/ferroptosis activation and reversal of immune-suppressed tumor microenvironment. Biomaterials 290, 121832 (2022).
Tang, L. et al. Extracellular vesicles-derived hybrid nanoplatforms for amplified CD47 blockade-based cancer immunotherapy. Adv. Mater. 35, e2303835 (2023).
Rao, L. et al. Activating macrophage-mediated cancer immunotherapy by genetically edited nanoparticles. Adv. Mater. 32, e2004853 (2020).
Logtenberg, M. E. W. et al. Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPα axis and a target for cancer immunotherapy. Nat. Med. 25, 612–619 (2019).
Wu, Z. et al. Identification of Glutaminyl Cyclase isoenzyme isoQC as a regulator of SIRPα-CD47 axis. Cell Res 29, 502–505 (2019).
Weiss, S. et al. 389 Phase II of CD40 agonistic antibody sotigalimab (APX005M) in combination with nivolumab in subjects with metastatic melanoma with confirmed disease progression on anti-PD-1 therapy. J. Immunother. Cancer 9, A422 (2021).
Ko, A. H. et al. 1229P A multicenter phase II study of sotigalimab (CD40 agonist) in combination with neoadjuvant chemoradiation for resectable esophageal and gastroesophageal junction (GEJ) cancers. Ann. Oncol. 33, S1111 (2022).
Barlesi, F. et al. 291 Phase Ib study of selicrelumab (CD40 agonist) in combination with atezolizumab (anti-PD-L1) in patients with advanced solid tumors. J. Immunother. Cancer 8, A318 (2020).
Fidler, I. J. et al. Involvement of macrophages in the eradication of established metastases following intravenous injection of liposomes containing macrophage activators. Cancer Res 42, 496–501 (1982).
Fidler, I. J. Inhibition of pulmonary metastasis by intravenous injection of specifically activated macrophages. Cancer Res 34, 1074–1078 (1974).
Fidler, I. J. & Poste, G. Macrophage-mediated destruction of malignant tumor cells and new strategies for the therapy of metastatic disease. Springe. Semin. Immunopathol. 5, 161–174 (1982).
Henry, T. D. et al. Safety and efficacy of ixmyelocel-T: an expanded, autologous multi-cellular therapy, in dilated cardiomyopathy. Circ. Res. 115, 730–737 (2014).
Patel, A. N. et al. Ixmyelocel-T for patients with ischaemic heart failure: a prospective randomised double-blind trial. Lancet Lond. Engl. 387, 2412–2421 (2016).
Chen, Y. et al. The application of HER2 and CD47 CAR-macrophage in ovarian cancer. J. Transl. Med. 21, 654 (2023).
Shah, Z. et al. Human anti-PSCA CAR macrophages possess potent antitumor activity against pancreatic cancer. Cell Stem Cell 31, 803–817.e6 (2024).
Abdin, S. M. et al. Scalable generation of functional human iPSC-derived CAR-macrophages that efficiently eradicate CD19-positive leukemia. J. Immunother. Cancer 11, e007705 (2023).
Jin, G., Chang, Y. & Bao, X. Generation of chimeric antigen receptor macrophages from human pluripotent stem cells to target glioblastoma. Immuno-Oncol. Technol. 20, 100409 (2023).
Patra, J. K. et al. Nano based drug delivery systems: recent developments and future prospects. J. Nanobiotechnol. 16, 71 (2018).
Shi, Y. & Lammers, T. Combining nanomedicine and immunotherapy. Acc. Chem. Res. 52, 1543–1554 (2019).
Bobo, D., Robinson, K. J., Islam, J., Thurecht, K. J. & Corrie, S. R. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm. Res. 33, 2373–2387 (2016).
Waheed, S. et al. Engineering nano-drug biointerface to overcome biological barriers toward precision drug delivery. J. Nanobiotechnol. 20, 395 (2022).
Zhang, T.-T., Li, W., Meng, G., Wang, P. & Liao, W. Strategies for transporting nanoparticles across the blood-brain barrier. Biomater. Sci. 4, 219–229 (2016).
Hu, C.-M. J. & Zhang, L. Nanoparticle-based combination therapy toward overcoming drug resistance in cancer. Biochem. Pharmacol. 83, 1104–1111 (2012).
Plana, D., Palmer, A. C. & Sorger, P. K. Independent drug action in combination therapy: implications for precision oncology. Cancer Discov 12, 606–624 (2022).
Wei, Q. et al. The promise and challenges of combination therapies with antibody-drug conjugates in solid tumors. J. Hematol. Oncol. 17, 1 (2024).
Xia, T. et al. Advances in the study of macrophage polarization in inflammatory immune skin diseases. J. Inflamm. Lond. Engl. 20, 33 (2023).
Wang, X. et al. Macrophage-related therapeutic strategies: regulation of phenotypic switching and construction of drug delivery systems. Pharmacol. Res. 199, 107022 (2024).
Wu, Y. et al. Macrophage cell membrane-based nanoparticles: a new promising biomimetic platform for targeted delivery and treatment. J. Nanobiotechnol 20, 542 (2022).
Meng, Q.-F. et al. Inhalation delivery of dexamethasone with iSEND nanoparticles attenuates the COVID-19 cytokine storm in mice and nonhuman primates. Sci. Adv. 9, eadg3277 (2023).
Meng, Q.-F. et al. Genetically programmable fusion cellular vesicles for cancer immunotherapy. Angew. Chem. Int. Ed. Engl. 60, 26320–26326 (2021).
Huang, L., Xu, X. & Hao, Y. The possible mechanisms of tumor progression via CSF-1/CSF-1R pathway activation. Rom. J. Morphol. Embryol. 55, 501–506 (2014).
Hu, Y. et al. Tumor-microenvironment-activatable nanoparticle mediating immunogene therapy and M2 macrophage-targeted inhibitor for synergistic cancer immunotherapy. ACS Nano 18, 3295–3312 (2024).
Sun, Y., Cronin, M. F., Mendonça, M. C. P., Guo, J. & O’Driscoll, C. M. Sialic acid-targeted cyclodextrin-based nanoparticles deliver CSF-1R siRNA and reprogram tumour-associated macrophages for immunotherapy of prostate cancer. Eur. J. Pharm. Sci. 185, 106427 (2023).
Gu, C. et al. Engineered macrophage membrane-coated nanoparticles with enhanced CCR2 expression promote spinal cord injury repair by suppressing neuroinflammation and neuronal death. Small 20, e2305659 (2024).
Zhu, Y. et al. Two-dimensional Mg2 Si nanosheet-enabled sustained hydrogen generation for improved repair and regeneration of deeply burned skin. Adv. Healthcare Mater. 12, e2201705 (2023).
Zhang, F. et al. Dominolike’ barriers elimination with an intratumoral adenosine-triphosphate-supersensitive nanogel to enhance cancer chemoimmunotherapy. ACS Nano 17, 18805–18817 (2023).
Shi, J. et al. Engineering CXCL12 biomimetic decoy-integrated versatile immunosuppressive nanoparticle for ischemic stroke therapy with management of overactivated brain immune microenvironment. Small Methods 6, e2101158 (2022).
Kühne, M. et al. Biocompatible valproic acid-coupled nanoparticles attenuate lipopolysaccharide-induced inflammation. Int. J. Pharm. 601, 120567 (2021).
Liu, L., Wang, Y., Guo, X., Zhao, J. & Zhou, S. A biomimetic polymer magnetic nanocarrier polarizing tumor-associated macrophages for potentiating immunotherapy. Small 16, e2003543 (2020).
Rodell, C. B. et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2, 578–588 (2018).
Li, H., Somiya, M. & Kuroda, S. Enhancing antibody-dependent cellular phagocytosis by Re-education of tumor-associated macrophages with resiquimod-encapsulated liposomes. Biomaterials 268, 120601 (2021).
Jiang, S. et al. Cathepsin B-Responsive programmed brain targeted delivery system for chemo-immunotherapy combination therapy of glioblastoma. ACS Nano 18, 6445–6462 (2024).
Zhang, M. et al. Anti-CD47 treatment stimulates phagocytosis of glioblastoma by M1 and M2 Polarized macrophages and promotes M1 polarized macrophages in vivo. PloS ONE 11, e0153550 (2016).
Zhou, Y. et al. Co-delivery of phagocytosis checkpoint and STING agonist by a Trojan horse nanocapsule for orthotopic glioma immunotherapy. Theranostics 12, 5488–5503 (2022).
Guo, Y., Bao, Q., Hu, P. & Shi, J. Nanomedicine-based co-delivery of a calcium channel inhibitor and a small molecule targeting CD47 for lung cancer immunotherapy. Nat. Commun. 14, 7306 (2023).
Gong, M. et al. A nanodrug combining CD47 and sonodynamic therapy efficiently inhibits osteosarcoma deterioration. J. Control. Release 355, 68–84 (2023).
Gao, L. et al. Convection-enhanced delivery of nanoencapsulated gene locoregionally yielding ErbB2/Her2-specific CAR-macrophages for brainstem glioma immunotherapy. J. Nanobiotechnol. 21, 56 (2023).
Jaynes, J. M. et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl. Med. 12, eaax6337 (2020).
Wang, J. et al. Nanoparticle delivery of CD40 siRNA suppresses alloimmune responses by inhibiting activation and differentiation of DCs and macrophages. Sci. Adv. 8, eabq3699 (2022).
Ding, D. et al. Multifunctional nanodrug mediates synergistic photodynamic therapy and MDSCs-targeting immunotherapy of colon cancer. Adv. Sci. 8, e2100712 (2021).
Song, Y. et al. Albumin nanoparticle containing a PI3Kγ inhibitor and paclitaxel in combination with α-PD1 induces tumor remission of breast cancer in mice. Sci. Transl. Med. 14, eabl3649 (2022).
Chen, Y. et al. Nanodrugs mediate TAMs-related arginine metabolism interference to boost photodynamic immunotherapy. J. Control. Release 367, 248–264 (2024).
Gao, Q. et al. In situ mannosylated nanotrinity-mediated macrophage remodeling combats Candida albicans infection. ACS Nano 14, 3980–3990 (2020).
Tian, M. et al. Adipose-derived biogenic nanoparticles for suppression of inflammation. Small 16, e1904064 (2020).
Shi, L. et al. Transforming a toxic drug into an efficacious nanomedicine using a lipoprodrug strategy for the treatment of patient-derived melanoma xenografts. J. Control. Release 324, 289–302 (2020).
Gan, C. et al. Niclosamide-loaded nanoparticles (Ncl-NPs) reverse pulmonary fibrosis in vivo and in vitro. J. Adv. Res. 51, 109–120 (2023).
Turk, B. R. et al. Dendrimer-N-acetyl-L-cysteine modulates monophagocytic response in adrenoleukodystrophy. Ann. Neurol. 84, 452–462 (2018).
Mishra, M. K. et al. Dendrimer brain uptake and targeted therapy for brain injury in a large animal model of hypothermic circulatory arrest. ACS Nano 8, 2134–2147 (2014).
Nance, E. et al. Dendrimer-mediated delivery of N-acetyl cysteine to microglia in a mouse model of Rett syndrome. J. Neuroinflamm. 14, 252 (2017).
Kannan, S. et al. Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci. Transl. Med. 4, 130ra46 (2012).
Niño, D. F. et al. Cognitive impairments induced by necrotizing enterocolitis can be prevented by inhibiting microglial activation in mouse brain. Sci. Transl. Med. 10, eaan0237 (2018).
Gusdon, A. M. et al. Dendrimer nanotherapy for severe COVID-19 attenuates inflammation and neurological injury markers and improves outcomes in a phase2a clinical trial. Sci. Transl. Med. 14, eabo2652 (2022).
Xue, J. et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288 (2014).
Mosser, D. M. & Edwards, J. P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008).
Dolgin, E. The tangled history of mRNA vaccines. Nature 597, 318–324 (2021).
Makkouk, A. & Weiner, G. J. Cancer immunotherapy and breaking immune tolerance: new approaches to an old challenge. Cancer Res 75, 5–10 (2015).
Oberdörster, G., Oberdörster, E. & Oberdörster, J. Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ. Health Perspect. 113, 823–839 (2005).
Couvreur, P. Nanoparticles in drug delivery: past, present and future. Adv. Drug Deliv. Rev. 65, 21–23 (2013).
Andreu, V. & Arruebo, M. Current progress and challenges of nanoparticle-based therapeutics in pain management. J. Control. Release 269, 189–213 (2018).
Roszer, T. Understanding the Mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm 2015, 816460 (2015).
Xu, T., Yu, S., Zhang, J. & Wu, S. Dysregulated tumor-associated macrophages in carcinogenesis, progression and targeted therapy of gynecological and breast cancers. J. Hematol. Oncol. 14, 181 (2021).
Krishna, C. et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 39, 662–677.e6 (2021).
Hao, B. et al. Single-cell RNA sequencing analysis revealed cellular and molecular immune profiles in lung squamous cell carcinoma. Transl. Oncol. 27, 101568 (2023).
Zhou, J. et al. Exosomes released from tumor-associated macrophages transfer miRNAs that induce a Treg/Th17 cell imbalance in epithelial ovarian cancer. Cancer Immunol. Res. 6, 1578–1592 (2018).
Song, G. et al. Single-cell transcriptomic analysis suggests two molecularly subtypes of intrahepatic cholangiocarcinoma. Nat. Commun. 13, 1642 (2022).
Korbecki, J., Olbromski, M. & Dziegiel, P. CCL18 in the progression of cancer. Int. J. Mol. Sci. 21, 7955 (2020).
Abdou, Y. et al. A phase 1, first-in-human (FIH) study of autologous macrophages engineered to express an anti-HER2 chimeric antigen receptor (CAR) in participants with HER2-overexpressing solid tumors. J. Clin. Oncol. 41, TPS2666 (2023).
Abdou, Y. et al. A phase 1, first-in-human study of autologous monocytes engineered to express an anti-HER2 chimeric antigen receptor (CAR) in participants with HER2-overexpressing solid tumors. J. Clin. Oncol. 42, TPS2682-TPS2682 (2024).
Annunziata, C. M. et al. Feasibility and preliminary safety and efficacy of first-in-human intraperitoneal delivery of MCY-M11, anti-human-mesothelin CAR mRNA transfected into peripheral blood mononuclear cells, for ovarian cancer and malignant peritoneal mesothelioma. J. Clin. Oncol. 38, 3014 (2020).
Chernykh, E. R. et al. Safety and therapeutic potential of M2 macrophages in stroke treatment. Cell Transplant 25, 1461–1471 (2016).
Sawitzki, B. et al. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet 395, 1627–1639 (2020).
Patel, A. N. et al. Ixmyelocel-T for patients with ischaemic heart failure: a prospective randomised double-blind trial. Lancet 387, 2412–2421 (2016).
Henry, T. D. et al. Safety and efficacy of ixmyelocel-T: an expanded, autologous multi-cellular therapy, in dilated cardiomyopathy. Circ. Res. 115, 730–737 (2014).
Brana, I. et al. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol 10, 111–123 (2015).
Pienta, K. J. et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs 31, 760–768 (2013).
Raghu, G. et al. CC-chemokine ligand 2 inhibition in idiopathic pulmonary fibrosis: a phase 2 trial of carlumab. Eur. Respir. J. 46, 1740–1750 (2015).
Sandhu, S. K. et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother. Pharmacol. 71, 1041–1050 (2013).
Colombo, A. et al. A double-blind randomised study to evaluate the efficacy and safety of bindarit in preventing coronary stent restenosis. EuroIntervention 12, e1385–e1394 (2016).
Venturini, N. J. et al. 170P Targeting myeloid cells in non-small cell lung cancer and hepatocellular carcinoma: A window-of-opportunity trial of nivolumab with BMS-813160 (CCR2/5i) or BMS-986253 (anti-IL8). IOTECH 20, 100629 (2023).
Behrens, F. et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann. Rheum. Dis. 74, 1058–1064 (2015).
Genovese, M. C. et al. MRI of the joint and evaluation of the granulocyte-macrophage colony-stimulating factor-CCL17 axis in patients with rheumatoid arthritis receiving otilimab: a phase 2a randomised mechanistic study. Lancet Rheumatol. 2, e666–e676 (2020).
Buckley, C. D. et al. Efficacy, patient-reported outcomes, and safety of the anti-granulocyte macrophage colonystimulating factor antibody otilimab (GSK3196165) in patients with rheumatoid arthritis: a randomised, phase 2b, dose-ranging study. Lancet Rheumatol. 2, e677–e688 (2020).
Fleischmann, R. M. et al. Anti-GM-CSF otilimab versus tofacitinib or placebo in patients with active rheumatoid arthritis and an inadequate response to conventional or biologic DMARDs: two phase 3 randomised trials (contRAst 1 and contRAst 2). Ann. Rheum. Dis. 82, 1516–1526 (2023).
Taylor, P. C. et al. Anti-GM-CSF otilimab versus sarilumab or placebo in patients with rheumatoid arthritis and inadequate response to targeted therapies: a phase III randomised trial (contRAst 3). Ann. Rheum. Dis. 82, 1527–1537 (2023).
Schett, G. et al. Anti-granulocyte-macrophage colony-stimulating factor antibody otilimab in patients with hand osteoarthritis: a phase 2a randomised trial. Lancet Rheumatol. 2, e623–e632 (2020).
Constantinescu, C. S. et al. Randomized phase 1b trial of MOR103, a human antibody to GM-CSF, in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2, e117 (2015).
Patel, J. et al. A randomised trial of anti-GM-CSF otilimab in severe COVID-19 pneumonia (OSCAR). Eur. Respir. J. 61, 2101870 (2023).
Molfino, N. A. et al. Phase 2, randomised placebo-controlled trial to evaluate the efficacy and safety of an anti-GMCSF antibody (KB003) in patients with inadequately controlled asthma. BMJ Open 6, e007709 (2016).
Patnaik, M. M. et al. Phase 1 study of lenzilumab, a recombinant anti–human GM-CSF antibody, for chronic myelomonocytic leukemia. Blood 136, 909–913 (2020).
Oluwole, O. O. et al. ZUMA-19: A Phase 1/2 Study of Axicabtagene Ciloleucel Plus Lenzilumab in Patients With Relapsed or Refractory Large B-Cell Lymphoma. Blood 140, 10318–10320 (2022).
Tanaka, S. et al. Randomized, double-blind, placebo-controlled, phase I study of the safety and pharmacokinetics of namilumab in healthy Japanese and Caucasian men. Int. J. Clin. Pharmacol. Ther. 56, 507–517 (2018).
Huizinga, T. W. et al. Phase 1b randomized, double-blind study of namilumab, an anti-granulocyte macrophage colony-stimulating factor monoclonal antibody, in mild-to-moderate rheumatoid arthritis. Arthritis. Res. Ther. 19, 53 (2017).
Taylor, P. C. et al. Efficacy and safety of namilumab, a human monoclonal antibody against granulocyte-macrophage colony-stimulating factor (GM-CSF) ligand in patients with rheumatoid arthritis (RA) with either an inadequate response to background methotrexate therapy or an inadequate response or intolerance to an anti-TNF (tumour necrosis factor) biologic therapy: a randomized, controlled trial. Arthritis. Res. Ther. 21, 101 (2019).
Papp, K. A. et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) as a therapeutic target in psoriasis: randomized, controlled investigation using namilumab, a specific human anti-GM-CSF monoclonal antibody. Br. J. Dermatol. 180, 1352–1360 (2019).
Kivitz, A. et al. Morab-022, an anti-granulocyte macrophage-colony stimulating factor (GM-CSF) monoclonal antibody (MAB): results of the first study in patients with mild-to-moderate rheumatoid arthritis (RA). Ann. Rheum. Dis. 75, 507 (2016).
Burmester, G. R. et al. Mavrilimumab, a human monoclonal antibody targeting GM-CSF receptor-α, in subjects with rheumatoid arthritis: a randomised, double-blind, placebo-controlled, phase i, first-in-human study. Ann. Rheum. Dis. 70, 1542–1549 (2011).
Burmester, G. R. et al. A randomised phase iib study of mavrilimumab, a novel GM-CSF receptor alpha monoclonal antibody, in the treatment of rheumatoid arthritis. Ann. Rheum. Dis. 76, 1020–1030 (2017).
Cid, M. C. et al. Efficacy and safety of mavrilimumab in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 81, 653–661 (2022).
Kuemmel, S. et al. A randomized phase ii study of anti-csf1 monoclonal antibody lacnotuzumab (MCS110) combined with gemcitabine and carboplatin in advanced triple-negative breast cancer. Clin. Cancer Res. 28, 106–115 (2022).
Tap, W. D. et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomized phase 3 trial. Lancet 394, 478–487 (2019).
Manji, G. A. et al. A phase i study of the combination of pexidartinib and sirolimus to target tumor-associated macrophages in unresectable sarcoma and malignant peripheral nerve sheath tumors. Clin. Cancer Res. 27, 5519–5527 (2021).
Cassier, P. A. et al. MEDIPLEX: A phase 1 study of durvalumab (D) combined with pexidartinib (P) in patients (pts) with advanced pancreatic ductal adenocarcinoma (PDAC) and colorectal cancer (CRC). J. Clin. Oncol. 37, 2579–2579 (2019).
Lee, J. H. et al. A phase I study of pexidartinib, a colony-stimulating factor 1 receptor inhibitor, in Asian patients with advanced solid tumors. Invest New Drugs 38, 99–110 (2020).
Rosenbaum, E. et al. A phase i study of binimetinib (MEK162) combined with pexidartinib (PLX3397) in patients with advanced gastrointestinal stromal tumor. Oncologist 24, 1309–e983 (2019).
Lin, C.-C. et al. Abstract CT171: Phase I study of BLZ945 alone and with spartalizumab (PDR001) in patients (pts) with advanced solid tumors. Cancer Res. 80, 16_Supplement: CT171 (2020).
Johnson, M. et al. ARRY-382 in combination with pembrolizumab in patients with advanced solid tumors: results from a phase 1b/2 study. Clin. Cancer Res. 28, 2517–2526 (2022).
Siddiqui, B. et al. 352 Target modulation within the tumor microenvironment (TME) by daratumumab (anti-CD38) but not edicotinib (CSF-1R inhibitor) in men with high-risk localized prostate cancer. J. Immunother. Cancer 9, https://doi.org/10.1136/jitc-2021-SITC2021.352 (2021).
Dowlati, A. et al. LY3022855, an anti-colony stimulating factor-1 receptor (CSF-1R) monoclonal antibody, in patients with advanced solid tumors refractory to standard therapy: phase 1 dose-escalation trial. Invest New Drugs 39, 1057–1071 (2021).
Autio, K. A. et al. Phase 1 study of LY3022855, a colony-stimulating factor-1 receptor (CSF-1R) inhibitor, in patients with metastatic breast cancer (MBC) or metastatic castration-resistant prostate cancer (MCRPC). J. Clin. Oncol. 37, 2548–2548 (2019).
Falchook, G. S. et al. A phase 1a/1b trial of CSF-1R inhibitor LY3022855 in combination with durvalumab or tremelimumab in patients with advanced solid tumors. Invest New Drugs 39, 1284–1297 (2021).
Sankhala, K. K. et al. A phase I/II dose escalation and expansion study of cabiralizumab (cabira; FPA-008), an anti-CSF1R antibody, in tenosynovial giant cell tumor (TGCT, diffuse pigmented villonodular synovitis D-PVNS). J. Clin. Oncol. 35, 11078–11078 (2017).
Davis, A. A. et al. Abstract P3-06-07: Phase Ib/II study to evaluate safety and tolerability of cabiralizumab in combination with nivolumab and neoadjuvant chemotherapy in patients with localized triple-negative breast cancer. Cancer Res. 83, 5_Supplement: P3-06-07 (2023).
Wang-Gillam, A. et al. A randomized phase II study of cabiralizumab (cabira) + nivolumab (nivo) ± chemotherapy (chemo) in advanced pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 37, TPS465–TPS465 (2019).
Gomez-Roca, C. et al. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naïve or experienced for immune checkpoint blockade. J. Immunother. Cancer 10, e004076 (2022).
Machiels, J. P. et al. Phase Ib study of anti-CSF-1R antibody emactuzumab in combination with CD40 agonist selicrelumab in advanced solid tumor patients. J. Immunother. Cancer 8, e001153 (2020).
Gomez-Roca, C. A. et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann. Oncol. 30, 1381–1392 (2019).
Rahim, M. K. et al. Dynamic CD8+ T cell responses to cancer immunotherapy in human regional lymph nodes are disrupted in metastatic lymph nodes. Cell 186, 1127–1143.e18 (2023).
Ko, A. H. et al. Atezolizumab plus pegph20 versus chemotherapy in advanced pancreatic ductal adenocarcinoma and gastric cancer: morpheus phase ib/ii umbrella randomized study platform. Oncologist 28, 553–e472 (2023).
Hong, D. S. et al. Abstract P225: Preliminary interim data of elzovantinib (TPX-0022), a novel inhibitor of MET/SRC/CSF1R, in patients with advanced solid tumors harboring genetic alterations in MET: Update from the Phase 1 SHIELD-1 trial. Mol. Cancer Ther. 20, P225 (2021).
Rosenbaum, E. et al. A phase 1b study of avelumab plus DCC-3014, a potent and selective inhibitor of colony stimulating factor 1 receptor (CSF1R), in patients with advanced high-grade sarcoma. J. Clin. Oncol. 39, 11549–11549 (2021).
Gelderblom, H. et al. CSF1R inhibition in patients with advanced solid tumors or tenosynovial giant cell tumor: a phase i study of vimseltinib. Clin. Cancer Res. 30, 3996–4004 (2024).
Choi, B. J. et al. Abstract 2255: Patient pharmacodynamic biomarker and pk evaluation results from an ongoing phase I dose-escalation study of q702, an axl, mer and csf1r kinase inhibitor in patients with advanced solid tumors. Cancer Res. 83, 2255 (2023).
Baretti, M. et al. A phase II study of durvalumab (MEDI4736) in combination with a CSF-1R inhibitor (SNDX-6352) following chemotherapy or radio-embolization for patients with intrahepatic cholangiocarcinoma. J. Clin. Oncol. 41, e16217 (2023).
Patel, K. et al. CD47-blocker TTI-622 shows single-agent activity in patients with advanced relapsed or refractory lymphoma: update from the ongoing first-in-human dose escalation study. Blood 138, 3560 (2021).
Strati, P. et al. Interim results from the first clinical study of CC-95251, an anti-signal regulatory protein-alpha (SIRPα) antibody, in combination with rituximab in patients with relapsed and/or refractory non-hodgkin lymphoma (R/R NHL). Blood 138, 2493 (2021).
Champiat, S. et al. Safety, pharmacokinetics, efficacy, and preliminary biomarker data of first-in-class BI 765063, a selective SIRPα inhibitor: Results of monotherapy dose escalation in phase 1 study in patients with advanced solid tumors. J. Clin. Oncol. 39, 2623–2623 (2021).
Narkhede, M. et al. A phase 1 first-in-human study of GS-0189, an anti-signal regulatory protein alpha (SIRPα) monoclonal antibody, in patients with relapsed/refractory (R/R) non-Hodgkin lymphoma (NHL). EJHaem 4, 370–380 (2023).
Sikic, B. I. et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J. Clin. Oncol. 37, 946–953 (2019).
Brierley, C. K. et al. The effects of monoclonal anti-CD47 on RBCs, compatibility testing, and transfusion requirements in refractory acute myeloid leukemia. Transfusion 59, 2248–2254 (2019).
Roschewski, M. et al. Phase I study of acalabrutinib plus danvatirsen (AZD9150) in relapsed/refractory diffuse large b-cell lymphoma including circulating tumor dna biomarker assessment. Clin. Cancer Res. 29, 3301–3312 (2023).
Fisher, G. A. et al. A phase Ib/II study of the anti-CD47 antibody magrolimab with cetuximab in solid tumor and colorectal cancer patients. J. Clin. Oncol. 38, 114–114 (2020).
Lakhani, N. J. et al. A phase Ib study of the anti-CD47 antibody magrolimab with the PD-L1 inhibitor avelumab (A) in solid tumor (ST) and ovarian cancer (OC) patients. J. Clin. Oncol. 38, 18–18 (2020).
Daver, N. et al. Phase I/II study of azacitidine (AZA) with venetoclax (VEN) and magrolimab (Magro) in patients with newly diagnosed (ND) older/unfit or high-risk acute myeloid leukemia (AML) and relapsed/refractory (R/R) AML. Blood 140, 141–144 (2022).
Drakaki, A. et al. Phase Ib/II umbrella trial to evaluate the safety and efficacy of multiple 2L cancer immunotherapy (CIT) combinations in advanced/metastatic urothelial carcinoma (mUC): MORPHEUS-mUC. J. Clin. Oncol. 38, TPS591–TPS591 (2020).
Maakaron, J. et al. Magrolimab in combination with rituximab + chemotherapy in patients with relapsed or refractory (R/R) diffuse large b-cell lymphoma (DLBCL). Blood 140, 3728–3730 (2022).
Yardley, D. A. et al. Abstract OT2-06-04: MORPHEUS: A phase Ib/II trial platform evaluating the safety and efficacy of multiple cancer immunotherapy combinations in patients with hormone receptor–positive and triple-negative breast cancer. Cancer Res. 79, OT2–06–04 (2019).
Temesgen, Z. et al. Lenzilumab in hospitalised patients with COVID-19 pneumonia (LIVE-AIR): a phase 3, randomised, placebo-controlled trial. Lancet. Respir. Med. 10, 237–246 (2022).
Burmester, G. R. et al. Efficacy and Safety of Mavrilimumab in Subjects with Rheumatoid Arthritis. Ann. Rheum. Dis. 72, 1445–1452 (2013).
Weiss, S. A. et al. A Phase I Study of APX005M and Cabiralizumab with or without Nivolumab in Patients with 1170. Melanoma, Kidney Cancer, or Non-Small Cell Lung Cancer Resistant to Anti-PD-1/PD-L1. Clin. Cancer Res. 27, 4757–4767 (2021).
Acknowledgements
Biorender supported the material in graphics in this article. This work was supported by the National Natural Science Foundation of China (82372943, 82303610, 82073893), Hunan Provincial Natural Science Foundation of China (2022JJ20095), Health Research Project of Hunan Provincial Health Commission (202204044869), China Postdoctoral Science Foundation (2023MD734131), Chongqing Postdoctoral Science Foundation (CSTB2023NSCQBHX0002), Chongqing Postdoctoral Research Special Funding Project (2023CQBSHTB3095), Hunan Youth Science and Technology Talent Project (2023RC3074), Science Foundation of the AMHT Group (NO.2022YK04).
Author information
Authors and Affiliations
Contributions
Q.C., Z.X., and H.Z. conceived and designed the manuscript. F.G., R.W., P.L., and Z.Y. drafted the manuscript and designed the figures. F.G., R.W., Z.Y., W.L., Y.X., Z.X., Z.L., Q.C., H.Z., and P.L. revised the manuscript. Q.C., H.Z., and Z.X. supervised the manuscript and provided funding support. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All authors have agreed on the contents of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guan, F., Wang, R., Yi, Z. et al. Tissue macrophages: origin, heterogenity, biological functions, diseases and therapeutic targets. Sig Transduct Target Ther 10, 93 (2025). https://doi.org/10.1038/s41392-025-02124-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-025-02124-y
This article is cited by
-
Multi-omics data reveal that SAA1 + fibroblasts exacerbate periodontitis by regulating macrophage inflammation and chemotaxis
Journal of Translational Medicine (2025)
-
Advances and obstacles of T cell-based immunotherapy in gynecological malignancies
Molecular Cancer (2025)
-
FOLR2+ macrophages in cancer: allies or enemies
Cell Communication and Signaling (2025)
-
Macrophage polarization in diabetic vascular complications: mechanistic insights and therapeutic targets
Journal of Translational Medicine (2025)
-
Exploring promising biomarkers based on pathogenic mechanisms in interstitial cystitis/bladder pain syndrome
Nature Reviews Urology (2025)
