
Abstract
The infantile neuronal ceroid lipofuscinosis, also called CLN1 disease, is a fatal neurodegenerative disease caused by mutations in the CLN1 gene encoding palmitoyl protein thioesterase 1 (PPT1). Identifying the depalmitoylation substrates of PPT1 is crucial for understanding CLN1 disease. In this study, we found that GABAAR, the critical synaptic protein essential for inhibitory neurotransmission, is a substrate of PPT1. PPT1 depalmitoylates GABAAR α1 subunit at Cystein-260, while binding to Cystein-165 and -179. Mutations of PPT1 or its GABAAR α1 subunit binding site enhanced inhibitory synaptic transmission and strengthened oscillations powers but disrupted phase coupling in CA1 region and impaired learning and memory in 1- to 2-months-old PPT1-deficient and Gabra1em1 mice. Our study highlights the critical role of PPT1 in maintaining GABAAR palmitoylation homeostasis and reveals a previously unknown molecular pathway in CLN1 diseases induced by PPT1 mutations.
Similar content being viewed by others

Introduction
Palmitoylation is a posttranslational modification in which a long-chain 16-carbon fatty acid (palmitate) is attached to the thiol groups of cysteine residues in substrate proteins [1]. This modification influences the cellular distribution and function of substrate proteins [2]. The enzymes responsible for this process are palmitoyl acyltransferases that contain a zinc-finger domain with a conserved DHHC (Asp-His-His-Cys) motif (zDHHCs) [3, 4]. Depalmitoylation is a reverse process of palmitoylation and is catalyzed by palmitoyl-protein thioesterases (PPTs), acyl-protein thioesterases, and the α/β hydrolase domain-containing proteins [5, 6].
Neuronal ceroid lipofuscinoses (NCLs) are worldwidely distributed diseases with an incidence of 1–3/100,000 and a prevalence of 2–4/1,000,000 [7,8,9]. Different forms of NCL are typically classified according to the age of onset, with infantile NCL (INCL) exhibiting the earliest onset and the most rapid course [10]. Previous studies indicate that 25% of the patients worldwide and 40% in the United States harbor nonsense mutations in the CLN1 gene encoding PPT1, consequently causing PPT1 deficiency [11]. Patients with CLN1 disease undergo progressive retinal degeneration, leading to blindness, learning and memory loss, and seizures [12, 13].
Despite the severity of PPT1 mutations during early brain development, the mechanisms underlying the PPT1 deficiency-induced clinical profile of CLN1 disease remain poorly understood. A large-scale proteomic screen has shown that more than 100 synaptic proteins are PPT1 substrates [14], which depicts the complexity of PPT1 function in the central nervous system. Other studies have indicated that PPT1 plays important roles in synapses, such as regulating axonal growth [15] and neurite extension [16, 17]. In the presynaptic area, PPT1 modifies the synaptic protein synaptobrevin 2 (VAMP2), the synaptosomal-associated protein of 25kD (SNAP 25), and the cysteine string protein α (CSPα), thereby PPT1 deficiency causes vesicle recycling failure in both mice and humans [18]. In the postsynaptic area, GluN2B and Fyn kinases are hyperpalmitoylated in PPT1-/- neurons, which hinder the developmental N-methyl-D-aspartate receptor (NMDAR) subunit switch from GluN2B to GluN2A [19]. Studies have shown that postsynaptic density protein-95 (PSD-95) and GluN2A are not PPT1 substrates [19, 20]. To better understand the pathogenesis of CLN1 disease, we try to identify the key synaptic depalmitoylation substrates of PPT1, such as GluN1 and GABAAR, and others.
GABAAR plays a central role in synchronizing network neuronal activity, which are tightly associated with higher brain functions such as learning and memory [21]. PPT1-KI mice carrying nonsense mutations in the CLN1/PPT1 gene exhibited the reduced expression of GABAAR along with impaired gamma oscillation and seizure onset [22]. These findings indicate the importance of GABAAR in CLN1 disease.
In the present work, we undertook multilevel functional and biochemical approaches to investigate both inhibitory and excitatory synaptic transmission, and generated transgenic mice Gabra1em1 with a double mutation of GABAAR α1 binding sites to PPT1, aiming to elucidate the postsynaptic mechanisms of NCL. We demonstrated that the GABAAR α1 subunit is a substrate of PPT1. In 1- to 2-month-old PPT1-deficient (PPT1-KI) mice, we found inhibitory synaptic transmission was enhanced, GABAAR was hyperpalmitoylated, which resulted in membrane retention of GABAAR. Gabra1em1 mice show disrupted neural network oscillations similar to those observed in PPT1-deficient mice. These results indicated that GABAAR, as a PPT1 substrate, via alteration of its palmitoylation which affect function of neural circuits in the early stage and provides a novel synaptic mechanism for NCL pathogenesis.
Methods and materials
Animals
C57BL/6N-Gabra1em1Cin(p.C165S,p.C179S)/Cya (Gabra1em1) mutant mice
The mouse Gabra1 gene (GenBank accession number: NM_010250.5; Ensembl: ENSMUSG00000010803) is located on chromosome 11. Ten exons were identified, with an ATG start codon in exon 2 and a TAG stop codon in exon 10. p.C165 and p.C179 are located in exon 6. Exon 6 was selected as the target site. We designed a gRNA targeting vector and donor oligo (with a targeting sequence flanked by 240 bp homologous sequences combined on both sides) (Table 1).
The gRNA to the mouse Gabra1 gene, the donor oligo containing p.C165S (TGC to AGC) and p.C179S (TGC to AGC) mutations, two synonymous mutations (p.A163 = (GCT to GCG) and p.P180 = (CCA to CCT)), and Cas9 were co-injected into fertilized mouse eggs, followed by implantation of the eggs into surrogate mothers to obtain offspring to generate targeted knock-in offspring. F0 founder animals were identified by PCR, followed by sequence analysis, and were bred with WT mice to test germline transmission and F1 animal generation. F2 homozygotes were generated by F1 autocopuation. The target region of the mouse Gabra1 locus was amplified by PCR using specific primers. The PCR products were sequenced to confirm the targeting. Primer sequence: Primer-F: 5′-GGATTAGTGGGTGCTCTCTGTTAT-3′; Primer-R: 5′-GCCTGTGGATCTTCTACCCTAAC-3′.
PPT1-KI (CLN1 c.451C>T (p.R151X)) mutant mice
The strategy for point mutation of the CLN1 gene to generate PPT-KI mice has been described as before. As previous studies, PPT1-KI mice carrying c.451C>T/c.451C>T mutations are deficient in PPT1-protein as well as PPT1 enzyme activity. And the PPT1-KI mice did not show structural deterioration of the retinal layers until 6 months of age [11]. C57BL/6N mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd, Beijing, China (animal licence number:2016-0006) and used as the WT controls. All animals were housed and maintained in the specific pathogen-free animal facility of the Animal Experiment Center of the Institute of Psychiatry and Neuroscience of Xinxiang Medical University (XXMU) under a 12-h light/dark cycle. Animals had ad libitum access to food and water, except during food and water deprivation periods. A previous study demonstrated that N-(tert-Butyl)hydroxylamine hydrochloride (BuHA) can easily cross the blood-brain barrier in the mouse brain [23]. PPT1-KI mice and their littermates were orally administered 0.5 mM BuHA for 1 month before mating to breast-feed pups with BuHA after parturition. Young pups had free access to food and water (containing 0.5 mM BuHA) throughout the experiments. All efforts were made to minimize animal suffering and reduce the number of animals used. Experiments were performed in accordance with the guidelines published in the NIH Guide for the Care and Use of Laboratory Animals (NIH publication no. 86-23, revised 1987). All reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise stated.
Electrophysiology
In vivo electrophysiological recording
Stereotaxic surgery and electrode implantation
Mice were anesthetised with an intraperitoneal injection of 1% pentobarbital sodium (0.45–0.5 mL/100 g). Under anesthesia, mice were secured in a stereotaxic frame with ear bars. The head was shaved with a razor, and a midline 5-mm incision was made using a sterile scalpel. The subcutaneous tissue was removed from the skull, and a craniotomy (~1.5 × 0.5 mm) was drilled (AP: 1.82 mm, ML:1.25 mm, DV:1.5 mm, right hemisphere) in the CA1 region. Two steel screws are anchored to the anterior and posterior edges of the surgical site to secure the implant. After the endocranium was removed, a 4 × 2 micro wire electrode (KD-MWA-8, 25 μm nitinol wire, Kedou (Suzhou) Brain-Computer Technology Co., Ltd.) with 3 μm polyethylene glycol coating was implanted into the pyramidal cell layer of CA1. The craniotomy site was sealed with a sterile silicone elastomer (Kwik-Sil WPI) to prevent brain injury. After surgery, the implanted electrodes and screws were cemented integrally to the skull using a denture base resin type II (Shanghai Medical Instruments Co., Ltd). After surgery, the animals were housed individually on a reversed 12/12 h day/night schedule.
Signal acquisition
Following one week of recovery, wideband signals were recorded using an OmniPlex Neural Recording Data Acquisition System (Plexon Inc., Dallas, TX, USA) with an 8 kHz global low-pass filter. A continuous spike was sampled at 4 kHz, followed by a 300 Hz low-cut filter. The FP was set to a 200 low-pass filter and down-sampled to 1 kHz. Diazepam (Sanchine #H23021885) at a dosage of 3 mg/kg dissolved in 10% DMSO in saline [24] was intraperitoneally injected to PPT1-KI mice with epilepsy, and the electrophysiological signals were subsequently observed. After recording, the hippocampus was post-fixed for Nissl staining to verify the proper placement of the electrodes in the target region.
Spike sorting
Spikes were sorted using Offline Sorter (Plexon Inc., Dallas, TX, USA) to classify the electrical activity of individual neurons, based on the first to third principal components [25]. Spike units were excluded when the absolute refractory period of single-unit autocorrelation was <1 ms. Cross-channel artefacts identified by their time coincidence across channels were also invalidated [26].
Spectral analysis
The power spectrum density and spectrogram of continuous FP were computed using NeuroExplorer (Nex Technologies, Colorado Springs, CO, USA) with 1024 frequency values and 25% window overlap. Before this process, the FP signal values were multiplied by the coefficients of the Hann window and discrete fast Fourier transformations of the results were calculated using formulas defined previously [27]. Theta/gamma waves were filtered by bandpass filtering of the FP data using NeuroExplorer software with Digital Filtering of Continuous Variables function.
Phase locking analysis
To evaluate the wave phase locking, waves were further processed through Hilbert transformation using built-in or custom-built MATLAB scripts (R2016a, MathWorks, USA). Only neurons with ≥50 spikes during the period analyzed were used for phase locking estimation. We tested the significance of spike-FP phase locking using circular statistics (CirStat toolbox in Matlab) [28,29,30]. Rayleigh’s test was employed to assess the circular distribution of the mean phase angle and to test the nonuniformity of each neuron’s spike phase distribution to theta or gamma. Neurons were considered significantly phase-locked at P < 0.05. The mean phase angle was computed in the circular direction of the mean resultant vector. The Watson-Williams F-test was performed to compare the mean phase angles of neurons recorded from multiple groups.
We evaluated the phase locking of neurons by calculating the mean resultant vector (MRL, range 0–1) length of the spike phase angle distribution. An MRL value of one indicates exact phase synchrony, whereas a value of zero indicates no phase synchrony. The non-parametric Kruskal–Wallis test was performed to test the differences in the MRL values between multiple groups. P < 0.05 was considered statistically significant. Moreover, the valley of the wave timestamp was identified as a reference event using Find Oscillation function to plot the peri-event raster. Data and MATLAB scripts supporting the findings of this study are available from the corresponding author upon request.
The potential palmitoylation sites (C165, C179, C260, C319) of the GABAAR α1 subunit were forecast using the CSS-Palm Online Service webset. All mutants (GABAARα1 C165A, C179A, C165A&179A, C260A&C319A) with Myc tags were purchased from Fenghui Biology (Hu’nan, China). The ABE assay was performed as described previously.
Other experimental procedures are provided in the Supplementary Materials and Methods:
S1 In vitro electrophysical recording
Slice preparation
Patch clamp recording
LTP induction
S2 Biochemical analysis
Cell culture and Immunostaining
STED image
ABE assay
Cell membrane/cytoplasmic protein extraction
Co-immunoprecipitation (Co-IP)
Western blot
Quantitative polymerase chain reaction (qPCR)
S3 Behavioral studies
Morris water maze (MWM)
Y maze
Statistics
All data were acquired and analyzed by researchers who were blinded to the genotype of the mice and acute slices. All data were analyzed using SPSS Statistics 20 (IBM, Armonk, NY, USA). We confirmed the homogeneity of variances using Levene’s test and the equality of means using the Brown–Forsythe test. Data were statistically analyzed using Student’s t-test for comparisons between two groups and one-way analysis of variance (ANOVA) for comparisons among multiple groups. Data that did not follow a normal distribution were estimated by the non-parametric Mann–Whitney U-test for two-group comparisons and the Kruskal–Wallis test for multi-group comparisons. Cumulative probability distribution analysis was performed using the Kolmogorov-Smirnov test. Western blot results were analyzed using the t-test for two-group comparisons and one-way ANOVA for multi-group comparisons. All tests were conducted as two-sided tests to assess differences without assuming a specific direction.
Results
PPT1-KI enhances GABAAR α1 membrane expression and GABAergic neurotransmission
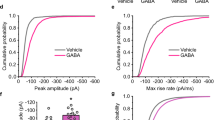
To investigate the function of postsynaptic receptors in PPT1-deficiency induced diseases, we first assessed the role of GABAAR α1 subunit hyperpalmitoylation in regulating inhibitory neurotransmission. Consequently, we investigated GABAergic neurotransmission in 1- to 2-month-old PPT1-KI mice to simulate the early stages of pathogenesis of CLN1 disease. Evoked inhibitory postsynaptic currents (eIPSCs) were recorded by holding pyramidal cells in the CA1 region at +40 mV, as shown in Fig. 1A. Compared with wildtype (WT) neurons, PPT1-KI neurons showed an increased amplitude (Fig. 1B) and a decreased fast tau value without affecting the slow tau by double exponential fitting of the repolarization phase (Fig. 1C). Miniature inhibitory postsynaptic currents (mIPSCs) amplitude and frequency were both enhanced in PPT1-KI neurons, which is similar to Gabra1em1mice (Fig. 1D–F), as described below. Next, we incubated hippocampal slices of PPT1-KI mice with 1 μM BuHA, a thioesterase mimetic that selectively cleaves thioester linkage in palmitoylated proteins and compensates for the molecular defect caused by PPT1 mutations [23], both the amplitude and frequency of mIPSCs were recovered to the control level (Fig. 1D–F). These results demonstrated that hyperpalmitoylation of the GABAAR α1 subunit by PPT1 deficiency strengthened GABAergic neurotransmission.

A Sample traces showing evoked IPSCs recorded at +40 mV from CA1 pyramidal cells in 1- to 2-month-old WT (black trace) and PPT1-KI mice (red trace). Amplitude (B), fast tau, and slow tau (C) of IPSCs from (A). WT: n = 8 neurons from six slices; PPT1-KI: n = 8 neurons from five slices. T-test, **P < 0.01. Sample traces (D) showing mIPSCs amplitude (E) and frequency (F) are enhanced in PPT1-KI mice, Gabra1em1 mice, compared to WT mice, and partially recovered by pre-incubation with 1 μM BuHA. WT: n = 12 neurons from 9 slices; PPT1-KI: n = 11 neurons from 7 slices; Gabra1em1: n = 11 neurons from 10 slices, PPT1-KI with BuHA: n = 11 neurons from 6 slices. Histogram: One-way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001; cumulative curve: Kolmogorov–Smirnov test, **P < 0.01, ***P < 0.001. G Sample traces of evoked EPSCs recorded from WT (black trace) and PPT1-KI mice (red trace) hippocampal pyramidal neurons at +40 mV (upper traces) and -70 mV (bottom traces). Amplitudes of AMPAR (H), NMDAR (I), and AMPAR/NMDA ratio (J) from the evoked EPSCs of hippocampal pyramidal neurons. WT: n = 11 neurons from 10 slices; PPT1-KI: n = 8 neurons from 6 slices; t-test: no significant difference. K–M Representative traces and analyses of mEPSCs recorded from CA1 pyramidal neurons of WT (black trace) and PPT1-KI (red trace) mice. WT: n = 12 neurons from 10 slices; PPT1-KI: n = 9 neurons from 9 slices; PPT1-KI with 5 μM BuHA: n = 8 neurons from 7 slices. A one-way ANOVA showed no significant differences. Data are represented as mean ± SEM.
Furthermore, we examined whether postsynaptic glutamatergic receptors could be affected by PPT1; AMPAR-and NMDAR-mediated currents (Fig. 1G–I), as well as the AMPA/NMDA current ratio (Fig. 1J) showed no difference between WT and PPT1-KI mice, miniature excitatory postsynaptic currents (mEPSCs) amplitude and frequency were also comparable in WT and PPT1-KI mice with and without BuHA treatment (Fig. 1K–M). Western blot analysis of AMPAR, NMDAR 2a/2b, and their scaffold proteins (PSD95/93 and SAP102) showed no changes in membrane protein expression in WT and PPT1-KI mice with and without BuHA treatment (Fig. S1). These data suggest that PPT1-KI did not affect glutamatergic transmission.
GABAAR α1 subunit membrane expression is developmentally regulated in PPT1-KI mice
We tested the membrane expression of GABAAR α1, β2, and γ2 subunits at different developmental stages by performing membrane fraction analysis using western blot. In PPT1-KI mice, we observed a distinct age-dependent pattern for the α1 subunit: membrane expression was increased at 1 and 2 months but decreased at 7 months (Fig. 2A, B). Notably, the membrane expression levels of the α1 subunit in PPT1-KI mouse brains at ages 5 and 15 days were found to be equivalent to those in the WT brain (as shown in Fig. 2A, B), even though PPT1 expression is documented during embryonic stage. We propose that during early developmental stages, neurons are immature, and the expression levels of the α1 subunit are inherently low, as indicated by the faint bands in Fig. 2A, B. Although PPT1 is expressed during embryogenesis, the regulation of the α1 subunit by PPT1 may not be prominent until neurons mature. The immature α1 subunit might not be efficiently palmitoylated or recognized by PPT1 at these stages. While, qPCR results showed no differences in the total mRNA expression of all GABAAR subunits until 7 months (Fig. 2C). Furthermore, the membrane expression level of GABAAR α1 in PPT1-KI mice was higher than that of the WT, which could be mitigated by in vitro incubation with different concentrations of BuHA (Fig. 2D, E). The membrane expression of β2 and γ2 subunits remained unchanged compared to the WT group (Fig. S2). These results suggest that the increased membrane level of the α1 subunit in 1- to 2-month-old PPT1-KI mice is initiated by post-translational modification rather than changes in gene expression. Further, the reduced membrane level of the α1 subunit in 7-month-old mice is likely due to neuron loss [31].

Representative immunoblot (A) and quantification (B) of GABAAR α1 subunits levels on the cellular membrane in WT and PPT1-KI mice at indicated ages. N = 3 for each group, two-way ANOVA, *P < 0.05. C Quantitative PCR showing developmental changes in the mRNA expression of GABAAR α1 subunit at the indicated ages. N = 3, two-way ANOVA, *P < 0.05. Representative immunoblotting (D) and quantification (E) of GABAAR α1 subunit levels on the cellular membrane in WT, PPT1-KI, and PPT1-KI mice treated with BuHA. N = 3 for each group, one-way ANOVA; *P < 0.05, **P < 0.01. F, G Increased levels of the palmitoylated GABAAR α1 subunit in cultured PPT1-KI neurons, which were recovered by incubation with BuHA. Representative images (H) and colocalization analysis (I) of GABAAR α1 subunit and PPT1 enzyme in cultured hippocampal neurons from WT mice. Fluorescence intensity was measured using the red line across the cell body. N = 8 cells. +HA, with hydroxylamine; N = 3 for each group, one-way ANOVA, *P < 0.05; **P < 0.01. Data are represented as mean ± SEM.
PPT1 depalmitoylates GABAAR α1 subunit at cysteine residues
We performed an immunocytochemical assay to examine the subcellular distributions of the GABAAR α1 subunit and the PPT1 enzyme in hippocampal neurons. We found that the endogenous GABAAR α1 subunit colocalized with PPT1 in neuronal soma, neurites and spines with super-resolution stimulated emission depletion (STED) microscopy (neurites and spines: Pearson’s coefficient: 0.79 ± 0.0091, overlap coefficient: 0.83 ± 0.0060; soma: Pearson’s coefficient: 0.80 ± 0.0077, overlap coefficient: 0.88 ± 0.0041, n = 8 cells) (Fig. 2H, I). The close localization of the GABAAR α1 subunit and PPT1 prompts the investigation of whether the GABAAR α1 subunit can be palmitoylated by PPT1. We employed the ABE assay to analyze the palmitoylation level of membrane fractions; the GABAAR α1 subunit’s palmitoylation level is upregulated in PPT1-KI mice, which was suppressed by in vitro incubation with BuHA (Fig. 2F, G). In contrast, the palmitoylation status of its postsynaptic scaffold protein gephyrin was not affected (Fig. S3), suggesting that an enzyme other than PPT1 regulates the depalmitoylation of gephyrin.
Moreover, an in vitro co-immunoprecipitation (co-IP) assay revealed the biochemical interaction between PPT1 and the GABAAR α1 subunit (Fig. 3A), indicating that GABAAR is a substrate of PPT1. Bioinformatics analysis predicted four potential palmitoylated cysteine residues (C165, C179, C260, and C319) (Fig. 3B). Mutation of Cys260 to alanine dramatically reduces palmitoylation levels, indicating it is a major palmitoylation site (Fig. 3C, D). However, the residual palmitoylation suggests that other cysteine residues may also be palmitoylated, contributing to the band observed in the +HA group.

(A) Interaction between PPT1 and GABAAR α1 subunit verified by in vitro CO-IP using mouse hippocampal tissue. IB: immunoblotting. (B) Bioinformatic prediction of palmitoylation site (Cys). (C, D) Identification of palmitoylation sites within GABAAR α1 subunit. Mutation of Cys-260 residues to alanine reduce palmitoylation level of GABAAR α1 in 293 T cell line. N = 3, one-way ANOVA, *P < 0.05. (E-H) Representative blots and quantification of immunoprecipitation show that mutations of Cys-165 and Cys-179 (E, F), but not Cys-260 or Cys-319 (G, H), to alanine block GABAAR α1 subunit binding to PPT1 in 293 T cell line. N = 3, one-way ANOVA, **P < 0.01.
To identify the binding site between PPT1 and GABAAR α1, we mutated different GABAAR α1 cysteine residues to alanine in the 293 T cell line. Co-IP results showed that single mutation of the cysteine residues did not affect PPT1-GABAAR α1 binding; however, this binding was strongly reduced when we double-mutated 165 and 179 cysteines to alanine (C165A& C179A) (Fig. 3E, F). In contrast, no reduction was detected when cysteine residues (260 and 319) were mutated to alanine residues (C260A and C319A) (Fig. 3G, H). These results indicate that C165 and C179 are the binding sites of GABAAR α1 to PPT1.
We also investigated the potential substrate PPT1 in excitatory synapses. The ABE results showed that the palmitoylation status of ionotropic glutamate receptors (iGluRs), such as AMPAR and NMDA 2a/2b, as well as their scaffold proteins PSD95/93 and SAP102, were comparable between PPT1-KI and WT mice (Fig. S4A–G). QPCR analysis showed no changes in mRNA expression at an early stage (Fig. S4H). Our data suggest that these excitatory synaptic proteins are unlikely to be PPT1 substrates.
Disruption of PPT1 or GABAAR α1 subunit impairs neural network oscillations
A previous study reported that CLN3−/− mice, as young as 2 months old, exhibited a widespread decrease in network coordination in most of the hippocampus and fast beta frequency power with decreased slow delta activity by 18 months of age [32]. Using in vivo field potential (FP) recordings of the hippocampal CA1 region, we observed epileptiform discharges in 6- to 7-month-old PPT1-KI mice [33], which can be effectively supressed by intraperitoneal administration of diazepam (Figure S5). Further analysis of the neural circuitry of 1- to 2-month-old PPT1-KI mice showed that power spectral density (PSD) had an increased power of theta (3–8 Hz) and gamma (30–80 Hz) bands compared to in WT mice, which could be suppressed by oral treatment with BuHA (Fig. 4A–C, E–G). Given the abnormal inhibitory neurotransmission, neural network activity, and altered expression of GABAAR in the PPT1-KI mice, we further explored the function of GABAAR as a PPT1 substrate. By genetically mutating 165 and 179 cysteines of GABAAR α1 to serine, we generated Gabra1em1 mutant mice (Figs. S6 and S7). Remarkably, Gabra1em1 mice also exhibited enhanced power in theta and gamma bands (Fig. 4D, H, I–K) and disrupted phase locking (Figs. 4O, S, T and S8D, H, I) similar to that observed in PPT1-KI mice (Fig. 4M, Q). BuHA treatment partially rescued the disrupted phase locking of the theta (Fig. 4) and gamma rhythms (Fig. S8) in PPT1-KI mice. This implies that an interruption of GABAAR α1 subunit palmitoylation status disturbed temporal alignment.

FP signals recorded in the CA1 region of WT (A), PPT1-KI (B), PPT1-KI treated with BuHA (C), and Gabra1em1 mice (1 to 2 months old) (D). Lower traces show filtered theta (3–8 Hz) and gamma oscillation (30–80 Hz) from the FP signals. Spectrograms of the FP signals recorded from WT (E), PPT1-KI (F), BuHA-treated PPT1-KI (G), and Gabra1em1 (H) mice. I PSD of FP recorded from WT (black), PPT1-KI (red), BuHA-treated PPT1-KI (blue), and Gabra1em1 (green) mice. Analysis of the theta (J) and gamma (K) PSD. WT: n = 8 mice; PPT1-KI: n = 8 mice; PPT1-KI treated with BuHA: n = 8 mice; Gabra1em1: n = 6 mice; one-way ANOVA, ***P < 0.001. Sample traces show theta phase locking in the CA1 region. Peri-event raster (upper panel) and histogram (lower panel) displaying phase coupling of spike units and theta waves recorded from WT (L), PPT1-KI (M), BuHA-treated PPT1-KI (N), and Gabra1em1 (O) mice. Tips display the timestamps of the spike units. Dotted lines indicate valleys of theta waves. Bin width is 2 ms. Circular distribution of mean-spike theta phase angles (15° bin width) (upper panel) recorded from the CA1 area of WT (P), PPT1-KI (Q), BuHA-treated PPT1-KI (R), and Gabra1em1 (S) mice. The red bars represent the direction and magnitude (length) of the MRL of the population. T Comparison of mean MRL values between multiple groups. WT: n = 31 neurons from 6 mice; PPT1-KI: n = 35 neurons from 5 mice; BuHA-treated PPT1-KI: n = 29 neurons from 5 mice; Gabra1em1: n = 40 neurons from 6 mice. Kruskal–Wallis test, ***P < 0.001. Data are represented as mean ± SEM.
Research has shown that spike-phase coupling can predict memory strength [34], with a recent study demonstrating that spike–phase locking is the most sensitive index for the detection of early pathological AD [35]. The polar histograms indicate that most phase-locked neurons fire during the descending phase of the theta oscillations (Fig. 4P–S). The length of the mean resultant vector (MRL) is calculated to evaluate the degree of phase locking. As shown in Fig. 4T, the MRL was shorter in PPT1-KI and Gabra1em1 mice than in WT mice. Similarly, the gamma band polar histograms showed dramatic changes in the preferred phase and shorter MRL in PPT1-KI hippocampal CA1 neurons (Fig. S8).
These results indicate that the interruption of the GABAAR palmitoylation status alters theta/gamma band oscillations and uncouples the temporal relationships between neuronal discharge and theta/gamma rhythms. Our data provide pivotal evidence that normal neural activity is maintained through the PPT1-GABAAR α1 subunit axis, and dysfunction of such an axis may lead to neural network disorders.
Interruption of GABAAR palmitoylation homeostasis impairs learning and memory in mice
Disruption of PPT1 or its binding site to the GABAAR α1 subunit causes massive synaptic and neural network disorders. It would be interesting to understand how learning and memory are affected in these mice. A recent study reported that PPT1-KO mice exhibit impaired long- and short-term potentiation at 3-6 months [16]. We found that long-term potentiation (LTP) induced in the hippocampal SC-CA1 region was normal in 1- to 2-month-old mice, but impaired in 6- to 7-month-old PPT1-KI mice likely due to the hippocampal NMDAR 2a/b and PSD95 expression decrease (Figure S9). Consistently, the activation and expression of voltage-gated calcium channels was not altered in 1- to 2-month-old PPT1-KI mice, which sustained normal LTP formation (Fig. S10).
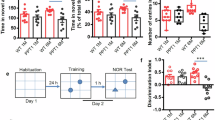
Despite normal LTP formation, PPT1-KI mice began to suffer from impaired spatial learning and memory as early as 2 months. As shown in Fig. 5A, PPT1-KI and Gabra1em1mice took more time to find the hidden platform and spent less time in the target entrance area in the water maze test on training days 2-5 compared to their WT littermates, and oral administration of BuHA (0.5 mM) improved their learning ability (Fig. 5A, B). No obvious differences in motor activity were detected between all the groups because swimming speed to locate the submerged escape platform during the 5 days of testing were the same (Fig. 5C). On the test day (day 6), the entry time and distance spent in the target arena were strongly reduced in PPT1-KI mice, which could be recovered by BuHA treatment (Fig. 5D–F). In addition, our Y-maze assay showed that 1- to 2-month-old PPT1-KI mice had impaired short-term spatial working memory as they entered and spent less time in the novel arm zone (Fig. 5G–I). Gabra1em1 mice showed similar behavioral results compared to PPT1-KI mice (Fig. 5).

Average latency to reach the hidden platform (A), number of times entering the platform area (B), averaged swimming speed (C) in five days. WT (n = 14), PPT1-KI (n = 9), PPT1-KI mice treated with BuHA (n = 10), and Gabra1em1 mice (n = 12), two-way ANOVA, WT vs. PPT1-KI: *P < 0.05, **P < 0.01, ***P < 0.001; PPT1-KI vs. PPT1-KI mice treated with BuHA: #P < 0.05, ##P < 0.01; WT vs. Gabra1em1: †P < 0.05, †††P< 0.001. D Representative paths of WT, PPT1-KI, PPT1-KI mice treated with BuHA, and Gabra1em1 mice were recorded on the spatial probe test day. Times entering the platform area (E) and total distance (F) in the test trial (day 6). WT mice: n = 14; PPT1-KI mice: n = 9; PPT1-KI mice treated with BuHA: n = 10; Gabra1em1 mice: n = 12, one-way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001. G Path taken in the Y-maze test by WT, PPT1-KI, PPT1-KI mice treated with BuHA and Gabra1em1 mice. Times of novel arm entries in test trial (H) and time spent in the novel arm zone (I). WT (n = 11), PPT1-KI (n = 11), PPT1-KI mice treated with BuHA (n = 11), and Gabra1em1 mice (n = 7); one-way ANOVA, *P < 0.05, ***P < 0.001. Data are represented as mean ± SEM.
Our results indicate that PPT1-KI and Gabra1em1mice begin to exhibit impaired spatial learning and memory formation during early development, likely due to the interruption of GABAAR α1 subunit palmitoylation and its resulting neural network disorders.
Discussion
Our study identified the GABAAR α1 subunit as a substrate of PPT1. The palmitoylation status of the GABAAR α1 subunit regulates inhibitory synaptic transmission, neuronal oscillation, and learning and memory ability. Our study sheds light on the potential role of postsynaptic GABAARs in the early onset of CLN1 disease.
GABAergic neurotransmission dysfunction and neuronal apoptosis
As a fatal infantile neurodegenerative disorder, CLN1 disease causes severe nervous system deficits, such as complete retinal degeneration and blindness, cognitive and motor deficits, seizures, and flat electroencephalograms [10, 36]. In PPT1-deficient mice, the loss of GABAergic interneurons in the cortical region occurs before the onset of seizure activity [19, 31]. Other studies have shown that GABAAR dysfunction is closely linked to neuronal apoptosis [37, 38].
Similarly, we observed considerable neuron loss in the hippocampus of 6- to 7-month-old PPT1-KI mice [22], as well as reduced expression of NMDAR 2a/b and PSD95, which may be the underlying mechanism for impaired LTP in old PP1-KI mice (Figure S9). Neuron loss was not apparent in 1-2-month-old PPT1-KI mice; however, overactivation of GABAAR was detected. To our surprise, newly generated Gabra1em1 mice exhibited severe developmental deficits with ~ 80% embryonic lethality, median lifespan of 40 days, and prominent weight loss irrespective of gender (Fig. S6B, C). Therefore, it is likely that the palmitoylation status change of GABAAR triggers chronic neuron loss in PPT1 deficiency and Gabra1em1mice, which further develops into severe symptoms, such as seizures. It would be of great interest to verify this proposal in the future.
Palmitoylation status of GABAAR α1 as a regulator of synaptic function
More than 600 palmitoylated proteins have been identified, a high percentage of which are localized to synapses [39,40,41]. Approximately 10% of palmitoylated synaptic proteins are the substrates of PPT1 [14]. PPT1 deficiency causes abnormal and persistent membrane accumulation of presynaptic vesicle proteins such as VAMP 2 and syntaxin-1 [18]; however, none of them are critical in the genesis of CLN1 disease. Proteomic studies have also predicted that many proteins involved in inhibitory GABAergic transmission, such as GAD 1/2, GABA transporter 1, and dynamin 1, are putative substrates of PPT1 [14]. GAD1 and GAD2 are critical enzymes for GABA synthesis at the presynaptic terminal [42, 43]. While essential for inhibitory neurotransmission, our study focuses on postsynaptic mechanisms, specifically the palmitoylation of the GABAAR α1 subunit. We have not examined the status of GAD1/2 in PPT1-KI mice in this study.
Numerous studies have demonstrated that both GABAAR [44,45,46] and gephyrin [47, 48] are palmitoylated proteins (Figure S3). Palmitoylation regulates the clustering and cell surface stability of GABAAR [44]. We discovered that PPT1 can depalmitoylate GABAAR α1 subunit at C260 (Fig. 3C, D). The lack of PPT1 caused abnormal membrane accumulation of GABAAR and enhancement of the amplitude and frequency of mIPSCs (Figs. 1A-F and 2D-E).
GABAAR α1 was demonstrated in this study as a new substrate for PPT1, which is also supported by the recent palmitoylome analyses that showed the implication of GABA receptor signaling in palmitoylation regulation [14]. The evidence about the palmitoylation modification of the GABAAR γ1 subunit have not been found, which may require further study. The GABAAR γ2 and gephyrin were reported to be palmitoylated by DHHC3 [49] and DHHC12 [47, 48]. Our experiments showed that γ2 membrane distribution (Fig. S2C) and gephyrin palmitoylation levels (Figure S3E) are not altered in PPT1 deficiency, and imply that GABAAR depalmitoylation is subunit-specific. Interestingly, PPT1 is also palmitoylated by DHHC3 at Cys6 which decreases PPT1 activity [50], raising a question that the altered palmitoylation level of PPT1 level may be implicated in the disease process of CLN1 disease, given that the deficiency of PPT1 activity in this disease, such a question may need to be solved in the future study.
In contrast to inhibitory synapses, in PPT1-KI mice, there was no change in excitatory synaptic transmission (Fig. 1G-M). We detected no retention of iGluRs or their scaffold proteins in the membranes of the hippocampal neurons of PPT1-KI mice at an early stage (Figure S1). The ABE results indicated that excitatory glutamate receptors are not depalmitoylated by PPT1 and are unlikely to be key molecules involved in the early onset of PPT1-related disease. It is also noteworthy that the mRNA expression of ABDH17B, another depalmitoylase in the central nervous system [20, 51,52,53], was lower in the hippocampus of PPT1-KI mice (Fig. S4H). Similarly, a previous study indicated that GluN2A and PSD-95 are not PPT1 substrates [19]. Nevertheless, they demonstrated that PPT1 controls the developmental NMDAR subunit switch from GluN2B to GluN2A, probably by the depalmitoylation of Fyn kinase or GluN2B, and thus, the surface retention of GluN2B. However, it is not clear whether GluN2B is a PPT1 substrate, and why GluN2A and PSD-95 expression levels decrease from P33 to P60, whereas GluN2B remains the same [19]. In addition, the rise time of NMDAR-mediated EPSCs did not show difference in PPT1-/- mice compared to that of the EPSC trace [19]. Thus, PPT1 can dramatically alter synaptic strength through GABAAR rather than through excitatory synaptic proteins.
Palmitoylation homeostasis of GABAAR regulates neural circuitry oscillation and brain function
GABAAR plays a critical role in generating neural network oscillations such as theta and gamma oscillations [54,55,56,57,58,59], which are key to cognitive processes [60, 61] and are associated with cognitive related behaviors such as place coding [62, 63], memory formation [64, 65], exploratory locomotion [66], attention, object recognition, and working memory [54, 56, 58, 59]. The enhanced theta and gamma oscillations power but the loss of theta or gamma phase locking in the CA1 area of 1- to 2-month-old PPT1-deficit mice indicate that GABAAR function is affected by PPT1. Interestingly, when we generated Gabra1em1 mice by mutating the binding sites between PPT1 and GABAAR α1 at C165 and C179, in vivo data also showed the enhanced theta and gamma oscillations in the CA1 area, as well as poor phase-locking in theta or gamma waves, which is similar to PPT1-KI mice (Fig. 4 and S8), provides the evidence that the GABAAR α1 subunit is likely the targeting molecule in PPT1 mutation. Moreover, PPT1 deficiency induces serious epilepsy in both humans suffering from NCLs [67,68,69] and animals [70]. In PPT1-/- mice, seizures can be detected after 7 months using electroencephalogram recordings [31]. Consistently, we observed epileptiform discharges at 6–7 months old but not in 1- to 2-month-old PPT1-KI mice (Figure S5).
Neural circuitry oscillations are fundamental to higher brain functions. One study found that older CLN1−/− mice (5–6 months) exhibited impairments in spatial learning and memory [71]. We demonstrated the impaired learning and memory even in 1- to 2-month-old PPT1-KI and Gabra1em1 mice (Fig. 5), which strongly correlates with the disrupted oscillation waves. These memory deficits in PPT1-KI mice could be partially rescued by feeding the mice with BuHA, indicating the crucial role of PPT1 in disease pathology.
In summary, we found that the absence of depalmitoylation by PPT1 leads to an accumulation of palmitoylated α1 subunits at the synaptic membrane, thereby strengthening GABAergic neurotransmission. The GABAAR α1 subunit is a substrate for PPT1-mediated depalmitoylation, and its palmitoylation homeostasis is crucial for GABAergic neurotransmission, neuronal network oscillations, and high brain functions in mice. Mutation at the GABAAR and PPT1 binding sites mimicked the phenotype of PPT1-deficient mice, underscoring the importance of GABAAR in the pathogenesis of NCL. Those findings position GABAAR as a potential therapeutic target that can be leveraged not only for CLN1 disease but also for other GABAAR dysfunction-induced neurodegenerative disorders.
Data availability
Data are made available from the corresponding author upon request.
References
Camp LA, Verkruyse LA, Afendis SJ, Slaughter CA, Hofmann SL. Molecular cloning and expression of palmitoyl-protein thioesterase. J Biol Chem. 1994;269:23212–9.
Jin J, Zhi X, Wang X, Meng D. Protein palmitoylation and its pathophysiological relevance. J Cell Physiol. 2021;236:3220–33.
Lemonidis K, Werno MW, Greaves J, Diez-Ardanuy C, Sanchez-Perez MC, Salaun C, et al. The zDHHC family of S-acyltransferases. Biochem Soc Trans. 2015;43:217–21.
Mitchell DA, Vasudevan A, Linder ME, Deschenes RJ. Protein palmitoylation by a family of DHHC protein S-acyltransferases. J Lipid Res. 2006;47:1118–27.
Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, et al. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. 1995;376:584–7.
Lin DT, Conibear E. Enzymatic protein depalmitoylation by acyl protein thioesterases. Biochem Soc Trans. 2015;43:193–8.
Santorelli FM, Garavaglia B, Cardona F, Nardocci N, Bernardina BD, Sartori S, et al. Molecular epidemiology of childhood neuronal ceroid-lipofuscinosis in Italy. Orphanet J Rare Dis. 2013;8:19.
Sleat DE, Gedvilaite E, Zhang Y, Lobel P, Xing J. Analysis of large-scale whole exome sequencing data to determine the prevalence of genetically-distinct forms of neuronal ceroid lipofuscinosis. Gene. 2016;593:284–91.
Williams RE. 361Appendix 1: NCL incidence and prevalence data. In: Mole S, Williams R, Goebel H, editors. The neuronal ceroid lipofuscinoses (Batten Disease). Oxford University Press; 2011.
Santavuori P, Haltia M, Rapola J, Raitta C. Infantile type of so-called neuronal ceroid-lipofuscinosis. 1. A clinical study of 15 patients. J Neurol. Sci. 1973;18:257–67.
Bouchelion A, Zhang Z, Li Y, Qian H, Mukherjee AB. Mice homozygous for c.451C>T mutation in Cln1 gene recapitulate INCL phenotype. Ann Clin Transl Neurol. 2014;1:1006–23.
Hobert JA, Dawson G. Neuronal ceroid lipofuscinoses therapeutic strategies: past, present and future. Biochim Biophys Acta. 2006;1762:945–53.
Mukherjee AB, Appu AP, Sadhukhan T, Casey S, Mondal A, Zhang Z, et al. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol Neurodegener. 2019;14:4.
Gorenberg EL, Massaro Tieze S, Yücel B, Zhao HR, Chou V, Wirak GS, et al. Identification of substrates of palmitoyl protein thioesterase 1 highlights roles of depalmitoylation in disulfide bond formation and synaptic function. PLoS Biol. 2022;20:e3001590.
Chu-LaGraff Q, Blanchette C, O’Hern P, Denefrio C. The batten disease palmitoyl protein thioesterase 1 gene regulates neural specification and axon connectivity during Drosophila embryonic development. PLoS ONE. 2010;5:e14402.
Sapir T, Segal M, Grigoryan G, Hansson KM, James P, Segal M, et al. The interactome of palmitoyl-protein thioesterase 1 (PPT1) affects neuronal morphology and function. Front Cell Neurosci. 2019;13:92.
Lange J, Haslett LJ, Lloyd-Evans E, Pocock JM, Sands MS, Williams BP, et al. Compromised astrocyte function and survival negatively impact neurons in infantile neuronal ceroid lipofuscinosis. Acta Neuropathol Commun. 2018;6:74.
Kim SJ, Zhang Z, Sarkar C, Tsai PC, Lee YC, Dye L, et al. Palmitoyl protein thioesterase-1 deficiency impairs synaptic vesicle recycling at nerve terminals, contributing to neuropathology in humans and mice. J Clin Investig. 2008;118:3075–86.
Koster KP, Francesconi W, Berton F, Alahmadi S, Srinivas R, Yoshii A. Developmental NMDA receptor dysregulation in the infantile neuronal ceroid lipofuscinosis mouse model. eLife. 2019;8:e40316.
Yokoi N, Fukata Y, Sekiya A, Murakami T, Kobayashi K, Fukata M. Identification of PSD-95 depalmitoylating enzymes. J Neurosci. 2016;36:6431–44.
Nakazono T, Jun H, Blurton-Jones M, Green KN, Igarashi KM. Gamma oscillations in the entorhinal-hippocampal circuit underlying memory and dementia. Neurosci Res. 2018;129:40–46.
Zhang X, Wang M, Feng B, Zhang Q, Tong J, Wang M. et al. Seizures in PPT1 knock-in mice are associated with inflammatory activation of microglia. Int J Mol Sci. 2022;23:5586.
Sarkar C, Chandra G, Peng S, Zhang Z, Liu A, Mukherjee AB. Neuroprotection and lifespan extension in Ppt1(-/-) mice by NtBuHA: therapeutic implications for INCL. Nat Neurosci. 2013;16:1608–17.
Bleakley LE, McKenzie CE, Reid CA. Efficacy of antiseizure medication in a mouse model of HCN1 developmental and epileptic encephalopathy. Epilepsia. 2023;64:e1–e8.
Adamos DA, Kosmidis EK, Theophilidis G. Performance evaluation of PCA-based spike sorting algorithms. Comput Methods Prog Biomed. 2008;91:232–44.
Xu H, Liu L, Tian Y, Wang J, Li J, Zheng J, et al. A disinhibitory microcircuit mediates conditioned social fear in the prefrontal cortex. Neuron. 2019;102:668–682.e665.
Sfondouris JL, Quebedeaux TM, Holdgraf C, Musto AE. Combined process automation for large-scale EEG analysis. Comput Biol Med. 2012;42:129–34.
Fisher NI. Statistical analysis of circular data || Descriptive methods. 1993p. 15–38. https://doi.org/10.1017/CBO9780511564345
Gregoire TG. Statistical analysis of circular data. Forest Science. 1996;42:515–16.
Berens P. CircStat: a MATLAB toolbox for circular statistics. J Stat Softw. 2009;31:1–21.
Kielar C, Maddox L, Bible E, Pontikis CC, Macauley SL, Griffey MA, et al. Successive neuron loss in the thalamus and cortex in a mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. 2007;25:150–62.
Ahrens-Nicklas RC, Tecedor L, Hall AF, Lysenko E, Cohen AS, Davidson BL. et al. Neuronal network dysfunction precedes storage and neurodegeneration in a lysosomal storage disorder. JCI insight. 2019;4:e131961.
Musto AE, Walker CP, Petasis NA, Bazan NG. Hippocampal neuro-networks and dendritic spine perturbations in epileptogenesis are attenuated by neuroprotectin d1. PLoS ONE. 2015;10:e0116543.
Rutishauser U, Ross IB, Mamelak AN, Schuman EM. Human memory strength is predicted by theta-frequency phase-locking of single neurons. Nature. 2010;464:903–7.
Arroyo-García LE, Isla AG, Andrade-Talavera Y, Balleza-Tapia H, Loera-Valencia R, Alvarez-Jimenez L, et al. Impaired spike-gamma coupling of area CA3 fast-spiking interneurons as the earliest functional impairment in the App(NL-G-F) mouse model of Alzheimer’s disease. Mol Psychiatry. 2021;26:5557–67.
Mole SE, Williams RE, Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107–26.
Qiu J, Shi P, Mao W, Zhao Y, Liu W, Wang Y. Effect of apoptosis in neural stem cells treated with sevoflurane. BMC Anesthesiol. 2015;15:25.
Hann UA, Hann SR. The a1 subunit of GABAA receptor is repressed by c-Myc and is pro-apoptotic. J Cell Biochem. 2006;97:1094–1103.
Collins MO, Woodley KT, Choudhary JS. Global, site-specific analysis of neuronal protein S-acylation. Sci Rep. 2017;7:4683.
Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, et al. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456:904–9.
Segal-Salto M, Hansson K, Sapir T, Kaplan A, Levy T, Schweizer M, et al. Proteomics insights into infantile neuronal ceroid lipofuscinosis (CLN1) point to the involvement of cilia pathology in the disease. Hum Mol Genet. 2017;26:1678.
Erlander MG, Tillakaratne NJ, Feldblum S, Patel N, Tobin AJ. Two genes encode distinct glutamate decarboxylases. Neuron. 1991;7:91–100.
Bu DF, Erlander MG, Hitz BC, Tillakaratne NJ, Kaufman DL, Wagner-McPherson CB, et al. Two human glutamate decarboxylases, 65-kDa GAD and 67-kDa GAD, are each encoded by a single gene. Proc Natl Acad Sci USA. 1992;89:2115–9.
Rathenberg J, Kittler JT, Moss SJ. Palmitoylation regulates the clustering and cell surface stability of GABAA receptors. Mol Cell Neurosci. 2004;26:251–7.
Fang C, Deng L, Keller CA, Fukata M, Fukata Y, Chen G, et al. GODZ-mediated palmitoylation of GABA(A) receptors is required for normal assembly and function of GABAergic inhibitory synapses. J Neurosci. 2006;26:12758–68.
Keller CA, Yuan X, Panzanelli P, Martin ML, Alldred M, Sassoè-Pognetto M, et al. The gamma2 subunit of GABA(A) receptors is a substrate for palmitoylation by GODZ. J Neurosci. 2004;24:5881–91.
Shen ZC, Wu PF, Wang F, Xia ZX, Deng Q, Nie TL, et al. Gephyrin palmitoylation in basolateral amygdala mediates the anxiolytic action of benzodiazepine. Biol Psychiatry. 2019;85:202–13.
Dejanovic B, Semtner M, Ebert S, Lamkemeyer T, Neuser F, Lüscher B, et al. Palmitoylation of gephyrin controls receptor clustering and plasticity of GABAergic synapses. PLoS Biol. 2014;12:e1001908.
Kilpatrick CL, Murakami S, Feng M, Wu X, Lal R, Chen G, et al. Dissociation of Golgi-associated DHHC-type zinc finger protein (GODZ)- and sertoli cell gene with a zinc finger domain-β (SERZ-β)-mediated palmitoylation by loss of function analyses in knock-out mice. J Biol Chem. 2016;291:27371–86.
Segal-Salto M, Sapir T, Reiner O. Reversible cysteine acylation regulates the activity of human palmitoyl-protein thioesterase 1 (PPT1). PLoS ONE. 2016;11:e0146466.
Won SJ, Cheung See Kit M, Martin BR. Protein depalmitoylases. Crit Rev Biochem Mol Biol. 2018;53:83–98.
Kanadome T, Yokoi N, Fukata Y, Fukata M. Systematic screening of depalmitoylating enzymes and evaluation of their activities by the Acyl-PEGyl exchange gel-shift (APEGS) assay. Methods Mol Biol. 2019;2009:83–98.
Lin DT, Conibear E. ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. eLife. 2015;4:e11306.
Garner HL, Whittington MA, Henderson Z. Induction by kainate of theta frequency rhythmic activity in the rat medial septum-diagonal band complex in vitro. J Physiol. 2005;564:83–102.
Lu CB, Henderson Z. Nicotine induction of theta frequency oscillations in rodent hippocampus in vitro. Neuroscience. 2010;166:84–93.
Lu CB, Li CZ, Li DL, Henderson Z. Nicotine induction of theta frequency oscillations in rodent medial septal diagonal band in vitro. Acta Pharmacol Sin. 2013;34:819–29.
Soltesz I, Deschênes M. Low- and high-frequency membrane potential oscillations during theta activity in CA1 and CA3 pyramidal neurons of the rat hippocampus under ketamine-xylazine anesthesia. J Neurophysiol. 1993;70:97–116.
Colgin LL, Moser EI. Gamma oscillations in the hippocampus. Physiology. 2010;25:319–29.
Buzsáki G, Wang XJ. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35:203–25.
Kahana MJ, Sekuler R, Caplan JB, Kirschen M, Madsen JR. Human theta oscillations exhibit task dependence during virtual maze navigation. Nature. 1999;399:781–4.
Belchior H, Lopes-Dos-Santos V, Tort AB, Ribeiro S. Increase in hippocampal theta oscillations during spatial decision making. Hippocampus. 2014;24:693–702.
Jensen O, Lisman JE. Position reconstruction from an ensemble of hippocampal place cells: contribution of theta phase coding. J Neurophysiol. 2000;83:2602–9.
Buzsáki G. Theta rhythm of navigation: link between path integration and landmark navigation, episodic and semantic memory. Hippocampus. 2005;15:827–40.
Winson J. Loss of hippocampal theta rhythm results in spatial memory deficit in the rat. Science. 1978;201:160–3.
Jones MW, Wilson MA. Theta rhythms coordinate hippocampal-prefrontal interactions in a spatial memory task. PLoS Biol. 2005;3:e402.
Buzsáki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–40.
Kravljanac R, Sims K. A case of juvenile CLN1- challenge in diagnosis and epilepsy treatment. Neurocase. 2021;27:165–8.
Maeser S, Petre BA, Ion L, Rawer S, Kohlschütter A, Santorelli FM, et al. Enzymatic diagnosis of neuronal lipofuscinoses in dried blood spots using substrates for concomitant tandem mass spectrometry and fluorimetry. J Mass Spectrom. 2021;56:e4675.
Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases - clinical perspectives. Biochim Biophys Acta. 2013;1832:1801–6.
Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, et al. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc Natl Acad Sci USA. 2001;98:13566–71.
Dearborn JT, Harmon SK, Fowler SC, O’Malley KL, Taylor GT, Sands MS, et al. Comprehensive functional characterization of murine infantile Batten disease including Parkinson-like behavior and dopaminergic markers. Sci Rep. 2015;5:12752.
Acknowledgements
This work was supported by funds from the Open Project Program of the Third Affiliated Hospital of Xinxiang Medical University KFKTYB202111 (JT), Scientific and Technological Project in He’nan Province 232102311227 (JT), Startup Grants of Xinxiang Medical University 300-505481 (LHG), National Natural Science Foundation 81771517 (CBL), Hubei 3551 Talented Fund 3551-T82 (SYP). We thank Hai Huang, Wenyan Han, and Jisen Huai for helpful comments on this manuscript.
Author information
Authors and Affiliations
Contributions
JT, JG, YWQ, SYP and LHG designed experiments; JT, JG, YWQ, ZYG, QQW, YL, CBL, SYP and LHG performed experiments and analyses; JT, XZ, MLR, JMX, YYL and LHG performed electrophysiological data analyses; JG, YWQ, ZYG, QQW, YL, TGYGXY, ZYL, JL, HYS and YHM conducted behavior test, biochemical analysis and cell line culture; JT, SYP, CBL and LHG obtained the funding; JT, CBL, SYP and LHG supervised the study and cowrote the paper. All the authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The experiments did not involve tissues from humans. Animal use and procedures were performed according to the regulations and requirements of XXMU Animal Ethics Committee (No. XYLL2021053).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tong, J., Gao, J., Qi, Y. et al. GABAAR-PPT1 palmitoylation homeostasis controls synaptic transmission and circuitry oscillation. Transl Psychiatry 14, 488 (2024). https://doi.org/10.1038/s41398-024-03206-1
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-024-03206-1
This article is cited by
-
Dysfunction of GABAergic interneurons underlies altered neural network oscillations associated with epileptiform activity in PPT1-deficient mice
Translational Psychiatry (2026)
-
Neuronal ceroid lipofuscinosis: underlying mechanisms and emerging therapeutic targets
Nature Reviews Neurology (2025)
