
Abstract
Major Depressive Disorder (MDD) is increasingly recognized as a neuroinflammatory condition characterized by dysregulated cytokine networks. This comprehensive review examines the immunomodulatory effects of antidepressant medications, revealing their significant impact on Th1/Th2 cytokine balance beyond their classical neurotransmitter actions. Clinical data show that diverse antidepressant classes consistently demonstrate immunomodulatory properties that extend beyond their classical neurotransmitter effects. These medications reduce pro-inflammatory markers (IFN-γ, TNF-α, IL-6) while enhancing anti-inflammatory cytokines (IL-10, TGF-β), effects particularly relevant for treatment-resistant cases with elevated baseline inflammation. The therapeutic potential of these immunoregulatory effects is supported by emerging interventions, including low-dose IL-2 immunotherapy, vagus nerve stimulation, and microbiota-targeted therapies, which show promise for specific depression subtypes. Importantly, these approaches appear most effective when guided by inflammatory biomarkers, suggesting a path toward personalized treatment strategies. By integrating findings from clinical studies and translational research, this work establishes immune modulation as a fundamental component of antidepressant action. The review provides a framework for developing next-generation treatments that target neuroimmune pathways in MDD, with particular emphasis on practical applications for treatment-resistant cases. These insights bridge the gap between neuropharmacology and clinical psychiatry, offering new therapeutic possibilities for patients with inflammation-associated depression.
Similar content being viewed by others


Introduction
Major depressive disorder (MDD), commonly referred to as clinical depression, is a mood disorder characterized by a spectrum of persistent sense of sadness, loss of interest or pleasure in activities, and a variety of physical and other emotional symptoms [1, 2]. MDD is the second leading cause of years lived with disability (YLDs) globally [3, 4]. As a severe condition, MDD can profoundly affect an individual’s thoughts, feelings, and behavior, often necessitating professional treatment that may include therapy, pharmacological interventions [2], and lifestyle modifications [5].
Current models of MDD pathogenesis propose that it is a multifactorial condition, with at least six interrelated hypotheses that have been extensively reviewed elsewhere [6]. These include a) the hypothalamic-pituitary-adrenal (HPA) axis dysregulation hypothesis, which suggests that MDD is associated with an overactive HPA axis, leading to cortisol dysregulation; b) the monoamine hypothesis, which posits that MDD is caused by a deficiency of neurotransmitters such as serotonin (5-HT), norepinephrine (NE), and dopamine (DA) in the brain; c) the genetic and epigenetic anomaly hypothesis, which implicates inherited and acquired changes in gene expression in the development of MDD; d) the brain circuit hypothesis, which suggests that MDD results from alterations in brain structure and function, particularly in areas responsible for mood regulation and cognitive function; e) the social psychological hypothesis, which considers the role of personal relationships, social support, and other psychosocial factors in the onset and course of MDD; and f) the inflammatory hypothesis, which links chronic inflammation with the pathophysiology of MDD [6].
Among these hypotheses, the involvement of the immune system in the pathogenesis of MDD represents a growing research field. Increasing evidence suggests that neuroinflammation, driven by glial cell activation and the release of pro-inflammatory cytokines, plays a central role in depressive symptomatology [6]. Elevated levels of cytokines such as IL-1β, IL-6, and TNF-α have been consistently observed in individuals with MDD, implicating immune dysregulation in disease onset and progression [7, 8] (Fig. 1). Astrocytes and microglia, key regulators of the brain’s immune response, respond to various stressors by producing reactive oxygen species (ROS) and inflammatory mediators. Oxidative stress not only damages neuronal structures but also serves as a trigger for inflammasome activation, particularly the NLRP3 inflammasome, which amplifies inflammatory signaling through caspase-1 activation and subsequent release of IL-1β and IL-18 [8,9,10,11,12]. These immune-related mechanisms are further linked to a reduction in neurotrophic support, notably brain-derived neurotrophic factor (BDNF), which is essential for synaptic plasticity and neuronal survival [10, 11]. Experimental models demonstrate that inhibition of astrocytic or microglial inflammatory responses, or targeting of ROS production, can reverse depressive-like behaviors, reinforcing the notion that MDD may, in part, result from or cause chronic neuroinflammation and immune dysfunction [13,14,15].

This figure depicts the interplaybetween the immune system and central nervous system (CNS) homeostasis. Disruption of the blood-brain barrier (BBB) increases brain access to immune cells and cytokines, activating microglia and promoting neuroinflammation. Similarly, peripheral inflammation, driven by immune cell activation and the release of peripheral cytokines (e.g., IL-1β, IL-6, TNF-α, IFN-γ) further contribute by altering vascular permeability and crossing or signaling to the brain. IFN-γ also activates IDO, diverting tryptophan from serotonin to neurotoxic kynurenine metabolites (e.g., 3-HK, QA), which are implicated in depressive symptoms. IDO Indoleamine 2,3-dioxygenase, IFN-γ Interferon-gamma, TNF-α Tumor Necrosis Factor-alpha, IL Interleukin, QA Quinolinic Acid, 3-HK 3-Hydroxykynurenine, Treg Regulatory T cells, CREB cAMP response element-binding protein, Htr1b 5-Hydroxytryptamine Receptor 1B. Created with BioRender (www.BioRender.com).
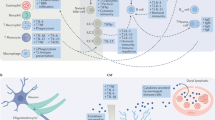
Currently, the primary pharmacological treatments for depression target synaptic neurotransmitter levels [16]. These treatments include tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), serotonin-noradrenaline reuptake inhibitors (SNRIs), as well as mood stabilizers and anticonvulsants like lamotrigine and valproic acid. Antipsychotics such as quetiapine, olanzapine, and amisulpride [16], which bind to neuronal receptors, are also used (Fig. 2). However, in addition to their well-recognized mechanisms of action on neurotransmitter transporters [6, 17] and on pre- and postsynaptic receptors (e.g., serotonergic, dopaminergic, and adrenergic receptors) [17, 18], there is emerging evidence suggesting that these drugs also exert neuroimmunomodulatory effects [19,20,21], biding to neuronal receptors expressed by several immune cells [6, 17,18,19,20,21,22] (Fig. 3).

This figure illustrates the mechanisms by which different classes of antidepressants modulate neurotransmitter activity, as well as an overview of relevant neurotransmitter receptor groups and immune cell interactions. Monoamine Oxidase Inhibitors (MAOIs) inhibit the breakdown of neurotransmitters at nerve terminals, increasing the storage and subsequent release of norepinephrine (NE) and serotonin (5-HT), thereby enhancing their synaptic activity. Tricyclic Antidepressants (TCAs) prevent the reuptake of NE and 5-HT at the post-synaptic receptors, prolonging their effects. Selective Serotonin Reuptake Inhibitors (SSRIs) specifically inhibit the serotonin transporter (SERT), increasing serotonin levels while sparing norepinephrine transporters and minimizing interaction with peripheral receptors. Serotonin and Norepinephrine Reuptake Inhibitors (SNRIs) target both SERT and norepinephrine transporters (NET), increasing the availability of both neurotransmitters in the synapse, with minimal action on peripheral receptors. Created with BioRender.com.

Schematic representation of neurotransmitter receptors expressed on immune cells, including serotonin (5-HT), dopamine (DA), norepinephrine (NE), gamma-aminobutyric acid (GABA), acetylcholine (ACh), and histamine (H1). Different classes of antidepressants interact with these receptors, modulating immune function: SARIs (mast cells, B-cells), SSRIs (natural killer cells, dendritic cells, eosinophils), SNRIs (macrophages, dendritic cells), and NASSAs (monocytes). These interactions contribute to neuroimmune regulation, influencing inflammatory pathways and immune cell activity. SARIs serotonin antagonist and reuptake inhibitors, SSRIs selective serotonin reuptake inhibitors, SNRIs serotonin-norepinephrine reuptake inhibitors, NASSAs noradrenergic and specific serotonergic antidepressants, GABA(A)R/GABA(B)R gamma-aminobutyric acid receptors type A and B, AChR acetylcholine receptor. Created with BioRender.com.
In this context, this article reviews the neuroimmunological aspects of MDD, focusing on the impact of antidepressants on cytokines, contributing to a better understanding of the interplay between these drugs and the immune system [23,24,25]. By synthesizing interdisciplinary knowledge from psychiatry, immunology, and neuroscience, this review aims to map key trends and gaps in the field, offering insights into the role of antidepressants in regulating T Helper 1 and 2 (Th1/Th2) cytokines and their broader implications for neuroimmune modulation.
Cytokines, neuroendocrinoimmunology, and depression
The first cytokine discovered was interferon, identified in 1957 by Alick Isaacs and Jean Lindenmann [26, 27]. Following the discovery of interferons, other cytokines were identified, including several interleukins, tumor necrosis factor (TNF), and growth factors. These cytokines play essential roles in immune response and cell signaling [28]. However, cytokines are now recognized to have broader roles beyond immune regulation. For instance, they can shape the nervous system’s function and control the manifestation of host behavior [29]. Various cell types, including those in the CNS, such as astrocytes, microglia, and even neurons, which typically communicate through neurotransmitters and neuropeptides, can also produce cytokines [24, 30, 31] (Fig. 4).

This figure illustrates the dynamic cross-talk between the nervous and immune systems, highlighting four primary interaction types: neuronal-neuronal, immune-neuronal, neuronal-immune, and immune-immune. Neurotransmitters, neuropeptides, and cytokines are shown as key chemical messengers facilitating these interactions, while their respective receptors are indicated for each communication pathway. The diagram emphasizes how these systems influence and regulate one another, providing insight into the complex mechanisms underlying neuroimmune modulation. Created with BioRender.com.
In turn, immune cells primarily communicate through cytokine signaling via cytokine receptors, but they can also secrete and respond to molecules commonly used by the CNS, such as neurotransmitters and neuropeptides [32]. Thus, the intricate interplay between the CNS and the immune system exemplifies the bidirectional communication essential for maintaining homeostasis and responding to physiological challenges. Extended exposure to environmental stress activates molecular pathways, enhancing cytokine secretion and signaling [33], resulting in dysregulated inflammation within the CNS.
This theory of depression implicates both the innate and adaptive arms of the immune network [34]. IL-1 [35] and IL-6 [36] stimulate the release of corticotropin-releasing hormone (CRH) from the hypothalamus, which in turn promotes the secretion of adrenocorticotropic hormone (ACTH) from the pituitary gland, leading to cortisol release from the adrenal glands [37]. These cytokines act as key mediators of the immune response to stress and inflammation, and their activation of the HPA axis illustrates the intricate crosstalk between immune and endocrine systems under such conditions [38].
The HPA axis consists of intricate loops of stimulation and inhibition involving the hypothalamus, brain, pituitary gland, and adrenal glands, which play a role in controlling the production of glucocorticoids [39]. Cortisol, released by the adrenal glands, binds to mineralocorticoid receptors (MRs) and glucocorticoid receptors (GRs) in the brain and immune cells [40, 41]. Exposure to stress also stimulates a series of afferent neural pathways that induce the release of CRH and arginine vasopressin (AVP) from parvocellular neurons of the paraventricular nucleus (PVN) of the hypothalamus [42].
CRH and AVP reach the pituitary gland, where they bind to their specific receptors and stimulate the release of ACTH from corticotropin cells. ACTH stimulates the adrenal gland to produce and secrete glucocorticoid hormones into general circulation. Glucocorticoids operate on their target tissues, including the brain, by binding primarily to the GR and MR, thereby increasing or reducing neuroendocrine secretion throughout the axis [43].
The development of MDD involves an imbalance or malfunction of MR and/or GR within the HPA axis [39]. The excessive release of glucocorticoids and pro-inflammatory cytokines (e.g., IFNs) disrupts noradrenergic and serotonergic neurotransmission in the brain, contributing to depression and fatigue [30, 33, 44,45,46] This disruption resembles the sickness syndrome observed in humans and animals [47, 48], which is characterized by changes in sleep, appetite, activity, mood, energy, and sociability [49].
Cytokines and brain function: direct and indirect mechanisms
Cytokines can influence brain function through two complementary mechanisms. Peripherally produced cytokines may enter the brain by crossing the blood-brain barrier (BBB), either via passive diffusion (for lipophilic molecules) or through active transport systems [50]. Concurrently, locally produced cytokines within the CNS play a direct role in modulating neural activity. During neuroinflammatory states, increased cytokine production, whether peripheral or central, disrupts the balance between excitatory and inhibitory signaling, impairs synaptic plasticity, and contributes to synapse loss and network dysfunction [51].
Peripheral cytokines can cross the BBB through passive diffusion (if lipophilic) or active transport mechanisms, acting on neurons and supporting cells such as astrocytes and microglia. Additionally, cytokines can modulate brain function through peripheral immune signaling pathways, such as afferent vagus nerve activation, which triggers neuroinflammatory responses within the central nervous system [52].
There are four primary mechanisms by which cytokines can influence brain activity: 1) passive transport at circumventricular sites where the BBB is absent, 2) binding to cerebral vascular endothelial cells, generating secondary messengers such as prostaglandins and nitric oxide, 3) carrier-mediated transport across the BBB, and 4) activation of peripheral nerve terminals at the site of cytokine release [53]. Furthermore, cytokines are also synthesized and released within the CNS, primarily by astrocytes and microglia, with some evidence suggesting that neurons may produce them under specific conditions [54]. Once in the brain, cytokines have various effects, including immunological, neurochemical, neuroendocrine, and behavioral changes [55].
Inflammation and the systemic neuroimmunological interactions in MDD
As systemic inflammation intensifies in severity and during systemic infections, the BBB permeability to solutes escalates, accompanied by increased serum levels of cytokines, heightened leukocyte trafficking, and infiltration of immune cells in the brain, all contributing to neuroinflammation [56]. Aggressive immunotherapies, such as checkpoint inhibitor treatments and T-cell therapies, can exacerbate these processes, leading to cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) [57].
Injection of lipopolysaccharide (LPS) induces depression-like behavior in animals. This inflammatory response is associated with increased production of pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, which result in behavioral alterations, including anhedonia and reduced locomotor activity [58]. Additionally, LPS administration has been shown to cause splenomegaly, reflecting peripheral immune activation and further contributing to neuroinflammation, a process mediated by the activation of indoleamine 2,3-dioxygenase (IDO) [59]. IDO reduces the availability of tryptophan, a precursor of serotonin, leading to decreased serotonin levels and contributing to depressive symptoms [59]. This process is also induced by pro-inflammatory cytokines, such as IFN-γ [60], which explains why clinical treatments with IFNs, used for viral infections such as chronic hepatitis B and C, often have depression as a side effect [61].
Additionally, conditions like COVID-19 can cause severe pneumonia and neurological complications, including stroke, neurovascular damage, blood-brain barrier disruption, and elevated intracranial pro-inflammatory cytokines, which contribute to endothelial cell damage in the brain [62]. Studies suggest that COVID-19-induced microbiome dysbiosis can disrupt mucosal immune responses, increase intestinal permeability, and lead to secondary bloodstream infections, with potentially severe consequences [63]. Furthermore, persistent dysbiosis of the gut microbiome following SARS-CoV-2 infection has been linked to impaired signaling along the microbiota-gut-brain (MGB) axis, which may contribute to chronic neuroinflammation and cognitive impairments seen in postacute sequelae of COVID-19 (PASC) [64,65,66]. This altered communication between the gut and the brain, exacerbated by reduced levels of beneficial butyrate-producing bacteria and increased pro-inflammatory taxa, can lead to activation of microglia and elevation of cytokines like CCL11, impairing neurogenesis and contributing to the development of symptoms such as brain fog and memory deficits [67,68,69]. These findings support the hypothesis that gut dysbiosis and subsequent MGB axis disruption represent a potential mechanistic link between gastrointestinal pathology and COVID-19-related neuropsychiatric manifestations.
Th cell differentiation and its relationship with depression
The differentiation of T helper (Th) cells is influenced by cytokines, and their chronic production in patients with MDD may affect the fate of these cells [70]. Evidence suggests that depressed patients have elevated levels of Th1 and Th2 cytokines [71] and an increased Th1/Th2 (IFN-γ/IL-4) ratio [72], which can be reduced by antidepressants [73].
Th1 cytokines are primarily pro-inflammatory and include IFN-γ, TNF-α, IL-2, IL-1β, and IL-6 [74]. Elevated levels of Th1 cytokines are associated with depression [75]. Chronic inflammation can alter neurotransmitter metabolism, neuroendocrine function, and synaptic plasticity, contributing to the development of depressive symptoms [52]. TNF-α [76] and IFN-γ [77] can induce neurotoxic effects, contributing to neuronal damage and dysfunction [31]. This event can negatively impact brain regions involved in mood regulation, such as the hippocampus and prefrontal cortex [78]. Additionally, Th1 cytokines can stimulate the HPA axis, resulting in increased cortisol production, further immune dysregulation, and mood disturbances [79].
Th2 cytokines are primarily anti-inflammatory and include IL-4, IL-10, and IL-13 [80]. They counteract the effects of Th1 cytokines, reducing inflammation. Adequate levels of anti-inflammatory cytokines may help protect against depression by mitigating the harmful effects of chronic inflammation and maintaining a healthy brain environment conducive to normal mood regulation [75]. IL-10, for example, has neuroprotective properties that can help preserve neuronal function and integrity [81].
Additionally, MDD patients show elevated levels of Th17 cells in the blood [82], with the highest levels observed in individuals at high risk of suicide [83]. In vitro activation of CD4+ cells from patients with generalized anxiety disorder also induces a Th17 phenotype [84, 85], and autoimmune diseases associated with increased Th17 cells frequently coexist with depression [86, 87]. The interleukin IL-17A, characteristic of Th17 cells, has been found elevated in some studies of depressed patients [88], although this finding has not been consistently replicated across other studies [89,90,91]. Furthermore, IL-17A may predict the response to certain antidepressants [92]. Animal model studies further support the link between Th17/IL-17A and depression, demonstrating that IL-17A administration induces depressive-like behaviors [93], while stress increases its levels [94, 95] and promotes the accumulation of Th17 cells in the brain [96] and spleen after social stress [97].
Antidepressants
Selective serotonin reuptake inhibitors (SSRIs)
SSRIs selectively block the 5-hydroxytryptamine (5-HT or serotonin) transporter [98], progressively increasing extracellular 5-HT levels, also in circulation, and influencing the immune response in a dose-dependent manner [99]. Evidence indicates that the enhancement of 5-HT activity mediated by SSRIs has immunomodulatory effects on blood cytokine levels [100]. A clinical study showed that treatment with SSRIs (Fluoxetine 20 mg/day, Paroxetine 20 mg/day, Sertraline 100 mg/day, or Escitalopram 10 mg/day) reduced IFN-γ levels in MDD patients. In turn, IL-4 levels, undetectable in healthy individuals, increased in MDD patients. Although SSRI treatment reduced the levels of this cytokine, some pro-inflammatory cytokines remained elevated in MDD patients [99]. Consistently, Sutcigil and coauthors [101] found that MDD patients exhibited increased levels of pro-inflammatory cytokines (IL-2, IL-12, and TNF-α) and monocyte chemoattractant protein-1 (MCP-1) compared to healthy controls, along with reduced levels of IL-4 and TGF-β1 [101].
SSRIs have anti-inflammatory effects not only through cytokines. SSRIs (e.g., fluvoxamine) also exhibit the capacity to modulate endothelial cell expression of vascular cell adhesion molecule (VCAM-1) and intracellular adhesion molecule (ICAM-1), typically upregulated during inflammatory events [102]. Furthermore, SSRIs have downregulated the expression of inflammatory mediators, including cyclo-oxygenase 2 (COX-2) and inducible nitric oxide synthase (iNOS) [102]. SSRIs also diminish polymorphonuclear chemotaxis and T cell proliferation [103].
Sigma-1 receptors (Sig-1R) have emerged as important regulators in the pathophysiology of MDD, bridging neuroinflammation, synaptic plasticity, and antidepressant efficacy. Chronic stress and depression are associated with elevated pro-inflammatory cytokines (e.g., IL-6, TNF-α), which disrupt the excitatory/inhibitory (E/I) balance in the prefrontal cortex (PFC) by impairing GABAergic transmission and enhancing glutamatergic signaling [104, 105]. Sig-1R agonists, such as fluvoxamine and cutamesine (SA-4503), counteract these effects by suppressing neuroinflammation (via NF-κB/NLRP3 inhibition) and restoring GABA/glutamate homeostasis [106]. For instance, fluvoxamine reduces IL-6 levels by 40% in depressed patients, correlating with improved mood [107], while Sig-1R activation enhances GABA uptake and modulates 5-HT1A receptor signaling, which is critical for maintaining E/I balance [108]. Additionally, Sig-1R agonists promote neurosteroid synthesis and BDNF release, further mitigating cytokine-induced synaptic damage [109]. Despite mixed clinical trial outcomes (e.g., igmesine’s phase III failure), recent evidence underscores Sig-1R’s role in rapid serotonergic modulation [110] and microglial regulation, positioning it as a promising target for inflammation-resistant depression. Future therapies may combine Sig-1R ligands (e.g., YL-0919) with cytokine inhibitors to address both monoaminergic deficits and neuroimmune dysfunction [111].
Serotonin and norepinephrine reuptake inhibitors (SNRIs)
Venlafaxine, a serotonin and norepinephrine reuptake inhibitor (SNRI), has an intricate pharmacodynamics. At lower doses (75 mg), venlafaxine’s effects on neurotransmission and receptor expression mirror those of selective SSRIs [112]. However, at higher doses (375 mg), it functions as a true SNRI, equally inhibiting the reuptake of serotonin and norepinephrine (NE) [113].
Venlafaxine has demonstrated the ability to decrease IFN-γ production in whole-blood cells from patients with treatment-resistant depression while increasing anti-inflammatory cytokines like IL-10 [73]. Moreover, venlafaxine also demonstrated anti-inflammatory effects in an astroglia-microglia co-culture model of neuroinflammation [114]. Key findings include the reversal of inflammation-induced depolarization of astrocytic membrane resting potential, increased release of the anti-inflammatory cytokine TGF-beta, and reduced levels of pro-inflammatory cytokines IL-6 and IFN-gamma. Likewise, Vollmar et al. [115] reported venlafaxine’s effects on IFN-γ and IL-12 p40, with an overall reduction in cytokine secretion by 50% [115]. Venlafaxine also decreased the expression levels of CCL5, IL-6, and TNF-α in a dose-dependent manner. Peritoneal macrophages released fewer pro-inflammatory cytokines IL-6 and TNF-α when treated with venlafaxine [115] compared to untreated cells.
In contrast, duloxetine, another SNRI, has a greater affinity for the NE transporter than venlafaxine, blocking the reuptake of both NE and 5-HT [116]. Fornaro and coauthors [117] reported that MDD patients who responded to 60 mg/day duloxetine over six weeks had lower baseline levels of IL-6, potentially predicting a favorable treatment response. This suggests that lower IL-6 levels might be associated with hypo-noradrenergic activity, benefiting from duloxetine’s pro-norepinephrinergic action, which normalizes IL-6 levels through a Th2 shift mediated by norepinephrine [117]. Thus, although distinct classes of antidepressants modulate cytokine levels, the mechanisms underlying these effects remain to be elucidated.
Serotonin modulators: serotonin receptor antagonists with serotonin reuptake inhibitor (SARI)
The serotonin modulators known as serotonin receptor antagonists with serotonin reuptake inhibitor (SARI) exhibit the pharmacological characteristic of moderate to strong serotonin receptor antagonism along with weak inhibition of the serotonin reuptake transporter (SERT). Their primary pharmacodynamic effects and mechanisms of action are not solely due to SERT inhibition [118]. In the case of trazodone (TDZ), its structure contains a triazole moiety believed to contribute to its antidepressant effects. Its primary metabolite, m-chlorophenyl piperazine (m-cpp), acts as a potent 5-HT2 antagonist [119].
Daniele and coauthors [120] findings suggest that TDZ can mitigate inflammation and promote neuroprotection by modulating critical inflammatory and signaling pathways in neuronal-like cells. The authors investigated the impact of TDZ on neuronal-like cells in an in vitro model of inflammation induced by LPS and TNF-α. TDZ enhances the expression of BDNF and cAMP response element-binding protein (CREB) while reducing the production of the pro-inflammatory cytokine IFN-γ. Under inflammatory conditions, the expression of BDNF and CREB decreased, while NF-κB and pro-inflammatory cytokines such as IL-6 and IFN-γ increased. Pre-treatment with TDZ reversed these effects, restoring normal BDNF and CREB levels, inhibiting the release of inflammatory mediators, and returning IL-10 production to control levels. Further analysis revealed that TDZ activated extracellular signal-regulated kinase (ERK) signaling and inhibited p38 and c-Jun N-terminal kinase (JNK) activation, suggesting its neuroprotective action is attributed to the modulation of these pathways [120].
Tricyclic antidepressants (TCAs)
Despite the general mechanism of tricyclics involving the blockade of NE and, to a lesser extent, 5-HT reuptake, the individual molecules within this class exhibit heterogeneity [121]. They enhance noradrenergic and serotonergic transmission across different spectrums, and each molecule blocks M1, α1, and H1 receptors with varying affinities, potentially influencing immunomodulation. This complexity makes it challenging to distinguish the effects of individual neurotransmitter systems on cytokine production within this class of drugs [121].
Notably, desipramine, a potent TCA, has been shown to inhibit TNF-α production and increase the release of IL-10, favoring a Th2 response [121]. Studies on TCAs (imipramine, clomipramine) indicate that this class of molecules inhibits the secretion of pro-inflammatory cytokines such as IL-1β, IL-2, TNF-α, and IFNγ [122, 123]. Likewise, Obuchowicz et al. [124] demonstrated that amitriptyline and nortriptyline similarly reduced the release of IL-1β and TNF-α by rat glial cells when stimulated in vitro by LPS [124, 125].
However, the effects on IL-6 seem to vary, with one clinical study reporting an increase in IL-6 following TCA administration [125], while other preclinical studies show reductions [126, 127], and others indicating no change [128, 129]. On the other hand, TCAs stimulate the production of the anti-inflammatory cytokine IL-10 [125].
Monoamine oxidase inhibitors (MAOIs)
Monoamine Oxidase Inhibitors (MAOIs) constitute a class of antidepressants that hinder the activity of one or both monoamine oxidase enzymes, specifically monoamine oxidase A (MAO-A) and monoamine oxidase B (MAO-B). With a longstanding history of use, physicians prescribe MAOIs for the treatment of depression [130].
Whole blood stimulated with LPS and phytohemagglutinin, in the presence of moclobemide, a reversible MAO-A inhibitor, exhibits immunoregulatory effects. Specifically, moclobemide markedly reduces the basal production of pro-inflammatory cytokines such as TNF-α and IL-8, while simultaneously enhancing the inducible production of IL-10, a key anti-inflammatory cytokine. However, the production of IL-6, IL-1RA, and IFN-γ remains unaffected under both unstimulated and stimulated conditions. These findings suggest that moclobemide exerts negative immunoregulatory effects by modulating the inflammatory response system [131].
Similarly, additional data indicate that MAO-A and MAO-B inhibition shifts the immune response towards a Th2 profile by enhancing catecholamine release [121]. For instance, phenelzine, an irreversible inhibitor of MAO-A and MAO-B, decreased TNF-α production [132]. Meanwhile, selegiline, a reversible inhibitor of MAO-B, reduced the production of TNF-α and stimulated the biosynthesis of IL-6 and IL-1β [132].
Selective norepinephrine reuptake inhibitors (NRIs)
Currently, no clinical studies examine the interaction between the selective norepinephrine reuptake inhibitors (NRIs) reboxetine and atomoxetine and cytokine production. However, preclinical investigations indicate that reboxetine treatment enhances the production of IL-10 and IL-1β in the cortex of rats [133]. Another study observed that pre-treatment with reboxetine inhibited the increase of IL-6 induced by IFN-γ in murine cells [134]. Similarly, atomoxetine treatment reduced the gene expression of IL-1β and TNF-α in the cortex of rats [59]. While the available data is limited, it suggests that this class of molecules may potentially shift the immune response towards a Th2 profile.
Norepinephrine-dopamine reuptake inhibitor (NDRI)
Norepinephrine Dopamine Reuptake Inhibitors (NDRIs), such as bupropion, inhibit the reuptake of the neurotransmitters DA and NE, with a slightly greater potency at the DA transporter [135]. This inhibition leads to increased concentrations of DA and NE in the prefrontal cortex and elevated DA concentrations in the nucleus accumbens. Notably, the impact on 5-HT reuptake is minimal [136]. Bupropion and its metabolites do not exhibit significant affinity for various postsynaptic receptors, including histamine, α- or β-adrenergic, 5-HT, or DA receptors [136]. Recent evidence suggests that bupropion behaves as a non-competitive antagonist of several nicotinic acetylcholine (ACh) receptors [137, 138].
Huang et al. [139] demonstrated that four weeks of bupropion treatment (150 mg/d) in patients with MDD significantly increased the levels of pro-inflammatory cytokines (IL-1β, IL-7, IL-8) and anti-inflammatory cytokines (IL-4, IL-5) [139]. Notably, the percentage change in most cytokines, including anti-inflammatory factors like IL-4, IL-5, IL-10, and IL-13, was significantly elevated post-treatment [140]. These findings suggest that the therapeutic efficacy of bupropion in MDD may involve a delicate balance between pro- and anti-inflammatory responses, with a potential shift towards predominance of anti-inflammatory cytokines. This shift could contribute to the observed clinical improvements in patients receiving bupropion. Furthermore, additional studies indicate that bupropion decreases blood levels of inflammatory cytokines, including TNF-α, IFN-γ, and IL-1β, while increasing levels of anti-inflammatory cytokines such as IL-10 [135, 141].
N-Methyl-D-Aspartate-Glutamatergic ionoreceptor antagonist/inverse agonist/partial agonist
Ketamine is a racemic mixture composed of two enantiomers: S-(+)-ketamine (esketamine) and R-(–)-ketamine (arketamine), each displaying distinct pharmacological profiles. Preclinical studies suggest that R-ketamine produces longer-lasting antidepressant-like effects compared to S-ketamine, despite being a less potent NMDAR antagonist [142,143,144,145]. Several preclinical and clinical studies indicate that ketamine can decrease levels of pro-inflammatory cytokines, highlighting its anti-inflammatory properties [146,147,148]. This anti-inflammatory action particularly relevant given the established link between inflammation and depressive symptoms [70, 149].
Ketamine treatment in depressed patients has been shown to modulate kynurenine pathway metabolites, frequently reducing kynurenine levels [150, 151]. Therefore, ketamine’s anti-inflammatory effects may inhibit activation of the kynurenine pathway, providing neuroprotection and supporting neurogenesis.
In peripheral blood mononuclear cells (PBMCs) from healthy males, ketamine suppresses the differentiation of Th1/Th2 cells. However, these antagonists can also increase the Th1/Th2 ratio following stimulation with phorbol 12-myristate 13-acetate (PMA) and ionomycin. This dual effect may result from the ability of NMDA antagonists to regulate the activities of T-bet and GATA3, which are transcription factors that play crucial roles in Th1 and Th2 cell differentiation, respectively [152].
Mood stabilizers
Mood stabilizers, including lithium, valproate, and lamotrigine, have intricate mechanisms of action, all influencing neurotransmission in the brain [153, 154]. A meta-analysis has revealed elevated levels of pro-inflammatory and anti-inflammatory cytokines, including TNF-α, IL-1β, IL-6, IL-4, and IL-10 in patients with bipolar disorder compared to controls at baseline [155]. Studies indicate that lithium and lamotrigine decreased the levels of IL-6, IL-10, IFN-γ, and IL-1β, as well as C-reactive protein (CRP) [156,157,158]. In cultures of human microglia, lithium reduced IFN-γ and diminished signaling through the STAT1/STAT3 pathways [159]. Valproic acid exhibited similar effects on cytokines as lithium, also reducing the differentiation of Th17 and dendritic cells, chemotaxis migration of dendritic cells, lymphocyte proliferation, NF-κB activation, and nuclear levels of IFN regulatory factors [160, 161].
Challenges and therapeutic opportunities
The interplay between depression and inflammation presents both challenges and therapeutic opportunities in treating MDD. Growing evidence highlights the complex relationship between mood disorders and immune dysregulation, particularly in the context of autoimmune conditions. As inflammation becomes increasingly recognized as a contributing factor to depression, understanding this bidirectional relationship opens the door for new treatment strategies. Addressing systemic inflammation, particularly in patients who do not respond to conventional antidepressants, holds promise for improving mental health outcomes. This section explores the intersection of depression with autoimmune disorders and the potential of anti-inflammatory therapies as innovative treatment options.
Bidirectional relationship between depression and autoimmune disorders
The connection between immune dysregulation and depression has increasingly been recognized in neurologic disorders, particularly in neurologic autoimmune diseases [162]. Studies have shown that immune activation and the production of inflammatory cytokines, such as IL-1 and IL-6, contribute to the development of depressive symptoms by triggering cell-mediated immunity and promoting the release of these pro-inflammatory cytokines [163, 164].
Some immunomodulatory drugs, such as IFN-beta and corticosteroids, have also been linked to the development of depression. IFN-beta, commonly used in multiple sclerosis (MS) to reduce relapse rates and delay disability progression, has been associated with depression in several cases [165]. A study by Patten et al. showed that the incidence of depression was significantly higher in MS patients who used IFN-beta compared to those who received a placebo, with depression rates in the treatment group being twice as high [166].
Corticosteroids, especially oral corticosteroids, have also been implicated in the development of depression in patients with myasthenia gravis (MG). A study by Suzuki et al. [167] found that corticosteroid dosage was a significant factor in the emergence of depressive symptoms in MG patients, with higher doses correlating with more severe depressive symptoms [167, 168]. Exogenous corticosteroids have also been associated with depression in the general population, although the exact nature of this relationship remains unclear [169].
There is evidence indicating a higher occurrence of depressive symptoms among individuals diagnosed with other autoimmune disorders, including type 1 diabetes, rheumatoid arthritis, ankylosing spondylitis, Crohn’s disease, psoriasis, primary sclerosing cholangitis, chronic inflammatory bowel disease, and autoimmune thyroiditis [170]. Furthermore, there is a possible bidirectional association between depression and systemic inflammation [171], which is a crucial component in the pathophysiology of autoimmune disorders [172].
Epidemiological data from a nationwide cohort study conducted in Denmark indicate that approximately 32% of individuals diagnosed with mood disorders had a prior hospital contact for infections, and about 5% had been hospitalized for autoimmune diseases before receiving a psychiatric diagnosis [173]. These findings suggest that at least one-third of mood disorder cases may be associated with detectable immune alterations, reinforcing the existence of a subgroup of patients in whom immune activation may play a relevant role in the pathophysiology of and onset of depression [173,174,175].
Notably, randomized controlled trials involving 2370 participants demonstrated that anti-cytokine therapies, especially anti-TNF drugs like adalimumab, etanercept, and infliximab, reduced depressive symptoms compared to placebo. These findings suggest that targeting cytokines could be a promising approach for treating depression, particularly in patients with high levels of inflammation [176].
The study conducted by Poletti et al. [177] explores immune-inflammatory mechanisms as promising targets in antidepressant pharmacology. The researchers investigated the use of low-dose IL-2 to potentiate the antidepressant response in patients with mood disorders, such as MDD and bipolar disorder (BD). IL-2, a T-cell growth factor with anti-inflammatory properties, has proven effective in autoimmune conditions (Type 1 Diabetes, Systemic Lupus Erythematosus, Rheumatoid Arthritis, Multiple Sclerosis) and may correct defects observed in T cells of patients with these disorders. In a randomized, double-blind, placebo-controlled clinical trial with 36 patients, a significant improvement in antidepressant response was observed, with expansion of regulatory T cells, T helper 2 cells, and an increase in naïve CD4 + /CD8 + T cells, with changes in cell frequencies detected within the first five days of treatment. This study provides important evidence that strengthening the T-cell system may correct immune-inflammatory abnormalities associated with mood disorders and improve the response to antidepressant treatment [177].
Depression, quality of life, physical comorbidities, and autoimmunity
The detrimental impact of MDD on Quality of Life (QoL) is exacerbated in autoimmune diseases of the nervous system (ADNS), where neuroinflammation and cytokine dysregulation (e.g., elevated TNF-α, IL-6) may underlie both psychiatric symptoms and disease progression [178, 179]. In conditions like MS, neuromyelitis optica (NMO), and myasthenia gravis (MG), depression correlates with worse Health-Related Quality of Life (HRQL), fatigue, and cognitive dysfunction, creating a vicious cycle that amplifies disability [180,181,182]. Notably, psychosocial symptoms (e.g., hopelessness, guilt) and somatic manifestations (e.g., sleep disturbances) further impair the physical and mental domains of QoL [183], mirroring the bidirectional relationship between depression and metabolic comorbidities like diabetes and cardiovascular disease [179].
Antidepressants, particularly SSRIs, may mitigate these effects by modulating inflammatory pathways [178], though their efficacy in ADNS remains understudied. Lifestyle interventions (e.g., exercise, diet) could complement pharmacotherapy by reducing systemic inflammation and improving HRQL, as seen in general MDD populations [184, 185]. However, treatment challenges persist in ADNS due to overlapping symptoms (e.g., fatigue, cognitive deficits) and the need for integrated care models [162]. Collaborative approaches that combine immunomodulatory therapies, antidepressants, and psychosocial interventions are critical to addressing the dual burden of autoimmunity and depression [186].
Non-pharmacological therapies
Physical exercise also stands out for its anti-inflammatory properties and its ability to enhance the effects of antidepressants [70]. A meta-analysis demonstrated that moderate-intensity exercise performed three times a week for approximately nine weeks significantly reduced depressive symptoms [187]. These therapeutic effects span different age groups and are comparable to traditional pharmacological treatments, whether used as monotherapy, adjunctive therapy, or in combination [188]. One proposed mechanism underlying these benefits is the anti-inflammation induced by chronic physical exercise, which has been demonstrated to involve a differential cytokine response, characterized by increased circulating IL-6 levels followed by a rise in IL-1ra and IL-10 levels and a suppression of TNF production, contributing to its mood-enhancing properties [189, 190].
Evidence indicates that combining physical activity with cognitive-behavioral therapy can increase plasma levels of the anti-inflammatory cytokine IL-10 while decreasing CRP concentrations in individuals at heightened cardiovascular risk [191]. Additionally, a study among college students found that moderate-intensity exercise was more effective than high-intensity interval training in reducing stress, depressive symptoms, and systemic TNF-α levels [192]. However, it is important to recognize that high-intensity exercise may trigger acute stress responses, highlighting the need for personalized and well-structured exercise regimens [193].
Non-pharmacological therapies, such as psychotherapy, transcranial magnetic stimulation, and electroconvulsive therapy, may also influence inflammation and immune function, opening new treatment possibilities [70]. Additionally, identifying immune biomarkers may enable personalized treatments by using algorithms that combine multiple parameters, such as cytokines and microbiota composition, to predict the risk of depression and treatment response [70].
Psychedelics
The growing understanding of the interaction between classical psychedelics and the immune system points to a potential antidepressant effect mediated by immune modulation. These compounds can significantly interfere with the cytokine profiles produced by immune cells, leading to suppression of antigen presentation, reduction in inflammatory cytokine and chemokine secretion, and an increase in anti-inflammatory cytokines in the tissue microenvironment [194]. Psychedelics (such as ayahuasca and psilocybin) primarily act on serotonergic receptors 5-HT1A, 5-HT2A, 5-HT2B, and 5-HT2C, whose activation regulates the production of cytokines such as IL-1β, TNF-α, IL-10, and TGF-β, promoting an immune profile that may attenuate inflammatory processes associated with depression [195, 196].
The study by Palhano-Fontes et al. [197] investigated the antidepressant effects of ayahuasca in patients with treatment-resistant depression. The results showed a significant reduction in the severity of depression after a single dose of ayahuasca, with greater improvements compared to the placebo. Clinical response was observed as early as the first day and persisted until day seven, with the response rate significantly higher in the ayahuasca group. The study suggests that ayahuasca may have fast-acting antidepressant properties in treatment-resistant patients, similar to ketamine, but with a different temporal response profile [197].
Microbiota-targeted interventions
Serotonin (5-HT) serves as a critical neurotransmitter and hormonal signaling molecule, with dual roles in both the central (CNS) and peripheral nervous systems (PNS). While 95% of the body’s serotonin resides in the gastrointestinal tract, primarily produced by enterochromaffin cells (ECs, 90%) and enteric neurons (10%), research has predominantly focused on its CNS functions in mood and anxiety regulation [198]. This historical emphasis is now being reconsidered as emerging evidence reveals the gut 5-HT system’s significant contribution to SSRI therapeutic mechanisms through microbiota-gut-brain axis signaling.
Recent findings by Jiang et al. [199] demonstrate that SSRI treatment response in MDD patients correlates strongly with specific gut microbial profiles. Their comprehensive metabolomic analysis revealed distinct microbial signatures in treatment non-responders versus responders, suggesting microbiota-derived metabolites may modulate serotonergic pathways to influence SSRI efficacy [199].
Gut-brain communication appears to be mediated through multiple interconnected pathways. First, gut microbiota metabolites play a crucial regulatory role in cytokine networks implicated in depression pathophysiology. Microbial tryptophan metabolites like tryptophol have been shown to suppress TNF-α responses [200, 201], while palmitoleic acid, whose levels are modulated by gut microbiota, selectively inhibits monocyte-derived cytokines (TNF-α, IL-1β, IL-6) without affecting lymphocyte cytokines (IFN-γ, IL-17, IL-22) [202]. Importantly, depression-associated dysbiosis, characterized by reductions in beneficial bacteria like Coprococcus comes, may disrupt these regulatory mechanisms, leading to elevated pro-inflammatory cytokines (IL-1β, IL-6) and subsequent neuroinflammation [203].
Vagus nerve stimulation
In addition, the vagus nerve serves as a crucial bidirectional communication pathway between the gut and the brain, offering additional therapeutic opportunities. Through the cholinergic anti-inflammatory pathway, vagal activation suppresses macrophage cytokine production via α7nAChR receptors, effectively dampening systemic inflammation [204, 205]. Recent studies demonstrate that vagus nerve stimulation (VNS) not only reduces pro-inflammatory cytokines but also enhances inflammation resolution through increased production of specialized pro-resolving mediators (SPMs) and improved efferocytosis [206]. These mechanisms may explain VNS’s efficacy in treatment-resistant depression, particularly in patients with elevated inflammatory markers [207, 208].
While further research is needed to fully elucidate the relationship between VNS, immune modulation, and depression, current evidence supports its potential as an adjunctive therapy for inflammatory subtypes of the disorder [209].
Adjuvant anti-inflammatory therapy
The adjunctive use of anti-inflammatory agents has gained increasing attention as a promising therapeutic strategy. These treatments aim to directly target inflammatory pathways that may not be fully addressed by traditional antidepressants. Several compounds have demonstrated antidepressant potential through their anti-inflammatory or immunoregulatory properties. For example, NSAIDs reduce prostaglandin E2 (PGE2) synthesis, thereby suppressing IDO activation and limiting tryptophan degradation to kynurenine, a pathway associated with depressive symptoms [210].
Additional agents, including omega-3 fatty acids [211], pioglitazone [212], statins [213], and monoclonal antibodies [214], have shown efficacy in reducing cytokine-induced neuroinflammation and neuronal damage. Moreover, minocycline inhibits microglial activation [215], and N-acetylcysteine enhances antioxidant capacity, modulates glutamate, and supports neurogenesis [216,217,218]. These findings support the potential of targeting inflammation directly targeting inflammation, particularly in patients with inflammatory biomarkers or poor responses to standard treatment, as a valuable step toward personalized approaches in MDD therapy.
Conclusion and future perspectives
In conclusion, antidepressants have a broader impact beyond neurotransmitter modulation, significantly influencing immune system function by regulating Th1 and Th2 cytokines. This indicates that antidepressants may not only alleviate mood symptoms in MDD but also help correct the underlying immune dysregulation commonly associated with the condition. By reducing pro-inflammatory cytokines such as IFN-γ and TNF-α while increasing anti-inflammatory cytokines like IL-4 and IL-10, these medications assist in restoring a more balanced immune response. Understanding this dual action of antidepressants paves the away for developing holistic and personalized treatment strategies for MDD. Further research is needed to explore how these immune-modulating properties influence long-term treatment outcomes and to better understand the intricate interplay between neurotransmitters, immune function, and neuroimmunological dysregulation.
Although many antidepressants exert immunomodulatory effects and can partially restore immune balance, their efficacy remains limited in a subset of patients, particularly those with elevated baseline inflammation [193]. This suggests that, in these individuals, conventional antidepressants may be insufficient to counteract persistent immune dysregulation or may act too late in the inflammatory cascade to reverse its full impact on neurobiology. Given that MDD is a multifactorial condition involving complex interactions among psychological, environmental, genetic, and immunological factors [2, 219], immune dysfunction likely plays a central role in treatment resistance for some patients.
Additionally, a broad spectrum of factors should be incorporated into the treatment of MDD to achieve optimal mental well-being. These factors include therapies that promote physical health, foster healthy social connections [220], encourage self-awareness [221], build self-esteem [222], and embrace spirituality [223], generosity [224], and connection to nature. Addressing how these factors influence neuroimmunological dysregulation will be essential for improving MDD treatments.
References
Spijker J, Graaf R, Bijl RV, Beekman ATF, Ormel J, Nolen WA. Functional disability and depression in the general population. Results from the Netherlands mental health survey and incidence study (NEMESIS). Acta Psychiatr Scand. 2004;110:208–14.
Kessler RC, Bromet EJ. The epidemiology of depression across cultures. Annu Rev Public Health. 2013;34:119–38.
Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2012;380:2163–96.
Becker AE, Kleinman A. Mental health and the global agenda. N Engl J Med. 2013;369:66–73.
Aguilar-Latorre A, Pérez Algorta G, Navarro-Guzmán C, Serrano-Ripoll MJ, Oliván-Blázquez B. Effectiveness of a lifestyle modification programme in the treatment of depression symptoms in primary care. Front Med. 2022;9:954644. https://doi.org/10.3389/fmed.2022.954644.
Cui L, Li S, Wang S, Wu X, Liu Y, Yu W, et al. Major depressive disorder: hypothesis, mechanism, prevention and treatment. Signal Transduct Target Ther. 2024;9:30.
de Morais H, de Souza CP, da Silva LM, Ferreira DM, Werner MF, Andreatini R, et al. Increased oxidative stress in prefrontal cortex and hippocampus is related to depressive-like behavior in streptozotocin-diabetic rats. Behav Brain Res. 2014;258:52–64.
Early JO, Menon D, Wyse CA, Cervantes-Silva MP, Zaslona Z, Carroll RG, et al. Circadian clock protein BMAL1 regulates IL-1β in macrophages via NRF2. Proc Natl Acad Sci USA. 2018;115:E8460–E8468. https://doi.org/10.1073/pnas.1800431115.
Beckwith KS, Beckwith MS, Ullmann S, Sætra RS, Kim H, Marstad A, et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat Commun. 2020;11:2270.
Alcocer‐Gómez E, Cordero MD. <scp>NLRP</scp> 3 inflammasome: a new target in major depressive disorder. CNS Neurosci Ther. 2014;20:294–5.
Sun L, Ma W, Gao W, Xing Y, Chen L, Xia Z, et al. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis. 2019;10:542.
Greaney JL, Saunders EFH, Alexander LM. Short-term salicylate treatment improves microvascular endothelium-dependent dilation in young adults with major depressive disorder. Am J Physiol Heart Circ Physiol. 2022;322:H880–H889.
Maes M, Landucci Bonifacio K, Morelli NR, Vargas HO, Barbosa DS, Carvalho AF, et al. Major differences in neurooxidative and neuronitrosative stress pathways between major depressive disorder and types I and II bipolar disorder. Mol Neurobiol. 2019;56:141–56.
Chen B, Zhang M, Ji M, Zhang D, Chen B, Gong W, et al. The neuroprotective mechanism of lithium after ischaemic stroke. Commun Biol. 2022;5:105.
Bélanger M, Magistretti PJ. The role of astroglia in neuroprotection. Dialogues Clin Neurosci. 2009;11:281–95.
Szałach ŁP, Lisowska KA, Cubała WJ. The influence of antidepressants on the immune system. Arch Immunol Ther Exp (Warsz). 2019;67:143–51.
Diniz LRL, Souza MTS, Barboza JN, Almeida RN, Sousa DP. Antidepressant potential of cinnamic acids: mechanisms of action and perspectives in drug development. Molecules. 2019;24:4469.
Kim J, Kim T-E, Lee S-H, Koo JW. The role of glutamate underlying treatment-resistant depression. Clin Psychopharmacol Neurosci. 2023;21:429–46.
Xiao L, Li X, Fang C, Yu J, Chen T. Neurotransmitters: promising immune modulators in the tumor microenvironment. Front Immunol. 2023;14:1118637. https://doi.org/10.3389/fimmu.2023.1118637.
Imamdin A, van der Vorst EPC. Exploring the role of serotonin as an immune modulatory component in cardiovascular diseases. Int J Mol Sci. 2023;24:1549.
Roumier A, Béchade C, Maroteaux L. Serotonin and the immune system. In: Serotonin. Paris: Elsevier; 2019. pp. 181–96.
Roman M, Irwin MR. Novel neuroimmunologic therapeutics in depression: a clinical perspective on what we know so far. Brain Behav Immun. 2020;83:7–21.
Das UN. Vagus nerve stimulation, depression, and inflammation. Neuropsychopharmacology. 2007;32:2053–4.
Rustenhoven J, Kipnis J. Brain borders at the central stage of neuroimmunology. Nature. 2022;612:417–29.
Zipp F, Bittner S, Schafer DP. Cytokines as emerging regulators of central nervous system synapses. Immunity. 2023;56:914–25.
Isaacs A, Lindenmann J, Valentine RC. Virus interference. II. Some properties of interferon. Proc R Soc Lond B Biol Sci. 1957;147:268–73.
Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147:258–67.
Zlotnik A. Perspective: insights on the nomenclature of cytokines and chemokines. Front Immunol. 2020;11:908. https://doi.org/10.3389/fimmu.2020.00908.
Salvador AF, de Lima KA, Kipnis J. Neuromodulation by the immune system: a focus on cytokines. Nat Rev Immunol. 2021;21:526–41.
Novakovic MM, Korshunov KS, Grant RA, Martin ME, Valencia HA, Budinger GRS, et al. Astrocyte reactivity and inflammation-induced depression-like behaviors are regulated by Orai1 calcium channels. Nat Commun. 2023;14:5500.
Zhang W, Xiao D, Mao Q, Xia H. Role of neuroinflammation in neurodegeneration development. Signal Transduct Target Ther. 2023;8:267.
Franco R, Pacheco R, Lluis C, Ahern GP, O’Connell PJ. The emergence of neurotransmitters as immune modulators. Trends Immunol. 2007;28:400–7.
Fries GR, Saldana VA, Finnstein J, Rein T. Molecular pathways of major depressive disorder converge on the synapse. Mol Psychiatry. 2023;28:284–97.
Dey A, Giblin PAH. Insights into macrophage heterogeneity and cytokine-induced neuroinflammation in major depressive disorder. Pharmaceuticals. 2018;11:64. https://doi.org/10.3390/ph11030064.
Ohgo S, Nakatsuru K, Ishikawa E, Matsukura S. Interleukin-1 (IL-1) stimulates the release of corticotropin-releasing factor (CRF) from superfused rat hypothalamo-neurohypophyseal complexes (HNC) independently of the histaminergic mechanism. Brain Res. 1991;558:217–23.
Bethin KE, Vogt SK, Muglia LJ. Interleukin-6 is an essential, corticotropin-releasing hormone-independent stimulator of the adrenal axis during immune system activation. Proc Natl Acad Sci USA. 2000;97:9317–22.
Lightman SL, Birnie MT, Conway-Campbell BL. Dynamics of ACTH and cortisol secretion and implications for disease. Endocr Rev. 2020;41:bnaa002. https://doi.org/10.1210/endrev/bnaa002.
Renner V, Schellong J, Bornstein S, Petrowski K. Stress-induced pro- and anti-inflammatory cytokine concentrations in female PTSD and depressive patients. Transl Psychiatry. 2022;12:158.
Keller J, Gomez R, Williams G, Lembke A, Lazzeroni L, Murphy GM, et al. HPA axis in major depression: cortisol, clinical symptomatology and genetic variation predict cognition. Mol Psychiatry. 2017;22:527–36.
Oakley RH, Cruz-Topete D, He B, Foley JF, Myers PH, Xu X, et al. Cardiomyocyte glucocorticoid and mineralocorticoid receptors directly and antagonistically regulate heart disease in mice. Sci Signal. 2019;12:eaau9685. https://doi.org/10.1126/scisignal.aau9685.
Chantong B, Kratschmar DV, Nashev LG, Balazs Z, Odermatt A. Mineralocorticoid and glucocorticoid receptors differentially regulate NF-kappaB activity and pro-inflammatory cytokine production in murine BV-2 microglial cells. J Neuroinflammation. 2012;9:260.
Douglass AM, Resch JM, Madara JC, Kucukdereli H, Yizhar O, Grama A, et al. Neural basis for fasting activation of the hypothalamic–pituitary–adrenal axis. Nature. 2023;620:154–62.
Hantsoo L, Jagodnik KM, Novick AM, Baweja R, di Scalea TL, Ozerdem A, et al. The role of the hypothalamic-pituitary-adrenal axis in depression across the female reproductive lifecycle: current knowledge and future directions. Front Endocrinol. 2023;14:1295261. https://doi.org/10.3389/fendo.2023.1295261.
Felger JC, Li L, Marvar PJ, Woolwine BJ, Harrison DG, Raison CL, et al. Tyrosine metabolism during interferon-alpha administration: association with fatigue and CSF dopamine concentrations. Brain Behav Immun. 2013;31:153–60.
Leonard BE. The immune system, depression and the action of antidepressants. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:767–80.
Miller AH, Haroon E, Raison CL, Felger JC. Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety. 2013;30:297–306.
Vismari L, Jussilane Alves G, Palermo-Neto J Revisão da literatura depressão, antidepressivos e sistema imune: um novo olhar sobre um velho problema depression, antidepressants and immune system: a new look to an old problem. 2008.
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56.
Dantzer R. Evolutionary aspects of infections: inflammation and sickness behaviors. Curr Top Behav Neurosci. 2022;61:1–14.
Kronfol Z. Cytokines and the brain: implications for clinical psychiatry. Am J Psychiatry. 2000;157:683–94.
Benarroch E. What is the role of cytokines in synaptic transmission? Neurology. 2024;103:e209928. https://doi.org/10.1212/WNL.0000000000209928.
Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16:22–34.
Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. 2011;130:226–38.
Malau IA, Chang JP-C, Lin Y-W, Chang C-C, Chiu W-C, Su K-P. Omega-3 fatty acids and neuroinflammation in depression: targeting damage-associated molecular patterns and neural biomarkers. Cells. 2024;13:1791.
Correa SG, Sotomayor CE, Rodrĺguez-Galán MC Cytokines and the immune–neuroendocrine network. 2010. pp 79–90.
Galea I. The blood–brain barrier in systemic infection and inflammation. Cell Mol Immunol. 2021;18:2489–501.
Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383:2255–73.
Kong Y, He G, Zhang X, Li J. The role of neutrophil extracellular traps in lipopolysaccharide-induced depression-like behaviors in mice. Brain Sci. 2021;11:1514.
O’Sullivan JB, Ryan KM, Curtin NM, Harkin A, Connor TJ. Noradrenaline reuptake inhibitors limit neuroinflammation in rat cortex following a systemic inflammatory challenge: implications for depression and neurodegeneration. Int J Neuropsychopharmacol. 2009;12:687.
Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G, et al. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-α: relationship to CNS immune responses and depression. Mol Psychiatry. 2010;15:393–403.
Franscina Pinto E, Andrade C. Interferon-related depression: a primer on mechanisms, treatment, and prevention of a common clinical problem. Curr Neuropharmacol. 2016;14:743–8.
Kempuraj D, Selvakumar GP, Ahmed ME, Raikwar SP, Thangavel R, Khan A, et al. COVID-19, mast cells, cytokine storm, psychological stress, and neuroinflammation. Neuroscientist. 2020;26:402–14.
Bernard-Raichon L, Venzon M, Klein J, Axelrad JE, Zhang C, Sullivan AP, et al. Gut microbiome dysbiosis in antibiotic-treated COVID-19 patients is associated with microbial translocation and bacteremia. Nat Commun. 2022;13:5926.
Bostick JW, Schonhoff AM, Mazmanian SK. Gut microbiome-mediated regulation of neuroinflammation. Curr Opin Immunol. 2022;76:102177.
Cryan JF, O’Riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, et al. The microbiota-gut-brain axis. Physiol Rev. 2019;99:1877–2013.
Liu Q, Mak JWY, Su Q, Yeoh YK, Lui GC-Y, Ng SSS, et al. Gut microbiota dynamics in a prospective cohort of patients with post-acute COVID-19 syndrome. Gut. 2022;71:544–52.
Yeoh YK, Zuo T, Lui GC-Y, Zhang F, Liu Q, Li AY, et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut. 2021;70:698–706.
Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–94.
Fernández-Castañeda A, Lu P, Geraghty AC, Song E, Lee M-H, Wood J. et al. Mild respiratory COVID can cause multi-lineage neural cell and myelin dysregulation. Cell. 2022;185:2452–68.e16.
Beurel E, Toups M, Nemeroff CB. The bidirectional relationship of depression and inflammation: double trouble. Neuron. 2020;107:234–56.
Myint A, Leonard B, Steinbusch H, Kim Y. Th1, Th2, and Th3 cytokine alterations in major depression. J Affect Disord. 2005;88:167–73.
Maes M, Stevens W, Peeters D, DeClerck L, Scharpe S, Bridts C, et al. A study on the blunted natural killer cell activity in severely depressed patients. Life Sci. 1992;50:505–13.
Kubera M, Lin A-H, Kenis G, Bosmans E, van Bockstaele D, Maes M. Anti-inflammatory effects of antidepressants through suppression of the interferon-γ/Interleukin-10 production ratio. J Clin Psychopharmacol. 2001;21:199–206.
Kisuya J, Chemtai A, Raballah E, Keter A, Ouma C. The diagnostic accuracy of Th1 (IFN-γ, TNF-α, and IL-2) and Th2 (IL-4, IL-6 and IL-10) cytokines response in AFB microscopy smear negative PTB- HIV co-infected patients. Sci Rep. 2019;9:2966.
Beckett CW, Niklison-Chirou MV. The role of immunomodulators in treatment-resistant depression: case studies. Cell Death Discov. 2022;8:367.
Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, et al. Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–8.
Mizuno T, Zhang G, Takeuchi H, Kawanokuchi J, Wang J, Sonobe Y, et al. Interferon‐γ directly induces neurotoxicity through a neuron specific, calcium‐permeable complex of IFN‐γ receptor and AMPA GluRl receptor. FASEB J. 2008;22:1797–806.
Han K-M, Ham B-J. How inflammation affects the brain in depression: a review of functional and structural MRI studies. J Clin Neurol. 2021;17:503.
Hassamal S. Chronic stress, neuroinflammation, and depression: an overview of pathophysiological mechanisms and emerging anti-inflammatories. Front Psychiatry. 2023;14:1130989. https://doi.org/10.3389/fpsyt.2023.1130989.
Espinosa Gonzalez M, Volk-Draper L, Bhattarai N, Wilber A, Ran S. Th2 cytokines IL-4, IL-13, and IL-10 promote differentiation of pro-lymphatic progenitors derived from bone marrow myeloid precursors. Stem Cells Dev. 2022;31:322–33.
Lobo-Silva D, Carriche GM, Castro AG, Roque S, Saraiva M. Balancing the immune response in the brain: IL-10 and its regulation. J Neuroinflammation. 2016;13:297.
Chen Y, Jiang T, Chen P, Ouyang J, Xu G, Zeng Z, et al. Emerging tendency towards autoimmune process in major depressive patients: a novel insight from Th17 cells. Psychiatry Res. 2011;188:224–30.
Schiweck C, Valles-Colomer M, Arolt V, Müller N, Raes J, Wijkhuijs A, et al. Depression and suicidality: a link to premature T helper cell aging and increased Th17 cells. Brain Behav Immun. 2020;87:603–9.
Ferreira TB, Kasahara TM, Barros PO, Vieira MMM, Bittencourt VCB, Hygino J, et al. Dopamine up-regulates Th17 phenotype from individuals with generalized anxiety disorder. J Neuroimmunol. 2011;238:58–66.
Vieira MMM, Ferreira TB, Pacheco PAF, Barros PO, Almeida CRM, Araújo-Lima CF, et al. Enhanced Th17 phenotype in individuals with generalized anxiety disorder. J Neuroimmunol. 2010;229:212–8.
Olivier C, Robert PD, Daihung D, Urbà G, Catalin MP, Hywel W, et al. The risk of depression, anxiety, and suicidality in patients with psoriasis. Arch Dermatol. 2010;146:891–5. https://doi.org/10.1001/archdermatol.2010.186.
Patten SB, Marrie RA, Carta MG. Depression in multiple sclerosis. Int Rev Psychiatry. 2017;29:463–72.
Davami M, Baharlou R, Ahmadi Vasmehjani A, Ghanizadeh A, Keshtkar M, Dezhkam I, et al. Elevated IL-17 and TGF-β serum levels: a positive correlation between T-helper 17 cell-related pro-inflammatory responses with major depressive disorder. Basic Clin Neurosci. 2016;7:137–42. https://doi.org/10.15412/J.BCN.03070207.
Kim J-W, Kim Y-K, Hwang J-A, Yoon H-K, Ko Y-H, Han C, et al. Plasma levels of IL-23 and IL-17 before and after antidepressant treatment in patients with major depressive disorder. Psychiatry Investig. 2013;10:294–9.
Liu Y, Ho RC, Mak A. The role of interleukin (IL)‐17 in anxiety and depression of patients with rheumatoid arthritis. Int J Rheum Dis. 2012;15:183–7.
Griffiths CEM, Fava M, Miller AH, Russell J, Ball SG, Xu W, et al. Impact of ixekizumab treatment on depressive symptoms and systemic inflammation in patients with moderate-to-severe psoriasis: an integrated analysis of three phase 3 clinical studies. Psychother Psychosom. 2017;86:260–7.
Jha MK, Minhajuddin A, Gadad BS, Greer TL, Mayes TL, Trivedi MH. Interleukin 17 selectively predicts better outcomes with bupropion-SSRI combination: novel T cell biomarker for antidepressant medication selection. Brain Behav Immun. 2017;66:103–10.
Nadeem A, Ahmad SF, Al-Harbi NO, Fardan AS, El-Sherbeeny AM, Ibrahim KE, et al. IL-17A causes depression-like symptoms via NFκB and p38MAPK signaling pathways in mice: implications for psoriasis associated depression. Cytokine. 2017;97:14–24.
Gu M, Li Y, Tang H, Zhang C, Li W, Zhang Y, et al. Endogenous omega (n)-3 fatty acids in Fat-1 mice attenuated depression-like behavior, imbalance between microglial M1 and M2 phenotypes, and dysfunction of neurotrophins induced by lipopolysaccharide administration. Nutrients. 2018;10:1351. https://doi.org/10.3390/nu10101351.
Cheng Y, Desse S, Martinez A, Worthen RJ, Jope RS, Beurel E. TNFα disrupts blood brain barrier integrity to maintain prolonged depressive-like behavior in mice. Brain Behav Immun. 2018;69:556–67.
Beurel E, Lowell JA. Th17 cells in depression. Brain Behav Immun. 2018;69:28–34.
Ambrée O, Ruland C, Zwanzger P, Klotz L, Baune BT, Arolt V, et al. Social defeat modulates T helper cell percentages in stress susceptible and resilient mice. Int J Mol Sci. 2019;20:3512.
Nemeroff CB, Owens MJ. Pharmacologic differences among the SSRIs: focus on monoamine transporters and the HPA axis. CNS Spectr. 2004;9:23–31.
Hernández ME, Mendieta D, Martínez-Fong D, Loría F, Moreno J, Estrada I, et al. Variations in circulating cytokine levels during 52 week course of treatment with SSRI for major depressive disorder. Eur Neuropsychopharmacol. 2008;18:917–24.
Kubera M, Maes M, Kenis G, Kim YK, Lasoń W. Effects of serotonin and serotonergic agonists and antagonists on the production of tumor necrosis factor α and interleukin-6. Psychiatry Res. 2005;134:251–8.
Sutcigil L, Oktenli C, Musabak U, Bozkurt A, Cansever A, Uzun O, et al. Pro- and anti-inflammatory cytokine balance in major depression: effect of sertraline therapy. Clin Dev Immunol. 2007;2007:76396. https://doi.org/10.1155/2007/76396.
Rafiee L, Hajhashemi V, Javanmard SH. Fluvoxamine inhibits some inflammatory genes expression in LPS/stimulated human endothelial cells, U937 macrophages, and carrageenan-induced paw edema in rat. Iran J Basic Med Sci. 2016;19:977–84.
Diamond M, Kelly JP, Connor TJ. Antidepressants suppress production of the Th1 cytokine interferon-γ, independent of monoamine transporter blockade. Eur Neuropsychopharmacol. 2006;16:481–90.
Della Vecchia A, Arone A, Piccinni A, Mucci F, Marazziti D. GABA system in depression: impact on pathophysiology and psychopharmacology. Curr Med Chem. 2022;29:5710–30.
Page CE, Coutellier L. Prefrontal excitatory/inhibitory balance in stress and emotional disorders: evidence for over-inhibition. Neurosci Biobehav Rev. 2019;105:39–51.
Pozdnyakova N, Krisanova N, Dudarenko M, Vavers E, Zvejniece L, Dambrova M, et al. Inhibition of sigma-1 receptors substantially modulates GABA and glutamate transport in presynaptic nerve terminals. Exp Neurol. 2020;333:113434.
Rosen DA, Seki SM, Fernández-Castañeda A, Beiter RM, Eccles JD, Woodfolk JA, et al. Modulation of the sigma-1 receptor–IRE1 pathway is beneficial in preclinical models of inflammation and sepsis. Sci Transl Med. 2019;11:eaau5266. https://doi.org/10.1126/scitranslmed.aau5266.
Zhang Y, Ye L, Li T, Guo F, Guo F, Li Y, et al. New monoamine antidepressant, hypidone hydrochloride (YL-0919), enhances the excitability of medial prefrontal cortex in mice via a neural disinhibition mechanism. Acta Pharmacol Sin. 2022;43:1699–709.
Ishima T, Fujita Y, Hashimoto K. Interaction of new antidepressants with sigma-1 receptor chaperones and their potentiation of neurite outgrowth in PC12 cells. Eur J Pharmacol. 2014;727:167–73.
Robichaud M, Debonnel G. Modulation of the firing activity of female dorsal raphe nucleus serotonergic neurons by neuroactive steroids. J Endocrinol. 2004;182:11–21.
Sałaciak K, Pytka K. Revisiting the sigma-1 receptor as a biological target to treat affective and cognitive disorders. Neurosci Biobehav Rev. 2022;132:1114–36.
Béı̈que J-C, de Montigny C, Blier P, Debonnel G. Effects of sustained administration of the serotonin and norepinephrine reuptake inhibitor venlafaxine: II. In vitro studies in the rat. Neuropharmacology. 2000;39:1813–22.
Debonnel G, Saint-André É, Hébert C, De Montigny C, Lavoie N, Blier P. Differential physiological effects of a low dose and high doses of venlafaxine in major depression. Int J Neuropsychopharmacol. 2007;10:51–61.
Vollmar P, Haghikia A, Dermietzel R, Faustmann PM. Venlafaxine exhibits an anti-inflammatory effect in an inflammatory co-culture model. Int J Neuropsychopharmacol. 2008;11:111–7. https://doi.org/10.1017/S1461145707007729.
Vollmar P, Nessler S, Kalluri SR, Hartung H-P, Hemmer B. The antidepressant venlafaxine ameliorates murine experimental autoimmune encephalomyelitis by suppression of pro-inflammatory cytokines. Int J Neuropsychopharmacol. 2009;12:525.
Invernizzi RW, Garattini S. Role of presynaptic α2-adrenoceptors in antidepressant action: recent findings from microdialysis studies. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:819–27.
Fornaro M, Martino M, Battaglia F, Colicchio S, Perugi G. Increase in IL-6 levels among major depressive disorder patients after a 6-week treatment with duloxetine 60 mg/day: a preliminary observation. Neuropsychiatr Dis Treat. 2011;7:51–6.
Fasipe O. Neuropharmacological classification of antidepressant agents based on their mechanisms of action. Arch Med Health Sci. 2018;6:81.
Odagaki Y, Toyoshima R, Yamauchi T. Trazodone and its active metabolite m-chlorophenylpiperazine as partial agonists at 5-HT1A receptors assessed by [35S]GTPγS binding. J Psychopharmacol. 2005;19:235–41.
Daniele S, Da Pozzo E, Zappelli E, Martini C. Trazodone treatment protects neuronal-like cells from inflammatory insult by inhibiting NF-κB, p38 and JNK. Cell Signal. 2015;27:1609–29.
Martino M, Rocchi G, Escelsior A, Fornaro M. Immunomodulation mechanism of antidepressants: interactions between serotonin/norepinephrine balance and Th1/Th2 balance. Curr Neuropharmacol. 2012;10:97–123.
Kenis G, Maes M. Effects of antidepressants on the production of cytokines. Int J Neuropsychopharmacol. 2002;5:S1461145702003164.
San L, Arranz B. Agomelatine: A novel mechanism of antidepressant action involving the melatonergic and the serotonergic system. Eur Psychiatry. 2008;23:396–402.
Obuchowicz E, Kowalski J, Labuzek K, Krysiak R, Pendzich J, Herman ZS. Amitriptyline and nortriptyline inhibit interleukin-1 release by rat mixed glial and microglial cell cultures. Int J Neuropsychopharmacol. 2005;9:27.
Kubera M, Kenis G, Bosmans E, Kajta M, Basta-Kaim A, Scharpe S, et al. Stimulatory effect of antidepressants on the production of IL-6. Int Immunopharmacol. 2004;4:185–92.
Huang YY, Peng CH, Yang YP, Wu CC, Hsu WM, Wang HJ, et al. Desipramine activated Bcl-2 expression and inhibited lipopolysaccharide- induced apoptosis in hippocampus-derived adult neural stem cells. J Pharmacol Sci. 2007;104:61–72.
Tai YH, Wang YH, Wang JJ, Tao PL, Tung CS, Wong CS. Amitriptyline suppresses neuroinflammation and up-regulates glutamate transporters in morphine-tolerant rats. Pain. 2006;124:77–86.
Budziszewska B, Basta-Kaim A, Kubera M, Jaworska L, Leśkiewicz M, Tetich M, et al. Effect of lipopolysaccharide and antidepressant drugs on glucocorticoid receptor-mediated gene transcription. Pharmacol Rep. 2005;57:540–4.
Wang W, Danielsson A, Svanberg E, Lundholm K. Lack of effects by tricyclic antidepressant and serotonin inhibitors on anorexia in MCG 101 tumor-bearing mice with eicosanoid-related cachexia. Nutrition. 2003;19:47–53.
Thase ME. MAOIs and depression treatment guidelines. J Clin Psychiatry. 2012;73:e24.
Lin A, Song C, Kenis G, Bosmans E, De Jongh R, Scharpé S, et al. The in vitro immunosuppressive effects of moclobemide in healthy volunteers. J Affect Disord. 2000;58:69–74.
Ekuni D, Firth JD, Nayer T, Tomofuji T, Sanbe T, Irie K, et al. Lipopolysaccharide-induced epithelial monoamine oxidase mediates alveolar bone loss in a rat chronic wound model. Am J Pathol. 2009;175:1398–409.
McNamee EN, Ryan KM, Griffin ÉW, González-Reyes RE, Ryan KJ, Harkin A, et al. Noradrenaline acting at central β-adrenoceptors induces interleukin-10 and suppressor of cytokine signaling-3 expression in rat brain: implications for neurodegeneration. Brain Behav Immun. 2010;24:660–71.
Hashioka S, Klegeris A, Monji A, Kato T, Sawada M, McGeer PL, et al. Antidepressants inhibit interferon-γ-induced microglial production of IL-6 and nitric oxide. Exp Neurol. 2007;206:33–42.
Brustolim D, Ribeiro-dos-Santos R, Kast RE, Altschuler EL, Soares MBP. A new chapter opens in anti-inflammatory treatments: the antidepressant bupropion lowers production of tumor necrosis factor-alpha and interferon-gamma in mice. Int Immunopharmacol. 2006;6:903–7.
Stahl SM, Pradko J, Haight BR, Modell JG, Rockett CB, Learned-Coughlin S. A review of the neuropharmacology of bupropion, a dual norepinephrine and dopamine reuptake inhibitor. Prim Care Companion CNS Disord. 2004;6:159–66. https://doi.org/10.4088/PCC.v06n0403.
Arias HR. Is the inhibition of nicotinic acetylcholine receptors by bupropion involved in its clinical actions? Int J Biochem Cell Biol. 2009;41:2098–108.
Arias HR, Gumilar F, Rosenberg A, Targowska-Duda KM, Feuerbach D, Jozwiak K, et al. Interaction of bupropion with muscle-type nicotinic acetylcholine receptors in different conformational states. Biochemistry. 2009;48:4506–18.
Huang C-C, Chu H-T, Lin Y-K, Tsai C-K, Liang C-S, Yeh T-C. Bupropion associated immunomodulatory effects on peripheral cytokines in male with major depressive disorder. J Med Sci. 2024;44:66–73.
Kokkinou M, Ashok AH, Howes OD. The effects of ketamine on dopaminergic function: meta-analysis and review of the implications for neuropsychiatric disorders. Mol Psychiatry. 2018;23:59–69.
Eller T, Vasar V, Shlik J, Maron E. Effects of bupropion augmentation on pro-inflammatory cytokines in escitalopram-resistant patients with major depressive disorder. J Psychopharmacol. 2009;23:854–8.
Yang C, Shirayama Y, Zhang J, Ren Q, Yao W, Ma M, et al. R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry. 2015;5:e632.
Fukumoto K, Toki H, Iijima M, Hashihayata T, Yamaguchi J, Hashimoto K, et al. Antidepressant potential of (R)-ketamine in rodent models: comparison with (S)-ketamine. J Pharmacol Exp Ther. 2017;361:9–16.
Chang L, Zhang K, Pu Y, Qu Y, Wang S, Xiong Z, et al. Comparison of antidepressant and side effects in mice after intranasal administration of (R,S)-ketamine, (R)-ketamine, and (S)-ketamine. Pharmacol Biochem Behav. 2019;181:53–59.
Jelen LA, Young AH, Stone JM. Ketamine: a tale of two enantiomers. J Psychopharmacol. 2021;35:109–23.
Beilin B, Rusabrov Y, Shapira Y, Roytblat L, Greemberg L, Yardeni IZ, et al. Low-dose ketamine affects immune responses in humans during the early postoperative period. Br J Anaesth. 2007;99:522–7.
Loix S, De Kock M, Henin P. The anti-inflammatory effects of ketamine: state of the art. Acta Anaesthesiol Belg. 2011;62:47–58.
Nikkheslat N. Targeting inflammation in depression: ketamine as an anti-inflammatory antidepressant in psychiatric emergency. Brain Behav Immun Health. 2021;18:100383.
Pitharouli MC, Hagenaars SP, Glanville KP, Coleman JRI, Hotopf M, Lewis CM, et al. Elevated C-reactive protein in patients with depression, independent of genetic, health, and psychosocial factors: results from the UK biobank. Am J Psychiatry. 2021;178:522–9.
Kopra E, Mondelli V, Pariante C, Nikkheslat N. Ketamine’s effect on inflammation and kynurenine pathway in depression: a systematic review. J Psychopharmacol. 2021;35:934–45.
Zhou Y, Zheng W, Liu W, Wang C, Zhan Y, Li H, et al. Antidepressant effect of repeated ketamine administration on kynurenine pathway metabolites in patients with unipolar and bipolar depression. Brain Behav Immun. 2018;74:205–12.
Gao M, Jin W, Qian Y, Ji L, Feng G, Sun J. Effect of N-methyl-D-aspartate receptor antagonist on T helper cell differentiation induced by phorbol-myristate-acetate and ionomycin. Cytokine. 2011;56:458–65.
Rapoport SI, Basselin M, Kim H-W, Rao JS. Bipolar disorder and mechanisms of action of mood stabilizers. Brain Res Rev. 2009;61:185–209.
Chiu C-T, Wang Z, Hunsberger JG, Chuang D-M. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev. 2013;65:105–42.
Modabbernia A, Taslimi S, Brietzke E, Ashrafi M. Cytokine alterations in bipolar disorder: a meta-analysis of 30 studies. Biol Psychiatry. 2013;74:15–25.
Queissner R, Lenger M, Birner A, Dalkner N, Fellendorf F, Bengesser S, et al. The association between anti-inflammatory effects of long-term lithium treatment and illness course in bipolar disorder. J Affect Disord. 2021;281:228–34.
Shi S, Li L, Song L, Wang X. Effect of lamotrigine on cognitive function and serum inflammatory factors in patients with depression of recurrent bipolar disorder. Pak J Pharm Sci. 2018;31:2775–8.
Boufidou F, Nikolaou C, Alevizos B, Liappas IA, Christodoulou GN. Cytokine production in bipolar affective disorder patients under lithium treatment. J Affect Disord. 2004;82:309–13.
Göttert R, Fidzinski P, Kraus L, Schneider UC, Holtkamp M, Endres M, et al. Lithium inhibits tryptophan catabolism via the inflammation‐induced kynurenine pathway in human microglia. Glia. 2022;70:558–71.
Nencioni A, Beck J, Werth D, Grünebach F, Patrone F, Ballestrero A, et al. Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin Cancer Res. 2007;13:3933–41.
Leu S, Yang Y, Liu H, Cheng C, Wu Y, Huang M, et al. Valproic acid and lithium meditate anti‐inflammatory effects by differentially modulating dendritic cell differentiation and function. J Cell Physiol. 2017;232:1176–86.
Liu Y, Tang X. Depressive syndromes in autoimmune disorders of the nervous system: prevalence, etiology, and influence. Front Psychiatry. 2018;9:451. https://doi.org/10.3389/fpsyt.2018.00451.
Gold SM, Irwin MR. Depression and immunity: inflammation and depressive symptoms in multiple sclerosis. Immunol Allergy Clin North Am. 2009;29:309–20.
Wright CE, Strike PC, Brydon L, Steptoe A. Acute inflammation and negative mood: mediation by cytokine activation. Brain Behav Immun. 2005;19:345–50.
Patten SB, Metz LM. Interferon β1a and depression in secondary progressive MS: data from the SPECTRIMS trial. Neurology. 2002;59:744–6.
Patten SB, Barbui C. Drug-induced depression: a systematic review to inform clinical practice. Psychother Psychosom. 2004;73:207–15.
Suzuki Y, Utsugisawa K, Suzuki S, Nagane Y, Masuda M, Kabasawa C, et al. Factors associated with depressive state in patients with myasthenia gravis: a multicentre cross-sectional study. BMJ Open. 2011;1:e000313.
Mourão AM, Gomez RS, Barbosa LSM, da Silva Freitas D, Comini-Frota ER, Kummer A, et al. Determinants of quality of life in Brazilian patients with myasthenia gravis. Clinics. 2016;71:370–4.
Patten SB. Exogenous corticosteroids and major depression in the general population. J Psychosom Res. 2000;49:447–9.
Orsolini L, Pompili S, Tempia Valenta S, Salvi V, Volpe U. C-reactive protein as a biomarker for major depressive disorder? Int J Mol Sci. 2022;23:1616.
Matthews KA, Schott LL, Bromberger JT, Cyranowski JM, Everson-Rose SA, Sowers M. Are there bi-directional associations between depressive symptoms and C-reactive protein in mid-life women? Brain Behav Immun. 2010;24:96–101.
Smolen JS, Aletaha D, Koeller M, Weisman MH, Emery P. New therapies for treatment of rheumatoid arthritis. Lancet. 2007;370:1861–74.
Benros ME. Autoimmune diseases can be associated with depression. Evid Based Ment Health. 2016;19:e27.
Benros ME, Waltoft BL, Nordentoft M, Østergaard SD, Eaton WW, Krogh J, et al. Autoimmune diseases and severe infections as risk factors for mood disorders. JAMA Psychiatry. 2013;70:812.
Lee C-H, Giuliani F The role of inflammation in depression and fatigue. Front Immunol 2019;10. https://doi.org/10.3389/fimmu.2019.01696.
Kappelmann N, Lewis G, Dantzer R, Jones PB, Khandaker GM. Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol Psychiatry. 2018;23:335–43.
Poletti S, Zanardi R, Mandelli A, Aggio V, Finardi A, Lorenzi C, et al. Low-dose interleukin 2 antidepressant potentiation in unipolar and bipolar depression: safety, efficacy, and immunological biomarkers. Brain Behav Immun. 2024;118:52–68.
Tully PJ, Ang SY, Lee EJ, Bendig E, Bauereiß N, Bengel J, et al. Psychological and pharmacological interventions for depression in patients with coronary artery disease. Cochrane Database Syst Rev. 2021;12:CD008012. https://doi.org/10.1002/14651858.CD008012.pub4.
Grenard JL, Munjas BA, Adams JL, Suttorp M, Maglione M, McGlynn EA, et al. Depression and medication adherence in the treatment of chronic diseases in the United States: a meta-analysis. J Gen Intern Med. 2011;26:1175–82.
Basta IZ, Pekmezović TD, Perić SZ, Kisić-Tepavčević DB, Rakočević-Stojanović VM, Stević ZD, et al. Assessment of health-related quality of life in patients with myasthenia gravis in Belgrade (Serbia). Neurol Sci. 2012;33:1375–81.
Ozakbas S, Turkoglu R, Tamam Y, Terzi M, Taskapilioglu O, Yucesan C, et al. Prevalence of and risk factors for cognitive impairment in patients with relapsing-remitting multiple sclerosis: multi-center, controlled trial. Mult Scler Relat Disord. 2018;22:70–76.
Shi Z, Chen H, Lian Z, Liu J, Feng H, Zhou H. Factors that impact health-related quality of life in neuromyelitis optica spectrum disorder: anxiety, disability, fatigue and depression. J Neuroimmunol. 2016;293:54–58.
Renwick L, Jackson D, Foley S, Owens E, Ramperti N, Behan C, et al. Depression and quality of life in first-episode psychosis. Compr Psychiatry. 2012;53:451–5.
Marx W, Manger SH, Blencowe M, Murray G, Ho FY-Y, Lawn S, et al. Clinical guidelines for the use of lifestyle-based mental health care in major depressive disorder: world federation of societies for biological psychiatry (WFSBP) and australasian society of lifestyle medicine (ASLM) taskforce. World J Biol Psychiatry. 2023;24:333–86.
Malhi GS, Bell E, Bassett D, Boyce P, Bryant R, Hazell P, et al. The 2020 royal australian and new zealand college of psychiatrists clinical practice guidelines for mood disorders. Aust N Z J Psychiatry. 2021;55:7–117.
Bagert B, Camplair P, Bourdette D. Cognitive dysfunction in multiple sclerosis. CNS Drugs. 2002;16:445–55.
Morres ID, Hatzigeorgiadis A, Stathi A, Comoutos N, Arpin-Cribbie C, Krommidas C, et al. Aerobic exercise for adult patients with major depressive disorder in mental health services: a systematic review and meta-analysis. Depress Anxiety. 2019;36:39–53.
Xie Y, Wu Z, Sun L, Zhou L, Wang G, Xiao L et al. The effects and mechanisms of exercise on the treatment of depression. Front Psychiatry 2021;12. https://doi.org/10.3389/fpsyt.2021.705559.
Metsios GS, Moe RH, Kitas GD. Exercise and inflammation. Best Pract Res Clin Rheumatol. 2020;34:101504.
da Scheffer DL, Latini A. Exercise-induced immune system response: anti-inflammatory status on peripheral and central organs. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165823.
Euteneuer F, Dannehl K, del Rey A, Engler H, Schedlowski M, Rief W. Immunological effects of behavioral activation with exercise in major depression: an exploratory randomized controlled trial. Transl Psychiatry. 2017;7:e1132.
Paolucci EM, Loukov D, Bowdish DME, Heisz JJ. Exercise reduces depression and inflammation but intensity matters. Biol Psychol. 2018;133:79–84.
Yin Y, Ju T, Zeng D, Duan F, Zhu Y, Liu J, et al. “Inflamed” depression: a review of the interactions between depression and inflammation and current anti-inflammatory strategies for depression. Pharmacol Res. 2024;207:107322.
Rogers TJ. The molecular basis for neuroimmune receptor signaling. J Neuroimmune Pharmacol. 2012;7:722–4.
de las Casas-Engel M, Domínguez-Soto A, Sierra-Filardi E, Bragado R, Nieto C, Puig-Kroger A, et al. Serotonin skews human macrophage polarization through HTR2B and HTR7. J Immunol. 2013;190:2301–10.
Cloëz-Tayarani I, Changeux J-P. Nicotine and serotonin in immune regulation and inflammatory processes: a perspective. J Leukoc Biol. 2007;81:599–606.
Palhano-Fontes F, Barreto D, Onias H, Andrade KC, Novaes MM, Pessoa JA, et al. Rapid antidepressant effects of the psychedelic ayahuasca in treatment-resistant depression: a randomized placebo-controlled trial. Psychol Med. 2019;49:655–63.
Bellini M, Fornai M, Usai Satta P, Bronzini F, Bassotti G, Blandizzi C, et al. The role of serotonin and its pathways in gastrointestinal disorders. In: The complex interplay between gut-brain, gut-liver, and liver-brain axes. France: Elsevier; 2021. pp 67–94.
Jiang Y, Qu Y, Shi L, Ou M, Du Z, Zhou Z, et al. The role of gut microbiota and metabolomic pathways in modulating the efficacy of SSRIs for major depressive disorder. Transl Psychiatry. 2024;14:493.
Moffett JR, Namboodiri MA. Tryptophan and the immune response. Immunol Cell Biol. 2003;81:247–65.
Nowak EC, de Vries VC, Wasiuk A, Ahonen C, Bennett KA, Le Mercier I, et al. Tryptophan hydroxylase-1 regulates immune tolerance and inflammation. J Exp Med. 2012;209:2127–35.
Welters HJ, Tadayyon M, Scarpello JHB, Smith SA, Morgan NG. Mono-unsaturated fatty acids protect against beta-cell apoptosis induced by saturated fatty acids, serum withdrawal or cytokine exposure. FEBS Lett. 2004;560:103–8.
Li Y, Oosting M, Smeekens SP, Jaeger M, Aguirre-Gamboa R, Le KTT. et al. A functional genomics approach to understand variation in cytokine production in humans. Cell. 2016;167:1099–110.e14
Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–96.
Bonaz B, Bazin T, Pellissier S. The vagus nerve at the interface of the microbiota-gut-brain axis. Front Neurosci. 2018;12:49. https://doi.org/10.3389/fnins.2018.00049.
Caravaca AS, Gallina AL, Tarnawski L, Shavva VS, Colas RA, Dalli J, et al. Vagus nerve stimulation promotes resolution of inflammation by a mechanism that involves Alox15 and requires the α7nAChR subunit. Proc Natl Acad Sci USA. 2022;119:e2023285119. https://doi.org/10.1073/pnas.2023285119.
Bonaz B, Sinniger V, Hoffmann D, Clarençon D, Mathieu N, Dantzer C, et al. Chronic vagus nerve stimulation in Crohn’s disease: a 6‐month follow‐up pilot study. Neurogastroenterol Motil. 2016;28:948–53.
Pavlov VA, Tracey KJ. Bioelectronic medicine: preclinical insights and clinical advances. Neuron. 2022;110:3627–44.
Falvey A. Vagus nerve stimulation and inflammation: expanding the scope beyond cytokines. Bioelectron Med. 2022;8:19.
Braun D, Longman RS, Albert ML. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood. 2005;106:2375–81.
Borsini A, Nicolaou A, Camacho-Muñoz D, Kendall AC, Di Benedetto MG, Giacobbe J, et al. Omega-3 polyunsaturated fatty acids protect against inflammation through production of LOX and CYP450 lipid mediators: relevance for major depression and for human hippocampal neurogenesis. Mol Psychiatry. 2021;26:6773–88.
Munhoz CD, García-Bueno B, Madrigal JLM, Lepsch LB, Scavone C, Leza JC. Stress-induced neuroinflammation: mechanisms and new pharmacological targets. Braz J Med Biol Res. 2008;41:1037–46.
Köhler-Forsberg O, Otte C, Gold SM, Østergaard SD. Statins in the treatment of depression: hype or hope? Pharmacol Ther. 2020;215:107625.
Lapadula G, Marchesoni A, Armuzzi A, Blandizzi C, Caporali R, Chimenti S, et al. Adalimumab in the treatment of immune-mediated diseases. Int J Immunopathol Pharmacol. 2014;27:33–48.
De Marco R, Barritt AW, Cercignani M, Cabbai G, Colasanti A, Harrison NA. Inflammation-induced reorientation of reward versus punishment sensitivity is attenuated by minocycline. Brain Behav Immun. 2023;111:320–7.
Dean O, Giorlando F, Berk M. N-acetylcysteine in psychiatry: current therapeutic evidence and potential mechanisms of action. J Psychiatry Neurosci. 2011;36:78–86.
Deepmala, Slattery J, Kumar N, Delhey L, Berk M, Dean O, et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci Biobehav Rev. 2015;55:294–321.
Erickson MA, Hansen K, Banks WA. Inflammation-induced dysfunction of the low-density lipoprotein receptor-related protein-1 at the blood–brain barrier: protection by the antioxidant N-acetylcysteine. Brain Behav Immun. 2012;26:1085–94.
Beumer W, Gibney SM, Drexhage RC, Pont-Lezica L, Doorduin J, Klein HC, et al. The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. J Leukoc Biol. 2012;92:959–75.
Xue S, Massazza A, Akhter-Khan SC, Wray B, Husain MI, Lawrance EL. Mental health and psychosocial interventions in the context of climate change: a scoping review. npj Ment Health Res. 2024;3:10.
Nonweiler J, Vives J, Barrantes-Vidal N, Ballespí S. Emotional self-knowledge profiles and relationships with mental health indicators support value in ‘knowing thyself’. Sci Rep. 2024;14:7900.
Barbalat G, Plasse J, Gauthier E, Verdoux H, Quiles C, Dubreucq J, et al. The central role of self-esteem in the quality of life of patients with mental disorders. Sci Rep. 2022;12:7852.
Basu P. Spirituality’s rising role in medicine stirs debate. Nat Med. 2005;11:1259–1259.
Park SQ, Kahnt T, Dogan A, Strang S, Fehr E, Tobler PN. A neural link between generosity and happiness. Nat Commun. 2017;8:15964.
Acknowledgements
We thank the São Paulo Research Foundation (FAPESP grants 2018/18886-9 to OCM, 2023/12268-0 to ASA; 2020/16246-2 and 2023/07806-2 to ISF; 2023/06086-6 to PM; 2019/14526-0 and 2020/04667-3 to GC-M, and 2018/149332 to HIN) for financial support. We acknowledge the National Council for Scientific and Technological Development (CNPq) Brazil (grants: 140481/2025-7 to RGN; 309482/2022-4 to OCM; 102430/2022-5 to LFS and YLGC to 130027/2023-5). We thank Coordination for the Improvement of Higher Education Personnel (CAPES) (88887.801068/2023-00 ALN) for financial support.
Author information
Authors and Affiliations
Contributions
RGN and OCM wrote the manuscript. ALN, ISF, ALN, OCM, PMB, YLGC, SFO, GCM, HDD, LFS, RAG, RJSD, RGN, HIN and RMR provided scientific insights. HDD, LFS, RAG, RJSD, HIN and RMR revised and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nava, R.G., Adri, A.S., Filgueiras, I.S. et al. Modulation of neuroimmune cytokine networks by antidepressants: implications in mood regulation. Transl Psychiatry 15, 314 (2025). https://doi.org/10.1038/s41398-025-03532-y
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03532-y
This article is cited by
-
Methylphenidate modulates the ınflammatory response and PI3K signaling in macrophages
Naunyn-Schmiedeberg's Archives of Pharmacology (2026)
-
Diet and gut microbiota during depression and stress
Discover Food (2026)
-
Alterations in serum IL-10 and IL-19 levels in drug-naïve first-episode adolescent-onset schizophrenia and their associations with clinical symptoms
Schizophrenia (2025)
