
Abstract
Monocyte chemoattractant protein-1 (MCP-1), a cytokine involved in peripheral inflammation, has been shown to modulate established Alzheimer’s disease (AD) loci. In this study, we hypothesized that blood MCP-1 levels may impact the associations of other genetic variants with AD risk beyond the well-established AD loci. We performed a genome-wide association study (GWAS) using logistic regression with the generalized estimating equation (GEE) and Cox proportional hazards models to examine the combined effects of single nucleotide polymorphisms (SNPs) and blood MCP-1 levels on AD. Three datasets were used: the Framingham Heart Study (FHS), Religious Orders Study/Memory and Aging Project (ROSMAP), and Alzheimer’s Disease Neuroimaging Initiative (ADNI). We identified SNPs in two genes in the meta-analysis, namely, neuron navigator 3 (NAV3, also named unc-53 homolog 3, rs696468) (p < 7.55 × 10−9) and the homolog unc-5 netrin receptor c (UNC5C rs72659964) (p < 1.07 × 10−8), which are modified by blood MCP-1 concentration for AD risk. Elevated blood MCP-1 concentrations increased AD risk and brain AD pathology in individuals with NAV3 (rs696468-CC) and UNC5C (rs72659964-AT + TT) genotypes. Given that NAV3 and UNC5C are involved in regulating neurite outgrowth and guidance, increased MCP-1 levels may disturb the functions of vulnerable gene carriers to increase AD risk.
Similar content being viewed by others


Introduction
The relationship between peripheral proinflammatory factors and genetic vulnerability to late-onset Alzheimer’s disease (AD) remains largely unclear [1]. Peripheral chronic inflammation causes a cascade of cytokines and chemokines to surge, and some of them may interact with inherited genetic factors to modulate the risk for AD. One such peripheral inflammatory factor is monocyte chemoattractant protein-1 (MCP-1), also known as C-C motif chemokine ligand 2 (CCL2). Our previous study revealed that blood MCP-1 levels modulated the genetic risk of AD for two gene loci, APOE and HLA-DRB1, i.e., an elevated blood MCP-1 level is associated with AD risk in individuals carrying HLA-DRB1 rs9271192-AC/CC and APOE ε4 genotype; in contrast, among HLA-DRB1 rs9271192-AA and APOE ε4 noncarriers, blood MCP-1 levels are not associated with the risk [2]. We hypothesized that there may be other genetic loci beyond APOE and HLA-DRB1 that interact with blood MCP-1 levels to affect AD risk.
The main function of MCP-1 as a chemoattractant is to drive leukocyte migration, especially monocyte/macrophage migration into damaged or infected tissues, including AD brains [3,4,5], indicating that MCP-1 may play a role in activating microglia in the brain and thus leading to cognitive decline [6,7,8,9,10,11,12]. Peripheral MCP-1 is also implicated in blood-brain barrier (BBB) disruption and astrocyte activation in the brain [13]. The MCP-1 level in human cerebrospinal fluid (CSF) is associated with a faster rate of cognitive decline during the early stages of AD [9], and its overexpression in the brain promotes glial activation and accelerates tau pathology in a mouse model [14].
How peripheral MCP-1 impacts genetic susceptibility factors for the risk of developing AD is unclear [1]. In particular, no genome-wide search has been performed for interactions between blood MCP-1 levels and genetic variants for AD risk. In this study, we conducted a gene-by-environment (G×E) genome-wide association study (GWAS) by applying logistic regression utilizing generalized estimating equations (GEEs) to two datasets: the Framingham Heart Study (FHS) and the Alzheimer’s Disease Neuroimaging Initiative (ADNI). The top findings were further tested with Cox proportional hazards models for the incidence of AD. For the first time, a meta-analysis of summary statistics from the FHS and ADNI at the genome-wide significance level (p < 5.0 × 10−8) showed that elevated blood MCP-1 levels increase AD risk and neuropathology only in individuals carrying specific genotypes of variants in two gene loci: neuron navigator 3 (NAV3 rs696468) and unc-5 netrin receptor c (UNC5C rs72659964). Interestingly, both the NAV3 and UNC5C genes have been identified in C. elegans to be involved in neuron outgrowth and guidance in C. elegans, and both are implicated in AD pathogenesis in humans [15, 16]. Further multi-omics integration analyses of the Religious Orders Study/Memory and Aging Project (ROSMAP) cohort provided molecular mechanism evidence for the significant associations between AD and NAV3 variants at DNA methylation and gene expression levels.
Materials and methods
Participants
Framingham heart study
The Framingham Heart Study (FHS) is a single-site, multigeneration, community-based, prospective cohort study in Framingham, Massachusetts [17]. This study focused on offspring cohort (Generation 2) participants who had genome-wide genotyping and serum MCP-1 measurement data from Exam 7 (baseline) and who were followed up on AD/dementia outcomes up to 2019 (Figure S1). Informed consent was obtained from all study participants, and the study protocol was approved by the Institutional Review Board (IRB) of Boston University Medical Campus. Subjects with mismatched reported sex and biological sex were excluded.
Alzheimer’s disease neuroimaging initiative (ADNI)
To validate the findings of the FHS, we used data from the ADNI-1, a longitudinal multicenter study launched in 2003 as a public-private partnership. It was designed to test whether neuroimaging, biological markers in CSF and blood, and clinical and neuropsychological assessments can be combined to predict the diagnosis and progression of AD [18]. Participants with blood MCP-1 levels measured at baseline were included in this analysis. Subjects with mismatched reported sex and biological sex were excluded. After filtering out self-reported non-European ancestry subjects and those without MCP-1 and genotype information, 398 ADNI-1 participants were included in the analysis (Figure S1).
Religious orders study/memory and aging project (ROSMAP)
ROSMAP data were further used to investigate whether cis-regulatory variants affect DNA methylation and gene expression in the brain and their relationships with AD (Figure S1). The ROS is a longitudinal, epidemiologic, clinicopathological study of memory, motor, and functional problems in older Catholic nuns, priests, and brothers aged 65 years and older across the United States. Since 1994, approximately 1200 older persons have been enrolled. The MAP is a longitudinal, epidemiologic clinical-pathologic study of dementia and other chronic diseases of aging. Older persons were recruited from approximately 40 continuous care retirement communities and senior subsidized housing facilities around the Chicago metropolitan area. MAP began in 1997, and more than 1600 older adults were enrolled. ROSMAP participants have yearly blood draws, which result in the storage of serum, plasma, and cells. Clinical evaluation, self-reports, and medication inspection are used to document medical conditions. Subjects are evaluated neurologically every year, and a review of all antemortem data at the time of death leads to a final clinical diagnosis for each participant.
Diagnoses of Alzheimer’s disease in three cohorts
Overall, all three cohorts had longitudinal monitoring of cognition and consensus diagnoses to diagnose AD and other dementia following the guidelines created by the National Institute on Aging and Alzheimer’s Association [19]. In the FHS study, surveillance for incident AD/dementia was initiated in 1975, and diagnoses of AD and other causes of dementia were made through a consensus panel [20]. The ADNI participants underwent longitudinal in-depth neuropsychological evaluations [21], and consensus diagnoses of cognitive normal (CN), mild cognitive impairment (MCI), and AD were assigned based on established research diagnostic criteria [22]. In the ROSMAP study, each individual receives a diagnosis of syndromic AD, MCI, or no cognitive impairment (NCI), and the details have been previously described [23].
Blood MCP-1 measurements
MCP-1 levels (Exam 7) in FHS participants were measured using enzyme-linked immunosorbent assay (ELISA) with a Dade Behring BN100 nephelometer [24] from fasting blood samples that were collected at Exam 7 from the antecubital vein (details have been previously described) [25]. In the ADNI-1, MCP-1 in plasma samples was measured using the Human Discovery MAP Panel and measurement platform [26, 27]. The MCP-1 level was log-transformed in the downstream analysis as a continuous variable. The median MCP-1 level was also used to categorize participants into low versus high MCP-1 groups as a categorical variable.
We also used FHS data on plasma AD biomarkers, including plasma phosphorylated Tau181 (pTau181) [28], total Tau (t-Tau), Aβ40 and Aβ42 [29]. These variables were log-transformed to enable normal distribution. Details of blood sample collection, processing and quality control for measurements of Aβ, t-Tau [30], and pTau181 [28] in the FHS have been previously reported.
Genome-wide association studies of FHS data
Genome-wide association study (GWAS) was performed using the Framingham analytical pipeline for common autosomal single nucleotide polymorphisms (SNPs) imputed using MACH with the 1000 G European Ancestry reference panel from March 2012. In brief, a total of 412,053 genotyped SNPs were used as input to the MACH program (http://genome.sph.umich.edu/wiki/minimac) for phasing. The final genotype set was obtained by following the GIANT protocol for imputation [31] for the November 2010 release of the 1000 G EUR panel based only on individuals of European descent. The relationship between AD and the interaction term for MCP-1 and SNP dosage was tested using GEEPACK (logistic regression utilizing generalized estimating equations; accounts for relatedness in the FHS) while adjusting for age, sex, years of education, and the first 10 principal components (PCs). Additive models were assumed. Only autosomal SNPs (chr 1-22) with a minor allele frequency (MAF) ≥ 5% were chosen for the analysis to reduce false positives due to the sample size of the FHS. Additionally, samples with a genotyping rate of less than 97% and an excess number of heterozygote observations (P < 10−6) or Mendelian errors were removed. Manhattan plots, QQ plots, and genomic controls [32] were used for visualization, quality control, and to account for genomic inflation. LocusZoom [33] was used to present the regional information. P values < 5.0 × 10−8 were considered to indicate genome-wide statistical significance for the interaction between SNPs and MCP-1 for AD.
Brain AD neuropathology in the FHS
A subset of the FHS offspring participants (n = 105) donated their brains to the Boston University AD Research Center (BU-ADRC), which was used for neuropathological characterization. For the data analysis, four routine neuropathology variables were selected: the Braak stage for neurofibrillary degeneration, the CERAD score for the density of neocortical neuritic plaques, the CERAD semiquantitative score for diffuse plaques and the presence of microinfarcts. Neuropathological brain evaluation was performed by neuropathologists blinded to all demographic and clinical information at the BU-ADRC [34]. A total of 78 participants with the above four neuropathology variables and blood MCP-1 measurements were included in the analysis (Figure S1).
DNA methylation and gene expression analyses
ROSMAP data were used to investigate whether cis-regulatory variants (SNPs within 5 kb for mQTLs and 1 Mb for eQTLs of neighboring genes) affect DNA methylation (mQTLs) and gene expression (eQTLs) in the brain as well as their relationships with AD. The mQTL summary results for selected variants were obtained from Brain xQTLServe for ROSMAP (http://mostafavilab.stat.ubc.ca/xQTLServe/). Genotype data were generated from 2093 individuals of European descent. For these individuals, DNA methylation array (Illumina Infinium HumanMethylation 450 K BeadChip) data were derived from fresh frozen postmortem brain samples collected from the dorsolateral prefrontal cortex (DLPFC) region of 468 subjects. Gene expression RNA-seq (Illumina HiSeq) data were generated from the DLPFC of 494 individuals with an average sequencing depth of 90 million reads. A detailed description was previously provided [35]. Next, we studied the associations between the specific CpG site methylation levels and (1) the gene expression levels, and (2) AD status, by Pearson correlation and logistic regression tests, respectively. Furthermore, we examined the differential gene expression patterns across different brain regions using the Agora database (https://agora.adknowledgeportal.org/genes), which includes the cerebellum (CBE), DLPFC, frontal pole (FP), inferior frontal gyrus (IFG), parahippocampal gyrus (PHG), superior temporal gyrus (STG) and temporal cortex (TCX). This database was initially developed by the NIA-funded AMP-AD consortium that shared evidence in support of AD target discovery.
Statistical analysis
Analyses were performed using the R statistical environment (R 3.6.2). Several variables, including sample size, age at baseline, sex, years of education, APOE ε4 status, and incident AD status, were summarized as the baseline characteristics stratified by MCP-1 levels. Group differences were assessed using analysis of variance (ANOVA) for normally distributed continuous variables, the Kruskal–Wallis rank sum test for continuous variables with skewed distributions, and the chi-square test of independence for categorical variables.
In the genotype-stratified analysis, logistic regression was performed for AD status (prevalent + incident AD) using the MCP-1 level (log-transformed continuous or high vs. low binary variable) as the predictor and adjusting for covariates (age at MCP-1 measurement, sex, years of education, and the first 5 PCs). Additionally, both Cox proportional hazards regression and Kaplan‒Meier survival analyses were conducted for AD incidence. Heterozygous and homozygous minor allele genotypes were combined in this section due to the relatively low MAF of the two SNPs (0.14 for rs696468 and 0.06 for rs72659964).
For plasma AD biomarkers, linear regression was performed to examine whether the interactions between SNPs and MCP-1 were associated with log-transformed pTau181, total tau, Aβ40, and Aβ42, adjusting for age, sex, and PCs.
For neuropathology variables, ordinal regression was performed to examine whether the interactions between SNPs and MCP-1 were associated with AD neuropathology traits, including Braak stage, diffuse plaque CERAD score, neuritic plaque CERAD score, and microinfarcts (with logistic regression), adjusting for age, sex, and time to death.
To support the findings in the FHS, the associations between AD cases and blood MCP-1 in ADNI-1 cohort were analyzed using logistic regression models adjusted for age at baseline, sex, and years of education in both interactive analyses and stratification analyses. The results were then combined with FHS results in an inverse-variance weighted meta-analysis using METAL [36]. Code will be made available upon request.
Results
Characteristics of the FHS participants
The 2916 FHS generation 2 participants with data from Exam 7 included in this study had an average follow-up period of 18.46 years (Table 1). Among these participants, there are 190 cases, including prevalent (at the basement when MCP-1 was measured, n = 19) and incident (after the measurement of MCP-1, n = 171) AD cases. Subjects were divided into four quartiles based on their serum MCP-1 concentrations measured at baseline (Exam 7). Participants with the lowest MCP-1 concentration were younger (p < 0.001) and had more years of education (p < 0.001) than participants with higher MCP-1 quartiles. Sex and APOE ɛ4 genotype did not differ across MCP-1 quartiles. Although there was an overall increase in the incidence of AD with increasing MCP-1 quartiles, the relationship was not linear (3.57% vs. 6.31% vs. 4.66% vs. 8.92%, p < 0.001) (Table 1). After adjusting for age, sex, and education, the associations between continuous MCP-1 levels and AD were not significant in the whole sample (data not shown).
Interactions between SNPs and blood MCP-1 for AD risk in the FHS
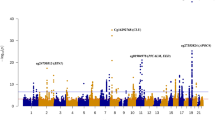
To increase the number of AD cases, we included both prevalent and incident AD cases in a logistic regression model to identify novel genetic variants that interact with blood MCP-1 levels in AD patients by conducting a G×E GWAS with an interaction term between MCP-1 and SNPs as a predictor and AD as an outcome adjusting for age at baseline, sex, years of education, and PCs (Fig. 1 and Figure S2). As shown in the Manhattan plot, 21 SNPs showed suggestive significance (p < 1.0 × 10−7) (Fig. 1A). Notably, one locus on chromosome 12 reached genome-wide significant interactions with MCP-1 for AD association (p < 5.0 × 10−8) (Table S1), including 19 SNPs in the intronic region of the NAV3 gene. Among the SNPs that exhibited genome-wide significance (p < 5.0 × 10−8), rs696468 (minor allele=T, MAF = 0.14) in NAV3 was selected as a sentinel SNP for further analyses (Fig. 1B).

G×E GWAS analyses between MCP-1 (log-transformed) and common SNPs (MAF > 5%) for AD were performed using logistic regression adjusted for age, sex, years of education and PCs. A Manhattan plot is presented with the following highlighted SNPs: NAV3 rs696468 and UNC5C rs72659964 (A). The genomic regional plots present the locations, LD information, p values, recombination rates, nearby gene information and DNA methylation CpG sites associated with the tagged SNPs of NAV3 (B) and UNC5C (C).
Another locus on chromosome 4 reached suggestive significance (p < 1.0 × 10−7), including two SNPs in the intronic region of UNC5C, and rs72659964 (minor allele=T, MAF = 0.06) was chosen as a sentinel SNP in Fig. 1C. Although the UNC5C results did not reach genome-wide significance (p < 5 × 10−8) in FHS cohort, we still included it for further characterization because of the following reasons: (1) Fig. 1 and Table S1 are results from the discovery FHS cohort, where the two UNC5C SNPs reach a near genome-wide significance (p = 8.3 × 10−8, Table S1). (2) NAV3 and UNC5C have similar functions [15, 16]; (3) UNC5C gene is reportedly a susceptibility locus for familial and sporadic AD [37,38,39].; (4) The UNC5C locus includes two SNPs (rs72659964, rs72659961) in perfect LD (p = 8.3 × 10−8, Table S1), along with additional SNPs in moderate LD (r2 = 0.6-0.8, Fig. 1C), indicating a regional signal. Notably, these SNPs have a lower minor allele frequency (MAF = 0.06) compared to those at NAV3 (MAF = 0.14, Table S1), which may explain the fewer tightly linked variants observed.
Specifically, we found the SNP main effects of both NAV3 and UNC5C variants alone were not significant for AD risk (p-adjusted ≥ 0.05). However, the interactive effects of the two SNPs and MCP-1 levels on AD risk were significant: NAV3 rs696468 (p = 1.64 × 10−8) and UNC5C rs72659964 (p = 8.36 × 10−8) (Table S1).
Stratified analysis of NAV3 and UNC5C variants and their interactions with blood MCP-1 for AD risk in the FHS
We next performed SNP-stratified analysis to examine the association between blood MCP-1 levels and AD within each genotype group using logistic regression models with the same covariates included. Individuals with heterozygous and homozygous minor allele genotypes were combined into one group for comparison with individuals with the homozygous major allele genotype group. We found that the relationships between blood MCP-1 levels and AD were significantly different between the two genotype groups of NAV3 and UNC5C, respectively (Table S2). Specifically, the MCP-1 concentration was positively associated with AD risk among NAV3 rs696468-CC carriers (OR = 3.09, 95% CI = 1.75, 5.52; p = 1.20 × 10−4). In contrast, the MCP-1 concentration was negatively associated with AD risk among NAV3 rs696468-CT + TT carriers (OR = 0.20, 95% CI = 0.08, 0.53; p = 0.001). Similarly, the MCP-1 concentration was positively associated with AD incidence among UNC5C rs72659964-AT + TT carriers (OR = 32.84, 95% CI = 4.15, 380.14; p = 0.002), but this trend was largely attenuated and did not reach statistical significance for the UNC5C rs72659964-AA carriers (p = 0.39).
To investigate if blood MCP-1 promotes AD development in different genotypes, we exclusively analyzed AD incidence (n = 171) using Cox proportional hazards regression models. Similar to the logistic regression result, we found that the MCP-1 concentration was positively associated with AD risk among NAV3 rs696468-CC carriers (HR = 2.68, 95% CI = 1.55, 4.62; p = 3.9 × 10−4). In contrast, the MCP-1 concentration was negatively associated with AD risk among NAV3 rs696468-CT + TT carriers (HR = 0.20, 95% CI = 0.10, 0.60; p = 0.002). Similarly, the MCP-1 concentration was positively associated with AD incidence among UNC5C rs72659964-AT + TT carriers (HR = 80.74, 95% CI = 8.32, 783.28; p = 1.5 × 10−4), but this trend was largely attenuated and did not reach statistical significance for the UNC5C rs72659964-AA carriers (p = 0.64) (Table 2). Similar associations were observed if the variants were stratified by three genotypes (Tables S3).
Participants were also divided into different groups based on percentile cutoffs of MCP-1 concentrations, such as <50% and ≥ 50%, <60% and ≥60%, and <75% and ≥75%. Cox regression analyses were subsequently conducted to compare the relatively dose-dependent risk of developing AD over time between the high MCP-1 group and the low MCP-1 group (e.g., MCP-1 ≥ 50% vs. <50%) within each genotype group. As shown in Fig. 2A, increasing MCP-1 concentration cutoffs were positively associated with a greater incidence of AD among NAV3 rs696468-CC carriers (i.e., NAV3-) but negatively associated with AD risk among NAV3 rs696468-CT + TT carriers (i.e., NAV3 + ). Similarly, increasing MCP-1 cutoffs were positively associated with AD incidence among UNC5C rs72659964-AT + TT carriers, but this increasing trend for AD risk was not observed in UNC5C rs72659964-AA carriers (Fig. 2B). Consistent with these findings, when individuals were categorized based on <50% or ≥ 50% of the MCP-1 concentration according to Kaplan‒Meier analyses, we observed that high MCP-1 levels were associated with increased AD risk among NAV3 rs696468-CC carriers (p = 1.1 × 10−4) and UNC5C rs72659964-AT + TT carriers (p = 0.03). In contrast, among persons with their counterpart genotypes, elevated MCP-1 showed no (NAV3 rs696468-CT + TT, p = 0.15) or attenuated (UNC5C rs72659964-AA, p = 0.04) associations with AD risk (Fig. 2C, D).

The FHS Gen2 participants were divided into different genotype groups: NAV3\(-\) (rs696468 CC, n = 2189) and NAV3\(+\) (rs696468 CT/TT, n = 695) or UNC5C\(-\) (rs72659964 AA, n = 2552) and UNC5C\(+\) (rs72659964 AT/TT, n = 332). Within each genotype group, individuals were further divided into low and high MCP-1 groups based on different cutoff MCP-1 levels: <50% and ≥ 50%, <60% and ≥ 60%, and <75% and ≥ 75%. Cox proportional hazards regression analyses were applied to examine the effect of different levels of MCP-1 on the incidence of AD within each genotype group after adjusting for age, sex, years of education and PCs. HRs and p values are illustrated (A and B). In addition, participants were divided into low and high groups based on the median MCP-1 concentration. Kaplan‒Meier survival plots were then used to show the probability of AD-free survival within each genotype group. The differences between the low vs. high groups for each genotype are illustrated with p values (C and D).
Associations of blood MCP-1 levels and plasma AD biomarkers among different genotypes in the FHS
To further study the impact of blood MCP-1 on the associations of the NAV3 variant and UNC5C variant for AD risk, we examined their relationships with AD biomarkers, including plasma pTau181, t-Tau, Aβ40, and Aβ42, within each genotype group. Consistent with AD risk, using linear regression analysis after adjusting for age, sex, and PCs, we found that increasing MCP-1 concentration was positively associated with plasma pTau181 among NAV3 rs696468-CC carriers (n = 1467, β = 0.09, p = 0.02), but no such relationship was noted among NAV3 rs696468-CT + TT carriers (Fig. 3A). Similarly, increasing MCP-1 concentrations were positively associated with plasma pTau181 among UNC5C rs72659964-AT + TT carriers (n = 240, β = 0.23, p = 0.04), but this increasing trend was not observed in UNC5C rs72659964-AA carriers (Fig. 3B). There was no significant association for plasma t-Tau regardless of genotype status (Fig. 3C, D). Regarding plasma Aβ40 or Aβ42 as an outcome (Fig. 3E–H), we found that increasing MCP-1 concentrations were positively associated with plasma Aβ40 levels only among UNC5C rs72659964-AT + TT carriers (n = 342, β = 0.08, p = 0.03) (Fig. 3F) and with plasma Aβ42 levels only among NAV3 rs696468-CT + TT carriers (n = 716, β = 0.06, p = 0.04) (Fig. 3G). Of note, adjusting for APOE ε4 in the models does not change our conclusion (Figure S3).

The FHS Gen2 participants were divided into subgroups based on the genotype of NAV3 rs696468 CC (n = 1467 for pTau181) and NAV3 rs696468 CT/TT (n = 465 for pTau181) (A); UNC5C rs72659964 AA (n = 1692 for pTau181) and UNC5C rs72659964 AT/TT (n = 240 for pTau181) (B). The data on plasma levels of pTau181 (A and B), t-Tau (C and D), Aβ40 (E and F) and Aβ42 (G and H) were used. The linear regression models were performed between the AD biomarkers (log-transformed) and continuous blood MCP-1 concentration (log-transformed) after adjusting for age, sex, years of education and PCs. Bar charts showing regression estimates (β)±se of the MCP-1 effect for each biomarker within each genotype with p values.
Associations of blood MCP-1 and brain AD neuropathology among different genotypes in the FHS
To further investigate the impact of blood MCP-1 levels on AD pathology in the brains with different genotypes, we used the FHS Brain Bank dataset, in which 78 participants donated their brains after death. Due to the small number of brain donors with blood MCP-1 measurements when they were alive, we first divided blood MCP-1 levels based on the median ( < 50% vs. ≥ 50%). The relationships between categorized blood MCP-1 levels and the four neuropathological features within each genotype were evaluated using Fisher’s exact test. Consistent with AD risk (Table 2 and Fig. 2), Fig. 4 shows that in NAV3 rs696468-CC carriers, but not in rs696468-CT + TT carriers, the group with high MCP-1 levels had a greater average Braak score (Fig. 4A, p = 0.04) and was significantly associated with a higher CERAD score (Fig. 4B, p = 0.05), a higher CERAD semiquantitative score (Fig. 4C, p = 0.02) and a greater percentage of microinfarcts (Fig. 4D, p = 0.007) than those with low MCP-1 levels. The UNC5C rs72659964 results were not conclusive for the neuropathology comparisons because there were only 5 subjects in the rs72659964-AT + TT genotype group (Fig. 4E–H).

The FHS participants with neuropathology data (n = 78) were divided into NAV3\(-\) (rs696468 CC, n = 56) and NAV3\(+\) (rs696468 CT/TT, n = 22) genotype groups (A–D) or UNC5C\(-\) (rs72659964 AA, n = 73) and UNC5C\(+\) (rs72659964 AT/TT, n = 5) genotype groups (E–H). Due to the small sample size, brain neuropathology scores were divided into low- vs. high-score groups, including Braak stage (low = 0, 1, 2, 3 vs. high = 4, 5, 6) (A and E), CERAD (neocortical neuritic plaque, low = 0 vs. high = 1, 2, 3) (B and F), CERAD (diffuse plaques, low = 0,1 vs. high = 2, 3) (C and G) and microinfarcts (no vs. yes) (D and H). MCP-1 levels were also divided into low ( < 50%) and high ( ≥ 50%) groups. Bar charts showing the percentages of patients with categorized neuropathology scores within each MCP-1 group for each genotype of NAV3 and UNC5C. The number of participants, percentage, and Fisher exact test p values are shown.
Ordinal regression and logistic regression analyses were also conducted using neuropathology traits as outcomes after adjusting for age, sex, and time to death. We found that Braak stage and CERAD score were associated with the interaction between the two SNPs and blood MCP-1 levels. The interaction of NAV3 rs696468 with blood MCP-1 concentration significantly influenced the Braak stage (p = 0.02), CERAD score (neocortical neuritic plaque) (p = 0.03), and CERAD semiquantitative score (diffuse plaques) (p = 0.03). UNC5C rs72659964 significantly interacted with blood MCP-1 to influence the CERAD semiquantitative score (p < 0.001) (Table S4).
Integrative analysis of variants in NAV3 and UNC5C with DNA methylation and gene expression in AD
Consistent with the neuropathology findings, in the Agora, an AMP-AD database, there was a significantly lower level of NAV3 gene expression in the DLPFC (p = 0.02) and temporal cortex (TCX) (p = 8.7 × 10−6) of AD patients than in cognitively normal controls (Fig. 5A). To investigate the possible regulatory mechanism of SNP rs696468 in NAV3 and whether rs696468 cis-regulates NAV3 gene expression levels, we studied mQTL and eQTL mapping by leveraging the available genotype, DNA methylation and RNA-seq gene expression data measured from the same DLPFC brain region in the ROSMAP (http://mostafavilab.stat.ubc.ca/xQTLServe/). Interestingly, NAV3 rs696468 was negatively associated with the methylation level of the CpG site cg20521863, which is located within the intron region of NAV3 (p = 0.003) (Figs. 5B and 1B), and the cg20521863 methylation level was negatively associated with NAV3 gene expression (r = -0.19, p = 1.2 × 10−5) (Fig. 5C). Notably, there was a greater methylation level of cg20521863 in AD patients (p = 0.01) than in normal controls (Fig. 5D), which may have led to lower NAV3 expression (Fig. 5B) and further interaction with MCP-1 to influence AD risk (Fig. 5E).

Associations between AD and NAV3 gene expression levels across different brain regions. CBE: cerebellum; DLPFC: dorsolateral frefrontal cortex; FP: frontal pole; IFG: inferior frontal gyrus; PHG: parahippocampal gyrus; STG: superior temporal gyrus; TCX: temporal cortex (A). The associations between the NAV3 SNP rs696468 and the DNA methylation CpG site cg20521863 (B), between the CpG site cg20521863 and NAV3 gene expression (C), and between the CpG site cg20521863 and cognitive status in the DLPFC region of the ROSMAP cohort (D). The interaction effect between NAV3 rs696468 and blood MCP-1 on AD risk in the FHS (E).
On the other hand, using the same datasets, we did not observe significant associations for UNC5C rs72659961/rs72659964 in any of the molecular QTL mapping databases we studied (data not shown). The regulatory mechanism of UNC5C variants in AD is unclear.
Replication analysis of the interactive effects between blood MCP-1 and variants in NAV3 and UNC5C on AD risk in the ADNI cohort
To replicate the findings in the FHS, we analyzed the ADNI-1 cohort using logistic regression models and adjusted for the same covariates as in the FHS. The interaction effects between the SNPs and MCP-1 on AD risk did not reach statistical significance according to the ADNI, probably due to the small sample size (n = 398) (Table S5). However, the same directions of associations with AD risk for NAV3 rs696468 and UNC5C rs72659964 were observed in the ADNI as in the FHS.
We conducted a meta-analysis using the summary statistics results from the FHS and ADNI-1 for the 21 suggestive SNPs (p < 1.0 × 10−7). The meta-analysis increased the significance of the interaction effects of both NAV3 rs696468 (p = 7.6 × 10−9) and UNC5C rs72659964 (p = 1.1 × 10−8) with MCP-1 on AD risk to meet the genome-wide threshold, especially for UNC5C (Table S6). In the SNP stratified meta-analysis, we observed the same effect direction of elevated blood MCP-1 for increasing AD risk in the rs696468-CC (Z = 3.87, p = 1.1 × 10−4) and UNC5C rs72659964-AT + TT (Z = 3.35, p = 8.0 × 10−4) genotypes in both the FHS and ADNI datasets (Table S5).
Discussion
In this study, we conducted a genome-wide G×E analysis to investigate the interactive influence of genetic and internal environmental factors (blood MCP-1 levels) on AD development. We discovered that elevated blood MCP-1 levels were associated with a greater risk of AD among individuals who carried the NAV3-rs696468-CC and UNC5C-rs72659964-AT + TT alleles, but not among the carriers of NAV3-rs696468-CT + TT and UNC5C-rs72659964-AA (Fig. 2, Table 2 and Table S2). It is worth noting that the genotypes alone for AD risk are NAV3-rs696468 T allele carriers (main effect, p = 0.05-nominally significant, Table S1). In the interaction analysis, we found that NAV3-rs696468-CC, but not T allele, carriers are the ones where higher MCP-1 levels lead to more AD risk. These results indicated that the CC carriers are likely to be influenced by peripheral inflammatory factors like MCP-1 to develop AD, while T allele carriers may be predisposed to AD probably through other mechanisms or pathways. Consistent with our findings, it is reported that a higher plasma MCP-1 level is associated with greater severity of AD and MCI as well as faster cognitive decline [10]. Cherry and Stein et al [12]. reported that MCP-1 protein levels in the DLPFC are correlated with the density of Iba1+ cells and CD68+ cells and with increased chronic traumatic encephalopathy (CTE) severity and are correlated with pTau independent of age at death and Aβ42 in AD and CTE patients.
The relationship between peripheral MCP-1 and AD biomarkers varied by the genotypes of NAV3-rs696468 and UNC5C-rs72659964, i.e., positive associations with plasma pTau181 in NAV3-rs696468-CC and UNC5C-rs72659964-AT + TT carriers (Fig. 3). This finding is supported by AD neuropathology in the brain (Fig. 4). Further explorations in the ROSMAP cohort revealed that NAV3 rs696468 reduces AD risk by interacting with blood MCP-1 levels through regulating the level of DNA methylation, which subsequently regulates NAV3 gene expression in the brain (Fig. 5). Importantly, we observed negative associations between NAV3 expression and AD in the DLPFC and temporal cortex region. Blood MCP-1 levels may act as a moderator for the association between AD and NAV3 expression, which has been shown to be impacted by NAV3 genetic variants. NAV3 exhibits cellular function in the nervous system in C. elegans [40]. It has been reported that some microRNAs, e.g., miR-29c, regulate NAV3 protein expression in an AD mouse model [41]. In a recent study, the association between AD and the NAV3 gene was further highlighted using an AI-based approach with data from the ROSMAP and Mayo RNAseq studies (https://adknowledgeportal.org) [42].
The two SNPs are relatively common, with MAFs of 0.14 for NAV3 rs696468 and of 0.06 for UNC5C rs72659964. In contrast to the NAV3 variant, our study suggested that UNC5C-rs72659964 genotypes were influenced by peripheral MCP-1 and reached statistical significance only in the meta-analysis of the FHS and ADNI results (Fig. 1 and Table S6), which is probably due to a relatively lower MAF of the SNP rs72659964. The UNC5C variant is a genetic risk factor associated with AD in humans [15, 16, 43, 44]. Furthermore, UNC5C and NAV3 are homologs that play crucial roles in neuron outgrowth and guidance in C. elegans [15, 16, 43, 44]. Mechanistic studies using cell and animal models revealed that aberrant UNC5C might contribute to AD by activating death-associated protein kinase 1 (DAPK1), which has been implicated in AD neuropathogenesis due to its extensive involvement in aberrant tau, Aβ and neuronal apoptosis/autophagy [16, 45]. In addition, deleting UNC5C from netrin-1-depleted mice can mitigate AD pathologies and reduce cognitive disorders. δ-Secretase truncates UNC5C and increases its neurotoxicity, contributing to AD pathogenesis as well [37]. In brain imaging genetic studies, the UNC5C locus has been reported to be associated with temporal volume and to alter the atrophy of strategic regions of AD, such as the hippocampus and precuneus [46].
Additionally, our study revealed a correlation between increased blood MCP-1 concentrations and older age (Table 1). This finding aligns with previous research indicating that advancing age is linked to peripheral chronic inflammation leading to neurodegeneration [47]. Based on the findings in this study and others [17, 48], it is possible that proinflammatory factors such as MCP-1 in the peripheral nervous system may heighten the risk of neurodegenerative changes in the brain, particularly in vulnerable genetic carriers of UNC5C and NAV3.
This study has several limitations. First, due to the small sample size of the ADNI cohort and the age difference in the FHS participants, we only observed a trend of the interactive genotype-MCP-1 effects on AD without statistical significance in ADNI. Second, the low MAF (0.06) for UNC5C rs72659964) and the small number of samples with available brain neuropathology data in the FHS (n = 78) limited the genotype stratification analysis for AD neuropathological features, which results in a wide 95% CI. Third, without cell and animal model studies, the mechanism by which increased peripheral MCP-1 during the aging process may influence the ability of UNC5C and NAV3 variants to increase AD risk is unclear. Fourth, we picked the lead SNP with the smallest p-value for further analyses, and did not perform any fine mapping analysis to identify the potential causal SNP--this decision was based on the study’s primary focus on G×E interactions rather than SNP main effects. In the current study, we have performed some standard functional analyses, such as mQTL by associating SNPs with DNA methylation, eQTL by associating SNPs with gene expression, and differential expression analysis, to obtain additional supporting evidence for the implicated SNPs and genes. However, we recognize the importance of causal inference and are planning to conduct analyses, including fine mapping of interactive SNPs, in the future. Fifth, when performing the analyses, only 1000 G imputation was available in our FHS GWAS pipeline, and the TOPMed imputation will be used in the future when we finish updating the whole pipeline. Future studies are needed to replicate the effect of blood MCP-1 levels on AD and AD-related endophenotypes in larger postmortem cohorts, especially those with multiethnic representation, to assess the generalizability of our findings.
Notably, our 2023 study [2] (i.e., MCP-1×APOE/HLA-DRB1 interactions) mainly examined a targeted set of 10 well-established AD SNPs located in inflammatory-related genes, including APOE ε4 (P = 0.001) and HLA-DRB1 (rs9271192, P = 0.004). Because that was a candidate gene study, we applied a Bonferroni correction specific to the number of SNPs tested (0.05/10 = 0.005), rather than a genome-wide significance threshold (P < 5 × 10−8). As a result, these SNPs do not appear as top in our Manhattan plot here, which is based on genome-wide significance criteria. In this study, our goal is to perform a genome-wide screening to identify novel loci that may interact with MCP-1 levels to influence AD risk, including SNPs outside of canonical AD pathways.
By applying the G×E GWAS in multiple cohorts and assessing clinical outcomes, plasma AD biomarkers and AD brain pathology, our study identified two novel loci and revealed that these two genetic loci were impacted and probably modified by elevated blood MCP-1 levels for AD risk. This finding implies the importance of considering individual genotypes for precision medicine/prevention when assessing the impact of specific peripheral inflammatory factors on AD risk. As multiple MCP-1 inhibitors are currently being developed and tested in clinical trials for cancer and autoimmune diseases [49], these medications could be repurposed for AD intervention and prevention in individuals who carry the NAV3 rs696468-CC and UNC5C rs72659964-AT + TT alleles and who suffer from peripheral inflammation.
Data availability
The FHS data are available at dbGaP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000007.v33.p14) upon reasonable request/application. The ADNI data are available at https://adni.loni.usc.edu/ upon reasonable request/application. The ROSMAP data are available at the AD Knowledge Portal (https://adknowledgeportal.org).
References
Shen XN, Niu LD, Wang YJ, Cao XP, Liu Q, Tan L, et al. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: a meta-analysis and systematic review of 170 studies. J Neurol Neurosurg Psychiatry. 2019;90:590–8.
Huang J, Stein TD, Wang Y, Ang TFA, Tao Q, Lunetta KL, et al. Blood levels of MCP-1 modulate the genetic risks of Alzheimer’s disease mediated by HLA-DRB1 and APOE for Alzheimer’s disease. Alzheimers Dement. 2023;19:1925–37.
Yadav A, Saini V, Arora S. MCP-1: chemoattractant with a role beyond immunity: a review. Clin Chim Acta. 2010;411:1570–9.
Gschwandtner M, Derler R, Midwood KS. More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front Immunol. 2019;10:2759.
Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature. 2020;577:399–404.
Fenoglio C, Galimberti D, Lovati C, Guidi I, Gatti A, Fogliarino S, et al. MCP-1 in Alzheimer’s disease patients: A-2518G polymorphism and serum levels. Neurobiol Aging. 2004;25:1169–73.
Galimberti D, Fenoglio C, Lovati C, Venturelli E, Guidi I, Corra B, et al. Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol Aging. 2006;27:1763–8.
Galimberti D, Schoonenboom N, Scheltens P, Fenoglio C, Bouwman F, Venturelli E, et al. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch Neurol. 2006;63:538–43.
Westin K, Buchhave P, Nielsen H, Minthon L, Janciauskiene S, Hansson O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS One. 2012;7:e30525.
Lee WJ, Liao YC, Wang YF, Lin IF, Wang SJ, Fuh JL. Plasma MCP-1 and cognitive decline in patients with Alzheimer’s disease and mild cognitive impairment: a two-year follow-up study. Sci Rep. 2018;8:1280.
Bettcher BM, Neuhaus J, Wynn MJ, Elahi FM, Casaletto KB, Saloner R, et al. Increases in a pro-inflammatory chemokine, MCP-1, are related to decreases in memory over time. Front Aging Neurosci. 2019;11:25.
Cherry JD, Meng G, Daley S, Xia W, Svirsky S, Alvarez VE, et al. CCL2 is associated with microglia and macrophage recruitment in chronic traumatic encephalopathy. J Neuroinflammation. 2020;17:370.
Yao Y, Tsirka SE. Monocyte chemoattractant protein-1 and the blood-brain barrier. Cell Mol Life Sci. 2014;71:683–97.
Joly-Amado A, Hunter J, Quadri Z, Zamudio F, Rocha-Rangel PV, Chan D, et al. CCL2 overexpression in the brain promotes glial activation and accelerates tau pathology in a mouse model of tauopathy. Front Immunol. 2020;11:997.
Shioya M, Obayashi S, Tabunoki H, Arima K, Saito Y, Ishida T, et al. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol Appl Neurobiol. 2010;36:320–30.
Wetzel-Smith MK, Hunkapiller J, Bhangale TR, Srinivasan K, Maloney JA, Atwal JK, et al. A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat Med. 2014;20:1452–7.
Tao Q, Ang TFA, DeCarli C, Auerbach SH, Devine S, Stein TD, et al. Association of chronic low-grade inflammation with risk of Alzheimer disease in ApoE4 carriers. JAMA Netw Open. 2018;1:e183597.
Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, et al. Alzheimer’s disease neuroimaging initiative (ADNI): clinical characterization. Neurology. 2010;74:201–9.
Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–62.
Weinstein G, Wolf PA, Beiser AS, Au R, Seshadri S. Risk estimations, risk factors, and genetic variants associated with Alzheimer’s disease in selected publications from the Framingham Heart Study. J Alzheimers Dis. 2013;33:S439–45. Suppl 1
Aisen PS, Petersen RC, Donohue MC, Gamst A, Raman R, Thomas RG, et al. Clinical core of the Alzheimer’s disease neuroimaging initiative: progress and plans. Alzheimers Dement. 2010;6:239–46.
Huang K, Lin Y, Yang L, Wang Y, Cai S, Pang L, et al. A multipredictor model to predict the conversion of mild cognitive impairment to Alzheimer’s disease by using a predictive nomogram. Neuropsychopharmacology. 2020;45:358–66.
Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. J Alzheimers Dis. 2018;64:S161–S89.
Wilson PW, Nam BH, Pencina M, D’Agostino RB Sr., Benjamin EJ, O’Donnell CJ. C-reactive protein and risk of cardiovascular disease in men and women from the Framingham Heart Study. Arch Intern Med. 2005;165:2473–8.
McDermott DH, Yang Q, Kathiresan S, Cupples LA, Massaro JM, Keaney JF Jr., et al. CCL2 polymorphisms are associated with serum monocyte chemoattractant protein-1 levels and myocardial infarction in the Framingham Heart Study. Circulation. 2005;112:1113–20.
Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007;13:1359–62.
Trojanowski JQ, Vandeerstichele H, Korecka M, Clark CM, Aisen PS, Petersen RC, et al. Update on the biomarker core of the Alzheimer’s disease neuroimaging initiative subjects. Alzheimers Dement. 2010;6:230–8.
McGrath ER, Beiser AS, O’Donnell A, Yang Q, Ghosh S, Gonzales MM, et al. Blood phosphorylated Tau 181 as a biomarker for amyloid burden on brain PET in cognitively healthy adults. J Alzheimers Dis. 2022;87:1517–26.
Romero JR, Demissie S, Beiser A, Himali JJ, DeCarli C, Levy D, et al. Relation of plasma beta-amyloid, clusterin, and tau with cerebral microbleeds: Framingham Heart Study. Ann Clin Transl Neurol. 2020;7:1083–91.
Chouraki V, Beiser A, Younkin L, Preis SR, Weinstein G, Hansson O, et al. Plasma amyloid-beta and risk of Alzheimer’s disease in the Framingham Heart Study. Alzheimers Dement. 2015;11:249–57 e1.
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–9.
Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004.
Boughton AP, Welch RP, Flickinger M, VandeHaar P, Taliun D, Abecasis GR, et al. LocusZoom.js: interactive and embeddable visualization of genetic association study results. Bioinformatics. 2021;37:3017–8.
Au R, Seshadri S, Knox K, Beiser A, Himali JJ, Cabral HJ, et al. The Framingham Brain Donation Program: neuropathology along the cognitive continuum. Curr Alzheimer Res. 2012;9:673–86.
Ng B, White CC, Klein HU, Sieberts SK, McCabe C, Patrick E, et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci. 2017;20:1418–26.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
Chen G, Kang SS, Wang Z, Ahn EH, Xia Y, Liu X, et al. Netrin-1 receptor UNC5C cleavage by active delta-secretase enhances neurodegeneration, promoting Alzheimer’s disease pathologies. Sci Adv. 2021;7:eabe4499.
Velez JI, Samper LA, Arcos-Holzinger M, Espinosa LG, Isaza-Ruget MA, Lopera F, et al. A comprehensive machine learning framework for the exact prediction of the age of onset in familial and sporadic Alzheimer’s disease. Diagnostics (Basel). 2021;11:887.
Chen G, Ahn EH, Kang SS, Xia Y, Liu X, Zhang Z, et al. UNC5C receptor proteolytic cleavage by active AEP promotes dopaminergic neuronal degeneration in Parkinson’s disease. Adv Sci (Weinh). 2022;9:e2103396.
Maes T, Barcelo A, Buesa C. Neuron navigator: a human gene family with homology to unc-53, a cell guidance gene from Caenorhabditis elegans. Genomics. 2002;80:21–30.
Zong Y, Yu P, Cheng H, Wang H, Wang X, Liang C, et al. miR-29c regulates NAV3 protein expression in a transgenic mouse model of Alzheimer’s disease. Brain Res. 2015;1624:95–102.
Raschka T, Sood M, Schultz B, Altay A, Ebeling C, Frohlich H. AI reveals insights into link between CD33 and cognitive impairment in Alzheimer’s Disease. PLoS Comput Biol. 2023;19:e1009894.
Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci USA. 2008;105:6415–20.
Li Q, Wang BL, Sun FR, Li JQ, Cao XP, Tan L. The role of UNC5C in Alzheimer’s disease. Ann Transl Med. 2018;6:178.
Hashimoto Y, Toyama Y, Kusakari S, Nawa M, Matsuoka M. An Alzheimer disease-linked rare mutation potentiates netrin receptor uncoordinated-5C-induced signaling that merges with amyloid beta precursor protein signaling. J Biol Chem. 2016;291:12282–93.
Sun JH, Wang HF, Zhu XC, Yu WJ, Tan CC, Jiang T, et al. The impact of UNC5C genetic variations on neuroimaging in Alzheimer’s disease. Mol Neurobiol. 2016;53:6759–67.
Cao W, Zheng H. Peripheral immune system in aging and Alzheimer’s disease. Mol Neurodegener. 2018;13:51.
Tao Q, Alvin Ang TF, Akhter-Khan SC, Itchapurapu IS, Killiany R, Zhang X, et al. Impact of C-Reactive protein on cognition and Alzheimer disease biomarkers in homozygous apolipoprotein E varepsilon4 carriers. Neurology. 2021; 97:e1243–52.
Bianconi V, Sahebkar A, Atkin SL, Pirro M. The regulation and importance of monocyte chemoattractant protein-1. Curr Opin Hematol. 2018;25:44–51.
Acknowledgements
We want to express our thanks to the FHS/ADNI/ROSMAP participants for their decades of dedication and to the FHS/ADNI/ROSMAP staff for their hard work in collecting and preparing the data. The sponsor institutes did not play any role in the design or execution of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; or the decision to submit the manuscript for publication. For the Alzheimer’s Disease Neuroimaging Initiative (ADNI): Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Funding
This work was supported by National Institute on Aging grants U19-AG068753, RF1-AG057519, and R01-AG048927. Data collection and sharing for this project were funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). The ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following companies: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC. ; Johnson & Johnson Pharmaceutical Research & Development LLC. ; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC. ; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The data available in the AD Knowledge Portal would not be possible without the participation of research volunteers and the contribution of data by collaborating researchers. The results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.org). Study data were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding from NIA grants P30AG10161 (ROS), R01AG15819 (ROSMAP; genomics and RNAseq), R01AG17917 (MAP), R01AG30146, R01AG36042 (5hC methylation, ATACseq), RC2AG036547 (H3K9Ac), R01AG36836 (RNAseq), R01AG48015 (monocyte RNAseq), RF1AG57473 (single nucleus RNAseq), U01AG32984 (genomic and whole exome sequencing), U01AG46152 (ROSMAP AMP-AD, targeted proteomics), U01AG46161 (TMT proteomics), U01AG61356 (whole-genome sequencing, targeted proteomics, ROSMAP AMP-AD), the Illinois Department of Public Health (ROSMAP), and the Translational Genomics Research Institute (genomic).
Author information
Authors and Affiliations
Contributions
JH: Formal analysis, Methodology, Software, Writing - original draft. YW: Formal analysis, Validation, Writing - review & editing. TS: Writing - review & editing. TFAA: Data curation, Resources, Writing - review & editing. YZ: Formal analysis, Validation. QT: Data curation, Resources, Writing - review & editing. KL: Writing - review & editing. JM: Writing - review & editing. RA: Writing - review & editing, Funding acquisition. LF: Writing - review & editing, Funding acquisition. WQ: Conceptualization, Methodology, Project administration, Supervision, Writing - original draft. XZ: Conceptualization, Methodology, Project administration, Supervision, Writing - original draft.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The FHS was approved by the Institutional Review Board of Boston University, and all participants provided written informed consent. The ROS and MAP studies were approved by the Institutional Review Board of Rush University Medical Center. All participants signed informed consent, the Anatomic Gift Act for brain donation, and repository consent forms to allow their data and biospecimens to be shared.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, J., Wang, Y., Stein, T.D. et al. The impact of blood MCP-1 levels on Alzheimer’s disease with genetic variation at the NAV3 and UNC5C loci. Transl Psychiatry 15, 296 (2025). https://doi.org/10.1038/s41398-025-03542-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03542-w
