
Abstract
We have previously shown that general deletion of the gene encoding the p53-inducible Mir34a microRNA enhances the number and invasion of colitis-associated colorectal cancers (CACs) in mice. Since the p53-pathway has been implicated in tumor-suppression mediated by cells in the tumor microenvironment (TME) we deleted Mir34a in myeloid cells and characterized CACs in these with scRNA-Seq (single cell RNA sequencing). This revealed an increase in specific macrophage subtypes, such as Cdk8+ macrophages and Mrc1+, M2-like macrophages. The latter displayed elevated expression of 21 known Mir34a target mRNAs, including Csf1r, Axl, Foxp1, Ccr1, Nampt, and Tgfbr2, and 32 predicted Mir34a target mRNAs. Furthermore, Mir34a-deficient BMDMs showed enhanced migration, elevated expression of Csf1r and a shift towards M2-like polarization when compared to Mir34a-proficient BMDMs. Concomitant deletion of Csf1r or treatment with a Csf1r inhibitor reduced the CAC burden and invasion in these mice. Notably, loss of myeloid Mir34a function resulted in a prominent, inflammatory CAC cell subtype, which displayed epithelial and macrophage markers. These cells displayed high levels of the EMT transcription factor Zeb2 and may therefore enhance the invasiveness of CACs. Taken together, our results provide in vivo evidence for a tumor suppressive role of myeloid Mir34a in CACs which is, at least in part, mediated by maintaining macrophages in an M1-like state via repression of Mir34a targets, such as Csf1r. Collectively, these findings may serve to identify new therapeutic targets and approaches for treatment of CAC.
Similar content being viewed by others


Introduction
Chronic inflammation of the intestine, as observed in ulcerative colitis (UC) and inflammatory bowel disease (IBD), often results in colitis-associated colorectal cancers (CAC; [1]). Patients with CACs have a poorer prognosis than patients with sporadic or inherited colorectal cancers (CRCs; [1,2,3,4]). The genetic alterations that contribute to sporadic CRC and CACs are similar but occur in a different order [2]. For example, the mutational inactivation of p53 is an early event in CAC, whereas it occurs late in sporadic CRC [2].
Among the p53-induced microRNAs (miRNAs), miR-34a stands out as the most strongly induced miRNA according to numerous comprehensive expression studies [5]. We had previously observed that germline deletion of Mir34a enhances tumor initiation, progression, and invasion in a murine model of colitis-associated colon cancer [6]. However, it remained unknown whether the effects of the Mir34a inactivation were tumor cell autonomous, or whether Mir34a-deficient cells within the tumor stroma were also involved.
The tumor microenvironment plays an important role in tumor progression [7], as it contributes to invasion and therapy resistance by modulating cell-cell interactions and altering cytokine and chemokine signaling (reviewed in ref. [8]). Macrophages are an important part of the tumor microenvironment. It has been shown ex vivo that miR-34a inhibits polarization towards M2-macrophages [9, 10], which are typically alternatively activated, tumor-associated macrophages (TAM) and are known to enhance tumor growth, angiogenesis, invasion, metastasis and therapy resistance [11,12,13]. Furthermore, M2-polarization of macrophages is promoted by growth factors, such as the colony-stimulating factor (CSF-1), and cytokines, like IL-4, IL-13 and IL-10, released by cells in the tumor-microenvironment (reviewed in ref. [11]).
Colony-stimulating factor 1 receptor (CSF1R) is the receptor for CSF1, also known as macrophage colony-stimulating factor (M-CSF), which, among other functions, is involved in the differentiation of hematopoietic stem cells into macrophages [14,15,16] and is secreted by epithelial tumor cells after p53 inactivation [17]. Recently, we showed that CSF1R is a direct, conserved target of miR-34a in human CRC cells [18] and in a murine model of inherited CRC [19]. Deletion of Mir34a in Apcmin/+ mice resulted in increased adenoma formation, increased immune cell infiltration and decreased survival of mice. Concomitant intestinal-epithelial-cell (IEC) specific deletion of Csf1r reversed these effects [19]. Targeting CSF1R appears to be a promising anti-tumor strategy [11] and the presence of CSF1R-positive macrophages is associated with poor survival [20, 21].
Here, we determined that myeloid cell-specific deletion of Mir34a resulted in more invasive CACs, which displayed increased infiltration by macrophages and neutrophils. A comprehensive characterization of CACs and their tumor microenvironment (TME) by scRNA-Seq identified an increase of specific macrophage subtypes in Mir34aΔMye CACs as well as elevated levels of Csf1r expression in Mir34aΔMye macrophages. Deletion or therapeutic inhibition of Csf1r reduced the tumor burden in these mice. In addition, numerous Mir34a mRNA targets were significantly upregulated in Mir34a-deficient macrophages and neutrophils. Mir34a-deficient bone-marrow derived macrophages (BMDMs) showed increased migration, elevated levels of Csf1r as well as upregulation of M2 markers whereas the expression levels of M1 markers decreased. Finally, scRNA-Seq revealed a prominent population of inflammatory tumor cells with high expression of chemokines and cytokines as well as macrophage markers in Mir34aΔMye CACs.
Materials and methods
Breeding and handling of mice
The generation of Mir34afl/fl mice was previously described [22]. LysM-Cre (B6.129P2-Lyztm1(cre)Ifo/J) mice and Csf1rfl/fl (B6.Cg-Csf1rtm1Jwp/J) were purchased from Jackson Laboratories (Stock: 004781 and Stock: 021212) [23, 24]. To delete Mir34a in myeloid cells (Mir34aΔMye), Mir34afl/fl mice were crossed with LysM-Cre mice. Mir34aΔMye mice were crossed with Csf1rfl/fl mice to obtain Mir34a; Csf1rΔMye mice. For generation of Csf1rΔMye mice, LysM-Cre mice were crossed with Csf1rfl/fl mice. All mice were backcrossed on C57Bl/6 background for at least five generations. Co-housed Mir34a-proficient Mir34afl/fl littermates were used as controls in all experiments. Mice at 6–10 weeks of age were intraperitoneally (i.p.) injected with azoxymethane (AOM, 10 mg/kg) 5 days prior to the first and second dextran sodium sulfate (DSS) induced colitis period. The first and second colitis period were induced by giving 2.5% DSS dissolved in drinking water for a period of 5 days, followed by 16 days of regular water. Subsequently, the third colitis period was induced by giving 2% DSS for 5 days. All mice were sacrificed 110 days after the first AOM injection. For experiments with additional conditional deletion of Csf1r and inhibitor treatment, the dosage of DSS was reduced in second cycle to 2% and mice were sacrificed 100 days after the first injection. The Csf1r inhibitor GW2580 (Sigma-Aldrich) was given in chow (1%) with ad libidum access from day 60 onwards. GW2580-supplemented food was prepared by Sniff Special Diets (Soest, Germany). Mice were kept on a 12 h light/dark cycle in individually ventilated cages in a specific pathogen-free facility at Institute of Pathology, Ludwig-Maximilians-University Munich, with chow (standard formulation) and water supply ad libitum. All animal protocols were approved by the local authorities (Regierung von Oberbayern, AZ: 55.2-1-54-2532-201-2014; AZ:55.2-2532.Vet_02-21-111). Genotyping primers are listed in Table S3.
Single cell isolation from murine CACs
6–8 CACs from 3 female mice per genotype were isolated and dissociated according to the 10× Genomics protocol “Tumor Dissociation for Single Cell RNA Sequencing” (CG000147 Rev B). In brief, isolated CACs were lysed using the Mouse Tumor Dissociation kit in gentleMACS C tubes using the gentleMACS Octo Dissociator with heaters (Miltenyi Biotec). The lysis of red blood cells was performed using 1× Red Blood Cell Lysis Solution (Miltenyi Biotec). To increase the percentage of viable cells the protocol “Removal of Dead Cells for Single Cell RNA Sequencing” (10x Genomics, CG000093 Rev C) was employed according to manufacturer’s instructions. For this, the MACS Dead Cell Removal kit and MS Columns with the OctoMACS separator on the MACS multistand (Miltenyi Biotec) were used.
scRNA-Seq analysis
Single cells were loaded on the 10× Chromium system and labeled with the 3′-CellPlex kit set A (10× Genomics) according to the manufacturer’s recommendations. For each genotype 30,000 cells were loaded. Barcoded sequencing libraries were prepared with the Chromium Single Cell 3′ v.3.1 Gene Expression Kit (10× Genomics). Paired-end sequencing (2 × 100 bp) was performed on Illumina NovaSeq 6000 (Illumina) to obtain 90,000 reads/cell using the standard workflow.
scRNA-Seq data analysis
Cellranger v7.2.0 software (10× Genomics) was used to align reads to the refdata-gex-mm10-2020-A mouse reference transcriptome and generate a gene-cell unique molecular identifier count matrix for each cell. Cells with more than 200 expressed mRNAs and less than 25% mitochondrial RNA content were selected for further analysis. Moreover, cell doublets were excluded by applying the Scrublet package [25]. The gene expression matrices were normalized using the LogNormalize function of Seurat v5.01 [26]. Next, the Seurat FindVariableFeatures, FindIntegrationAnchors, and IntegrateData functions were used to obtain 2000 of the most variant mRNAs and perform integration with batch correction. Cell clustering was performed by using the Seurat RunPCA, RunUMAP, and Findclusters functions with resolutions from 0.5 to 1.5 for best cluster representation. Cell-type-specific mRNAs and differentially expressed mRNAs between genotypes were identified by the Seurat function FindMarkers. Gene set enrichment analyses were performed using clusterProfiler [27]. The significance of differences in the abundance of cell types/clusters in CACs derived from Mir34aΔMye vs. Mir34aF/F mice was calculated using a single-cell specific method based on the mixed-effect modeling of associations on single cells (MASC) [28]. Detailed results are provided in Table S4.
Analysis of intercellular ligand-receptor communications
Intercellular communication was analyzed using CellChat [29]. Clustered UMAPs generated by Seurat were imported to CellChat and converted to CellChat objects. Results are presented as Circo plots using the netVisual_aggregate CellChat function.
Cell differentiation trajectory analysis
Cell differentiation trajectories were estimated based on pseudotime calculated by three different algorithms: Palantir [30], Monocle3 [31], and SlingShot [32]. The naive state of lineages was used as the root node/early cell. The trajectories are shown on the UMAPs calculated with Seurat as described above. To analyze RNA velocity, velocyto [33] was first used to separate unspliced and spliced mRNAs in each cell. Next, scVelo [34] was used to calculate RNA velocity vectors. To determine cell lineages, initial and terminal macrostates were determined using CellRank [35]. For the visualization of gene expression trends for each lineage the expression data was imputed with MAGIC (Markov Affinity-based Graph Imputation of Cells).
Bioinformatics analysis of public datasets
Expression and clinical data of the TCGA colon adenocarcinoma (COAD) and rectal adenocarcinoma (READ) cohorts was obtained from the MD Anderson standardized data browser (http://bioinformatics.mdanderson.org/TCGA/databrowser/). The RNA-Seq by Expectation-Maximization (RSEM) normalized expression values from the Illumina RNASeqV2 (genes) datasets were used. Expression and clinical data of other CRC patient datasets was downloaded from NCBI GEO (www.ncbi.nlm.nih.gov/geo). The statistics for survival analysis was calculated by log-rank test. For binary classification of cases (high/low expression), the Survminer R-package (https://CRAN.R-project.org/package=survminer) was used to determine optimal cut-off values. The CMS classification of public datasets was obtained from Guiney et al. [36].
Statistical analysis
GraphPad Prism 10.1.0 was used to determine significant differences between groups via two-tailed unpaired Students t-test. Values are represented as mean ± SEM. P-values < 0.05 were regarded as statistically significant (p < 0.05*; p < 0.01**; p < 0.001*** and p < 0.0001****).
Results
We had previously reported that germline deletion of Mir34a enhances tumor initiation, progression and invasion in a murine model of colitis-associated colon cancer [6]. However, it remained unknown whether the effects of the Mir34a inactivation were tumor cell autonomous, or whether Mir34a-deficient cells within the tumor stroma were also involved. In a mouse model of intestinal tumorigenesis, it was observed that p53 is a critical regulator of tumorigenesis during inflammation in macrophages [37]. Furthermore, it was shown that p53 is activated during inflammation [38] and may have non-tumor-cell-autonomous tumor suppressive functions [39]. Since Mir34a is directly induced by p53, it may mediate these effects of p53. To determine, which cell type mediates the effects of Mir34a inactivation on CAC formation, we generated Villin-Cre/Mir34aF/F and LysM-Cre/Mir34aF/F mice, to obtain mice with intestinal epithelial cell-specific or myeloid lineage-specific deletion of Mir34a, designated as Mir34aΔIEC and Mir34aΔMye mice, respectively. When CACs were induced as described in Fig. 1A, Mir34aΔIEC and Mir34aΔMye mice formed more and larger CACs than Mir34aF/F mice (Fig. 1B–D). The tumor load of Mir34aΔIEC and Mir34aΔMye mice was less than in Mir34a–/– mice (Fig. 1C). Mir34aΔIEC, Mir34aΔMye and Mir34a–/– mice displayed invasive CACs, whereas CACs in Mir34aF/F mice were non-invasive (Fig. 1E). However, Mir34aΔIEC and Mir34aΔMye mice displayed less invasive CRCs than Mir34a–/– mice (Fig. 1E, F). Surprisingly, tumor invasiveness was increased to a larger extent by inactivation of Mir34a in myeloid cells than by its deletion in intestinal epithelial cells. Taken together, these results suggested a tumor suppressive role of Mir34a in both, the intestinal epithelial cells and in TAMs. As the CAC-promoting effect of myeloid-specific inactivation of Mir34a was unexpected, we decided to characterize it in more detail.

A Treatment scheme of the CAC model induced by AOM/DSS. B Histology of AOM/DSS challenged mice with indicated genotypes, scale bars are 1 mm. C CAC incidence in Mir34aF/F, Mir34aΔIEC, Mir34aΔMye and Mir34a–/– mice (n ≥ 7 mice per group). D CAC size of mice with indicated genotypes, CAC areas measured by computerized image analysis (n ≥ 7 mice per group). E Percentage of invasive CACs of indicated genotypes. F Number of mice with invasive CACs/total number of mice of indicated genotype. (C–F) Students t-test was used to determine significance. Values represent the mean ± SEM with p < 0.05*; p < 0.01**; p < 0.001*** and p < 0.0001****.
scRNA-Seq analysis of CACs in Mir34a ΔMye mice
To comprehensively characterize cell types and differential gene expression in CACs and their TME after myeloid-specific Mir34a-inactivation, we performed single-cell RNA sequencing (scRNA-Seq) analysis of CACs derived from Mir34aΔMye mice and Mir34aF/F mice. We isolated 6–8 tumors from three mice for each genotype and combined these for scRNA-Seq analyses. After quality control, 38,883 single-cell transcriptomes were obtained (18,571 from Mir34aΔMye CACs and 20,312 from Mir34aF/F CACs). Cell-types were clustered based on the scRNA-Seq results using a uniform manifold approximation and projection (UMAP) plot (Fig. 2A). By utilizing cell-type-specific marker-based annotations, eleven major cell-type subtypes were identified (Fig. 2A and Supplementary Fig. S1A, B). CACs from Mir34aΔMye mice showed an increase of macrophages and neutrophils, and a decrease of tumor epithelial cells when compared to Mir34aF/F CACs (Fig. 2B). As determined by Cellchat, Csf1/Csf1r signaling, which plays an important role in macrophage recruitment and polarization [40], was elevated in Mir34aΔMye CACs (Fig. 2C). This increase is presumably based on the elevated expression of the Csf1 ligand in neutrophils, mast cells, fibroblasts and basophils as well as on the elevated levels of Csf1r in macrophages, neutrophils, and dendritic cells in Mir34aΔMye CACs (Fig. 2D). Moreover, Mir34aΔMye macrophages displayed elevated expression of Socs3, which is a target of Stat3 signaling that is activated by Csf1r [40] (Fig. 2D). Furthermore, Cellchat analyses showed that Cxcr2 signaling, which plays an important role in neutrophil recruitment [41, 42] was elevated in Mir34aΔMye CACs (Fig. 2E), which may be due to the elevated expression of Cxcr2 receptor in neutrophils and increased expression of Cxcl1 and Cxcl3 in basophils (Fig. 2F). Moreover, Mir34aΔMye neutrophils show elevated expression of Socs3 (Fig. 2D), which can be induced by Cxcr2-mediated activation of Stat3 [43]. These results imply that the elevated Csf1r and Cxcr2 signaling observed in Mir34aΔMye CACs could be responsible for a stronger recruitment and a consequent increase of macrophages and neutrophils.

A Combined UMAP plot showing subtypes of all cells (38,883 cells) from CACs, derived from Mir34aΔMye and Mir34aF/F mice. B Proportions of major cell types in Mir34aΔMye and Mir34aF/F CACs. C CSF1 signaling networks between indicated cell types in Mir34aΔMye and Mir34aF/F CACs. The edge width represents the communication probability. D Expression of CSF1 signaling components in indicated cell types in Mir34aΔMye and Mir34aF/F CACs. E CXCR2 signaling networks between indicated cell types in Mir34aΔMye and Mir34aF/F CACs. The edge width represents the communication probability. F Expression of CXCR2 signaling components in indicated cell types in Mir34aΔMye and Mir34aF/F CACs. Significance was determined as described in the methods section scRNA-Seq data analysis (p < 0.05*; p < 0.01**; p < 0.001*** and p < 0.0001****). The mRNAs not marked with asterices did not reach significance in their differential expression.
scRNA-Seq analysis of CAC cells
Next, we divided CAC cells (7848 cells from Mir34aΔMye CACs and 8823 cells from Mir34aF/F CACs) into six subtypes (Krt20+, Peak1+, Lgr5+, Inflammatory, Ki67+, and Ribosomal) (Fig. 3A, B). These included a subtype characterized by high expression of the colonic stem cell marker Lgr5 and elevated Wnt signaling, a subtype with high levels of the pan-differentiation marker Krt20, and a subtype of proliferating cells, which were positive for Ki67 (Fig. 3B). These 3 subtypes were more abundant in Mir34aF/F CACs (Fig. 3C). In contrast, a subtype characterized by inflammatory chemokine and cytokine signaling was increased in Mir34aΔMye CACs (Fig. 3C, D). These cells expressed pro-inflammatory genes, such as Cd74, S100a8, and S100a9, which have been shown to contribute to the progression of various cancer types [44, 45] (Fig. S2). Beside epithelial markers, such as Epcam, these cells also expressed macrophage marker genes, including Csf1r and Adgre1 (F4/80), as well as the macrophage fusion receptor Tyrobp (DAP12) (Fig. 3E). We also analyzed the expression of Tyrobp mRNA in the publicly available RNA-Seq data (SRP097890) obtained with the iKAP mouse model of metastatic CRC. The iKAP mouse model harbors conditional alleles of the most common mutations found in human CRC allowing to recapitulate CRC progression, i.e., K-RAS, APC and p53 [46]. The expression of Tyrobp was lowest in normal colon (NC), increased in non- and low-invasive tumors (TA and T1) and reached its highest level in tumors that invade through the serosa and the outer intestinal wall (T4) and was still elevated in the corresponding liver metastases (Fig. S3A). In human CRC patients, high expression of Tyrobp was significantly associated with poor survival in the majority of ten analyzed CRC patient cohorts (Fig. S3B). Since Mir34aΔMye CACs showed increased invasion (Fig. 1E), we analyzed the expression of EMT-associated genes, which play an important role in tumor invasion [47]. The expression of epithelial cell state-associated genes Cdh1, Rbm47, Krt8, Cldn7, and Cldn4 was decreased in Mir34aΔMye CAC cells, whereas the expression of the mesenchymal cell state-associated gene Zeb2 was increased (Fig. 3F). However, the expression of the mesenchymal cell state associated genes Vim, Snai1, Snai2, and Zeb1 did not differ (Fig. 3F), suggesting that CAC cells in Mir34aΔMye tumors underwent a partial EMT. Epithelial cell state-associated genes including Cdh1 were pre-dominantly expressed in the more differentiated, Krt20+ CAC cells and decreased in the inflammatory CAC cells (Fig. 3G). In contrary, the mesenchymal cell state associated gene Zeb2 was mainly expressed in the inflammatory CAC cells (Fig. 3G). In the iKAP mouse model the expression of Zeb2 increased from normal colon (NC) towards non- and low-invasive tumors (TA and T1) and reached its highest level in tumors that invade through the serosa and the outer intestinal wall (T4) (Fig. S3C). In human CRC patients, high expression of Zeb2 was significantly associated with poor survival in the majority of ten analyzed CRC patient cohorts (Fig. S3D). Taken together, myeloid Mir34a-deficient CACs show a prominent increase of cancer cells with enhanced inflammatory signaling, which display a partially mesenchymal signature and may therefore represent cancer cells with enhanced migratory and invasive capacities.

A UMAP plot showing subtypes of tumor epithelial cells from CACs, derived from Mir34aΔMye and Mir34aF/F mice. B Heatmap of top ten differentially expressed genes in each tumor epithelial cell subtype. C Proportions of tumor epithelial cell subtypes in Mir34aΔMye and Mir34aF/F CACs. D Enrichment of indicated MsigDB Hallmark gene sets in differentially expressed genes for each tumor epithelial cell subtype. E UMAP plots of tumor epithelial cells colored by the expression of indicated mRNAs. F Expression of indicated EMT-related mRNAs in Mir34aΔMye and Mir34aF/F tumor epithelial cells. G UMAP plots of tumor epithelial cells colored by the expression of indicated EMT-related mRNAs. Significance was determined as described in the methods section scRNA-Seq data analysis (p < 0.001***). The mRNAs not marked with asterices did not reach significance in their differential expression.
scRNA-Seq analysis of neutrophils in the TME
Sub-clustering of neutrophils (1128 cells from Mir34aΔMye CACs and 1115 cells from Mir34aF/F CACs) revealed nine subtypes (Fig. 4A, B). Differentiation trajectory and RNA velocity analyses predicted three lineages of neutrophil differentiation, which initiate at subtype Neutro1, continue via subtypes Neutro2 and Neutro3, and terminate at subtypes TAN1, TAN2, and TAN3 (Fig. 4C–F). Recently, a “neutrotime” transcriptional signature has been characterized, which includes mRNAs encoding factors relevant for differentiation of pre-neutrophils residing in the bone marrow into mature neutrophils in blood and spleen [48]. Our results show that during neutrophil differentiation in Mir34aF/F and Mir34aΔMye CACs the expression of early neutrotime genes decreases, whereas the expression of late neutrotime genes initially increases and then also decreases (Fig. 4G). These results suggest that the differentiation of neutrophils from subtype Neutro1 to subtypes Neutro2 and Neutro3 recapitulates the neutrotime lineage in healthy neutrophil populations, whereas other subtypes might represent tumor associated neutrophils (TAN). The abundance of neutrophil subtypes between Mir34aF/F and Mir34aΔMye mice was similar except for subtype TAN5, which was present primarily in Mir34aΔMye mice (Fig. 4H). These neutrophils showed an enrichment of IL6/STAT3 and reactive oxygen species (ROS) signaling (Fig. 4I). Finally, we determined differential mRNA expression between Mir34aF/F and Mir34aΔMye neutrophils (Fig. 4J and extended data 1) and showed that 40 (14 expressed in at least 50% of the cells) published miR-34a targets, such as Mmp9, Notch2, Nampt, Cd44, Foxp1 and Csf1r, were among the mRNAs upregulated in Mir34aΔMye neutrophils (Fig. 4K). Moreover, 59 significantly upregulated genes in Mir34aΔMye neutrophils represent potential, not yet published miR-34a targets, with 26 of them expressed in at least 50% of the cells (extended data 1). Among these miR-34a targets the increased levels of Matrix metalloproteinase 9 (Mmp9) might play an important role in the enhanced progression of Mir34aΔMye CACs, because it has been shown that neutrophil-secreted Mmp9 can break down collagen and remodel the ECM to promote tumor cell invasion and metastasis [49]. Mmp9 was primarily expressed in Neutro1-3 neutrophil subtypes (Fig. S4). Taken together, Mir34aΔMye neutrophils show an increased expression of the miR-34a target Mmp9, which could contribute to the promotion of tumor progression and invasion. Furthermore, the TAN5 subtype, which showed enrichment in IL6/STAT3 and ROS signaling, was almost exclusively present in Mir34aΔMye mice.

A UMAP plot showing subtypes of neutrophils in CACs derived from Mir34aΔMye and Mir34aF/F mice. B Heatmap of the top ten differentially expressed genes in each neutrophil subtype. C UMAP plot showing the Palantir pseudotime of neutrophils. The color indicates pseudotime directionality from the earliest (blue) to the latest (red). D UMAP plot showing the Monocle analysis of neutrophils. The color indicates pseudotime directionality from the earliest (blue) to the latest (yellow). E UMAP plot showing the Slingshot analysis of neutrophils. Terminal cell lineages are indicated. F RNA velocity analysis of neutrophil subtypes with vector fields representing RNA velocity projected onto the UMAP plot. G Expression trends of indicated Neutrotime mRNAs for the indicated neutrophil lineages. H Proportions of neutrophil subtypes in Mir34aΔMye and Mir34aF/F CACs. I Enrichment of indicated MsigDB Hallmark gene sets in differentially expressed genes for each neutrophil subtype. J Volcano plot of genes differentially expressed between Mir34aΔMye and Mir34aF/F neutrophils. Predicted and published miR-34a targets are indicated in blue and red, respectively. K Expression of indicated miR-34a target mRNAs in Mir34aΔMye and Mir34aF/F neutrophils. Significance was determined as described in the methods section scRNA-Seq data analysis (p < 0.001***). The mRNAs not marked with asterices did not reach significance in their differential expression.
scRNA-Seq analysis of macrophages in the TME of CACs
Sub-clustering analysis of macrophages and monocytes (2508 cells from Mir34aΔMye CACs and 2090 cells from Mir34aF/F CACs) identified one monocyte (Ly6c2+) and six macrophage subtypes (Mrc1+, Cdk8+, Spp1+, Mitochondrial+, Nr4a+, and Nos2+) (Fig. 5A, B). Differentiation trajectory analyses using three algorithms (Palantir, Monocle3, and Slingshot) revealed three major lineages of macrophage differentiation: from monocytes towards Mrc1+, Nos2+, and Cdk8+ macrophages (Fig. 5C–E). Similar results were obtained by RNA velocity analysis, which also showed a directional flow of vector fields from monocytes towards the Mrc1+, Nos2+, and Cdk8+ macrophage lineages (Fig. 5F). The Spp1+, Mitochondrial+, and Nr4a+ subtypes presumably represent intermediate stages of macrophages. High expression of Mrc1 is typical for M2-polarized macrophages, which exhibit pro-tumorigenic and anti-inflammatory features [50], whereas high levels of Nos2 characterizes M1-polarized macrophages with anti-tumorigenic and pro-inflammatory characteristics [51]. The Mrc1+ macrophages were more abundant in Mir34aΔMye CACs (Fig. 5G), while Nos2+ macrophages were more abundant in Mir34aF/F CACs (Fig. 5G). These results suggest that the loss of Mir34a shifts the polarization of macrophages towards the M2-like state. Mir34aΔMye CACs also showed a strong increase of the Cdk8+ macrophage subtype (Fig. 5G). Cdk8 was shown to enhance β-catenin and MYC activity [52]. Consistently, these macrophages also expressed high levels of MYC target genes (Fig. 5H). Furthermore, Cdk8+ macrophages expressed elevated levels of mRNAs encoding enzymes involved in oxidative phosphorylation, which has been associated with M2 macrophage polarization [53]. Next, we determined, which mRNAs display differential expression in Mir34aF/F vs. Mir34aΔMye macrophages (Fig. 5I and extended data 1). Among the mRNAs significantly upregulated in Mir34aΔMye macrophages were 21 published miR-34a targets, five of them expressed in at least 50% of the cells: Csf1r, Axl, Foxp1, Ccr1, Nampt, and Tgfbr2 (Fig. 5J). The expression of these miR-34a targets was highest in Mrc1+ M2-like macrophages (Fig. 5K) and upregulated during differentiation into this macrophage lineage as determined by pseudotime analysis (Fig. S5A). Recently, Zhang and colleagues identified two TAM subtypes expressing Maf and Mgl2 which were sensitive to anti-Csf1r treatment [54]. Here, Maf and Mgl2 were predominantly expressed in the Mrc1+ macrophage subtype, which also expresses Csf1r but not Nos2 (Fig. S5B). According to our recent miR-34a target meta-analysis [55], 32 of the significantly upregulated mRNAs in Mir34aΔMye macrophages represent potential, not yet published miR-34a targets, with seven of them expressed in at least 50% of the cells: Btg2, Mbln1, Ell2, Prkcb, Cap1, Nf1, and Synj1 (extended data 1). The increased levels of Csf1r expression might play an important role in the enhanced progression of Mir34aΔMye CACs, because Csf1r plays an important role in macrophage recruitment and polarization [40]. Moreover, several other published and potential miR-34a targets that were upregulated in Mir34aΔMye macrophages, such as Axl, have been associated with M2-polarization and pro-tumorigenic functions in TAMs and might therefore also be involved in the progression of Mir34aΔMye CACs [56,57,58]. In addition, Pdgfc, which promotes angiogenesis (reviewed in ref. [59]) and the immunosuppressive cytokine Interleukin 10 (Il10), that contributes to tumor immune evasion (reviewed in ref. [60]), were upregulated in Mir34aΔMye macrophages, particularly in the Mrc1+ subtype (Fig. S6A, S6B). Finally, we analyzed which factors, that are secreted by macrophages, might promote tumor cell invasiveness. CellChat analyzes showed that the semaphorin signaling, which has been previously implicated in tumor progression (reviewed in ref. [61]), is elevated in Mir34aΔMye CACs (Fig. S6C). The Sema4d ligand was expressed in macrophages, neutrophils, mast cells, T-cells, and B-cells (Fig. S6D, S6E) and upregulated in Mir34aΔMye CACs (Fig. S6F). Notably, Sema4d is a potential miR-34a target according to our recent miR-34a target meta-analysis [55]. The Plxnb2, which is a receptor for Sema4d, showed the highest expression in tumor epithelial cells (Fig. S6D), suggesting that the elevated levels of Sema4d in Mir34aΔMye CACs might have an impact on tumor cells towards increased invasiveness. Taken together, these results show that myeloid Mir34a-deficiency leads to an increase of macrophages harboring pro-tumorigenic capacities (Mrc1+), presumably by upregulation of Mir34a target mRNAs, such as Csf1r and Axl.

A UMAP plot showing subtypes of macrophages/monocytes from CACs, derived from Mir34aΔMye and Mir34aF/F mice. B Heat-map of top ten differentially expressed genes in each macrophage/monocyte subtype. C UMAP plot showing the Palantir pseudotime of monocytes and macrophages. The color indicates pseudotime directionality from the earliest (blue) to the latest (red). D UMAP plot showing the Monocle analysis of monocytes and macrophages. The color indicates pseudotime directionality from the earliest (blue) to the latest (yellow). E UMAP plot showing the Slingshot analysis of monocytes and macrophages. Terminal cell lineages are indicated. F RNA velocity analysis of monocyte/macrophage subtypes with vector fields representing RNA velocity projected onto the UMAP plot. G Proportions of monocyte/macrophage subtypes in Mir34aΔMye and Mir34aF/F CACs. H Enrichment of indicated MsigDB Hallmark gene sets in differentially expressed genes for each monocyte/macrophage cluster. I Volcano plot of differentially expressed genes between Mir34aΔMye and Mir34aF/F monocyte/macrophage. Predicted and published miR-34a targets are indicated in blue and red, respectively. J Expression of indicated miR-34a target mRNAs in Mir34aΔMye and Mir34aF/F monocytes/macrophages. K UMAP plots of monocytes/macrophages colored by the expression of indicated miR-34a target mRNAs. Significance was determined as described in methods section scRNA-Seq data analysis with p < 0.05*; p < 0.01** and p < 0.001***.
Mir34a-deficiency promotes migration and M2 polarization in BMDMs
On the functional level, deletion of Mir34a in BMDMs resulted in increased migration when compared to Mir34a-proficient BMDMs (Figs. 6A and S7A). When murine CT-26 CRC cells were co-cultured with Mir34a-deficient BMDMs their migration was increased when compared to co-culture with Mir34a-proficient BMDMs (Figs. 6B and S7B).

A Fold change of migrated BMDMs after 4 and 24 h (n = 3). Assay was performed in triplicate inserts. B Percentage of wound closure of CT-26 cells after co-culture with Mir34a-proficient or -deficient BMDMs for 24 h (n = 4). C pri-Mir34a expression in Mir34a-proficient BMDMs after treatment with 0.5 mM NO-donor (n = 3). D pri-miR34a expression after treatment of HCT116 p53 wildtype and p53 knockout cells (n = 3) with a 1 mM NO-donor for 24 h. Expression was normalized to GAPDH. E Mrc1; Arginase-1 and Nos2 expression in Mir34a-proficient and Mir34a-deficient BMDMs (n = 3). F Expression of Mrc1; (G) Arginase-1 and (H) Nos2 in Mir34a-proficient and Mir34a-deficient BMDMs after treatment with IL-4 or LPS + INFγ for 4 h (n = 3). I Immunohistochemical analysis of Arginase-1 (Arg-1) and Nos2 in CACs of Mir34aF/F, Mir34aΔIEC and Mir34aΔMYE mice (n = 3 mice per genotype). Scale bar represent 40 µm. (A–I) Students t-test was used to determine significance. Values represent the mean ± SEM with p < 0.05*; p < 0.01**; p < 0.001*** and p < 0.0001****.
Next, we hypothesized that Mir34a expression might be induced by NO, which is released by different immune cells during inflammation [62], as it has been reported that p53 is activated by NO in a CAC model [38]. Indeed, treatment of BMDMs with the NO–donor spermine nonoate induced the expression of pri-Mir34a (Fig. 6C). Furthermore, the induction of pri-miR-34a by NO is p53-dependent, since p53-proficient HCT116 cells, but not p53-deficient HCT116 CRC cells showed an induction of pri-miR-34a after addition of an NO-donor (Fig. 6D). These results provide a model as to how myeloid Mir34a expression is induced under physiological conditions.
Next, we aimed to confirm the increase in M2-like macrophages in Mir34a-deficient CACs detected by scRNA-Seq (see Fig. 5G). In Mir34a-deficient BMDMs a significant elevation of Arginase-1 and Mrc1 and a significant decrease of Nos2 mRNA levels was detected when compared to Mir34a-proficient BMDMs (Fig. 6E). Treatment of BMDMs with IL-4 led to significantly elevated levels of Arg-1 and Mrc1 in BMDMs compared to their untreated control (Fig. 6F–G). BMDMs treated with LPS + INFγ showed increased Nos2 expression compared to untreated control BMDMs (Fig. 6H). Unexpectedly, after IL4-treatment, the induction of Mrc1 was significantly less pronounced in Mir34a-deficient BMDMs when compared to Mir34a-proficient BMDMs, whereas the repression of Nos2 was more pronounced in Mir34a-deficient BMDMs (Fig. S7C). After LPS/IFNγ treatment Nos2 was more induced in Mir34-deficient BMDMs, whereas the minor induction of Mrc1 and Arg1 was not affected by Mir34a deletion (Fig. S7D). In addition, using immunohistochemical detection of protein markers, we observed, that the number of stromal cells displaying expression of the M2-marker Arginase-1 was significantly elevated in Mir34aΔMye CACs, whereas stromal cells positive for the M1 marker Nos2 were decreased when compared to control CACs (Fig. 6I). In the iKAP mouse model the expression of M2 markers Arg1 and Mrc1 increased from normal colon (NC) towards non- and low-invasive tumors (TA and T1) and reached its highest level in tumors that invade through the serosa and the outer intestinal wall (T4) and liver metastases (Fig. S8A, S8B), whereas the M1 marker Nos2 showed the opposite expression pattern (Fig. S8C). In human CRC patients, high expression of M2 makers ARG1 and MRC1 was significantly associated with poor survival, whereas high expression of the M1 marker NOS2 was significantly associated with good survival in the majority of ten analyzed CRC patient cohorts (Fig. S8D–F). Overall, these results imply that Mir34a may prevent CAC migration and invasion by inhibiting polarization of macrophages towards M2.
Reduction of tumor burden in Mir34a ΔMye CACs by concomitant deletion of Csf1r
The receptor tyrosine kinase Csf1r plays an important role in macrophage recruitment and has been validated as a miR-34a target in CRC cells relevant for the formation of murine intestinal adenomas and human CRCs [18, 19]. As shown above, our scRNA-Seq analysis detected increased expression of Csf1r and its ligand CSF1 in Mir34aΔMye vs. Mir34aF/F macrophages and neutrophils (Figs. 4K and 5J). In Mir34a-deficient BMDMs, we could observe an increased expression of Csf1r on mRNA and protein levels when compared to Mir34a-proficient BMDMs (Fig. 7A, B; uncropped Western blot membrane: see Supplementary Material). The number of cells positive for the macrophage marker F4/80 was significantly increased (Fig. S9A) in CACs of Mir34aΔMye mice when compared to Mir34aΔIEC and Mir34aF/F mice. Csf1r positive, stromal cells were also more abundant in the CAC stroma of Mir34aΔMye mice when compared to Mir34aΔIEC and Mir34aF/F mice (Fig. S9B), suggesting that the Mrc1+ macrophage subtype identified by scRNA-Seq might contribute to this cell population (Fig. 5K). Furthermore, an increase of Csf1r mRNA expression in CACs of Mir34aΔMye mice was observed (Fig. 7C). In addition, CSF1R expression was highest in the CMS4 and CRISB molecular subtypes of CRCs in cohorts of CRC patients (Fig. S9C). These subtypes are associated with EMT, metastasis and poor prognosis [36, 63]. Furthermore, scRNA-Seq analysis of human CRCs [54] (GSE81861) revealed that CSF1R is predominantly expressed in myeloid cells (Fig. S9D). In the iKAP mouse model the expression of Csf1r was lowest in normal colon (NC), increased in non- and low-invasive tumors (TA and T1), and reached its highest level in tumors that invade through the serosa and the outer intestinal wall (T4) and was still elevated in the corresponding liver metastases (Fig. S9E). In addition, we analyzed the expression of Csf1r in publicly available RNA-Seq data from the KPN model of metastatic CRC [64], which combines mutant K-Ras (K), p53 (P) deletion and NOTCH1 (N) activation. KP and KPN mice develop lymph node and liver metastases, whereas AP and APN mice do not. The expression of Csf1r was elevated in KP and KPN mice when compared to AP and APN mice (Fig. S9F). Taken together, these results imply that the elevated expression of Csf1r caused by Mir34a-deficiency may play a major role in tumor progression via TME—tumor cell interactions in the mouse model analyzed here.

A Csf1r expression in Mir34a-proficient and Mir34a-deficient bone-marrow derived macrophages (BMDMs) (n = 3 mice per genotype). B Western blot analysis of Csf1r in Mir34a-proficient and Mir34a-deficient BMDMs. C Csf1r expression in CACs of Mir34aF/F, Mir34aΔIEC and Mir34aΔMYE mice (n = 3 mice per genotype). D Histology of CACs with indicated genotypes, scale bars are 1600 µm. E CAC incidence in Mir34aF/F, Csf1rΔMye, Mir34aΔMye, Mir34aΔMyeCsf1rΔMye/+ and Mir34aΔMyeCsf1rΔMye mice (n = 8 mice per group). F CAC size of mice with indicated genotypes, CAC areas measured by computerized image analysis (n = 8 mice per group). G Example of CAC invasion in Mir34aΔMye mice. Scale bars are 100 µm. H Percentage of invasive CACs of indicated genotypes. I Number of mice with invasive CACs/total number of mice of indicated genotype. J Immunohistochemical quantification of Ki-67, (K) Cleaved-Caspase 3, (L) Arginase-1 (Arg-1) and (M) Nos2 in CACs of indicated genotypes (n = 3 per genotype). (A, B, C, E, F, H, I, J, K, L, M) Students t-test was used to determine significance. Values represent the mean ± SEM with p < 0.05*; p < 0.01**; p < 0.001*** and p < 0.0001****.
In order to determine whether the observed upregulation of Csf1r in macrophages is required for the effects of Mir34a-deficiency we generated Mir34a-deficient mice with concomitant heterozygous and homozygous deletion of Csf1r in myeloid cells. Csf1rΔMye mice were used as additional controls. Interestingly, concomitant Csf1r deletion—heterozygous as well as homozygous—reduced the number of CACs when compared to Mir34aΔMye mice (Fig. 7D, E). Csf1r deletion alone had no effect when compared to Mir34afl/fl mice. Furthermore, the increased size of CACs with a myeloid Mir34a deletion was reduced by concomitant Csf1r inactivation (Fig. 7F). In Mir34aΔMye mice we observed in almost every mouse at least one invasive CAC. Therefore, the percentage of invasive CACs as well as mice with invasive CACs was significantly increased in Mir34aΔMye mice when compared to Mir34afl/fl or Csf1rΔMye mice, and was significantly decreased by concomitant deletion of Csf1r when compared to Mir34aΔMye mice (Figs. 7G–I and S10A).
Next, we determined whether inactivation of Csf1r in this context also influences proliferation and apoptosis. Concomitant homozygous deletion of Csf1r and Mir34a indeed reduced the number of Ki-67-positive cells significantly, whereas the number of Cleaved-Caspase 3 positive cells increased (Figs. 7J, K and S10B) indicating that deletion of Csf1r in Mir34a-deficient myeloid cells reduces proliferation and increases apoptosis. Concomitant myeloid-specific deletion of Csf1r with Mir34a resulted in a reduction of cells expressing the M2 marker Arginase-1 in CACs when compared with CACs in Mir34aΔMye mice. Nos2, a M1 marker, showed the opposite pattern of expression. Concomitant homozygous deletion of Csf1r and Mir34a increased the number of Nos2-positive cells significantly to the level present in CACs of Mir34afl/fl and Csf1rΔMye mice (Figs. 7L, M and S10B). Taken together, these results demonstrate that the increase in Csf1r expression is a required mediator of the effects of Mir34a inactivation on myeloid cells. The results imply that Mir34a inhibits CAC progression and M2-like polarization, at least in part, by decreasing Csf1r expression levels.
Inhibition of Csf1r in Mir34a ΔMye mice reduces tumor burden
Since CAC-associated macrophages with Mir34a-deficiency displayed an elevated level of Csf1r expression (Fig. 5J, K), they may be dependent on Csf1r signaling and therefore especially sensitive to its inhibition. Therefore, we asked whether therapeutic inhibition of Csf1r has a preferential effect on Mir34a-deficient CACs. To induce CACs, mice were subjected to AOM/DSS as described above and received the small molecule Csf1r inhibitor GW2580 from day 60 onwards. In Mir34a-deficient mice treatment with GW2580 significantly decreased the number of CACs and invasiveness (Figs. 8A, B, D and E; S11A). In wild-type mice treatment with GW2580 significantly reduced CAC size although the number of CACs showed a minor increase (Fig. 8B, C). GW2580 had no effect on invasion of CACs in wild-type mice (Fig. 8D, E). Treatment of Mir34aΔMye mice with the GW2580 resulted in decreased proliferation and increased apoptosis in CACs, whereas it had no effect in Mir34afl/fl mice (Fig. 8F and G; S11B). GW2580 significantly decreased the number of Arg-1-positive cells in CACs in wild-type mice, but in CACs of Mir34aΔMye mice no significant effect was evident (Fig. 8H). For both genotypes the number of Nos2-positive cells increased with GW2580 treatment by ~2%. However, this effect did not reach significance (Figs. 8I and S11B). Taken together, these results demonstrate that CACs with Mir34a-deficient myeloid cells and therefore increased Csf1r expression levels are sensitive to treatment with a Csf1r inhibitor. In addition, these results partially agree with the observations obtained by genetic inactivation of Csf1r in Mir34a-deficient myeloid cells and further confirm Csf1r-inhibition as an important mediator of non-tumor-cell autonomous suppression of CACs by miR-34a.

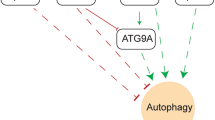
A Histology of CACs with indicated genotypes, scale bars are 1600 µm. B CAC incidence in Mir34aF/F, Mir34aF/F + GW2580, Mir34aΔMye and Mir34aΔMye + GW2580 mice (n = 7–8 mice per group). C CAC size of mice with indicated genotypes, CAC areas measured by computerized image analysis (n = 7-8 mice per group). D Percentage of invasive CACs of indicated genotypes. E Number of mice with invasive CACs/total number of mice of indicated genotype. F Immunohistochemical quantification of Ki-67, (G) Cleaved-Caspase 3, H Arginase-1 (Arg-1) and (I) Nos2 in CACs of indicated genotypes (n = 3 per genotype). J Model summarizing the results and main conclusions. (B, C, E–I) Students t-test was used to determine significance. Values represent the mean ± SEM with p < 0.05* and p < 0.01**.
Discussion
Here we show that deletion of Mir34a in myeloid cells promotes CAC formation and progression (see summarizing model in Fig. 8J). Therefore, Mir34a functions as a non-cell autonomous suppressor of CAC formation in tumor-associated myeloid cells. Notably, myeloid-specific inactivation of Mir34a increased CAC invasiveness to a larger extent than intestinal epithelial cell-specific deletion of Mir34a. Among other Mir34a target mRNAs, Csf1r was upregulated in Mir34a-deficient myeloid cells in CACs. In mice with inactivation of Mir34a in myeloid cells concomitant Csf1r deletion as well as therapeutic inhibition of Csf1r resulted in decreased proliferation, elevated apoptosis, less invasiveness of CACs and decreased macrophage polarization towards M2. Therefore, Csf1r is a required mediator of the effects of Mir34a inactivation in tumor-associated myeloid cells.
We found that deletion of Mir34a in myeloid cells increased the number of macrophages and neutrophils. In particular, the number of Mrc1+ and CDK8+ macrophages was highly increased in Mir34aΔMye CACs. Since these subtypes of macrophages are known to promote cancer [65, 66], their increased abundance may also promote progression of CACs. In a study of lung cancer, it was shown that under hypoxic stress tumor cells suppress exosomal miR-101 [65]. This suppression of miR-101 enhanced CDK8 in macrophages which promoted the expression of pro-inflammatory cytokines IL1A and IL6 in macrophages. Impairment of those cytokines suppressed tumor cell growth in vitro and in vivo. Interestingly, in a study by Zhang and colleagues, two TAM subtypes expressing Maf and Mgl2 were identified which are sensitive to anti-Csf1r treatment [54]. These genes were predominantly expressed in the Mrc1+ subtype of TAMs, which also expressed Csf1r and therefore might also be especially sensitive to Csf1r treatment. Additionally, Zhang et al. compared the scRNA-Seq data from murine and human CRCs and concluded that they are similar [54] suggesting that the new therapeutics or druggable pathways identified here in murine CRCs may be transferred to human.
We showed that Csf1r deletion or inhibition reprogrammed the macrophages from M2-like towards M1-like, especially in Mir34aΔMye mice. Lv et al. showed that the treatment of human M2-like BMDMs with the CSF1R inhibitor PBX17 also reprograms them to M1-like macrophages [67]. They co-cultured human CRC organoids with M2- macrophages and CD8+ T-cells and showed that CSF1R inhibition reduces the viability of CRC organoids. However, CSF1R inhibition had no effect on CRC organoid viability without co-culture. These findings indicate that CSF1R inhibition promotes an anti-tumor immune response of CD8+ T cells by facilitating the reprogramming of M2-like macrophages towards M1-like macrophages. Consistently, CSF1R inhibition enhanced the therapeutic activity of a PD-1-specific mAb in subcutaneous xenograft mouse models of CRC by improving the infiltration and activation of CD8+ T cells in tumors [67]. Since PD-L1 is a miR-34a target [68, 69], which is expressed in macrophages, multimodal therapy studies with Csf1r and PD-1 inhibitors in Mir34aΔMye CACs should be performed in the future.
It has been reported that p53-deficient mice display an increased risk of developing severe chronic inflammation ([70]; reviewed in ref. [71]) and CACs when exposed to DSS ([72], reviewed in ref. [73]) and that p53 inhibits M2-like polarization [74]. In the APCmin mouse model of FAP/familial adenomatous polyposis deletion of p53 in myeloid cells leads to a higher level of inflammatory cytokines, increased M2-like polarization of TAMs and tumor progression, whereas p53 activation had the opposite effects [37]. Therefore, p53 functions as a non-cell autonomous tumor suppressor in macrophages (also reviewed in refs. [38, 73]). Here, inactivation of the p53 target gene miR-34a in myeloid cells associated with CACs had similar effects. Therefore, our results imply that Mir34a presumably represents an important mediator of the effects of p53 on macrophages and of the suppression of CAC formation by p53.
During inflammation NO is released from macrophages. Notably, NO was shown to activate p53 [38]. Therefore, Mir34a is presumably activated by p53 during inflammation and may direct macrophage polarization towards an M1-like state. In contrast, loss of p53 or, as shown here, Mir34a inactivation in macrophages enhances their polarization towards an M2-like state. We recently showed that miR-34a expression is induced by ROS donors, such as curcumin and H202, via the KEAP/NRF2 pathway in a p53-independent manner [75]. Since inflammation is known to generate ROS (reviewed in ref. [76]), it is conceivable that miR-34a is induced by ROS under such circumstances. Therefore, miR-34a may also function to suppress M2-like-polarization in the absence of p53 activation.
Here, the expression of Csf1r, which is a conserved miR-34a target mRNA [18, 19], was elevated in macrophages, neutrophils and dendritic cells in Mir34aΔMye CACs. Consistently, the presence of CSF1R-positive macrophages is associated with poor survival [11]. We could show that Csf1r inhibition or deletion in myeloid cells decreases the growth and progression of CACs. Therefore, Csf1r-positive macrophages presumably represent important therapeutic targets. Furthermore, CACs harboring Mir34a-deficient myeloid cells with increased Csf1r expression were more sensitive to treatment with the Csf1r-inhibitor GW2580 than CACs of wild-type mice. This presumably resulted from an increased dependency on Csf1r signaling. Inhibitors of CSF1R or its ligand CSF1 interfere with tumor-promoting TAMs or other myeloid cells in the TME and are currently being tested in clinical trials for treatment of several types of malignancies, such as CRC, melanoma or ovarian cancer (reviewed in refs. [11, 77]). Here we did not study the effect of myeloid Csf1r deletion or Csf1r inhibition on overall survival of mice, because we focused on the differences in CAC characteristics at a specific time point. The effect on overall survival should be determined in the future.
Notably, our scRNA-Seq analysis revealed a CAC cell subtype, designated as inflammatory cancer cells, which was almost exclusively present in Mir34aΔMye CACs. These cells expressed pro-inflammatory genes, such as Cd74, which contributes to inflammation-driven epithelial cell regeneration in IBD [78]. Furthermore, CD74 was shown to promote a pro-inflammatory tumor microenvironment in pancreatic cancer by inducing the secretion of S100A8/A9 via activation of TRAF6-NF-κB [44]. S100a8 and S100a9 were also highly expressed in the inflammatory cluster of Mir34a-deficient CACs. These proteins contribute to the progression of inflammatory diseases and cancer [45]. Interestingly, the inhibition of S100A8/A9 with chemical inhibitors decreased tumor burden in the AOM/DSS-induced mouse model of CAC [79]. Therefore, increased levels of Cd74 and S100a8/9 in the inflammatory CAC cell subtype may drive migration and invasion in myeloid Mir34a-deficient CACs.
Furthermore, the inflammatory cancer cell subtype identified here expressed high levels of the EMT transcription factor Zeb2, particularly in Mir34aΔMye CACs. Interestingly, it has been shown that mice with intestinal epithelial cell-specific, ectopic expression of Zeb2 develop invasive CRCs, which are driven by myeloid cell-induced inflammation [80]. Thus, the increased expression of Zeb2 may also play an important role in the enhanced invasion of Mir34aΔMye CACs.
Among the M2-like macrophage-derived factors with a known role in tumor progression, Pdgfc and Il10 were upregulated in Mir34a-deficient macrophages, particularly in the Mrc1+ subtype. Pdgfc promotes angiogenesis (reviewed in ref. [59]) and Il10 contributes to tumor immune evasion (reviewed in ref. [59]), suggesting that these factors might contribute to the enhanced progression of Mir34aΔMye CACs. Our results also demonstrate that the semaphorin signaling is elevated in Mir34aΔMye CACs. The Sema4d ligand, which represents a potential miR-34a target was upregulated in Mir34a-deficient macrophages and neutrophils. It has been shown that TAM-derived Sema4d promotes angiogenesis, invasion, and metastasis in breast and gastric cancer [81, 82]. Furthermore, elevated expression of Sema4D in macrophages was associated with poor prognosis in ovarian cancer and facilitated the differentiation of macrophages toward the M2-like lineage [83]. Therefore, the elevated semaphorin signaling might also contribute to the enhanced progression of Mir34aΔMye CACs. However, to achieve a better understanding of the crosstalk between myeloid Mir34a-deficient stromal cells and CAC cells and to identify myeloid cell-secreted factors that promote tumor progression, further investigations are needed.
Matrix metalloproteinase 9 (Mmp9) showed elevated expression levels in Mir34aΔMye neutrophils and represents a known miR-34 target [84]. Neutrophils release Mmp9, which degrades collagen and thereby remodels the ECM to promote tumor cell invasion and metastasis [49]. Moreover, Mmp9 promotes VEGF activation and tumor angiogenesis [85]. Interestingly, neutrophils are the only cells that can release Mmp9 from its endogenous inhibitors, the tissue inhibitors of metallo-proteinases [86]. Thus, Mmp9 in neutrophils may represent a potential therapeutic target in cancer.
Taken together, our study provides evidence that myeloid Mir34a prevents CAC progression by inhibiting M2-like polarization of TAM which is, at least in part, mediated by the downregulation of Csf1r signaling. Furthermore, we identified multiple, additional hallmarks of CACs harboring Mir34a-deficient myeloid cells, such as increased expression of Mmp9 in neutrophils and elevated expression of Zeb2 in an inflammatory subtype of CAC tumor cells. In Mir34a-deficient macrophages the miR-34a target mRNA encoding the Axl receptor-tyrosine-kinase, an already approved drug target [87], was upregulated. Also, engulfment and cell motility protein 1 (ELMO1) displayed increased expression in Mir34a-deficient macrophages. As shown previously, elevated ELMO1 expression in stromal cells may promote CRC progression and is associated with poor survival [88]. Furthermore, the predicted Mir34a target protein kinase C beta type (Prkcb) was elevated in macrophages and neutrophils of Mir34aΔMye CACs. Interestingly, Prkcb was reported to have tumor-promoting properties in breast cancer [89]. This may also apply to CACs. Targeting these and additional, deregulated factors in myeloid cells within the TME identified in this study may improve therapy of CAC in the future.
Data availability
Single cell RNA-Seq data from this study has been deposited at NCBI GEO GSE253907. Codes were implemented in R (version 4.1.2) and python (version 3.10.0) and are deposited in https://github.com/MatjazRokavec/miR34a_MYE_KO_scRNA-seq. The RNA-Seq data from the iKAP mouse model [46] is available at https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP097890. The RNA-Seq data from the KPN mouse model [64] is available at https://www.ebi.ac.uk/ena/browser/view/PRJEB38364.
References
Zhang M, Li X, Zhang Q, Yang J, Liu G. Roles of macrophages on ulcerative colitis and colitis-associated colorectal cancer. Front Immunol. 2023;14:1103617.
Porter RJ, Arends MJ, Churchhouse AMD, Din S. Inflammatory bowel disease-associated colorectal cancer: translational risks from mechanisms to medicines. J Crohns Colitis. 2021;15:2131–41.
Watanabe T, Konishi T, Kishimoto J, Kotake K, Muto T, Sugihara K. Ulcerative colitis-associated colorectal cancer shows a poorer survival than sporadic colorectal cancer: a nationwide Japanese study. Inflamm Bowel Dis. 2011;17:802–8.
Leowardi C, Schneider ML, Hinz U, Harnoss JM, Tarantino I, Lasitschka F, et al. Prognosis of ulcerative colitis-associated colorectal carcinoma compared to sporadic colorectal carcinoma: a matched pair analysis. Ann Surg Oncol. 2016;23:870–6.
Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer. 2012;12:613–26.
Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2014;124:1853–67.
Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20:840.
Tsai MJ, Chang WA, Huang MS, Kuo PL. Tumor microenvironment: a new treatment target for cancer. ISRN Biochem. 2014;2014:351959.
Essandoh K, Li Y, Huo J, Fan GC. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock. 2016;46:122–31.
Jiang P, Liu R, Zheng Y, Liu X, Chang L, Xiong S, et al. MiR-34a inhibits lipopolysaccharide-induced inflammatory response through targeting Notch1 in murine macrophages. Exp Cell Res. 2012;318:1175–84.
Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5:53.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61.
Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. 2015;36:229–39.
Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. 2006;18:39–48.
Hume DA, MacDonald KP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119:1810–20.
Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–59.
Blagih J, Zani F, Chakravarty P, Hennequart M, Pilley S, Hobor S, et al. Cancer-specific loss of p53 leads to a modulation of myeloid and T cell responses. Cell Rep. 2020;30:481–96.e6.
Shi X, Kaller M, Rokavec M, Kirchner T, Horst D, Hermeking H. Characterization of a p53/miR-34a/CSF1R/STAT3 feedback loop in colorectal cancer. Cell Mol Gastroenterol Hepatol. 2020;10:391–418.
Liu F, Bouznad N, Kaller M, Shi X, König J, Jaeckel S, et al. Csf1r mediates enhancement of intestinal tumorigenesis caused by inactivation of Mir34a. Int J Biol Sci. 2022;18:5415–37.
Pedersen MB, Danielsen AV, SJ Hamilton-Dutoit, Bendix K, Norgaard P, Moller MB, et al. High intratumoral macrophage content is an adverse prognostic feature in anaplastic large cell lymphoma. Histopathology. 2014;65:490–500.
Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE. 2012;7:e50946.
Cheng CY, Hwang CI, Corney DC, Flesken-Nikitin A, Jiang L, Oner GM, et al. miR-34 cooperates with p53 in suppression of prostate cancer by joint regulation of stem cell compartment. Cell Rep. 2014;6:1000–7.
Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–77.
Li J, Chen K, Zhu L, Pollard JW. Conditional deletion of the colony stimulating factor-1 receptor (c-fms proto-oncogene) in mice. Genes 2006;44:328–35.
Wolock SL, Lopez R, Klein AM. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019;8:281–91.e9.
Hao Y, Stuart T, Kowalski MH, Choudhary S, Hoffman P, Hartman A, et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat Biotechnol. 2024;42:293–304.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–7.
Fonseka CY, Rao DA, Teslovich NC, Korsunsky I, Hannes SK, Slowikowski K, et al. Mixed-effects association of single cells identifies an expanded effector CD4(+) T cell subset in rheumatoid arthritis. Sci Transl Med. 2018;10:eaaq0305.
Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 2021;12:1088.
Setty M, Kiseliovas V, Levine J, Gayoso A, Mazutis L, Pe’er D. Characterization of cell fate probabilities in single-cell data with Palantir. Nat Biotechnol. 2019;37:451–60.
Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502.
Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018;19:477.
La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. RNA velocity of single cells. Nature 2018;560:494–8.
Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol. 2020;38:1408–14.
Lange M, Bergen V, Klein M, Setty M, Reuter B, Bakhti M, et al. CellRank for directed single-cell fate mapping. Nat Methods. 2022;19:159–70.
Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–6.
He XY, Xiang C, Zhang CX, Xie YY, Chen L, Zhang GX, et al. p53 in the Myeloid Lineage Modulates an Inflammatory Microenvironment Limiting Initiation and Invasion of Intestinal Tumors. Cell Rep. 2015;13:888–97.
Hofseth LJ, Saito S, Hussain SP, Espey MG, Miranda KM, Araki Y, et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc Natl Acad Sci USA. 2003;100:143–8.
Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170:1062–78.
Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. 2014;6:a021857.
Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–75.
SenGupta S, Hein LE, Parent CA. The recruitment of neutrophils to the tumor microenvironment is regulated by multiple mediators. Front Immunol. 2021;12:734188.
Korbecki J, Kupnicka P, Chlubek M, Gorący J, Gutowska I, Baranowska-Bosiacka I. CXCR2 receptor: regulation of expression, signal transduction, and involvement in cancer. Int J Mol Sci. 2022;23:2168.
Hong WC, Lee DE, Kang HW, Kim MJ, Kim M, Kim JH, et al. CD74 promotes a pro-inflammatory tumor microenvironment by inducing S100A8 and S100A9 secretion in pancreatic cancer. Int J Mol Sci. 2023;24:12993.
Gebhardt C, Németh J, Angel P, Hess J. S100A8 and S100A9 in inflammation and cancer. Biochem Pharm. 2006;72:1622–31.
Boutin AT, Liao WT, Wang M, Hwang SS, Karpinets TV, Cheung H, et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev. 2017;31:370–82.
De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110.
Grieshaber-Bouyer R, Radtke FA, Cunin P, Stifano G, Levescot A, Vijaykumar B, et al. The neutrotime transcriptional signature defines a single continuum of neutrophils across biological compartments. Nat Commun. 2021;12:2856.
Bekes EM, Schweighofer B, Kupriyanova TA, Zajac E, Ardi VC, Quigley JP, et al. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol. 2011;179:1455–70.
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–95.
Isidro RA, Appleyard CB. Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am J Physiol Gastrointest Liver Physiol. 2016;311:G59–73.
Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455:547–51.
He L, Jhong JH, Chen Q, Huang KY, Strittmatter K, Kreuzer J, et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep. 2021;37:109955.
Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell. 2020;181:442–59.e29.
Rokavec M, Huang Z, Hermeking H. Meta-analysis of miR-34 target mRNAs using an integrative online application. Comput Struct Biotechnol J. 2023;21:267–74.
Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol Cancer. 2019;18:94.
Wang T, Chen Y, Gao R, Shui J, Xie B. Overexpression of AXL on macrophages associates with disease severity and recurrence in chronic rhinosinusitis with nasal polyps. Int Immunopharmacol. 2023;121:110449.
Pei JP, Wang Y, Ma LP, Wang X, Liu L, Zhang Y, et al. AXL antibody and AXL-ADC mediate antitumor efficacy via targeting AXL in tumor-intrinsic epithelial-mesenchymal transition and tumor-associated M2-like macrophage. Acta Pharm Sin. 2023;44:1290–303.
Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66:1–9.
Salkeni MA, Naing A. Interleukin-10 in cancer immunotherapy: from bench to bedside. Trends Cancer. 2023;9:716–25.
Ch’ng ES, Kumanogoh A. Roles of Sema4D and Plexin-B1 in tumor progression. Mol Cancer. 2010;9:251.
Mintz J, Vedenko A, Rosete O, Shah K, Goldstein G, Hare JM, et al. Current advances of nitric oxide in cancer and anticancer therapeutics. Vaccines 2021;9:94.
Isella C, Brundu F, Bellomo SE, Galimi F, Zanella E, Porporato R, et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat Commun. 2017;8:15107.
Jackstadt R, van Hooff SR, Leach JD, Cortes-Lavaud X, Lohuis JO, Ridgway RA, et al. Epithelial NOTCH signaling rewires the tumor microenvironment of colorectal cancer to drive poor-prognosis subtypes and metastasis. Cancer Cell. 2019;36:319–36.e7.
Li J, Xu P, Wu D, Guan M, Weng X, Lu Y, et al. Hypoxic stress suppresses lung tumor-secreted exosomal miR101 to activate macrophages and induce inflammation. Cell Death Dis. 2021;12:776.
Shi JH, Liu LN, Song DD, Liu WW, Ling C, Wu FX, et al. TRAF3/STAT6 axis regulates macrophage polarization and tumor progression. Cell Death Differ. 2023;30:2005–16.
Lv Q, Zhang Y, Gao W, Wang J, Hu Y, Yang H, et al. CSF1R inhibition reprograms tumor-associated macrophages to potentiate anti-PD-1 therapy efficacy against colorectal cancer. Pharm Res. 2024;202:107126.
Wang X, Li J, Dong K, Lin F, Long M, Ouyang Y, et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. 2015;27:443–52.
Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst. 2016;108:djv303.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr., Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992;356:215–21.
Gudkov AV, Komarova EA. p53 and the carcinogenicity of chronic inflammation. Cold Spring Harb Perspect Med. 2016;6:a026161.
Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, et al. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell. 2013;23:634–46.
Cooks T, Harris CC, Oren M. Caught in the cross fire: p53 in inflammation. Carcinogenesis. 2014;35:1680–90.
Li L, Ng DS, Mah WC, Almeida FF, Rahmat SA, Rao VK, et al. A unique role for p53 in the regulation of M2 macrophage polarization. Cell Death Differ. 2015;22:1081–93.
Liu C, Rokavec M, Huang Z, Hermeking H. Curcumin activates a ROS/KEAP1/NRF2/miR-34a/b/c cascade to suppress colorectal cancer metastasis. Cell Death Differ. 2023;30:1771–85.
Lei Y, Wang K, Deng L, Chen Y, Nice EC, Huang C. Redox regulation of inflammation: old elements, a new story. Med Res Rev. 2015;35:306–40.
Lin CC. Clinical development of colony-stimulating factor 1 receptor (CSF1R) inhibitors. J Immunother Precis Oncol. 2021;4:105–14.
Farr L, Ghosh S, Jiang N, Watanabe K, Parlak M, Bucala R, et al. CD74 signaling links inflammation to intestinal epithelial cell regeneration and promotes mucosal healing. Cell Mol Gastroenterol Hepatol. 2020;10:101–12.
Cho E, Mun SJ, Kim HK, Ham YS, Gil WJ, Yang CS. Colon-targeted S100A8/A9-specific peptide systems ameliorate colitis and colitis-associated colorectal cancer in mouse models. Acta Pharm Sin. 2024;45:581–93.
Slowicka K, Petta I, Blancke G, Hoste E, Dumas E, Sze M, et al. Zeb2 drives invasive and microbiota-dependent colon carcinoma. Nat Cancer. 2020;1:620–34.
Sierra JR, Corso S, Caione L, Cepero V, Conrotto P, Cignetti A, et al. Tumor angiogenesis and progression are enhanced by Sema4D produced by tumor-associated macrophages. J Exp Med. 2008;205:1673–85.
Li H, Wang JS, Mu LJ, Shan KS, Li LP, Zhou YB. Promotion of Sema4D expression by tumor-associated macrophages: Significance in gastric carcinoma. World J Gastroenterol. 2018;24:593–601.
Chen Y, Zhang L, Lv R, Zhang WQ. Overexpression of Semaphorin4D indicates poor prognosis and prompts monocyte differentiation toward M2 macrophages in epithelial ovarian cancer. Asian Pac J Cancer Prev. 2013;14:5883–90.
Jia LF, Wei SB, Mitchelson K, Gao Y, Zheng YF, Meng Z, et al. miR-34a inhibits migration and invasion of tongue squamous cell carcinoma via targeting MMP9 and MMP14. PLoS ONE. 2014;9:e108435.
Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci USA. 2006;103:12493–8.
Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci USA. 2007;104:20262–7.
Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer. 2017;116:415–23.
Wen B, Li S, Ruan L, Yang Y, Chen Z, Zhang B, et al. Engulfment and cell motility protein 1 fosters reprogramming of tumor-associated macrophages in colorectal cancer. Cancer Sci. 2023;114:410–22.
Wallace JA, Pitarresi JR, Sharma N, Palettas M, Cuitiño MC, Sizemore ST, et al. Protein kinase C Beta in the tumor microenvironment promotes mammary tumorigenesis. Front Oncol. 2014;4:87.
Acknowledgements
We thank Bert Vogelstein for p53-deficient and p53-proficient HCT116 cells, Anne Krug for reading the manuscript draft and advice, Ursula Götz for technical support and Liliana Mochmann for access to the tissue dissociator and experimental advice.
Funding
This study was funded by a grant of the Deutsche Forschungsgemeinschaft (He2701/13-3) to HH. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
JK: designed and performed experiments, analyzed results, wrote the manuscript; MR: performed bioinformatics analysis of scRNA-Seq data and experimental assistance with scRNA-Seq analysis, wrote the manuscript; MGOZ: initial mouse analysis presented in Fig. 1.; YF: treated BMDMs and HCT116 cells with NO-donor and performed qPCR analysis; HH: conceived and supervised the study, planned experiments and wrote the manuscript. All authors read and corrected the final draft of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
All methods were performed in accordance with the relevant guidelines and regulations. Animal experimentations and analyses were approved by the local authorities (Regierung von Oberbayern, AZ: 55.2-1-54-2532-201-2014; AZ:55.2-2532.Vet_02-21-111).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
König, J., Rokavec, M., Öner-Ziegler, M.G. et al. Myeloid Mir34a suppresses colitis-associated colon cancer: characterization of mediators by single-cell RNA sequencing. Cell Death Differ 32, 225–241 (2025). https://doi.org/10.1038/s41418-024-01380-9
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41418-024-01380-9
