
Abstract
The abnormal osteogenic differentiation of bone marrow mesenchymal stem cells (BMSCs) is an important cause of senile osteoporosis (SOP). Glutamine synthetase (GLUL) is a key enzyme in glutamine biosynthesis; however, its functional role in SOP remains unclear. Here, we found that GLUL expression was downregulated in the BMSCs of SOP patients. Mice with BMSC-specific Glul-knockout (KO) exhibited dysplasia of the skull and phalanges and osteoporosis due to disordered osteogenic differentiation. Mechanistically, GLUL competitively bound to the Tripartite Motif Containing 25 (TRIM25) SPRY subunit, reduced the ubiquitin-mediated degradation of UDP-N-acetylglucosamine pyrophosphorylase 1 (UAP1) and increased the synthesis of uridine 5-diphosphate N-acetylglucosamine (UDP-GlcNAc), thereby regulating the O-linked β-N-acetylglucosamine modification (O-GlcNAcylation) of serine 296 residues and increasing Forkhead Box O3 (FOXO3) stability to reduce oxidative stress. Moreover, blocking the O-GlcNAcylation of FOXO3 at Ser296 inhibited osteogenic differentiation. Finally, GLUL supplementation specifically in BMSCs slowed bone loss in SOP model mice. Overall, our study suggests that GLUL plays an important role in regulating osteogenic differentiation and bone development, which may have implications for SOP treatment.

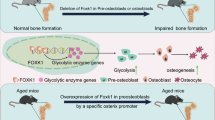
Schematic illustration of the molecular mechanism by which GLUL mediates FOXO3 O-GlcNAcylation to regulate the osteogenic differentiation of BMSCs and senile osteoporosis. The graphical abstract was created by figdraw2.0.
Similar content being viewed by others


Introduction
Population aging is a global trend, and SOP-related health problems will become increasingly prominent. SOP is characterized by decreased bone mass, destruction of the bone microstructure, and susceptibility to fracture [1, 2]. SOP generally refers to osteoporosis in individuals over the age of 70, and SOP-induced fractures increase disability and mortality in elderly individuals [3, 4]. Changes in the quantity and function of BMSCs, especially decreases in their osteogenic differentiation, are among the key causes of SOP [5, 6]. Thus, understanding how BMSC osteogenic differentiation is regulated will provide insights for the development of novel therapeutic strategies for SOP.
GLUL is responsible for converting glutamate and ammonia into glutamine, and it plays important roles in various physiological and pathological processes [7,8,9]. Studies have shown that when glutamine is present at normal physiological levels, the glutamine synthesis activity of GLUL in vascular endothelial cells is almost negligible. However, the absence of GLUL significantly affects the growth of vascular buds during vascular development [10]. In an Alzheimer’s disease mouse model, GLUL expression decreases with age, which may lead to defects in glutamate transmission and affect cognitive function [11]. Nevertheless, the role of GLUL in SOP has not been characterized.
O-linked β-N-acetylglucosamine modification (O-GlcNAcylation) is a posttranslational modification of proteins in which N-acetylglucosamine is added to serine or threonine residues [12]. The hexosamine biosynthesis pathway (HBP) provides the UDP-GlcNAc that is necessary for this modification [13, 14]. UAP1 is one of the key enzymes that regulates the HBP pathway to generate substrates [13, 15]. O-GlcNAcylation is closely associated with a variety of physiological processes, including signal transduction, transcriptional regulation, protein stability, and cell survival [16,17,18]. Moreover, this modification is closely related to osteoblast metabolism and bone formation [19, 20]. Additionally, the O-GlcNAcylation of key transcription factors modulates the osteogenic/lipogenic differentiation of BMSCs [21]. Although studies have shown that GLUL is involved in protein O-GlcNAcylation [22], its regulatory role in this process has not been fully characterized.
Protein ubiquitination is a dynamic posttranslational modification that participates in almost all aspects of eukaryotic biology, including osteogenic differentiation [23,24,25]. E3 ubiquitin ligases determine the precise substrate specificity of ubiquitination [26, 27]. However, the detailed mechanism by which ubiquitination regulates UDP-GlcNAc generation is still poorly understood.
In this study, we investigated the role of GLUL in the progression of SOP. We revealed the molecular mechanism by which GLUL increases UDP-GlcNAc biosynthesis and induces protein O-GlcNAcylation, and we described the importance of FOXO3 O-GlcNAcylation for BMSC osteogenic differentiation. Importantly, our study reveals a new link between GLUL- and HBP-mediated O-GlcNAcylation modification, and we propose a novel strategy for treating SOP.
Results
GLUL plays an important role in the osteogenic differentiation of BMSCs and the progression of SOP
We first used the Gene Expression Omnibus database to examine the expression of GLUL during the osteogenic differentiation of stem cells. Compared with that in the control group, the expression of GLUL was upregulated during the osteogenic differentiation of BMSCs (Figs. 1A and S1A). Then, we induced the osteogenic differentiation of BMSCs from C57BL/6 mice and measured the expression of GLUL at different time points. We found that the expression of GLUL increased with time (Fig. 1B). Then, immunohistochemical staining was performed on femoral sections from the mice on the first day and in the first week, second week, and fourth week after birth. The results revealed that GLUL expression gradually increased during bone development (Fig. 1C).

A Heatmap of the metabolic and transcriptional changes that occur during the osteogenic differentiation of human mesenchymal stem cells according to the GSE191136 dataset. B Western blotting analysis of GLUL expression during the osteogenic differentiation of BMSCs. C Immunohistochemical staining analysis of GLUL expression in mouse femur sections at different times after birth. D Heatmap of the effects of SOP on human mesenchymal stem cells according to the GSE35956 dataset. E Immunohistochemical staining analysis of GLUL expression in femur sections from young and old samp6 mice (n = 3). F Quantification of GLUL expression in bone samples from patients with SOP. G Western blotting analysis of GLUL expression in BMSCs from patients in the SOP group (n = 6) and the control group (n = 7). ** P < 0.01 vs. other groups; *P < 0.05 vs. other groups.
Owing to the close correlation between the disordered osteogenic differentiation of BMSCs and the occurrence of osteoporosis, we analysed data related to animal models and human osteoporosis; we found that GLUL expression differed in the BMSCs from animals and humans with different types of osteoporosis. That is, GLUL was upregulated in a postmenopausal osteoporosis animal model (Fig. S1B). However, GLUL expression was downregulated in the BMSCs of elderly patients with osteoporosis (SOP) (Fig. 1D). To confirm this conclusion, we measured the expression of GLUL in the femoral tissues of SAM-P6 mice at 4 and 12 months of age, and we found that GLUL expression was downregulated in aged SAM-P6 mice (Figs. 1E and S1C). GLUL expression also tended to decrease in bone and BMSCs from SOP patients (Figs. 1F, G and S1D). These data suggest that GLUL may play an important role in the osteogenic differentiation of aged BMSCs and the pathogenesis of SOP.
Mesenchymal stem cell-specific Glul deficiency leads to abnormal bone formation
To explore the role of GLUL in the commitment of mesenchymal stem cells to the osteoblast lineage, we generated Prx1Cre and Glul flox/flox mice (referred to as GlulPrx1 mice) (Figs. 2A and S2A), in which limb and craniofacial mesenchymal cells were the sites of Prx1Cre activity. We used Western blotting to confirm that Glul was knocked out in limb BMSCs (Fig. S2B). The survival and fertility of the GlulPrx1 mice were normal after birth. However, compared with Glulfl/fl mice, GlulPrx1 mice presented delayed bone development, especially in limb bones (Fig. 2B–D), and these effects were independent of the sex of the mice. To evaluate the role of GLUL in BMSC osteogenic differentiation, BMSCs from Glulfl/fl and GlulPrx1 mice were cultured with osteogenic medium to induce differentiation, and sufficient glutamine was added to eliminate the effects of insufficient GLUL enzyme activity. The results indicate that Glul knockout (KO) led to a significant decrease in cellular ALP activity and mineralization (Fig. 2E–H). The results of haematoxylin‒eosin staining indicated a decrease in trabecular bone in the femurs of GlulPrx1 mice (Fig. S2D). Moreover, the Western blotting and immunohistochemical staining results suggested that Glul-KO reduced the expression of osteogenic marker genes (RUNX2, CoL1A1, OSX, and SPP1) (Fig. S2C and S2E).

A Construction strategies for BMSC-specific Glul-KO (GlulPrx1) mice. B Representative images of Alizarin Red-Alcian Blue double staining of tissues from GlulPrx1 and Glulfl/fl mice(n = 5). C Representative images of Alizarin Red-Alcian Blue double staining of the skull, upper limb, and lower limb from GlulPrx1 and Glulfl/fl mice (n = 5). D Quantitative analysis of Alizarin Red-Alcian Blue double staining. E Representative images of ARS and ALP staining of BMSCs from GlulPrx1 and Glulfl/fl mice. F Quantitative analysis of ARS and ALP staining. G Representative images of ARS and ALP staining of BMSCs from GlulPrx1 and Glulfl/fl mice after treatment with glutamine. H Quantitative analysis of ARS and ALP staining. I Representative micro-CT images of trabecular bone from the femoral metaphysis of GlulPrx1 and Glulfl/fl mice (n = 5). J Quantitative analysis of cancellous bone volume (BV/TV, %), trabecular thickness (Tb.Th). K Quantitative analysis of trabecular number (Tb.N), and trabecular separation (Tb.Sp). L Representative images of VON KOSSA staining (n = 3). M Calcein double-label staining image. N Quantitative analysis of the MAR and BFR/BS in GlulPrx1 and Glulfl/fl mice (n = 3). **P < 0.01 vs. other groups; *P < 0.05 vs. other groups.
To further determine the function of GLUL in the skeletal system, we compared the static bone parameters of femurs from Glulfl/fl and GlulPrx1 mice with a microquantitative computed tomography (micro-CT) system. We found that the bone mass per unit volume (BV/TV), trabecular number (Tb.N) and trabecular thickness (Tb.Th) of 16-week-old GlulPrx1 mice decreased (Fig. 2I–K) and that the trabecular separation (Tb.Sp) increased. Von Kossa staining revealed a decrease in bone mineral deposition in 16-week-old GlulPrx1 mice (Fig. 2L), and the calcium double-labelling experiment suggested that the bone formation rate of GlulPrx1 mice decreased (Fig. 2M), the bone mineral apposition rate (MAR) decreased, and the same to bone formation rate/bone surface (BFR/BS) (Fig. 2N). Overall, there are obstacles to bone development and BMSC osteogenic differentiation in conditional Glul-KO mice.
GLUL regulates HBP metabolism and affects UDP-GlcNAc generation
Next, we investigated the specific mechanism by which GLUL regulates osteogenic differentiation. We performed proteomic and metabolomic assays on BMSCs from GlulPrx1 and Glulfl/fl mice after osteogenic differentiation. Component analysis revealed that Glul KO significantly altered the intracellular metabolic characteristics of BMSCs (Fig. S3A-C). The results of liquid chromatography‒mass spectrometry (LC‒MS/MS) revealed a significant decrease in the levels of UDP-GlcNAc (HBP metabolic end product) in Glul-KO cells (Figs. 3A and S3B). Surprisingly, the levels of Fruc-6P, GlcNAc-6P, and GlcNAc-1P, which are intermediate products of HBP metabolism, exhibited opposite trends. These findings indicate that the key regulatory point that affects the generation of UDP-GlcNAc in Glul-KO cells is the final step of the HBP metabolic pathway (Fig. 3A, B), in which UAP1 catalyses the generation of UDP-GlcNAc from GlcNAc-1P.

A Heatmap of metabolomic assays of the BMSCs from GlulPrx1 and Glulfl/fl mice (n = 6). B Schematic diagram of the hexosamine biosynthetic pathway. C Heatmap of the proteomics analysis of BMSCs from GlulPrx1 and Glulfl/fl mice (n = 3). D Western blotting analysis of O-GlcNAcylation levels in BMSCs from GlulPrx1 and Glulfl/fl mice at different osteogenic differentiation timepoints. The numbers on the line graph represent the P -values comparing the relative protein expression levels between the two groups. E Western blotting analysis of UAP1 expression in BMSCs from GlulPrx1 and Glulfl/fl mice at different osteogenic differentiation timepoints. The numbers on the line graph represent the P -values comparing the relative UAP1 expression levels between the two groups. F Western blotting analysis of UAP1 expression in BMSCs from patients in the SOP group (n = 6) and the control group (n = 7). G Western blotting analysis of OGT levels in BMSCs from GlulPrx1 and Glulfl/fl mice at different osteogenic differentiation timepoints. H Representative images of ARS and ALP staining of BMSCs from GlulPrx1 and Glulfl/fl mice after transfection with the UAP1 overexpression or control plasmid. **P < 0.01 vs. other groups; *P < 0.05 vs. other groups.
By analysing proteomic data, we found that UAP1 expression was downregulated in Glul-KO cells (Fig. 3C), and this result was confirmed by Western blotting (Fig. 3E). UAP1 expression also tended to decrease in BMSCs from SOP patients (Fig. 3F). Moreover, there was no significant change in OGT expression (Fig. 3G). Therefore, GLUL affects HBP flux by regulating UAP1 expression. Research has shown that changes in the UDP-GlcNAc concentration can directly affect the level of protein O-GlcNAcylation [22]. We found that during the osteogenic differentiation of BMSCs, the overall level of protein O-GlcNAcylation increased (Fig. 3D), and the expression level of UAP1 also tended to increase (Fig. 3E). To some extent, supplementation with UAP1 rescued the disordered osteogenic differentiation of Glul-KO cells (Figs. 3H and S3E). Therefore, GLUL regulates UDP-GlcNAc biosynthesis by affecting the UAP1 expression level, thereby regulating protein O-GlcNAcylation and affecting BMSC osteogenic differentiation.
GLUL regulates UAP1 expression by interacting with Tripartite Motif Containing 25 (TRIM25)
To explore the mechanism by which GLUL regulates UAP1 expression, we consulted the NCBI database and found that TRIM25 and TRIM67 are potential interacting proteins between GLUL and UAP1 (Fig. S4A). The molecular docking results suggest that TRIM25 has potential binding sites with GLUL and UAP1 (Fig. 4A). Biacore experiments confirmed the interaction between the TRIM25 protein and GLUL or UAP1 (Fig. 4B). Moreover, co-immunoprecipitation (co-IP) experiments were conducted with the HEK293T cell line and BMSCs. The results revealed that TRIM25 coimmunoprecipitated with exogenous and endogenous GLUL and UAP1 (Figs. 4C and S4C). The bimolecular fluorescence complementation (BiFC) results suggested that, owing to the interaction of TRIM25 with GLUL or UAP1, the two molecular fragments of the YFP carried by TRIM25 reformed active fluorescent groups (Fig. 4D and Videos 1 and 2). The interactions of the three molecules in living cells were also recorded (Fig. 4E and Video 3). In addition, endogenous TRIM25 was shown to colocalize with GLUL and UAP1 in the cytoplasm of BMSCs (Fig. 4F). The molecular structure of TRIM25 includes a RING Finger domain (aa 1–54), a coiled coil domain (aa 217–307), and a SPRY domain (aa 389–630) (Fig. 4G). We conducted co-IP experiments on different TRIM25 mutants, which are shown in the figure. The absence of the SPRY domain caused the binding affinity of TRIM25 for GLUL or UAP1 to be lost (Fig. 4H). Therefore, the SPRY domain is necessary for the interaction of TRIM25 with GLUL or UAP1. We hypothesize that the competitive binding of GLUL to the TRIM25 SPRY domain inhibits its interaction with UAP1.

A Molecular docking image of the TRIM25 protein and the GLUL/UAP1 protein. B The results of the Biacore assay to assess the binding strength of GLUL/UAP1 to TRIM25. C Representative co-IP images of TRIM25 with GLUL/UAP1 in HEK-293T cells. D Representative images of bimolecular fluorescence complementation experiments between TRIM25 carrying YFP (aa 1–154) and GLUL/UAP1 carrying YFP (aa 155–238). E Representative images of live-cell imaging after cotransfection of GFP-TRIM25, RFP-GLUL, and BFP-UAP1 into HEK293T cells. F Representative images of immunofluorescence staining for TRIM25, GLUL and UAP1 in BMSCs. G Schematic diagram of TRIM25 truncation. H Representative images of co-IP experiments conducted with Myc-UAP1, HA-GLUL, and Flag-TRIM25 full-length and truncated samples. **P < 0.01 vs. other groups; *P < 0.05 vs. other groups.
Research has shown that TRIM25 promotes substrate degradation through the ubiquitin proteasome pathway. We investigated whether TRIM25 regulates the degradation of UAP1. The results indicate that the level of TRIM25 was inversely proportional to the level of the UAP1 protein (Figs. 5A, B and S4B). In addition, TRIM25 significantly reduced UAP1 expression in a dose-dependent manner (Fig. 5A). When the proteasome inhibitor MG132 was added, the level of the UAP1 protein in HEK293T cells was significantly increased (Fig. 5C), but the lysosome inhibitor chloroquine did not restore UAP1 expression (Fig. 5D). These results indicate that TRIM25 promotes UAP1 degradation through the proteasome pathway. Lys48-linked polyubiquitin chains target proteins for degradation via proteasome-dependent processes [28]. The interaction between TRIM25 and UAP1 led to the induction of UAP1 Lys48 ubiquitination (Fig. 5E), whereas during BMSC osteogenic differentiation, UAP1 Lys48 ubiquitination gradually decreased (Fig. 5F). In this process, GLUL reduces TRIM-mediated the Lys48 ubiquitination of UAP1 (Fig. 5G), which is the specific mechanism by which TRIM25 regulates UAP1 expression.

A Representative Western blotting images of HEK-293T cells cotransfected with the TRIM25 overexpression vector and UAP1 in a dose-dependent manner. B Representative image of a Western blotting experiment showing UAP1 expression levels after cell transfection with si-TRIM25. C Representative image of a Western blotting experiment showing UAP1 expression levels after cell transfection with si-TRIM25. D Representative images of Western blotting analysis of proteins that were extracted from HEK293T cells that were transfected with the Myc-UAP1 and Flag-TRIM25 overexpression plasmids and treated with MG132. E Representative images of Western blotting analysis of proteins that were HEK293T cells that were transfected with the Myc-UAP1 and Flag-TRIM25 overexpression plasmids and treated with MG132 and chloroquine. F A Myc-UAP1 overexpression plasmid was transfected into BMSCs, and osteogenic differentiation was induced. Proteins were extracted from the cells at different timepoints, and representative images were obtained after co-IP experiments with anti-UAP1 antibodies. G The V5 Ub overexpression plasmid was transfected into HEK293T cells, which were then cotransfected with Flag-TRIM25, Flag-TRIM25+Myc-UAP1 + HA-GLUL, or Myc-UAP1 + HA-GLUL overexpression plasmids. Subsequent Western blot images and quantitative analysis revealed the K48-linked ubiquitination of UAP1 in different cell groups. **P < 0.01 vs. other groups; *P < 0.05 vs. other groups.
O-GlcNAcylation of FOXO3-Ser296, which is regulated by GLUL, promotes the osteogenic differentiation of BMSCs
To further explore how GLUL-mediated regulation of protein O-GlcNAcylation affects the osteogenic differentiation of BMSCs, we used 4D DIA O-GlcNAcylation omics analysis (PTM BIO, Hangzhou, China) to identify O-GlcNAcylation-modified proteins specifically regulated by GLUL. A total of 73 candidate proteins and 129 modification sites were identified (Figs. 6A, B and S5A -F). We subsequently focused on FOXO3, which is closely associated with processes such as cellular ageing and osteogenic differentiation. The Flag-Foxo3 overexpression plasmid was transfected into BMSCs (Glulfl/fl) and BMSCs (GlulPrx1), anti-Flag antibody was used for co-IP to assess O-GlcNAcylation levels [29, 30]. The result confirmed that the O-GlcNAcylation level of FOXO3 in BMSCs was reduced upon Glul knockout or Uap1 knockdown (Figs. 6C and S5H-I). The omics analysis suggested that Ser296 (S296) is the main O-GlcNAcylation modification site in FOXO3. S296 and surrounding amino acid sequences are highly conserved in vertebrates (Fig. S5G). Motif analysis of O-GlcNAcylation modification sites revealed that the polar residue (SADDSPSTSSKWPGSPTSRSSDE), in which FOXO3-Ser296 is located, conforms to the XxxxxxxSxxSRxxxxxxxxxxxx motif (Fig. S5B-C, Supplementary Table 1). Next, we generated a site-specific mutant of FOXO3 (S296V) and screened BMSCs with CRISPR/Cas9 to knock out Foxo3 (Fig. S5J). We conducted a series of actinomycete ketone tracking experiments to evaluate the stability of FOXO3. Compared with that in cells that were transfected with the empty vector, the half-life of FOXO3 in Glul-KO cells was shorter (Fig. 6D). Compared with wild-type cells, the S296V mutation shortened the half-life of FOXO3 from 24 hours to 12 hours (Fig. 6E). These results indicate that O-GlcNAcylation of S296 can stabilize FOXO3.

A Volcano plots of BMSCs from GlulPrx1 (n = 3) and Glulfl/fl (n = 3) mice subjected to 4D DIA O-GlcNAcylation profiling analysis. B Mass spectrometry image of the O-GlcNAcylation modification of the FOXO3 Ser296 residue. C The results of the co-IP experiment on the effect of Glul knockout on the level of O-GlcNAcylation modification of FOXO3 in BMSCs. D Representative images of target protein decay at different time points in BMSCs from GlulPrx1 and Glulfl/fl mice. E Representative images of target protein attenuation at different time points were obtained by transfecting wild-type or S296V-mutant FOXO3 plasmids into Foxo3-KO BMSCs. F Representative images of ARS and ALP staining of Foxo3-KO BMSCs after transfection with wild-type or S296V-mutant FOXO3 plasmids. G Quantitative analysis of ARS and ALP staining. H Representative images of FOXO3-Ser297 O-GlcNAcylation levels as determined by Western blotting analysis of BMSCs from patients in the SOP and control groups. I Western blotting analysis of the expression of osteogenesis-related genes in Foxo3-KO BMSCs after transfection with wild-type or S296V-mutant FOXO3 plasmids. J Representative images of ROS detection. **P < 0.01 vs. other groups; *P < 0.05 vs. other groups.
To further investigate whether the absence of O-GlcNAcylation in FOXO3-S296 affects BMSC osteogenic differentiation, we transiently overexpressed WT Foxo3 or the S296V mutant in Foxo3-KO BMSCs and induced osteogenic differentiation. Alizarin Red and ALP staining indicated that the Foxo3 S296V mutation weakened the osteogenic differentiation potential of the BMSCs, and this effect was not reversed by UAP1 (Fig. 6F, G). Correspondingly, the expression of osteogenic marker genes (RUNX2 and SPP1) was downregulated (Fig. 6I). Foxo3 S296V mutation leads to increased levels of reactive oxygen species (ROS) in BMSCs (Fig. 6J). We generated anti-O-GlcNAcylation antibodies targeting the corresponding site in human FOXO3 (S297) and performed Western blotting to measure the levels of this modification in BMSCs from SOP patients. The results showed that in BMSCs from SOP patients, the levels of the FOXO3-S297O-GlcNAcylation modification were decreased (Fig. 6H). Overall, the O-GlcNAcylation of FOXO3-Ser296, which is regulated by GLUL, stabilizes FOXO3 and promotes BMSC osteogenic differentiation in vitro, and dysregulation of this pathway occurs in SOP patients.
Supplementation of GLUL can alleviate bone loss in SOP model mice
To evaluate the potential role of GLUL in bone loss in SOP, Glul-overexpressing adenovirus (aav) was injected into 18-month-old Prx1-Cre mice via the tail vein. Micro-CT analysis revealed that Glul overexpression reduced bone loss in SOP model mice (Fig. 7A–C), and the P1NP serum concentration increased (Fig. 7D), indicating increased osteoblast activity. Furthermore, immunohistochemical staining suggested that the expression of osteogenic marker genes was increased (Fig. 7E). Dual-labelling experiments with calcein confirmed that Glul overexpression increased bone formation (Fig. 7F). Similarly, after Glul overexpression, the number and surface area of osteoblasts significantly increased (Fig. 7G). To evaluate the effect of GLUL supplementation on BMSC osteogenic differentiation, BMSCs were isolated from the femurs of Prx1-Cre mice in the control, SOP, and Glul-OE aav groups. The results revealed that GLUL expression was downregulated in the BMSCs of SOP model mice, whereas Glul-OE aav promoted BMSC osteogenic differentiation and bone formation (Fig. 7H–J). Moreover, GLUL overexpression increased osteogenic marker genes expression in the BMSCs of SOP model mice (Fig. 7K, L). In summary, these results indicate that supplementation with GLUL can alleviate bone loss in SOP model mice.

A Schematic diagram of in vivo experiment. B Representative micro-CT images of trabecular bone from the femoral metaphysis of mice(n = 4). C Quantitative analysis of BV/TV, Tb.Th, Tb.N and Tb.Sp. D ELISA detection results of serum P1NP levels in each group of mice (n = 4). E Immunohistochemical staining of osteogenesis-related genes in mice femur sections. (n = 3). F Calcein double-label staining image and quantitative analysis of the MAR and BFR/BS in each group of mice (n = 3). G Analysis of Osteoblast Density (N.O/BS) and Osteoblast Surface Area (O.S/BS) Data. H Representative images of ARS and ALP staining of BMSCs from mice of each group. I Quantitative analysis of ARS and ALP staining. J Representative images of VON KOSSA staining (n = 3). K Western blot analysis of the expression of osteogenesis-related genes in BMSCs from mice of each group. L Quantitative data analysis of Western blot results. **P < 0.01 vs. other groups; *P < 0.05 vs. other groups.
Discussion
New evidence suggests that the lack of GLUL is one of the characteristics of BMSCs from SOP patients and that the weakened osteogenic differentiation of BMSCs is an important cause of the occurrence and development of SOP [31]. Promoting the osteogenic differentiation of BMSCs is considered a promising strategy for ameliorating the symptoms of SOP [32]. Our research indicates that the absence of GLUL in BMSCs leads to abnormal development of the skull and phalanges of mice, as well as reduced bone mass in adult mice. Moreover, targeted GLUL supplementation in BMSCs alleviated bone loss in SOP model mice. Therefore, GLUL plays an indispensable role in the osteogenic differentiation of BMSCs. In summary, these data provide the first example of GLUL regulating lineage allocation and bone homeostasis in BMSCs.
Although GLUL is a key enzyme in glutamine synthesis, roles for GLUL independent of metabolic enzymes in gastric cancer, lung cancer, epilepsy, and angiogenesis have been revealed by multiple studies [7, 33,34,35]. We discovered the unique role of GLUL in regulating UDP-GlcNAc synthesis. Previous studies have shown that in hepatocellular carcinoma, the mechanism by which GLUL increases the HBP flux is that GLUL increases glutamine synthesis via its enzymatic activity, providing a nitrogen source for UTP synthesis and thereby increasing UDP-GlcNAc generation [22]. During the osteogenic differentiation of BMSCs, knocking out Glul and supplementing glutamine cannot rescue the abnormal differentiation phenotype. By analysing the synthesis of various intermediate products in the HBP pathway, we ultimately identified and revealed that UAP1 is the regulatory point for GLUL synthesis in BMSCs. Additionally, in this process, the role of GLUL is independent of its catalytic activity.
O-GlcNAcylation is a posttranslational modification that is involved in important functions such as glucose metabolism, transcriptional regulation, subcellular localization, enzyme activity, and immune regulation, and it plays a regulatory role in determining the differentiation fate of bone marrow stromal cells [36,37,38]. During this process, OGT mediates the O-GlcNAcylation of the transcription factor RUNX2 and supports osteogenic differentiation [21]. In this study, there was no difference in the expression of OGT between Glul-KO and control cells. Although OGT is the only enzyme that catalyses this modification, our results suggest that other mechanisms are involved in the decrease in protein O-GlcNAcylation caused by GLUL downregulation. We found that the UAP1-catalysed synthesis of glycosylation substrates plays a more significant regulatory role in this process. However, how UAP1 is regulated by ubiquitination is largely unknown.
FOXO3 is a key factor in the cellular defence against oxidative stress, and it is equally important for the osteogenic differentiation of BMSCs [39,40,41]. During the differentiation of BMSCs, the increase in mitochondrial respiration leads to an increase in endogenous reactive oxygen species level [42,43,44]. The instability or downregulation of FOXO3 causes the ability of BMSCs to control ROS levels to be lost, thereby impairing differentiation [45, 46]. Here, we first describe the important role of GLUL in regulating the O-GlcNAcylation of FOXO3 at the S296 site during SOP progression. The modification of this site helps to stabilize FOXO3, thereby participating in the regulation of BMSC differentiation.
This study has certain limitations. The reason for the downregulation of GLUL in the BMSCs of SOP patients has not been revealed. The mechanism by which GLUL specifically regulates the O-GlcNAcylation of FOXO3 is also unclear. Moreover, the mechanism by which S296 O-GlcNAcylation mediates FOXO3 stability has not been extensively explored.
Overall, we revealed a link between disordered BMSC osteogenic differentiation and O-GlcNAcylation in SOP, and we demonstrated that GLUL can promote HBP-mediated UDP-GlcNAc biosynthesis by reducing UAP1 ubiquitination-mediated degradation. In addition, GLUL promotes O-GlcNAcylation at Ser296 through the mechanism described above to stabilize FOXO3 and maintain its control over ROS during BMSC osteogenic differentiation. In addition, these findings increase our understanding of the role of O-GlcNAcylation in SOP pathogenesis and progression and suggest that targeting GLUL in BMSCs may be a strategy for treating SOP.
Materials and methods
Reagents and antibodies
The Disodium beta-glycerol phosphate pentahydrate (S27785) was purchased from Shanghai yuanye Bio-Technology Co., Ltd (Shanghai, China). Ascorbic acid (A92902) and dexamethasone (D4902) were obtained from Merck (Darmstadt, Germany). BCIP/NBT Alkaline Phosphatase Color Development Kit and Alkaline Phosphatase Assay Kit were obtained from Beyotime Biotechnology (Shanghai, China). Cy3 goat anti-mouse IgG (A22210), DyLight 488 goat anti-rabbit IgG (A23220) and DyLight 549 goat anti-mouse IgG (A23310) were obtained from Abbkine Scientific (Wuhan China). Cycloheximide (GC17198) were obtained from GLPBIO (Montclair, USA), and MG-132 (HY-13259), Thiamet G (HY-12588), Chloroquine (HY-17589A) were obtained from MedChemExpress LLC (New Jersey, USA). TRIM25 (NM_005082) Human Recombinant Protein, Glutamine Synthetase (GLUL) (NM_002065) Human Recombinant Protein and UAP1 (NM_003115) Human Recombinant Protein were obtained from OriGene (Maryland, USA). Biacore Series S Sensor Chip CM5 were obtained from Cytiva (Washington, D.C. USA).
The information on the primary antibodies applied in this research are listed in Supplementary Table 2.
Mice
Conditional Glul-KO (Glulfl/fl) mouse (S-CKO-02655) and Prx1-Cre mouse (C001029) strains were obtained from Cyagen Biotechnology Co., Ltd. These strains were generated as previously described [47,48,49]. Glulfl/fl mice were crossed with the Prx1-Cre strain to generate GlulPrx1 mice. During the breeding process, the mice were maintained on the C57BL/6 background. The mice were fed in an air-conditioned room at 23–25 °C with 12-hour light‒dark cycles and free access to water and food. The animal experiments were carried out in strict accordance with protocols approved by the Medical Science Research Ethics Committee of the Affiliated Hospital of Jining Medical University.
Alizarin Red-Alcian Blue double staining
On the first day after birth, neonatal mice were euthanized via the injection of pentobarbital and fixed with 95% alcohol for at least 48 hours. After termination of fixation, the skin and internal organs of the neonatal mice were carefully removed and fixed in acetone for one week. Alizarin Red Alizarin Blue staining solution was prepared as follows: 0.3% Alizarin Blue, 0.1% Alizarin Red, glacial acetic acid, and 70% ethanol were mixed in a 1:1:1:17 ratio. The samples were then placed in the staining solution and incubated for 24–48 hours. The samples were then washed with pure water, soaked in 1% KOH solution for 48 hours, soaked in 20% glycerol for one week, transferred to 50% glycerol, and photographed with a Zeiss Axio Zoom. V16 microscope.
Bone dynamic analysis
The mice were intraperitoneally injected with calcein (20 mg/kg) 10 and 3 days before sacrifice. After sampling, the femur was embedded, and nondecalcified sections were prepared. These sections were observed and photographed using a fluorescence microscope (Zeiss Axio Zoom V16).
Micro-CT
After the mice were euthanized, the femurs were separated and fixed with 4% paraformaldehyde. A micro-CT imaging system (PerkinElmer; Quantum GX2) was used to scan femoral tissue. The scanning parameters were set as follows: voltage of 90 kV, 88 μA, 14 minutes, and resolution of 36 μm pixels. Skyscan NRecon software was used to reconstruct the images, and CTVox software was used to analyse the sample parameters.
Bone histomorphometry and immunohistochemistry
All the procedures were carried out as previously reported [50]. The static reconstruction parameters in Goldner three-colour stained sections were quantified using Osteomeasure (OsteoMetrics, Inc., US) software. All the histomorphological parameters were measured in accordance with the recommendations of the ASBMR Nomenclature Committee.
Lentivirus-mediated KO and aav-mediated overexpression
The Foxo3-KO lentivirus based on CRISPR/Cas9 technology was provided by Shanghai Genechem Co., Ltd. Glul- aav was also purchased from the same company. The Glul-aav construction strategy was as follows: CAG-FLEX-EGFP-FT2A-NM_008131-3Flag-SV40 Polya. The Foxo3-KO lentiviral construction strategy was as follows: GV392-U6-sgRNA-Foxo3-EF1a-Cas9-FLAG-P2A-puro. For lentivirus transfection, 80% confluent cells were incubated with lentivirus and polybrene (8 μg/mL), puromycin (P8230, Solarbio) was used for screening, and target gene expression was measured by Western blotting.
Plasmid and siRNA transfection
For a single well of a six-well plate, 4 µg of plasmid and 10 μL of Lipofectamine 2000 (11668019; Thermo Fisher Scientific) were mixed with 250 μL of Opti-MEM I (31985070; Thermo Fisher Scientific), and the mixture was incubated for 5 minutes at room temperature. During transfection, 1.5 mL of Opti-MEMI and transfer solution were added to a single well of a six-well plate. After 4 hours, the transfection mixture was removed and replaced with fresh medium. Analysis of overexpression efficiency and other experiments were performed 48 hours after transfection. Plasmids overexpressing Flag-TRIM25, Myc-UAP1, HA-GLUL, V5-Ubiquitin, Flag-Uap1, Flag-Foxo3-WT, Flag-Foxo3-S296V, Flag-FOXO3-WT, Flag-FOXO3-S297V, Flag-TRIM25(Δ13-54aa), Flag-TRIM25(Δ217-307aa), Flag-TRIM25(Δ439-630aa), GFP-TRIM25, RFP-GLUL, and BFP-UAP1, were synthesised by WZ Biosciences Inc. Plasmids overexpressing YFP(1-154aa)-TRIM25, YFP(155-238aa)-UAP1, YFP(155-238aa)-GLUL were synthesised by GenePharma Corporation.
For TRIM25-siRNA (GenePharma) transfection, the procedure was the same as that for plasmid transfection. The amounts of plasmid and Lipofectamine 2000 that were used were 100 pmol and 5 μL, respectively.
Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (RT-qPCR) was performed as previously described [51].
Western blotting and co-immunoprecipitation
Western blotting was performed according to previously described methods [52]. After electrophoresis and membrane transfer, the PVDF membranes were shaken and incubated overnight with the primary antibody at 4 °C. The next day, the PVDF membranes were incubated at room temperature with HRP-labelled Goat Anti Rabbit IgG or Goat anti Mouse IgG (1:5000) for 1 hour. The PVDF membrane was subsequently washed thoroughly. A Tanon-5200 chemiluminescence imaging system (Shanghai, China) and ImageJ software (National Institutes of Health, USA) were used to record and quantify the signal intensities. The protein levels were normalized to those of β-actin (1:3000) as a control.
For co-IP, whole-cell extracts were lysed with Pierce IP lysis buffer (87787; Thermal Science). Then, the extraction solution and the corresponding antibodies were added and incubated with the extracts overnight at 4 °C. Then, Protein A&G beads (Bersinbio) were added, and the solutions were incubated at 4 °C for 4 hours. The coprecipitated proteins were washed with SDS loading buffer at 95 °C for 5 minutes. The subsequent results were obtained using Western blotting, as described above. The difference was that a Mouse anti-Rabbit IgG (Conformation Specific) mAb (HRP Conjugate) (#5127, CST) was used as a secondary antibody.
Generation of an anti-O-GlcNAc antibody targeting the S297 site of human FOXO3 (FOXO3 S297-O-GlcNAc)
Two modified peptide sequences were designed and synthesized, ensuring their immunogenicity and specificity. A negative unmodified peptide was designed for antibody detection. FOXO3 S297 O-GlcNAc-modified peptide A: C WPGSPT-(O-GlcNAc)S-RSSDEL 291-303; FOXO3 S297 O-GlcNAc-modified peptide B: PGSPT-(O-GlcNAc)S-RSSDE C 292-302; FOXO3 unmodified peptide: C WPGSPTSRSSDEL 291-303. The peptide samples were separately purified. These peptides were subsequently conjugated with KLH. Two New Zealand white rabbits were immunized with each peptide, for a total of four rabbits. The immunization method involved injecting the antigens into two subcutaneous points on both shoulders and two muscle points on both hind legs. A total of six immunizations were performed before and after the fourth immunization, and blood samples were collected four times. The antibody titres in the serum were measured by ELISA, and Western blotting was performed on the serum. When the serum (OD450 > 1.0) reached 1:10000 (after the third or fourth immunization), it was considered to meet the standard, and subsequent purification was carried out. On the basis of the ELISA and Western blotting results, 2-4 rabbit serum samples were selected for two-step affinity purification of Protein A and antigen peptides. The specificity and viability of the antibodies were confirmed by Western blotting with FOXO3- and S297-mutant FOXO3-overexpressing HEK293T cells.
Surface plasmon resonance (SPR) binding assay
BiacoreT200 was used for the SPR experiments. The recombinant TRIM25 protein was fixed on the S-series sensor chip CM5 (Cytiva). The protein was injected into the sample channel at 25 °C at a flow rate of 30 μL/min (180 seconds for association, 300 seconds for dissociation) to measure the binding of the recombinant proteins UAP1, GLUL and TRIM25. After each interaction analysis cycle, the sensor chip surface was completely regenerated for 10 s with 10 mM glycine-HCl as the injection buffer at a flow rate of 150 μL/min to remove the analyte.
Immunofluorescence
All the procedures were carried out as previously reported [53], and images were captured with an LSM800 laser confocal microscope (Zeiss). To stain TRIM25, GLUL, and UAP1 simultaneously, on the first day, the cells were incubated with a rabbit anti-UAP1 antibody and a mouse anti-TRIM25 antibody. On the second day, the cells were incubated with Cy3-conjugated goat anti-mouse IgG and DyLight 488-conjugated goat anti-rabbit IgG. Then, the cells were incubated with an Alexa Fluor 647-conjugated anti-GLUL antibody again as the primary antibody.
Statistics and reproducibility
Statistical analysis was performed using the unpaired two-tailed Student’s t-test, one-way ANOVA or two-way ANOVA, followed by post-hoc analysis using Tukey’s or Dunnet’s methods, were employed using Prism9 (GraphPad Software Inc., San Diego, CA, USA). All the data are shown as the means ± standard deviations (SDs). All the experiments were repeated at least three times unless otherwise indicated. A p value < 0.05 was considered statistically significant.
Data availability
The datasets generated during the current study are available in the Figureshare repository. https://doi.org/10.6084/m9.figshare.28105184.v1. All data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
Compston JE, McClung MR, Leslie WD. Osteoporosis. Lancet. 2019;393:364–76.
Vilaca T, Eastell R, Schini M. Osteoporosis in men. Lancet Diab Endocrinol. 2022;10:273–83.
Resnick NM. Greenspan SL. ‘Senile’ osteoporosis reconsidered. JAMA. 1989;261:1025–9.
Boonen S, Dejaeger E, Vanderschueren D, Venken K, Bogaerts A, Verschueren S, et al. Osteoporosis and osteoporotic fracture occurrence and prevention in the elderly: a geriatric perspective. Best Pr Res Clin Endocrinol Metab. 2008;22:765–85.
Zhang Y, Chen CY, Liu YW, Rao SS, Tan YJ, Qian YX, et al. Neuronal Induction of Bone-Fat Imbalance through Osteocyte Neuropeptide Y. Adv Sci (Weinh). 2021;8:e2100808.
Qadir A, Liang S, Wu Z, Chen Z, Hu L, Qian A. Senile osteoporosis: the involvement of differentiation and senescence of bone marrow stromal cells. Int J Mol Sci. 2020;21:349.
Zhao JS, Shi S, Qu HY, Keckesova Z, Cao ZJ, Yang LX, et al. Glutamine synthetase licenses APC/C-mediated mitotic progression to drive cell growth. Nat Metab. 2022;4:239–53.
Dai W, Shen J, Yan J, Bott AJ, Maimouni S, Daguplo HQ, et al. Glutamine synthetase limits β-catenin-mutated liver cancer growth by maintaining nitrogen homeostasis and suppressing mTORC1. J Clin Invest. 2022;132:e161408.
Villar VH, Allega MF, Deshmukh R, Ackermann T, Nakasone MA, Vande Voorde J, et al. Hepatic glutamine synthetase controls N(5)-methylglutamine in homeostasis and cancer. Nat Chem Biol. 2023;19:292–300.
Eelen G, Dubois C, Cantelmo AR, Goveia J, Brüning U, DeRan M, et al. Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature. 2018;561:63–69.
Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: mechanism for deficient glutamatergic transmission. Mol Neurodegener. 2011;6:55.
Ma J, Wu C, Hart GW. Analytical and biochemical perspectives of protein O-GlcNAcylation. Chem Rev. 2021;121:1513–81.
Chatham JC, Zhang J, Wende AR. Role of O-Linked N-acetylglucosamine protein modification in cellular (patho)physiology. Physiol Rev. 2021;101:427–93.
Alhusban S, Nofal M, Kovacs-Kasa A, Kress TC, Koseoglu MM, Zaied AA, et al. Glucosamine-mediated hexosamine biosynthesis pathway activation uses ATF4 to promote “exercise-like” angiogenesis and perfusion recovery in PAD. Circulation. 2024;150:1702–19.
Yang S, Jin S, Xian H, Zhao Z, Wang L, Wu Y, et al. Metabolic enzyme UAP1 mediates IRF3 pyrophosphorylation to facilitate innate immune response. Mol Cell. 2023;83:298–313.e8.
Ma J, Hou C, Wu C. Demystifying the O-GlcNAc code: a systems view. Chem Rev. 2022;122:15822–64.
Yu SB, Wang H, Sanchez RG, Carlson NM, Nguyen K, Zhang A, et al. Neuronal activity-driven O-GlcNAcylation promotes mitochondrial plasticity. Dev Cell. 2024;59:2143–2157.e9.
Very N, Staels B, Eeckhoute J. Increased O-GlcNAcylation connects metabolic to transcriptional reprogramming during pathophysiological cell activation. Trends Cell Biol. 2024;34:988–91.
You C, Shen F, Yang P, Cui J, Ren Q, Liu M, et al. O-GlcNAcylation mediates Wnt-stimulated bone formation by rewiring aerobic glycolysis. EMBO Rep. 2024;25:4465–87.
Li S, Ren W, Zheng J, Li S, Zhi K, Gao L. Role of O-linked N-acetylglucosamine protein modification in oxidative stress-induced autophagy: a novel target for bone remodeling. Cell Commun Signal. 2024;22:358.
Zhang Z, Huang Z, Awad M, Elsalanty M, Cray J, Ball LE, et al. O-GlcNAcglycosylation orchestrates fate decision and niche function of bone marrow stromal progenitors. Elife. 2023;12:e85464.
Zhou P, Chang WY, Gong DA, Xia J, Chen W, Huang LY, et al. High dietary fructose promotes hepatocellular carcinoma progression by enhancing O-GlcNAcylation via microbiota-derived acetate. Cell Metab. 2023;35:1961–1975.e6.
Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26:399–422.
Liu F, Chen J, Li K, Li H, Zhu Y, Zhai Y, et al. Ubiquitination and deubiquitination in cancer: from mechanisms to novel therapeutic approaches. Mol Cancer. 2024;23:148.
Dikic I, Schulman BA. An expanded lexicon for the ubiquitin code. Nat Rev Mol Cell Biol. 2023;24:273–87.
Cruz Walma DA, Chen Z, Bullock AN, Yamada KM. Ubiquitin ligases: guardians of mammalian development. Nat Rev Mol Cell Biol. 2022;23:350–67.
Zhao Z, Song X, Wang Y, Yu L, Huang G, Li Y, et al. E3 ubiquitin ligase TRIM31 alleviates dopaminergic neurodegeneration by promoting proteasomal degradation of VDAC1 in Parkinson’s Disease model. Cell Death Differ. 2024;31:1410–21.
Kulathu Y, Komander D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–23.
Kronlage M, Dewenter M, Grosso J, Fleming T, Oehl U, Lehmann LH, et al. O-GlcNAcylation of histone deacetylase 4 protects the diabetic heart from failure. Circulation. 2019;140:580–94.
Li Y, An W, Lu L, Yuan J, Wu D, Yang Q, et al. O-GlcNAc of STING mediates antiviral innate immunity. Cell Commun Signal. 2024;22:157.
He Q, Liu Z, Xia X, Zeng J, Liu Y, Xun J, et al. Amlexanox enforces osteogenic differentiation and bone homeostasis through inhibiting ubiquitin-dependent degradation of β-catenin. Int J Biol Sci. 2024;20:5254–71.
Li M, Tang Q, Liao C, Wang Z, Zhang S, Liang Q, et al. Extracellular vesicles from apoptotic BMSCs ameliorate osteoporosis via transporting regenerative signals. Theranostics. 2024;14:3583–602.
Tsai PY, Lee MS, Jadhav U, Naqvi I, Madha S, Adler A, et al. Adaptation of pancreatic cancer cells to nutrient deprivation is reversible and requires glutamine synthetase stabilization by mTORC1. Proc Natl Acad Sci USA. 2021;118:e2003014118.
Owji AP, Yu K, Kittredge A, Wang J, Zhang Y, Yang T. Bestrophin-2 and glutamine synthetase form a complex for glutamate release. Nature. 2022;611:180–7.
Kim GW, Lee DH, Jeon YH, Yoo J, Kim SY, Lee SW, et al. Glutamine synthetase as a therapeutic target for cancer treatment. Int J Mol Sci. 2021;22:1701.
Sun K, Zhi Y, Ren W, Li S, Zheng J, Gao L, et al. Crosstalk between O-GlcNAcylation and ubiquitination: a novel strategy for overcoming cancer therapeutic resistance. Exp Hematol Oncol. 2024;13:107.
Yan X, Zheng J, Ren W, Li S, Yang S, Zhi K, et al. O-GlcNAcylation: roles and potential therapeutic target for bone pathophysiology. Cell Commun Signal. 2024;22:279.
Ye L, Ding W, Xiao D, Jia Y, Zhao Z, Ao X, et al. O-GlcNAcylation: cellular physiology and therapeutic target for human diseases. MedComm (2020). 2023;4:e456.
Rodriguez-Colman MJ, Dansen TB, Burgering B. FOXO transcription factors as mediators of stress adaptation. Nat Rev Mol Cell Biol. 2024;25:46–64.
Orea-Soufi A, Paik J, Bragança J, Donlon TA, Willcox BJ, Link W. FOXO transcription factors as therapeutic targets in human diseases. Trends Pharm Sci. 2022;43:1070–84.
Calissi G, Lam EW, Link W. Therapeutic strategies targeting FOXO transcription factors. Nat Rev Drug Discov. 2021;20:21–38.
Ning K, Liu S, Yang B, Wang R, Man G, Wang DE, et al. Update on the effects of energy metabolism in bone marrow mesenchymal stem cells differentiation. Mol Metab. 2022;58:101450.
Lee WC, Guntur AR, Long F, Rosen CJ. Energy Metabolism of the Osteoblast: Implications for Osteoporosis. Endocr Rev. 2017;38:255–66.
Zhang C, Li H, Li J, Hu J, Yang K, Tao L. Oxidative stress: A common pathological state in a high-risk population for osteoporosis. Biomed Pharmacother. 2023;163:114834.
Gómez-Puerto MC, Verhagen LP, Braat AK, Lam EW, Coffer PJ, Lorenowicz MJ. Activation of autophagy by FOXO3 regulates redox homeostasis during osteogenic differentiation. Autophagy. 2016;12:1804–16.
Bai H, Fang CW, Shi Y, Zhai S, Jiang A, Li YN, et al. Mitochondria-derived H(2)O(2) triggers liver regeneration via FoxO3a signaling pathway after partial hepatectomy in mice. Cell Death Dis. 2023;14:216.
He Y, Hakvoort TB, Vermeulen JL, Labruyère WT, De Waart DR, Van Der Hel WS, et al. Glutamine synthetase deficiency in murine astrocytes results in neonatal death. Glia. 2010;58:741–54.
Ben Haim L, Schirmer L, Zulji A, Sabeur K, Tiret B, Ribon M, et al. Evidence for glutamine synthetase function in mouse spinal cord oligodendrocytes. Glia. 2021;69:2812–27.
Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80.
Li C, Qiu M, Chang L, Qi J, Zhang L, Ryffel B, et al. The osteoprotective role of USP26 in coordinating bone formation and resorption. Cell Death Differ. 2022;29:1123–36.
Zhang L, Jiao G, Ren S, Zhang X, Li C, Wu W, et al. Exosomes from bone marrow mesenchymal stem cells enhance fracture healing through the promotion of osteogenesis and angiogenesis in a rat model of nonunion. Stem Cell Res Ther. 2020;11:38.
Zhang L, Jiao G, You Y, Li X, Liu J, Sun Z, et al. Arginine methylation of PPP1CA by CARM1 regulates glucose metabolism and affects osteogenic differentiation and osteoclastic differentiation. Clin Transl Med. 2023;13:e1369.
He B, Bie Q, Zhao R, Yan Y, Dong G, Zhang B, et al. Arachidonic acid released by PIK3CA mutant tumor cells triggers malignant transformation of colonic epithelium by inducing chromatin remodeling. Cell Rep. Med. 2024;5:101510.
Acknowledgements
We thank Xiaobei Zhang for her guidance on microscopic image shooting. We thank the Translational Medicine Core Facility of Shandong University, Medical Research Center of Affiliated Hospital of Jining Medical University, Key laboratory of cell and biomedical Technology of Shandong Province, Jining Key Laboratory of Spinal and Spinal Cord Diseases for consultation and instrument availability that supported this work. The Graphical Abstract and Fig. 7A were drawn using Figdraw2.0.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) (8237092097), China Postdoctoral Science Foundation (2024M761146), the Tai Shan Young Scholar Foundation of Shandong Province (tsqn202408394), Natural Science Foundation of Shandong Province (ZR2023MH369, ZR2020QH080), Shandong Postdoctoral Innovation Project (SDCX-ZG-202400011), Postdoctoral Fund of Affiliated Hospital of Jining Medical University (JYFY363226) and Shandong Society of Geriatric Science and Technology Research Project (LKJGG2024W040).
Author information
Authors and Affiliations
Contributions
LZ and CM designed the research and provided financial support. LZ conducted the experiments, analyzed the data, and drafted the manuscript. BQ, YL, XL, and TY participated in both in vitro and in vivo studies. SJ, XG, and SC performed metabolomics and proteomics analyses. ZZ and YL(Yanming Li) provided clinical specimens. GJ performed statistical data analysis. YL(Yangyang Li) carried out tissue specimen sectioning and staining analyses. HZ supervised the research and verified the data. YC, BZ, and GL provided essential resources and technical support for the study. All authors contributed to manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This experimental plan was approved by the Medical Science Research Ethics Committee of the Affiliated Hospital of Jining Medical University (Approval No. 2023-11-B001). The skeletal tissues that were used for the experiment were all discarded after surgical resection, and the collection of the tissues did not interfere with the treatment plan. BMSCs were extracted from the outflow fluid of the medullary cavity during joint replacement surgery. The collection of surgical specimens was informed to the patients and their families and obtained their consent.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, L., Qi, B., Li, Y. et al. GLUL mediates FOXO3 O-GlcNAcylation to regulate the osteogenic differentiation of BMSCs and senile osteoporosis. Cell Death Differ (2025). https://doi.org/10.1038/s41418-025-01543-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41418-025-01543-2
