
Abstract
RhoA (ras homolog family member A) is a small G-protein that transduces intracellular signaling to regulate a broad range of cellular functions such as cell growth, proliferation, migration, and survival. RhoA serves as a proximal downstream effector of numerous G protein-coupled receptors (GPCRs) and is also responsive to various stresses in the heart. Upon its activation, RhoA engages multiple downstream signaling pathways. Rho-associated coiled-coil-containing protein kinase (ROCK) is the first discovered and best characterized effector or RhoA, playing a major role in cytoskeletal arrangement. Many other RhoA effectors have been identified, including myocardin-related transcription factor A (MRTF-A), Yes-associated Protein (YAP) and phospholipase Cε (PLCε) to regulate transcriptional and post-transcriptional processes. The role of RhoA signaling in the heart has been increasingly studied in last decades. It was initially suggested that RhoA signaling pathway is maladaptive in the heart, but more recent studies using cardiac-specific expression or deletion of RhoA have revealed that RhoA activation provides cardioprotection against stress through various mechanisms including the novel role of RhoA in mitochondrial quality control. This review summarizes recent advances in understanding the role of RhoA in the heart and its signaling pathways to prevent progression of heart disease.

Similar content being viewed by others

Facts
-
RhoA functions as a signaling hub that senses a variety of upstream signals such as GPCRs, mechanical force and oxidative stress.
-
RhoA engages various downstream effectors such as ROCK, YAP, MRTF-A and PLCε to regulate cardiac pathophysiology.
-
Activation of RhoA is not inherently pathological but provides protection in the heart against stress, such as pressure-overload, ischemia and aging, but induces fibrosis.
-
Mitochondrion is a critical point of convergence for RhoA-mediated cardioprotection against ischemic stress.
Open questions
-
Does RhoA regulate other mitochondrial quality control mechanisms besides PINK1/Parkin mitophagy to provide cardioprotection?
-
Is general autophagy also regulated by RhoA in the heart?
-
How does RhoA signaling pathway regulate cardiac inflammation?
-
Is RhoA activity protective in diabetic cardiomyopathy and sepsis-induced cardiac dysfunction?
Introduction
Heart failure is a clinical syndrome characterized by structural or functional impairment of ventricular filling or ejection of blood resulting in insufficient perfusion to meet metabolic demands and is often the end-stage manifestation of cardiovascular diseases. Despite extensive research, heart failure remains a leading cause of morbidity and mortality globally [1]. There are two main etiologies of heart failure, ischemic and non-ischemic cardiomyopathy. Coronary artery disease is the most common type of heart disease, causing impaired blood flow to the heart and irreversible ischemic damage (myocardial infarction: MI) [1]. Re-establishing blood flow is critical to salvage the ischemic myocardium, but paradoxically induces and exacerbates tissue injury (ischemia/reperfusion injury: I/R injury) [1]. Nonischemic heart failure is not a well-defined diagnostic entity and includes all causes of decreased heart function other than those caused by blockages in the coronary arteries. Long-lasting hypertension can eventually lead to heart failure and it is established that diabetes is associated with heart failure (diabetic cardiomyopathy) [1]. Although aging is not a disease, it is a significant risk factor for heart failure: The prevalence of heart failure in the young adult population is less than 1%, but this rises to 8.4% after the age of 75 [2].
Rat sarcoma virus (Ras) homolog family member A (RhoA) is a member of the Ras superfamily of small GTPases that plays a central role in a range of biological processes. RhoA was discovered by Madaule and Axel as a first Ras homolog in 1985 [3] and subsequent studies established the paradigm that RhoA functions as a molecular switch for actin reorganization regulating cell shape, migration, and contraction [4,5,6]. Since then RhoA signaling research has expanded and it is now appreciated as a signaling hub for induction of transcriptional and post-transcriptional responses to regulate diverse cellular functions including cell growth, proliferation and survival [7,8,9,10,11]. Extensive studies have been devoted to understanding the role of RhoA signaling pathway in the heart and it has been increasingly clear that RhoA and its downstream effectors play significant roles in the heart. In this review, we summarize regulation of RhoA signaling pathways in the heart and discuss their roles in cardiac pathophysiology.
RhoA signaling pathway
Activation of RhoA
Activation of RhoA is mediated by Rho guanine nucleotide exchange factors (RhoGEFs), which catalyze the exchange of GDP for GTP on RhoA, leading to RhoA activation [4,5,6, 12] (Fig. 1). RhoGEFs comprise approximately 80 members in humans [13, 14]. Termination of RhoA activity is regulated by GTPase activating proteins (GAPs) and Rho GDP dissociation inhibitors (RhoGDIs). RhoGAPs increase the rate of hydrolysis of GTP by RhoA leading to its inactivation and RhoGDIs inhibit the release of GDP to keep RhoA inactive [12].

The heterotrimeric Gα12/13 protein is the most established G-protein that couples receptors to RhoGEFs to activate RhoA. In some systems, Gq protein also stimulates GEFs to activate RhoA. RhoA is also activated in response to mechanical stretch and oxidative stress. Termination of RhoA activity is regulated by GAPs and RhoGDIs. GAPs accelerate the rate of hydrolysis of GTP by RhoA and GDIs inhibit the release of GDP.
GPCRs
The seminal article of Ridley and Hall in 1992 demonstrated that RhoA is activated in response to serum and that a major RhoA activating factor in serum is lysophosphatidic acid (LPA), a G-protein coupled receptor (GPCR) ligand [15]. It was subsequently established that ligands for GPCRs such as sphingosine-1-phosphate (S1P), thrombin, and thromboxane A2 are also efficacious activators of RhoA [7, 16,17,18]. In neonatal rat ventricular myocytes (NRVMs), RhoA has been shown to be activated by S1P, LPA, angiotensin-II (Ang-II), and endothelin-1 (ET-1) but not by adrenoceptor agonists (isoproterenol, phenylephrine) or muscarinic agonist (carbachol) [19,20,21,22,23]. In response to stimulation of GPCRs, the α subunits of heterotrimeric G-proteins (Gαq/11, Gα12/13, Gαs, Gαi) are activated and transduce their signals to downstream effectors. The mechanism by which GPCR signaling activates RhoA was first discovered by the Sternweis laboratory in 1998: The α-subunit of G13 (Gα13) binds to and activates p115-RhoGEF to elicit RhoA activation [24]. Subsequently Gα12 was also shown to regulate p115-RhoGEF, indicating that RhoA activity is regulated by Gα12/13 family proteins (Fig. 1) [25, 26]. Additional work further identified other RhoGEFs such as PDZRhoGEF, LARG and Lbc-RhoGEF regulated by Gα12/13 proteins [13, 14, 27]. In cardiomyocytes, we previously demonstrated that S1P-induced activation of RhoA is mediated by S1P3 receptor subtype-dependent Gα13 activation [21]. Although endothelin receptor-A (ETA) and angiotensin-II receptor-1 (AT1) are generally considered as Gαq/11 coupled receptors, it has been shown that inactivation of Gα13, but not of Gαq/11, abrogates RhoA activation induced by ET-1 and Ang-II in NRVMs and in adult mouse ventricular myocytes (AMVMs) [28]. A recent large-scale functional interaction study analyzed and characterized GPCRs coupling to specific heterotrimeric G-proteins and expanded the list of GPCRs that couple to Gα12/13 proteins such as prostaglandin F receptor and prostaglandin E2 receptor 3 [29]. These GPCRs would thus be regulators of RhoA in the heart. Although phospholipase Cβ (PLCβ) is the principal effector of Gαq signaling, there is evidence that, in some systems, RhoGEFs (p63RhoGEF and TRIO) can be activated by Gαq [30, 31]. The role of these RhoGEFs in cardiomyocytes has not, however, been well established.
Mechanical stress
RhoA signaling represents a major mechanosensitive pathway (Fig.1). Mechanical force applied to cell adhesion receptors (e.g. integrins or cadherins) activates RhoA through activation of RhoGEFs (e.g. LARG, p115RhoGEF and GEF-H1) and inhibition of RhoGAPs (e.g. DLC1 and p190RhoGAP), converting mechanical stretch into biochemical signals [32, 33]. RhoA is activated in NRVMs in response to stretch [23, 34]. Mechanical stress due to hemodynamic overload, one of primary triggers of cardiac remodeling, activates RhoA in the heart, in which integrin β1-mediated activation of LARG has been suggested to play an essential role [27, 28, 35].
Oxidative stress
RhoA is also activated in response to reactive oxygen species (ROS) through oxidation of RhoA [36,37,38]. Direct activation of RhoA by ROS requires two critical cysteine residues, Cys16 and Cys20 [36,37,38]. We have previously shown that RhoA is activated by hydrogen peroxide in cardiomyocytes and suggested that this could be involved in RhoA activation induced by I/R observed in the perfused heart [10, 39, 40].
RhoA effectors
As a downstream of many GPCRs and stress signals, RhoA activation is involved in regulation of diverse biological processes that are mediated by multiple effectors of RhoA. We discuss here RhoA effectors that have been suggested to regulate cardiac pathophysiology (Fig. 2).

A RhoA regulates actin polymerization through ROCK, mDia and PLCε. ROCK phosphorylates and activates LIMK, leading to phosphorylation and inactivation of the actin-severing protein cofilin. mDia accelerates actin nucleation while interacting with actin filament fast-growing ends. PLCε is activated by RhoA and leads to PKD activation. PKD, in turn, phosphorylates and inhibits SSH1L, a cofilin phosphatase. B MRTF-A associates with G-actin and is thus sequestrated in the cytoplasm under resting conditions. Activation of RhoA induces polymerization of G-actin to form F-actin filaments, freeing MRTF-A to translocate to the nucleus. C YAP is activated by RhoA through actin polymerization-dependent inhibition of LATS. YAP inhibition is also mediated by PA produced by PLD and by AMOT binding to YAP. RhoA-induced F-actin accumulation displaces AMOT from YAP and promotes YAP nuclear entry.
Rho-associated coiled-coil-containing kinase (ROCK)
Earlier work on RhoA signaling focused on how activation of RhoA controls actin cytoskeleton to regulate cell shape, migration and contraction and identified ROCK as the first downstream effector of RhoA [41,42,43]. ROCK is a serine/threonine kinase and binding of active RhoA to the Rho-binding domain in ROCK leads to its activation. There are two isoforms of ROCK, ROCK1 and 2, that share overall 65% homology at the amino acid level [44,45,46]. ROCK1 is ubiquitously expressed, whereas ROCK2 is enriched in some tissues such as colon, eyes, kidney, brain, and heart [44, 47]. Among the best studied and physiologically important contractile targets of ROCK is the myosin phosphatase target subunit 1 (MYPT1). MYPT1 is one of the regulatory subunits of myosin-light-chain phosphatase (MLCP) that dephosphorylates the myosin light chain (MLC) [48]. The functional role of MYPT1 in cardiomyocytes has not been known since MYPT1 is most abundantly expressed in smooth muscle cells which are also a component of the heart [49]. ROCK phosphorylates and inhibits MYPT1 which leads to increase in phosphorylation of myosin light chain and thereby increases calcium sensitivity of the contractile elements of smooth muscle [50, 51]. ROCK stabilizes actin filaments through LIM kinases (LIMK) activation. LIMK phosphorylates cofilin and inhibits its actin depolymerization activity (Fig. 2A) [52]. Y-27632 is the first small molecule ROCK inhibitor and shown to lower blood pressure in a rat model of hypertension [51]. Fasudil (HA-1077) was originally reported to inhibit PKA and PKC [53], but later found to be more potent for inhibiting ROCK [54].
Mammalian homolog of diaphanous (mDia)
mDia was identified as a Rho effector that induces actin filaments upon activation in 1997 [55]. Besides its Rho binding domain, mDia has two formin homology domains and belongs to the formin family and there are three mDia isoforms in mammals, mDia1, mDia2 and mDia3 [5, 56]. mDia accelerates actin nucleation while interacting with actin filament fast-growing ends, inducing actin polymerization (Fig. 2A). mDia cooperatively works with ROCK to regulate the formation of actin stress fiber [5, 57].
Phospholipase Cε (PLCε) and protein kinase D (PKD)
PLCε was discovered in Caenorhabditis elegans [58] and the human form of PLCε was cloned in 2001 [59]. PLCε is the only isoform of PLC that has a GTP-RhoA binding insertion and that acts as a direct RhoA effector [60,61,62,63]. In addition, PLCε contains other regulatory domains, two Ras association homology (RA) domains and a CDC25 homology GEF domain that are not found in other PLC isoforms [64]. Because of its regulation by small G-proteins, unlike the other PLCs, PLCε mediates sustained signaling. Specifically, it has been suggested that PLCβ predominantly accounts for acute and PLCε for sustained agonist-stimulated phosphatidylinositol (PI) hydrolysis [63, 65, 66]. Sustained generation of diacylglycerol and activation of its regulated kinases PKC mediates sustained activation of PKD [63, 66]. PKD phosphorylates and inhibits Slingshot 1 L (SSH1L), a cofilin phosphatase, leading to increase in phosphorylated and inactivated cofilin (Fig. 2A) [10, 39].
Myocardin-related transcription factor-A (MRTF-A)
RhoA activation mediated by GPCR agonists regulates gene transcription. An earlier study demonstrated that LPA activates serum response factor (SRF) through RhoA activation and that this is not mediated by the previously described transcriptional coactivator, ternary complex factor (TCF) [67]. The transcriptional coactivator downstream of RhoA was identified as a member of the myocardin family of proteins, MRTF-A (also known as MAL or MKL) [68, 69]. MRTF-A associates with G-actin and is thus sequestrated in the cytoplasm under resting conditions (Fig. 2B). Activation of RhoA induces polymerization of G-actin to form F-actin filaments, freeing MRTF-A to translocate to the nucleus, and this triggers activation of SRF target genes that are involved in a variety of cellular processes such as migration, fibrosis, proliferation and differentiation [68,69,70].
Yes-associated-protein (YAP)
YAP was first discovered as a tyrosine kinase Yes-associated protein in chicken in 1994 and the human and mouse homologs were identified in 1995 [71, 72]. YAP is a transcriptional coactivator and plays an important role in the regulation of organ size, proliferation, and cell survival [73,74,75,76]. The mammalian Hippo pathway composes core kinases, MST1/2 (macrophage stimulating 1 and 2) and LATS1/2 (large tumor suppressor kinase 1 and 2), inhibiting YAP. MST1/2 phosphorylate and activate LATS1/2, which in turn, phosphorylate and inhibit YAP. (Fig. 2C) [73,74,75,76]. When the Hippo pathway is turned off, dephosphorylated YAP translocates into the nucleus and primarily binds to TEAD, a transcription factor, to regulate expression of a wide range of genes [73,74,75,76]. YAP is activated by diverse cellular signals such as mechanical stress, extracellular matrix stiffness and GPCR signaling [8, 77,78,79]. LPA, S1P and thrombin activate YAP through Gα12/13 and RhoA signaling, and this is shown to be mediated by actin polymerization-dependent LATS1/2 inhibition [8, 77]. A recent study further demonstrated that phospholipase-D (PLD) activation and resultant phosphatidic acid (PA) production are involved in RhoA-mediated LATS1/2 inhibition and YAP activation [80]. In addition to the inhibition of LATS1/2, RhoA-mediated actin polymerization results in sequestration of angiomotin (AMOT), an inhibitory binding protein of YAP, thereby promoting YAP nuclear translocation [81, 82].
RhoA in the heart
Basal effect of RhoA in the heart
An earlier study using transgenic mice expressing constitutively active RhoA (L63RhoA) under the control of the cardiac-specific α-myosin heavy chain promoter revealed that cardiac expression of L63RhoA from birth results in the development of dilated cardiomyopathy and bradycardia [83]. Overexpression of L63RhoA in NRVMs for 48 h induces apoptosis through regulation of BAX, an apoptotic BCL2 family protein [84], suggesting that supraphysiological and prolonged activation of RhoA is deleterious in the heart. On the contrary, tetracycline-off inducible expression of L63RhoA (2-5 fold) in the adult heart (from the age of 11 weeks) does not alter cardiac structure, dimensions and contractility nor induce cardiac remodeling such as hypertrophy and fibrosis [10]. Notably, this study also demonstrated that chronic expression of active RhoA in the heart beginning in early development induces cardiac hypertrophy at the age of 4 months and suggests that the induction of hypertrophy in response to RhoA activation reflects signaling events that occur during heart development but not in adulthood [10]. Furthermore, cardiac-specific RhoA KO (RhoAfl/fl x αMHC-Cre; RhoA cKO) mice are born at the expected Mendelian ratio and do not have an overt phenotype [9]. Specifically, cardiac structure, size, and contractility are similar in RhoA cKO and control mice [9]. Thus the basal level of RhoA activity is not indispensable in the heart, and physiological levels of activation of RhoA in the adult mouse heart is not inherently pathological. Instead, accumulating evidence reveals that the modest activation of RhoA is protective rather than maladaptive in the heart under stress conditions, as discussed below.
RhoA and pressure-overload induced heart failure
Cardiac hypertrophy is considered to be an initial adaptive response to hemodynamic stress and necessary for normalizing ventricular wall stress and maintaining cardiac output. However, when stress is sustained, the heart transitions from compensated hypertrophy to dilated heart failure. Transverse aortic constriction (TAC) is widely used to study pressure overload-induced heart failure in mice. RhoA is activated in the heart in response to TAC through stretch-dependent and/or Gα13-mediated signaling mechanisms [27, 28, 35]. Since the discovery of ROCK inhibitors, many studies have focused on examining the role of ROCK in the heart and suggested that ROCK contributes to the development of heart failure induced by pressure overload [85,86,87,88,89,90,91], as discussed in detail later, and this lead to the hypothesis that activation of RhoA/ROCK pathway is maladaptive in the heart. Surprisingly, a study using RhoA cKO mice, however, demonstrated that cardiac deletion of RhoA leads to greater dilation, with thinner ventricular walls and larger chamber dimensions, and aggravates contractile dysfunction induced by TAC, suggesting that RhoA is required for compensatory hypertrophy and important in maintaining cardiac dimensions and contractility against TAC (Fig. 3) [9]. The salutary effects are associated with activation of cardioprotective kinases such as Akt and ERK [9]. However, cardiac deletion of RhoA inhibits cardiac fibrosis induced by TAC, indicating the contribution of cardiomyocyte RhoA to the development of fibrosis in the heart [9]. These results suggest that cardiac hypertrophy and fibrosis are independently regulated processes and that it is important to delineate the downstream effectors of RhoA in the heart.

RhoA is activated in response to pressure overload and contributes to the development of compensatory hypertrophy and protects the heart against cardiac dilation and contractile dysfunction. The protective effects of RhoA are mediated by YAP, ROCK1, and MRTF-A-mDia pathways. However, RhoA facilitates cardiac fibrosis possibly through activation of ROCK2 and MRTF-A.
YAP activity is increased in the mouse heart in response to TAC as well as in human hypertrophic cardiomyopathy [35, 92] and this response has been suggested to be mediated by RhoA [35]. Cardiac-specific heterozygous YAP KO mice exhibit no basal phenotype, with respect to mouse survival, heart size, or contractile function [93], but show attenuated compensatory hypertrophy and exaggerated cardiac dysfunction induced by TAC [35, 94], demonstrating the protective role of YAP. Akt activation through downregulation of PTEN and transcriptional upregulation of aerobic glycolysis are demonstrated to be involved in YAP-mediated cardioprotection against TAC (Fig. 3) [35, 94]. Pharmacological inhibition of ROCK has been shown to attenuate cardiac hypertrophy, dilation, contractile dysfunction and fibrosis induced by pressure overload [85,86,87,88,89,90,91], suggesting that systemic inhibition of ROCKs (ROCK1 and ROCK2) is overall cardioprotective. However, recent seminal studies have demonstrated that ROCK1 and ROCK2 are functionally different, playing the opposing roles in cardiomyocytes. Specifically, TAC-induced hypertrophy, fibrosis, cardiac dilation and contractile dysfunction are augmented in cardiac-specific ROCK1 KO (ROCK1 cKO) mice, whereas these responses are attenuated in ROCK2 cKO mice [95, 96]. Decreased hypertrophy and fibrosis were also observed in ROCK2 cKO mice subjected to Ang-II infusion [96]. These results clearly indicate that ROCK1 protects but ROCK2 jeopardizes heart failure induced by pressure overload and that ROCK1 in cardiomyocytes could contribute to the cardioprotective effects of RhoA. Mechanistically, ROCK1 exerts anti-oxidative effects through inhibition of expression of cyclophilin A (CyPA) and basigin (Bsg), both of which augment oxidative stress [95]. Given that ROCK1 and ROCK2 share 65% homology in their amino acid sequence and 92% homology in their kinase domain [45], it is intriguing that ROCK1 and ROCK2 can play such opposite roles in the heart. Earlier studies in non-cardiomyocytes reported the distinct functions of ROCK isoforms [97, 98]. For instance, a study using mouse embryonic fibroblasts (MEFs) derived from ROCK1 KO and ROCK2 KO mice demonstrates that phosphorylation of myosin light chain 2 is mediated by ROCK1 and ROCK2, while phosphorylation of cofilin is regulated mainly by ROCK2 [97]. Differential substrate specificity of ROCK isoforms in the heart has, however, not been determined. The activation kinetics of the isoforms in response to stress have not also been analyzed in the heart. Thus further studies using genetically modified mouse lines will be required to understand the underlying mechanisms of the functional differences of ROCK1 and ROCK2 in the stressed heart.
A recent study suggested that mDia1 is also involved in the development of compensatory hypertrophy in response to pressure overload through mechanotransduction-dependent activation of ERK, focal adhesion kinase (FAK), and MRTF-A, preventing the progression to heart failure [56]. MRTF-A is, however, also suggested to be responsible for enhanced fibrosis by RhoA in the TAC heart [9, 88]. Thus MRTF-A activation appears to play both protective and deleterious roles in the heart under pressure-overload stress. It is worth to note that ROCK regulates diverse cellular functions in many types of cells, not only cardiac pathophysiology, but also contractility of vascular smooth muscle cells, endothelial barrier function, myofibroblast differentiation and regulation of immune cell differentiation and function [5, 99, 100]. This might be, at least in part, responsible for conflicting data on the effect of global vs. cardiac-specific inhibition of ROCK on cardiac pathophysiology.
Together, these studies suggest that RhoA activation in cardiomyocytes plays a significant role in the development of compensatory hypertrophy induced by pressure overload, maintaining cardiac dimension and contractility. YAP, ROCK1, and mDia1 in cardiomyocytes are likely to contribute to the RhoA-mediated salutary effects (Fig. 3). Activation of RhoA in cardiomyocytes, however, increases fibrotic responses induced by TAC, which might be regulated by ROCK2/MRTF-A pathway. Further studies will be required to fully determine the involvement of these molecules in RhoA-mediated responses in the TAC heart. It would also be of importance to examine the role of RhoA in other pressure-overload models (e.g., angiotensin-II infusion model), using genetically modified mouse lines.
RhoA and Ischemic Injury
Adult cardiomyocytes are highly differentiated cells and their regenerative capacity is limited thus cell death plays a critical role in the development of heart failure induced by ischemic stress [101,102,103]. Mitochondria are the main site of ATP generation in cardiomyocytes, but in response to stress, mitochondria become damaged and play a major role in cell death. Mitochondrial apoptosis is induced by release of apoptotic factors such as cytochrome c from mitochondria and regulated by apoptotic BCL-2 family proteins, such as BAK and BAX. This process is counteracted by anti-apoptotic BCL-2 proteins, such as BCL-2 and BCL-xL, but enhanced by BH3-only proteins such as BAD and t-BID [101,102,103]. The critical event in the induction of mitochondrial necrosis is the opening of the mitochondrial permeability transition pore (mPTP), a high-conductance and non-specific channel spanning both outer and inner mitochondrial membranes. mPTP is activated by mitochondrial Ca2+ overload and increased ROS levels, leading to dissipation of the mitochondrial membrane potential, swelling and eventual rupture of the mitochondrial membranes [101,102,103].
RhoA is activated in response to ischemic stress in the heart [10, 40, 104] and this response could occur through activation of GPCRs in response to released mediators and/or as a direct result of oxidative stress. Studies in NRVMs demonstrated that modest levels of RhoA activation inhibit cell death induced by oxidative stress [23, 105]. Importantly, conditional, and cardiac-specific L63RhoA TG mice show reduced infarct size after I/R in vivo, while cardiac-specific RhoA KO mice show increased I/R damage in the Langendorff heart [10]. These studies strongly suggest that RhoA activation in cardiomyocytes confers protection against I/R (Fig. 4). Activation of RhoA has also been demonstrated to contribute to the protective effects of GPCR agonists against I/R. For instance, S1P, a lysophospholipid that is generated and released at sites of tissue injury, activates RhoA to provide cardioprotection against I/R [10, 19, 21, 39, 106, 107]. Our recent work further demonstrated that AAV9-mediated expression of L63RhoA in the mouse heart reduces infarct size induced by ischemia [11], thus activation of RhoA in cardiomyocytes is protective against I/R and MI.

RhoA is activated by ischemic stress and engages multiple downstream molecules such as YAP, ROCK, MRTF-A and PLCε to activate protective kinase Akt, ERK and PKD, leading to inhibition of mitochondrial death pathways. RhoA also increases mitochondrial fission through regulation of Drp1 and enhances mitophagy through inhibition of PINK1 protein degradation to control mitochondrial quality in the ischemic heart.
It has been demonstrated that RhoA engages multiple downstream effectors to regulate cell survival. Akt signaling inhibits the mPTP opening as well as BAD activation to prevent mitochondrial death pathways [103, 108,109,110] and there are some evidences suggesting that activation of Akt contributes to the RhoA-mediated cardioprotection. Mechanistically, ROCK-dependent activation of FAK is suggested to link RhoA signal to Akt activation [23] and ROCK-MRTF-A dependent expression of CCN1, a matricellular protein, is also suggested to lead to Akt activation through the integrin signaling pathway [19]. A study using cardiac-specific heterozygous deletion of YAP demonstrated that Akt activity is also regulated by YAP in the I/R heart, contributing to YAP-mediated protection [93]. The protective effect of YAP was supported by subsequent studies demonstrating that cardiac-specific activation of YAP ameliorates cardiac damage after I/R and MI, in which cardiomyocyte proliferation and inhibition of inflammatory responses are suggested to contribute to the YAP-mediated cardioprotection [111, 112]. Global inhibition of ROCK has been established to reduce infarct size in the I/R heart [91, 104, 113,114,115,116,117], suggesting the deleterious role of ROCK. As discussed above, however, the site of inhibition of ROCK signaling responsible for its salutary effects may not be the cardiomyocyte and ROCK1 and ROCK2 play the opposing roles in the heart subjected to pressure overload. It would be of importance to determine if ischemic injury is also differently regulated by ROCK1 and ROCK2. PKD is another recently identified downstream molecule involved in RhoA-mediated cardioprotection. RhoA activates PKD through PLCε, and PKD in turn phosphorylates and inhibits SSH1L, leading to inactivation of cofilin (Fig.2A). This results in prevention of cofilin-mediated active BAX translocation to mitochondria and resultant cell death (Fig. 4) [10, 39].
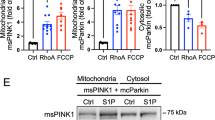
In addition to inhibition of mitochondrial death pathways, mitochondrial integrity is also preserved by mitochondrial quality control mechanisms. Mitochondria-specific autophagy (mitophagy) removes damaged mitochondria through lysosomal degradation, inhibiting the accumulation of toxic mitochondria and resultant cell death [118,119,120,121]. One of the best-established mechanisms of tagging damaged mitochondria for autophagic removal is the mitochondrial membrane-depolarization-dependent PTEN-induced kinase 1 (PINK1)/Parkin pathway. Dissipation of the mitochondrial membrane potential induced by stress stabilizes and accumulates PINK1 at the mitochondrial outer membrane. PINK1, in turn, recruits Parkin, an E3 ubiquitin ligase, which ubiquitinates and tags damaged mitochondria for removal [118, 119]. Although PINK1/Parkin-mediated mitophagy is generally protective, full depolarization of the mitochondrial membrane potential occurs just before cell death, and additional intracellular mechanisms that enhance the PINK1/Parkin pathway at early stages would be important for prolonging cell survival but such a mechanism has not been fully explored. We recently discovered that RhoA signaling increases mitophagy by stabilizing PINK1 protein at mitochondria without concomitantly diminishing mitochondrial membrane potential and provides cardioprotection against MI (Fig. 4) [11, 122]. Regulation of PINK1/Parkin-mediated mitophagy by RhoA was further confirmed by a recent study [123]. Mechanistically, we demonstrated that RhoA, upon its activation, translocates to mitochondria and interacts with PINK1 and that the molecular interaction of RhoA with PINK1 prevents PINK1 degradation, ensuing sequelae of events culminating in mitophagy. Interestingly, RhoA translocation to mitochondria is regulated by PKD [11, 122]. Although the precise mechanism by which RhoA prevents PINK1 degradation at mitochondria remains to be elucidated, our results suggest that receptor agonists and interventions that activate RhoA represent a novel means of stabilizing PINK1 to stimulate mitophagy without compromising mitochondrial functions to promote cell survival. Activation of RhoA-ROCK pathway leads to phosphorylation of Drp1 and its translocation to mitochondria to induce mitochondrial fission [105], a cellular process that segregates damaged mitochondria from intact mitochondrial network and facilitates autophagic removal of damaged mitochondria (Fig. 4) [124]. In addition to mitochondrial fission, recent studies have revealed that Drp1 can directly regulate ULK1-Rab9-dependent alternative mitophagy in the heart [125]. Whether RhoA regulates alternative mitophagy to protect the heart is an interesting unanswered question.
Taken together, these studies suggest that mitochondria are a convergence point for RhoA-mediated cellular protection, with RhoA signaling through multiple pathways to preserve mitochondrial integrity by preventing mitochondrial death pathways and by facilitating removal of damaged mitochondria during ischemic stress.
RhoA and other heart diseases
Aging is a significant risk factor for heart failure and results in progressive deterioration in the structure and function of the heart [2]. Dysfunctional proteins and mitochondria are accumulated in the aging heart and enhancing autophagic clearance exerts beneficial effects [126]. A recent study using RhoA cKO mice demonstrated that RhoA-mediated mitophagy provides cardioprotection against aging-induced adverse cardiac remodeling, including cardiac hypertrophy, fibrosis, dilation, and contractile dysfunction [123]. ROCK-dependent transcriptional upregulation of Parkin through inhibition of N-Myc, a transcriptional repressor of Parkin, is suggested to contribute to RhoA-mediated mitophagy in the aging heart (Fig. 5) [127]. An earlier study also noted that Parkin mRNA levels are decreased in ROCK1 cKO and in ROCK2 cKO mice [95]. YAP has also been demonstrated to regulate Parkin gene expression to protect the heart against doxorubicin-induced cardiotoxicity [128]. Thus it appears that RhoA regulates PINK1/Parkin-mediated mitophagy through early regulation of PINK1 protein stabilization [11, 122] and late transcriptional upregulation of Parkin [95, 123, 128] to ensure cell survival against acute and sustained stress.

PINK1 protein stability is regulated by the molecular interaction of RhoA with PINK1. Parkin expression is transcriptionally regulated by ROCK through inhibition of N-Myc as well as YAP.
There is some evidence that RhoA signaling can regulate general autophagy in the heart.
For instance, inducible ROCK1 and ROCK2 double cKO mice show no overt basal phenotype but decreased fibrosis by aging and this fibrotic effect of ROCK is suggested to be attributed to inhibition of autophagy [129]. However, the inhibitory effect of ROCK on autophagy was not evident when ROCK1 or ROCK2 was individually deleted from adult cardiomyocytes [129]. The role of RhoA/ROCK signaling in autophagy in the heart requires further studies.
Diabetic cardiomyopathy describes diabetes-associated changes in the structure and function of the myocardium that occur independently of a recognized cause such as hypertension and coronary artery disease. RhoA expression and activity in the heart has been demonstrated to be increased in streptozotocin-diabetic rats [130,131,132]. However, whether this response is adaptive or deleterious has not been determined. Pharmacological studies have shown that ROCK inhibition ameliorates diabetes-induced cardiac dysfunction [131,132,133] and a study using heterozygous ROCK2 KO mice demonstrated that ROCK2 phosphorylates ryanodine receptors, impairing cardiac Ca2+ homeostasis [134]. Inhibition of MST1, an upstream negative regulator of YAP, alleviates diabetic cardiomyopathy [135,136,137], implying the possibility that YAP plays a protective role in diabetic hearts. MST1, however, not only inhibits YAP but also induces apoptosis through inhibition of BCL-xL [138] and it would be of importance to further elucidate the roles of RhoA and its downstream effectors in diabetic cardiomyopathy.
Sepsis is a systemic inflammatory response triggered by bacterial infection. Cardiac dysfunction is an important component of multi-organ failure and resultant mortality induced by severe sepsis. Several studies have shown that ROCK inhibitors provide salutary effects in the heart against septic shock [130, 139, 140]. In contrast to ROCK, YAP activity has been shown to be protective against lipopolysaccharide (LPS)-induced cardiac dysfunction [141, 142]. YAP activation is also shown to suppress inflammatory responses in NRVMs treated with LPS [111] and the anti-inflammatory effects of YAP in the heart have also been reported in ischemic heart models [111, 143, 144]. On the contrary, YAP has been reported to enhance inflammatory responses in fibroblast and macrophages [145, 146].
Conclusion
It was originally discovered that RhoA plays a major role in regulation of cytoskeletal function in smooth muscle and other cells and that ROCK is the main downstream effector of RhoA. It is now evident that RhoA can regulate diverse cellular functions and plays an important role in the heart in response to stress. In addition to ROCK, various signaling molecules such as YAP, MRTF-A and PLCε have been discovered to play an important role in RhoA-mediated transcriptional and post-transcriptional regulation. Recent evidence demonstrates that RhoA in cardiomyocytes prevents the development of heart failure induced by stress, such as pressure overload, ischemia and aging. RhoA-mediated transcriptional regulation contributes to the development of compensatory hypertrophy but also enhances fibrotic responses in the heart. The protective effect of RhoA is derived, at least in part, from preservation of mitochondrial integrity through inhibition of mitochondrial cell death pathways as well as through regulation of mitochondrial quality control.
Although our understanding of the role of RhoA signaling pathway in the heart has expanded greatly in recent years, there are unsolved questions that warrant future research. Further studies of the role of RhoA signaling pathway in regulation of mitochondrial quality control mechanisms including PINK1/Parkin independent mitophagy, mitochondrial dynamics and mitochondrial biogenesis will be required. General autophagy also plays a critical role in maintaining cardiac homeostasis. There is, however, only limited data available with regard to regulation of general autophagy by RhoA in the in vivo heart. Cardiac inflammation is evident in the failing heart [147] and now considered to be a driver of the adverse cardiac remodeling [148, 149], but the role of RhoA signaling in regulation of inflammation in the heart is still emerging and many more research will be required to determine how RhoA signaling regulates cardiac inflammation through regulation of inflammatory gene expression and through regulation of cell survival in the heart.
RhoA controls many different biological functions in different types of cells, thus a deeper understanding of the intracellular signaling molecules regulated by RhoA in cardiac cells as well as in non-cardiac cells is obligatory for identifying precise targets for modulating RhoA signaling pathway to provide a new therapeutic strategy for prevention or treatment of heart disease.
References
Tanai E, Frantz S. Pathophysiology of heart failure. Compr Physiol. 2015;6:187–214.
Triposkiadis F, Xanthopoulos A, Butler J. Cardiovascular aging and heart failure: JACC review topic of the week. J Am Coll Cardiol. 2019;74:804–13.
Madaule P, Axel R. A novel ras-related gene family. Cell. 1985;41:31–40.
Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–35.
Narumiya S, Thumkeo D. Rho signaling research: history, current status and future directions. FEBS Lett. 2018;592:1763–76.
Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem. 1999;68:459–86.
Yu OM, Brown JH. G protein-coupled receptor and RhoA-stimulated transcriptional responses: links to inflammation, differentiation, and cell proliferation. Mol Pharm. 2015;88:171–80.
Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–91.
Lauriol J, Keith K, Jaffre F, Couvillon A, Saci A, Goonasekera SA, et al. RhoA signaling in cardiomyocytes protects against stress-induced heart failure but facilitates cardiac fibrosis. Sci Signal. 2014;7:ra100.
Xiang SY, Vanhoutte D, Del Re DP, Purcell NH, Ling H, Banerjee I, et al. RhoA protects the mouse heart against ischemia/reperfusion injury. J Clin Investig. 2011;121:3269–76.
Tu M, Tan VP, Yu JD, Tripathi R, Bigham Z, Barlow M, et al. RhoA signaling increases mitophagy and protects cardiomyocytes against ischemia by stabilizing PINK1 protein and recruiting Parkin to mitochondria. Cell Death Differ. 2022;29:2472–86.
Mosaddeghzadeh N, Ahmadian MR. The RHO family GTPases: mechanisms of regulation and signaling. Cells. 2021;10:1831.
Eckenstaler R, Hauke M, Benndorf RA. A current overview of RhoA, RhoB, and RhoC functions in vascular biology and pathology. Biochem Pharm. 2022;206:115321.
Cervantes-Villagrana RD, Garcia-Jimenez I, Vazquez-Prado J. Guanine nucleotide exchange factors for Rho GTPases (RhoGEFs) as oncogenic effectors and strategic therapeutic targets in metastatic cancer. Cell Signal. 2023;109:110749.
Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–99.
Siehler S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br J Pharmacol. 2009;158:41–9.
Post GR, Collins LR, Kennedy ED, Moskowitz SA, Aragay AM, Goldstein D, et al. Coupling of the thrombin receptor to G12 may account for selective effects of thrombin on gene expression and DNA synthesis in 1321N1 astrocytoma cells. Mol Biol Cell. 1996;7:1679–90.
Ishii I, Friedman B, Ye X, Kawamura S, McGiffert C, Contos JJ, et al. Selective loss of sphingosine 1-phosphate signaling with no obvious phenotypic abnormality in mice lacking its G protein-coupled receptor, LP(B3)/EDG-3. J Biol Chem. 2001;276:33697–704.
Zhao X, Ding EY, Yu OM, Xiang SY, Tan-Sah VP, Yung BS, et al. Induction of the matricellular protein CCN1 through RhoA and MRTF-A contributes to ischemic cardioprotection. J Mol Cell Cardiol. 2014;75:152–61.
Aoki H, Izumo S, Sadoshima J. Angiotensin II activates RhoA in cardiac myocytes: a critical role of RhoA in angiotensin II-induced premyofibril formation. Circ Res. 1998;82:666–76.
Yung BS, Brand CS, Xiang SY, Gray CB, Means CK, Rosen H, et al. Selective coupling of the S1P3 receptor subtype to S1P-mediated RhoA activation and cardioprotection. J Mol Cell Cardiol. 2017;103:1–10.
Zeidan A, Gan XT, Thomas A, Karmazyn M. Prevention of RhoA activation and cofilin-mediated actin polymerization mediates the antihypertrophic effect of adenosine receptor agonists in angiotensin II- and endothelin-1-treated cardiomyocytes. Mol Cell Biochem. 2014;385:239–48.
Del Re DP, Miyamoto S, Brown JH. Focal adhesion kinase as a RhoA-activable signaling scaffold mediating Akt activation and cardiomyocyte protection. J Biol Chem. 2008;283:35622–9.
Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, et al. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science. 1998;280:2112–4.
Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, et al. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science. 1998;280:2109–11.
Tanabe S, Kreutz B, Suzuki N, Kozasa T. Regulation of RGS-RhoGEFs by Galpha12 and Galpha13 proteins. Methods Enzymol. 2004;390:285–94.
Takefuji M, Kruger M, Sivaraj KK, Kaibuchi K, Offermanns S, Wettschureck N. RhoGEF12 controls cardiac remodeling by integrating G protein- and integrin-dependent signaling cascades. J Exp Med. 2013;210:665–73.
Takefuji M, Wirth A, Lukasova M, Takefuji S, Boettger T, Braun T, et al. G(13)-mediated signaling pathway is required for pressure overload-induced cardiac remodeling and heart failure. Circulation. 2012;126:1972–82.
Inoue A, Raimondi F, Kadji FMN, Singh G, Kishi T, Uwamizu A, et al. Illuminating G-protein-coupling selectivity of GPCRs. Cell. 2019;177:1933–47 e25.
Shankaranarayanan A, Boguth CA, Lutz S, Vettel C, Uhlemann F, Aittaleb M, et al. Galpha q allosterically activates and relieves autoinhibition of p63RhoGEF. Cell Signal. 2010;22:1114–23.
Vaque JP, Dorsam RT, Feng X, Iglesias-Bartolome R, Forsthoefel DJ, Chen Q, et al. A genome-wide RNAi screen reveals a Trio-regulated Rho GTPase circuitry transducing mitogenic signals initiated by G protein-coupled receptors. Mol Cell. 2013;49:94–108.
Burridge K, Monaghan-Benson E, Graham DM. Mechanotransduction: from the cell surface to the nucleus via RhoA. Philos Trans R Soc Lond B Biol Sci. 2019;374:20180229.
Abiko H, Fujiwara S, Ohashi K, Hiatari R, Mashiko T, Sakamoto N, et al. Rho guanine nucleotide exchange factors involved in cyclic-stretch-induced reorientation of vascular endothelial cells. J Cell Sci. 2015;128:1683–95.
Kawamura S, Miyamoto S, Brown JH. Initiation and transduction of stretch-induced RhoA and Rac1 activation through caveolae: cytoskeletal regulation of ERK translocation. J Biol Chem. 2003;278:31111–7.
Byun J, Del Re DP, Zhai P, Ikeda S, Shirakabe A, Mizushima W, et al. Yes-associated protein (YAP) mediates adaptive cardiac hypertrophy in response to pressure overload. J Biol Chem. 2019;294:3603–17.
Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One. 2009;4:e8045.
Heo J, Campbell SL. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J Biol Chem. 2005;280:31003–10.
Hurst M, McGarry DJ, Olson MF. Rho GTPases: Non-canonical regulation by cysteine oxidation. Bioessays. 2022;44:e2100152.
Xiang SY, Ouyang K, Yung BS, Miyamoto S, Smrcka AV, Chen J, et al. PLCepsilon, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci Signal. 2013;6:ra108.
Miyamoto S, Del Re DP, Xiang SY, Zhao X, Florholmen G, Brown JH. Revisited and revised: is RhoA always a villain in cardiac pathophysiology? J Cardiovasc Transl Res. 2010;3:330–43.
Leung T, Chen XQ, Manser E, Lim L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol Cell Biol. 1996;16:5313–27.
Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, et al. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 1996;15:1885–93.
Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996;15:2208–16.
Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392:189–93.
Shimizu T, Liao JK. Rho kinases and cardiac remodeling. Circ J. 2016;80:1491–8.
Wei L, Surma M, Shi S, Lambert-Cheatham N, Shi J. Novel insights into the roles of Rho kinase in cancer. Arch Immunol Ther Exp (Warsz). 2016;64:259–78.
Julian L, Olson MF. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases. 2014;5:e29846.
Pfitzer G. Invited review: regulation of myosin phosphorylation in smooth muscle. J Appl Physiol. 2001;91:497–503.
Oslin K, Reho JJ, Lu Y, Khanal S, Kenchegowda D, Prior SJ, et al. Tissue-specific expression of myosin phosphatase subunits and isoforms in smooth muscle of mice and humans. Am J Physiol Regul Integr Comp Physiol. 2022;322:R281–R91.
Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 1996;273:245–8.
Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–4.
Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–8.
Asano T, Suzuki T, Tsuchiya M, Satoh S, Ikegaki I, Shibuya M, et al. Vasodilator actions of HA1077 in vitro and in vivo putatively mediated by the inhibition of protein kinase. Br J Pharmacol. 1989;98:1091–100.
Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105.
nabe N, Madaule P, Reid T, Ishizaki T, Watanabe G, Kakizuka A, et al. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997;16:3044–56.
Abe I, Terabayashi T, Hanada K, Kondo H, Teshima Y, Ishii Y, et al. Disruption of actin dynamics regulated by Rho effector mDia1 attenuates pressure overload-induced cardiac hypertrophic responses and exacerbates dysfunction. Cardiovasc Res. 2021;117:1103–17.
Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–43.
Shibatohge M, Kariya K, Liao Y, Hu CD, Watari Y, Goshima M, et al. Identification of PLC210, a Caenorhabditis elegans phospholipase C, as a putative effector of Ras. J Biol Chem. 1998;273:6218–22.
Lopez I, Mak EC, Ding J, Hamm HE, Lomasney JW. A novel bifunctional phospholipase c that is regulated by Galpha 12 and stimulates the Ras/mitogen-activated protein kinase pathway. J Biol Chem. 2001;276:2758–65.
Hains MD, Wing MR, Maddileti S, Siderovski DP, Harden TK. Galpha12/13- and rho-dependent activation of phospholipase C-epsilon by lysophosphatidic acid and thrombin receptors. Mol Pharm. 2006;69:2068–75.
Seifert JP, Wing MR, Snyder JT, Gershburg S, Sondek J, Harden TK. RhoA activates purified phospholipase C-epsilon by a guanine nucleotide-dependent mechanism. J Biol Chem. 2004;279:47992–7.
Wing MR, Snyder JT, Sondek J, Harden TK. Direct activation of phospholipase C-epsilon by Rho. J Biol Chem. 2003;278:41253–8.
Smrcka AV, Brown JH, Holz GG. Role of phospholipase Cepsilon in physiological phosphoinositide signaling networks. Cell Signal. 2012;24:1333–43.
Kelley GG, Reks SE, Smrcka AV. Hormonal regulation of phospholipase Cepsilon through distinct and overlapping pathways involving G12 and Ras family G-proteins. Biochem J. 2004;378:129–39.
Kelley GG, Kaproth-Joslin KA, Reks SE, Smrcka AV, Wojcikiewicz RJ. G-protein-coupled receptor agonists activate endogenous phospholipase Cepsilon and phospholipase Cbeta3 in a temporally distinct manner. J Biol Chem. 2006;281:2639–48.
Dusaban SS, Purcell NH, Rockenstein E, Masliah E, Cho MK, Smrcka AV, et al. Phospholipase C epsilon links G protein-coupled receptor activation to inflammatory astrocytic responses. Proc Natl Acad Sci USA. 2013;110:3609–14.
Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 1995;81:1159–70.
Cen B, Selvaraj A, Burgess RC, Hitzler JK, Ma Z, Morris SW, et al. Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of SRF target genes. Mol Cell Biol. 2003;23:6597–608.
Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–42.
Guettler S, Vartiainen MK, Miralles F, Larijani B, Treisman R. RPEL motifs link the serum response factor cofactor MAL but not myocardin to Rho signaling via actin binding. Mol Cell Biol. 2008;28:732–42.
Sudol M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene 1994;9:2145–52.
Sudol M, Bork P, Einbond A, Kastury K, Druck T, Negrini M, et al. Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J Biol Chem. 1995;270:14733–41.
Ma S, Meng Z, Chen R, Guan KL. The Hippo pathway: biology and pathophysiology. Annu Rev Biochem. 2019;88:577–604.
Wang J, Liu S, Heallen T, Martin JF. The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nat Rev Cardiol. 2018;15:672–84.
Ikeda S, Sadoshima J. Regulation of myocardial cell growth and death by the Hippo pathway. Circ J. 2016;80:1511–9.
Meng F, Xie B, Martin JF. Targeting the Hippo pathway in heart repair. Cardiovasc Res. 2022;118:2402–14.
Mo JS, Yu FX, Gong R, Brown JH, Guan KL. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev. 2012;26:2138–43.
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–83.
Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development. 2011;138:3907–14.
Han H, Qi R, Zhou JJ, Ta AP, Yang B, Nakaoka HJ, et al. Regulation of the Hippo Pathway by Phosphatidic Acid-Mediated Lipid-Protein Interaction. Mol Cell. 2018;72:328–40 e8.
Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, et al. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011;25:51–63.
Mana-Capelli S, Paramasivam M, Dutta S, McCollum D. Angiomotins link F-actin architecture to Hippo pathway signaling. Mol Biol Cell. 2014;25:1676–85.
Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW 2nd, Ross J Jr, et al. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Investig 1999;103:1627–34.
Del Re DP, Miyamoto S, Brown JH. RhoA/Rho kinase up-regulate Bax to activate a mitochondrial death pathway and induce cardiomyocyte apoptosis. J Biol Chem. 2007;282:8069–78.
Phrommintikul A, Tran L, Kompa A, Wang B, Adrahtas A, Cantwell D, et al. Effects of a Rho kinase inhibitor on pressure overload induced cardiac hypertrophy and associated diastolic dysfunction. Am J Physiol Heart Circ Physiol. 2008;294:H1804–14.
Higashi M, Shimokawa H, Hattori T, Hiroki J, Mukai Y, Morikawa K, et al. Long-term inhibition of Rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: effect on endothelial NAD(P)H oxidase system. Circ Res. 2003;93:767–75.
Kobayashi N, Horinaka S, Mita S, Nakano S, Honda T, Yoshida K, et al. Critical role of Rho-kinase pathway for cardiac performance and remodeling in failing rat hearts. Cardiovasc Res. 2002;55:757–67.
Yu B, Sladojevic N, Blair JE, Liao JK. Targeting Rho-associated coiled-coil forming protein kinase (ROCK) in cardiovascular fibrosis and stiffening. Expert Opin Ther Targets. 2020;24:47–62.
Satoh S, Ueda Y, Koyanagi M, Kadokami T, Sugano M, Yoshikawa Y, et al. Chronic inhibition of Rho kinase blunts the process of left ventricular hypertrophy leading to cardiac contractile dysfunction in hypertension-induced heart failure. J Mol Cell Cardiol. 2003;35:59–70.
Fukui S, Fukumoto Y, Suzuki J, Saji K, Nawata J, Tawara S, et al. Long-term inhibition of Rho-kinase ameliorates diastolic heart failure in hypertensive rats. J Cardiovasc Pharmacol. 2008;51:317–26.
Li Q, Xu Y, Li X, Guo Y, Liu G. Inhibition of Rho-kinase ameliorates myocardial remodeling and fibrosis in pressure overload and myocardial infarction: role of TGF-beta1-TAK1. Toxicol Lett. 2012;211:91–7.
Wang P, Mao B, Luo W, Wei B, Jiang W, Liu D, et al. The alteration of Hippo/YAP signaling in the development of hypertrophic cardiomyopathy. Basic Res Cardiol. 2014;109:435.
Del Re DP, Yang Y, Nakano N, Cho J, Zhai P, Yamamoto T, et al. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J Biol Chem. 2013;288:3977–88.
Kashihara T, Mukai R, Oka SI, Zhai P, Nakada Y, Yang Z, et al. YAP mediates compensatory cardiac hypertrophy through aerobic glycolysis in response to pressure overload. J Clin Invest. 2022;132:e150595.
Sunamura S, Satoh K, Kurosawa R, Ohtsuki T, Kikuchi N, Elias-Al-Mamun M, et al. Different roles of myocardial ROCK1 and ROCK2 in cardiac dysfunction and postcapillary pulmonary hypertension in mice. Proc Natl Acad Sci USA. 2018;115:E7129–E38.
Okamoto R, Li Y, Noma K, Hiroi Y, Liu PY, Taniguchi M, et al. FHL2 prevents cardiac hypertrophy in mice with cardiac-specific deletion of ROCK2. FASEB J. 2013;27:1439–49.
Shi J, Wu X, Surma M, Vemula S, Zhang L, Yang Y, et al. Distinct roles for ROCK1 and ROCK2 in the regulation of cell detachment. Cell Death Dis. 2013;4:e483.
Newell-Litwa KA, Badoual M, Asmussen H, Patel H, Whitmore L, Horwitz AR. ROCK1 and 2 differentially regulate actomyosin organization to drive cell and synaptic polarity. J Cell Biol. 2015;210:225–42.
Shimokawa H, Sunamura S, Satoh K. RhoA/Rho-kinase in the cardiovascular system. Circ Res. 2016;118:352–66.
Bros M, Haas K, Moll L, Grabbe S. RhoA as a key regulator of innate and adaptive immunity. Cells. 2019;8:733.
Jia XF, Liang FG, Kitsis RN. Multiple cell death programs contribute to myocardial infarction. Circ Res. 2021;129:397–9.
Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. 2019;99:1765–817.
Yu JD, Miyamoto S. Molecular signaling to preserve mitochondrial integrity against ischemic stress in the heart: rescue or remove mitochondria in danger. Cells. 2021;10:3330.
Bao W, Hu E, Tao L, Boyce R, Mirabile R, Thudium DT, et al. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res. 2004;61:548–58.
Brand CS, Tan VP, Brown JH, Miyamoto S. RhoA regulates Drp1 mediated mitochondrial fission through ROCK to protect cardiomyocytes. Cell Signal. 2018;50:48–57.
Sattler KJ, Elbasan S, Keul P, Elter-Schulz M, Bode C, Graler MH, et al. Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res Cardiol. 2010;105:821–32.
Theilmeier G, Schmidt C, Herrmann J, Keul P, Schafers M, Herrgott I, et al. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation. 2006;114:1403–9.
Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15:521–9.
Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem. 2013;288:23798–806.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41.
Chen J, Ma Q, King JS, Sun Y, Xu B, Zhang X, et al. aYAP modRNA reduces cardiac inflammation and hypertrophy in a murine ischemia-reperfusion model. Life Sci Alliance. 2020;3:e201900424.
Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res. 2014;115:354–63.
Hattori T, Shimokawa H, Higashi M, Hiroki J, Mukai Y, Tsutsui H, et al. Long-term inhibition of Rho-kinase suppresses left ventricular remodeling after myocardial infarction in mice. Circulation. 2004;109:2234–9.
Li Y, Zhu W, Tao J, Xin P, Liu M, Li J, et al. Fasudil protects the heart against ischemia-reperfusion injury by attenuating endoplasmic reticulum stress and modulating SERCA activity: the differential role for PI3K/Akt and JAK2/STAT3 signaling pathways. PLoS ONE. 2012;7:e48115.
Hamid SA, Bower HS, Baxter GF. Rho kinase activation plays a major role as a mediator of irreversible injury in reperfused myocardium. Am J Physiol Heart Circ Physiol. 2007;292:H2598–606.
Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, et al. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/- haploinsufficient mice. Circulation. 2005;112:2959–65.
Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, et al. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol. 2004;24:1842–7.
Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170–R85.
Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 2016;26:733–44.
Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2023;24:167–85.
Gustafsson AB, Dorn GW 2nd. Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol Rev. 2019;99:853–92.
Tu M, Miyamoto S. RHOA, a small G-protein, signals to mitophagy through regulation of PINK1 protein stability and protects cardiomyocytes against ischemia. Autophagy. 2022;19:1–2.
Soh JEC, Shimizu A, Molla MR, Zankov DP, Nguyen LKC, Khan MR, et al. RhoA rescues cardiac senescence by regulating Parkin-mediated mitophagy. J Biol Chem. 2023;299:102993.
Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 2011;14:1939–51.
Saito T, Nah J, Oka SI, Mukai R, Monden Y, Maejima Y, et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Investig. 2019;129:802–19.
Miyamoto S. Autophagy and cardiac aging. Cell Death Differ. 2019;26:653–64.
West AB, Kapatos G, O’Farrell C, Gonzalez-de-Chavez F, Chiu K, Farrer MJ, et al. N-myc regulates parkin expression. J Biol Chem. 2004;279:28896–902.
Xiao D, Chang W, Ding W, Wang Y, Fa H, Wang J. Enhanced mitophagy mediated by the YAP/Parkin pathway protects against DOX-induced cardiotoxicity. Toxicol Lett. 2020;330:96–107.
Shi J, Surma M, Yang Y, Wei L. Disruption of both ROCK1 and ROCK2 genes in cardiomyocytes promotes autophagy and reduces cardiac fibrosis during aging. FASEB J. 2019;33:7348–62.
Soliman H, Craig GP, Nagareddy P, Yuen VG, Lin G, Kumar U, et al. Role of inducible nitric oxide synthase in induction of RhoA expression in hearts from diabetic rats. Cardiovasc Res. 2008;79:322–30.
Lin G, Craig GP, Zhang L, Yuen VG, Allard M, McNeill JH, et al. Acute inhibition of Rho-kinase improves cardiac contractile function in streptozotocin-diabetic rats. Cardiovasc Res. 2007;75:51–8.
Lai D, Gao J, Bi X, He H, Shi X, Weng S, et al. The Rho kinase inhibitor, fasudil, ameliorates diabetes-induced cardiac dysfunction by improving calcium clearance and actin remodeling. J Mol Med. 2017;95:155–65.
Zhou H, Li YJ, Wang M, Zhang LH, Guo BY, Zhao ZS, et al. Involvement of RhoA/ROCK in myocardial fibrosis in a rat model of type 2 diabetes. Acta Pharmacol Sin. 2011;32:999–1008.
Soliman H, Nyamandi V, Garcia-Patino M, Zhang PC, Lin E, Jia ZP, et al. ROCK2 promotes ryanodine receptor phosphorylation and arrhythmic calcium release in diabetic cardiomyocytes. Int J Cardiol. 2019;281:90–8.
Wang S, Zhao Z, Fan Y, Zhang M, Feng X, Lin J, et al. Mst1 inhibits Sirt3 expression and contributes to diabetic cardiomyopathy through inhibiting Parkin-dependent mitophagy. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1905–14.
Xiong Z, Li Y, Zhao Z, Zhang Y, Man W, Lin J, et al. Mst1 knockdown alleviates cardiac lipotoxicity and inhibits the development of diabetic cardiomyopathy in db/db mice. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165806.
Feng X, Wang S, Yang X, Lin J, Man W, Dong Y, et al. Mst1 knockout alleviates mitochondrial fission and mitigates left ventricular remodeling in the development of diabetic cardiomyopathy. Front Cell Dev Biol. 2020;8:628842.
Del Re DP, Matsuda T, Zhai P, Maejima Y, Jain MR, Liu T, et al. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol Cell. 2014;54:639–50.
Preau S, Delguste F, Yu Y, Remy-Jouet I, Richard V, Saulnier F, et al. Endotoxemia engages the RhoA kinase pathway to impair cardiac function by altering cytoskeleton, mitochondrial fission, and autophagy. Antioxid Redox Signal. 2016;24:529–42.
Zhu Y, Wu H, Wu Y, Zhang J, Peng X, Zang J, et al. Beneficial effect of intermedin 1-53 in septic shock rats: contributions of Rho kinase and BKCA pathway-mediated improvement in cardiac function. Shock. 2016;46:557–65.
Gao Y, Sun Y, Ercan-Sencicek AG, King JS, Akerberg BN, Ma Q, et al. YAP/TEAD1 complex is a default repressor of cardiac Toll-like receptor genes. Int J Mol Sci. 2021;22:6649.
Yu W, Mei X, Zhang Q, Zhang H, Zhang T, Zou C. Yap overexpression attenuates septic cardiomyopathy by inhibiting DRP1-related mitochondrial fission and activating the ERK signaling pathway. J Recept Signal Transduct Res. 2019;39:175–86.
Ramjee V, Li D, Manderfield LJ, Liu F, Engleka KA, Aghajanian H, et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J Clin Investig. 2017;127:899–911.
Liu S, Tang L, Zhao X, Nguyen B, Heallen TR, Li M, et al. Yap promotes noncanonical wnt signals from cardiomyocytes for heart regeneration. Circ Res. 2021;129:782–97.
Francisco J, Zhang Y, Nakada Y, Jeong JI, Huang CY, Ivessa A, et al. AAV-mediated YAP expression in cardiac fibroblasts promotes inflammation and increases fibrosis. Sci Rep. 2021;11:10553.
Mia MM, Cibi DM, Abdul Ghani SAB, Song W, Tee N, Ghosh S, et al. YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction. PLoS Biol. 2020;18:e3000941.
Markousis-Mavrogenis G, Baumhove L, Al-Mubarak AA, Aboumsallem JP, Bomer N, Voors AA, et al. Immunomodulation and immunopharmacology in heart failure. Nat Rev Cardiol. 2023;21:119–49.
Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015;15:117–29.
Suetomi T, Miyamoto S, Brown JH. Inflammation in nonischemic heart disease: initiation by cardiomyocyte CaMKII and NLRP3 inflammasome signaling. Am J Physiol Heart Circ Physiol. 2019;317:H877–H90.
Funding
This work was supported by American Heart Association Grant 23TPA1141722 to SM.
Author information
Authors and Affiliations
Contributions
SM planned and wrote the manuscript and drew figures.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Sergio Lavandero
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miyamoto, S. Untangling the role of RhoA in the heart: protective effect and mechanism. Cell Death Dis 15, 579 (2024). https://doi.org/10.1038/s41419-024-06928-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-024-06928-8
This article is cited by
-
The gut-heart axis: a correlation between Paneth cells’ dysfunction, microbiome dysbiosis, and cardiovascular diseases
Cell Communication and Signaling (2025)
-
Emerging functions of Plakophilin 4 in the control of cell contact dynamics
Cell Communication and Signaling (2025)
-
A cell type-specific expression atlas of small and total RNA in the heart after myocardial infarction
Scientific Data (2025)
-
Role of Neural Circuits in Cognitive Impairment
Neurochemical Research (2025)
-
Inhibition of myocyte-specific enhancer factor 2A (MEF2A) attenuates cardiac fibrosis and improves heart function by regulating the Snail1/RhoA/α-SMA pathway
Journal of Bioenergetics and Biomembranes (2025)
