
Abstract
Subcellular compartmentalization of metabolic enzymes establishes a unique metabolic environment that elicits specific cellular functions. Indeed, the nuclear translocation of certain metabolic enzymes is required for epigenetic regulation and gene expression control. Here, we show that the nuclear localization of the mitochondrial enzyme methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) ensures mitosis progression. Nuclear MTHFD2 interacts with proteins involved in mitosis regulation and centromere stability, including the methyltransferases KMT5A and DNMT3B. Loss of MTHFD2 induces severe methylation defects and impedes correct mitosis completion. MTHFD2 deficient cells display chromosome congression and segregation defects and accumulate chromosomal aberrations. Blocking the catalytic nuclear function of MTHFD2 recapitulates the phenotype observed in MTHFD2 deficient cells, whereas restricting MTHFD2 to the nucleus is sufficient to ensure correct mitotic progression. Our discovery uncovers a nuclear role for MTHFD2, supporting the notion that translocation of metabolic enzymes to the nucleus is required to meet precise chromatin needs.
Similar content being viewed by others


Introduction
Enzymes of central metabolism can translocate to the nucleus and influence chromatin remodeling, epigenetics, and transcription regulation1,2,3,4,5. A clear example of this in loco regulatory activity of metabolic enzymes is the synthesis of acetylation and methylation cofactors required for histone modification directly on chromatin6,7,8,9.
One-carbon folate metabolism is a pivotal pathway, being indispensable for the de novo synthesis of nucleotides, amino acid homeostasis, DNA and histone methylation, and the maintenance of the cellular redox state10. Folate metabolism is compartmentalized between the cytosol and the mitochondria, and the mitochondrial one-carbon metabolism enzyme methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) emerged as the most consistently overexpressed metabolic gene in cancer11. While expressed during embryonic development and cancer, MTHFD2 is barely expressed in adult tissues11, making it a very attractive anticancer target12,13,14. High levels of MTHFD2 are associated with a worse outcome in several cancer types, including breast15, colon16, and lung17 cancer. MTHFD2 has been shown to support cancer cell proliferation and survival in vitro and tumor growth in vivo16,17,18,19,20, and to promote metastatic features such as cell migration and invasion18,21. MTHFD2 can localize in the nucleus22,23. However, its nuclear function remains poorly characterized and elusive.
During cell division, cells duplicate their genetic material, which is divided between two daughter cells during mitosis. Mitotic defects lead to the accumulation of genetic aberrations that can result in cellular transformation or lead to cell death24. Several mechanisms ensure faithful chromosome segregation, and centromeres are important pillars in this process. Centromeres orchestrate the chromosomal attachment to spindle fibers through the assembly of the kinetochore complex25,26,27. Centromeric and peri-centromeric DNA is compacted into heterochromatin28 and heterochromatin marks H3K9me3, H3K27me3, and H4K20me1 decorate the centromere, contributing to the formation of the kinetochore29,30,31,32. Moreover, DNA methylation at centromeres is abundant and maintains chromatin structure, preventing errors in chromosome segregation and genomic instability33,34,35,36,37. The expression of the centromeric region regulates the loading of the centromeric histone variant CENP-A and contributes to the recruitment of inner kinetochore proteins to the centromeric region38.
Here, we discovered that MTHFD2 localizes in the nucleus to regulate DNA and centromeric histone methylation, centromeric expression, and proper mitotic progression. MTHFD2 nuclear partners are mostly cell cycle regulators and methyltransferases responsible for depositing methylation marks at centromeres, such as KMT5A29,39, DNMT3B40, and PRMT141. The absence of MTHFD2 leads to a drastic reduction of DNA and centromeric histone methylation, increased centromeric alpha-satellite expression, and accumulation of genomic aberrations. Consequently, cell cycle progression is also impaired when cells lack MTHFD2, with a significant reduction in mitotic events. The absence of total MTHFD2 or its nuclear inhibition alone induces chromosome congression and segregation defects, as well as micronuclei accumulation, indicating that the nuclear localization of MTHFD2 plays an active enzymatic role in controlling centromeric heterochromatin maintenance and correct mitotic cell division.
Results
MTHFD2 localizes within the nucleus in proliferating cells
A meta-analysis comprising microarray expression data covering 19 types of tumors highlighted the mitochondrial folate enzyme MTHFD2 as the top-scoring upregulated metabolic enzyme in cancer11. To corroborate this finding, we retrieved RNA-sequencing data from The Cancer Genome Atlas (TCGA) database42. We filtered for solid tumor types where paired normal tissue data were available (Supplementary Fig. 1a) and confirmed that MTHFD2 was significantly upregulated in 13 out of 15 evaluated tumor types. Among these, breast carcinoma, colon adenocarcinoma, lung adenocarcinoma and lung squamous cell carcinoma showed the most significantly increased MTHFD2 levels (Fig. 1a). Then, we asked whether MTHFD2 expression alone could be used to predict the status of a sample (tumor versus healthy) in breast, lung and colon cancer. Training a tree-based algorithm with a subset of the TCGA expression data yielded a prediction accuracy (true positive rate) over 0.84 and AUC (area under the curve) values between 0.77 and 0.88 on unseen data, confirming MTHFD2 expression’s predictive usefulness (Supplementary Fig. 1b–d).

a Comparison between Transcripts per Million (TPM) expression values of MTHFD2 in healthy and tumor tissues; paired two-tailed Wilcoxon test (n indicates the number of patients with paired samples for each cancer type). BLDA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; ESCA, esophageal carcinoma; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PRAD, prostate adenocarcinoma; STAD, stomach adenocarcinoma; THCA, thyroid carcinoma; UCEC, uterine corpus endometrial carcinoma. b Western blot of cytosolic (cyt) and chromatin (chr) fractions after subcellular fractionation in a panel of cancer cell lines. Vinculin, Histone H3 and FDX1 are used as cytosolic, nuclear and mitochondrial markers, respectively. c Immunofluorescence of MTHFD2 in MCF7 (up), H358 (middle) and HCT116 (down) cells. MTHFD2 is shown in green (left) or royal (right) and DAPI in gray; confocal mode, scale bar 10 μm. d MTHFD2 and DAPI mean +/- s.d. intensity line profiles of the cell diameter of the respective cell lines, along with their corresponding backgrounds (n indicates number of individual cells).
Although MTHFD2 primarily localizes within the mitochondria, its nuclear23 and chromatin43 localizations have been previously reported in some cancer cell lines. Therefore, we asked whether MTHFD2 chromatin localization is a generalizable event. We selected a panel of breast, colon and lung cancer cell lines in which MTHFD2 was highly expressed (CCLE data44; Supplementary Fig. 1e, f), but not essential (Supplementary Fig. 1g). Subcellular fractionation of chromatin (hereafter, the chromatome) revealed the presence of MTHFD2 on chromatin in all the tested cell lines (Fig. 1b). MTHFD2 nuclear localization was also confirmed by confocal immunofluorescence in cell lines (Fig. 1c, d) and patient-derived colon cancer sections (Supplementary Fig. 1h). Since MTHFD2 is expressed not only in cancer but also during embryonic development, we asked whether we could observe differences in its nuclear localization between healthy multipotent cells and cancer cells. To this end, we performed confocal microscopy on either patient-derived colon cancer organoids or patient-derived multipotent healthy colon organoids. Strikingly, we observed that MTHFD2 was weakly localized to the nucleus in colon cancer organoids, whereas its nuclear localization was clear in multipotent healthy colon organoids (Fig. 2a). To test whether the nuclear localization of MTHFD2 was associated with a proliferative phenotype typical of multipotent progenitors, we cultured the multipotent healthy colon organoids in differentiation media for one week to induce terminal differentiation. We observed that while the differentiation marker Mucin-2 (MUC2) appeared, the nuclear staining of MTHFD2 was drastically reduced, together with reduced levels of the proliferation marker Ki67 (Fig. 2b).

a Immunofluorescence of colon cancer (left) and multipotent healthy colon (right) patient-derived organoids. MTHFD2 is shown in green, VDAC1 (mitochondrial marker) in red and DAPI in gray; confocal mode; scale bar 50 μm. b Immunofluorescence of the multipotent healthy colon (left) and differentiated healthy colon (right) patient-derived colon organoids. MTHFD2 is shown in green, DAPI in gray, the proliferation marker Ki67 in yellow, and the differentiation marker Mucin-2 (MUC2) in magenta; confocal mode; scale bar 50 μm.
Collectively, our data suggest that MTHFD2 nuclear localization is associated with an active proliferative cellular state and is not restricted to cancer.
Nuclear MTHFD2 interacts with cell division players
To elucidate the nuclear function of MTHFD2, we characterized the nuclear MTHFD2 interactome using the HCT116 cell line as a model. We chose this cell line because it shows a very high level of MTHFD2 on chromatin (Fig. 1b) and because it is a diploid cell line with a stable karyotype that does not accumulate aberrations. We coupled mass spectrometry (MS) analysis with a pull-down of MTHFD2, either from the cytosolic or chromatin fractions, obtained after subcellular fractionation (Supplementary Data 1). We verified the purity of the fractions by checking the relative abundance of the detected proteins in the different subcellular compartments using Human Protein Atlas (HPA)45 annotation. As expected, cytosolic MTHFD2 and IgG immunoprecipitations (IP) spanned a wider range of cell compartments, with the cytosol representing the highest proportion (41% and 43%, respectively). Conversely, chromatin MTHFD2 and IgG IPs contained predominantly nuclear proteins (80% and 85%, respectively) (Supplementary Fig. 2a). To streamline our analysis, we prioritized a list of interactors using the Significance Analysis of INTeractome (SAINT) software46 (v. 3.2.0). We identified 127 and 43 potential MTHFD2 interactors in the cytosolic and chromatin fractions, respectively (Supplementary Fig. 2b, Supplementary Data 2). We categorized these MTHFD2 interactors according to their HPA nuclear localization status, shortlisting only those considered to be either completely or partially localized to the nucleus (Supplementary Fig. 2c, Supplementary Data 2).
To get insights into the role of nuclear MTHFD2, we functionally categorized its top nuclear interactors (Fig. 3a, Supplementary Fig. 2d). The most abundant functional category in our interactome was cell cycle and mitosis, containing 23% of the top interactors, followed by epigenetics and transcription regulation (Supplementary Fig. 2e). IntAct47 network analysis showed a high degree of overall connectivity, with previously described interactions among 57% of our hits (Fig. 3b). With respect to the nuclear interactors of MTHFD2, previously only Microsomal Glutathione S-Transferase 3 (MGST3) has been described. The top MTHFD2 nuclear interactor was Nucleoprotein TPR, a nucleopore complex protein that stabilizes the mitotic spindle assembly checkpoint proteins MAD1L1 and MAD2L1 during mitosis, contributing to the activation of the spindle assembly checkpoint48. MAD1L1 was also among the MTHFD2 interactors (Fig. 3a, b). Additionally, we retrieved all the members of the AMP-activated protein kinase (AMPK) complex (PRKAA1, PRKAB1, PRKAG1, and PRKAG2), which is known to be involved in mitosis progression49, anaphase spindle length determination50 and spindle orientation51. Kinesin KIF4A, a substrate of AMPK50 that participates in chromosome condensation and segregation52, and Protein Arginine N-Methyltransferase 1 (PRMT1), which methylates Inner Centromere Protein (INCENP) triggering the centromeric recruitment and activation of AURKB41, also scored as significant MTHFD2 nuclear interactors. Furthermore, we identified N-Lysine Methyltransferase KMT5A, which deposits H4K20me1 at centromeric CENP-A containing nucleosomes enabling kinetochore assembly29,39,53, and DNA Methyltransferase 3B (DNMT3B), which is recruited to the centromeric and pericentromeric regions by CENP-C for DNA methylation40, as putative MTHFD2 nuclear interactors. MTHFD2 pull-down experiments performed in HCT116 and H358 nuclear extracts (Supplementary Fig. 2f) validated most of these interactions (Supplementary Fig. 2g, h).

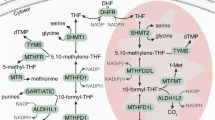
a Volcano plot of top nuclear MTHFD2 interactors identified in HCT116 cells. Interactors with a log2 fold change >= 2.3 and Bonferroni False Discovery Rate (BFDR) <= 0.2 are colored according to their functional category. IP, immunoprecipitation. b Network of top nuclear MTHFD2 interactors. The color indicates the functional category, the size represents the fold change and the width of the edges shows the interaction score. c Biological Process gene ontologies enriched in MTHFD2 core co-expressed genes. d One-carbon folate metabolism pathway with enzymes colored by the enrichment of mitotic terms in the Gene Ontology enrichment analysis performed with their core co-expressed genes. Gray indicates the absence of data. DHF, dihydrofolate; THF, tetrahydrofolate. Scheme adapted from Lin et al. 4. Two-dimensional density plots of 5% top cells with the highest nuclear (e, g) or cytosolic (f, h) MTHFD2 signal obtained from the adapted FUCCI4 MCF7 (e, f) and U2OS (g, h) cells along with the cell cycle phase; turquoise and clover mean intensities (x and y axis, respectively) are in log10 scale.
Since co-expression can be used to infer functionality, we exploited the TCGA RNA-sequencing data to identify genes that are co-expressed with MTHFD2. We selected positively correlated genes as those co-expressed with MTHFD2 in at least 10 different cancer types, and performed an over-representation analysis of Gene Ontology (GO) Biological Processes (Supplementary Fig. 3a, b). Top-scoring terms were mostly related to mitosis and cell cycle (Fig. 3c), showing a particular enrichment for terms associated with chromosome segregation. We performed a similar analysis with all the enzymes of the folate pathway (Supplementary Data 3, 4) and observed that MTHFD2 was, by far, the folate enzyme with the highest proportion of mitotic-related (Fig. 3d and Supplementary Fig. 3c, d) and cell cycle-related terms (Supplementary Fig. 3e). A similar analysis was done at the protein level with ProteomeHD54, which uses proteomics data in response to biological perturbations to perform co-regulation analysis using unsupervised machine learning. Over-representation analysis of GO Biological Processes using the top 5% proteins co-regulated with MTHFD2 also revealed several cell cycle and mitotic terms (Supplementary Fig. 3f).
Given the possible implication of MTHFD2 in cell cycle regulation, we investigated whether its nuclear localization is cell cycle dependent. A publicly available cell cycle proteomics analysis55 allowed us to determine that MTHFD2 protein levels increase significantly in the G2 phase (Supplementary Fig. 4a). We then used a FUCCI456 adapted MCF7 (Supplementary Fig. 4b) and U2OS (Supplementary Fig. 4c) cell line reporters57 that can track cell cycle phases in immunofluorescence. We observed that MTHFD2 localized to the nucleus across all cell cycle phases and that cells in the G2-M phase transition had the highest levels of nuclear MTHFD2 (Fig. 3e, g). Cells with the highest cytosolic MTHFD2 levels were instead in the S-G2 phase of the cell cycle (Fig. 3f, h). Since the FUCCI4 system does not allow the detection of mitotic cells, we performed stochastic optical reconstruction microscopy (STORM), which showed that only a small fraction of MTHFD2 is retained on mitotic chromosomes (Supplementary Fig. 4d, e), suggesting that MTHFD2 may be excluded from chromatin once it condenses into chromosomes.
In conclusion, MTHFD2 interacts with methyltransferases that are required for methylation of centromeric DNA and centromeric histones, and kinetochore proteins whose function is critical for proper cell division.
Centromeric DNA is overexpressed in the absence of MTHFD2
To explore the consequences of MTHFD2 loss, we generated MTHFD2 knock-out (KO) HCT116 cells by CRISPR-Cas9. Two MTHFD2 KO cell lines were validated by sequencing (Supplementary Fig. 5a), Western blot (Fig. 4a) and immunofluorescence (Fig. 4b, Supplementary Fig. 5b, c). Both KO cell lines showed decreased proliferation rates (Supplementary Fig. 5d), invasion (Supplementary Fig. 5e) and clonogenic (Supplementary Fig. 5f) capacities, as previously reported17,19,21.

a Western blot of HCT116 MTHFD2 knock-out (KO1, KO2) and wild-type (WT) cells. Vinculin is used as loading control. b Immunofluorescence of MTHFD2 WT, KO1 and KO2 cells. MTHFD2 is shown in green and DAPI in blue; non-confocal mode, scale bar 10 μm. c Percentage of centromeric intersects normalized by the total mapped reads in WT and KO1 cells (n = 6); unpaired two-tailed Wilcoxon test. d Relative mRNA expression of 20 chromosomal centromeres in KO1 cells normalized to WT cells. The dashed line indicates fold change = 1; means + s.e.m. (n = 5), one-sample two-tailed t-test (P values for each centromere are indicated on the top). e Comparison of the log2 mean intensity of nuclear levels of histone marks H4K20me1, H3K9me3, and H3K27me3 in WT and KO1 cells; unpaired two-tailed Wilcoxon test. Representative images are shown above; non-confocal mode, scale bar 10 μm. f–h Fold enrichment of H4K20me1 (f), H3K9me3 (g), and H3K27me3 (h) signal normalized to IgG in centromeric regions of 4 independent chromosomes in WT and KO1 cells; means + s.d. (n = 3), one-sample two-tailed t-test. i Whole-genome scheme showing the hypermethylated CpG sites in red and hypomethylated CpG sites in blue. Regions shown in pale purple correspond to the centromeres and peri-centromeres. The height of the bars is proportional to the degree of hyper- or hypomethylation. j Chromosomes 6 and 14 showing the peri-centromeric alterations found in KO1 and KO2 cells in red. Number of alterations (insertions, deletions, duplications or inversions) found in WT and KO1 cells in the whole-genome (k) or at the centromeric regions (l). Source data are provided as a Source Data file. The n indicates the number of biological sample replicates for c, d, f–h, and number of individual cells for e. For e, individual cells from 4 biological replicates were pooled.
We performed transcriptomic analysis to investigate whether the loss of MTHFD2 could transcriptionally affect cell cycle progression. RNA sequencing was performed at an early time point (immediately after KO isolation) and at a late time point (after two months of culture) to determine whether the transcriptome is stable following MTHFD2 KO. We compared HCT116 MTHFD2 wild-type (WT) cells with MTHFD2 KO cells at both time points. Principal component analysis (PCA) revealed that the primary segregating factor was the presence of MTHFD2, with no time-dependent effects on transcription (Supplementary Fig. 5g and Supplementary Data 5), suggesting a rapid and stable transcriptional rewiring upon MTHFD2 KO. We then asked whether the transcriptional rewiring driven by MTHFD2 absence could affect the expression of mitotic genes. Overrepresentation analysis of GO Biological Processes for downregulated genes revealed terms related to cell projection and cell-matrix interactions, consistent with the reduced invasive ability of MTHFD2 KO cells, while for upregulated genes we found terms related to metabolic processes involving aminoglycans or fatty acids, suggesting metabolic rewiring following MTHFD2 loss (Supplementary Fig. 5h, i). However, we did not retrieve any terms related to mitosis or cell cycle. Likewise, Gene Set Enrichment Analysis (GSEA) did not identify any gene sets associated with mitosis or cell cycle, although we observed a negative enrichment of the PI3K-Akt-mTOR signaling in the KO condition (Supplementary Fig. 5j), which has been previously reported58, validating the quality of our dataset. Finally, we did not observe changes of proteins composing the centromere-kinetochore structure59 following MTHFD2 loss (Supplementary Fig. 5k). Only ZWINT and CENP-C were respectively significantly up- and downregulated upon MTHFD2 KO. However, there was no change in CENP-C or CENP-A protein levels in MTHFD2 KO cells (Supplementary Fig. 5l).
Since the MTHFD2 pull-down showed that MTHFD2 is associated with proteins involved in centromeric functions, we specifically analyzed the expression of centromeres in the absence of MTHFD2. Centromeres consist of alpha-satellite repeats that are transcribed into non-coding RNA and allow the centromeric recruitment of the histone H3 variant CENP-A and inner kinetochore protein CENP-C38. Therefore, an altered centromeric transcription results in defective centromere and kinetochore assembly38,60,61,62. To assess centromeric expression, we calculated the percentage of reads that aligned to centromeric regions normalized by the total number of mapped reads. We observed that the percentage of reads intersecting with the centromeres was significantly higher in the KO condition compared to the WT counterpart (Fig. 4c). To validate this observation, we performed retro-transcription quantitative PCR of 20 chromosomal centromeres, which showed a trend towards upregulation of centromeric expression in the absence of MTHFD2 in both asynchronized cells (Fig. 4d) and cells synchronized in late G2 (Supplementary Fig. 5m). Given the role of centromere expression in recruiting CENP-A, we quantified CENP-A recruitment at centromeric regions by high-throughput immunofluorescence. Centromeres were identified by anti-centromere (CREST) staining. MTHFD2 KO cells showed increased levels of CENP-A at centromere/kinetochore sites (Supplementary Fig. 5n), which was consistent throughout the cell cycle (Supplementary Fig. 5o), likely due to increased centromere expression.
Collectively, these results show that the transcriptional switch induced by MTHFD2 loss does not have a drastic effect on mitosis or the cell cycle. However, centromeres are overexpressed in the absence of MTHFD2, which leads to aberrant CENP-A recruitment.
Loss of MTHFD2 decreases centromeric histone and DNA methylation
We hypothesized that the upregulation of centromeric expression may arise from an aberrant increase in accessibility of centromeric chromatin upon MTHFD2 loss. Given the fact that our interactome analysis revealed MTHFD2 nuclear interactions with centromeric histone and DNA methyltransferases (KMT5A and DNMT3B), we queried whether the absence of MTHFD2 could impair centromere methylation. We observed a significant decrease of global nuclear H4K20me1 levels, a specific centromeric histone mark deposited by KMT5A29,39,53, upon MTHFD2 absence, with minor changes in H3K9me3 and an overall mild increase in H3K27me3 (Fig. 4e). Additionally, we measured the coefficient of variation of the three histone marks, which assesses the heterogeneity in the nuclear density distribution of the staining. Only H4K20me1 showed a clear deviation towards less variation (more homogenous distribution) in the absence of MTHFD2 (Supplementary Fig. 6a). Similar results were also observed irrespective of cell cycle stages, which were examined either by cell synchronization (Supplementary Fig. 6b), or by cell cycle classification using DAPI staining (Supplementary Fig. 6c, d). These results indicate that the methylation defect was maintained throughout the cell cycle. Finally, chromatin immunoprecipitation (ChIP) coupled with quantitative PCR revealed considerably reduced levels of H4K20me1, H3K9me3 and H3K27me3 at centromeres (Fig. 4f–h, Supplementary Fig. 6e). This supports the hypothesis that MTHFD2 depletion leads to a demethylation-induced increase in chromatin accessibility, which facilitates transcriptional upregulation of centromeric regions.
Beyond centromeres, we observed a more homogeneous DAPI staining in MTHFD2 KO cells, suggesting a global chromatin reorganization, which was cell cycle-independent (Supplementary Fig. 6f, g). Given the fact that DNMT3B was among MTHFD2 nuclear interactors, we investigated whether the loss of DNA methylation could explain the observed changes in DAPI staining upon MTHFD2 KO. We performed Nanopore whole-genome sequencing and conducted a comparative methylation analysis of HCT116 MTHFD2 WT and KO cells (Supplementary Fig. 7a–d, Supplementary Data 6). We classified the differentially methylated sites into hypermethylated or hypomethylated if they were significantly more or less methylated, respectively, in the MTHFD2 KO condition compared to the WT. MTHFD2 KO cells showed strong hypomethylation either when looking at CpG sites located at CpG islands and shores (Fig. 4i) or considering all CpG sites (Supplementary Fig. 7e). Centromeres and the genome in general were equally impacted. Annotation of the genomic location of the hyper- and hypomethylated sites showed that they largely fell in intergenic regions (61% and 65%, respectively) (Supplementary Fig. 7f, g), and were equally distributed into up- and downregulated genes (Supplementary Fig. 7h). Likewise, differentially expressed genes were also equally distributed into hyper- and hypomethylated regions (Supplementary Fig. 7i), suggesting a lack of correlation between changes in DNA methylation and gene expression following MTHFD2 loss.
Finally, we asked whether the loss of centromeric histone and DNA methylation could affect the recruitment of phosphorylated Aurora kinases to mitotic chromosomes, key factors required for kinetochore and chromosome stability63,64 and proper mitotic progression. Phosphorylated Aurora kinases (A, B, C; serine/threonine) levels on mitotic chromosomes were significantly reduced in MTHFD2 KO cells as quantified by high-throughput immunofluorescence (Supplementary Fig. 7j, k).
Collectively, our data indicate that MTHFD2 fine-tunes the recruitment of CENP-A and phosphorylated Aurora kinases by modulating centromeric expression through methylation-dependent chromatin compaction, suggesting a direct role for MTHFD2 in setting the stage for proper mitotic progression.
MTHFD2 loss leads to accumulation of chromosomal alterations
To understand if MTHFD2 loss leads to macroscopic genetic aberrations, we karyotyped the HCT116 MTHFD2 WT and KO cells. HCT116 cells are well-suited for this type of analysis since they are near diploid and have a stable karyotype. We observed that the karyotype remained largely unchanged between the three cell lines, as expected, although both KO cell lines each presented an additional alteration. The MTHFD2 KO1 had a deletion in the small arm of chromosome 6 (del(6)(p12)), while the MTHFD2 KO2 showed an insertion in the small arm of chromosome 14 (add(14)(p11)) (Fig. 4j, Supplementary Fig. 8a–c). Of note, both alterations were located very close to the centromeric region.
Given the limitation of the karyotype technique in identifying smaller genomic alterations, we queried the Nanopore whole-genome sequencing data presented above to investigate whether the loss of MTHFD2 was associated with an accumulation of genomic structural variation. Although MTHFD2 WT and KO cells shared a considerable number of genetic variants (Supplementary Fig. 8d), MTHFD2 KO cells diverged from the WT population by showing a higher number of each type of variant (Fig. 4k). The size distribution of the variants, which could span from a few bases to 100 kb, was similar in both conditions (Supplementary Fig. 8e). We excluded that deletions could be the reason of the observed hypomethylation by checking the proportion of hypomethylated and hypermethylated sites located at deleted regions, which did not change across conditions (Supplementary Data 7). Finally, we performed a centromere-focused analysis and observed that the absence of MTHFD2 induced a higher number of alterations (Fig. 4l).
Taken together, our data show that MTHFD2 loss leads to genomic aberrations, which may be the result of mitotic defects arising from centromeric and genomic instability.
MTHFD2 is required for proper chromosome segregation
Next, we investigated whether loss of MTHFD2 leads to mitotic defects, which could link centromere expression and methylation defects to the accumulation of genomic variants. Quantification of the mitotic marker histone H3 phospho-Ser10 (H3PS10) by high-throughput immunofluorescence showed a reduced mitotic index in MTHFD2 KO cells (Fig. 5a and Supplementary Fig. 9a). In agreement, we observed that the number of G2/M cells were decreased in both KO cell lines (Supplementary Fig. 9b). In addition, we synchronized MTHFD2 WT and KO cells at the G2-M transition by treating them with the CDK1 inhibitor RO-3306 for 20 hours and quantified the percentage of cells in G1 at different time points after drug withdrawal. While MTHFD2 WT cells progressed rapidly through mitosis, with ~70% of cells in G1 after 2.5 hours, both MTHFD2 KO cell lines showed a mitotic delay, with only ~50% of cells in G1 at the same time point (Fig. 5b and Supplementary Fig. 9c). We reasoned that the observed delay could be due to a defect in mitotic progression. Indeed, classifying mitotic cells into different phases showed an imbalance in their proportion in comparison to the WT counterpart. MTHFD2 KO caused an increase in cells in early mitotic phases (prophase and prometaphase), suggesting a defect in chromosome congression at the metaphase plate, and a decreased proportion of cells in telophase and cytokinesis, which could result from chromosome segregation defects in anaphase (Fig. 5c). To validate these hypotheses, we analyzed metaphases in both conditions and found that the loss of MTHFD2 increased the percentage of uncongressed metaphase plates as shown by CREST staining (Fig. 5d, e). In addition, MTHFD2 KO cells exhibited a higher number of DNA anaphase bridges (Supplementary Fig. 9d, e), which clearly involved the centromeric region as identified by CREST staining (Fig. 5f, g). As a consequence, MTHFD2 KO cells showed more CREST-positive micronuclei than their WT parental population (Supplementary Fig. 9f, g).

a Percentage of mitotic cells in HCT116 MTHFD2 wild-type (WT) and knock-out (KO1, KO2) conditions, measured with the mitotic marker histone H3 phospho-Ser10 by high-throughput immunofluorescence; means + s.d. (n = 5), unpaired two-tailed t-test. b Percentage of WT, KO1, and KO2 cells in G1 phase at 0, 0.5, 1, 1.5, 2, and 2.5-hour release after RO-3306 drug treatment for 20 hours; means + s.d. (n = 3), at indicated times, unpaired two-tailed t-test. c Percentage of WT, KO1, and KO2 cells at different mitotic phases; means + s.d. (n = 3), a minimum of 150 mitotic cells per replicate were analyzed, unpaired two-tailed t-test. Representative images (d) and quantification (e) of uncongressed chromosomes in metaphase in WT, KO1 and KO2 cells. CREST is shown in ICA (left) or magenta (right) and DAPI in gray; non-confocal mode, scale bar 10 μm. For the quantification, means + s.d. (n = 3), a minimum of 10 metaphase cells were analyzed, unpaired two-tailed t-test. Representative images (f) and quantification (g) of anaphase defects in WT, KO1 and KO2 cells. CREST is shown in ICA (left) or magenta (right) and DAPI in gray; non-confocal mode, scale bar 10 μm. For the quantification, means + s.d. (n = 3), a minimum of 25 anaphase cells were analyzed, unpaired two-tailed t-test. h Difference between the beta score of all genes in KO and WT cells. Synthetic lethal hits with MTHFD2 KO with a beta score < −1 are shown in red, and synthetic viable hits with MTHFD2 KO with a beta score > 1 are shown in blue. Shared hits between both KOs are indicated with a black stroke. i Percentage of survivor WT, KO1, and KO2 cells normalized to DMSO after etoposide treatment with indicated concentrations for 72 hours; means + s.d. (n = 3), unpaired two-tailed t-test. Inside the graph, a representative scanned image of one replicate (4 technical replicates are shown per condition). Source data are provided as a Source Data file. The n indicates the number of biological sample replicates for a–c, e, g, i.
Since nuclear MTHFD2 interacts with the key spindle regulators TPR and MAD1L1, we queried whether it might be functionally related to the spindle assembly checkpoint. Correlation analysis using TCGA patient expression data showed that MTHFD2 positively correlated with 11 of 12 genes involved in the spindle assembly checkpoint59 (AURB, BUB1, BUB1B, BUB3, CCNB1, CDC20, MAD1L1, MAD2L1, MD2BP, TTK, ZW10 and ZWILC) (Supplementary Fig. 10a–l). However, MTHFD2 KO cells did not show spindle assembly checkpoint defects upon treatment with increasing concentration of nocodazole (Supplementary Fig. 10m).
Finally, we performed a genome-wide CRISPR-Cas9 genetic screen by transducing MTHFD2 WT and KO cells. Cells were selected with puromycin for 8 days, where the initial population was harvested, and then kept in culture for three more weeks, where the final population was harvested. For the three samples, DNA from the initial and final population was extracted and sequenced. The average coverage of the initial population for all the samples was higher than 500X (Supplementary Fig. 11a–c), and the Gini index was lower than 0.1 in all initial samples (Supplementary Fig. 11d), ensuring the evenness of sgRNA read counts65. A cell-cycle normalization was performed to compensate for cell cycle differences among conditions (Supplementary Fig. 11e, f). Enriched and depleted sgRNAs were identified by comparing each final population with their corresponding initial population, and enriched and depleted sgRNAs in each KO cell line were compared with those in the WT to identify synthetic lethal genes or synthetic viable genes upon MTHFD2 loss. Genes detected in both KO were further considered (Fig. 5h, Supplementary Data 8). In support of the role of MTHFD2 in mitosis regulation, we have recovered key mitotic players as either synthetically lethal or viable with MTHFD2 KO. For example, the loss of WAPL, which enables chromosome condensation by binding Cohesin to chromatin66, was found to be synthetically viable with MTHFD2 KO (Fig. 5h). On the other hand, TOP2A was found to be among the top synthetic lethal genes in MTHFD2 KO (Fig. 5h). In contrast to WAPL, TOP2A is required for mitotic chromosome condensation67. Given the methylation defects observed in the MTHFD2 KO cells, we hypothesized that any additional impairment in chromatin condensation could be detrimental. Treatment with the Topoisomerase II inhibitor etoposide confirmed the increased sensitivity of MTHFD2 KO cells to the loss of TOP2A (Fig. 5i). Furthermore, by integrating CCLE data68 we observed that near-euploid cells have the highest MTHFD2 essentiality while highly-aneuploid cells the lowest (Supplementary Fig. 11g). Moreover, near-euploid cell lines had the highest MTHFD2 expression, while highly-aneuploid cell lines had the lowest MTHFD2 expression (Supplementary Fig. 11h). In agreement, cells with high MTHFD2 expression (top 25%) showed a lower aneuploidy score compared to low MTHFD2 expression (bottom 25%) cells (Supplementary Fig. 11i).
Overall, our results show that MTHFD2 depletion causes a delay in chromosome congression, defects in chromosome segregation, and an accumulation of centromere-containing micronuclei, confirming that MTHFD2 is a mitotic regulator.
Nuclear MTHFD2 catalytic activity is required for mitosis progression
Finally, we wanted to show whether the nuclear localization of MTHFD2 is essential for mitotic progression. We generated a nuclear-exclusive-MTHFD2 (NLS) HCT116 cell line by introducing a triple nuclear localization signal in the MTHFD2 locus by CRISPR knock-in (Supplementary Fig. 12a, b). We compared the mitotic index of MTHFD2 NLS cells to those of either MTHFD2 WT or KO cells and observed that the nuclear MTHFD2 alone was sufficient to ensure a mitotic index comparable to that in WT cells (Fig. 6a). Additionally, MTHFD2 NLS cells also exhibited similar proliferation rates in comparison to MTHFD2 WT, which was significantly higher than in MTHFD2 KO populations (Supplementary Fig. 12c). Similarly, the mitotic phase distribution and the rate of anaphase bridges observed in the MTHFD2 NLS cells were similar to the ones of the MTHFD2 WT cells (Fig. 6b, c). We also assessed global H4K20me1 levels, which indicated that the MTHFD2 NLS cells do not show the histone methylation defect present in the KO condition and had slightly higher levels of H4K20me1 than the MTHFD2 WT cells (Fig. 6d, Supplementary Fig. 12d). In line with this result, in the MTHFD2 NLS cells the coefficient of variation of DAPI, which was decreased upon MTHFD2 loss, was even higher than in WT cells (Supplementary Fig. 12e). We reasoned that if nuclear MTHFD2 regulates the methylation of centromeric histones and DNA required for proper mitotic progression, supplementation with the universal methyl group donor S-adenosylmethionine (SAM) or folate derivatives could potentially restore the mitotic index in MTHFD2-deficient cells. We treated MTHFD2 WT and KO cells with two near plasma physiological level concentrations of formate, folate, 5,10-methylenetetrahydrofolate (MTHFD2 product) or SAM for 72 hours. We observed no recovery in the mitotic index in the KO cell lines (Supplementary Fig. 12f). This result suggests two scenarios: nuclear MTHFD2 might have a structural role necessary for DNA and histone methylation, or it might have an enzymatic function crucial for maintaining a specific nuclear metabolic environment. To rule out whether the nuclear enzymatic function of MTHFD2 is required for mitosis control, we treated HCT116 cells using a recently published MTHFD2 inhibitor TH961914 that cannot enter the mitochondria69, in essence, a nuclear MTHFD2 inhibitor. When we treated the cells with non-toxic concentrations of TH9619 (Supplementary Fig. 12g) for 96 hours, we observed a dose-response decrease in the mitotic index (Fig. 7a). Additionally, the treatment induced an imbalance of the mitotic phases (Fig. 7b), anaphase defects (Fig. 7c, d), and increased CREST-positive micronuclei (Fig. 7e). Finally, H4K20me1 levels were significantly decreased in the presence of the treatment (Fig. 7f, Supplementary Fig. 12h), although the coefficient of variation of DAPI did not noticeably change (Supplementary Fig. 12i).

a Percentage of mitotic cells in HCT116 MTHFD2 wild-type (WT), nuclear (NLS), and knock-out (KO1, KO2) conditions; means + s.d. (n = 3), unpaired two-tailed t-test. b Percentage of WT, NLS and KO1 cells at different mitotic phases; means + s.d. (n = 3), a minimum of 100 mitotic cells per replicate were analyzed, unpaired two-tailed t-test. c Quantification of anaphase bridges in WT, NLS and KO1 cells; means + s.d. (n = 3), a minimum of 10 anaphases per replicate were analyzed, unpaired two-tailed t-test. d Comparison of the log2 mean intensity of nuclear levels of H4K20me1 in WT, NLS, KO1 and KO2 cells; unpaired two-tailed Wilcoxon test. Representative images are shown on the right; non-confocal mode, scale bar 10 μm. Source data are provided as a Source Data file. The n indicates the number of biological sample replicates for a–c; and the number of individual cells for d, where 3 biological replicates were pooled.

a Proportion of HCT116 mitotic cells treated with the indicated concentrations of TH9619 inhibitor for 96 hours normalized to DMSO condition; means + s.d. (n = 3), one-sample two-tailed t-test. b Percentage of HCT116 cells treated with DMSO or 63 nM TH9619 inhibitor for 96 hours at different mitotic phases; means + s.d. (n = 3), a minimum of 300 mitotic cells per replicate were analyzed, unpaired two-tailed t-test. c Quantification of anaphase bridges in HCT116 cells treated with DMSO or 63 nM TH9619 inhibitor for 96 hours; means + s.d. (n = 3), a minimum of 40 anaphase cells per replicate were analyzed, unpaired two-tailed t-test. d Representative images of anaphase defects in HCT116 cells treated with DMSO or 63 nM TH9619 inhibitor for 96 hours. DAPI is shown in gray and H3 phospho-Ser10 (H3PS10) is shown in red (second row) or ICA (third row); non-confocal mode, scale bar 10 μm. e Quantification (left) and representative images (right) of micronuclei in HCT116 cells treated with DMSO or 63 nM TH9619 inhibitor for 96 hours. CREST is shown in magenta and DAPI in gray; non-confocal mode, scale bar 10 μm. For the quantification, means + s.d. (n = 3), a minimum of 3500 cells per replicate were analyzed, unpaired two-tailed t-test. f Comparison of the log2 mean intensity of nuclear levels of H4K20me1 in HCT116 cells treated with DMSO or the indicated concentrations of TH9619 inhibitor for 96 hours; unpaired two-tailed Wilcoxon test. Representative images are shown on the right; non-confocal mode, scale bar 10 μm. Source data are provided as a Source Data file. The n indicates the number of biological sample replicates for a–c, e; and the number of individual cells for f, where 3 biological replicates were pooled.
These results indicate that nuclear MTHFD2 enzymatic activity is required for proper centromeric histone methylation and successful mitotic completion.
Discussion
Nuclear metabolism is an emerging field of biology1,2,3,4,5. The evidence that metabolic reactions can happen in unexpected cellular compartments, such as the nucleus, is revolutionizing the classical idea of central metabolism and suggests an on-demand production of metabolites to satisfy precise cellular needs. Clear examples of nuclear metabolism events are the production of nuclear acetyl-CoA6,8,9, ATP70, SAM7, and nucleotides43.
In the last decade, the folate enzyme MTHFD2 has become a promising therapeutic target, since it is upregulated and supports cancer progression in a wide range of cancer types11,15,16,17,19,20,21. Consequently, several MTHFD2 inhibitors have been already developed with favorable pre-clinical results12,13,14. The canonical function of MTHFD2 appears to contribute to tumorigenesis by providing substrates for de novo synthesis of purines and pyrimidines71, or by maintaining the redox state of the cell16,72. However, MTHFD2 does not perform an exclusive enzymatic reaction within the folate pathway and its catalytic activities are shared with other members of the MTHFD family10. Therefore, it remains unclear why cancer cells show increased expression levels of MTHFD2.
Recent publications have shown that MTHFD2 can localize within the nuclear environment22,23,43,73, where it might regulate RNA translation and metabolism74, DNA damage repair73 or even bind to chromatin43. However, a detailed understanding of the cellular processes determining the need for nuclear MTHFD2, and whether its enzymatic function is necessary, has not yet been achieved. Here, we demonstrated that enzymatically active MTHFD2 is required to maintain methylation of centromeres, thus preventing their transcriptional upregulation. Genetic or pharmacological disruption of this safeguard mechanism leads to aberrant mitosis, resulting in chromosomal instability that compromises cancer cell survival.
We characterized the MTHFD2 nuclear interactome by coupling the pull-down of the enzyme following subcellular fractionation to mass spectrometry. Classification of the nuclear MTHFD2 interactors into functional categories revealed a possible role for nuclear MTHFD2 regulating cell division (Fig. 3a). Transcriptomics and proteomics co-expression analyses (Fig. 3c, Supplementary Fig. 3f) not only supported the connection between MTHFD2 and cell division, but also pointed towards specific mitotic processes involving chromosome segregation in anaphase. Interestingly, a potential cell cycle-related role of MTHFD2 has been previously suggested23,75, although never fully addressed.
The transcriptomics analysis showed that MTHFD2 loss provokes overexpression of the centromeric regions (Fig. 4c, d). Given that alterations of centromere expression are associated with kinetochore malfunctioning and mitotic aberrations38,60,61,62, we reasoned that centromere overexpression may be a result of chromatin decompaction following MTHFD2 KO. The methyltransferase KMT5A is a nuclear MTHFD2 interactor (Fig. 3a) and the writer of H4K20me139, whose deposition at the CENP-A centromeric nucleosome is essential for kinetochore assembly29,39,53. Nuclear levels of H4K20me1 significantly decreased in the absence of MTHFD2 (Fig. 4e, Supplementary Fig. 6b, d). When specifically checking the centromeric regions, we observed that the methylation defect goes beyond H4K20me1 and affects the levels of the repressive marks H3K9me3 and H3K27me3 (Fig. 4f–h), supporting the hypothesis of a chromatin demethylation-mediated centromeric upregulation. Besides, we observed an impaired signal of CENP-A at centromeric regions (Supplementary Fig. 5n, o). Upregulation of CENP-A has previously been associated with centromeric instability63,76. Given that the expression of alpha-satellite regions at centromeres determines CENP-A recruitment38, we speculate that their overexpression may lead to CENP-A centromeric enrichment. In addition, we observed a decrease in phosphorylated Aurora kinases on mitotic chromosomes (Supplementary Fig. 7j, k), confirming that the absence of MTHFD2 promotes centromeric and kinetochore defects.
DNA methylation was also largely reduced in the absence of MTHFD2 (Fig. 4i, Supplementary Fig. 7e), and this reduction was general rather than centromere specific. DNA hypomethylation has been widely linked to chromosomal instability33,34,35,36. Interestingly, the loss of function of DNMT3B, a nuclear interactor of MTHFD2 (Fig. 3a), drives chromosomal instability through DNA hypomethylation77,78.
A karyotype analysis showed that MTHFD2 KO cells had pericentromeric chromosomal defects (Fig. 4j, Supplementary Fig. 8a–c), and whole-genome Nanopore sequencing revealed that they accumulate more structural variants than their WT counterpart (Fig. 4k, l). Structural variants are incorporated in the DNA during DNA replication and are cell division dependent. MTHFD2 KO cells showed reduced cell proliferation (Supplementary Fig. 5d) when compared to the WT cells. Hence, correcting for the proliferation rate of each cell population may even increase the divergence in structural variation accumulation observed between the MTHFD2 WT and KO cells.
Supporting the role of MTHFD2 in centromere and chromosomal stability, in the absence of MTHFD2, we observed concrete mitotic defects, such as defects in chromosome congression (Fig. 5d, e) and segregation (Fig. 5f, g, Supplementary Fig. 9d, e), along with an increased number of centromere-containing micronuclei (Supplementary Fig. 9f, g). With an orthogonal functional genomics approach, we discovered that the mitotic players WAPL and TOP2A are, respectively, synthetically viable and synthetically lethal with MTHFD2 KO (Fig. 5h). WAPL depletion induces chromatin condensation by locking Cohesin on the DNA66. Thus, the absence of WAPL could compensate for the MTHFD2 loss-driven chromatin decompaction caused by DNA and histone methylation defects. On the contrary, TOP2A loss induces chromosome structural defects and mitotic delay67, which would exacerbate the hypomethylation and chromosomal instability phenotype observed in the absence of MTHFD2, explaining the synthetic lethality. However, TOP2A also has key functions in DNA replication and repair, which cannot be excluded as part of the synthetic lethality. Indeed, MTHFD2 is also crucial for DNA replication and repair, since it provides metabolites for the de novo pyrimidine synthesis14, suggesting a profound role of MTHFD2 in regulating DNA replication, DNA damage repair, and mitosis progression, which are inherently interrelated processes.
To show that the nuclear pool of MTHFD2 is crucial for the proper progression of mitosis, we demonstrated that nuclear-exclusive MTHFD2 is sufficient to prevent mitotic index defects (Fig. 6a), maintain WT levels of H4K20me1 (Fig. 6d), and prevent anaphase defects (Fig. 6c). Interestingly, these results suggest that the mitochondrial function of MTHFD2 is dispensable for correct mitosis progression.
To test whether the enzymatic activity of MTHF7D2 is required in the nucleus, we used a recently published MTHFD2 inhibitor14 that cannot pass the mitochondrial membrane, thus behaving as a nuclear MTHFD2 inhibitor69. The compound can also inhibit MTHFD1, the cytoplasmic MTHFD2 homolog, and induces cell death by provoking an imbalance of cytoplasmic folate derivatives69. When cells were cultured in the presence of thymidine, which counteracts MTHFD1 inhibition, we observed a reduction in the cellular mitotic index (Fig. 7a), an increase in mitotic defects (Fig. 7c–e) and a decrease in H4K20me1 levels (Fig. 7f), recapitulating the phenotype triggered by the loss of MTHFD2.
Intriguingly, the product of MTHFD2, 5,10-methylenetetrahydrofolate, per se cannot directly contribute to DNA or histone methylation, which requires SAM. 5,10-methylenetetrahydrofolate needs to be converted into SAM by two sequential catalytic reactions performed by Methylenetetrahydrofolate Reductase (MTHFR) and Methionine Synthase (MTR)10, which were not found among our MTHFD2 interactors, nor on chromatin43. This apparent discrepancy suggests that the nuclear enzymatic function of MTHFD2 may be required to keep a suitable metabolic environment that ensures adequate centromeric methylation and stability, rather than to methylate centromeres directly. Additionally, the treatment with MTHFD2-downstream metabolites did not rescue the mitotic index defect observed in MTHFD2 KO cells (Supplementary Fig. 12f). These results suggest that MTHFD2 might fulfill a precise local metabolic requirement that cannot be fully met by increasing absolute metabolite levels, underlying the fundamental importance of nuclear metabolism compartmentalization. Our findings extend the role of MTHFD2 to regulate methylation not only at the mRNA level, as previously described in kidney cancer79, but also at the histone and DNA levels. Most likely, MTHFD2 fulfills compartmentalized metabolic demands required for specific biological processes, such as methylation reactions of macromolecules.
While nuclear MTHFD2 levels are low in cancer cells, its nuclear localization is evident in healthy proliferating colon organoids (Fig. 2a, b). Given that MTHFD2 maintains self-renewal of mouse embryonic stem cells and facilitates reprogramming of induced pluripotent stem cells80, it would be very interesting to further investigate the role of MTHFD2 in healthy proliferating cells through the lens of subcellular compartments. In this regard, it would be intriguing to explore the possibility that nuclear MTHFD2 controls the nuclear redox balance using NAD+/NADH to favor either proliferation or quiescence, or that it may be involved in the maintenance of telomeres, which dictate a cell’s ability to proliferate.
Methods
This research complies with all relevant ethical and biosecurity regulations. Organoid lines, from either colorectal cancer tissue or of healthy colon tissue, were established by Tissue Engineering Unit at CRG following the approved PMAR Ethics and Scientific Committees (CEIC) n 2021/9796 from surgery biopsies provided by MARbiobanc (CarlosIII Health/FEDER PT20/00023 and XBTC). Experiments involving lentiviral particles were conducted following the protocols CBS19_005A and CBS24_006A approved by the Barcelona Biomedical Research Park biosafety Committee (CBS-PRBB).
Cell culture
A549 (ATCC; #CCL-185), BT-549 (ATCC; #HTB-122), EBC-1 (Cellosaurus; #CVCL_2891), HCT 116 (ATCC; #CCL-247), HEK293T (ATCC; #CRL-3216), HT-29 (ATCC; #HTB-38), MCF7 (ATCC; #HTB-22), MDA-MB-231 (ATCC; #HTB-26), RKO (ATCC; #CRL-2577), SK-BR-3 (ATCC; #HTB-30), SW480 (ATCC; #CCL-228), SW620 (ATCC; #CCL-227), T-47D (ATCC; #HTB-133) and U-2 OS (ATCC; #HTB-96) cells were cultured in DMEM (Gibco; #11966025) supplemented with 10% fetal bovine serum (FBS) (Gibco; #10270106) and 1% penicillin/streptomycin (Gibco; #15140122) at 37 °C in 5% CO2. H1437 (ATCC; #CRL-5872), H226 (ATCC; #CRL-5826) and H358 (ATCC; #CRL-5807) cells were cultured in RPMI GlutaMAX (Gibco; #61870036) supplemented with 10% FBS (Gibco; #10270106) and 1% penicillin/streptomycin (Gibco; #15140122) at 37 °C in 5% CO2. Cell cultures were tested every month for mycoplasma contamination.
Plasmids and primers
The plasmids and primers (including gene blocks) used in this study are listed in Supplementary Methods Table 1 and 2, respectively.
To generate the plasmids needed for CRISPR-Cas9 KO/knock-in, Brand and Winter protocol was followed81. Briefly, to obtain the cutting vectors sgMTHFD2ex4_GW223 and sgMTHFD2int1-2_GW223, primers with sense and antisense sgRNA sequences (primers 1-4) were designed to generate the sgRNA. The cutting vector GW223_pX330A_sgX_sgPITCh (2 μg) was digested with BbsI-HF (New England Biolabs; #R3539) in Cutsmart Buffer (New England Biolabs; #B6004) for 1 hour, dephosphorylated with Shrimp Alkaline Phosphatase (rSAP) (New England Biolabs; #M0371) and gel purified with the QIAquick PCR & Gel Cleanup Kit (Qiagen; #28506). Sense and antisense oligos were annealed with T4 Polynucleotide Kinase (PNK) (New England Biolabs; #M0201) and ligated with the digested cutting plasmid with T4 DNA ligase (New England Biolabs; #M0202). The ligated fragments were transformed into DH5α E. coli competent cells (Thermo Fisher Scientific; #18265017) and single colonies were analyzed with Sanger sequencing (Eurofins) to select positive clones (primer 5).
To obtain the repair vector 3xNLS-dTAG-GFP-NEO_GW209 several steps were followed. To obtain first the 1xNLS-dTAG-GFP_GW209 plasmid, the repair vector GW209_pCRIS-PITChv2-C-dTAG-Puro (BRD4) (2 μg) was digested with MluI-HF (New England Biolabs; #R3198) in Cutsmart Buffer for 1 hour, dephosphorylated with rSAP and gel purified with the QIAquick PCR & Gel Cleanup Kit. A gene block (Integrated DNA Technologies) with the repair sequence was designed (dTAG_GFP_NLS_MTHFD2) and cloned into the digested repair vector using the Gibson reaction approach for 2 hours at 50 °C, followed by DH5α E. coli cells transformation. Single clones were Sanger sequenced (primers 6-8). This plasmid 1xNLS-dTAG-GFP_GW209 was used as a template to amplify the sequence of FKBP-V-GFP-NLS (primers 9-10) and the plasmid pcDNA3-hLOXL2 was used to amplify the SV40Pr-Neo-term sequence (primers 11-12) using the Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific; #F530). These two PCR products were cloned into the digested repair vector GW209_pCRIS-PITChv2-C-dTAG-Puro (BRD4) using the Gibson reaction approach for 2 hours at 50 °C, followed by DH5α E. coli cells transformation, generating the vector 1xNLS-dTAG-GFP-NEO_GW209. To introduce a 3xNLS sequence, the vector 1xNLS-dTAG-GFP-NEO_GW209 was sequentially cut with AarI (Thermo Fisher Scientific; #ER1581) and BsmBI (New England Biolabs; #R0580) and gel purified. The obtained backbone was ligated via custom Gibson reaction with the gene block 3xNLS (Integrated DNA Technologies) for 2 hours at 50 °C. Afterward, the product was transformed in DH5α competent cells to obtain 3xNLS-dTAG-GFP-NEO_GW209. Single clones were Sanger sequenced (primers 6-7, 13-15).
Chromatome fractionation
1^107 cells were first lysed in CHAPS (3-cholamidopropyl dimethylammonium 1-propane sulfonate) Buffer (0.5-10% CHAPS in phosphate-buffered saline (PBS)) for 15 minutes to break the cytosolic membrane and centrifuged for 5 min at 720 g at 4 °C. The concentration of CHAPS was optimized for each cell line. The supernatant was harvested as the cytosolic fraction. The nuclear pellet was resuspended in Cytoplasmic Lysis Buffer (IGEPAL 0.1%, NaCl 150 mM, Tris-HCl 10 mM pH 7 in H2O), placed on the top of a Sucrose Gradient Buffer (NaCl 150 mM, sucrose 25%, Tris-HCl 10 mM pH 7 in H2O) and centrifuged for 5 min at 10000 g at 4 °C. Purified nuclei were then washed 3 times by resuspending in Nuclei Washing Buffer (EDTA 1 mM, IGEPAL 0.1% in PBS) and centrifuged for 5 min at 1200 g at 4 °C. Then, the washed nuclear pellet was resuspended in Nuclei Resuspension Buffer (EDTA 1 mM, NaCl 75 mM, 50% sucrose, Tris-HCl 20 mM pH 8 in H2O) and the nuclear membrane was lysed by adding Nuclei Lysis Buffer (EDTA 0.2 mM, HEPES 20 mM pH 7.5, IGEPAL 0.1%, NaCl 300 mM in H2O), vortexing and incubating for 5 min. After centrifugation for 2 min at 16,000 g at 4 °C, the resulting chromatin was resuspended in Benzonase Digestion Buffer (15 mM HEPES pH 7.5, 0.1% IGEPAL, TPCK 5 μg/mL) and sonicated on a Bioruptor Pico (Diagenode) for 15 cycles 30 sec ON/30 sec OFF in 1.5 mL Diagenode tubes (Diagenode; #C30010016). Finally, sonicated chromatin was digested with benzonase enzyme (VWR; #706643; 2.5U) for 30 min at room temperature, and the resulting sample was harvested as chromatome fraction. All the steps were performed on ice and all buffers were supplemented with proteinase inhibitors (Roche; #4693132001). Cytosolic and chromatome extracts were quantified with Pierce BCA Protein Assay Kit (Thermo Scientific; #PIER23225).
SDS-electrophoresis and Western blot
Samples were mixed with 4X Laemmli sample buffer (Bio-Rad; #1610747) and boiled at 95 °C for 5 min. Proteins were separated by sodium dodecyl-sulfate (SDS)–polyacrylamide gel electrophoresis, and transferred to a nitrocellulose membrane by wet transfer (10% Transfer buffer, 20% methanol in H2O). Membranes were blocked in 5% milk (Millipore; #70166) in 0.05% Tween20 in PBS for 1 hour at room temperature. Membranes were incubated with primary antibodies prepared in 0.05% Tween20 in PBS overnight at 4 °C. Fluorescent secondary antibodies rabbit-800 (Thermo Fisher Scientific; #A32735; 1:10000) and mouse-680 (Thermo Fisher Scientific; #A21058; 1:10000) were also prepared in 0.05% Tween20 in PBS and incubated for 1 hour at room temperature. Three washes after the primary and secondary antibodies were performed with 0.05% Tween20 in PBS. Detection was achieved with Odyssey CLx (Li-Cor) and analyzed with Image Studio Lite (version 5.2.5).
For the chromatome and nuclear enrichment experiments, the following primary antibodies were used: MTHFD2 (Abcam; #ab151447; 1:1000), Vinculin (Cell Signaling; #13901S; 1:1000), FDX1 (Thermo Fisher Scientific; #PA559653; 1:1000) and H3 (Cell Signaling; #14269S; 1:1000).
For the immunoprecipitation and co-immunoprecipitation experiments, the following primary antibodies were used: MTHFD2 (Abcam; #ab151447; 1:1000), MTHFD2 (Abcam; #ab56772; 1:700), TPR (Santa Cruz Biotechnology; #sc-101294; 1:1000), KIF4A (Santa Cruz Biotechnology; #sc-365144; 1:1000), PRMT1 (Santa Cruz Biotechnology; #sc-166963; 1:1000), KMT5A (Thermo Fisher Scientific; #PA5-31467; 1:2000), H4K20me1 (Diagenode; #C15410034; 1:1000), H3K9me3 (Diagenode; #C15410193; 1:1000), H3K27me3 (Diagenode; #C15410195; 1:1000), H3 (Cell Signaling; #14269S; 1:1000) and H4 (Cell Signaling; #13919; 1:1000).
For experiments with whole-cell extracts, these were obtained using an SDS lysis buffer (2% SDS, 50 mM Tris-HCl pH 7, 10% glycerol in H2O), and the following primary antibodies were used: MTHFD2 (Abcam; #ab151447; 1:1000), Vinculin (Cell Signaling; #13901S; 1:1000), CENPA (Thermo Fisher Scientific; #PA5-17194; 1:1000) and CENPC (Thermo Fisher Scientific; #MA5-34742; 1:1000). All the raw western blot images are provided in the Source Data file.
Immunofluorescence
Immunofluorescence experiments were performed by seeding cells on clear flat-bottom 96-well plates (Perkin Elmer; #6055302) and fixing them with 4% formaldehyde (Thermo Fisher Scientific; #28908) for 15 min at room temperature. Permeabilization was performed using 0.2% Triton X-100 in PBS for 30 min, followed by blocking with 2% bovine serum albumin (BSA) in PBS for 45 min. Cells were incubated first with primary antibodies for 1 hour at room temperature. The following antibodies were used: MTHFD2 (Abcam; #ab151447; 1:500), H4K20me1 (Diagenode; #C15410034; 1:500), H3K9me3 (Diagenode; #C15410193; 1:1000), H3K27me3 (Diagenode; #C15410195; 1:200), CREST (AntibodiesInc; #15-234; 1:500), CENPA (Thermo Fisher Scientific; #PA5-17194; 1:200), phospho-Aurora A/B/C (Cell Signaling; #2914; 1:150), H3PS10 (Sigma-Aldrich; #06-570; 1:500) and α-tubulin (Sigma-Aldrich; #T9026; 1:500). After washing, cells were incubated with secondary antibodies for 1 hour at room temperature in the dark. The following antibodies were used: Alexa Fluor 488 donkey anti-rabbit (Thermo Fisher Scientific; #A21206; 1:1000), Alexa Fluor 647 goat anti-rabbit (Thermo Fisher Scientific; #A21244; 1:1000), Alexa Fluor 555 goat anti-mouse (Thermo Fisher Scientific; #A21058; 1:1000) and Alexa Fluor 647 goat anti-human (Thermo Fisher Scientific; #A21445; 1:1000). Finally, cells were incubated with DAPI (4,6-diamidino-2-phenylindole) (Sigma-Aldrich; #MBD0015; 1:1000) for 5 min at room temperature in the dark (except FUCCI cells). After the incubations with antibodies and DAPI, cells were washed twice with 0.05% Tween20 in PBS and once with PBS. Images were taken with the Operetta High Content Screening System (PerkinElmer) using a 10X, 40X or 63X objective and non-confocal or confocal mode. Images were quantified using the Harmony software (version 4.9), first by identifying the nuclei and/or cytosol and then quantifying their properties (mean intensity, integrated intensity and/or coefficient of variation).
For the quantification of the epigenetic marks and DAPI, the individual mean intensity and coefficient of variation of H4K20me1, H3K9me3, H3K27me3 and DAPI were considered. In the unsynchronized cultures, mitotic cells were excluded by filtering by nuclear roundness and DAPI intensity. For identifying the cell cycle phase in unsynchronized cultures, DAPI integrated intensity was used. Epigenetic marks and DAPI were also measured in synchronized cultures after treating HCT116 WT and KO cells with either DMSO (PanReac AppliChem; #A3672), thymidine 2 mM for 24 hours (MedChem Express; #HY-N1150) or RO-3306 9 μM for 20 hours (MedChem Express; #HY-12529). For the centromeric analysis, CENPA and CREST spots were obtained and quantified using a Harmony pipeline for spots identification, and DAPI integrated intensity was used to identify the cell cycle phase. For the quantification of phospho-Aurora A/B/C, prometaphase cells were analyzed and identified as those with the 1% highest phospho-Aurora A/B/C signal. The signal was further normalized to the mean phospho-Aurora A/B/C signal of all cells to eliminate antibody background bias.
For calculating the mitotic index, individual cells with a signal higher than 3 standard deviations of the average H3PS10 mean intensity were considered mitotic cells. The classification of mitotic cells and the identification and quantification of mitotic defects were performed with ImageJ (version 1.52q) using the DAPI, H3PS10, CREST, and α-tubulin staining.
To quantify the percentage of survival of HCT116 cells after TH9619 treatment, cells were fixed and permeabilized as previously indicated and directly stained with DAPI for 5 minutes. Using the Harmony software, the number of nuclei in each well was quantified and used as a proxy for cell survival.
STORM – Immunolabeling and imaging
HCT116 WT cells were seeded at a density of 60,000 cells per well per 350 µl onto borosilicate glass bottom 8-well chamber slides (Lab-Tek; #155411) overnight. Cells were fixed with formaldehyde 4% (Thermo Fisher Scientific; #28908) diluted in PBS for 10 min at room temperature and washed three times with PBS for 5 min each. Permeabilization was performed with 0.3% Triton X-100 in PBS, followed by blocking with 10% BSA–0.01% Triton X-100 in PBS for 1 hour at room temperature. Cells were incubated with primary antibody MTHFD2 (Abcam; #ab56772; 1:50) in blocking buffer overnight at 4 °C. Cells were washed three times with wash buffer 2% BSA–0.01% Triton X-100 in PBS for 5 min each and were then incubated with secondary antibody Alexa Fluor 647 Goat anti-Mouse IgG (H + L) (Invitrogen; #A-21235; 1:250) in blocking buffer for 45 minutes at room temperature in the dark. Picogreen® (Invitrogen; #P11495; 1:10000) in blocking buffer was added and incubated for 10 min at room temperature. Cells were washed three times with wash buffer for 5 min each.
STORM imaging was performed on an N-STORM 4.0 microscope (Nikon) equipped with a CFI HP Apochromat TIRF 100 × 1.49 oil objective and an iXon Ultra 897 camera (Andor), and using Highly Inclined and Laminated Optical (HILO) sheet illumination.
Before STORM imaging, conventional images were taken for MTHFD2 signal and for DNA signal (Picogreen labeled). STORM imaging for MTHFD2 was performed with continuous imaging acquisition (i.e. simultaneous stimulation with 405 and 647 nm lasers) with 10 ms exposure time for 60,000 frames. 647 nm laser was used at constant ~2 kW/cm2 power density and 405 nm was gradually increased over the imaging. The composition of the imaging buffer for STORM imaging was 100 mM Cysteamine MEA (Sigma-Aldrich; #30070), 5% Glucose (Sigma-Aldrich; #G8270) and 1% Glox Solution (0.5 mg/mL glucose oxidase, 40 mg/mL catalase (Sigma-Aldrich; #G2133 and #C100)) in PBS.
STORM imaging analysis and quantifications
STORM images were analyzed and rendered with Insight3 software (kind gift of Huang et al.82) as previously described83,84. Localizations were identified based on an intensity threshold (900) and fit to a simple Gaussian to determine the x and y positions. After generating MTHFD2 localization lists for each STORM image, cell masks were generated to segment the obtained localizations overlapping to DNA (Picogreen positive regions) to the ones that do not overlap (Picogreen negative but MTHDF2 positive regions). Masks generation, quantification of masks’ areas and segmentation of STORM localizations were performed in Fiji/ImageJ (version 1.52q). Masks were generated by applying an automatic threshold on conventional images based on Picogreen intensity signal and MTHFD2 intensity signal. Masks were visually inspected individually and adjusted manually in cases where dim signal or noise compromised the identification of the mask. The mask based on the MTHFD2 signal defined the cell area, while the mask based on the Picogreen signal defined the areas occupied by DNA (corresponding to the mitotic chromosomes). The number of localizations corresponding to Picogreen negative regions were derived by the subtraction of localizations corresponding to Picogreen positive regions from the total number of localizations of the cell. The same was done for the calculation of the size of the areas corresponding to Picogreen negative regions. Finally, the density of localizations (number of localizations/area of the mask) for the areas inside and outside DNA masks was calculated. MatLab software used for imaging data analysis can be found at: https://www.mathworks.com/products/matlab.html.
Patient-derived tissue and organoids immunofluorescence
Patient and ethics statement
Colonic cancer resections were provided by MARBiobanc (CarlosIII Health/FEDER PT20/00023 and XBTC) from surgery biopsies following approved PMAR Ethics and Scientific Committees (CEIC) n 2021/9796 and patient written informed consent. Patient (female, woman; white, caucasian) was diagnosed with rectum-sigmoid adenocarcinoma. From the resected colon segment, normal tissue as well as tumor was isolated.
Organoid culture
Organoid lines, from either colorectal cancer tissue or healthy colon tissue, were established by the Tissue Engineering Unit at CRG. Tumor specimens were obtained from Hospital del Mar and promptly transferred to cold PBS with a cocktail of 2.5 µg/mL Amphotericin B (Thermo Fisher Scientific; #15290-018), 100 µg/mL penicillin/streptomycin (Thermo Fisher Scientific; #15140-122) and 0.1 mg/mL primocin (Invivogen; #ant-pm-05). The isolation of healthy crypts and tumor epithelium was performed as described in Sato et al.85. Briefly, biopsy specimens were chopped into 5-mm pieces and digested by incubating in 5 mg/mL Collagenase IV (Thermo; #17104019) for 60 minutes at 37 °C. After digestion, isolated intestinal crypts were embedded in 40 µL of Matrigel Growth Factor Reduced (Corning; #354230) and seeded in 24-well plates (1000 crypts/40 µL Matrigel dome). Matrigel was polymerized for 10 minutes at 37 °C and 700 µL of either healthy or tumor culture medium was overlaid on the dome. For healthy patient-derived organoids (PDOs), Human IntestiCult Organoid Growth Medium (OGM) (STEMCELL Technologies; #06010) was used. For tumor PDOs, Oncopro Tumorid Culture Medium Kit (Thermo; #A5701201) was used.
Organoid differentiation
Healthy colon organoids were differentiated in 3D using Human IntestiCult Organoid Differentiation Medium (ODM) (STEMCELL Technologies; #100-0214) supplemented with 5 µM DAPT (Selleck Chem; #S2215) for 5 days, performing a full-medium change every 2 days.
Immunofluorescence
Patient-derived organoids at 10 days of culture, including 5 days of differentiation for differentiated organoids, were collected and incubated in ice-cold Corning Cell Recovery Solution (Corning; #345235) for 1 hour at 0 °C for Matrigel to dissolve. Organoids were then fixed in 4% paraformaldehyde (Sigma; #P6148) for 1 hour at 4 °C. After fixation, cells were washed twice with PBS and permeabilized with 1% Triton X-100 for 30 min at room temperature. Organoids were blocked 1 hour at 4 °C in PBS + 0.1% BSA (Sigma; #A2058), 0.2% Triton X-100 + 0.1% Tween20 (Applichem; #A13890500). Organoids were stained for 48 h at 4 °C for primary antibodies and 24 h for secondary antibodies. The following primary antibodies were used: MTHFD2 (Abcam; #ab151447; 1:200), VDAC1/Porin (Santa Cruz Biotechnology; #sc-390996; 1:50), MUC2 (Novus Bio; #NB120-11197; 1:50) and Ki67 (Abcam; #ab254123; 1:100). The following secondary antibodies were used: goat anti-rabbit 488 (Thermo Fisher Scientific; #A-11008; 1:1000), goat anti-rabbit 555 (Thermo Fisher Scientific; #A-21428; 1:1000), goat anti-mouse 555 (Thermo Fisher Scientific; #A-28180; 1:1000), goat anti-mouse 647 (Thermo Fisher Scientific; #A-21463; 1:1000) and goat anti-chicken 488 (Thermo Fisher Scientific; #A-11039; 1:1000). Nuclei were visualized with DAPI (Biogen Cientifica; #BT-40043; 1:1000) and samples mounted with Vectashield Antifade Mounting Medium (Vector Lab; #H1200). Imaging was performed with the Leica SP8 confocal microscope.
Pull-down – Mass Spectrometry
Sample preparation
For pull-down experiments after chromatome fractionation, Protein G Dynabeads (Thermo Scientific; #10004D) were incubated for 6 hours on a rotating wheel at 4 °C with primary antibodies MTHFD2 (Abcam; #ab151447; 5 μg) or negative control IgG (Sigma-Aldrich; #I5006; 5 μg). Then, antibody-bound beads were incubated with 2 mg of cytosolic or chromatome extracts overnight on a rotating wheel at 4 °C. The complexes were then washed three times with Nuclei Wash Buffer (EDTA 1 mM, IGEPAL 0.1% in PBS). Beads used in the immunoprecipitations were washed three times with 200 mM Ammonium Bicarbonate (ABC) and resuspended in Urea 6M-ABC. Samples were then reduced with dithiothreitol in ABC (30 nM, 37 °C, 60 min), alkylated in the dark with iodoacetamide in ABC (60 nM, 25 °C, 30 min), and diluted to 1 M Urea with 200 mM ABC for trypsin digestion (1 μg, 37 °C, overnight shaking, Promega, #V5113). On the next day, beads were separated from the digested extract with a magnet and the peptide mix was acidified with formic acid and desalted with a MicroSpin C18 column (The Nest Group; #SUM SS18V) before LC-MS/MS analysis. Three independent biological replicates for each immunoprecipitation were processed.
Chromatographic and mass spectrometric (MS) analysis
Samples were analyzed using an LTQ-Orbitrap Eclipse mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) coupled to an EASY-nLC 1200 (Thermo Fisher Scientific (Proxeon), Odense, Denmark). Peptides were loaded directly onto the analytical column and were separated by reversed-phase chromatography using a 50-cm column with an inner diameter of 75 μm, packed with 2 μm C18 particles.
Chromatographic gradients started at 95% buffer A and 5% buffer B with a flow rate of 300 nL/min and gradually increased to 25% buffer B and 75% A in 79 min and then to 40% buffer B and 60% A in 11 min. After each analysis, the column was washed for 10 min with 100% buffer B. Buffer A: 0.1% formic acid in water. Buffer B: 0.1% formic acid in 80% acetonitrile.
The mass spectrometer was operated in positive ionization mode with nanospray voltage set at 2.4 kV and source temperature at 305 °C. The acquisition was performed in data-dependent acquisition (DDA) mode and full MS scans with 1 micro scans at a resolution of 120,000 were used over a mass range of m/z 350-1400 with detection in the Orbitrap mass analyzer. Auto gain control (AGC) was set to ‘standard’ and injection time to ‘auto’. In each cycle of data-dependent acquisition analysis, following each survey scan, the most intense ions above a threshold ion count of 10000 were selected for fragmentation. The number of selected precursor ions for fragmentation was determined by the ‘Top Speed’ acquisition algorithm and a dynamic exclusion of 60 sec. Fragment ion spectra were produced via high-energy collision dissociation (HCD) at normalized collision energy of 28% and they were acquired in the ion trap mass analyzer. AGC and injection time were set to ‘Standard’ and ‘Dynamic’, respectively, and an isolation window of 1.4 m/z was used.
Digested bovine serum albumin (New England Biolabs; #P8108S) was analyzed between each sample to avoid sample carryover and to assure the stability of the instrument, and QCloud86 was used to control instrument longitudinal performance during the project.
Data Analysis
Acquired spectra were analyzed using the Proteome Discoverer software suite v1.4 (Thermo Fisher Scientific) and the Mascot search engine87 (version 2.6, Matrix Science). The data were searched against a Swiss-Prot human database (as in June 2020) plus a list88 of common contaminants and all the corresponding decoy entries. For peptide identification, a precursor ion mass tolerance of 7 ppm was used for the MS1 level, trypsin was chosen as the enzyme and up to three missed cleavages were allowed. The fragment ion mass tolerance was set to 0.5 Da for MS2 spectra. Oxidation of methionine was set as a variable modification whereas carbamidomethylation on cysteine was set as a fixed modification. False discovery rate (FDR) in peptide identification was set to a maximum of 5%. To check the quality of the fractionation, the relative protein abundance of all proteins identified in all the different subcellular compartments was obtained for all the immunoprecipitations. The subcellular location of the proteins was retrieved from Human Protein Atlas45 (www.proteinatlas.org). The Significance Analysis of INTeractome (SAINT) express algorithm46 (v. 3.2.0) was used to score protein-protein interactions. We considered as MTHFD2 potential interactors those with a fold change >= 5 and a Bonferroni False Discovery Rate (BFDR) <= 0.2. The network analysis with MTHFD2 nuclear interactors was performed with Cytoscape89 (version 3.9.1) using IntAct47 to retrieve protein-protein interactions.
Immunoprecipitation – Western blot
For immunoprecipitation and co-immunoprecipitation experiments, cells were washed twice with cold PBS and lysed in Soft-salt Lysis Buffer (10 mM EDTA, 10% glycerol, 0.1% IGEPAL, 50 mM Tris-HCl pH 8 in H2O) for 10 minutes on ice. After centrifugation at 800 g for 15 min at 4 °C, the supernatant was harvested as the cytosolic fraction, and the nuclear pellet was lysed in High-salt Lysis Buffer (20 mM HEPES pH 7.5, 10% glycerol, 1 mM MgCl2, 350 mM NaCl, 0.5% Triton X-100 in H2O) for 10 minutes on ice. Nuclear lysates were centrifuged at 15,700 g at 4 °C for 10 min. Balance Buffer (20 mM HEPES pH 7.5, 10 mM KCl, 1 mM MgCl2 in H2O) was then added to the resulting nuclear supernatant to reach a final NaCl concentration of 150 mM. Nuclear extracts were quantified using Pierce BCA Protein Assay Kit (Thermo Scientific; #PIER23225) and 1–2 mg of nuclear extract was incubated overnight on a rotating wheel at 4 °C with primary antibodies MTHFD2-rabbit (Abcam; #ab151447; 4 μg), MTHFD2-mouse (Abcam; #ab56772; 4 μg), H4K20me1 (Diagenode; #C15410034; 3 μg), H3K9me3 (Diagenode; #C15410193; 3 μg) and H3K27me3 (Diagenode; #C15410195; 3 μg), and the corresponding negative controls IgG-rabbit (Sigma-Aldrich; #I5006; 4 μg) or IgG-mouse (Sigma-Aldrich; #I5381; 4 μg). Before the addition of the antibodies, 10% of the sample was reserved as input. The next day, the samples were incubated with Protein G Dynabeads (Thermo Scientific; #10004D) or Protein A Dynabeads (Thermo Scientific; #10002D) for 2 hours at 4 °C. The complexes were then washed three times with Wash Buffer (20 mM HEPES pH 7.5, 10% glycerol, 1 mM MgCl2, 150 mM NaCl, 0.5% Triton X-100 in H2O) and eluted with 2X Laemmli buffer after boiling at 95 °C for 5 min.
Lentiviral production and transfection
HEK293T cells at 70% confluency were used to produce lentiviral particles. Cells were transfected with polyethyleneimine (PEI) (Polysciences; #23966-1) with pCMV-dR8_91 and pVSV-G packaging plasmids, along with the vector of interest, in OptiMeM (Gibco; #11058021). The mixtures with the vectors and PEI were incubated for 5 minutes separately and then mixed and incubated together for 20 minutes to allow complex formation. HEK293T media was changed for serum-free media and the transfection mixture was added dropwise on top. 6 hours after transfection, transfection media was replaced with fresh media. After 48 hours and 72 hours, the viral supernatant was collected and filtered with a 0.45 μm filter unit (Merck Millipore; #051338), and viral aliquots were stored at −80 °C until use.
FUCCI cells generation
To generate stable U2OS and MCF7 cell lines with a Fluorescent Ubiquitination-based Cell Cycle Indicator (FUCCI) system, U2OS and MCF7 cells were transduced with viral particles containing the vectors pLL3.7m-mTurquoise2-SLBP(18-126)-Neomycinin and pLL3.7m-Clover-Geminin(1-110)-IRES-mKO2-Cdt(30-120)-Hygromycin in the presence of polybrene (Sigma-Aldrich; #TR1003G; 10 μg/mL). Since these vectors contained neomycin- and hygromycin-resistance cassettes, respectively, 24 hours after transduction, the media was replaced with fresh media containing 200 μg/mL Geneticin (Thermo Scientific; #10131035) or 150 μg/mL hygromycin (Sigma-Aldrich; #H3274), respectively. Antibiotic selection lasted 4–7 days. Transduced cells were further selected through FACS sorting (BD Influx) to keep cells that showed proper activation and degradation of the FUCCI system. The FUCCI system used is an adaptation of FUCCI456 to show 3 cell cycle-regulated fusion proteins: Clover-Geminin, SLBP-Turquoise2, and Cdt1-mKO257, which allows to distinguish the G1, S, G2, and G2-M phases as described in Bajar et al.56. FUCCI cells were used for immunofluorescence as previously described. Mitotic cells after the nuclear envelope breakdown were excluded by filtering by nuclear roundness. Cells with the highest MTHFD2 staining (top 5%) in the nucleus or the cytosol were filtered and its cell cycle phase distribution was represented by a density plot.
MTHFD2 KO and 3xNLS-MTHFD2 knock-in generation
HCT116 cells were nucleofected using the Lonza Amaxa Kit V (Lonza; #VCA-1003) and Amaxa Nucleofector (Lonza) following the HCT116 protocol. Briefly, 2^106 trypsinized cells were resuspended in a supplemented nucleofector solution and nucleofected using the D-32 program.
For the MTHFD2 KO, cells were nucleofected with 12 μg of the sgMTHFD2ex4_GW223 cutting vector, which contains Cas9. Two days after nucleofection, single cells were seeded in 96-well plates for isolating single clones. Single clones were tested by Western blot. From the clones tested, we kept two clones that were homozygous KO. These two KO clones were further validated by immunofluorescence and by Sanger sequencing (Eurofins, primers 16-17).
For the 3xNLS-MTHFD2 knock-in, cells were nucleofected with 6 μg of the sgMTHFD2int1-2_GW223 cutting vector (containing Cas9) and 6 μg of the 3xNLS-dTAG-GFP-NEO_GW209 repair vector, following the intron-tagging strategy described by Serebrenik et al.90. Since the repair vector contained a neomycin-resistance cassette, 3 days post-nucleofection cells were treated with 800 μg/mL Geneticin (Thermo Scientific; #10131035) for 7 days. After antibiotic selection, single cells were seeded in 96-well plates for isolating single clones. Single clones were tested by immunofluorescence, and one clone with a homozygous knock-in was kept after Sanger validation (primers 18-19).
Cellular assays: growth rate, invasion, and clonogenic assays
For the growth rate assay, HCT116 MTHFD2 WT, KO and NLS cells were seeded in 12-well plates, fixed with formalin (Sigma-Aldrich; #HT501128) at days 0, 2, 4, and 6, and stained with 0.1% crystal violet solution (Sigma-Aldrich; #HT90132). Wells were then solubilized with 10% acetic acid and measured at 590 nm in a TECAN Infinite M200 Plate Reader.
For the invasion assay, Corning 24-well plates with transwells were used (Corning; #3422). Matrigel (Corning; #354230; 0.3 mg/mL) was seeded in the transwells and left to solidify. After gel formation, HCT116 MTHFD2 WT and KO cells were seeded on the top of the matrigel in serum-free media. After 48 hours, transwells were fixed in 4% formaldehyde (Thermo Fisher Scientific; #28908), permeabilized in 0.1% Triton X-100, 2% BSA in PBS for 15 minutes and stained with DAPI (Sigma-Aldrich; #MBD0015; 1:1000) for 5 minutes. The bottom of the transwells was imaged with a Leica DMI6000B microscope and DAPI mean intensity was used to quantify the number of invading cells.
For the clonogenic assay, HCT116 MTHFD2 WT and KO cells were seeded at a low dilution in 24-well plates. After 15 days, colonies were fixed with formalin (Sigma-Aldrich; #HT501128) and stained with 0.1% crystal violet solution (Sigma-Aldrich; #HT90132). The plates were scanned, and the area covered by colonies was obtained with the ImageJ plugin ColonyArea91.
RNA-sequencing
Sample preparation
Three biological replicates were obtained from 2.5^106 HCT116 MTHFD2 WT and KO1 cells at an early time-point and after two months of cell culture. RNA was extracted using the PureLink RNA mini kit (Thermo Fisher Scientific; #12183018 A). Libraries were prepared from 500 ng of RNA using the TruSeq stranded mRNA Library Prep (Illumina; #20020594) according to the manufacturer’s protocol, to convert total RNA into a library of template molecules of known strand origin and suitable for subsequent cluster generation and DNA sequencing. Final libraries were analyzed using the Bioanalyzer DNA 1000 (Agilent; #5067-1504) to estimate the quantity and validate the size distribution and were then quantified by qPCR using the KAPA Library Quantification Kit KK4835 (Roche; #07960204001) before the amplification with Illumina’s cBot. Libraries were sequenced on the Illumina HiSeq 2500 machine using single-read 50 bp sequencing.
Data Analysis
Quality control was performed with FastQC92 (version 0.11.9). Single-end, 50-bp-long reads were aligned to the GRCh38.p13 Homo sapiens reference genome using the STAR Aligner93 (version 2.7.6a). Gene level counts were obtained with featureCounts from subread94 (version 2.0.1), using gene annotations downloaded from Gencode (Release 38 GRCh38.p13). Differential expression analysis was performed in R (version 4.1.1) using the DESeq2 package95 (version 1.32.0). Principal Component analysis was performed with the function prcomp. The lfcShrink function from DESeq2 with the apeglm method96 was used for visualization purposes. Genes with adjusted p-value < 0.05 were considered differentially expressed. Those differentially expressed genes that were shared in both time points, and with an absolute log2 Fold Change > 0.58 in at least one time point were further considered for functional enrichment analysis. Gene Ontology enrichment analysis was performed using the ClusterProfiler97 package (version 4.0.5). Gene Set Enrichment Analysis (GSEA) was performed with GSEA software98 (version 4.1.0) using the ‘hallmark’ gene set from the MSigDB collection. For the centromeric analysis, centromeric coordinates of the assembly GRCh38 were retrieved from the Table Browser of UCSC (University of California Santa Cruz) and the intersect was obtained with bedtools (v. 2.30.0)99.
RNA extraction and RT-qPCR
For unsynchronized cell populations or cells synchronized at late G2 after treatment with 9 μM RO-3306 (MedChem Express; #HY-12529) for 20 hours, RNA was extracted using the PureLink RNA Mini Kit (Thermo Fisher Scientific; #12183018 A) and converted into cDNA using the High-Capacity RNA-to-DNA kit (Applied Biosystems; #4387406). Quantitative PCR was performed using the Power SYBR Green PCR Master Mix (Applied Biosystems; #4367659) in a ViiA 7 Real-Time PCR System (Thermo Fisher Scientific). Results were analyzed with the Design and Analysis Software QuantStudio 6/7 Pro systems (Thermo Fisher Scientific, version 2.6). Primers 20-59 (Supplementary Methods Table 2) were used, obtained from Contreras-Galindo et al.100.
Chromatin Immunoprecipitation (ChIP)-qPCR
20^106 HCT116 MTHFD2 WT and KO1 cells were crosslinked by adding on the plate culture media with 1% formaldehyde (Thermo Fisher Scientific; #28908) for 10 min at room temperature shaking. Crosslinking was stopped by adding glycine at a final concentration of 0.125 M for 5 min at room temperature shaking. Crosslinked cells were washed twice with PBS, scrapped in PBS with proteinase inhibitors (Roche; #4693132001), and collected by centrifugation for 5 min at 1500 g 4 °C. Cell pellets were lysed in Nuclei Lysis Buffer (10 mM EDTA, 1% SDS, 50 mM Tris-HCl pH 8 in H2O) supplemented with proteinase inhibitors. Nuclear extracts were sonicated on a Bioruptor Pico (Diagenode) for 10 cycles 30 sec ON/30 sec OFF in 1.5 mL Diagenode tubes (Diagenode; #C30010016) to generate 100–400 bp DNA fragments. Sonicated extracts were centrifuged for 15 minutes at 15,700 g 4 °C to remove insoluble material, and the supernatant was kept and diluted 1:10 in Immunoprecipitation Buffer (1.2 mM EDTA, 167 mM NaCl, 16.7 mM Tris-HCl pH 8, 1.1% Triton X-100 in H2O) supplemented with proteinase inhibitors. 20 μg of chromatin were used for each immunoprecipitation, which was incubated overnight on a rotatory wheel at 4 °C with the primary antibodies H4K20me1 (Diagenode; #C15410034; 2 μg), H3K9me3 (Diagenode; #C15410193; 2 μg), H3K27me3 (Diagenode; #C15410195; 2 μg) and IgG-rabbit (Sigma-Aldrich; #I5006; 2 μg) as a negative control. Before the addition of the antibodies, 10% of each sample was reserved as input. The next day, the samples were incubated with Protein A Dynabeads (Thermo Scientific; #10002D) for 2 hours at 4 °C. The complexes were then washed four times with four wash buffers: Wash Buffer 1 (2 mM EDTA, 150 mM NaCl, 0.1% SDS, 20 mM Tris-HCl pH 8, 1% Triton X-100 in H2O), Wash Buffer 2 (2 mM EDTA, 400 mM NaCl, 0.1% SDS, 20 mM Tris-HCl pH 8, 1% Triton X-100 in H2O), Wash Buffer 3 (1 mM EDTA, 1% IGEPAL, 0.25 M LiCl, 1% NaDOC, 10 mM Tris-HCl in H2O) and TE Buffer (1 mM EDTA, 10 mM Tris-HCl in H2O). Beads were resuspended in 200 μL of freshly-prepared ChIP Elution Buffer (0.1 M NaHCO3, 1% SDS in H2O) and incubated at 65 °C for 1 hour. Then, beads were removed from the chipped chromatin with the magnet, and the immunoprecipitated chromatin was de-crosslinked by adding 200 mM NaCl and incubating at 65 °C overnight. On the next day, RNA and protein were digested in 40 mM Tris-HCl pH 6 and 10 mM EDTA by adding first RNAse A (Qiagen; #19101, 1.5 hours, 37 °C), followed by Proteinase K (Thermo Fisher Scientific; #EO0491, 1 hour, 45 °C). Inputs were incubated in parallel during the de-crosslinking step. Finally, DNA was purified using a MiniElute PCR Purification Kit (Qiagen; #28006) and eluted in nuclease-free water, and quantitative PCR was performed using the Power SYBR Green PCR Master Mix (Applied Biosystems; #4367659) in a ViiA 7 Real-Time PCR System (Thermo Fisher Scientific). Results were analyzed with the Design and Analysis Software QuantStudio 6/7 Pro systems (Thermo Fisher Scientific, version 2.6). Primers 20-23,36-39 (Supplementary Methods Table 2) were used, obtained from Contreras-Galindo et al.100.
Nanopore whole-genome sequencing
Genomic DNA extraction
3 × 10^6 HCT116 MTHFD2 WT and KO1 frozen cell pellets were used to extract high molecular weight genomic DNA (HMW gDNA) following the manufacturer’s protocol of Nanobind tissue kit (Circulomics). The HMW gDNA eluate was quantified by Qubit DNA BR Assay kit (Thermo Fisher Scientific) and the DNA purity was evaluated using Nanodrop 2000 (Thermo Fisher Scientific) UV/Vis measurements. The HMW gDNA samples were stored at 4 °C.
Long Read Whole-genome library preparation and sequencing
After quality control of the HMW gDNA for purity, quantity, and integrity for long-read sequencing, the libraries were prepared using the 1D Sequencing kit SQK-LSK110 from Oxford Nanopore Technologies (ONT). Briefly, 4.0 μg of the DNA were DNA-repaired and DNA-end-repaired using the NEBNext FFPE DNA Repair Mix (New England Biolabs; #M6630) and the NEBNext UltraII End Repair/dA-Tailing Module (New England Biolabs; #E7546), and followed by the sequencing adaptors ligation, purified by 0.4X AMPure XP beads (Agencourt, Beckman Coulter) and eluted in Elution Buffer. The sequencing runs were performed on PromethIon 24 (ONT) using a flow cell R9.4.1 FLO-PRO 002 (ONT) and the sequencing data was collected for 110 hours. The quality parameters of the sequencing runs were monitored by the MinKNOW platform (version 21.11.7) in real-time and basecalled with modified basecalling for 5mC using Guppy (version 5.1.13).
Basecalling and mapping
Raw Nanopore data in the format of fast5 files were analyzed using Master of Pores 2101 suite performing the basecalling with Guppy (version 6.1.1) and the modified base model “dna_r9.4.1_450bps_modbases_5mc_cg_hac_prom”. The unaligned bam outputs were first converted to fastq files keeping the information about the modified bases in the header, filtered using nanoq102 for removing the reads with average coverage lower than 7, and then aligned to the human T2T (telomere to telomere) genome using Winnowmap103 (v. 2.03 parameters -y -ax map-ont), a minimap-derived aligner specifically designed for repetitive regions. Final alignment files were sorted, indexed and filtered with samtools104 for further analysis. Only primary alignments were retained.
Methylation analysis
From the final filtered alignment bam files, methylation data (counts of reads of methylated and unmethylated CpG) were obtained with modbam2bed (parameters -extended, -aggregate, -cpg) in bedmethyl format. R package methylKit105 (version 1.20.0) and custom scripts were used to analyze methylation data and transform between different formats. The significance of methylation level change between the conditions, for every CpG site, was calculated with methylKit. Those sites with the largest methylation variation were extracted, by filtering on methylation difference >= 15% and q-value < 0.05. Centromeric coordinates for each chromosome were acquired from Altemose et al.106.
Structural variation analysis
Aligned reads were fed to CuteSV tool107 (v. 2.0.0) for predicting large structural variations. These predicted variants were annotated using Ensembl Variant Effect Predictor (VEP)108 (v. 95.3). Variants were crossed with methylation information using bedtools (v. 2.30.0)99. Plots were made using scripts written with R statistical language (version 4.1.1).
Chromosome Banding Analysis of cell lines
Cytogenetic studies were performed in HCT116 MTHFD2 WT and KO cells. Cell cultures were incubated for 3-4 days at 37 °C/5% CO2 up to the time of its extraction, when the cell culture was 80% confluent. 10 μg/mL of colcemid was added to the cultures and incubated for 4 hours for arresting the cells in metaphase. After trypsinization, cells were swollen hypotonically and fixed with Carnoy (methanol:acetic acid). Then, the cells were dropped on 4-5 slides. For chromosome banding, slides with metaphase spreads were treated in a slide warmer at 100 °C for 1 hour and stained with Wright’s solution to create characteristic light and dark bands. Metaphase spreads were captured using an automated imaging system for cytogenetics (CytoInsight GSL, Leica Biosystems) and karyotyped with Cytovision Software 7.0 (Applied Imaging Corporation). Karyotypes were described following the International System for Human and Cytogenetic Nomenclature (2020). A minimum of 20 metaphases were analyzed.
Cell cycle analysis
For evaluating mitotic progression, HCT116 MTHFD2 WT and KO cells were seeded on 6-well plates and treated the day after either with dimethyl sulfoxide (DMSO) (PanReac AppliChem; #A3672) or CDK1 inhibitor RO-3306 9 μM (MedChem Express; #HY-12529) for 20 hours to synchronize cells at the end of G2 phase. Treated cells were then washed three times with PBS and released in normal media. For cells treated with DMSO, cells were harvested immediately after release. For cells treated with RO-3306, cells were harvested at 0, 0.5, 1, 1.5, 2 and 2.5 hours after release.
For evaluating the spindle assembly checkpoint functionality, HCT116 MTHFD2 WT and KO cells were seeded on 6-well plates and treated the day after either with DMSO or 100 nM, 200 nM, 500 nM or 1 uM of nocodazole (MedChem Express; #HY-13520) for 18 hours, where cells were harvested.
Cells were then fixed with 1 mL of 70% cold ethanol in PBS, added dropwise while vortexing, left on ice for 2 hours, and kept overnight at 4 °C. Cells were then washed four times with 5 mM EDTA in PBS, and stained with propidium iodide solution (15 μg/mL propidium iodide (Life Technologies; #P3566), 1 mM sodium citrate, 30 μg/mL RNAse A (Qiagen; #19101)) overnight at 4 °C in the dark. Cells were analyzed in the flow cytometry analyzer LSRII (BD Biosciences) and plotted with FlowJo (version 10.8.2).
Whole-genome CRISPR genetic screening
Sample preparation
The human CRISPR knockout pooled (Brunello) library109 consists of 76,441 sgRNAs targeting 19,114 genes (whole genome), with ~4 sgRNA/gene, as well as 1000 intergenic control sgRNAs in a Cas9-expressing lentiviral vector. The Brunello library was amplified with the QIAGEN Plasmid Plus Mega Kit (Qiagen; #12981). For virus production, HEK293T cells were transfected with the Brunello lentiCRISPRv2 plasmid as previously described. HCT116 MTHFD2 WT and KO cells were infected with the lentiviral particles containing the Brunello library at a multiplicity of infection (MOI) of 0.4 in the presence of polybrene (Sigma-Aldrich; #TR1003G; 10 μg/mL). The next day, puromycin-containing medium (Sigma-Aldrich; #P8833; 2 μg/mL) was added to select transductants. At 8 days post-selection, half of the cells were harvested as the initial population and the other half were reseeded, maintaining a 500X coverage. Cells were kept in culture for three weeks when the final population was harvested. Genomic DNA from all samples was extracted using the QIAmp DNA Blood Midi kit (Qiagen; #51106) and treated with RNAse A (Qiagen; #19101). The sgRNA library was prepared by a first PCR with the Phusion High Fidelity DNA Polymerase (Thermo Fisher Scientific; #F530) with a mixture of P5 forward primers with staggers from 3 to 6 bp and a P7 reverse primer (Primers 60-64, Supplementary Methods Table 2). The number of cycles was optimized for each sample to prevent over-amplification, and the DNA input for each sample corresponded to a coverage of ~500X. A second PCR was performed using NEBNext Q5 Hot Start HiFi PCR Master Mix (New England Biolabs; #M0543) and Index 1 (i7) and Index 2 (i5) to complete the adaptors and to add the barcodes. Final libraries were analyzed using Agilent Bioanalyzer (Agilent; #5067-4626) to estimate the quantity and check size distribution, and were then quantified by qPCR using the KAPA Library Quantification Kit (KapaBiosystems; #KK4835) prior to amplification with Illumina’s cBot. Samples were sequenced on the Illumina HiSeq 2500 machine using single-read 50 bp sequencing.
Data Analysis
MAGeCK (v. 0.5.9.5) was used for alignment, sgRNA count, copy number variation (CNV) correction, and for the obtention of gene-level depletion and enrichment scores65. MAGeCKFlute110 (v. 1.14) was additionally used to correct for cell cycle-related effects between MTHFD2 WT and KO cells. Gene log2 fold change was calculated by taking the average of the log2 fold change for all sgRNAs targeting the same gene.
Etoposide treatment
HCT116 MTHFD2 WT and KO cells were seeded in 96-well plates and, after 24 hours, cells were treated with either DMSO (PanReac AppliChem; #A3672) or different doses of etoposide (MedChem Express; #HY-13629): 0.25, 0.5, 1, 2 and 4 μM for 72 hours. Afterward, cells were fixed with formalin (Sigma-Aldrich; #HT501128) and stained with 0.1% crystal violet solution (Sigma-Aldrich; #HT90132). Cells were then solubilized with 10% acetic acid and measured at 590 nm in a TECAN Infinite M200 Plate Reader.
Folate metabolites supplementation
HCT116 MTHFD2 WT and KO cells were seeded in 96-well plates and, after 24 hours, cells were supplemented with either DMSO (PanReac AppliChem; #A3672) or different metabolites: formic acid (low dose 0.25 mM, high dose 1 mM, Thermo Fisher Scientific; #A11750), folate (low dose 0.125 μM, high dose 50 μM, MedChem Express; #HY-1663), 5,10-methylenetetrahydrofolate (low dose 0.25 μM, high dose 2.5 μM, MedChem Express; #HY-14769) or SAM (low dose 0.375 μM, high dose 50 μM, MedChem Express; #HY-B0617A) for 72 hours. Afterward, cells were fixed for immunofluorescence to measure the mitotic index as previously described.
TH9619 treatment
HCT116 WT cells were seeded in 96-well plates and, after 24 hours, cells were treated with DMSO (PanReac AppliChem; #A3672) or 25, 62.5, and 156.25 nM TH9619 inhibitor (One-Carbon Therapeutics) for 96 hours. After TH9619 inhibitor treatment, cells were fixed for immunofluorescence to measure the mitotic index and H4K20me1 levels. To measure the sensitivity of HCT116 cells to TH9619, cells were seeded in 96-well plates and, after 24 hours, cells were treated with DMSO (PanReac AppliChem; #A3672) or 4 nM, 13 nM, 41 nM, 123 nM, 370 nM, 1.11 μM, 3.33 μM, 10 μM and 30 μM of TH9619 inhibitor for 96 hours. After TH9619 inhibitor treatment, cells were fixed for immunofluorescence to quantify DAPI staining.
TCGA RNA-sequencing data analysis
RNA-sequencing data from 31 tumor types belonging to 26 different primary sites from The Cancer Genome Atlas (TCGA) database were retrieved from the Genomic Data Commons (GDC) data portal from The National Cancer Institute42 using the command-line tool GDC Data Transfer Tool (v. 1.6.1). For assessing MTHFD2 expression in tumor versus healthy tissues, FPKM (fragments per kilobase million) values of the MTHFD2 gene were first converted to TPM (transcripts per million) values. 15 solid tumor types, where paired normal tissue data was available and with a minimum of 10 samples, were kept. In these tumor types, tumor and healthy MTHFD2 expression were compared with a paired two-tailed Wilcoxon test in R (version 4.1.1).
For the machine learning analysis, tumor and healthy MTHFD2 expression from breast (BRCA), lung (LUAD and LUSC) and colon (COAD) cancer, separately, were used to predict the sample status: healthy or tumor. Two-thirds of the data were used for training a tree-based machine learning algorithm with a number of trees of 500 using the randomForest111 package (version 4.7-1.1) in R. The resting one-third of the data was used to evaluate model performance and to obtain the ROC (receiver operating characteristic) curves and the corresponding AUC (area under the curve) values, using the packages caret112 (version 6.0-93) and ROCR113 (version 1.0-11).
For the co-expression analysis, for each cancer type, Pearson correlation was measured between the TPM expression values of each one of the enzymes of the folate metabolism (ATIC, DHFR, GART, MTHFD1, MTHFD1L, MTHFD2, MTHFD2L, MTHFR, MTR, SHMT1, SHMT2) and all the other genes. P-values were adjusted with the Benjamini & Hochberg correction. Positively correlated genes with a Pearson correlation coefficient (r) > 0.6 and p-adjusted values < 0.05 were further considered, and those which were positively correlated with the folate enzyme in >= 10 cancer types were selected for over-representation analysis. Gene Ontology enrichment analysis was performed using the ClusterProfiler97 package (version 4.0.5) in R.
CCLE data analysis
MTHFD2 RNA levels (from Expression Public 22Q4), MTHFD2 protein levels (from Proteomics) and MTHFD2 essentiality levels (from CRISPR DepMap Public 22Q4+Score) were retrieved from the Cancer Cell Line Encyclopedia (CCLE) data44 hosted on the DepMap Portal (https://depmap.org). The aneuploidy score and aneuploidy group classification from all CCLE cell lines were retrieved from Cohen-Sharir et al.68.
Proteome-HD data analysis
The ProteomeHD tool54, which employs proteomics data in response to biological perturbations to perform co-regulation analysis using unsupervised machine learning, was used to identify proteins that are co-regulated with MTHFD2. The 5% strongest co-regulated proteins were selected, along with their corresponding enriched Gene Ontology Biological Process terms.
MTHFD2 levels in cell cycle phases
The MTHFD2 SILAC ratios of three biological replicates in each of the cell cycle phases (G1, S, G2, M) were obtained from the supplementary file 2 from Ly et al.55. As mentioned in their publication, for each biological replicate, the ratios were normalized to the ratio measured in G1, and an offset was then added to the G1 ratio to account for the difference in time between cell division and an average G1 cell. Statistical significance was obtained using a one-sample two-tailed t-test.
Statistics and reproducibility
All the statistical parameters including the exact value of the number of replicates, number of cells, deviations, p-values, and type of statistical test are reported in their respective Figures. Statistical analysis was performed across biological replicates, by taking the average of the respective technical replicates, when appropriate. Statistical significance was analyzed using unpaired two-tailed Student’s t-test after testing for normality with Shapiro-Wilk test and equal variance with Levene test. If assumption of normality and homoscedasticity were not fulfilled, the unpaired (or paired, when relevant) non-parametric two-tailed Wilcoxon test was used. When boxplots were used, center line indicated the median; the box limits, the IQR (percentiles 25-75%); and the whiskers, the maximum/minimum, or if maximum/minimum exceeded +/−1.5*IQR, the last value within this range.
Western blots (Figs. 1b and 4a; Supplementary Fig. 2f–h, 5l, 6e) and immunofluorescence (Figs. 1c and 4b; Supplementary Fig. 12b) experiments with cell lines were repeated at least three independent times with similar results. Immunofluorescence experiments with colon cancer-derived tissue or organoids (Fig. 2a, b; Supplementary Fig. 1h) or STORM microscopy experiments (Supplementary Fig. 4d) were repeated two independent times with similar results.
No statistical method was used to predetermine sample size. For the mitotic cell analysis, we considered that the number of mitotic cells in a cycling population is usually between 2 and 5%, thus, the experiments were scaled up accordingly to reach a number of mitotic cells that would allow the statistical analysis. No data were excluded from the analyses. As the study is based on the comparison between MTHFD2 WT and KO cells, randomization does not apply here. However, whenever possible, technical replicates of the same biological replicate were randomized in the multiwell to avoid plate effect. The investigators were not blinded to group allocation during experiments but data analysis was, whenever possible, automatized and blind.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE114 partner repository with the dataset identifier PXD041821. Sequencing samples (raw data and processed files) are available at NCBI GEO. RNA-sequencing data is under the accession number GSE230750 and Nanopore whole-genome sequencing data is under the accession number GSE232307. Genome-wide CRISPR genetic screening raw data have been deposited to the European Nucleotide Archive (ENA) under the accession number PRJEB61666. High-throughput data from omics datasets or high-throughput microscopy data are publicly available in the SdelciLab GitHub repository [https://github.com/SdelciLab/MTHFD2_mitosis]115. Source data are provided with this paper.
Code availability
All the scripts used for this manuscript are publicly available in the SdelciLab GitHub repository [https://github.com/SdelciLab/MTHFD2_mitosis]115.
References
Reid, M. A., Dai, Z. & Locasale, J. W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 19, 1298–1306 (2017).
Wong, C. C., Qian, Y. & Yu, J. Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches. Oncogene 36, 3359–3374 (2017).
Crispo, F. et al. Metabolic dysregulations and epigenetics: a bidirectional interplay that drives tumor progression. Cells 8, 798 (2019).
Lin, J. M. G. et al. Metabolic modulation of transcription: The role of one-carbon metabolism. Cell Chem. Biol. 29, 1664–1679 (2022).
Schvartzman, J. M., Thompson, C. B. & Finley, L. W. S. Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 217, 2247–2259 (2018).
Li, W. et al. Nuclear localization of mitochondrial TCA cycle enzymes modulates pluripotency via histone acetylation. Nat. Commun. 13, 7414 (2022).
Li, S. et al. Serine and SAM responsive complex SESAME regulates histone modification crosstalk by sensing cellular metabolism. Mol. Cell 60, 408–421 (2015).
Wellen, K. E. et al. ATP-Citrate Lyase links cellular metabolism to histone acetylation. Science (1979) 324, 1076–1080 (2009).
Sutendra, G. et al. A nuclear pyruvate dehydrogenase complex is important for the generation of Acetyl-CoA and histone acetylation. Cell 158, 84–97 (2014).
Ducker, G. S. & Rabinowitz, J. D. One-Carbon metabolism in health and disease. Cell Metab. 25, 27–42 (2017).
Nilsson, R. et al. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 5, 3128 (2014).
Gustafsson, R. et al. Crystal structure of the emerging cancer target MTHFD2 in complex with a substrate-based inhibitor. Cancer Res. 77, 937–948 (2017).
Kawai, J. et al. Discovery of a Potent, Selective, and Orally Available MTHFD2 Inhibitor (DS18561882) with in Vivo Antitumor Activity. J. Med. Chem. 62, 10204–10220 (2019).
Bonagas, N. et al. Pharmacological targeting of MTHFD2 suppresses acute myeloid leukemia by inducing thymidine depletion and replication stress. Nat. Cancer 3, 156–172 (2022).
Liu, F., Liu, Y., He, C. & Tao, L. Increased MTHFD2 expression is associated with poor prognosis in breast cancer. Tumor Biol. 35, 8685–8690 (2014).
Ju, H. et al. Modulation of Redox Homeostasis by Inhibition of MTHFD2 in colorectal cancer: mechanisms and therapeutic implications. J. Natl Cancer Inst. 111, 584–596 (2019).
Nishimura, T. et al. Cancer stem-like properties and gefitinib resistance are dependent on purine synthetic metabolism mediated by the mitochondrial enzyme MTHFD2. Oncogene 38, 2464–2481 (2019).
Liu, X. et al. Methylenetetrahydrofolate dehydrogenase 2 overexpression is associated with tumor aggressiveness and poor prognosis in hepatocellular carcinoma. Digest. Liver Dis. 48, 953–960 (2016).
Pikman, Y. et al. Targeting MTHFD2 in acute myeloid leukemia. J. Exp. Med. 213, 1285–1306 (2016).
Koufaris, C., Gallage, S., Yang, T., Lau, C. & Valbuena, G. N. Suppression of MTHFD2 in MCF-7 breast cancer cells increases glycolysis, dependency on exogenous glycine, and sensitivity to folate depletion. J. Proteome Res. 15, 2618–2625 (2016).
Lehtinen, L. et al. High-throughput RNAi screening for novel modulators of vimentin expression identifies MTHFD2 as a regulator of breast cancer cell migration and invasion. Oncotarget 4, 48–63 (2013).
Liu, X. et al. Non-metabolic function of MTHFD2 activates CDK2 in bladder cancer. Cancer Sci. 112, 4909–4919 (2021).
Sheppard, N. G. et al. The folate-coupled enzyme MTHFD2 is a nuclear protein and promotes cell proliferation. Sci. Rep. 5, 15029 (2015).
Andor, N., Maley, C. C. & Ji, H. P. Genomic instability in cancer: Teetering on the limit of tolerance. Cancer Res 77, 2179–2185 (2017).
Cleveland, D. W., Mao, Y. & Sullivan, K. F. Centromeres and Kinetochores: From Epigenetics To Mitotic Checkpoint Signaling. Cell 112, 407–421 (2003).
Barra, V. & Fachinetti, D. The dark side of centromeres: types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 9, 4340 (2018).
Tanaka, T. U., Clayton, L. & Natsume, T. Three wise centromere functions: See no error, hear no break, speak no delay. EMBO Rep. 14, 1073–1083 (2013).
Bloom, K. S. Centromeric heterochromatin: The primordial segregation machine. Annu Rev. Genet 48, 457–484 (2014).
Hori, T. et al. Histone H4 Lys 20 monomethylation of the CENP-A Nucleosome is essential for kinetochore assembly. Dev. Cell 29, 740–749 (2014).
Martins, N. M. C. et al. Epigenetic engineering shows that a human centromere resists silencing mediated by H3K27me3/K9me3. Mol. Biol. Cell 27, 177–196 (2016).
Saksouk, N., Simboeck, E. & Déjardin, J. Constitutive heterochromatin formation and transcription in mammals. Epigenet. Chromatin 8, 3 (2015).
Yi, Q. et al. HP1 links centromeric heterochromatin to centromere cohesion in mammals. EMBO Rep. 19, e45484 (2018).
Nishiyama, A. & Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 37, 1012–1027 (2021).
Eden, A., Gaudet, F., Waghmare, A. & Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science (1979) 300, 455 (2003).
Vilain, A., Vogt, N., Dutrillaux, B. & Malfoy, B. DNA methylation and chromosome instability in breast cancer cell lines. FEBS Lett. 460, 231–234 (1999).
Rodriguez, J. et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 66, 8462–8468 (2006).
Scelfo, A. & Fachinetti, D. Keeping the centromere under control: A promising role for DNA methylation. Cells 8, 912 (2019).
McNulty, S. M., Sullivan, L. L. & Sullivan, B. A. Human Centromeres produce chromosome-specific and array-specific alpha satellite transcripts that are complexed with CENP-A and CENP-C. Dev. Cell 42, 226–240 (2017).
Shi, L., Huang, L., Long, H., Song, A. & Zhou, Z. Structural basis of nucleosomal H4K20 methylation by methyltransferase SET8. FASEB J. 36, e22338 (2022).
Gopalakrishnan, S., Sullivan, B. A., Trazzi, S., Della Valle, G. & Robertson, K. D. DNMT3B interacts with constitutive centromere protein CENP-C to modulate DNA methylation and the histone code at centromeric regions. Hum. Mol. Genet 18, 3178–3193 (2009).
Deng, X. et al. PRMT1 promotes mitosis of cancer cells through arginine methylation of INCENP. Oncotarget 6, 35173–35182 (2015).
Grossman, R. L. et al. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 375, 1109–1112 (2016).
Sdelci, S. et al. MTHFD1 interaction with BRD4 links folate metabolism to transcriptional regulation. Nat. Genet 51, 990–998 (2019).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019).
Thul, P. J. et al. A subcellular map of the human proteome. Science (1979) 356, eaal3321 (2017).
Teo, G. et al. SAINTexpress: Improvements and additional features in Significance Analysis of INTeractome software. J. Proteom. 100, 37–43 (2014).
Orchard, S. et al. The MIntAct project-IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 42, D358–D363 (2014).
Schweizer, N. et al. Spindle assembly checkpoint robustness requires Tpr-mediated regulation of Mad1/Mad2 proteostasis. J. Cell Biol. 203, 883–893 (2013).
Banko, M. R. et al. Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol. Cell 44, 878–892 (2011).
Li, Q. R., Yan, X. M., Guo, L., Li, J. & Zang, Y. AMPK regulates anaphase central spindle length by phosphorylation of KIF4A. J. Mol. Cell Biol. 10, 2–17 (2018).
Thaiparambil, J. T., Eggers, C. M. & Marcus, A. I. AMPK regulates mitotic spindle orientation through phosphorylation of myosin regulatory light chain. Mol. Cell Biol. 32, 3203–3217 (2012).
Mazumdar, M., Sundareshan, S. & Misteli, T. Human chromokinesin KIF4A functions in chromosome condensation and segregation. J. Cell Biol. 166, 613–620 (2004).
Arimura, Y. et al. The CENP-A centromere targeting domain facilitates H4K20 monomethylation in the nucleosome by structural polymorphism. Nat. Commun. 10, 572 (2019).
Kustatscher, G. et al. Co-regulation map of the human proteome enables identification of protein functions. Nat. Biotechnol. 37, 1361–1371 (2019).
Ly, T. et al. Proteomic analysis of cell cycle progression in asynchronous cultures, including mitotic subphases, using PRIMMUS. Elife 6, e27574 (2017).
Bajar, B. T. et al. Fluorescent indicators for simultaneous reporting of all four cell cycle phases. Nat. Methods 13, 993–996 (2016).
Moretton, A. et al. A metabolic map of the DNA damage response identifies PRDX1 in the control of nuclear ROS scavenging and aspartate availability. Mol. Syst. Biol. 19, e11267 (2023).
Sugiura, A. et al. MTHFD2 is a metabolic checkpoint controlling effector and regulatory T cell fate and function. Immunity 55, 65–81 (2022).
Musacchio, A. & Salmon, E. D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8, 379–393 (2007).
Rošić, S., Köhler, F. & Erhardt, S. Repetitive centromeric satellite RNA is essential for kinetochore formation and cell division. J. Cell Biol. 207, 335–349 (2014).
Cáceres-Gutiérrez, R. E. et al. Proteasome inhibition alters mitotic progression through the upregulation of centromeric α-Satellite RNAs. FEBS J. 289, 1858–1875 (2022).
Ichida, K. et al. Overexpression of satellite alpha transcripts leads to chromosomal instability via segregation errors at specific chromosomes. Int J. Oncol. 52, 1685–1693 (2018).
Sharma, A. B., Dimitrov, S., Hamiche, A. & Van Dyck, E. Centromeric and ectopic assembly of CENP-A chromatin in health and cancer: old marks and new tracks. Nucleic Acids Res 47, 1051–1069 (2019).
Mahlke, M. A. & Nechemia‐Arbely, Y. Guarding the genome: Cenp‐a‐chromatin in health and cancer. Genes (Basel) 11, 1–26 (2020).
Li, W. et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 16, 281 (2015).
Tedeschi, A. et al. Wapl is an essential regulator of chromatin structure and chromosome segregation. Nature 501, 564–568 (2013).
Nielsen, C. F., Zhang, T., Barisic, M., Kalitsis, P. & Hudson, D. F. Topoisomerase IIα is essential for maintenance of mitotic chromosome structure. PNAS 117, 12131–12142 (2020).
Cohen-Sharir, Y. et al. Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature 590, 486–491 (2021).
Green, A. C. et al. Formate overflow drives toxic folate trapping in MTHFD1 inhibited cancer cells. Nat. Metab. 5, 642–659 (2023).
Wright, R. H. G. et al. ADP-ribose-derived nuclear ATP synthesis by NUDIX5 is required for chromatin remodeling. Science (1979) 352, 1221–1225 (2016).
Ben-Sahra, I., Hoxhaj, G., Ricoult, S. J. H., Asara, J. M. & Manning, B. D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science (1979) 351, 728–733 (2016).
Wan, X. et al. Cisplatin inhibits SIRT3-deacetylation MTHFD2 to disturb cellular redox balance in colorectal cancer cell. Cell Death Dis. 11, 649 (2020).
Li, G., Wu, J., Li, L. & Jiang, P. p53 deficiency induces MTHFD2 transcription to promote cell proliferation and restrain DNA damage. Proc. Natl Acad. Sci. USA 118, e2019822118 (2021).
Koufaris, C. & Nilsson, R. Protein interaction and functional data indicate MTHFD2 involvement in RNA processing and translation. Cancer Metab. 6, 12 (2018).
Yu, C. et al. Down-regulation of MTHFD2 inhibits NSCLC progression by suppressing cycle-related genes. J. Cell Mol. Med 24, 1568–1577 (2020).
Schittenhelm, R. B., Althoff, F., Heidmann, S. & Lehner, C. F. Detrimental incorporation of excess Cenp-A/Cid and Cenp-C into Drosophilacentromeres is prevented by limiting amounts of the bridging factor Cal1. J. Cell Sci. 123, 3768–3779 (2010).
Karpf, A. R. & Matsui, S. I. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 65, 8635–8639 (2005).
Dodget, J. E. et al. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 280, 17986–17991 (2005).
Green, N. H. et al. MTHFD2 links RNA methylation to metabolic reprogramming in renal cell carcinoma. Oncogene 38, 6211–6225 (2019).
Yue, L. et al. Mthfd2 Modulates Mitochondrial Function and DNA Repair to Maintain the Pluripotency of Mouse Stem Cells. Stem Cell Reports 15, 529–545 (2020).
Brand, M. & Winter, G. E. Locus-specific knock-in of a degradable tag for target validation studies. in Methods in Molecular Biology vol. 1953 105–119 (Humana Press Inc., 2019).
Huang, B., Wang, W., Bates, M. & Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science (1979) 319, 810–813 (2008).
Bates, M., Huang, B., Dempsey, G. T. & Zhuang, X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science (1979) 317, 1749–1753 (2007).
Rust, M. J., Bates, M. & Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795 (2006).
Sato, T. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011).
Chiva, C. et al. QCloud: A cloud-based quality control system for mass spectrometry-based proteomics laboratories. PLoS One 13, e0189209 (2018).
Perkins, D. N., Pappin, D. J. C., Creasy, D. M. & Cottrell, J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 (1999).
Beer, L. A., Liu, P., Ky, B., Barnhart, K. T. & Speicher, D. W. Efficient quantitative comparisons of plasma proteomes using label-free analysis with MaxQuant. Methods Mol. Biol. 1619, 339–352 (2017).
Shannon, P. et al. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003).
Serebrenik, Y. V., Sansbury, S. E., Santhosh Kumar, S., Henao-Mejia, J. & Shalem, O. Efficient and flexible tagging of endogenous genes by homology-independent intron targeting. Genome Res 28, 1322–1328 (2019).
Guzmán, C., Bagga, M., Kaur, A., Westermarck, J. & Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One 9, e92444 (2014).
Simon A. FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Zhu, A., Ibrahim, J. G. & Love, M. I. Heavy-Tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 35, 2084–2092 (2019).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. PNAS 102, 15545–15550 (2005).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Contreras-Galindo, R. et al. Rapid molecular assays to study human centromere genomics. Genome Res. 27, 2040–2049 (2017).
Cozzuto, L., Delgado-Tejedor, A., Hermoso Pulido, T., Novoa, E. M. & Ponomarenko, J. Nanopore Direct RNA Sequencing Data Processing and Analysis Using MasterOfPores. in Methods Mol Biol (ed. Walker, J. M.) vol. 2624 185–205 (Springer Nature, Hatfield, Hertfordshire, UK, 2023).
Steinig, E., Coin, L. & Nanoq ultra-fast quality control for nanopore reads. J. Open Source Softw. 7, 2991 (2022).
Jain, C., Rhie, A., Hansen, N. F., Koren, S. & Phillippy, A. M. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat. Methods 19, 705–710 (2022).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Akalin, A. et al. MethylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, R87 (2012).
Altemose, N. et al. Complete genomic and epigenetic maps of human centromeres. Science (1979) 376, eabl4178 (2022).
Jiang, T. et al. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 21, 189 (2020).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Doench, J. G. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016).
Wang, B. et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc. 14, 756–780 (2019).
Liaw, A. & Wiener, M. Classification and Regression by randomForest. R. N. 2, 18–22 (2002).
Kuhn, M. Building predictive models in R Using the caret package. J. Stat. Softw. 28, 1–26 (2008).
Sing, T., Sander, O., Beerenwinkel, N. & Lengauer, T. ROCR: Visualizing classifier performance in R. Bioinformatics 21, 3940–3941 (2005).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Pardo-Lorente, N. Nuclear localization of MTHFD2 is required for correct mitosis progression. GitHub repository https://doi.org/10.5281/zenodo.13169749.
Acknowledgements
We acknowledge support of the Spanish Ministry of Science and Innovation through the Centro de Excelencia Severo Ochoa (CEX2020-001049-S, MCIN/AEI /10.13039/501100011033), and the Generalitat de Catalunya through the CERCA programme. We are grateful to the CRG Core Technologies Programme for their support and assistance in this work. We would like to thank the CRG and CNAG Sequencing Facilities (Barcelona, Spain) for all next-generation sequencing, the CRG Proteomics Facility (Barcelona, Spain) for the MTHFD2 interactome analysis, the CRG Flow Cytometry Facility (Barcelona, Spain) for the sorting, the IMIM Platform for molecular cytogenetics (Barcelona, Spain) for the karyotype analysis, and MARbiobank for provision of patient samples for organoid generation. The CRG/UPF Proteomics Unit is part of the Spanish Infrastructure for Omics Technologies (ICTS OmicsTech). We would like to thank Prof. Thomas Helleday for providing the MTHFD2 inhibitor TH9619 and for the fruitful discussions. N.P.L. was supported by a Boehringer Ingelheim Fonds Ph.D. fellowship. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 852343, ERC-StG-852343-EPICAMENTE) and from a Spanish Plan Estatal grant (Ministerio de Ciencia e Innovación, Project PID2019-110598GA-I00 funded by MICIU/AEI /10.13039/501100011033).
Author information
Authors and Affiliations
Contributions
S.S. and N.P.L. conceptualized and designed the project. N.P.L. performed and/or analyzed all the experiments shown in Figs. 1–7 and Supplementary Figs. 1–12. S.S. performed the mitotic phases and mitotic defects quantification shown in Figs. 5–7 and Supplementary Fig. 9. A.G. and L.C. performed the analysis of Nanopore whole-genome sequencing data shown in Fig. 4 and Supplementary Fig. 7. A.G.Z. performed the FUCCI experiments shown in Fig. 3 and Supplementary Fig. 4 and the CENPA immunofluorescence in Supplementary Fig. 5. L.E. cloned of all the de novo vectors described in this manuscript and performed the phospho-Aurora A/B/C immunofluorescence in Supplementary Fig. 7. R.G. modified the revised version of the manuscript and figures. J.S. performed the immunofluorescence of patient-derived colon tissue and organoids shown in Supplementary Fig. 1 and Fig. 2. L.G.L. contributed to the pull-down and ChIP-qPCR experiments shown in Supplementary Fig. 2 and Fig. 4. R.G.A. performed the qPCR experiments shown in Fig. 4. L.M. and M.V.N. performed the STORM microscopy experiments shown in Supplementary Fig. 4. E.D. contributed to the pull-down experiments shown in Supplementary Fig. 2. S.S., J.P., M.P.C. and L.B.M. supervised the study. S.S. acquired the funding. S.S. and N.P.L. wrote the manuscript with contributions from all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pardo-Lorente, N., Gkanogiannis, A., Cozzuto, L. et al. Nuclear localization of MTHFD2 is required for correct mitosis progression. Nat Commun 15, 9529 (2024). https://doi.org/10.1038/s41467-024-51847-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51847-z
This article is cited by
-
Hallmarks of nuclear metabolism: implications for genome integrity, nuclear signaling, and therapeutic targeting
npj Metabolic Health and Disease (2026)
-
K11- and K29-ubiquitination-mediated nuclear translocation of glycolytic enzyme aldolase A promotes pancreatic cancer progression by NF-κB activation
Cell Death & Differentiation (2025)
-
The centromere: the punching bag of the chromosome
Chromosome Research (2025)
-
MTHFD2 marks pemetrexed resistance in pulmonary adenocarcinoma with EGFR wild type
Discover Oncology (2025)
