
Abstract
The paraventricular hypothalamus (PVH) is crucial for food intake control, yet the presynaptic mechanisms underlying PVH neurons remain unclear. Here, we show that RUVBL2 in the PVH is significantly reduced during energy deficit, and knockout (KO) of PVH RUVBL2 results in hyperphagic obesity in mice. RUVBL2-expressing neurons in the PVH (PVHRUVBL2) exert the anorexigenic effect by projecting to the arcuate hypothalamus, the dorsomedial hypothalamus, and the parabrachial complex. We further demonstrate that PVHRUVBL2 neurons form the synaptic connections with POMC and AgRP neurons in the ARC. PVH RUVBL2 KO impairs the excitatory synaptic transmission by reducing presynaptic boutons and synaptic vesicles near active zone. Finally, RUVBL2 overexpression in the PVH suppresses food intake and protects against diet induced obesity. Together, this study demonstrates an essential role for PVH RUVBL2 in food intake control, and suggests that modulation of synaptic plasticity could be an effective way to curb appetite and obesity.
Similar content being viewed by others


Introduction
The brain maintains energy homeostasis by regulating food intake and energy expenditure after integrating both peripheral and central signals1,2,3. This regulation relies on a complex neural network composed of neurons within and outside of the hypothalamus4,5,6. Numerous studies has established the regulatory role for several hypothalamic regions in energy homeostasis including the arcuate hypothalamus (ARC), the paraventricular hypothalamus (PVH), the dorsomedial hypothalamus (DMH), the ventromedial hypothalamus (VMH), and the lateral hypothalamus (LH)7,8,9.
The PVH is a heterogeneous region in the hypothalamus and has been shown to regulate energy homeostasis through a variety of cell types expressing various markers10,11,12,13,14,15,16,17. Of interest, the vast majority of PVH neurons have been demonstrated to promote satiety, supporting an indispensable role for the PVH in satiety control18. The neural circuits underlying the anorexigenic effects of PVH neurons have also been extensively studied15,19,20,21. Some downstream target sites receiving projections from the PVH have been defined including the parabrachial complex (PB)15 and the VMH13. Nevertheless, given the prominence of the PVH in energy homeostasis, additional markers and/or downstream sites projected by PVH neurons need to be identified to better understand the regulation mechanisms underlying PVH control of satiety.
Synaptic plasticity is defined as a reshape of the strength and efficacy of synaptic transmission among neurons, which could last for milliseconds, hours, days, or even longer22,23. A growing body of evidence has indicated a close relationship between synaptic plasticity and energy homeostasis24,25,26. The initial evidence for a potential role of synaptic plasticity in energy homeostasis came from the observations that leptin treatment in ob/ob mice rapidly restored synaptic inputs to POMC-expressing neurons (ARCPOMC) and AgRP-expressing neurons (ARCAgRP) in the ARC before the occurrence of leptin’s anorexigenic effect27. Although an intimate relationship between synaptic plasticity and energy homeostasis has been further suggested by later studies over the past two decades28,29,30,31,32,33,34,35,36,37, whether modulating synaptic plasticity regulates appetite and body weight remains unclear.
RuvB-like 2 (RUVBL2) is a highly conserved AAA+ (ATPases associated with diverse cellular activities) ATPase that has been shown to participate in a variety of cellular processes38,39 including chromatin remodeling39, transcriptional regulation40, glucose transport41, and assembly of macromolecular complexes39,42. Interestingly, one recent study conducting exome-sequencing of type 2 diabetes (T2D) patients and controls indicates that RUVBL2 together with the melanocortin-4 receptor (MC4R) and others are the most significant T2D-associated genes according to gene-level analyses43, suggesting a potential role of RUVBL2 in metabolic control. Although RUVBL2 has been reported to be ubiquitously expressed in the brain44, and we observed its abundant expression in the hypothalamus, particularly in the PVH, the functional role of RUVBL2 in energy homeostasis is unknown.
In this study, we found that RUVBL2 expression in the PVH is significantly reduced during energy deficit. Using different mouse models combined with stereotaxic injection, chemo-/opto-genetics, viral tracing and circuit-specific chemo-/opto-genetics, we show that knockout (KO) of PVH RUVBL2 leads to hyperphagic obesity, and RUVBL2-expressing neurons in the PVH (PVHRUVBL2) exert the anorexigenic effect by projecting to the ARC, the DMH, and the PB. Interestingly, PVHRUVBL2 neurons project to both ARCPOMC and ARCAgRP neurons. Through electrophysiology, transmission electron microscopy, immunohistochemistry, and chromatin immunoprecipitation sequencing (ChIP-Seq) combined with RNA sequencing (RNA-Seq), we further reveal that RUVBL2 KO impairs the excitatory synaptic transmission by reducing presynaptic boutons and synaptic vesicles near the active zone (AZ), which is owing to RUVBL2 transcription control of genes related to neuron axonogenesis and synaptic vesicle. Finally, RUVBL2 overexpression increases synaptic vesicles near the AZ and the excitatory synaptic transmission, consequently suppressing food intake and protecting against diet-induced obesity, suggesting that modulation of synaptic plasticity could be a feasible way to regulate food intake and body weight.
Results
RUVBL2 KO in the adult PVH leads to massive obesity owing to hyperphagia
Using a monoclonal antibody against RUVBL2, we showed that RUVBL2 is broadly expressed in the brain, including the hypothalamus (Supplementary Fig. 1a, b). The PVH is an indispensable hypothalamic site regulating energy homeostasis18. We observed that RUVBL2 is expressed throughout the PVH, ranging from the anterior, the medial, to the posterior part (Supplementary Fig. 1c). Interestingly, fasting significantly reduces the expression of RUVBL2 in the PVH at both mRNA and protein levels (Fig. 1a,b), suggesting a potential role for RUVBL2 in control of energy homeostasis.

a, b Expression of Ruvbl2 at mRNA (a) or protein (b) levels in the PVH of 8-week-old male WT mice fed a regular chow diet or fasted for 48 h (n = 7 per group for mRNA; n = 6 per group for protein). c Schematic of AAV injection into the PVH. AAV2-Cre-GFP or control AAV2-GFP was bilaterally injected into the PVH of 8-week-old Ruvbl2fl/fl mice. Mouse body weight was recorded weekly for 12 weeks post AAV injection, followed by different analyses including measurement of food intake and body composition, metabolic cages, IPGTT and IPITT. d Representative image of AAV2-Cre-GFP infection in the PVH (n = 7 mice; scale bar, 100 μm). e Representative images showing PVH RUVBL2 KO efficiency. A monoclonal antibody against RUVBL2 was used to detect RUVBL2 in the PVH by IHC staining (n = 3 mice; scale bar, 100 μm). f, g Body weight of male (f) and female (g) mice after AAV injection. For males, AAV2-GFP group (n = 6 mice); AAV2-Cre-GFP group (n = 7 mice). For females, n = 7 mice per group. h, i Daily food intake of male (h) and female (i) mice on chow diet. For males, AAV2-GFP group (n = 6 mice); AAV2-Cre-GFP group (n = 7 mice). For females, n = 7 mice per group. j–l Metabolic rate by body weight (j), locomotor activity (k), and metabolic rate by analysis of covariance (ANCOVA) (l) of male mice. ANCOVA was performed using SPSS Version 26 with metabolic rate as dependent variable, genotype as fixed variable and body mass as covariate. AAV2-GFP group (n = 6 mice); AAV2-Cre-GFP group (n = 7 mice). m, n IPGTT and AUC (m), and IPITT and AUC (n) of male mice. Intraperitoneal glucose tolerance test (IPGTT) and intraperitoneal insulin tolerance test (IPITT) were performed in mice 12 weeks post AAV injection. AAV2-GFP group (n = 6 mice); AAV2-Cre-GFP group (n = 7 mice). Two-tailed unpaired Student’s t-test for (a, b, h–k) and (m, n) (AUC). Two-way ANOVA followed by Bonferroni’s test for (f, g, m, n). ns, no significance. All data are presented as means ± SEM. Source data are provided as a Source Data file.
To study the role of PVH RUVBL2 in energy homeostasis, we generated the floxed Ruvbl2 mouse strain (Ruvbl2fl/fl)42 (Supplementary Fig. 2a), and bilaterally injected adeno-associated virus (AAV) (AAV2-GFP or AAV2-Cre-GFP) into the PVH of Ruvbl2fl/fl mice (Fig. 1c, d and Supplementary Fig. 2b). Of interest, one recent study reported that injection of some batches of AAV-Cre in the mouse PVH resulted in obesity due to their neuronal toxicity45. Hence, we also examined the toxicity of AAV2-Cre by injecting AAV2-Cre-GFP or AAV2-GFP as control into the PVH of 8 week-old male C57BL/6 J mice and monitored their weekly body weight. It turned out that there was no difference in body weight of AAV2-GFP- and AAV2-Cre-GFP-injected mice (Supplementary Fig. 2c), excluding the possible neuronal toxicity of AAV2-Cre-GFP.
As shown by immunohistochemical (IHC) staining, AAV2-Cre-GFP infection drastically reduced the expression of RUVBL2 in the PVH (Fig. 1e). The mice injected with AAV2-Cre-GFP became massively obese 12 weeks post virus injection compared with those injected with AAV2-GFP (Fig. 1f, g and Supplementary Fig. 2d, e). Daily food intake of mice with AAV2-Cre-GFP injection was significantly increased (Fig. 1h, i). Using comprehensive lab animal monitoring system (CLAMS), we showed that mice injected with AAV2-Cre-GFP exhibited similar energy expenditure (metabolic rate) (Fig. 1j and Supplementary Fig. 2f), locomotor activity (Fig. 1k and Supplementary Fig. 2g), and respiratory exchange ratio (RER) (Supplementary Fig. 2h, i) in both light and dark cycles, compared with the mice injected with AAV2-GFP.
Note that our metabolic cages measurements were carried out in mice showing different body weights after 12 weeks of AAV injection. One study has suggested that analysis of covariance (ANCOVA) should be performed to evaluate the metabolic rate in animals with different body weights46. Hence, we performed ANCOVA using body weight as covariate, indicating similar metabolic rate between two groups (Fig. 1l and Supplementary Fig. 2j). In addition, mice with AAV2-Cre-GFP injection showed significantly reduced blood glucose control and insulin sensitivity as demonstrated by the intraperitoneal glucose tolerance tests (IPGTT) and the intraperitoneal insulin tolerance tests (IPITT), respectively (Fig. 1m, n and Supplementary Fig. 2k, l). Taken together, these data demonstrate that PVH RUVBL2 KO leads to hyperphagic obesity.
PVHRUVBL2 neurons constitute the majority of PVH neurons
We next characterized PVHRUVBL2 neurons by examining the co-localization of RUVBL2 with other known markers. To investigate the co-localization between RUVBL2 and SIM1, an essential molecule for PVH development47, Sim1-Cre transgenic mice were crossed with Ai14 (Rosa26-CAG-LSL-tdTomato) reporter mice48 to generate Sim1-Cre;Ai14 mice. As shown, the majority (79.2%) of PVHSIM1 neurons expresses RUVBL2 (Supplementary Fig. 3a), suggesting that PVHRUVBL2 neurons constitute a large part of PVH neurons.
MC4R-expressing neurons (PVHMC4R) and prodynorphin-expressing neurons (PVHPDYN) in the PVH are two well-characterized cell types that have been suggested to account for the whole satiety control by the PVH15. Therefore, we analyzed the co-localization of RUVBL2 with MC4R or PDYN in the PVH. Through Mc4r-2a-Cre mice49 and Ai14 mice, we showed that ~32.7% of PVHMC4R neurons co-express RUVBL2, while ~16.8% of PVHRUVBL2 neurons co-expresses MC4R (Supplementary Fig. 3b). On the other hand, the vast majority (~93%) of PVHPDYN neurons co-expresses RUVBL2, while ~55.8% of PVHRUVBL2 neurons co-expresses PDYN (Supplementary Fig. 3c). We also analyzed the co-expression of RUVBL2 with several other markers in the PVH including Oxytocin, CRH, TH, and TRH. As shown, most of neurons investigated co-expresses RUVBL2, while only a small part of PVHRUVBL2 neurons co-expresses these markers (Supplementary Fig. 3d–g). Together, these data indicate that PVHRUVBL2 neurons constitute the majority of PVH neurons, and also suggest that PVHRUVBL2 neurons may exert an anorexigenic effect, given an established role for the PVH in satiety control18.
Manipulation of PVHRUVBL2 neuron activity alters food intake
To explore the role of PVHRUVBL2 neurons in satiety control, designer receptors exclusively activated by designer drugs (DREADDs) and their specific ligand clozapine N-oxide (CNO)50 were applied to manipulate PVHRUVBL2 neuron activity. We first generated Ruvbl2-Cre knock-in mice by placing Cre sequence after the coding sequence of Ruvbl2 gene using CRISPR-Cas9 (Supplementary Fig. 4a), and bilaterally delivered the Cre-dependent AAV9-DIO-hM3D(Gq)-mCherry (stimulatory) or control AAV9-DIO-mCherry into the PVH of Ruvbl2-Cre mice (Fig. 2a). Administration of CNO robustly increased the PVHRUVBL2 neuron activity as revealed by c-Fos immunoreactivity (Fig. 2b). Chemogenetic activation of PVHRUVBL2 neurons significantly reduced food intake of fasted mice in both light and dark cycles (Fig. 2c, d and Supplementary Fig. 4b, c). Note that chemogenetic activation of PVHRUVBL2 neurons did not influence the energy expenditure of mice in both cycles as demonstrated by metabolic rate or oxygen consumption rate (VO2) (Supplementary Fig. 4d–g).

a Schematic of stereotaxic injection of AAV9-DIO-hM3D(Gq)-mCherry or AAV9-EF1α-DIO-mCherry. b Representative images showing AAV9-DIO-hM3D(Gq)-mCherry infection and c-Fos immunoreactivity in the PVH after CNO treatment (n = 3 mice; scale bar, 100 μm). c, d Food intake of male (c) and female (d) mice post CNO injection in light cycle. AAV9-DIO-hM3D(Gq)-mCherry or AAV9-EF1α-DIO-mCherry was bilaterally injected into the PVH of chow-fed 8 week-old Ruvbl2-Cre mice and hourly food intake was recorded for 4 h post CNO injection (n = 5 mice per group). e Schematic of stereotaxic injection of AAV9-DIO-hM4D(Gi)-mCherry or AAV9-EF1α-DIO-mCherry. f Representative images showing AAV9-DIO-hM4D(Gi)-mCherry infection and c-Fos immunoreactivity in the PVH after CNO treatment (n = 3 mice; scale bar, 100 μm). g, h Food intake of male (g) and female (h) mice post CNO injection in light cycle. Cre-dependent AAV9-DIO-hM4D(Gi)-mCherry or AAV9-DIO-mCherry was bilaterally injected into the PVH of chow-fed 8 week-old Ruvbl2-Cre mice and hourly food intake was recorded within 4 h post CNO injection (n = 5 mice per group). i Schematic of stereotaxic injection of AAV9-DIO-hChR2-mCherry or AAV9-DIO-mCherry. j Procedure of optogenetic stimulation. PVHRUVBL2 neurons were stimulated by a 465 nm blue laser with 10 ms pulse duration for 10 min. Food intake in 10 min of laser stimulation, and 10 min before or after laser stimulation was monitored, respectively. Stim, during laser stimulation; Pre, before laser stimulation; Post, after laser stimulation. k Representative image showing AAV9-DIO-hChR2-mCherry infection in the PVH (n = 5 mice; scale bar, 100 μm). l Food intake of fasted mice before and after optogenetic activation of PVHRUVBL2 neurons. ChR2 group (n = 5 mice); Control group (n = 3 mice). The experiment was repeated once using the same mice and all the data were included for quantitation. Two-tailed unpaired Student’s t-test for (c–l). ns, no significance. All data are presented as means ± SEM. Source data are provided as a Source Data file.
To investigate whether inhibition of PVHRUVBL2 neuron activity affects food intake, we bilaterally injected the Cre-dependent AAV9-DIO-hM4D(Gi)-mCherry (inhibitory) or control AAV9-DIO-mCherry into the PVH of Ruvbl2-Cre mice (Fig. 2e, f). Chemogenetic inhibition of PVHRUVBL2 neurons significantly increased food intake within 4 h after CNO administration (Fig. 2g, h and Supplementary Fig. 4h, i), without any effect on the energy expenditure of mice (Supplementary Fig. 4j–m).
We also performed optogenetics51 by injecting Cre-dependent AAV9-DIO-hChannelrhodopsin-2 (hChR2)-mCherry or control AAV9-DIO-mCherry into the PVH of male Ruvbl2-Cre mice that were simultaneously implanted with an optic fiber above the PVH (Fig. 2i). After recovery and acclimation, optogenetics was performed using a 465-nm blue laser with 10 ms pulse duration at 20 Hz for 10 min (Fig. 2j). Optogenetic stimulation significantly reduced food intake of overnight-starved mice injected with AAV9-ChR2 compared with AAV9-mCherry, while no change of food intake between these two groups was observed either before or after stimulation (Fig. 2k, l and Supplementary Movie 1), supporting that activation of PVHRUVBL2 neurons acutely suppresses food intake. Together, these chemogenetics and optogenetics studies have demonstrated the anorexigenic effect of PVHRUVBL2 neurons.
PVHRUVBL2 neurons project to several brain regions
To identify the brain regions receiving axonal projections from PVHRUVBL2 neurons, we carried out anterograde monosynaptic tracing52. The helper virus AAV8-DIO- EGFP-T2A-TK was first unilaterally injected into the PVH of 8 week-old male Ruvbl2-Cre mice that received the unilateral injection of the herpes simplex virus (HSV) (HSV-ΔTK-LSL-tdTomato) into the same site after 3 weeks of virus injection (Fig. 3a).

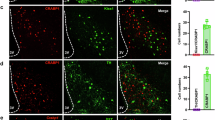
a Schematic of anterograde monosynaptic tracing of PVHRUVBL2 neurons. b Representative images showing PVHRUVBL2 neurons infected with AAV8-EF1α-DIO-EGFP-T2A-TK and HSV-ΔTK-LSL-tdTomato. 72 h after injection of HSV-ΔTK-LSL-tdTomato, the mice were euthanized and their brains were collected for histology analysis (n = 3 mice; scale bars, 100 μm). c Representative images indicating the brain regions receiving direct projections from PVHRUVBL2 neurons revealed by tdTomato fluorescence signals. These brains regions include the ventral lateral septum nucleus (LSv) (c1), the preoptic area (POA) (c2), the dorsomedial hypothalamus (DMH) (c3), the arcuate hypothalamus (ARC) (c4), the medial amygdala (MeA) (c5), the parabrachial complex (PB) (the parabrachial nucleus (PBN) and the pre-locus coeruleus (pLC)) (c6, c7), and the nucleus tractus solitarius (NTS) (c8) (n = 3 mice; scale bars, 100 μm). d Schematic of retrograde monosynaptic tracing of ARCPOMC neurons. e Representative image from three mice showing ARCPOMC neurons infected with AAV8-EF1α-DIO-EGFP-T2A-TVA, AAV8-EF1α-DIO-oRVG, and RV-EnVA-ΔG-LSL-DsRed as stated in d; 7 days after injection of RV-EnVA-ΔG-LSL-DsRed, the mice were euthanized and their brains were collected for coronal brain sectioning and subsequent histology analysis. Scale bar, 50 μm. f Representative images showing ARCPOMC-projecting neurons in the PVH and PVHRUVBL2 neurons (n = 3 mice; scale bar, 50 μm). g Schematic of retrograde monosynaptic tracing of ARCAgRP neurons. h Representative image from three mice showing ARCAgRP neurons infected with AAV8-EF1α-DIO-EGFP-T2A-TVA, AAV8-EF1α-DIO-oRVG, and RV-EnVA-ΔG-LSL-DsRed. Scale bar, 50 μm. i Representative images showing ARCAgRP-projecting neurons in the PVH and PVHRUVBL2 neurons (n = 3 mice; scale bar, 50 μm). Arrows denote PVHRUVBL2 neurons (green) containing DsRed fluorescence signals that are retrogradely derived from ARCPOMC or ARCAgRP neurons. Source data are provided as a Source Data file.
We observed the unilateral infection of both helper virus and HSV in the PVH (Fig. 3b), and that seven brain regions throughout the brain clearly showed tdTomato fluorescent signals, suggesting direct projections from PVHRUVBL2 neurons (Fig. 3c). These brain regions included the ventral lateral septum nucleus (LSv) (Fig. 3c1), the preoptic area (POA) (Fig. 3c2), the DMH (Fig. 3c3), the ARC (Fig. 3c4), the medial amygdala (MeA) (Fig. 3c5), the PB including the parabrachial nucleus (PBN) (Fig. 3c6) and the pre-locus coeruleus (pLC) (Fig. 3c7), and the nucleus tractus solitarius (NTS) (Fig. 3c8). Consistently, previous studies have indicated that several cell types in the PVH including PVHMC4R, PVHPDYN, PVHTrkB, and PVHTRH neurons regulate energy homeostasis by projecting to the PB (LPBN or pLC)15,49 or the ARC17,35.
We next performed retrograde tracing53 to demonstrate the synaptic connections between PVHRUVBL2 neurons and the above-mentioned seven brain regions (Supplementary Fig. 5a–h). These data together demonstrate the direct projections from PVHRUVBL2 neurons to the brain regions identified by anterograde monosynaptic tracing.
ARCPOMC and ARCAgRP neurons get involved in regulation of appetite as an important component of central melanocortin system18. Thus, we asked whether PVHRUVBL2 neurons regulate appetite by projecting to ARCPOMC and/or ARCAgRP neurons. To address this, Ruvbl2-Cre mice were crossed with Pomc-EGFP mice54 to generate Ruvbl2-Cre;Pomc-EGFP mice that were used for anterograde monosynaptic tracing (Supplementary Fig. 6a). We observed that many anterograde tdTomato signals from PVHRUVBL2 neurons are localized in ARCPOMC neurons (Supplementary Fig. 6b,c), indicating the monosynaptic projection from PVHRUVBL2 neurons to ARCPOMC neurons. To validate this synaptic connection, we performed retrograde monosynaptic tracing55 in Pomc-Cre mice56 (Fig. 3d), showing that almost all of the retrograde DsRed signals from ARCPOMC neurons are localized in PVHRUVBL2 neurons (Fig. 3e,f). Interestingly, PVHRUVBL2 neurons also form synaptic connections with ARCAgRP neurons as revealed by retrograde monosynaptic tracing in Agrp-Cre mice57 (Fig. 3g-i). Note that we observed that most of PVHRUVBL2 neurons projecting to ARCPOMC and ARCAgRP neurons are glutamatergic (Supplementary Fig. 6d,e). Taken together, these results demonstrate PVHRUVBL2 → ARCPOMC and PVHRUVBL2 → ARCAgRP neuronal circuits.
PVHRUVBL2 neurons exert their anorexigenic effect through PVHRUVBL2 → ARC, PVHRUVBL2 → DMH, and PVHRUVBL2 → PB neural circuits
To investigate which neural circuit (s) mediates the anorexigenic effect of PVHRUVBL2 neurons, we performed projection-specific chemogenetics. Control AAV5-Con/Fon-EGFP or Cre- and FLP-dependent AAV5-Con/Fon-hM4D(Gi)-EGFP were bilaterally injected into the PVH of Ruvbl2-Cre mice that simultaneously received bilateral injection of retroAAV-FLP into PVHRUVBL2-projected regions as identified above. We first investigated whether PVHRUVBL2 → ARC regulated food intake, given a well-established role for the ARC in energy homeostasis3. The injection sites of PVH and ARC were verified by histological analysis (Fig. 4a, b). Chemogenetic inhibition of ARC-projecting PVHRUVBL2 neurons significantly increased food intake in both light and dark cycles (Fig. 4c, d).

a Schematic of chemogenetic inhibition of ARC-projecting PVHRUVBL2 neurons. b Representative images from three mice showing viral infection in the PVH (b1) and axonal terminals labeled by EGFP in the ARC (b2, b3) as described in (a). c, d Food intake of mice in light (c) or dark (d) cycle after chemogenetic inhibition of ARC-projecting PVHRUVBL2 neurons. Hourly food intake was measured after CNO injection. n = 3 mice per group. e Schematic of chemogenetic inhibition of DMH-projecting PVHRUVBL2 neurons. f Representative images from three mice showing AAV infection in PVHRUVBL2 neurons (f1) and EGFP-labeled axonal terminals in the DMH (f2, f3). Arrows denote axonal terminals of PVHRUVBL2 neurons. g, h Food intake of mice in light (g) or dark cycle (h) after chemogenetic inhibition of DMH-projecting PVHRUVBL2 neurons. Hourly food intake was measured after CNO injection. n = 3 mice per group. i Schematic of optogenetic activation of ARC-projecting PVHRUVBL2 neurons. j Representative images from five mice showing AAV infection in PVHRUVBL2 neurons and ChR2-mCherry-labeled axonal terminals in the ARC. k Food intake of mice after optogenetic activation of ARC-projecting PVHRUVBL2 neurons. ChR2 group (n = 5 mice); control group (n = 3 mice). This experiment was repeated once using the same mice and all the data were included for quantitation. l Schematic of optogenetic activation of DMH-projecting PVHRUVBL2 neurons. m Representative images from five mice showing AAV infection in PVHRUVBL2 neurons and ChR2-mCherry-labeled axonal terminals in the DMH from PVHRUVBL2 neurons. n Food intake of mice after optogenetic activation of DMH-projecting PVHRUVBL2 neuron activity. ChR2 group (n = 5 mice); control group (n = 3 mice). This experiment was repeated once using the same mice and all the data were included for quantitation. ChR2 group (n = 5 mice); mCherry group (n = 3 mice). Scale bars, 100 μm. Two-tailed unpaired Student’s t-test for (c–n). ns, no significance. CNO,1 mg/kg BW. All data are presented as means ± SEM. Source data are provided as a Source Data file.
We next reasoned whether PVHRUVBL2 → DMH regulates food intake, considering that DMH is another hypothalamic region that is known to regulate energy homeostasis6. Likewise, we verified the injection sites of PVH and DMH (Fig. 4e, f). Chemogenetic inhibition of DMH-projecting PVHRUVBL2 neurons significantly increased food intake (Fig. 4g, h).
In addition, we tested the other PVHRUVBL2-projected structures including the PB, the LSv, the POA, the MeA, and the NTS. Chemogenetic inhibition of PB-projecting PVHRUVBL2 neuron activity significantly increased food intake in both light and dark cycles (Supplementary Fig. 7a–c). These data are consistent with previous studies demonstrating the critical role for the PB in mediating the anorexgenic effects of several known cell types in the PVH13,15,49. In comparison, chemogenetic inhibition of PVHRUVBL2 neurons projecting to other regions (LSv, MeA, POA, and NTS) had no effect on food intake (Supplementary Fig. 7d–o). Note that CNO alone has no effect on mouse food intake (Supplementary Fig. 7p, q). Taken together, these results indicate that PVHRUVBL2 neurons suppresses food intake through their projections to the ARC, the DMH, and the PB.
We also performed projection-specific optogenetics to examine the anorexigenic effect of PVHRUVBL2 → ARC and PVHRUVBL2 → DMH neural circuits. To examine the anorexigenic effect of PVHRUVBL2 → ARC neural circuit, control AAV9-DIO-mCherry or Cre-dependent AAV9-DIO-hChR2-mCherry were bilaterally injected into the PVH of Ruvbl2-Cre mice that simultaneously were implanted with an optic fiber above the ARC for neuron terminal activation (Fig. 4i,j). Optogenetic activation of ARC-projecting PVHRUVBL2 neurons triggered a marked decrease of food intake in overnight-fasted mice (Fig. 4k and Supplementary Movie 2).
To examine the anorexigenic effect of PVHRUVBL2 → DMH neural circuit, control AAV9-DIO-mCherry or Cre-dependent AAV9-DIO-hChR2-mCherry were bilaterally injected into the PVH of Ruvbl2-Cre mice that simultaneously received implantation of an optic fiber above the DMH for neuron terminal activation (Fig. 4l,m). Optogenetic activation of DMH-projecting PVHRUVBL2 neurons triggered a significant decrease of food intake in overnight-fasted mice (Fig. 4n). These data collectively demonstrate that PVHRUVBL2 → ARC, PVHRUVBL2 → DMH, and PVHRUVBL2 → PB neural circuits mediate the anorexigenic effect of PVHRUVBL2 neurons.
RUVBL2 KO in the PVH impairs the excitatory synaptic transmission
We next aimed to study the synaptic mechanisms underlying the anorexigenic effect of PVH RUVBL2 and PVHRUVBL2 neurons. First, we examined whether PVHRUVBL2 neurons are excitatory or inhibitory by analyzing the co-localization of RUVBL2 with VGLUT2 or VGAT as the excitatory or inhibitory neuron marker, respectively12,58. Accordingly, Vglut2-ires-Cre58 or Vgat-ires-Cre58 mice were crossed with Ai14 reporter mice to generate Vglut2-ires-Cre;Ai14 or Vgat-ires-Cre;Ai14 mice. We observed that ~49% of PVHRUVBL2 neurons are VGLUT2+ excitatory neurons, while ~8% of PVHRUVBL2 neurons are VGAT+ inhibitory neurons (Supplementary Fig. 8a, b), suggesting that most PVHRUVBL2 neurons are excitatory.
We then determined the valence of functional monosynaptic connections between PVHRUVBL2 neurons and the ARC or the DMH using ChR2-assisted circuit mapping (CRACM)59. To achieve this, we injected Cre-dependent AAV9-EF1a-DIO-hChR2-mCherry expressing the ChR2 into the PVH of Ruvbl2-Cre mice, and recorded the excitatory post-synaptic currents (EPSCs) and the inhibitory post-synaptic currents (IPSCs) in ARC or DMH neurons by optogenetically stimulating PVHRUVBL2 neuron terminals in the ARC or the DMH, respectively (Supplementary Fig. 8c, d). We recorded the excitatory postsynaptic currents (EPSCs) in 50% (6 out of 12 neurons) of ARC neurons, and the inhibitory post-synaptic currents (IPSCs) in 9.5% (2 out of 21 neurons) of ARC neurons (Supplementary Fig. 8e, f). In addition, 38.5% (5 out of 13 neurons) and 14.3% (3 out of 21 neurons) of DMH neurons displayed the EPSCs and the IPSCs, respectively (Supplementary Fig. 8g, h). These data demonstrate that PVHRUVBL2 neurons send both excitatory and inhibitory projections to the ARC and the DMH.
Considering that abnormalities in feeding behavior may arise from the disorder in synaptic transmission (excitatory or inhibitory)30,60, we reasoned that RUVBL2 KO in the PVH may impair synaptic transmission. To test this, we bilaterally injected AAV2-Cre-GFP into the PVH of 8 week-old Ruvbl2fl/fl mice to knock out RUVBL2. After 6 weeks of AAV infection, we first performed the voltage-clamp recordings in the brain slices to investigate the spontaneous EPSCs (sEPSCs) in ARC neurons (Fig. 5a). Both amplitude and frequency of sEPSC were significantly decreased in ARC neurons (Fig. 5b–d), suggesting a presynaptic effect of RUVBL2 KO on the excitatory synaptic transmission. Next, we investigated whether RUVBL2 KO affects the evoked EPSCs (eEPSCs) by comparing eEPSCs in ARC neurons under afferent fiber stimulation. Interestingly, the peak amplitude of eEPSCs was also significantly decreased after RUVBL2 KO (Fig. 5e, f), while the pair-pulse ratio (PPR) (interstimulus interval (ISI), 50 ms) reflecting the evoked vesicle release probability was significantly increased (Fig. 5g). Together, these results indicate a presynaptic role of RUVBL2 in regulating the excitatory synaptic transmission in PVHRUVBL2 → ARC circuit.

a Schematic of AAV injection into the PVH and electrophysiology recordings in the ARC. b Representative traces of sEPSCs recorded in the ARC in the presence of PTX. c, d Amplitude (c) and frequency (d) of sEPSCs recorded in ARC neurons. AAV2-GFP (n = 45 neurons/4 mice), AAV2-Cre-GFP (n = 42 neurons/4 mice). e Representative traces of eEPSCs recorded in ARC neurons in response to paired-pulse stimulations (50 ms interval) in the presence of PTX. f, g Amplitude of the first EPSCs (f) and PPR (g) recorded in ARC neurons. AAV2-GFP (n = 45 neurons/4 mice), AAV2-Cre-GFP (n = 42 neurons/4 mice). h Representative traces of sIPSCs recorded in ARC neurons in the presence of CNQX and D-APV. i, j Amplitude (i) and frequency (j) of sIPSCs recorded in ARC neurons. AAV2-GFP (n = 32 neurons/3 mice), AAV2-Cre-GFP (n = 38 neurons/3 mice). k Schematic of AAV injection into the PVH and electrophysiological recordings in DMH neurons. l Representative traces of sEPSCs recorded in DMH neurons in the presence of PTX. m, n Amplitude (m) and frequency (n) of sEPSCs recorded in DMH neurons. AAV2-GFP (n = 31 neurons/3 mice), AAV2-Cre-GFP (n = 32 neurons/3 mice). o Representative traces of eEPSCs recorded in DMH neurons in response to paired-pulse stimulations (50 ms interval) in the presence of PTX. p, q Amplitude of the first eEPSCs (p) and PPR (q) recorded in DMH neurons. AAV2-GFP (n = 30 neurons/3 mice), AAV2-Cre-GFP (n = 30 neurons/3 mice). r Representative traces of sIPSC recorded in DMH neurons in the presence of CNQX and D-APV. s, t Amplitude (s) and frequency (t) of sIPSCs recorded in DMH neurons. AAV2-GFP (n = 21 neurons/2 mice), AAV2-Cre-GFP (n = 20 neurons/2 mice). Two-tailed unpaired Student’s t-test for (c–t). ns, no significance. All data are presented as means ± SEM. Source data are provided as a Source Data file.
We also examined the potential effect of RUVBL2 KO on the inhibitory synaptic transmission in ARC neurons, but observed no differences in the spontaneous IPSCs (sIPSCs) (amplitude and frequency) between WT and RUVBL2 KO mice (Fig. 5h–j). Taken together, these results reveal that RUVBL2 KO impairs the excitatory synaptic transmission in PVHRUVBL2 → ARC circuit.
We next investigated whether PVH RUVBL2 KO affected synaptic transmission in PVHRUVBL2 → DMH circuit (Fig. 5k). RUVBL2 KO significantly decreased both amplitude and frequency of sEPSCs (Fig. 5l–n), and the amplitude of eEPSCs in DMH neurons (Fig. 5o, p). In addition, the pair-pulse ratio (PPR) (ISI, 50 ms) in DMH neurons was significantly increased after RUVBL2 KO (Fig. 5q). We further demonstrate that there were no changes in either amplitude or frequency of sIPSCs in DMH neurons after RUVBL2 KO (Fig. 5r–t). Collectively, these results demonstrate that PVH RUVBL2 KO impairs the excitatory synaptic transmission in both PVHRUVBL2 → ARC and PVHRUVBL2 → DMH circuits.
RUVBL2 KO impairs the excitatory synaptic transmission in PVHRUVBL2 → ARCPOMC and PVHRUVBL2 → ARCNPY circuits
Considering that PVH RUVBL2 KO impairs the excitatory synaptic transmission to ARC neurons (Fig. 5a–j), and PVHRUVBL2 neurons send projections to ARCPOMC and ARCAgRP neurons (Fig. 3d–i), we investigated whether PVH RUVBL2 KO impairs excitatory synaptic transmission to ARCPOMC or ARCAgRP neurons. To this end, we first generated Ruvbl2fl/fl;Pomc-EGFP mice by crossing Ruvbl2fl/fl mice with Pomc-EGFP mice, bilaterally injected AAV2-Cre-mCherry into the PVH of 8 week-old Ruvbl2fl/fl;Pomc-EGFP mice to knock out RUVBL2, and performed electrophysiological recordings in the brain slices containing ARCPOMC neurons after 6 weeks of AAV injection (Fig. 6a). As shown (Fig. 6b–d), both amplitude and frequency of sEPSCs in ARCPOMC neurons were significantly decreased after PVH RUVBL2 KO in the PVH. In addition, the peak amplitude of eEPSCs was also significantly decreased (Fig. 6e, f), while the PPR was significantly increased after PVH RUVBL2 KO (Fig. 6g). These results demonstrate a presynaptic role of RUVBL2 in the excitatory synaptic transmission to ARCPOMC neurons.

a Schematic of AAV injection into the PVH and electrophysiology recordings in ARCPOMC neurons. b Representative traces of sEPSCs recorded in ARCPOMC neurons in the presence of PTX. c, d Amplitude (c) and frequency (d) of sEPSCs recorded in ARCPOMC neurons. AAV2-mCherry (n = 30 neurons/3 mice), AAV2-Cre-mCherry (n = 25 neurons/3 mice). e Representative traces of eEPSCs recorded in ARCPOMC neurons in response to paired-pulse stimulations (50 ms interval) in the presence of PTX. f, g Amplitude of the first EPSCs (f) and PPR (g) recorded in ARCPOMC neurons. AAV2-mCherry (n = 38 neurons/4 mice), AAV2-Cre-mCherry (n = 23 neurons/3 mice). h Schematic of AAV injection into the PVH and electrophysiology recordings in ARCNPY neurons. i Representative traces of sEPSCs recorded in ARCNPY neurons in the presence of PTX. j, k Amplitude (j) and frequency (k) of sEPSCs recorded in ARCNPY neurons. AAV2-mCherry (n = 33 neurons/3 mice), AAV2-Cre-mCherry (n = 29 neurons/3 mice). l Representative traces of eEPSCs recorded in ARCNPY neurons in response to paired-pulse stimulations (50 ms interval) in the presence of PTX. m, n Amplitude of the first EPSCs (m) and PPR (n) recorded in ARCNPY neurons. n = 29 neurons/3 mice per group. o Schematic of simultaneously activating ARC-projecting PVHRUVBL2 neurons and ARCNPY neurons. p Food intake of mice within 1 h after different treatments as indicated (as in o). n = 8 mice per group. q Food intake of mice within 6 h after different treatments as indicated. SHU9119, 0.5 mg/kg. Stim, optical stimulation for 6 h when the laser light (465 nm) was switched on every other 30 min. For SHU9119+Stim, mice were first intraperitoneally injected with SHU9119, followed by optical stimulation for 6 h when the laser light (465 nm) was switched on every other 30 min. n = 6 mice per group. Two-tailed unpaired Student’s t-test for (c–n). ns, no significance. One-way ANOVA with Geisser-Greenhouse correction followed by Fisher’s LSD test for (p) and (q). All data are presented as means ± SEM. Source data are provided as a Source Data file.
To examine whether PVH RUVBL2 KO impacted the synaptic transmission to ARCAgRP neurons, we generated Ruvbl2fl/fl;Npy-GFP mice by crossing Ruvbl2fl/fl mice with Npy-GFP mice27 (Co-expression of Npy and AgRP in the ARC20), bilaterally injected AAV2-Cre-mCherry into the PVH of 8 week-old Ruvbl2fl/fl;Npy-GFP mice to knock out RUVBL2, and performed electrophysiological recordings in the brain slices containing ARCNPY neurons (Fig. 6h). PVH RUVBL2 KO did not change the amplitude of sEPSCs (Fig. 6i, j), but significantly decreased the frequency of sEPSCs in ARCNPY neurons (Fig. 6k). In addition, PVH RUVBL2 KO significantly decreased the peak amplitude of eEPSCs in ARCNPY neurons (Fig. 6l, m), but did not alter PPR (ISI, 50 ms) (Fig. 6n). Collectively, these data indicate that PVH RUVBL2 is required for excitatory synaptic transmission between PVHRUVBL2 neurons and ARCPOMC or ARCNPY neurons.
As ARCPOMC and ARCAgRP neurons exert an opposing role in feeding, and our projection-specific chemogenetics and optogenetics have demonstrated the anorexigenic effect of PVHRUVBL2 → ARC, we postulated that the anorexigenic effect of PVHRUVBL2 → ARCPOMC and other ARC neurons could outweigh the orexigenic effect of PVHRUVBL2 → ARCAgRP. To test this, we bilaterally injected Cre-dependent AAV9-DIO-hChR2-mCherry into the PVH of 8 week-old Ruvbl2-Cre mice that were implanted with an optic fiber above the ARC and also received bilateral injection of AAV9-Npy-FLP and FLP-dependent AAV9-fDIO-hM3D(Gq)-EGFP in the ARC (Fig. 6o). In this way, we were able to simultaneously activate ARC-projecting PVHRUVBL2 and ARCNPY neurons. As shown, activation of both ARC-projecting PVHRUVBL2 and ARCNPY neurons conferred a marked anorexigenic effect (Fig. 6p), supporting that the anorexigenic effect of PVHRUVBL2 → ARC outweighs the orexigenic effect of PVHRUVBL2 → ARCAgRP.
To demonstrate that activation of PVHRUVBL2 → ARCPOMC neural circuit partially, if not completely, mediates the anorexigenic effects by activation of PVHRUVBL2 → ARC projections, we bilaterally injected Cre-dependent AAV9-DIO-hChR2-mCherry into the PVH of 8 week-old Ruvbl2-Cre mice implanted with an optic fiber above the ARC. The mice were pre-treated with SHU9119, a potent antagonist of MC3/4R61, prior to optical activation of PVHRUVBL2 → ARC projections for 6 h. We observed that SHU9119 pretreatment significantly attenuated the anorexigenic effects induced by activation of PVHRUVBL2 → ARC projections (Fig. 6q), supporting the important contribution of the melanocortin pathway to anorexia by activation of PVHRUVBL2 → ARC.
RUVBL2 KO in the PVH reduces presynaptic boutons and alters synaptic vesicle distribution
Impairment in synaptic transmission could arise from aberrations of presynaptic inputs and/or synaptic vesicle distribution, which have been suggested to have direct impacts on synaptic plasticity22,62. Thus, we reasoned that RUVBL2 KO might impair presynaptic inputs and/or presynaptic vesicle distribution, consequently causing abnormalities in synaptic transmission. To address this, we bilaterally injected AAV2-GFP or AAV2-Cre-GFP into the PVH of WT or Ruvbl2fl/fl mice to knock out RUVBL2, and then analyzed presynaptic boutons as reflected by VGluT2-labeled puncta. As shown (Fig. 7a–d), VGluT2-labeled puncta in the ARC or DMH are significantly reduced after PVH RUVBL2 KO.

a Representative images for IHC staining of the excitatory synapses in the ARC (n = 3 mice; scale bar, 100 μm). b Quantitation of VGluT2+ puncta in the ARC. AAV2-GFP (n = 17 profiles/3 mice), AAV2-Cre-GFP (n = 20 profiles/3 mice). c Representative images for IHC staining of the excitatory synapses in the DMH. (n = 3 mice; scale bar, 100 μm). d Quantitation of VGluT2+ puncta in the DMH. AAV2-GFP (n = 22 profiles/3 mice), AAV2-Cre-GFP (n = 23 profiles/3 mice). e Representative transmission electron microscopy (TEM) micrographs of the ARC. (n = 3 mice; scale bar, 200 nm). f Length of the active zone (AZ) in the ARC. AAV2-GFP (n = 75 profiles/3 mice), AAV2-Cre-GFP (n = 65 profiles/3 mice). g, h Quantity of synaptic vesicles (SVs) within different distances (g) and quantity of SVs within 200 nm (h) from the AZ in the ARC. AAV2-GFP (n = 75 profiles/3 mice), AAV2-Cre-GFP (n = 65 profiles/3 mice). i Representative TEM micrographs of the DMH (n = 3 mice; scale bar, 200 nm). j AZ length in the DMH. AAV2-GFP (n = 70 profiles/2 mice), AAV2-Cre-GFP (n = 66 profiles/2 mice). k,l Quantity of synaptic vesicles (SVs) within different distances (k) and quantity of SVs within 200 nm (l) from the AZ in the DMH. AAV2-GFP (n = 70 profiles/2 mice), AAV2-Cre-GFP (n = 66 profiles/2 mice). m GO enrichment analysis of the genes based on chromatin immunoprecipitation sequencing (ChIP-Seq). The top canonical pathways associated with the central nervous system are shown (p < 0.05). n, o Genes related to synaptic vesicle (n), or neuron axonogenesis (o) identified by ChIP-Seq. p, q Heatmap showing down-regulated genes related to synaptic vesicle (p), or neuron axonogenesis (q) by RNA sequencing (RNA-Seq). The combined analyses of RNA-Seq and ChIP-Seq (as in n, o) identify a number of genes related to synaptic vesicle or neuron axonogenesis that are down-regulated after RUVBL2 KO in the PVH. Two-tailed unpaired Student’s t-test for (b, d, f, h, j, l). ns, no significance. Two-way ANOVA followed by Bonferroni’s test for (g) and (k). All data are presented as means ± SEM. Source data are provided as a Source Data file.
Next, we applied transmission electron microscopy (TEM) to examine the distribution of synaptic vesicles. Although RUVBL2 KO does not alter the length of active zone (AZ), it significantly reduces synaptic vesicles within 200 nm from the AZ in PVHRUVBL2 neuron terminals in the ARC or DMH (Fig. 7e-l). Given that PVH RUVBL2 is the only variable in these experiments, we suppose that the altered presynaptic boutons and synaptic vesicle distribution are ascribed to RUVBL2 KO in the PVH. Note that these results are also consistent with our electrophysiology analysis. Collectively, these data suggest that RUVBL2 KO-caused impairment in the excitatory synaptic transmission underlies food intake increase.
We next investigated how RUVBL2 KO reduces presynaptic boutons and alters synaptic vesicle distribution. As RUVBL2 has been shown to participate in transcriptional control by interacting with different transcription factors or forming various chromatin-modifying complexes40, we performed ChIP-Seq of the hypothalamus isolated from 8 week-old male WT mice. Our ChIP-Seq data show that RUVBL2 binds to the promoters of numerous genes that get involved in regulation of synaptic plasticity, in particular synaptic vesicle or neuron axonogenesis (Fig. 7m–o).
To further demonstrate the transcription control of genes related to synaptic vesicle or neuron axonognenesis by RUVBL2, we carried out RNA-Seq of the PVH from Ruvbl2fl/fl mice that received bilateral injection of AAV2-GFP or AAV2-Cre-GFP into the PVH to knock out RUVBL2. Our combined analyses of RNA-Seq and the above-described ChIP-Seq reveal the down-regulation of a number of genes related to synaptic vesicle or neuron axonognenesis after RUVBL2 KO in the PVH (Fig. 7p, q). Together, these data are in support of our findings that PVH RUVBL2 KO reduces presynaptic boutons and alters synaptic vesicle distribution.
RUVBL2 overexpression enhances the excitatory synaptic transmission and suppresses food intake
We finally asked whether increased expression of RUVBL2 in the PVH enhances the excitatory synaptic transmission and suppresses food intake. To address this, we made a Cre-dependent RUVBL2 overexpression mouse model by inserting mouse Ruvbl2 coding sequence into the Hipp11 (H11) locus63 (Ruvbl2KI/+) (Supplementary Fig. 8a), and crossed Ruvbl2KI/+ mice with Sim1-Cre transgenic mice10 to generate Ruvbl2KI/KI;Sim1-Cre mice showing RUVBL2 overexpression in the PVH (Supplementary Fig. 8b, c).
Using Ruvbl2KI/KI;Sim1-Cre mice, we examined the electrophysiological characteristics of ARC and DMH neurons receiving projections from PVHRUVBL2 neurons. We found that the amplitude of sEPSCs was significantly increased in ARC or DMH neurons of Ruvbl2KI/KI;Sim1-Cre mice compared with WT mice (Fig. 8a–d), although there was no difference in the frequency of sEPSCs (Fig. 8e, f). In addition, the amplitude of eEPSCs was significantly increased in ARC or DMH neurons of Ruvbl2KI/KI;Sim1-Cre mice (Fig. 8g–j), while no difference in PPR (ISI 50 ms) was observed (Fig. 8k, l). Note that no changes were observed in either amplitude or frequency of sIPSCs in ARC or DMH neurons after RUVBL2 overexpression (Supplementary Fig. 8d–i). Together, these results demonstrate that RUVBL2 overexpression through the Sim1-Cre transgene enhances the excitatory synaptic transmission in PVHRUVBL2 → ARC and PVHRUVBL2 → DMH circuits.

a, b Representative traces of sEPSCs recorded in ARC (a) and DMH (b) neurons in the presence of PTX. c–f Amplitude and frequency of sEPSCs recorded in ARC (c, e) and DMH (d, f) neurons. For ARC: WT (n = 30 neurons/3 mice), Ruvbl2KI/KI;Sim1-Cre (n = 31 neurons/3 mice). For DMH: WT (n = 28 neurons/4 mice), Ruvbl2KI/KI;Sim1-Cre (n = 25 neurons/3 mice). g, h Representative traces of eEPSCs recorded in ARC (g) and DMH (h) neurons in response to paired-pulse stimulations (50 ms interval) in the presence of PTX. i–l Amplitude of the first EPSCs recorded in ARC (i) and DMH (j) neurons. PPR in ARC (k) and DMH (l) neurons. For ARC: WT (n = 30 neurons/3 mice), Ruvbl2KI/KI;Sim1-Cre (n = 36 neurons/3 mice). For DMH: WT (n = 28 neurons/4 mice), Ruvbl2KI/KI;Sim1-Cre (n = 26 neurons/3 mice). m Representative TEM micrographs in the ARC. n-p AZ Length (n), SVs within different distances (o) and within 10 nm (p) from the AZ in the ARC. WT (n = 36 profiles/2 mice), Ruvbl2KI/KI;Sim1-Cre (n = 66 profiles/2 mice). q Representative TEM micrographs in the DMH. r-t AZ Length (r), SVs within different distances (s) and within 10 nm (t) from the AZ in the DMH. WT (n = 63 profiles/2 mice), Ruvbl2KI/KI;Sim1-Cre (n = 57 profiles/2 mice). u,v Body weight of male (u) and female (v) mice on chow. For males: WT (n = 7); Ruvbl2KI/KI;Sim1-Cre (n = 9). For females: WT (n =6); Ruvbl2KI/KI;Sim1-Cre (n = 8). w Daily chow intake of male and female mice. For males: WT (n = 7), Ruvbl2KI/KI;Sim1-Cre (n = 8). For females: n = 6 per group. x,y Body weight of male (x) and female (y) mice on HFD. For males: n = 8 per group. For females: WT (n = 8), Ruvbl2KI/KI;Sim1-Cre (n = 9). z Daily HFD intake of male and female mice. For males: n = 8 per group. For females: WT (n = 7), Ruvbl2KI/KI;Sim1-Cre (n = 8). Scale bar, 200 nm. Two-tailed unpaired Student’s t-test for (c–n, p, r, t, w, z). ns, no significance. Two-way ANOVA followed by Bonferroni’s test for (o, s, u, v, x, y). All data are presented as means ± SEM. Source data are provided as a Source Data file.
Our electrophysiological results implicate that RUVBL2 overexpression may influence the distribution of synaptic vesicles. Through TEM, we observed that synaptic vesicles within 10 nm from the AZ are significantly increased in PVHRUVBL2 neuron terminals in the ARC or DMH of Ruvbl2KI/KI;Sim1-Cre mice (Fig. 8m–t), suggesting that RUVBL2 overexpression alters the distribution of synaptic vesicles, thus enhancing the excitatory synaptic transmission.
When fed a regular chow diet, Ruvbl2KI/KI;Sim1-Cre mice showed significantly decreased body weight (Fig. 8u, v). The decrease of body weight in Ruvbl2KI/KI;Sim1-Cre mice is attributed to significantly decreased food intake (Fig. 8w), as there is comparable energy expenditure between these two genotypes as indicated by ANOVA (Supplementary Fig. 8j, k). Together, these data demonstrate that RUVBL2 overexpression suppresses food intake and this effect could be ascribed to an increase in the excitatory transmission.
To evaluate whether RUVBL2 overexpression confers any protection against diet-induced obesity (DIO), WT and Ruvbl2KI/KI;Sim1-Cre mice started HFD feeding at the age of 8 weeks, and their weekly body weight was monitored. Ruvbl2KI/KI;Sim1-Cre mice showed robust resistance to DIO (Fig. 8x, y), which is owing to the significantly reduced HFD intake (Fig. 8z), as no difference of energy expenditure was observed between these two genotypes as demonstrated by ANOVA (Supplementary Fig. 8l, m). We also observed significant improvement in blood glucose control and insulin sensitivity in Ruvbl2KI/KI;Sim1-Cre mice compared with WT mice after HFD (Supplementary Fig. 8n–q). These data collectively demonstrate that RUVBL2 overexpression protects mice against DIO by suppressing food intake, and indicate that PVH RUVBL2 controls food intake by modulating synaptic plasticity (synaptic vesicle distribution) and synaptic transmission
Given that Sim1 is expressed in multiple regions other than the PVH13, we sought to further confirm the functional role of PVH RUVBL2 in energy homeostasis by bilaterally injecting Cre-dependent AAV2-DIO-RUVBL2 into the PVH of 4 week-old Sim1-Cre mice (Supplementary Fig. 10a, b). As shown, RUVBL2 overexpression conferred resistance to DIO (Supplementary Fig. 10c, d), which was primarily attributed to decreased food intake (Supplementary Fig. 10e, f). RUVBL2 overexpression mice showed significantly improved glucose tolerance and insulin sensitivity (Supplementary Fig. 10g, h). Together, these results support that PVH RUVBL2 inhibits food intake and protects against body weight gain and DIO.
Discussion
In this study, we reveal that RUVBL2 in the PVH suppresses food intake by modulating synaptic plasticity, and demonstrate that PVHRUVBL2 neurons exert their anorexigenic effect through projecting to three brain regions. The elucidation of neural circuits between PVHRUVBL2 neurons and these brain regions should greatly advance our understanding of PVH control of satiety.
We found that RUVBL2 is abundantly expressed in the PVH and PVHRUVBL2 neurons constitute the majority of PVHSIM1 neurons. Previous studies have established the PVH as a crucial satiety control site in the brain1,18,20. Consistently, either KO of RUVBL2 in the PVH or chemogenetic inhibition of PVHRUVBL2 neurons increase food intake, whereas chemogenetic or optogenetic activation of PVHRUVBL2 neurons suppress food intake. Note that neither altering PVH RUVBL2 expression nor manipulating PVHRUVBL2 neuron activity caused any observed changes on energy expenditure, suggesting that RUVBL2 and PVHRUVBL2 neurons control energy balance primarily by regulating food intake. Nevertheless, it could be hard to exclude a potential effect of RUVBL2 on energy expenditure, given its abundant expression in the PVH and the reported role of PVHBDNF and PVHOxytocin neurons in energy expenditure in previous studies14,64.
Our study suggests three neural circuits in total mediating the anorexigenic effect of PVHRUVBL2 neurons: PVHRUVBL2 → PB, PVHRUVBL2 → ARC, and PVHRUVBL2 → DMH. We found that chemogenetic inhibition of PB-projecting PVHRUVBL2 neurons significantly increases food intake. Consistently, previous studies have shown that several types of PVH neurons including PVHMC4R, PVHPDYN, and PVHTrkB neurons exert their anorexigenic effects by projecting to the PB13,15,49. These studies together indicate that the PB is an important downstream brain structure mediating the anorexigenic effects of PVH neurons.
The anorexigenic effect of PVHRUVBL2 neurons could also be ascribed to their projections to the ARC that senses nutritional state and controls energy homeostasis5. On one hand, ARCPOMC and ARCAgRP neurons project to distinct downstream brain sites as two well-studied cell types and essential components of central melanocortin system8. One well-established neural pathway is composed of the projections from ARCPOMC and ARCAgRP neurons to the PVH, resulting in activation and inhibition of PVHMC4R neuron, respectively5. On the other hand, ARCPOMC or ARCAgRP neuron activity is also regulated by either peripheral signals21, or presynaptic inputs from a number of brain regions such as the brain stem65, the DMH34, the VMH28, and the PVH35. Interestingly, one study has suggested that leptin exerts it anorexigenic effects mainly through reducing the GABAergic inhibitory tone to POMC neurons58. In the present study, we show that ARCPOMC neurons receive dense projections from glutamatergic PVHRUVBL2 neurons, and PVH RUVBL2 KO impaired the excitatory synaptic transmission to ARCPOMC neurons, suggesting that PVHRUVBL2 → ARCPOMC circuit mediates the anorexigenic activity of PVHRUVBL2 neurons. Consistently, one study reported the synaptic connection between the PVH and ARCPOMC neurons, without elucidating the function66.
One previous study showed that activation of ARCPOMC neurons did not suppress food intake instantly, but significantly reduced food intake within 24 hr67, while our study showed that optogenetic activation of PVHRUVBL2 → ARC circuit instantly suppressed food intake. These suggest that projections of PVHRUVBL2 neurons to some non-POMC ARC neurons could exert an instant appetite-suppressing effect; yet the identities of these non-POMC ARC neurons remain to be defined in future studies. In addition, this study suggests a potential positive-feedback loop between the ARC and the PVH, thereby promoting a maximal or sustained satiety. Given the prominent role of ARCPOMC → PVHMC4R neural pathway in suppressing food intake and a part of PVHMC4R neurons (~33%) are RUVBL2-positive, we would suppose that ARCPOMC neurons and MC4R-positive PVHRUVBL2 neurons could form reciprocal circuits; or within the PVH there may exist a neuronal circuit between RUVBL2-negative PVHMC4R neurons and MC4R-negative PVHRUVBL2 neurons. These possible neural circuits connect the PVH with the ARC, forming an enclosed circuit that functions as a positive-feedback loop and thus promotes substantial satiety.
PVHRUVBL2 → DMH is another neural circuit mediating the anorexigenic effect of PVHRUVBL2 neurons. The DMH controls energy homeostasis through regulating food intake and energy expenditure68,69. Accumulative studies have demonstrated the stimulatory role for DMH neurons in adaptive thermogenesis70,71,72,73,74. Interestingly, however, DMH neurons have been shown to exert different roles in regulation of food intake depending on cell types75,76,77,78. In the present study, we found that alteration of PVH RUVBL2 expression or manipulation of PVHRUVBL2 neuron activity only affect food intake, but do not influence energy expenditure. Therefore, the identity of DMH neurons receiving axonal terminals from PVHRUVBL2 neurons remains to be defined.
Multiple factors are involved in modulating synaptic plasticity including quantity or activity of postsynaptic receptors, presynaptic input, and quantity or distribution of synaptic vesicles79,80,81,82. To date, most studies on synaptic plasticity mainly focused on postsynaptic mechanisms including the number or property of postsynaptic receptor; in contrast, the presynaptic mechanisms (e.g., presynaptic inputs, and quantity or distribution of synaptic vesicles) have been relatively underestimated. The presynaptic alterations including structural remodeling and reorganization of presynapses tend to last long, thereby underlying a long-term behavioral change22,83. Interestingly, one recent study showed that the increased axonal terminals in PVHTRH → ARCAgRP circuit promoted appetite and body weight (re)gain in the setting of weight loss, suggesting a role for structural synaptic plasticity in regulation of food intake35. Consistently, we demonstrated that PVH RUVBL2 KO decreases the presynaptic boutons of ARC-projecting PVHRUVBL2 neurons, impairs synaptic transmission, and increases food intake, implicating a causal role of structural synaptic plasticity in food intake control.
The quantity and distribution of synaptic vesicle could also affect synaptic plasticity in either a short-term (minutes to hours) or a long-term (days or more) manner, thereby altering synaptic strength and ultimately changing behaviors79. In agreement with this, we observed that RUVBL2 KO significantly reduces synaptic vesicles, whereas RUVBL2 overexpression significantly increases synaptic vesicles.
RUVBL2 has been shown to control transcription by forming different transcription-associated protein complexes40, or regulating the clustering of RNA polymerase II with different transcription factors84, which is in agreement with its abundant distribution in the nucleus41. Our data indicate that RUVBL2 modulates synaptic plasticity and synaptic transmission by regulating the expression of genes related to axonogenesis and synaptic vesicle, consistent with the role for RUVBL2 in transcription control40. In support of the role for PVH RUVBL2 in modulating synaptic plasticity, overexpression of RUVBL2 through the Sim1-Cre transgene increases synaptic vesicles within 10 nm near the AZ in PVHRUVBL2 neuron terminals in the ARC or DMH. In addition, we observed that PVH RUVBL2 is significantly reduced during energy deficit, suggesting that RUVBL2 regulates synaptic plasticity in a dynamic manner in response to changing energy state. Of interest, however, whether RUVBL2 modulates synaptic plasticity and controls food intake dependent of its ATPase activity remains unknown, given that hypothalamic AMP-activated protein kinase (AMPK), a cellular energy sensor, has been shown to regulate food intake through diverse mechanisms85,86,87,88.
In summary, this study reveals two neural circuits of PVHRUVBL2 → ARCPOMC and PVHRUVBL2 → DMH that mediate the anorexigenic effect of the PVH. Moreover, this study demonstrates an essential role for PVH RUVBL2 in food intake control by modulating synaptic plasticity, suggesting that modulation of synaptic plasticity could be an effective way to regulate food intake and body weight.
Methods
Animals
All animal care and use procedures were approved by the Animal Care and Use Committee (ACUC) at Chu Hsien-I Memorial Hospital & Tianjin Institute of Endocrinology, Tianjin Medical University (DXBYY-IACUC-2020036). The mice were euthanized with CO2, followed by cervical dislocation. Both male and female mice were used in the study.
All mice were maintained on C57BL/6 J background. Mice were housed at the animal facility harboring a stable temperature (~22 °C), humidity and 12/12 h light/dark cycle, and gained free access to food and water. C57BL/6 WT mice were purchased from GemPharmatech (Nanjing, China). Ai14 (Stock #:007914), Sim1-Cre (Stock #:006395), Mc4r-2a-Cre (Stock #:030759), Pomc-EGFP (Stock #:009593), Agrp-Cre (Stock #012899), Npy-GFP (Stock #008321), Vglut2-ires-cre (Stock #:016963), Vgat-ires-cre (Stock #:028862), and Pomc-Cre (Stock #:005965) mouse strains were from the Jackson Laboratory (Bar harbor, ME, USA).
Loxp-flanked Ruvbl2 (Ruvbl2fl/+) mice were generated by BIOCYTOGEN (Beijing, China). To generate Ruvbl2fl/+ mice, two loxp sequences were inserted to flank the exons E3-E6 of Ruvbl2 via CRISPR-Cas942.
Both Ruvbl2-Cre and Cre-dependent Ruvbl2 overexpression knock-in (Ruvbl2KI/+) mice were generated by GemPharmatech (Nanjing, China). To generate Ruvbl2-Cre mice, P2A-Cre sequence was inserted after the coding sequence of the Ruvbl2 gene via CRISPR-Cas9 system. To generate Ruvbl2KI/+ knock-in mice, a donor vector harboring the LoxP-Stop-LoxP (LSL) cassette and the mouse RUVBL2 coding sequence (CDS) that is in fusion with an HA tag was inserted into the ubiquitous H11 locus via CRISPR–Cas9 system.
Eight-week-old WT and Ruvbl2KI/KI;Sim1-Cre were fed a high-fat diet (HFD) and their weekly body weight was recorded. After 8 weeks of HFD feeding, analyses of daily food intake and metabolic cages, IPGTT and IPITT were carried out.
Stereotaxic Injection of AAV
Mice were deeply anesthetized with 1.2% avertin and placed in a stereotaxic apparatus. Body temperature was maintained at 37 °C using a heating pad, and AAV was injected into the PVH of mice using a 5 μl Hamilton syringe with a 33-gauge needle that was attached to a stereotaxic arm. Infusion of each virus (0.2 μl, 1.8 μl/h, 1012 viral particles/ml) was accomplished by a microsyringe pump into the PVH region using the following coordinates: AP, −0.65 mm; ML, ±0.28 mm; and DV,−5.05 mm relative to the bregma. After infusion, the needle was left in the brain for another 10 min to reduce backflow of viral injection along the needle track. The mice were recovered in a 37 °C incubator before returning to home cage. Mice with missed viral injections or unilateral injections were excluded from analysis (e.g., body weight and food intake measurement) after examining the virus-associated fluorescent protein expression.
The corresponding coordinates of other target brain regions in this study are as follows (calculated from the bregma): the ARC (AP, −1.85 mm; ML, ±0.27 mm; DV, −5.85 mm), the DMH (AP, −1.95 mm; ML, ±0.26 mm; DV, −5.10 mm), the POA (AP, −0.00 mm; ML, ±0.260 mm; DV, −5.15 mm), the MeA (AP,−1.46 mm; ML, ±1.95 mm; DV, −5.50 mm), the LSv (AP, 0.20 mm; ML, ±0.5 mm; DV, −3.50 mm), the NTS (AP, −7.0 mm; ML, ±0.7 mm; DV, −4.7 mm), and the PB (AP, −4.96; ML, ±1.4 mm; DV, −5.5 mm).
The following AAV were used: AAV2-GFP (HANBIO, Cat#57070645), AAV2-Cre-GFP (HANBIO, Cat#57070646), AAV8-EF1α-DIO-EGFP-T2A-TK (BrainVTA, Cat#PT-0087), HSV-ΔTK-LSL-tdTomato (BrainVTA, Cat#H03002), AAV2/retro-DIO-EGFP (OBIO, Cat#H6363), AAV8-EF1α-DIO-oRVG-WPRE (BrainVTA, #PT-0023), AAV8-EF1α-DIO-H2B-EGFP-T2A-TVA-WPRE (BrainVTA, #PT-0021), RV-ENVA-ΔG-DsRed (BrainVTA, Cat#R01002), AAV9-EF1a-DIO-hM3D(Gq)-mCherry (BrainVTA, Cat#PT-0042), AAV9-EF1a-DIO-hM4D(Gi)-mCherry (BrainVTA, Cat#PT-0043), AAV9-EF1a-DIO-mCherry (BrainVTA, Cat#PT-0013), AAV5-hSyn-Con/Fon-hM4D(Gi)-EYFP (BrainVTA, Cat#PT-1572), AAV2/retro-Flp (BrainVTA, Cat#PT-0341), AAV9-EF1a-DIO-hChR2-mCherry (BrainVTA, Cat#PT-0002), AAV9-Npy-FLP (BrainVTA, Cat#PT-2297), AAV9-fDIO-hM3D(Gq)-EGFP (BrainVTA, Cat#PT-1837), AAV2-mCherry (BrainVTA, Cat#PT-0407), AAV2-Cre-mCherry (BrainVTA, Cat#PT-0100), AAV2-DIO-GFP (OBIO, Cat#H15469), AAV2-DIO-RUVBL2-GFP (OBIO, Cat#H29352).
Isolation of the PVH
Mice were euthanized with CO2, followed by cervical dislocation. The whole mouse brain was rapidly dissected and placed in a brain mold (Reinwald, model: 68707). Slices at the thickness of 1 mm (bregma ~−0.4- ~ −1.4 mm) were obtained using a blade, and then placed under a dissecting microscope (Leica EZ4) to isolate the PVH region carefully using fine scissors and forceps. The collected samples were stored in Trizol reagent (Invitrogen, Cat# 15596018) at −80 °C for subsequent RNA extraction, cDNA synthesis and quantitative PCR (qPCR).
Quantitative real-time PCR (qPCR)
The extraction of RNA and qPCR were performed as previously described89. In brief, total RNA was extracted from the hypothalamic paraventricular nucleus (PVH) using Trizol reagent (Invitrogen, Cat# 15596018). Reverse transcription PCR was carried out using a RevertAid RT kit (thermoscientific, Cat#K1691), and quantitative PCR was conducted using SGExcel FastSYBR Mixture (Sangon Biotech, Cat# B532955) through QuantStudio 3 Real-Time PCR System (Applied Biosystems) following the manufacturer’s instructions. Hprt1 (forward: 5’-CAGTCCCAGCGTCGTGATTA-3’, reverse: 5’-TGGCCTCCCATCTCCTTCAT-3’), Ruvbl2 (forward: 5’-TTGGTGTCCGGATCAAGGAG-3’, reverse: 5’-CTTGAGGGTCAGTTTGCCCA-3’). The relative expression of target genes was calculated based on 2-ΔΔCt method with Hprt1 as the reference gene.
Western blotting (WB)
Eight week-old male C57BL/6 J mice fed a regular chow diet ad libitum or fasted for 48 h were euthanized with CO2, followed by cervical dislocation. The PVH was isolated and homogenized in 1 × lysis buffer containing 1% deoxycholic acid, 10 mM Na4P2O7, 1% Triton 100, 100 mM NaCl, 5 mM EDTA, 50 mM Tris-HCl, and 0.1% SDS. Protein concentrations were determined using a BCA protein assay kit (Biosharp, Cat# BL521). 20 μg of total protein was loaded in SDS-PAGE and transferred to PVDF membranes, followed by 1 h of blocking with 5% nonfat milk and incubation with primary antibodies against RUVBL2 (1:10000; proteintech #67851-1-Ig) or HSP90 (1:10000; proteintech #13171-1-AP) at 4 °C overnight. Finally, ECL detection systems were applied to develop signals. Densitometric analysis of WB bands was performed using ImageJ software.
Physiological measurements
Body weight was measured weekly after virus injection, or before and after HFD; daily food intake was measured after 12 weeks of virus injection or after 8 weeks of HFD as described previously90. Briefly, mice were singly housed for 1 week to minimize the stress before their daily food intake was measured. After measurement of weekly body weight and daily food intake, energy expenditure (metabolic rate or oxygen consumption rate VO2) was monitored using Promethion High-Definition Multiplexed Respirometry System (Sable Systems, North Las Vegas, NV, USA).
Immunohistochemistry
Mice were anesthetized with 1.2% avertin and perfused transcardially with ice-cold phosphate-buffered saline (PBS), followed by 4% paraformaldehyde (PFA) in PBS. Brain tissues were postfixed overnight at 4 °C and then immersed in 30% sucrose (in PBS) for 3 days at 4 °C before being sectioned at 30 μm thicknesses using a sliding microtome (Leica, SM2010R). Brain sections were rinsed once with PBS, permeabilized with 1% Triton X-100 in PBS for 10 min and washed with PBS for 5 min, then incubated with blocking buffer (0.2% Triton X-100, 5% goat serum or 1% bovine serum albumin and 10% goat serum in PBS) for 1 h at room temperature, and subsequently incubated with primary antibodies diluted in the blocking buffer overnight at 4 °C. The following primary antibodies were used: mouse anti-RUVBL2 (1:800; proteintech #67851-1-Ig), rabbit anti-RUVBL2 (1:400; abcam #ab36569), rabbit anti-prodynorphin (1:100; ABclonal #A5830), mouse anti-Oxytocin (1:1000; Millipore #MAB5296), rabbit anti-CRH (1:200; proteintech # 10944-1-AP), rabbit anti-tyrosine hydroxylase (1:1000; Sigma #AB152), rabbit anti-TRH (1:500; ThermoFisher #PA5-57331), rabbit anti-c-Fos (1:300; proteintech # 66590-1-Ig), rabbit anti-HA-Tag (1:1000; Cell Signaling Technology # 3724), rabbit anti-vGlut2 (1:1000, abcam #ab216463), rabbit anti-glutamate (1:500, Sigma #G6642). After three washes in PBST (PBS containing 0.2% Triton X-100) for 20 min each, sections were incubated with appropriate fluorescent secondary antibodies for 1 h at room temperature. Sections were washed in PBST for three times (20 min per time and a last wash in PBS for 10 min), incubated with 4′,6-diamidino-2- phenylindole (DAPI) for counter staining for 2 min, and then mounted onto slides for confocal image capture using a ZEISS confocal microscope (LSM 800).
An antibody against VGluT2 was used for IHC staining of the excitatory synapses in the ARC and DMH. AAV2-Cre-GFP or control AAV2-GFP was bilaterally injected into the PVH of 8 week-old Ruvbl2fl/fl mice that were euthanized for IHC staining 6 weeks later. To quantify vGlut2+ puncta, images were saved as CZI files and analyzed with NIH Image software. For each image, the quantity of VGlut2+ puncta in a selected area was counted.
To investigate the co-expression of RUVBL2 with PDYN, Oxytocin, or CRH, mice were treated with 20 μg of colchicine (Sigma, C3915) in 1 μL of H2O through intracerebraventricular injection with a microinjector at a slow rate (50 nl min−1) with customized controllers (Coordinates: 1.0 mm lateral, 0.4 mm back from the bregma, and 2.5 mm below the skull). Injection was begun 5 min after tissue penetration, and the microinjector was pulled out 10 min after injection. After suturing, mice were recovered in a warm bracket before they were transferred to housing cages for 48 h before perfusion. The IHC staining images were captured using a ZEISS confocal microscope (LSM 800).
Intraperitoneal glucose tolerance test (IPGTT) and intraperitoneal insulin tolerance test (IPITT)
IPGTT and IPITT were performed as previously described90. Briefly, as to IPGTT, mice were fasted overnight for 16 h with free access to water. The blood glucose levels were measured at baseline (0 min) from the tail vein using a glucometer before mice were intraperitoneally injected with glucose (1.5 g/kg body weight). After glucose injection, blood glucose levels were measured every 30 min until 2 h post injection. As to IPITT, mice were fasted for 6 h with free access to water. The fasting glucose levels were measured at baseline (0 min) before an intraperitoneal injection of insulin (0.5 U/kg body weight). Blood glucose levels were measured every 30 min until 2 h post injection of insulin. A portable glucometer (OneTouch Ultra) was used to measure blood glucose levels.
Anterograde monosynaptic tracing
The helper AAV8-EF1α-DIO-EGFP-T2A-TK and HSV-ΔTK-LSL-tdTomato were used to carry out anterograde monosynaptic tracing52. In the absence of the thymidine kinase (TK), the HSV-ΔTK does not replicates or spreads in neurons, but is capable of transmitting to the postsynaptic neurons in the presence of complementary TK expressed by the helper AAV8-EF1α-DIO-EGFP-T2A-TK, thereby labeling the postsynaptic neurons by tdTomato52. 8 week-old male Ruvbl2-Cre mice were deeply anesthetized with avertin and unilaterally injected with AAV8-DIO-EGFP-T2A-TK into the PVH. After infusion, the needle was left in the brain for another 10 min to reduce backflow of viral injection along the needle track before it was removed. The mice were recovered in a 37 °C incubator before returning to home cage. Three weeks later, HSV-ΔTK-LSL-tdTomato was injected into the same site of these mice as the previous helper AAV. Three days after HSV injection, mice were perfused and their brains were collected and sectioned for subsequent IHC analysis.
Retrograde tracing
Eight-week-old Ruvbl2-Cre mice were deeply anesthetized with 1.2% avertin and stereotaxically injected with AAV2/retro-DIO-EGFP into different brain regions identified by anterograde monosynaptic tracing studies including the LSv, the POA, the MeA, the DMH, the ARC, the PBN, and the NTS. After infusion, the needle was left in the brain for another 10 min to reduce backflow of the viral injection along the needle track. The wound was closed with sutures, and the mice were recovered in a 37 °C incubator before returning to a home cage. 2–3 weeks after viral injection, animals were perfused and the brains were harvested for IHC analysis.
Retrograde monosynaptic tracing
8 week-old Pomc-Cre and AgRP-Cre mice were deeply anesthetized with 1.2% avertin and unilaterally injected with Cre-dependent AAV8-EF1α-DIO-oRVG-WPRE and AAV8-EF1α-DIO-H2B-EGFP-T2A-TVA-WPRE into the ARC. After infusion, the needle was left in the brain for another 10 min to reduce the backflow of injected virus along the needle track. Three weeks later, these mice received a second injection of RV-ENVA-ΔG-DsRed into the same injection site. Seven days after viral injection, animals were deeply anesthetized and perfused.
Chemogenetics and projection-specific chemogenetics
For chemogenetics, 200 nL of AAV9-EF1a-DIO-hM3D(Gq)-mCherry, AAV9-EF1a-DIO-hM4D(Gi)-mCherry or AAV9-EF1a-DIO-mCherry were injected bilaterally into the PVH (AP, −0.65; ML, ±0.28: DV, −5.05) of 8 week-old Ruvbl2-Cre mice. For projection-specific chemogenetics, 200 nL AAV5-hSyn-Con/Fon-hM4D(Gi)-EYFP were injected bilaterally into the PVH of 8 week-old Ruvbl2-Cre mice that simultaneously received injection of 200 nL of AAV2/retro-Flp (into the various brain regions projected by PVHRUVBL2 neurons. These brain regions include the LSv, the POA, the MeA, the DMH, the ARC, the PB, and the NTS. After surgery, the mice were allowed to recover for at least two weeks and received daily handling to reduce stress prior to experiment.
To measure food intake after chemogenetic activation, mice were fasted overnight (for food intake during day time) or 12 h (for food intake during night time), and then injected with vehicle or CNO (1 mg/kg BW, Tocris, Cat#4936). Hourly food intake was measured within 4 h after CNO treatment. To measure food intake of mice after chemogenetic inhibition, mice were injected with vehicle or CNO (1 mg/kg BW) and hourly food intake was measured within 4 h after CNO administration.
Optogenetics and projection-specific optogenetics
For optogenetics, AAV9-EF1a-DIO-hChR2-mCherry or AAV9-EF1a-DIO-mCherry was bilaterally injected into the PVH (AP = −0.65, ML = ± 0.28, DV = −5.05) of Ruvbl2-Cre mice. A 200 μm diameter optic fiber was implanted into the target regions, including the PVH (AP, −0.65; ML, 0.28; DV, −4.95), the DMH (AP, −1.95; ML, 0.26; DV, −5.10), and the ARC (AP, −1.85; ML, 0.27; DV, −5.85), to activate RUVBL2 neurons in the PVH, or the axon terminals of PVHRUVBL2 neurons innervated the DMH or the ARC. Animals were singly housed for at least 2 weeks with daily handling prior to experiment.
Before optogenetics, mice were fasted overnight. For optogenetic activation, a 465 nm blue laser light with 20-ms pulse duration at 20 Hz for 10 min (constant 2 s on and 2 s interval alternating) was applied to activate the cell bodies or the axon terminals of PVHRUVBL2 neurons. Cumulative food intake during 10 min of laser stimulation, 10 min before or after laser stimulation was measured, respectively.
To simultaneously activate ARC-projecting PVHRUVBL2 neurons and ARCNPY neurons, Cre-dependent AAV9-DIO-ChR2-mCherry was injected into the PVH of Ruvbl2-Cre mice that received implantation of an optic fiber above the ARC and injection of mixture of AAV9-Npy-FLP and FLP-dependent AAV9-fDIO-hM3D(Gq)-EGFP (1:1 ratio) into the ARC. For simultaneous activation of ARC-projecting PVHRUVBL2 neurons and ARCNPY neurons, mice were injected with vehicle or CNO (2 mg/kg BW), followed by optical activation of the axon terminals of PVHRUVBL2 neurons in the ARC for 1 h using a 465-nm blue laser light. Cumulative food intake of mice within 1 h was measured.
To determine the contribution of the melanocortin pathways to PVHRUVBL2 → ARC projection-induced anorexia, Cre-dependent AAV9-DIO-ChR2-mCherry was bilaterally injected into the PVH of 8 week-old male Ruvbl2-Cre mice that received implantation of an optic fiber above the ARC. At night time, mice were first intraperitoneally injected with SHU9119 (0.5 mg/kg, MCE, HY-P0227), followed by optical activation of the axon terminals of PVHRUVBL2 neurons in the ARC using a 465-nm blue laser light for 6 h when the laser light was switched on every other 30 min. Cumulative food intake of mice within 6 h was measured.
Brain slice preparation
For electrophysiology recording, brain slices containing the PVH, the DMH, and the ARC were prepared from Ruvbl2fl/fl mice injected with AAV2-GFP or AAV2-Cre-GFP, or from Ruvbl2fl/fl;Pomc-EGFP and Ruvbl2fl/fl;Npy-GFP mice injected with AAV2-mCherry or AAV2-Cre-mCherry into the PVH for 8 weeks. For ChR2-assisted circuit mapping (CRACM), brain slices containing the PVH, the DMH, and the ARC were prepared from Ruvbl2-Cre mice injected with AAV9-EF1a-DIO-hChR2-mCherry for 4 weeks. Precise viral injection sites were confirmed by fluorescence in the PVH. Brain slices containing the DMH and the ARC were prepared from WT and Ruvbl2KI/KI;Sim1-Cre mice for electrophysiology recording of neurons after RUVBL2 overexpression.
Mice were euthanized with CO2, followed by cervical dislocation. Coronal brain slices (200 μm thickness) were cut in ice-cold cutting solution containing (mM): 72 sucrose, 83 NaCl, 26 NaHCO3, 22 glucose, 2.5 KCl, 1 NaH2PO4, 5 MgCl2 and 1 CaCl2 on a Leica vibratome (Leica). Brain slices were transferred to artificial cerebrospinal fluid (aCSF) containing (mM): 126 NaCl, 21.4 NaHCO3, 10 glucose, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2 and 2.4 CaCl2 and incubated at 34 °C for 45 min. Brain slices in aCSF were kept at room temperature (24–26 °C) for at least 30 min before electrophysiological recording.
Electrophysiology
Visually guided (infrared DIC videomicroscopy and water-immersion X40 objective) whole-cell recordings were obtained with patch pipettes (2.5-4.5 MΩ) pulled from borosilicate capillary glass with a Sutter P97 puller (Sutter Instrument, Novato, CA, USA). All recordings were obtained with a Multiclamp 700B amplifier, Digidata 1550B converter and pClamp 10.7 software (Axon Instruments), sampled at 20 or 50 kHz, and filtered at 2 kHz. All data were analyzed with Igor Pro-6.2 (WaveMetrics). During recordings, the chamber contained brain slice was continuously perfused with aCSF solution bubbled with 95% O2 and 5% CO2 at room temperature.
Whole-cell voltage-clamp recordings were made from DMH and ARC neurons with membrane potential clamped at −70 mV. sEPSCs and evoked EPSCs were recorded with an internal solution containing (in mM): 140 Cs-gluconate, 15 HEPES, 0.5 EGTA, 2 TEA-Cl, 2 MgATP, 0.3 NaGTP, 10 phosphocreatine, 2 QX 314-Cl, pH was adjusted to 7.2 with CsOH. 20 μM picotoxin (PTX) were applied in aCSF to block GABAergic synaptic transmission. sIPSCs and evoked IPSCs were recorded with an internal solution containing (in mM): 140 CsCl, 1 BAPTA, 10 HEPES, 5 MgCl2, 5 Mg-ATP, 0.3 Na3GTP, 10 QX 314-Cl, pH was adjusted to 7.2 with CsOH. 50 μM D-(-)−2-amino-5-phosphonopentanoic acid (D-APV) and 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) were applied in aCSF to block AMPAR- and NMDAR-mediated synaptic transmission. A pair of electrical stimulations (0.1 ms, 20–40 V) with 50 ms intervals was applied via a bipolar electrode positioned near the recorded cells. Electrical output was controlled by a programmable pulse stimulator (Master-8, A.M.P.I.) and pClamp 10.7 software.
ChR2-assisted circuit mapping (CRACM)
ChR2-assited circuit mapping (CARCM) was conducted as previously described59. In brief, Cre-dependent AAV9-EF1a-DIO-hChR2-mCherry was injected into the PVH of 8 week-old male Ruvbl2-Cre mice. Four weeks after surgery for recovery, the mice were ready to use. To photo-stimulate ChR2-expressing fibers, a blue light (473 nm wavelength, 5 ms, maximum) was applied to the field around the recorded cells in the ARC or the DMH via an optic fiber. The light output was controlled by a programmable pulse stimulator and pClamp 10.7 software.
Transmission electron microscopy (TEM)
Mice were euthanized with CO2, followed by cervical dislocation. Coronal brain sections (200 μm thickness) containing the ARC were prepared and transferred to freshly prepared solution containing 2.5% glutaraldehyde and 2% paraformaldehyde for at least 1 h at room temperature. The fixed sections were stored in 4 °C refrigerator overnight. On the next day, the sections were trimmed into ~ 0.2 mm3 cube containing the ARC under a stereoscope. The sections were post-fixed in 1% osmium tetroxide for 1.5 h, dehydrated in an ascending series of ethanol [30, 50, 70, 85, 95, and 100% (vol/vol)], and stored in epoxy (EPON 812) overnight. The sections were then embedded in epoxy resin at 60 °C for 48 h of polymerization. The semithin sections (500 nm thickness) were stained with Toluidine blue and the ARC was confirmed by bright-field microscope. Then the resin was further trimmed until only the ARC was included. The ultrathin sections (50 nm thickness) were cut by Leica EM UC6 tramicrotome and mounted on Formvar-coated slot grids. After poststaining with 2% (wt/vol) uranyl acetate and 8% (wt/vol) lead citrate, the ultrathin sections were examined on a Tecnai Spirit TEM (FEI) at 120 kV. Data were analyzed with IMOD 4.11 (University of Colorado).
Chromatin Immunoprecipitation Sequencing (ChIP-Seq)
ChIP-Seq library preparation
Eight-week-old male C57BL/6 J mice were euthanized with CO2, followed by cervical dislocation. The hypothalamus was isolated, cross-linked with 1% formaldehyde for 10 min at room temperature, and quenched with 125 mM glycine. The chromatin fragments were pre-cleared and then immunoprecipitated with Protein A + G Magnetic beads coupled with anti-RUVBL2 antibody (ABclonal, #A1905). After reverse crosslinking, CHIP and input DNA fragments were end-repaired and A-tailed using the NEBNext End Repair/dA-Tailing Module (E7442, NEB) followed by adapter ligation with the NEBNext Ultra Ligation Module (E7445, NEB). The DNA libraries were amplified for 15 cycles and sequenced using Illumina NovaSeq 6000 platform with paired-end 2 × 150 as the sequencing mode.
Data analysis
Raw reads were filtered to obtain high-quality clean reads by removing sequencing adapters, short reads (length < 35 bp) and low quality reads using Cutadapt v1.18 (https://doi.org/10.1093/bioinformatics/btu170) (non-default parameters: --max-n 0 --minimum-length 35) and Trimmomatic v0.38 (https://doi.org/10.1089/cmb.2017.0096) (non-default parameters: SLIDINGWINDOW:4:15 LEADING:10 TRAILING:10 MINLEN:35). Then FastQC (https://f1000research.com/articles/8-1874/v2) (with default parameters) was used to ensure high reads quality. The clean reads were mapped to the mouse genome (assembly GRCm38) using the Bowtie2 v2.3.4.1 (https://www.nature.com/articles/nmeth.1923) (with default parameters) software. Duplicate reads were then removed using picard MarkDuplicates. Peak detection was performed using the MACS v2.1.2 (https://doi.org/10.1186/gb-2008-9-9-r137) (non-default parameters: -f BAMPE -g hs/mm -p 0.01) peak finding algorithm with 0.01 set as the p-value cutoff. Annotation of peak sites to gene features was performed using the ChIPseeker R package (https://doi.org/10.1093/bioinformatics/btv145).
RNA sequencing (RNA-Seq)
AAV2-GFP and AAV2-Cre-GFP were injected into the PVH of 8 week-old male Ruvbl2fl/fl mice. 6 weeks after virus injection, mice were euthanized with CO2, followed by cervical dislocation. The PVH was isolated for total RNA extraction using Trizol reagent (Invitrogen), followed by RNA-Seq and data analysis completed by LC-Bio Technology CO. Ltd. (Hangzhou, China).
Quantitation and statistical analysis
All data are presented as means ± SEM. ImageJ was used to quantify WB images and VGluT2+ puncta of IHC images. Statistical analyses were performed using two-tailed unpaired or paired Student’s t-test (Excel 2011), or two-way ANOVA followed by Bonferroni’s test for multiple comparisons (GraphPad Prism 9). ANCOVA was performed using SPSS Version 26 (IBM SPSS Statistics, Chicago, IL, USA). p < 0.05 was considered statistically significant. Electrophysiology data were off-line analyzed with Igor 6.2 software (WaveMetrics, Portland, OR, USA). Prism 9 and Adobe Illustrator CS6 were used to generate and prepare all the figures.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the data generated or analyzed in this study are included in this paper or the Supplementary Information. The CHIP-Seq and RNA-Seq data generated in the study have been deposited in Gene Expression Omnibus (GEO) under accession code GSE249791 and GSE252308, respectively. Source data are provided with this paper.
References
Morton, G. J., Cummings, D. E., Baskin, D. G., Barsh, G. S. & Schwartz, M. W. Central nervous system control of food intake and body weight. Nature 443, 289–295 (2006).
Morton, G. J., Meek, T. H. & Schwartz, M. W. Neurobiology of food intake in health and disease. Nat. Rev. Neurosci. 15, 367–378 (2014).
Williams, K. W. & Elmquist, J. K. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat. Neurosci. 15, 1350–1355 (2012).
Gautron, L., Elmquist, J. K. & Williams, K. W. Neural control of energy balance: translating circuits to therapies. Cell 161, 133–145 (2015).
Myers, M. G. Jr, Affinati, A. H., Richardson, N. & Schwartz, M. W. Central nervous system regulation of organismal energy and glucose homeostasis. Nat. Metab. 3, 737–750 (2021).
Waterson, M. J. & Horvath, T. L. Neuronal regulation of energy homeostasis: beyond the hypothalamus and feeding. Cell Metab. 22, 962–970 (2015).
Xu, B. & Xie, X. Neurotrophic factor control of satiety and body weight. Nat. Rev. Neurosci. 17, 282–292 (2016).
Myers, M. G. Jr & Olson, D. P. SnapShot: neural pathways that control feeding. Cell Metab. 19, 732–732.e731 (2014).
Myers, M. G. Jr & Olson, D. P. Central nervous system control of metabolism. Nature 491, 357–363 (2012).
Balthasar, N. et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123, 493–505 (2005).
Shah, B. P. et al. MC4R-expressing glutamatergic neurons in the paraventricular hypothalamus regulate feeding and are synaptically connected to the parabrachial nucleus. Proc. Natl Acad. Sci. USA 111, 13193–13198 (2014).
Xu, Y. et al. Glutamate mediates the function of melanocortin receptor 4 on Sim1 neurons in body weight regulation. Cell Metab. 18, 860–870 (2013).
An, J. J. et al. TrkB-expressing paraventricular hypothalamic neurons suppress appetite through multiple neurocircuits. Nat. Commun. 11, 1729 (2020).
An, J. J., Liao, G. Y., Kinney, C. E., Sahibzada, N. & Xu, B. Discrete BDNF neurons in the ;araventricular hypothalamus control feeding and energy expenditure. Cell Metab. 22, 175–188 (2015).
Li, M. M. et al. The paraventricular hypothalamus regulates satiety and prevents obesity via two genetically distinct circuits. Neuron 102, 653–667.e656 (2019).
Li, C. et al. Defined paraventricular hypothalamic populations exhibit differential responses to food contingent on caloric state. Cell Metab. 29, 681–694.e685 (2019).
Krashes, M. J. et al. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 507, 238–242 (2014).
Andermann, M. L. & Lowell, B. B. Toward a wiring diagram understanding of appetite control. Neuron 95, 757–778 (2017).
Atasoy, D., Betley, J. N., Su, H. H. & Sternson, S. M. Deconstruction of a neural circuit for hunger. Nature 488, 172–177 (2012).
Cone, R. D. Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 8, 571–578 (2005).
Sohn, J. W., Elmquist, J. K. & Williams, K. W. Neuronal circuits that regulate feeding behavior and metabolism. Trend Neurosci. 36, 504–512 (2013).
Holtmaat, A. & Svoboda, K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 10, 647–658 (2009).
Zucker, R. S. & Regehr, W. G. Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405 (2002).
Zeltser, L. M., Seeley, R. J. & Tschop, M. H. Synaptic plasticity in neuronal circuits regulating energy balance. Nat. Neurosci. 15, 1336–1342 (2012).
Dietrich, M. O. & Horvath, T. L. Hypothalamic control of energy balance: insights into the role of synaptic plasticity. Trend Neurosci. 36, 65–73 (2013).
Hwang, E., Portillo, B., Grose, K., Fujikawa, T. & Williams, K. W. Exercise-induced hypothalamic neuroplasticity: Implications for energy and glucose metabolism. Mol. Metab. 73, 101745 (2023).
Pinto, S. et al. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science 304, 110–115 (2004).
Sternson, S. M., Shepherd, G. M. & Friedman, J. M. Topographic mapping of VMH –> arcuate nucleus microcircuits and their reorganization by fasting. Nat. Neurosci. 8, 1356–1363 (2005).
Gao, Q. et al. Anorectic estrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat. Med. 13, 89–94 (2007).
Yang, Y., Atasoy, D., Su, H. H. & Sternson, S. M. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 146, 992–1003 (2011).
Liu, T. et al. Fasting activation of AgRP neurons requires NMDA receptors and involves spinogenesis and increased excitatory tone. Neuron 73, 511–522 (2012).
Kong, D. et al. A postsynaptic AMPK–>p21-activated kinase pathway drives fasting-induced synaptic plasticity in AgRP neurons. Neuron 91, 25–33 (2016).
Suyama S. et al. Plasticity of calcium-permeable AMPA glutamate receptors in Pro-opiomelanocortin neurons. Elife 6, e25755 (2017).
Rau, A. R. & Hentges, S. T. GABAergic inputs to POMC neurons originating from the dorsomedial hypothalamus are regulated by energy state. J. Neurosci. 39, 6449–6459 (2019).
Grzelka, K. et al. A synaptic amplifier of hunger for regaining body weight in the hypothalamus. Cell Metab. 35, 770–785.e775 (2023).
Horvath, T. L. et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc. Natl Acad. Sci. USA 107, 14875–14880 (2010).
Liu, J. et al. Enhanced AMPA receptor trafficking mediates the anorexigenic effect of endogenous glucagon-like peptide-1 in the paraventricular hypothalamus. Neuron 96, 897–909 e895 (2017).
Grigoletto, A., Lestienne, P. & Rosenbaum, J. The multifaceted proteins Reptin and Pontin as major players in cancer. Biochim. Biophys. Acta 1815, 147–157 (2011).
Dauden, M. I., Lopez-Perrote, A. & Llorca, O. RUVBL1-RUVBL2 AAA-ATPase: a versatile scaffold for multiple complexes and functions. Curr. Opin. Struct. Biol. 67, 78–85 (2021).
Gallant, P. Control of transcription by pontin and reptin. Trend Cell Biol. 17, 187–192 (2007).
Xie, X. et al. RUVBL2, a novel AS160-binding protein, regulates insulin-stimulated GLUT4 translocation. Cell Res. 19, 1090–1097 (2009).
Javary, J. et al. Liver Reptin/RUVBL2 controls glucose and lipid metabolism with opposite actions on mTORC1 and mTORC2 signalling. Gut 67, 2192–2203 (2018).
Flannick, J. et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 570, 71–76 (2019).
Ju D. et al. Chemical perturbations reveal that RUVBL2 regulates the circadian phase in mammals. Sci. Transl. Med. 12, eaba0769 (2020).
Savani R. et al. Metabolic and behavioral alterations associated with viral vector-mediated toxicity in the paraventricular hypothalamic nucleus. Biosci. Rep. 44, https://doi.org/10.1042/BSR20231846 (2024).
Muller, T. D., Klingenspor, M. & Tschop, M. H. Revisiting energy expenditure: how to correct mouse metabolic rate for body mass. Nat. Metab. 3, 1134–1136 (2021).
Holder, J. L. Jr, Butte, N. F. & Zinn, A. R. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum. Mol. Genet. 9, 101–108 (2000).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010).
Garfield, A. S. et al. A neural basis for melanocortin-4 receptor-regulated appetite. Nat. Neurosci. 18, 863–871 (2015).
Alexander, G. M. et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39 (2009).
Zhang, F., Aravanis, A. M., Adamantidis, A., de Lecea, L. & Deisseroth, K. Circuit-breakers: optical technologies for probing neural signals and systems. Nat. Rev. Neurosci. 8, 577–581 (2007).
Zeng, W. B. et al. Anterograde monosynaptic transneuronal tracers derived from herpes simplex virus 1 strain H129. Mol. Neurodegener. 12, 38 (2017).
Tervo, D. G. et al. A designer AAV variant permits efficient retrograde access to projection neurons. Neuron 92, 372–382 (2016).
Cowley, M. A. et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484 (2001).
Wickersham, I. R. et al. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron 53, 639–647 (2007).
Balthasar, N. et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 42, 983–991 (2004).
Tong, Q., Ye, C. P., Jones, J. E., Elmquist, J. K. & Lowell, B. B. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat. Neurosci. 11, 998–1000 (2008).
Vong, L. et al. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154 (2011).
Petreanu, L., Huber, D., Sobczyk, A. & Svoboda, K. Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat. Neurosci. 10, 663–668 (2007).
Stachniak, T. J., Ghosh, A. & Sternson, S. M. Chemogenetic synaptic silencing of neural circuits localizes a hypothalamus–>midbrain pathway for feeding behavior. Neuron 82, 797–808 (2014).
Fan, W., Boston, B. A., Kesterson, R. A., Hruby, V. J. & Cone, R. D. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385, 165–168 (1997).
Qiu, X., Zhu, Q. & Sun, J. Quantitative analysis of vesicle recycling at the calyx of held synapse. Proc. Natl Acad. Sci. USA 112, 4779–4784 (2015).
Hippenmeyer, S. et al. Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron 68, 695–709 (2010).
Sutton, A. K. et al. Control of food intake and energy expenditure by Nos1 neurons of the paraventricular hypothalamus. J. Neurosci. 34, 15306–15318 (2014).
Aklan, I. et al. NTS catecholamine neurons mediate hypoglycemic hunger via medial hypothalamic feeding pathways. Cell Metab. 31, 313–326.e315 (2020).
Wang, D. et al. Whole-brain mapping of the direct inputs and axonal projections of POMC and AgRP neurons. Front Neuroanat. 9, 40 (2015).
Aponte, Y., Atasoy, D. & Sternson, S. M. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat. Neurosci. 14, 351–355 (2011).
Bellinger, L. L. & Bernardis, L. L. The dorsomedial hypothalamic nucleus and its role in ingestive behavior and body weight regulation: lessons learned from lesioning studies. Physiol. Behav. 76, 431–442 (2002).
Morrison, S. F., Madden, C. J. & Tupone, D. Central neural regulation of brown adipose tissue thermogenesis and energy expenditure. Cell Metab. 19, 741–756 (2014).
Cao, W. H., Fan, W. & Morrison, S. F. Medullary pathways mediating specific sympathetic responses to activation of dorsomedial hypothalamus. Neuroscience 126, 229–240 (2004).
Tan, C. L. et al. Warm-sensitive neurons that control body temperature. Cell 167, 47–59.e15 (2016).
Zhao, Z. D. et al. A hypothalamic circuit that controls body temperature. Proc. Natl Acad. Sci. USA 114, 2042–2047 (2017).
Kataoka, N., Hioki, H., Kaneko, T. & Nakamura, K. Psychological stress activates a dorsomedial hypothalamus-medullary raphe circuit driving brown adipose tissue thermogenesis and hyperthermia. Cell Metab. 20, 346–358 (2014).
Pinol, R. A. et al. Brs3 neurons in the mouse dorsomedial hypothalamus regulate body temperature, energy expenditure, and heart rate, but not food intake. Nat. Neurosci. 21, 1530–1540 (2018).
Liao, G. Y., Kinney, C. E., An, J. J. & Xu, B. TrkB-expressing neurons in the dorsomedial hypothalamus are necessary and sufficient to suppress homeostatic feeding. Proc. Natl Acad. Sci. USA 116, 3256–3261 (2019).
Caglar C. & Friedman, J. Restriction of food intake by PPP1R17-expressing neurons in the DMH. Proc. Natl Acad. Sci. USA 118, e2100194118 (2021).
Yang, L. et al. Role of dorsomedial hypothalamic neuropeptide Y in modulating food intake and energy balance. J. Neurosci. 29, 179–190 (2009).
Otgon-Uul, Z., Suyama, S., Onodera, H. & Yada, T. Optogenetic activation of leptin- and glucose-regulated GABAergic neurons in dorsomedial hypothalamus promotes food intake via inhibitory synaptic transmission to paraventricular nucleus of hypothalamus. Mol. Metab. 5, 709–715 (2016).
Monday, H. R., Younts, T. J. & Castillo, P. E. Long-term plasticity of neurotransmitter release: emerging mechanisms and contributions to brain function and disease. Annu Rev. Neurosci. 41, 299–322 (2018).
Malenka, R. C. & Bear, M. F. LTP and LTD: an embarrassment of riches. Neuron 44, 5–21 (2004).
Togashi, H. et al. Cadherin regulates dendritic spine morphogenesis. Neuron 35, 77–89 (2002).
Siegert, S. et al. The schizophrenia risk gene product miR-137 alters presynaptic plasticity. Nat. Neurosci. 18, 1008–1016 (2015).
Sudhof, T. C. Towards an understanding of synapse formation. Neuron 100, 276–293 (2018).
Wang, H. et al. The transcriptional coactivator RUVBL2 regulates Pol II clustering with diverse transcription factors. Nat. Commun. 13, 5703 (2022).
Minokoshi, Y. et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428, 569–574 (2004).
Dagon, Y. et al. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. 16, 104–112 (2012).
Hardie, D. G., Ross, F. A. & Hawley, S. A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 (2012).
Lopez, M., Nogueiras, R., Tena-Sempere, M. & Dieguez, C. Hypothalamic AMPK: a canonical regulator of whole-body energy balance. Nat. Rev. Endocrinol. 12, 421–432 (2016).
Bai, Z. et al. PRDX1 Cys52Ser variant alleviates nonalcoholic steatohepatitis by reducing inflammation in mice. Mol. Metab. 76, 101789 (2023).
Xie, X. et al. Activation of anxiogenic circuits instigates resistance to diet-induced obesity via increased energy expenditure. Cell Metab. 29, 917–931.e914 (2019).
Acknowledgements
We thank Profs. Dandan Wang (Fudan University, Shanghai, China), Cheng Zhan and Ji Liu (University of Science and Technology of China, Hefei, China), Juxue Li (Nanjing Medical University, Nanjing, China), and Yuanqing Gao (Nanjing Medical University, Nanjing, China) for sharing Ai14, Agrp-Cre and Npy-GFP, Mc4r-2a-Cre, Pomc-Cre, and Pomc-EGFP mouse models, respectively. We are grateful to Xueke Tan, Zhongshuang Lv, Xixia Li, and Qing Bian at the Center for Biological Imaging (CBI), Institute of Biophysics, Chinese Academy of Sciences, for help in sample preparation for electron microscopy and image capture. We thank Prof. Zhiping Pang (Rutgers University Robert Wood Johnson Medical School, USA) and Prof. Baoji Xu (UF Scripps Biomedical Research, University of Florida, USA) for critical reading of the manuscript. This work was supported by the National Key Research and Development Program of China (2019YFA0802500 to X.X. and W.J.Z.), the National Natural Science Foundation of China-Guangdong Joint Fund (U20A6005 to J.S.), ST2030-Majr Projects (2021ZD0202500 to J.S.), the National Natural Science Foundation of China (31971076 and 32271202 to X.X.), the Tianjin Municipal Science and Technology Commission (20JCJQJC00240 to X.X.), the R&D Program of Guangzhou National Laboratory (GZNL2023A02012 to X.X. and W.J.Z.), the Instrument Developing Project of the Chinese Academy of Sciences (YJKYYQ20180028 to J.S.), and the Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-032A).
Author information
Authors and Affiliations
Contributions
Conceptualization and methodology, X.X., S.Z., and Z.C.; Investigation, M.X., Y.L., Y.Z., J.Z., D.M., M.Z., M.T., T.O., and F.Z.; Supervision, X.X., S.Z., X.S., J.S., and W.J.Z.; Writing-Original Draft, X.X., S.Z., M.X., Y.L., Y.Z., and J.Z.; Resources, X.X., S.Z., and W.J.Z.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Qingchun Tong and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xing, M., Li, Y., Zhang, Y. et al. Paraventricular hypothalamic RUVBL2 neurons suppress appetite by enhancing excitatory synaptic transmission in distinct neurocircuits. Nat Commun 15, 8939 (2024). https://doi.org/10.1038/s41467-024-53258-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53258-6
