
Abstract
Neurotrophin receptors of the Trk family are involved in the regulation of brain development and neuroplasticity, and therefore can serve as targets for anti-cancer and stroke-recovery drugs, antidepressants, and many others. The structures of Trk protein domains in various states upon activation need to be elucidated to allow rational drug design. However, little is known about the conformations of the transmembrane and juxtamembrane domains of Trk receptors. In the present study, we employ NMR spectroscopy to solve the structure of the TrkB dimeric transmembrane domain in the lipid environment. We verify the structure using mutagenesis and confirm that the conformation corresponds to the active state of the receptor. Subsequent study of TrkB interaction with the antidepressant drug fluoxetine, and the antipsychotic drug chlorpromazine, provides a clear self-consistent model, describing the mechanism by which fluoxetine activates the receptor by binding to its transmembrane domain.
Similar content being viewed by others


Introduction
The tropomyosin receptor kinases (Trks) constitute a family of high-affinity neurotrophin receptors within the receptor tyrosine kinase (RTK) superfamily. These receptors play a crucial role in regulating neuronal survival and differentiation1,2, and are involved in a variety of diseases, including cancers and neurodegenerative disorders, which makes these proteins prospective drug targets3,4. The human Trk family includes three members: TrkA, TrkB, and TrkC, each specific to its own neurotrophin molecule: NGF, BDNF, and NT35. The structures and overall molecular mechanisms of Trk activation are mostly known. Extracellular domains of Trks have been studied separately and in complex with ligands by NMR and X-ray crystallography6,7,8,9,10, revealing a peculiar ligand-binding mode. These parts of Trks consist of five subdomains, and only the C-terminal Ig-like domain (d5) is involved in ligand binding, whereas the roles of the other four are still mostly elusive. Moreover, the ligands protrude from the d5 domains towards the cell membrane by approximately 2 nm and most likely interact with the disordered extracellular juxtamembrane regions11. The latter was found to play a significant role in regulating Trk activity12,13,14. The structures of the kinase domains of various Trks were also obtained by crystallography and revealed the fold typical for receptor tyrosine kinases15.
One of the most intriguing parts of Trk receptors is their transmembrane domains. Until recently, Trks were thought to be activated by ligand-induced dimerization. However, first, by cross-linking16,17 and later by single-molecule FRET approaches18, Trks were shown to exist as pre-formed dimers on the cell membrane, suggesting that dimerization itself does not induce receptor activity. This mechanism seems to be common for various kinds of receptor tyrosine kinases, and the TrkB receptor provides the highest population of pre-formed dimers among all the receptors tested19. Altogether, this implies that there are two dimeric states of Trk transmembrane domains (TMDs), corresponding to the active and inactive forms of the receptor. The structure of TrkA TMD in one of these states was previously solved by NMR spectroscopy20. The state is presumably inactive, according to the disulfide cross-linking experiments in the presence of a ligand. The second state was characterized by cross-linking, functional assays, and TOXRed assays20. Switching between the two conformations, induced by cross-linking in the juxtamembrane regions, has been directly observed in the most recent work11. However, the atomistic details of the TMD dimer, corresponding to the Trk active state, and the TMD conformations of other Trk members besides TrkA, remain elusive.
The functional role of Trk TMDs goes beyond receptor homodimerization or switching between the two conformations. TMDs can mediate protein-protein interactions, as demonstrated by the example of TrkA and p75 receptors21. TrkA can modulate the activity of the p75 neurotrophin receptor, and the TMDs of both proteins were shown to play an essential role in this phenomenon. The TMD of the TrkB receptor is the most intriguing, as it has been shown to bind several antidepressant drugs belonging to different chemical classes and is involved in neuroplasticity in response to antidepressant therapies22. Surprisingly, according to recent studies23, the binding of antidepressants occurs in the transmembrane part of the receptor and is closely related to the protein-cholesterol interactions in cell membranes. Moreover, the TrkB TMD was recently shown to be a high-affinity binding site for the psychedelic drugs LSD and psilocin, the active metabolite of psilocybin, which exhibit antidepressant potential in clinical trials24. This makes the TMD of TrkB an interesting drug target and highlights the need for atomistic structural data describing this region.
In this work we report the structural/functional investigation of the TrkB transmembrane domain in lipid bicelles, accompanied by direct analysis of its interactions with the environment and several commercial antidepressant drugs employing NMR spectroscopy in solution, optical microscopy and functional assays.
Results
Spatial structure of TrkB transmembrane domain in the lipid environment
It is well established that the structures of helical transmembrane dimers may be affected by the choice of membrane mimetic, and in some cases, switching between active and inactive states may occur upon transfer from one environment to another25,26,27. Additionally, the effects of cholesterol and antidepressant drugs must be studied in the presence of a lipid bilayer to ensure the proper position of these molecules with respect to the TM domain. For this purpose, in the current work, we synthesized the transmembrane domain of human TrkB (TrkBtm, residues 423-466, UniProt ID Q16620) and reconstituted it in the classic lipid-containing environment of DMPC/DHPC bicelles as described24. Under these conditions one can observe two sets of cross-peaks in the HSQC spectra of TrkBtm (S1 and S2 below), with intensities dependent on the lipid-to-protein ratio (LPR) (Fig. 1a, b, Supplementary Fig 1). This behavior is typical for oligomerization; therefore, we applied buffer/bicelle dilution and 1H/15N cross-correlated relaxation analysis28 to establish the order of the respective oligomers. Two samples, prepared at different protein concentrations (0.28 and 0.12 mM) were diluted by the bicelles solution, and the observed populations of S1 and S2 cross-peaks were analyzed assuming different oligomer orders k, aiming to find k with minimal root mean square deviation of points between the two concentrations on the plot, describing the dependence of apparent free energy (RTln([M]2/[D])) on the concentration of empty bicelles. This deviation was described as a penalty function χ2(k) (Fig. 1d). As revealed by titration, S2 is the dimer of S1, and the bicelle hydrodynamic radius (2.81 ± 0.16 nm) derived from the cross-correlated relaxation analysis suggests that S1 corresponds to the TrkBtm monomer, assuming the ideal bicelle model (radius should be equal to 2.82 nm29,30) (Fig. 1c, d). The S2 cross-peaks were observed until LPR ~ 200; therefore, we can state that TrkBtm forms moderately strong dimers in DMPC/DHPC bicelles, which are weaker than the dimers of VEGFR2 (dimers are still observed at LPR ~ 200031) but stronger than the dimers of EGFR or ErbB4 (dimers are observed until LPR 100-150)32,33; and dimer/monomer transitions are slow in the NMR chemical shift timescale. Peak splittings are observed mostly for residues 437-445, suggesting that the dimerization occurs at the N-terminus of the TM helix (Supplementary Figs. 1, 2).

a The signal of the A440 amide group in the 1H-15N-HSQC spectrum of TrkBtm reveals two forms, S1 and S2, whose populations depend on the LPR. b Relative intensities of the S1 (orange) and S2 (blue) backbone amide signals in the NMR spectra as a function of LPR. At LPR = 90 the sample was diluted 2.5x. TrkBtm overall concentration decreased from 0.28 to 0.11 mM. c the hydrodynamic radii (RH) of an empty q = 0.3 DMPC/DHPC bicelle (obtained from translational diffusion of lipids, error bar shows the error of measurement, gray) and S1- (orange) and S2-containing (blue) bicelles (obtained from the rotational diffusion of proteins, error bars show the SD among the n = 6 considered residues). * indicates the statistical significance of the difference according to the Mann-Whitney test, p = 0.0202. The dashed lines denote the theoretical RH values obtained using the ideal bicelle model, as described30. d the natural logarithm of the penalty function χ2(k) calculated for n = 12 pairs of signals at several tested oligomer orders k = 2–5 of the S2 state28. In the gray area, the penalty function was not determined (*** indicates the statistical significance of the difference according to the two-sided t-test, p = 3.08 × 10-6). Error bars show the SD among the tested pairs of residues. e the spatial structure of the dimeric TrkBtm. One of the helices is shown as a blue cartoon and the other - either as an orange cartoon or as a surface painted according to its hydrophobicity (using the White-Wimley scale71). f Accessible surface area (ASA) of TrkBtm residues buried in the helix dimerization interface. The gray dashed line indicates the 50% threshold. The orange bars indicate the contact residues. Error bars indicate the SD among the set of n = 10 best NMR structures. g Closer view of the presumed key interactions supporting the TrkBtm dimer: V438 CαH-S441 Oγ polar contact and F445 stacking. Source data are provided as a Source Data file.
To determine the structure of the TrkBtm dimer, we placed a 1:1 mixture of 13C/15N-labeled and unlabeled proteins into bicelles with perdeuterated acyl chains at LPR 40 and used 13C-filtered NOESY experiments to detect protein–protein interactions directly34 (Supplementary Fig. 3). The sample containing exclusively 13C/15N-labeled TrkBtm was used as a control to get rid of the effects of incomplete isotope labeling and filter leakage. This approach provided us with 13 intermolecular distance restraints, which allowed determining a high-resolution structure of the protein dimer (Fig. 1e–g, Table 1, Supplementary Figs. 4, 5). The protein chain forms an alpha-helix in the region H430-H459 (Supplementary Fig. 5), as was already shown previously24, and two helices engage into a parallel right-handed dimer, with a helix crossing angle equal to −33 ± 5o. The helix-helix interface is located at the center of the TM segment and is rather extended and nonpolar; the area of the contact surface is 493 ± 98 A2. Detailed analysis revealed that dimerization is mediated by a motif with a typical 4-residue periodicity: 437Vxxx441Sxxx445Fxxx449Vxx452FL (Fig. 1f). Most of the residues of the motif are hydrophobic and bulky, except for S441, whose side chain seems to form a non-canonical intermolecular H-bond to the CαH moiety of V438 (Fig. 1g). The CαH-O distance is relatively long (Fig. 1g), but the minimal values lie within the typical range of CɑH-O contacts in membrane proteins35. Of note, the introduction of a corresponding 2.7 Å restraint does not affect the structure substantially (Supplementary Fig. 6, backbone RMSD equals 0.47 Å). Thus the structure is compatible with the H-bond, but we cannot evaluate the strength of the contact within the experimental precision. In addition, the F445 aromatic rings are involved in face-to-face stacking interactions.
The conformation of TrkBtm dimer is unaffected by the environment
Since the cell membrane is mosaic36, and we worked with a model system, it was important to evaluate whether the parameters of the environment could affect the spatial structure of TrkBtm. In this regard, we sequentially changed the conditions of the sample and compared the 1H-15N-HSQC spectra of TrkBtm. We monitored the intensities of the monomer and dimer signals to assess the stability of the dimer, and the peak positions. The structure of TrkBtm was obtained at a relatively low pH (6.0), below the pKa of His residues; therefore, first we investigated the pH dependence of the TrkBtm spectra (Fig. 2a, Supplementary Fig. 7). The 1H-15N-HSQC spectra were almost identical after changing the pH from 6.0 to physiological 7.4 (Fig. 2a, Supplementary Fig. 7a), suggesting that the obtained dimer structure is relevant for neutral pH: significant changes in peak positions were observed for the H430 residue and its neighbors (Supplementary Fig. 7b) and should have been caused by the sidechain deprotonation of histidine. The monomer/dimer cross-peak intensity ratio did not change as well (Supplementary Fig. 7c, d), showing that the effect of pH on dimer stability is subtle or absent. The most noticeable change was the disappearance of signals corresponding to the unstructured terminal residues due to accelerated chemical exchange with the bulk water.

a Overlaid fragments of 1H-15N-HSQC spectra of TrkBtm in DMPC/DHPC q* = 0.3 bicelles at LPR = 80 and pH = 6.0 (black) and 7.4 (green). m and d denote the monomer and dimer signals, respectively. b Spectra of TrkBtm at pH = 7.4 in DMPC/DHPC bicelles formed with q* = 0.3 (green) and 0.5 (orange). c Spectra of TrkBtm at pH = 7.4 in DMPC/DHPC (orange) and DPPC/DHPC (blue) q* = 0.5 bicelles. d Spectra of TrkBtm at pH = 7.4 in DMPC/DHPC q*=0.5, with a bilayer containing 0 % (orange) and 7 % (black) cholesterol (n/n). Each spectrum represents a separately prepared sample of TrkBtm, spectra in (a, b) were recorded at 800 MHz, whereas spectra in (c, d) at 600 MHz.
Second, as we conducted our study in relatively small bicelles (q = 0.3), we investigated whether the size of the particles was sufficient to accommodate the dimeric TM domain. Thus, we varied the bicelle q ratio in the range of q = 0.3–0.5, which corresponds to a bicelle radius of 2.5–3.1 nm, by adding DMPC to the sample (Fig. 2b, Supplementary Fig. 8). As one can see, the increase of the bicelle size exerted some effects on the chemical shifts of certain residues located mostly at the N-terminus (Supplementary Fig. 8a, b). We also observed the increase in the monomer/dimer ratio with q (Supplementary Fig. 8c, d), but this is likely a result of the increase in LPR and different NMR relaxation of monomer and dimer signals and does not indicate changes in dimer stability.
Next, to take into account possible variations in the bilayer content, we studied the TrkBtm in thicker bicelles, composed of DPPC/DHPC37 (Fig. 2c, Supplementary Fig. 9). Under these conditions, cross-peaks in the NMR spectra of TrkBtm became broader because of the greater size of the particles; some subtle changes in peak positions were observed, however, the dimer/monomer ratio has not changed substantially (Supplementary Fig. 9).
Another aspect of TrkB interactions with the environment is the effect of cholesterol. According to Casarotto et al.23, the direct addition of cholesterol to neurons may affect the BDNF-induced phosphorylation of TrkB and enhance the PLCγ1 response. This effect was dependent on the Y434 sidechain. For this reason, we titrated the monomeric TrkBtm in bicelles with cholesterol (Fig. 2d, Supplementary Figs. 10, 11), introducing up to 7.0% cholesterol to the lipid vesicles, which corresponds to a 2:1 excess with respect to the protein. As a result, we observed subtle chemical shift alterations, which, however, cannot represent a high-affinity binding process because the changes are not saturated even at a 2:1 excess of cholesterol with respect to the protein. The addition of cholesterol did not influence the peak splitting pattern substantially; a statistically significant but slight decrease (7 ± 5%) in the monomer/dimer ratio was observed showing a subtle stabilization of the dimer (Supplementary Fig. 11f). On the other hand, the highest chemical shift changes were found at residues Y434 and V437 (Supplementary Fig. 11e), suggesting that Y434 participates in these weak interactions.
Whereas the overall chemical shift perturbations of particular NMR signals (CSP) most likely reflect the changes in the environment properties, peak splittings into monomer and dimer signals should serve as the fingerprint of dimer conformation (This statement is illustrated by the example of TrkA TM domain (TrkAtm) dimerization studied earlier11,20 in the Supplementary Fig. 12). Therefore, we plotted the dimer/monomer peak splittings observed in NMR spectra of TrkBtm under different conditions (Supplementary Fig. 13) and compared them. As one can see, the residue-wise splitting patterns were highly similar under all the conditions and the magnitudes and directions of splittings demonstrate a fair correlation for each particular change of the environment (R2 = 0.89–0.94), indicating the identity of dimer conformation. Altogether, while some environment-dependent alterations in the NMR spectra of TrkBtm are observed, the conformation of the transmembrane dimer remains unchanged.
To summarize, the found conformation of the TrkBtm dimer is highly insusceptible to the effects of the environment, which supports its relevance in the case of the full-length receptor.
The found state of the TrkB transmembrane dimer is important for TrkB activation
The structure of TrkBtm was obtained in the model environment and with the truncated protein construct, therefore it is necessary to show that the observed conformation is relevant. In addition, TrkB is known to form multiple dimeric states18,38, and it is necessary to understand the relationship between the spatial structure and the functional state of the receptor. To investigate the functional relevance of the observed binding mode, we selected several potentially critical residues and assessed their roles using site-directed mutagenesis. If the one-point mutation affects the receptor activation and is essential for the observed TMD dimerization mode, then one could state that the structure of the dimer corresponds to the active state of the protein. It is noteworthy that disruption of RTK TM helix-helix interaction through single site mutagenesis is not trivial. For instance, mutations of multiple residues in EGFR have no effect on EGFR phosphorylation39,40. Similarly, single-point TrkA mutations have no effect on TrkA function, with one notable exception: the A428I mutation which significantly perturbed TrkA TM helix dimerization20. Thus, we expect that only sites that are critically important for TrkB TM helix contacts will have an effect on TrkB interactions and function.
We used several types of functional assays: kinase phosphorylation at Y706/707 and Y516 (Figs. 3d, e, 4a–d, Supplementary Fig. 14), cell differentiation (Fig. 3c, Supplementary Fig. 15) and activation of downstream signaling cascades (Fig. 4b, e, f, Supplementary Fig. 16). Phosphorylation is the immediate event that follows the receptor activation, therefore such an analysis is at most straightforward. As an orthogonal approach, we studied the BDNF-induced cell differentiation in a specially designed cell line with the knockout of the TrkA gene41. The differentiation is the most final result of receptor activation and the readout of assay is entirely quantitative. Finally, we assessed the BDNF-induced activation of AKT and ERK, which represent the intermediate stages of TrkB signaling. As a negative control in both assays we used the readout in the absence of ligands and as a positive control - a readout for the wild-type TrkB after BDNF stimulation. We also controlled the cell membrane localization of the mutants by flow cytometry. TrkB mutants were expressed and reached the plasma membrane at the similar levels as wild type protein, as shown by immunofluorescence of impermeabilized transfected cells and by flow-cytometry, indicating they are correctly folded and trafficked to the membrane (Supplementary Figs. 17, 18).

a Spatial structure of the TrkBtm dimer, the critical residues of the interface (orange), and the positions of point mutations (green) are indicated. b Amino acid sequence of TrkBtm (highlighted in light blue), the critical residues of the interface (orange), and point mutations (green circles) to Ile (up) or Ala (down) are indicated. c Average length of dendrites upon stimulation of PC12 TrkA-KO cells with BDNF. Data are provided as mean values ± SD. n ≥ 3 biological replicates and n ≥ 27 cells were analyzed (each dot represents one cell). d, e Relative amounts of phosphorylated Y706 and Y707 before and 5 and 15 min after BDNF stimulation of wild-type TrkB and its single-point mutants. Data are provided as mean values ± SD. n ≥ 3 biological replicates (each dot represents one independent replicate). Statistical significance is provided according to the Multiple two-sided t-test with Bonferroni correction (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns denotes that changes with respect to the wt are not significant). Statistical analyses are listed in Supplementary Table 1. Source data are provided as a Source Data file.

a, c, d Dose-dependent phosphorylation of Y516 and Y706/607. b, e, f Activation of ERK and AKT cascades before, 5 and 15 min after BDNF stimulation. Data are shown as mean values ± SD. n ≥ 3 biological replicates (each dot represents one independent replicate). Statistical significance is provided according to the independent two-sided t-test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns denotes that changes with respect to the wt are not significant, if not otherwise indicated). g Analysis of oligomerization of TrkB-ECTM-mEYFP constructs in cells by Number&Brightness (see Methods section for more details). Statistical significance is determined using a non-parametric one-way ANOVA (Kruskal-Wallis test) followed by Dunn’s multiple comparisons test. n ≥ 2 replicates and n ≥ 37 points for each construct. The box plots were constructed with the upper box limit being the 75th percentile, the lower box being the 25th percentile, and the whiskers representing a 95% confidence interval. The mean is the open point, and the median is the horizontal line in the box. Statistical analysis results are shown in Supplementary Table 1. Source data are provided as a Source Data file.
For the obtained structure, we selected two key amino acids at the center of the interface: S441 and F445. Additionally, we tested all TrkBtm residues with small sidechains that are known to promote helix-helix interactions: S432, A435, A440, G444, and C446. Finally, we tested the role of Y434, which has previously been shown to be important for TrkB interactions with cholesterol and antidepressant drugs23,42. For most of the residues, we used two types of mutations - to the residues with small and bulky side chains (Ala and Ile), to check the effect of the side chain volume (Fig. 3a, b). Only one of the tested sites substantially inhibited TrkB phosphorylation: S441 (Fig. 3d, e, Supplementary Fig. 14). Even an almost neutral substitution, S441A, significantly decreased the TrkB kinase activity, and the effect of the S441I mutation was even more pronounced. S441I mutation entirely abolished the phosphorylation of TrkB over a broad range of BDNF concentrations from 0.01 to 100 ng/ml (Fig. 4a, c, d). This supports both the observed key position of S441 in the dimerization interface of TrkBtm and the formation of an intermolecular non-canonical polar contact, because the general volume and polarity of Ser and Ala sidechains do not differ significantly. In contrast, mutations of F445 to either Ala or Ile did not result in receptor inhibition, suggesting that ring stacking can be easily exchanged with tight van der Waals packing. A visible but not statistically significant decrease was also observed for S432I kinase activity. However, the position of the residue in the first turn of the TM helix suggests that changes in the TM helix structure may account for this effect. No effect was observed for the Y434 site at TrkB. Apparently, the latter residue, while being involved in the binding or sensing of lipid-like small molecules and translocation to lipid rafts, is not directly related to kinase activation and TM domain dimerization events, as previously suggested23.
Four of the mutants were further tested in the cell differentiations assay (Fig. 3c, Supplementary Fig. 15). Both S441A and mainly S441I substitutions significantly decreased the average length of dendrites, whereas two control mutations, C446I and G444I, did not. Noteworthy, while the effect of S441A was statistically significant but rather subtle, S441I mutation was able to completely block the BDNF-induced differentiation. To get the complete image, we further characterized the S441A and S441I mutants, by investigating the BDNF-induced activation of ERK and AKT, downstream participants of TrkB signaling cascades (Fig. 4b, e, f, Supplementary Fig. 16). As one can clearly see, S441A mutation inhibits substantially all the activities under investigation, while S441I blocks completely all kinds of ligand-induced responses.
The inhibitory effect of S441 mutations in the TrkB TMD can be attributed to two potential explanations. First, the mutation could block the formation of a receptor active state. Then the weaker effect of S441A is likely explained by the sidechain volume: alanines are less bulky than serines, therefore, the S441A mutation removes the favorable polar contact of CaH-O type, but does not disturb the overall dimer packing. On the contrary, isoleucine is one of the bulkiest residues, and S441I introduces the steric hindrances to the dimerization interface, which disturbs the overall packing and should destabilize the NMR-observed structure to a much higher extent than S441A. The second explanation implies that mutation disrupts the inactive dimeric state, thus preventing the receptor dimerization. To distinguish between the two possibilities, we used the Number&Brightness (N&B) technique43 in living CHO-K1 cells to assess the dimerization of TrkB. N&B detects co-diffusing proteins by comparing the variance of fluorescence intensity in a pixel over an image stack (see Methods for more details). A schematic of N&B is shown in Supplementary Fig. 19a. N&B experiments yield the molecular brightness, which is proportional to the oligomer order and is calculated as the ratio of the variance and the mean of the fluorescence intensity. We transfected the cells with mEYFP-tagged TrkB with deleted intracellular kinase domains (TrkB-ECTM-mEYFP) and measured the molecular brightness which was then scaled to a GPI anchored YFP monomer control to determine the apparent oligomer order. All the experiments consistently revealed the absence of statistically significant effects caused by the S441I mutation, both in the absence and in the presence of BDNF (Fig. 4g). This provides evidence that the mutations do not prevent TrkB dimerization.
Thus, we can state that substitutions of S441, a pivotal residue on the helix-helix interface of the reported TrkB dimer structure, inhibit BDNF-induced phosphorylation of TrkB, activation of downstream signaling cascades, and cell differentiation, while they do not affect the dimeric state of the receptor. This validates the reported conformation as corresponding to a receptor active state.
Fluoxetine interacts with the N-terminus of the TrkB transmembrane helix
Another important aspect of TrkB activity is its ability to interact with antidepressant drugs via the TM domain23. These properties have been studied in cells or computer simulations, but high-resolution techniques of structural biology have never been used. Here, we utilized NMR titration and chemical shift mapping to investigate the interactions between TrkBtm and drugs in lipid bicelles. We used four drugs: fluoxetine (FLX), which was previously shown to be a major drug acting on TrkB TMDs; chlorpromazine (CPR), which was shown to be inactive with respect to TrkB; and either RR or SS stereoisomers of hydroxynorketamine (RR-HNK and SS-HNK). RR-HNK was earlier shown to bind TrkBtm, while SS-HNK was reported as a non-binder.
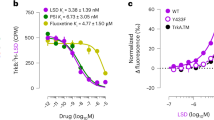
As a first observation, FLX and CPR partition into the membrane mimetic (up to 92% of drugs are membrane-bound at high drug contents; at low excess of FLX, this amount is close to 100%), while HNKs do not (Fig. 5f, Supplementary Fig. 20). In addition, FLX and CPR induced a noticeable CSPs in TrkBtm in a concentration-dependent manner (Fig. 5a, b). The maximal CSPs induced by FLX were comparable to the maximal peak splittings caused by the TrkBtm dimerization. They were also approximately twofold higher than the CSPs caused by CPR, therefore we compared the profiles of CSPs induced by 4:1 FLX and 8:1 CPR in the monomeric protein (Supplementary Fig. 20a, b). Both drugs provided maximal CSPs at the N-terminus of the monomeric TrkBtm within the region Y434-I439, while the CSP profiles in the rest of the TM helix were slightly dissimilar (Fig. 5a). The major differences were found in the central part of the TM helix (Supplementary Fig. 20b), indicating different modes of interaction with this region of the protein. In contrast to the monomer, the dimeric TrkBtm was affected by FLX several times higher than by CPR, especially in the V437-I439 region (Fig. 5a–d). Thus, the TrkBtm dimer interacts preferably with FLX.

a CSPs induced in TrkBtm by FLX (empty bars) and CPR (black bars). CSPs for the separate signals of the dimer are plotted at the bottom in a mirrored scale. Peak width (PW) of the TrkBtm signals is shown as a black dotted line. The maximal CSPs are shown by * symbols (See the Statistics subsection in the Methods section). b CSPs of V437 and Y434 plotted as a function of FLX (empty circles) and CPR (black circles) concentrations for the monomer and dimer forms of TrkBtm. The dashed lines indicate the results of a third degree polynomial approximation. TrkBtm dimer (left) and monomer (right) painted according to the amide CSP values induced by an 8:1 excess of FLX (c) or CPR (d). Residues with CSP higher than 0.05 ppm are shown in black. e The signal of the F445 backbone amide in the intact sample (left) and with 4:1 excess of either FLX (middle) or CPR (right). Slices along the 1H direction are shown on top with the corresponding signal to noise ratios (SNR) given. f Fraction of drugs residing in bicelles calculated from lateral diffusion at a drug:protein ratio of 8:1. Error bars indicate the error of the measurement. g Relative changes in the signal intensities of monomer (orange), dimer (blue), and overlapped (green) signals of TrkBtm upon drug addition. Error bars indicate the SD among the traced n = 5 NMR signals for each form. Star labels indicate statistically significant differences in the monomer and dimer populations according to the Mann-Whitney test (*p ≤ 0.05, **p ≤ 0.01), the p-values are provided in Supplementary Table 1. The relative intensities of the monomer and dimer are plotted until the signals overlap. Statistical analyses are listed in Supplementary Table 1. Source data are provided as a Source Data file.
The effects of FLX were similar in two tested lipid environments (DMPC and DPPC bicelles, Supplementary Fig. 20d). Notably, the highest CSPs were induced by FLX for residues Y434 and V437 in both the monomeric and dimeric states of TrkBtm, suggesting that these amino acids may be responsible for drug binding. This agrees with biochemical data that revealed the importance of tyrosine residues in FLX-modulated activation of TrkB23. In contrast, both HNK compounds induced only subtle perturbations, with no pronounced localization (Supplementary Fig. 20c). Together with the low bilayer-binding propensity of HNK (Fig. 5f), this suggests that R,R-HNK could bind elsewhere and not to the TM domain, and that the effects of HNK on FLX binding to TrkB23 might be allosteric. Thus, we can state that FLX exhibits specific interactions with the N-terminus of the TrkB TM helix in both the monomer and dimer states, while all other tested drugs do not.
Moreover, FLX stabilized the TrkBtm dimer. While the monomer intensities decreased upon the addition of FLX, the dimer cross-peaks remained unchanged, indicating an increased abundance of the dimeric state. This was not the case with all other tested drugs, which did not change the dimer/monomer intensity ratio (Fig. 5g). CPR may actually destabilize the TrkBtm dimer kinetically; the addition of the compound induces the merging of two peaks corresponding to the monomer and dimer states of TrkBtm (Fig. 5e, Supplementary Fig. 21). This may be interpreted as an acceleration of dimer/monomer transitions, which could become fast on the NMR chemical shift timescale.
Discussion
In summary, here we report the spatial structure of the TrkB dimeric transmembrane domain. The structure was obtained by solution NMR in the bicelle environment. Phospholipid bicelles, containing a patch of planar lipid bilayer30,44 are the best available environment to reproduce the properties of cell membranes for the high-resolution NMR studies. Since they do not contain other membrane proteins and some specific lipids, to make sure that the observed conformation is not driven by the lipid composition, we investigated the effects of particular lipids by embedding the protein into a thicker bilayer and mixed bicelles with cholesterol, which did not change the dimer structure, indicating that the obtained conformation is highly stable regardless of the protein surroundings.
Another important simplification that we made was to study a truncated protein construct that lacks the extramembrane domains. To show that the obtained TrkB dimer conformation is relevant, we assayed the activity of single-point TrkB mutants in living cells. We used three types of functional read-out, analyzing the kinase phosphorylation activity, which is the most immediate response, the phosphorylation of intracellular adaptor proteins (AKT, ERK), which is an intermediate consequence of ligand binding; and the cell differentiation, which is a much more general and late result of receptor activation. All tests indicate that the substitution of the sole small residue (S441), identified at the interface of TrkB TMDs, affects the kinase activation in the context of a full-length protein inside the cell membrane, unlike the other small residues, which are not involved in the TrkBtm dimerization. One could argue that a single mutation is not sufficient to confirm the structure, but as we mentioned above, inhibiting the RTKs by substitutions in their transmembrane helices may be a non-trivial task. In the works by Lu et al.39 and Bartzoka et al.40, the authors mutated multiple residues of EGFR transmembrane helix and observed no effects on EGFR phosphorylation, which shows that transmembrane domains can adapt to most of the substitutions, and the change should be indeed substantial to completely prohibit the helix-helix interaction. There is almost no mutagenesis data with regard to the TMDs of receptor tyrosine kinases, in which the single-point mutations were shown to inhibit the protein activity. On the other hand, there are multiple examples of stabilizing mutations, with charged residues at certain positions in TM domains triggering the ligand-independent activation of the kinase31,45,46. Therefore, stabilizing the certain dimeric conformation is much easier than destabilizing it. Thus, the effect observed for S441A and S441I in the present work is the strongest evidence in favor of the reported structure. With all aforesaid, we believe that the relevance of the reported structure is strongly supported by different types of biochemistry and molecular biology data.
The obtained structural data need to be discussed in the context of current information available for Trk receptors, which includes the structure of the TrkA transmembrane domain20 and model of TrkB TMD, obtained by computer simulations23. Notably, the aforementioned model is quite different from the spatial structure reported here. In that model, the dimer is formed via the canonical glycophorin GxxxG-like motif47 (440Axxx444G), which is supported by Y434 at certain cholesterol contents, and the dimer topology is left-handed. According to our data, the dimer is formed via an extended motif, with the key residue being S441, and with a right-handed mode of helix-helix interaction. The latter conformation is supported by functional assays; the S441A and S441I mutations drastically inhibited the BDNF-induced activation of TrkB, while the A440I and G440I substitutions did not demonstrate any pronounced biological effect. At the same time, the S441I mutation does not impact the dimerization of the receptor in living cells, as shown by fluorescence microscopy, whereas such effect would be expected if the obtained structure corresponded to the inactive dimeric state. Therefore, we conclude that the observed structure of the TrkBtm dimer most likely corresponds to the active state of the receptor. The alternative conformation, proposed by computer modeling, may in turn describe the dimeric but inactive state, which should also be a stage of TrkB signaling, according to the most recent findings18. The left-handed state may also be favored by the high cholesterol contents in the membrane, in agreement with the in silico data, which explains the inhibiting effects of high cholesterol excess on TrkB.
To further understand the transmembrane domain conformations that occur in the course of neurotrophin signaling, one could invoke data regarding TrkA, one of the closest TrkB relatives. The spatial structure of its TM domain most likely represents the inactive dimer state, as shown by cross-linking20 and further by NMR experiments11. The active state of TrkA TMDs can be obtained by cross-linking at the K410 position, but its exact conformation has not been reported. Comparing the two structures one could find out that they are drastically different (Fig. 6). First, the TM helix of TrkB is substantially (six residues) longer (Fig. 6b, c, Supplementary Fig. 22), which questions the appropriateness of such a comparison. This difference is not an artifact of membrane mimetics, because the length of the helix in TrkA does not depend on the choice of the environment and is the same in both micelles and bicelles11,20,30. It seems that while the structures of the ectodomains in complex with their ligands remained almost identical7,8, the transmembrane parts of these two related proteins diverged strongly during evolution. The sequence comparison of TMDs revealed almost no identity when looking at the distribution of small and aromatic residues that can promote dimerization, while a high identity level was found between the TM regions of TrkA and TrkC (Fig. 6c). This statement agrees with the results of functional studies. TrkA and TrkC were found to cause cell death, but the TrkB receptor was not, and the key role in this effect was played by the TM domain48,49. Thus, the parameters of transmembrane states may be not uniform within the Trk family.

The spatial structures of TrkA (a, PDB 2N90 and TrkB (b, this work) TM dimers obtained by NMR in DPC micelles and DMPC/DHPC bicelles, respectively. Orange color denotes the residues on the experimentally determined interface. Green color indicates the residues proposed for the alternative dimerization interfaces, assumed from the cross-linking experiments20 (a) or computer modeling23 (b). c Manual sequence alignment of Trk family members based on the Clustal Omega program (http://www.clustal.org/) results for alignment of the transmembrane and intracellular domains. Experimentally determined TM domains are indicated, blue background highlights the identical residues.
Next, one can find that the structures of the transmembrane domains are also essentially distinct. In the case of TrkA, the dimer is left-handed, supported by stacking interactions and van der Waals packing, whereas the TrkB dimer is right-handed and supported by a polar contact of a Ser sidechain. This difference may be ascribed to the different types of membrane mimetics that were used to accommodate the protein, and the ability of a dimeric TM domain conformation to switch between two states when the protein is studied in micelles and in bicelles25,26,27. Interestingly, the TrkBtm dimer structure obtained by computer simulations is quite similar to the TrkA structure (left-handed mutual arrangement of TM segments), supporting our hypothesis that the state could correspond to the inactive pre-formed dimer of TrkB. In this regard, one could assume that if the TM domain conformations are uniform within the family, signaling is accomplished by switching between the left-handed and right-handed conformations of Trk TM domains, which is accompanied by mutual rotations of the TM helices.
Finally, we analyzed the interactions between TrkBtm and antidepressant drugs as well as cholesterol. The effects of cholesterol are unclear. In our model system, we did not observe any significant events that could be interpreted as strong and specific interactions between the protein and cholesterol regardless of the presence of the CRAC motif50. The maximal chemical shift perturbations were twice lower than those induced by FLX at the same ligand/protein and ligand/lipid ratio but were comparable to the ones induced by CPR (Supplementary Fig. 20e, g). Unlike FLX, cholesterol stabilized neither the monomer nor the dimer of TrkBtm substantially (Supplementary Fig. 20f). Additionally, the changes were not saturated even under excess cholesterol. This is in agreement with the computer modeling data which suggests that the effects of cholesterol on TrkB are mostly due to variations in bilayer thickness23. However, weak interactions involving the Y434 sidechain were indeed detected. Taking into account the high overall cholesterol contents in membranes (30%), these weak contacts may become relevant at physiological cholesterol concentrations. Our data indicate that the effects of cholesterol on TrkB signaling are accomplished not via the highly specific interactions of cholesterol with the CRAC motif, but most likely by the effect of cholesterol on the physical properties of the cell membrane, its ability to maintain the liquid-ordered state of the bilayer and regulate the abundance of lipid microdomains. The partitioning of proteins between domains may be modulated by the CRAC domain, which explains both the effects of cholesterol in cells and the effect of the Y434 side chain. This also means that the mere presence of the CRAC/CARC motif does not ensure specific protein-cholesterol interactions. Alternatively, the CRAC motifs could become important when TrkB enters the lipid rafts of the cell membrane upon BDNF binding51, and the weak interactions that were found here may become strong within the liquid-ordered bilayer. Unfortunately, it seems hardly possible to mimic this state within lipid bicelles, and computer modeling is unable to reproduce it in systems with a low number of lipid molecules, leaving the question open for future studies.
As for drug interactions, our spatial structure helps us to understand the structural basis of their activity. Here, we show that FLX interacts with the N-terminus of the TrkB TM helix in both the dimer and monomer states, supporting all previous data obtained in cells23. The interaction does not change the structure of the TrkBtm dimer but rather stabilizes this state compared to the monomer. These features are somewhat different from the previously suggested mechanism, stating that FLX binds to the dimeric state of TrkB and alters the distance between the C-termini of the TMD helices23. However, these two hypotheses may be combined to provide a self-consistent view of the FLX mechanism, assuming that the AxxxG conformation of TrkB corresponds to the receptor inactive state. According to both modeling and NMR, the site of FLX binding to TrkB involves residues Y434 and V437, which interfere with the structure of the AxxxG TrkB dimer (inactive state). At the same time, FLX binding to TrkB does not conflict with the active dimeric state reported here, and, moreover, stabilizes it. Altogether, this would mean that FLX disturbs the formation of an inactive conformation and supports the formation of an active dimeric arrangement of TMDs, which agrees perfectly with the observed biological effects of FLX - the activation of the TrkB receptor. None of these features of FLX were observed for the antipsychotic CPR, which correlates with the absence of antidepressant effects. Besides, the absence of Y434 or similar residue in TrkA explains why the activity of this protein is not sensitive to FLX.
To summarize, we report here the spatial structure of the dimeric transmembrane domain of TrkB in lipid bicelles. The obtained conformation is stable in a variety of model environments and corresponds to the receptor’s active state, according to the directed mutagenesis experiments. Together with previously published results of computer simulations, our data helps to build a self-consistent model, explaining the activity of FLX with respect to the TrkB receptor.
Methods
Protein sample preparation
The details of protein synthesis, purification, and sample preparation for NMR analysis are provided in our most recent work24. The gene encoding the transmembrane domain of the human TrkB (M423TDKTGREHLSVYAVVVIASVVGFCLLVMLFLLKLARHSKFGMK466G UNIPROT Q16620), was synthesized by Cloning Facility (Russia) with codon optimization for E. coli and cloned into pGEMEX-1 vector using NdeI and HindII restriction sites. The protein was synthesized by a cell-free continuous exchange expression system. A standard cell-free reaction (2–3 ml of RM) was carried out in 50 ml tubes using a Pur-A-Lyzer Maxi dialysis kit (#PURX35050, Sigma). The TrkBtm was expressed as a precipitate. The algal mixture of 15N (#NLM-6695, CIL) or 13C-15N-labeled (#CCN070P1, CortecNet) amino acids and 15N (CN501P05, CortecNet) or 13C-15N-labeled (CCN500P1, CortecNet) cysteine were used to obtain a uniformly 15N or 13C-15N-labeled protein samples, respectively. The precipitate from the cell-free reaction was washed 3 times with water (milliQ), solubilized with detergent buffer (20 mM Tris 8.0, 150 mM NaCl, 10 mM bME, 1 mM EDTA and 1.5% lauryl sarcosine), purified using size-exclusion chromatography in SEC-buffer (20 mM Tris 8.0, 50 mM NaCl, 5 mM bME, 0.5% lauryl sarcosine) and target protein was precipitated by a TCA/acetone procedure52.
The dry powder of 15N or 13C-15N-labeled TrkBtm was dissolved in 450 μl of trifluoroethanol(TFE)-water mixture containing 45 μl deuterated TFE for frequency lock, 1 mM of TSP-d4 as a reference compound and 1 mM Tris(2-carboxyethyl)phosphine to prevent disulfide bonds between the transmembrane C451 residues. As TrkBtm contains no Trp residues and the optical concentration measurement is impossible, 1D 1H NMR spectra were acquired to estimate the concentration of protein using signals of the TSP methyl group at 0.0 ppm as a reference. Then, we solubilized TrkBtm in bicelles as described in our recent work53. Briefly, deuterated or protonated DMPC, DPPC, and DHPC were added in needed amounts to the sample, and water was added up to a TFE/water ratio of 1:1. Then the sample was freeze-dried for 12–15 h and dissolved in 450 or 330 μl 20 mM phosphate buffer pH 6.0 or 7.4 containing 5% D2O for frequency lock, 0.01% NaN3, 0.5 mM TSP-d4 as a chemical shift reference and 2 mM EDTA.
NMR spectroscopy and spatial structure calculation
NMR spectra were acquired at 40 °C on Bruker Avance III 600 MHz and 800 MHz spectrometers equipped with triple resonance cryogenic probes and Bruker Topspin 3.2 software. The 13C-15N-labeled samples (330 µl) for structure calculations were placed into a 5-mm Shigemi tube to maximize the protein concentration. The rest of the samples (450 µl) were placed into regular 5-mm glass tubes (Wilmad, USA).
To assign the 1H, 13C, and 15N resonances and resolve the structure of a TrkBtm monomer we recorded the set of 2D heteronuclear (1H-15N-HSQC, 1H-13C-HSQC aliphatic and 1H-13C-HSQC aromatic), 3D triple-resonance NMR spectra (HNCA, HNCO, HNcoCA, HNcaCO, HNCACB, NOESY-HSQC), 3D 13C- and 15N-NOESY-HSQC and 3D-HccH-TOCSY spectra. For all NOESY experiments the mixing time was set to 80 ms. Aromatic sidechains were assigned based on the 2D hC(CC)H and 3D (H)CCH-COSY experiments54,55. Spectra were recorded, where possible, using the BEST-TROSY pulse schemes56 and non-uniform sampling in indirect dimensions. Such spectra were processed with qMDD 2.0 using the “iterative soft thresholding” algorithm with virtual echo57. Resonance assignment was performed in the CARA software version 1.8.4.2 (www.http://cara.nmr.ch/)58.
The spatial structure of the TrkBtm dimer was calculated using the CYANA 3.98.13 program59. Backbone torsion angle restraints were obtained based on the NMR chemical shifts in TALOS-N software60. Sidechain angle restraints were found by manual analysis of NOESY spectra and 3JCGCO and 3JCGN vicinal J-couplings that were measured using the spin-echo difference constant-time HSQC NMR experiments61,62. To obtain the intermolecular distance restraints, we recorded two 3D 13C-filtered-NOESY-HSQC spectra63 (mixing time 80 ms) for the samples containing the 13C/15N-labeled TrkBtm and an equimolar mixture of isotope-labeled and unlabeled proteins (Supplementary Fig 3).
To measure the translational diffusion of DMPC, DHPC, and drugs we employed the 1H pulsed-gradient stimulated echo pulse sequence with convection compensation64. In this experiment, 16 1D spectra were recorded with the gradient power increasing from 13.9 to 53.0 Gs/cm and the diffusion delay of 400 ms. We fitted the peak intensity decays using the Wolfram Mathematica 5.0 software and obtained the diffusion coefficients to calculate the hydrodynamic radii as described in our previous work65.
The hydrodynamic radii of TrkBtm in bicelles were calculated from the rotational correlation time τc:
Here, kb is the Boltzmann constant, T is the absolute temperature and η is the dynamic viscosity of the solution. The τc values were derived from the cross-correlated relaxation rate measured as described elsewhere66 with the relaxation delay of 10.7526 ms, assuming that ps-ns range internal motions were negligible in the helical TrkBtm backbone. Cross-correlation relaxation rates were measured at LPR = 60.
Obtained structures were analyzed using MOLMOL67 and PyMOL (Schrödinger LLC) software.
Oligomer order determination
In the current work we used an NMR-based approach to describe complex oligomer equilibria of TMDs described earlier28,32. Briefly, we applied the so-called “micellar solvent” model (which can be applied to bicelles as well)33. According to this model, the equilibrium constant Keq of n monomers association into an n-mer depends on the concentrations of monomer [M], n-mer [N] and empty bicelles [bicE] in the following way:
Here γ is the effective order of reaction with respect to the concentration of empty bicelles. Thus, if we sequentially add bicelles to the sample, determine [M] and [N]/n according to the intensities, and plot [M]N/[N] as a function of [bicE] it would be a straight line in logarithmic coordinates with the tilt γ. The same is true for the intensities of monomer and n-mer signals, IM and IN, used in the calculations. In the titration series, we recorded the BEST-TROSY-1H-15N-HSQC spectra with a relaxation delay of 1 s for 8–16 h.
If we then dilute the sample and continue adding bicelles, the points after dilution would fit to the same line only if the oligomer order n was assumed correctly33. Thus, to determine the oligomer order we used a penalty function χ2(k) that measures the deviations of points after the dilution from the initial dependence28:
where k is the assumed oligomer order, pd is the number of experimental points after dilution, Δj(k) is the deviation of the jth point after dilution from the least-square-fitted line of points before dilution and D(k) is the mean dispersion of points before dilution from the fitted line. Penalty function is divided by D(k) to take into account the different magnitudes of IMk/IN obtained for different k and signals. Thus, if the experimental noise is low enough, the mean penalty function would reach a reliable minimum at k = kmin = n which allows determining the stoichiometry of the observed oligomer. The penalty function was evaluated for several best-resolved peaks of TrkB assuming different oligomer orders and the distributions were compared using the Student t-test.
Besides, we took into account the size of the bicelles, which was measured from the translational diffusion coefficients of lipids for the empty bicelles and from the rotational diffusion coefficients that were obtained based on the protein 1H,15N cross-correlated relaxation rates for the bicelles bearing the oligomers of TrkBtm as described30. Diffusion coefficients were then translated into the hydrodynamic radii using the Stokes-Einstein relations and then compared to the values predicted based on the “ideal bicelle model”, assuming that the bilayer thickness is 4.0 nm68 and bicelle surface area that is occupied by one TM helix is 1.4 nm230.
TrkB interactions with small molecules and the effect of environment
We performed a gradual titration of TrkBtm with 10 mM FLX and 40 mM CPR, RR- and SS-HNK solutions in a 20 mM phosphate buffer pH 7.4. These solutions contained DHPC at a concentration of 8.4 mM to keep the bicelle q* = 0.5 (effective lipid/detergent ratio taking into account that some detergent is present in the monomeric state) and size constant upon titration30. As CPR was insoluble at this pH, it was added as a well-shaken emulsion which turned transparent and solubilized well in the bicelles. The amount of added CPR was controlled by 1H NMR spectra.
To observe the partitioning of drugs between the solution and bicelles with TrkBtm we measured the diffusion coefficient of DMPC (DDMPC) and drugs (Ddrug). In a separate experiment, we measured the diffusion of 1 mM of each drug in a buffer containing 8.4 mM DHPC, which corresponds to the concentration of monomeric DHPC in the presence of q = 0.5 DMPC/DHPC bicelles (Ddrug,free)30. Assuming that DDMPC characterizes the whole bicelle, Ddrug depends on the distribution between bicelles and solution the following way:
Here α is a share of drug in bicelles and Ddrug,free is corrected to include the effect of volume fraction Ф occupied by bicelles69.
Finally, we can calculate α:
To characterize the interactions between the small molecules and TrkBtm, we measured the CSPs and populations of monomers and dimers. CSPs were calculated in the following way:
Here δCSH and δCSN are the changes of 1H and 15N chemical shifts, γH and γN are the gyromagnetic ratios of 1H and 15N nuclei, respectively (Supplementary Fig. 2a).
To observe the changes in monomer and oligomer populations, we measured the intensities of the separately standing cross-peaks of TrkBtm, corresponding to the monomeric and dimeric states in 1H,15N-HSQC spectra. All the intensities were normalized to the values measured in the absence of the drug and to the number of scans in each spectrum. The values for each drug ratio were averaged and the SD values were calculated.
Cholesterol was added to the sample as an 85 mM solution in ethanol-d5 (33 mg/ml) at 45 °C. We added cholesterol three times, monitoring its incorporation into the bicelles by following the signals of its methyl groups in the 1H spectra (Supplementary Fig. 11a). At each titration point some cholesterol precipitated, and at the 3rd point, no cholesterol was further retained in the solution. To measure the exact contents of cholesterol in bicelles we redissolved the supernatant of the sample after chol titration in CDCl3:CD3OD 1:1 with the addition of 1 mM TSP-d4 and applied the quantitative NMR.
To distinguish between the effects of cholesterol and ethanol we performed a separate experiment preceding the cholesterol titration. We reproduced the ethanol contents (2% v/v) at the last point of cholesterol titration and measured the CSP and intensity alterations. The ethanol-free CSP induced by cholesterol was calculated as a length of the vector difference between C2D5OH and C2D5OH+chol effects as follows:
DNA constructs for functional assays
A plasmid encoding rat TrkB with an N-terminal hemagglutinin (HA) tag was kindly provided by Dr. Y. Barde. All TrkB mutants and constructs were derived from this plasmid. Mutagenesis was done using the site-directed mutagenesis kit (Agilent) according to the manufacturer’s protocol. The oligonucleotide sequences of all of the constructs are listed in Supplementary Table 2. All DNA constructs were sequenced at Macrogen.
Cell culture and transfection
HeLa cells (obtained from ATCC, CCL-2), which do not express endogenous TrkB, were cultured in Dulbecco’s modified Eagle’s medium (Fisher) supplemented with 10% fetal bovine serum (Fisher), 1% Penicilin/Streptomicin and 1% L-Glutamine at 37 °C in a humidified atmosphere with 5% CO2. PC12-TrkA-KO cells were developed in-home as described in ref. 41 and were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, 1% Penicilin/Streptomicin and 1% L-Glutamine and 5% horse serum (Fisher). Transfection of cells was performed using Lipofectamine-2000 following the manufacturer’s protocol.
Cell signaling
HeLa cells were seeded in 60 mm dishes and transfected with 3 ng of DNA. Twenty-four hours after transfection, the cells were lifted and replated into 12-well plates at a density of 100,000 cells/well. Forty-eight hours after transfection, the cells were starved in serum-free medium for 2 h and were then stimulated with 50 ng/ml BDNF (Alomone) at the indicated time intervals. The cells were lysed with TNE buffer (Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA) supplemented with 1% Triton X-100 (Sigma), protease inhibitors (Roche), 1 mM phenylmethylsulfonyl fluoride (Sigma), 1 mM sodium orthovanadate (Sigma), and 1 mM sodium fluoride (Sigma). Lysates were kept on ice for 10 min and centrifuged at 12,000 × g for 15 min in a tabletop centrifuge. The protein level of the lysates was quantified using a Bradford kit (Pierce), and lysates were analyzed by SDS-PAGE. The proteins were resolved in SDS-PAGE gels and transferred to nitrocellulose membranes that were incubated overnight at 4 °C with one of the following antibodies: mouse monoclonal anti-HA (1:1000, Sigma, H9658); rabbit anti-P-TrkB-Tyr706/707 (1:1000, Cell Signaling, 4621S); rabbit anti-P-TrkB-Tyr516 (1:3000, Cell Signaling, 4619S); rabbit anti-ERK (1:1000, Cell Signaling, 4695 T); rabbit anti-P-ERK (1:1000, Cell Signaling, 4370 T); rabbit anti-AKT (1:1000, Cell Signaling, 4691S); rabbit anti-P-AKT (Ser473,1:1000, Cell Signaling, 4060S), mouse anti-actin (1:5000, Sigma, A5441I). Following incubation with the appropriate secondary antibody, anti-mouse Alexa Fluor680 (1:10000, Invitrogen, A32789), and anti-rabbit Alexa Fluor800 (1:10000, Invitrogen, A32802), the membranes were imaged using LICOR. Protein bands were quantified by the LOCOR lite software.
Differentiation of PC12-TrkA-KO cells
The generation of the PC12-TrkA-KO cells was described elsewhere41. Transfection in PC12-TrkA-KO cells was performed using Lipofectamine-2000 as previously described. Cells were plated into 24-well plates and transfected with 1 µg of plasmids encoding TrkB or TrkB mutants and 0,2 µg of GFP. Transfection was allowed for 24 h and cells were washed three times with serum-free medium and incubated for 48 h in a medium containing 1% fetal bovine serum and 20 ng/ml of BDNF (Alomone). At 48 h, the cells were washed with cold PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. DAPI was used for nuclei staining and samples were mounted using Mowiol medium. The cells were imaged using a Leica SP8 spectral confocal microscope. The length of the longest neurite was measured using ImageJ, data were analyzed and plotted with GraphPad Prism 8.
Cytometry and immunofluorescence
Membrane localization of TrkB mutants in HeLa cells was analyzed by Flow cytometry and by immunofluorescence in non-permeabilized cells. HeLa cells were transiently transfected with the indicated TrkB and GFP constructs. 48 h later, the cells were fixed with 4% paraformaldehyde, blocked with 10% FBS for an hour, and incubated with mouse monoclonal anti-HA antibody (1:200, Sigma, H9658) overnight. The following day, cells were washed with PBS and incubated with the secondary antibody anti-mouse Alexa555 (1:500, Invitrogen, A31570) for an hour. Nuclei were stained with DAPI and imaged using SP8 Leica confocal microscope. Alternatively, membrane localization of TrkB mutants in HeLa cells was analyzed by Flow Cytometry. Briefly, 48 h after transfection, HeLa cells were lifted and incubated with PBS with a mouse monoclonal anti-HA antibody (1:1000, Sigma, H9658) for an hour, followed by incubation with a secondary antibody anti-mouse Alexa633 (1:100, Invitrogen, A21052). GFP-positive cells were selected and the Alexa-633-positive cells were quantified using the Flow Cytometer MACS Quant 10 (Miltenyi).
Statistics
In Figs. 1c and 4g the error bars indicate the standard deviation produced by either multiple repeats, experimental noise or deviation in a set of tested signals. The significance of differences between the observed mean values for monomer and dimer were assessed by the Mann-Whitney statistical test performed in the Wolfram Mathematica software. Here we tested the null hypothesis of the equality of mean values. In the Fig. 1d we calculated the pairwise differences between the penalty function of k = 2 and k = 3 for each considered pair of peaks, i.e.,
Next, we performed the Student t-test in the Wolfram software with the null hypothesis that the mean difference is equal to zero, i.e., <Dij > =0.
In Fig. 3, the error bars indicate the standard deviation for multiple repeats (n ≥ 3). The significance of differences between the observed mean values for wild type and mutants were assessed by the multiple t-test with Bonferroni correction using Statannotations library in python 3. Here we tested the null hypothesis of the equality of mean values.
In all the figures, the probability of the null hypothesis (the p-value) was shown as * for p < 0.05, ** for p < 0.01, *** for p < 0.001 and **** for p < 0.0001, ns denotes that changes with respect to the control are insignificant.
To select the maximal CSPs we calculated the standard deviation from zero among all the measured CSPs (including monomer and dimer signals) and took the values lying outside the 95% interval (i.e., exceeding 1.96 SD). These values were excluded from the analysis and the new SD was calculated. This procedure was repeated until all the remaining CSP values were within the 95% interval. The outliers were considered as maximal.
As a reference for the CSPs we plotted the peak width (PW) equal to the halfwidth of Cys 446 backbone NH signal. C446 residue is located close to the center of the TM helix, so it should be representative of the whole TM region; moreover, the C446 NH signal in the 1H-15N-HSQC spectrum is not split at all the tested conditions and does not overlap (Supplementary Fig 1), so its halfwidth is easy to assess.
To determine the similarity of TrkBtm dimer conformations under various conditions, we evaluated the monomer/dimer peak splittings. We plotted the correlations between dimerization-induced changes in 1H and 15N chemical shifts of TrkBtm backbone at various sample conditions (Supplementary Figs. 12, 13). The splitting amplitudes along the 15N axis were multiplied by the ratio of gyromagnetic ratios (γN/γH) = -0.1 so that the changes along two dimensions have comparable amplitude. Thus, we obtained the scatter plots where each signal produced 2 points having the coordinates {Split1Hinit, Split1H} and {Split15Ninit, Split15N}. These plots describe both the magnitude and the “direction” of a peak splitting. Next, we calculated the R2 values for the linear correlation and compared them to each other, using the data on TrkA TMD as a proof-of-concept example (Supplementary Fig. 12).
Number and brightness experiments
Number and Brightness (N&B) is a fluorescence fluctuation technique that measures intensity fluctuations over time and calculates molecular brightness. Molecular brightness scales linearly with oligomer order. Our implementation of N&B was previously described in detail38,43.An overview of the N&B method is shown in Supplementary Fig. 19a. Receptors are tagged with fluorescent proteins and a series of images is taken. Intensity alone cannot determine oligomer order because several monomers have the same intensity as a single oligomer unit. The difference is that a monomer, diffusing in and out of a pixel, causes smaller fluctuations in the signal as compared to an oligomer. The variance in the signal, σ2, of a monomer and an oligomer will be different even if the average intensity, 〈I〉, is the same. By dividing the variance by the average intensity we obtain the apparent brightness, B
The apparent brightness contains two sources of variance in the signal, variance due to the diffusing fluorophores and variance due to the detector. The molecular brightness only contains the fluorophore variance. When a photon counting detector is used, the detector’s variance is equated to the average intensity. The molecular brightness, ε, is written as.
A brightness value was calculated for each pixel. Brightness values in a region of interest in the plasma membrane are averaged (Supplementary Fig. 19b). A representative trace of intensity vs. image number is shown in Supplementary Fig 19с. Bleaching and cell movement impact the brightness measurement and the average fluorescence decreases over time. These effects are corrected for using a boxcar averaging algorithm and image alignment using the MATLAB dftregistration function43. The red line in Supplementary Fig. 19c is an exponential decay fit of the average intensity, which is used for the correction.
The apparent oligomer order, OS, is determined by scaling the molecular brightness to a known monomer control, a monomeric YFP with a GPI anchor.
A non-parametric one-way ANOVA (Kruskal-Wallis) test was performed, comparing the mean oligomer order, followed by Dunn’s multiple comparison test, to compare the mean oligomer order across datasets consisting of 100-150 cells to test for significance.
The extracellular + transmembrane (ECTM) TrkB plasmid was cloned previously38. Both S441A & S441I mutations for the ECTM variants were cloned using Q5 Site-Directed Mutagenesis kit (E0554S) following the manufacturer’s protocol. The mEYFP-GPI plasmid was a kind gift from Barbara Baird’s lab described previously70.
Chinese hamster ovarian (CHO-K1) cells were obtained from ATTC (CCL-61) and were cultured in DMEM (31600034) adjusted to contain 1.8 g/L glucose (DX0145), 1X NEAA (11140050), and 10% FBS (35-011-CV). 48 h before imaging, cells were seeded onto collagen coated imaging dishes (P35GCOL1.514 C). 24 h before imaging, cells were transfected with 1–2 μg DNA using Lipofectamine 3000 (L3000015) following the manufacturer’s protocol. 4 h before imaging, the media was exchanged with serum free, starvation media (D2902) which is the same media used for imaging.
CHO cells were imaged in the starvation media described. Any ligands were added 10 min prior to imaging. BDNF (3897) was added at a saturating concentration of 5 μg/mL, or 357 nM. Imaging was performed on a LeicaSP8 microscope using the detector in a photon counting mode while focusing on the basolateral membrane of the cells. 150 images, each 256 by 256 pixels, were taken over about 90 s (5.4 μs pixel dwell time) with an 18x optical zoom (pixel size of 40.2 nm). For excitation a 488 nm laser set at 0.1% of its max power was used and the excitation window was set using the default YFP setting.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Spatial structure of TrkBtm and chemical shifts generated in this study were deposited to BMRB under the access code 34814 and to PDB, ID 8OYD. The reference PDB code used in this work is 2N90. Source Data is provided as a Source Data file. Additional data is available upon request to the corresponding authors. Source data are provided with this paper.
References
Bothwell, M. Recent advances in understanding neurotrophin signaling. F1000Research 5, 1885 (2016).
Friedman, W. J. & Greene, L. A. Neurotrophin signaling via Trks and p75. Exp. Cell Res. 253, 131–142 (1999).
Martin-Zanca, D., Hughes, S. H. & Barbacid, M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature 319, 743–748 (1986).
Cocco, E., Scaltriti, M. & Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 15, 731–747 (2018).
Segal, R. A. Selectivity in neurotrophin signaling: theme and variations. Annu. Rev. Neurosci. 26, 299–330 (2003).
Wehrman, T. et al. Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 53, 25–38 (2007).
Wiesmann, C., Ultsch, M. H., Bass, S. H. & De Vos, A. M. Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 401, 184–188 (1999).
Banfield, M. J. et al. Specificity in Trk Receptor:Neurotrophin Interactions. Structure 9, 1191–1199 (2001).
Ultsch, M. H. et al. Crystal structures of the neurotrophin-binding domain of TrkA, TrkB and TrkC 1 1Edited by I. A. Wilson. J. Mol. Biol. 290, 149–159 (1999).
Shoemark, D. K. et al. Design and Nuclear Magnetic Resonance (NMR) Structure Determination Of The Second Extracellular Immunoglobulin Tyrosine Kinase A (TrkAIg2) domain construct for binding site Elucidation in drug discovery. J. Med. Chem. 58, 767–777 (2015).
Kot, E. F. et al. Intrinsically disordered regions couple the ligand binding and kinase activation of Trk neurotrophin receptors. iScience 25, 104348 (2022).
Clary, D. O. & Reichardt, L. F. An alternatively spliced form of the nerve growth factor receptor TrkA confers an enhanced response to neurotrophin 3. Proc. Natl Acad. Sci. USA 91, 11133–11137 (1994).
Strohmaier, C., Carter, B. D., Urfer, R., Barde, Y. A. & Dechant, G. A splice variant of the neurotrophin receptor trkB with increased specificity for brain-derived neurotrophic factor. EMBO J. 15, 3332–3337 (1996).
Urfer, R. et al. High resolution mapping of the binding site of TrkA for nerve growth factor and TrkC for Neurotrophin-3 on the second immunoglobulin-like domain of the Trk Receptors. J. Biol. Chem. 273, 5829–5840 (1998).
Bertrand, T. et al. The crystal structures of TrkA and TrkB suggest key regions for achieving selective inhibition. J. Mol. Biol. 423, 439–453 (2012).
Shen, J. & Maruyama, I. N. Brain-derived neurotrophic factor receptor TrkB exists as a preformed dimer in living cells. J. Mol. Signal. 7, 2 (2012).
Shen, J. & Maruyama, I. N. Nerve growth factor receptor TrkA exists as a preformed, yet inactive, dimer in living cells. FEBS Lett. 585, 295–299 (2011).
Ahmed, F. & Hristova, K. Dimerization of the Trk receptors in the plasma membrane: effects of their cognate ligands. Biochem. J. 475, 3669–3685 (2018).
Paul, M. D. & Hristova, K. The transition model of RTK activation: a quantitative framework for understanding RTK signaling and RTK modulator activity. Cytokine Growth Factor Rev. 49, 23–31 (2019).
Franco, M. L. et al. Structural basis of the transmembrane domain dimerization and rotation in the activation mechanism of the TRKA receptor by nerve growth factor. J. Biol. Chem. 295, 275–286 (2020).
Franco, M. L. et al. Interaction between the transmembrane domains of neurotrophin receptors p75 and TrkA mediates their reciprocal activation. J. Biol. Chem. 297, 100926 (2021).
Castrén, E. & Antila, H. Neuronal plasticity and neurotrophic factors in drug responses. Mol. Psychiatry 22, 1085–1095 (2017).
Casarotto, P. C. et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184, 1299–1313.e19 (2021).
Moliner, R. et al. Psychedelics promote plasticity by directly binding to BDNF receptor TrkB. Nat. Neurosci. https://doi.org/10.1038/s41593-023-01316-5 (2023).
Bocharov, E. V. et al. Helix-helix interactions in membrane domains of bitopic proteins: Specificity and role of lipid environment. Biochim. Biophys. Acta 1859, 561–576 (2017).
Bragin, P. E. et al. HER2 transmembrane domain dimerization coupled with self-association of membrane-embedded cytoplasmic juxtamembrane regions. J. Mol. Biol. 428, 52–61 (2016).
Bocharov, E. V. et al. The conformation of the epidermal growth factor receptor transmembrane domain dimer dynamically adapts to the local membrane environment. Biochemistry 56, 1697–1705 (2017).
Kot, E. F. et al. Oligomerization analysis as a tool to elucidate the mechanism of EBV latent membrane protein 1 inhibition by pentamidine. Biochim. Biophys. Acta BBA - Biomembr. 1862, 183380 (2020).
Triba, M. N., Warschawski, D. E. & Devaux, P. F. Reinvestigation by phosphorus NMR of lipid distribution in bicelles. Biophys. J. 88, 1887–1901 (2005).
Mineev, K. S., Nadezhdin, K. D., Goncharuk, S. A. & Arseniev, A. S. Characterization of small isotropic bicelles with various compositions. Langmuir ACS J. Surf. Colloids 32, 6624–6637 (2016).
Manni, S. et al. Structural and functional characterization of alternative transmembrane domain conformations in VEGF receptor 2 activation. Structure 22, 1077–1089 (2014).
Bragin, P. E. et al. Probing the effect of membrane contents on transmembrane protein-protein interaction using solution NMR and computer simulations. Biochim. Biophys. Acta Biomembr. 1860, 2486–2498 (2018).
Mineev, K. S. et al. NMR-based approach to measure the free energy of transmembrane helix-helix interactions. Biochim. Biophys. Acta 1838, 164–172 (2014).
Bocharov, E. V. et al. Unique dimeric structure of BNip3 transmembrane domain suggests membrane permeabilization as a cell death trigger. J. Biol. Chem. 282, 16256–16266 (2007).
Senes, A., Ubarretxena-Belandia, I. & Engelman, D. M. The Calpha -H…O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc. Natl Acad. Sci. USA 98, 9056–9061 (2001).
Nicolson, G. L. The Fluid-Mosaic Model of Membrane Structure: still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim. Biophys. Acta 1838, 1451–1466 (2014).
Kornilov, F. D. et al. The architecture of transmembrane and cytoplasmic juxtamembrane regions of Toll-like receptors. Nat. Commun. 14, 1503 (2023).
Ahmed, F., Paul, M. D. & Hristova, K. The biophysical basis of receptor tyrosine kinase ligand functional selectivity: Trk-B case study. Biochem. J. 477, 4515–4526 (2020).
Lu, C. et al. Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor. Mol. Cell. Biol. 30, 5432–5443 (2010).
Bartzoka, F. et al. Activity of EGFR transmembrane region variants indicates specific transmembrane dimers are not required for EGFR activity. Biochem. J. 479, 2465–2475 (2022).
Testa, G. et al. Disentangling the signaling complexity of nerve growth factor receptors by CRISPR /Cas9. FASEB J. 36, e22498 (2022).
Cannarozzo, C. et al. Cholesterol‐recognition motifs in the transmembrane domain of the tyrosine kinase receptor family: The case of TRKB. Eur. J. Neurosci. 53, 3311–3322 (2021).
Paul, M. D., Rainwater, R., Zuo, Y., Gu, L. & Hristova, K. Probing membrane protein association using concentration‐dependent number and brightness. Angew. Chem. Int. Ed. 60, 6503–6508 (2021).
Kot, E. F., Goncharuk, S. A., Arseniev, A. S. & Mineev, K. S. Phase transitions in small isotropic Bicelles. Langmuir ACS J. Surf. Colloids 34, 3426–3437 (2018).
Ou, S.-H. I. et al. HER2 Transmembrane Domain (TMD) Mutations (V659/G660) That Stabilize Homo- and Heterodimerization Are Rare Oncogenic Drivers in Lung Adenocarcinoma That Respond to Afatinib. J. Thorac. Oncol. 12, 446–457 (2017).
Sarabipour, S. & Hristova, K. Mechanism of FGF receptor dimerization and activation. Nat. Commun. 7, 10262 (2016).
Russ, W. P. & Engelman, D. M. The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol. 296, 911–919 (2000).
Nikoletopoulou, V. et al. Neurotrophin receptors TrkA and TrkC cause neuronal death whereas TrkB does not. Nature 467, 59–63 (2010).
Dekkers, M. P. J., Nikoletopoulou, V. & Barde, Y.-A. Death of developing neurons: New insights and implications for connectivity. J. Cell Biol. 203, 385–393 (2013).
Fantini, J., Di Scala, C., Baier, C. J. & Barrantes, F. J. Molecular mechanisms of protein-cholesterol interactions in plasma membranes: Functional distinction between topological (tilted) and consensus (CARC/CRAC) domains. Chem. Phys. Lipids 199, 52–60 (2016).
Suzuki, S. et al. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts. J. Cell Biol. 167, 1205–1215 (2004).
Goncharuk, S. A. et al. Bacterial synthesis and purification of normal and mutant forms of human FGFR3 transmembrane segment. Acta Nat. 3, 77–84 (2011).
Motov, V. V. et al. Investigation of lipid/protein interactions in trifluoroethanol-water mixtures proposes the strategy for the refolding of helical transmembrane domains. J. Biomol. NMR 77, 15–24 (2023).
Pervushin, K., Riek, R., Wider, G. & Wüthrich, K. Transverse Relaxation-Optimized Spectroscopy (TROSY) for NMR studies of aromatic spin systems in 13C-Labeled Proteins. J. Am. Chem. Soc. 120, 6394–6400 (1998).
Löhr, F., Hänsel, R., Rogov, V. V. & Dötsch, V. Improved pulse sequences for sequence specific assignment of aromatic proton resonances in proteins. J. Biomol. NMR 37, 205–224 (2007).
Favier, A. & Brutscher, B. Recovering lost magnetization: polarization enhancement in biomolecular NMR. J. Biomol. NMR 49, 9–15 (2011).
Mayzel, M., Kazimierczuk, K. & Orekhov, V. Y. The causality principle in the reconstruction of sparse NMR spectra. Chem. Commun. Camb. Engl. 50, 8947–8950 (2014).
Keller, R. R. J. The Computer Aided Resonance Assignment Tutorial. (CANTINA Verlag, 2004).
Güntert, P. & Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 62, 453–471 (2015).
Shen, Y. & Bax, A. Protein structural information derived from NMR chemical shift with the neural network program TALOS-N. Methods Mol. Biol. Clifton NJ 1260, 17–32 (2015).
Vuister, G. W., Wang, A. C. & Bax, A. Measurement of three-bond nitrogen-carbon J couplings in proteins uniformly enriched in nitrogen-15 and carbon-13. J. Am. Chem. Soc. 115, 5334–5335 (1993).
Grzesiek, S., Vuister, G. W. & Bax, A. A simple and sensitive experiment for measurement of JCC couplings between backbone carbonyl and methyl carbons in isotopically enriched proteins. J. Biomol. NMR 3, 487–493 (1993).
Zwahlen, C. et al. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: application to a bacteriophage λ N-Peptide/boxB RNA complex. J. Am. Chem. Soc. 119, 6711–6721 (1997).
Zheng, G. & Price, W. S. Simultaneous convection compensation and solvent suppression in biomolecular NMR diffusion experiments. J. Biomol. NMR 45, 295–299 (2009).
Kot, E. F., Arseniev, A. S. & Mineev, K. S. Behavior of most widely spread lipids in isotropic bicelles. Langmuir 34, 8302–8313 (2018).
Chill, J. H., Louis, J. M., Baber, J. L. & Bax, A. Measurement of 15N relaxation in the detergent-solubilized tetrameric KcsA potassium channel. J. Biomol. NMR 36, 123–136 (2006).
Koradi, R., Billeter, M. & Wüthrich, K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55 (1996).
Kučerka, N., Nieh, M.-P. & Katsaras, J. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochim. Biophys. Acta 1808, 2761–2771 (2011).
Jóhannesson, H. & Halle, B. Solvent diffusion in ordered macrofluids: a stochastic simulation study of the obstruction effect. J. Chem. Phys. 104, 6807 (1996).
Keller, P., Toomre, D., Díaz, E., White, J. & Simons, K. Multicolour imaging of post-Golgi sorting and trafficking in live cells. Nat. Cell Biol. 3, 140–149 (2001).
Wimley, W. C. & White, S. H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3, 842–848 (1996).
Acknowledgements
The work was supported by the Russian Science Foundation grant #22-14-00130 to SAG (NMR studies), by the Spanish Ministry of Science and Innovation grant number PID2021/127600NB-I00 to M.V. A.B.M. is the recipient of an Investigo contract from the Generalitat Valenciana (INVEST/2022/456) and by EU funding within the NextGeneration EU-MUR PNRR TUSCANY HEALTH Ecosystem – THE (Project no. ECS_00000017)” spoke 1 and 8 to A.C. D.M.M. and K.H. were supported by NSF MCB 2106031. The work of K.S.M. was supported by the Collaborative Research Center #1507. We are thankful to Prof. Eero Castren for the fruitful discussion of the manuscript.
Author information
Authors and Affiliations
Contributions
E.F.K. conducted the NMR experiments and analyzed the data, S.A.G. synthesized the protein and analyzed the data, M.L.F. and A.B.M. constructed the TrkB mutants and performed the functional assays, D.M.M. performed the fluorescence microscopy experiments and analyzed the data, M.C. and A.C. created the knockout cell line, K.S.M., A.S.A., S.A.G., K.H., and M.V. designed and supervised the project, K.S.M. wrote the paper with the assistance from all the authors, S.A.G., A.B.M., M.V., and A.C. acquired the funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests
Peer review
Peer review information
Nature Communications thanks Eunhee Choi and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kot, E.F., Goncharuk, S.A., Franco, M.L. et al. Structural basis for the transmembrane signaling and antidepressant-induced activation of the receptor tyrosine kinase TrkB. Nat Commun 15, 9316 (2024). https://doi.org/10.1038/s41467-024-53710-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53710-7
This article is cited by
-
The therapeutic potential of psilocybin beyond psychedelia through shared mechanisms with ketamine
Molecular Psychiatry (2025)
