
Abstract
The cytokine TNF can trigger highly proinflammatory RIPK1-dependent cell death. Here, we show that the two adapter proteins, TANK and AZI2, suppress TNF-induced cell death by regulating the activation of TBK1 kinase. Mice lacking either TANK or AZI2 do not show an overt phenotype. Conversely, animals deficient in both adapters are born in a sub-Mendelian ratio and suffer from severe multi-organ inflammation, excessive antibody production, male sterility, and early mortality, which can be rescued by TNFR1 deficiency and significantly improved by expressing a kinase-dead form of RIPK1. Mechanistically, TANK and AZI2 both recruit TBK1 to the TNF receptor signaling complex, but with distinct kinetics due to interaction with different complex components. While TANK binds directly to the adapter NEMO, AZI2 is recruited later via deubiquitinase A20. In summary, our data show that TANK and AZI2 cooperatively sustain TBK1 activity during different stages of TNF receptor assembly to protect against autoinflammation.
Similar content being viewed by others


Introduction
Tumor necrosis factor (TNF) is a proinflammatory cytokine with a crucial role in mounting immune defenses. However, it is also involved in the progression of various autoimmune diseases, including rheumatoid arthritis, inflammatory bowel disease, or psoriasis. The inflammatory effects of TNF are mediated primarily by the ubiquitously expressed TNF receptor 1 (TNFR1). Stimulation of this receptor activates diverse signaling pathways, including nuclear factor-κB (NF-κB) or mitogen-activated protein kinases (MAPK), leading to the transcription of genes coding proinflammatory and anti-apoptotic proteins. However, TNFR1 stimulation can also directly trigger cell death mediated by receptor-interacting protein kinase 1 (RIPK1)1. Once recruited to the membrane-bound TNF receptor signaling complex (TNF-RSC), termed complex I, RIPK1 autophosphorylates itself and translocates to the cytoplasm, forming cell death-inducing complex II2,3. The interaction of RIPK1 with FADD and Caspase-8 triggers apoptosis. Alternatively, RIPK1 can bind and activate kinase RIPK3, which phosphorylates MLKL, leading to its oligomerization and pore formation in the plasma membrane and subsequent necroptosis4,5,6,7,8. In order to prevent complex II formation, RIPK1 is extensively modified by polyubiquitination and phosphorylation9,10,11,12,13,14.
Upon assembly of TNF-RSC, the adapter TRADD recruits non-degradative ubiquitin ligases TRAF2-cIAP1-cIAP2, generating K63-ubiquitin linkages, which in turn recruit linear ubiquitin chain assembly complex (LUBAC) that creates linear (M1) ubiquitin linkages15,16. The adapters TAB2/3 recruit kinase TAK1 to K63-ubiquitin chains, while the adapter NF-κB essential modulator (NEMO) enables the recruitment of IκB kinase α (IKKα) and IKKβ to M1 polyubiquitin linkages, leading to the activation of MAPK and NF-κB signaling pathways17,18. At the same time, activation of signaling responses is tightly regulated by deubiquitinases CYLD and A2019. Interestingly, NEMO also promotes the recruitment of TANK binding kinase 1 (TBK1), which has a dual function within TNF-RSC. On the one hand, TBK1 partially inhibits the activation of NF-κB and MAPK signaling pathways20,21. On the other, it phosphorylates and inhibits RIPK1 activity to prevent TNF-induced cell death20,22. Since the TBK1-mediated phosphorylation of RIPK1 functions as a separate cell death checkpoint from NF-κB-induced transcription of anti-apoptotic genes23, even enhanced activation of the NF-κB pathway cannot prevent TNF-induced cell death in TBK1-deficient cells20.
Tbk1−/− mice derived from the C57BL/6 strain die during embryonic development due to liver degeneration. This phenotype is rescued by ablation of TNFR1 or inactivation of RIPK1 kinase activity, indicating that it is driven by aberrant TNF-induced cell death20,24. Similarly, mice harboring enzymatically inactive TBK1 suffer from embryonic lethality caused by increased RIPK1-induced cell death25. Tbk1−/− mice on the 129S5 background are viable, but they are born at decreased Mendelian frequency and suffer from autoinflammation and immune infiltrates in multiple organs26. Human patients lacking TBK1 developed a severe autoimmune disease that could be treated by blocking TNF27. These findings collectively demonstrate the critical role of TBK1 in protecting against TNF-induced inflammation and cell death. TBK1 is also a potential target for cancer treatment, as its inhibition significantly potentiates anti-PD-L1 immunotherapy due to the enhanced apoptosis and necroptosis of tumor cells triggered by TNF and IFNγ28.
TBK1 is a pleiotropic kinase that modulates various processes, including metabolism, autophagy, interferon signaling, and cell death29. The broad range of TBK1 functions stems from its ability to interact with different adapters that connect it with distinct signaling complexes30,31. Furthermore, TBK1 can directly bind to immune receptors, such as the stimulator of interferon genes (STING) that detects cytosolic DNA derived from intracellular pathogens32. In the context of the TNF-RSC, TBK1 was proposed to be recruited via adapters TRAF family member associated NF-κB activator (TANK) and 5-azacytidine induced 2 (AZI2, also known as NAP1)22. However, the role of these two adapters in protecting against TNF-induced cell death has been enigmatic. Tank−/− mice are born at the expected Mendelian ratio and appear normal, although they develop glomerulonephritis over time, which is mediated by aberrant MyD88-signaling33. Azi2−/− mice show no overt phenotype34.
Here, we analyze whether TANK and AZI2 are partially redundant in protecting against TNF-driven autoinflammation. We show that combined deletion of TANK and AZI2 strongly suppresses TNF-induced activation of TBK1. This is associated with a modest increase in NF-κB signaling and a significant enhancement of TNF-induced cell death in TANK/AZI2-deficient cells. Consistently, mice lacking both proteins develop severe TNFR1-driven autoinflammatory illness. Altogether, TANK and AZI2 are crucial components of the TNF-RSC, protecting against aberrant TNF-induced signaling and cell death.
Results
TANK and AZI2 protect against TNF-induced cell death
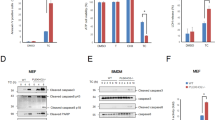
To investigate whether adapters TANK and AZI2 contribute to TNF-induced activation of TBK1, we generated murine stromal ST2 cell lines lacking TANK, AZI2, or both proteins using CRISPR/Cas9. We stimulated these cells with Strep-Flag tagged TNF (SF-TNF) for 15 minutes and isolated the TNF-RSC through Flag immunoprecipitation from cellular lysates. Compared to wild-type (WT) cells, the recruitment and phosphorylation of TBK1 within the TNF-RSC were reduced in Tank knockout (KO) and abolished in Tank/Azi2 double KO (DKO) cells (Fig. 1a, b).

a, b Immunoblot analysis of TNF-RSC isolated from ST2 WT, TankKO, Azi2KO, or Tank/Azi2DKO cell lines. Cells were stimulated with SF-TNF (500 ng/ml) as indicated, and lysates were subjected to anti-Flag immunoprecipitation. As a control, SF-TNF was added post-lysis to unstimulated samples. A representative experiment (a) and quantification of recruitment of indicated proteins to TNF-RSC from three independent experiments normalized to WT cells, mean + SEM (b). c, d Immunoblot analysis of lysates of ST2 WT, TankKO, Azi2KO, or Tank/Azi2DKO cells stimulated with TNF (100 ng/ml) as indicated. A representative experiment (c) and quantification of three independent experiments normalized to the maximal response of WT cells, mean ± SEM (d). Statistical analysis was based on the area under the curve (AUC) for particular cell lines in each experiment, one-way ANOVA (p-value shown) with Tukey´s post-tests. e, f Immunoblot analysis of lysates from ST2 WT or Tank/Azi2DKO cells that were unstimulated or stimulated for 24 h with TNF (250 ng/ml) and zVAD (20 µM) as indicated and subjected to RIPK1 immunoprecipitation. A representative experiment (e) and quantification of phosphorylated RIPK1 and cleaved Caspase-8 from three independent experiments normalized to WT cells stimulated with TNF alone, mean + SEM (f). g ST2 WT and Tank/Azi2DKO cells were unstimulated or stimulated with TNF (100 ng/ml), and the induction of cell death was monitored every 2 h using the Incucyte imaging system. The mean ± SEM from six separate experiments using two different Tank/Azi2DKO ST2 clones is shown. Statistical analysis was based on the AUC for different cell lines and treatments in each experiment, one-way ANOVA (p-value shown) with Tukey´s post-tests. h ST2 WT and Tank/Azi2DKO cells were unstimulated or stimulated with TNF (100 ng/ml) alone or in the presence of zVAD (20 µM), Nec-1s (20 µM), or both inhibitors. Induction of cell death after 48 h was measured using the Incucyte imaging system. The mean + SEM from six independent experiments using two different Tank/Azi2DKO ST2 clones is shown, two-way ANOVA with Tukey’s post-tests. n.s., not significant. #, nonspecific band.
Subsequently, we analyzed the role of the two adapters in TNF-induced activation of signaling pathways. We observed that TBK1 activating phosphorylation at serine 17235,36 was reduced in TankKO cells compared to wild-type (WT) or Azi2KO cells and strongly suppressed in Tank/Azi2DKO cells (Fig. 1c, d). In contrast, the induction of the NF-κB pathway, detected as activating phosphorylation of IKKα/β, was slightly but very reproducibly increased in cells lacking both adapters (Fig. 1c, d). Similarly, a detailed time-course analysis of TNF-induced signaling revealed that while TBK1 phosphorylation was suppressed, IKKα/β activating phosphorylation was both enhanced and prolonged in Tank/Azi2DKO cells compared to WT cells, which also correlated with prolonged phosphorylation of its downstream target p105 (Supplementary Fig. 1a, b). Activated IKKα/β phosphorylates IκBα, which is rapidly degraded37. Indeed, IκBα was barely detectable 15 minutes after TNF stimulation in both WT and Tank/Azi2DKO cells (Fig. 1c, d and Supplementary Fig. 1a, b). However, Tank/Azi2DKO cells had a markedly prolonged phase of IκBα phosphorylation and degradation detected at 30 minutes upon stimulation (Fig. 1c, d and Supplementary Fig. 1a, c), which correlated with enhanced and prolonged phosphorylation of IKKα/β. Altogether, TANK and AZI2 adapters are required for TBK1 activating phosphorylation, while they partially suppress IKKα/β activation and the induction of NF-κB pathway. These results align with previous studies showing enhanced TNF-induced activation of IKKα/β in TBK1-deficient cells20,21.
TBK1 is not required to prevent spontaneous activation of RIPK1 and induction of cell death in unstimulated cells. However, it inhibits the autophosphorylation of RIPK1 on Serine 166 and subsequent proteolytic activation of Caspase-8 upon TNF stimulation20,22. TNF-induced Caspase-8 cleavage was enhanced in Tank/Azi2DKO cells compared to WT cells (Fig. 1e, f). When cells were treated with TNF in the presence of the caspase inhibitor zVAD, which stabilizes complex II formation, we observed robust RIPK1 autophosphorylation in Tank/Azi2DKO cells (Fig. 1e, f). In accord, real-time cell imaging revealed that Tank/Azi2DKO cells were susceptible to TNF-induced cell death, which was partially inhibited by zVAD or the RIPK1 inhibitor Necrostatin-1s (Nec-1s) and rescued by simultaneous treatment with both inhibitors (Fig. 1g, h and Supplementary Fig. 1d). Similarly, the detection of dead cells by propidium iodide (PI) uptake via flow cytometry revealed that Tank/Azi2DKO cells are sensitive to TNF induced cell death (Supplementary Fig. 1e). This method detected even higher proportion of TNF-induced cell death in Tank/Azi2DKO cells, which likely reflects differences in sample preparation.
Regulation of RIPK1 activity and NF-κB-induced transcription of anti-apoptotic genes are two separate cell death checkpoints. Blocking NF-κB activation with IKKα/β inhibitor TPCA-1 further potentiated TNF-induced cell death in Tank/Azi2DKO ST2 cells (Supplementary Fig. 1f), indicating that IKKα/β-mediated cell death checkpoint is operating in Tank/Azi2DKO cells. We also observed strongly suppressed phosphorylation of TBK1, slightly enhanced phosphorylation of IKKα/β, and increased cell death upon TNF stimulation in human HeLa TANK/AZI2DKO cells (Supplementary Fig. 1g–j). Combined, these data suggest that TANK and AZI2 regulate TNF-induced signaling and cell death by enabling TBK1 recruitment to TNF-RSC.
TANK and AZI2 protect against autoinflammatory disease in mice
To investigate the potential redundant function of the adapters TANK and AZI2 in vivo, we generated Tank−/− and Azi2−/− mice using the CRISPR/Cas9 approach (Fig. 2a, b). As expected, Tank−/− and Azi2−/− mice were born in normal ratios and were indistinguishable from their WT littermates (Fig. 2c, d). In contrast, upon the breeding of Azi2−/−/Tank+/− mice, we observed that the Tank/Azi2DKO animals were born at a markedly sub-Mendelian ratio, indicating partial embryonic lethality (Fig. 2c). Tank/Azi2DKO pups exhibited delayed hair growth, and by four weeks of age, they completely lacked body hair except for the head and tail base regions (Fig. 2d). Their fur eventually grew, but the mice developed severe dermatitis within a few months (Fig. 2e). The median survival of Tank/Azi2DKO animals was only 25 weeks (Fig. 2f). These findings were confirmed in animal strains derived from independent founders harboring different mutations in Tank and Azi2 alleles. In this case, we crossed Tank−/−/Azi2+/− mice to obtain Tank/Azi2DKO animals, which were again born at a low ratio and displayed delayed hair growth and short lifespan (Supplementary Fig. 2a–e).

a, b Translations of DNA sequences (a) and an example of routine PCR genotyping (b) of the indicated Tank and Azi2 alleles. c Genotype and counts of pups born to parents of the indicated genotypes, two-tailed chi-square test. d, e Representative photographs of mice of the indicated genotype at the age of 4 weeks (d) and 20 weeks (e). f Survival curves of mice of the indicated genotype (n = 15 per group), log-rank Mantel-Cox test. g–i Tissue sections stained with H&E or the indicated dye. Skin (g), kidney (h), or liver (i) samples were collected from 20-24-week-old mice of the indicated genotype. Representative images are based on the analysis of five mice per group. j–n The concentration of different biochemical markers in the blood plasma collected from 20-24-week-old mice of the indicated genotype (n = 6 per group), mean + SEM, one-way ANOVA (p-value is shown) with Tukey’s post-tests. n.s., not significant. The scale bar is 100 µm.
To elucidate the likely cause of death of Tank/Azi2DKO animals, we analyzed organs isolated from 20-24-week-old mice by histology. No apparent differences were observed between WT, Tank−/− and Azi2−/− mice at this age. In contrast, histological examination of Tank/Azi2DKO mice tissue samples revealed a severe autoinflammatory disease affecting multiple organs. Hematoxylin and eosin (H&E) staining of skin sections from Tank/Azi2DKO mice revealed extensive immune cell infiltrations. The subdermal fat was completely missing, which was also demonstrated by staining with picrosirius red that binds to collagen fibers (Fig. 2g and Supplementary Fig. 2f). Kidney sections stained with H&E displayed glomerulonephritis characterized by swollen glomeruli, and Congo red staining identified massive amyloid deposits in the kidneys (Fig. 2h and Supplementary Fig. 2g). H&E staining of liver sections revealed numerous granulomas and picrosirius red staining showed collagen-filled scars and bile duct fibrosis (Fig. 2i and Supplementary Fig. 2h). The tissue damage in the skin, kidney, and liver in Tank/Azi2DKO animals was accompanied by enhanced apoptosis detected by TUNEL staining (Supplementary Fig. 2i). Additionally, we observed massive immune infiltrations in the pancreas, skeletal muscles, and white adipose tissues (Supplementary Fig. 2j).
The internal organ damage observed in Tank/Azi2DKO mice was further confirmed by biochemical analysis of blood plasma. Tank/Azi2DKO mice exhibited significantly elevated urea, phosphate, potassium, and magnesium levels, indicating severe kidney damage, which was not observed in WT, Tank−/− or Azi2−/− mice (Fig. 2j). Moreover, we detected substantial increases in serum levels of total bilirubin and aspartate aminotransferase (AST) in Tank/Azi2DKO mice compared to WT animals or mice lacking either adapter alone, indicating liver damage (Fig. 2k). In terms of lipid metabolism, Tank/Azi2DKO mice exhibited a strongly decreased level of plasma cholesterol concentration, while the concentration of triglycerides remained unchanged in Tank/Azi2DKO mice compared to WT animals (Supplementary Fig. 2k). High-density lipoprotein (HDL) concentration was lowered in Tank−/−, or Azi2−/− animals and further significantly decreased in Tank/Azi2DKO mice as compared to WT mice (Fig. 2l). In contrast, the plasma concentration of low-density lipoprotein (LDL), which contributes to cholesterol build-up in arteries, was substantially elevated in Tank/Azi2DKO mice. This was reflected by a very high ratio of LDL to HDL in Tank/Azi2DKO mice (Fig. 2l), indicating an increased risk for cardiovascular failure.
Plasma albumin levels were decreased in Tank/Azi2DKO mice (Fig. 2m), which is consistent with ongoing autoinflammatory disease and liver malfunction. These animals exhibited increased concentrations of total plasma proteins, suggesting an elevation in serum globulins and potentially an imbalance of the immune system and antibody production (Fig. 2n). In conclusion, the shortened lifespan of Tank/Azi2DKO animals seems to be caused by multiorgan inflammation. These findings provide strong evidence that TANK and AZI2 can compensate for each other in vivo to protect against severe autoinflammatory disease.
Tank/Azi2 DKO mice have dysregulated antibody production
The increased concentration of globulins in Tank/Azi2DKO mice prompted us to analyze the concentration of different antibody isotypes in plasma. Azi2−/− mice showed similar levels of plasma antibodies as WT animals. In contrast, Tank−/− mice displayed increased concentrations of all antibody isotypes, as reported previously33. Tank/Azi2DKO mice had similar plasma concentrations of IgM, IgG1, IgG2a, IgG2b, and IgE antibodies to Tank−/− animals, while IgA antibody levels were substantially increased (Fig. 3a).

a Titers of different antibody isotypes in the blood plasma from 20-24-week-old mice of the indicated genotype (n = 5 for WT and Azi2−/−, 6 for Tank−/− and Tank/Azi2DKO), mean + SEM, one-way ANOVA (p-value is indicated) with Tukey’s post-tests. b Representative photograph and weight of spleens isolated from 8-10-week-old WT and Tank/Azi2DKO mice (n = 4 per group), mean + SEM, unpaired two-tailed t-test. c H&E staining of spleen tissue sections from 20-24-week-old mice of the indicated genotype. Representative images are based on analyzing samples from five mice per group. d, e Flow cytometry measurement of cellularity and viability of splenocytes from 8-12-week-old mice of the indicated genotype (d), followed by the analysis of splenic myeloid cells (CD3−, CD19−, NK1.1−, CD11b+) separated into macrophages (CD11c−, Ly6G−), neutrophils (CD11c−, Ly6G+), and dendritic cells (DC) (CD11c+) (e) (n = 8 per group), mean + SEM, one-way ANOVA (p-value is indicated) with Tukey’s post-tests. f–i Flow cytometry analysis of B cells from the spleen (f–g) and lymph nodes (h, i) from 8-12-week-old mice (n = 8 per group). The proportion of B cells (CD19+) (f, h) and the percentage of isotype-switched B cells (IgM−, IgD−) in organs isolated from the indicated mouse strains, mean + SEM, one-way ANOVA (p-value is indicated) with Tukey’s post-tests (g, i). n.s., not significant. The scale bar is 100 µm.
Spleens from Tank/Azi2DKO mice were enlarged, especially when normalized to body weight (Fig. 3b). Their histological analysis by H&E staining revealed disrupted splenic architecture with poorly distinguishable boundaries between white and red pulp (Fig. 3c). Flow cytometry analysis of Tank/Azi2DKO spleen demonstrated increased cell counts with normal splenocytes viability (Fig. 3d). We detected massive splenic infiltration of macrophages and neutrophils, while the proportion of dendritic cells (DC) was decreased (Fig. 3e). The ratio of splenic B cells was similar in WT, Tank−/−, Azi2−/−, and Tank/Azi2DKO animals (Fig. 3f). Upon activation, mature IgD+ IgMLow B cells undergo isotype switching to produce antibodies of different isotypes. Tank/Azi2DKO mice exhibited a substantial increase in the proportion of isotype-switched B cells, characterized as CD19+ IgM− IgD−, which corresponds to the high concentration of antibodies observed in their plasma (Fig. 3g). Similarly, a highly increased percentage of isotype-switched B cells was observed in the lymph nodes (Fig. 3h, i).
To elucidate whether the increased proportion of isotype-switched B cells is driven by ongoing autoinflammatory disease or is a cell-intrinsic process, we performed competitive bone-marrow transfer. Bone marrow cells from WT and Tank/Azi2DKO mice were mixed in a 1:1 ratio and transplanted into lethally irradiated mice. The emergence of immune cells derived from Tank/Azi2DKO progenitors was significantly suppressed compared to cells derived from WT progenitors (Supplementary Fig. 3a, b). However, the percentage of isotype-switched B cells was substantially enhanced in Tank/Azi2DKO compared to WT B cells in both spleen and lymph nodes (Supplementary Fig. 3c–f). Altogether, our results indicate that TANK and AZI2 function as crucial but partially redundant regulators of hematopoiesis, isotype switching, and antibody production by B cells.
Enhanced signaling and cell death in cells isolated from Tank/Azi2 DKO mice
Mouse embryonic fibroblasts (MEFs) isolated from Tank−/− or Azi2−/− animals had decreased phosphorylation of TBK1, which was severely impaired in Tank/Azi2DKO compared to WT cells (Supplementary Fig. 4a, b). Similarly to Tbk1−/− MEFs20, Tank/Azi2DKO MEFs had slightly increased activation of IKKα/β, JNK, and p38 MAPK signaling pathways (Supplementary Fig. 4c, d). Moreover, the recruitment and phosphorylation of TBK1 within TNF-RSC were strongly reduced in Tank/Azi2DKO MEFs compared to WT or Tank−/− MEFs, and this correlated with enhanced NEMO recruitment (Supplementary Fig. 4e, f).
TNF stimulation of Tank/Azi2DKO bone marrow-derived macrophages (BMDMs) led to strongly suppressed phosphorylation of TBK1, which was accompanied by slightly enhanced activation of NF-κB and JNK MAPK signaling pathways (Fig. 4a, b). The stimulation of BMDMs with TNF triggers the expression of TNF-induced genes that can be identified by RNA sequencing (RNAseq) (Supplementary Fig. 4g and Supplementary Dataset 1). Tank/Azi2DKO BMDMs had enhanced basal expression of TNF-responsive genes, including transcription of anti-apoptotic genes Traf1, Cflar, Birc2, and Birc338,39, which likely reflects the increased basal expression of TNF in these cells. The transcription of TNF-induced genes was further enhanced in Tank/Azi2DKO compared to WT BMDMs upon TNF stimulation (Fig. 4c, d and Supplementary Fig. 4h). In contrast, we did not observe increased transcription of anti-apoptotic Bcl2 family members after two hours of TNF stimulation (Supplementary Fig. 4h).

a, b Immunoblot analysis of lysates of bone marrow-derived macrophages (BMDMs) isolated from WT or Tank/Azi2DKO mice and stimulated with TNF (100 ng/ml) as indicated. A representative experiment (a) and quantification of three independent experiments using BMDMs independently isolated from 3 animal pairs. Data were normalized to the maximal response of WT cells, mean ± SEM (b). Statistical analysis was based on the AUC for each cell line, unpaired two-tailed t-test. c, d RNAseq analysis of WT and Tank/Azi2DKO BMDMs stimulated for two hours with TNF (100 ng/ml). The BMDMs were independently isolated from 3 pairs of animals, and RNA was isolated in five separate experiments. Heatmap shows the expression of selected TNF-responsive genes (c). Gene set enrichment analysis (GSEA) shows the enrichment of 105 genes induced by TNF stimulation (log2 fold change > 1 and padj < 0.01; samples from WT and Tank/Azi2DKO BMDMs were analyzed together). The ranked gene list represents the contrast between unstimulated Tank/Azi2DKO and WT BMDMs (blue) or stimulated Tank/Azi2DKO and WT BMDMs (red). The p-value was estimated using an adaptive multi-level split Monte-Carlo scheme in the fgsea R package (d). e Analysis of cell death induced upon TNF (100 ng/ml) stimulation of WT or Tank/Azi2DKO BMDMs. Induction of cell death was detected via the Incucyte imaging system. Mean + SEM from eight separate experiments using samples isolated from 4 animal pairs, two-way ANOVA with Tukey’s post-tests. n.s., not significant. #, nonspecific band.
Tank/Azi2DKO BMDMs had increased basal level of cell death, and TNF stimulation of these cells showed enhanced induction of cell death in comparison to WT cells (Fig. 4e). Altogether, these data demonstrate that increased activation of the NF-κB pathway and induction of anti-apoptotic genes in Tank/Azi2DKO cells is not sufficient to prevent cell death caused by a block in TBK1 recruitment to TNF-RSC, which is required to control the activation of RIPK1.
TNFR1 is the primary driver of autoinflammation in Tank/Azi2 DKO mice
Since TBK1 activation is necessary to protect against TNF-induced lethality in vivo22, we aimed to investigate the role of TANK and AZI2 in safeguarding against a sublethal dose of TNF administered via tail-vein injection. While WT, Tank−/−, and Azi2−/− mice tolerated the injection well, Tank/Azi2DKO animals rapidly succumbed to the treatment (Supplementary Fig. 5a). Intriguingly, the analysis of plasma of 20-24-week-old Tank/Azi2DKO mice, which suffer from autoinflammation, revealed significantly elevated plasma levels of proinflammatory cytokines TNF, IFNγ, IL-1β, and IL-17, compared to WT mice (Supplementary Fig. 5b). The increased susceptibility of Tank/Azi2DKO mice to TNF-induced lethality, coupled with the increased levels of plasma TNF, prompted us to explore whether the autoinflammatory disease observed in Tank/Azi2DKO mice is driven by aberrant TNFR1 signaling.
We crossed Tank/Azi2DKO animals with a mouse strain deficient in TNFR1 (Tnfrsf1a−/−) to obtain Tank/Azi2/Tnfrsf1a triple KO (TKO) mice. These animals were born at a nearly Mendelian ratio, indicating a substantial rescue of the embryonic lethality phenotype (Fig. 5a). Tank/Azi2/Tnfrsf1aTKO mice did not experience the delayed hair growth observed at four weeks of age in Tank/Azi2DKO animals (Fig. 5b) and did not develop dermatitis by 30 weeks of age (Fig. 5c). Importantly, the survival of Tank/Azi2/Tnfrsf1aTKO mice was significantly improved compared to Tank/Azi2DKO mice, indicating a substantial rescue of the phenotype (Fig. 5d).

a Genotype and counts of pups born to Tank+/−/Azi2−/−/Tnfrsf1a−/− parents, two-tailed chi-square test. (b, c) Representative photographs of mice of the indicated genotype at the age of 4 weeks (b) and 30 weeks (c). d Survival curves of mice of the indicated genotype (n = 15 per group). The comparison with Tank/Azi2DKO mice (based on Fig. 2) is shown in gray, log-rank Mantel-Cox test. e–h Tissue sections stained with H&E or the indicated dye. Skin (e), kidney (f), liver (g), or spleen (h) samples were collected from 20-24-week-old mice of the indicated genotype. Representative images are based on the analysis of five mice per group. i–m The concentration of different biochemical markers in the blood plasma isolated from 20-24-week-old Azi2/Tnfrsf1a DKO or Tank/Azi2/Tnfrsf1a TKO mice (n = 6 per group). The comparison with Tank/Azi2DKO mice (based on Fig. 2) is shown in gray, mean + SEM, one-way ANOVA (p-value is indicated) with Tukey’s post-tests. n, o Flow cytometry analysis of the proportion of isotype-switched B cells (CD19+, IgM−, IgD−) in the spleen (n) and lymph nodes (o) isolated from 8-12-week-old mice of the indicated genotype (n = 8 per group). The comparison with Tank/Azi2DKO mice (based on Fig. 3) is shown in gray, mean + SEM, one-way ANOVA (p-value is indicated) with Tukey’s post-tests. n.s., not significant. The scale bar is 100 µm.
Histological analysis of organs isolated from five months old Tank/Azi2/Tnfrsf1aTKO mice demonstrated that they did not suffer from autoinflammation and appeared similar to control Azi2/Tnfrsf1aDKO animals. Notably, Tank/Azi2/Tnfrsf1aTKO mice did not exhibit aberrant immune cell infiltration in the skin and had normal subdermal fat deposition (Fig. 5e and Supplementary Fig. 5c). Furthermore, they did not show signs of glomerulonephritis or amyloid deposits in the kidneys, and the liver was devoid of granulomas and collagenous scars (Fig. 5f, g and Supplementary Fig. 5d, e). Additionally, no immune infiltrations were detected in the pancreas, skeletal muscle, or white adipose tissue samples isolated from these animals (Supplementary Fig. 5f). Finally, Tank/Azi2/Tnfrsf1aTKO mice displayed standard splenic architecture (Fig. 5h).
Further evidence supporting the rescue of Tank/Azi2DKO mice by TNFR1 ablation was obtained through plasma biochemical analysis. Normal plasma urea, phosphate, potassium, and magnesium levels indicated restored kidney function in Tank/Azi2/Tnfrsf1aTKO animals (Fig. 5i). Similarly, total plasma bilirubin and AST were at normal levels in these mice, indicating rescued liver function (Fig. 5j). Regarding lipid metabolism, the Tank/Azi2/Tnfrsf1aTKO mice exhibited comparable total cholesterol levels as control mice (Supplementary Fig. 5g). The concentration of HDL, LDL, and the ratio between LDL and HDL were rescued in Tank/Azi/Tnfrsf1aTKO mice (Fig. 5k). Finally, we observed no significant decrease in plasma albumin or increase in total protein concentration in Tank/Azi2/Tnfrsf1aTKO mice compared to control animals, indicating no significant increase in plasma globulins (Fig. 5l, m). Tank/Azi2/Tnfrsf1aTKO mice did not show significantly increased number of splenocytes compared to control animals (Supplementary Fig. 5h) and had a normal proportion of B cells in spleen, albeit it was mildly increased in the lymph nodes (Supplementary Fig. 5i). Importantly, Tank/Azi2/Tnfrsf1aTKO did not have significantly elevated proportion of isotype-switched B cells (Fig. 5n, o). These findings align with a previous report showing impaired isotype switching in B cells from Tnfrsf1a−/− mice40.
Interestingly, while Tank/Azi2DKO males could not produce offspring, Tank/Azi2/Tnfrsf1aTKO males were fertile (Supplementary Fig. 5j). Histological analysis of the testes revealed that seminiferous tubules of the Tank/Azi2DKO mice harbored spermatozoa (Supplementary Fig. 5k). However, their testes weight was decreased, and sperm count, sperm motility, and progressive sperm motility were substantially reduced compared to control Azi2−/− and Tank/Azi2/Tnfrsf1aTKO mice (Supplementary Fig. 5l, m). These results show that the autoinflammatory disease and male sterility observed in Tank/Azi2DKO mice are primarily driven by TNFR1 signaling.
RIPK1 activity partially mediates the phenotype of Tank/Azi2 DKO mice
Given the crucial role of RIPK1 as a mediator of TNF-induced cell death41, we aimed to investigate whether the expression of the kinase-dead form of RIPK1 (RIPK1KD) with the inactivating mutation D138A could rescue Tank/Azi2DKO mice. Introducing the inactivating mutation in only one RIPK1 allele in Tank/Azi2DKO/Ripk1KD/+ mice largely prevented embryonic lethality and rescued hair growth (Fig. 6a, b). Furthermore, male fertility was restored, and these mice were protected against the intravenous injection of a sublethal dose of TNF (Supplementary Fig. 6a, b). The median survival of these animals was limited to nine months, representing a significant improvement compared to Tank/Azi2DKO mice, but not reaching the level observed in Tank/Azi2/Tnfrsf1aTKO animals (Fig. 6c). Similarly, Tank/Azi2DKO/Ripk1KD/KD mice, in which both RIPK1 alleles contained kinase-dead mutation, were born at a Mendelian ratio and appeared grossly normal even at nine months of age (Supplementary Fig. 6c, d), but their median survival was not further improved (Fig. 6c).

a Genotype and counts of pups born to parents of the indicated genotype, two-tailed chi-square test. b Representative photographs of 4-week-old mice of the indicated genotype. c Survival curves of mice of the indicated genotype (n = 10 for Azi2−/− and 15 for other groups). The comparison with Tank/Azi2DKO mice (based on Fig. 2) is shown in gray, log-rank Mantel-Cox test. d–g Tissue sections stained with H&E or the indicated dye. Skin (d), kidney (e), liver (f), or spleen (g) samples were collected from 20-24-week-old mice of the indicated genotype. Representative images are based on the analysis of four mice per group. h–l The concentration of different biochemical markers in the blood plasma isolated from 20-24-week-old Azi2−/−/Ripk1KD/KD (n = 5) or Tank/Azi2DKO/Ripk1KD/KD mice (n = 6). The comparison with Tank/Azi2DKO mice (based on Fig. 2) is shown in gray, mean + SEM, one-way ANOVA (p-value is indicated) with Tukey’s post-tests. m, n Flow cytometry analysis of the proportion of isotype-switched B cells (CD19+, IgM−, IgD−) in the spleen (m) and lymph nodes (n) isolated from 8-12-week-old mice of the indicated genotype (n = 6 per group). The comparison with Tank/Azi2DKO mice (based on Fig. 3) is shown in gray, mean + SEM, one-way ANOVA (p-value is indicated) with Tukey’s post-tests. n.s., not significant. The scale bar is 100 µm.
Histological analysis from five months old Tank/Azi2DKO/Ripk1KD/KD mice revealed that the skin inflammation was fully rescued by ablation of RIPK1 kinase activity, and we observed typical deposition of subdermal fat that was not distinguishable from control Azi2−/−/Ripk1KD/KD animals (Fig. 6d and Supplementary Fig. 6e). Staining of the kidney sections did not show signs of glomerulonephritis or amyloid deposits (Fig. 6e and Supplementary Fig. 6f). Similarly, no immune cell infiltrations were detected in samples from the pancreas, skeletal muscle, or white adipose tissue (Supplementary Fig. 6g). However, to our surprise, we noticed a strong formation of granulomas in the liver of Tank/Azi2DKO/Ripk1KD/KD mice, accompanied by the formation of fibrotic scars (Fig. 6f and Supplementary Fig. 6h). Furthermore, we detected disrupted splenic architecture in these mice (Fig. 6g).
Biochemical analysis of the plasma from 20-24-week-old Tank/Azi2DKO/Ripk1KD/KD mice revealed a slight but not significant increase in urea concentration. At the same time, the ionic balance was not changed compared to control Azi2−/−/Ripk1KD/KD animals, suggesting that kidney function was not significantly impacted (Fig. 6h). Markers of liver damage, total bilirubin, and AST were increased in Tank/Azi2DKO/Ripk1KD/KD mice as compared to control animals, although we noticed a substantial variation between animals, and the differences in AST did not reach statistical significance (Fig. 6i). Interestingly, Tank/Azi2DKO/Ripk1KD/KD mice exhibited impaired lipid metabolism, characterized by decreased levels of plasma cholesterol, HDL, and LDL (Fig. 6j and Supplementary Fig. 6i), which accompanies liver disease in human patients and mouse models42,43.
We observed a slight but not statistically significant decrease in albumin and an increase in total plasma protein concentration in Tank/Azi2DKO/Ripk1KD/KD mice, which indicated only a small rise in globulin production (Fig. 6k, l). The number of splenocytes and their viability was normal in Tank/Azi2DKO/Ripk1KD/KD mice (Supplementary Fig. 6j) and the percentage of B cells in the lymph nodes and in the spleen was not significantly different from control animals (Supplementary Fig. 6k). Interestingly, a high proportion of B cells in the spleen and lymph nodes of Tank/Azi2DKO/Ripk1KD/KD mice exhibited an isotype-switched phenotype compared to control animals, although not to the same extent as in Tank/Azi2DKO mice (Fig. 6m, n). Overall, our data indicate that inactivation of RIPK1 in Tank/Azi2DKO/Ripk1KD/KD mice rescues most of the autoinflammatory phenotype observed in Tank/Azi2DKO animals. However, these mice still develop liver inflammation, accompanied by increased activation of B cells. Therefore, these observations suggest that the TNFR1-driven autoinflammation observed in Tank/Azi2DKO mice is primarily, but not entirely, driven by RIPK1 kinase activity.
TANK and AZI2 are differentially recruited to TNF-RSC
Our results demonstrate that TANK and AZI2 have overlapping roles in recruiting TBK1 to TNF-RSC and protecting against TNF-induced cell death. However, although both TANK and AZI2 bind TBK1, they lack substantial sequential or structural homology44, suggesting that they might have distinct functions in regulating TBK1 recruitment to TNF-RSC. To reveal the differences between these two adapters, we analyzed the kinetics of their association with TNF-RSC. We stimulated ST2 cells with SF-TNF for various durations and subsequently isolated TNF-RSC via Flag immunoprecipitation. TANK was strongly recruited at 15 minutes and was dissociated from the complex 30 minutes after TNF stimulation (Fig. 7a, b). The kinetics of TANK recruitment closely mirrored the recruitment of the adapter NEMO, which supports previous reports that these two proteins directly interact45,46. In contrast, AZI2 recruitment to TNF-RSC peaked only after 30 minutes of stimulation, and AZI2 subsequently remained associated with the complex. Interestingly, the recruitment kinetics of AZI2 were similar to those of the deubiquitinase A20 (Fig. 7a, b), which can remain associated with the complex for several hours due to the strong binding of A20 to M1-polyubiquitin chains while cleaving other ubiquitin linkage types19,47,48. The analysis of TNF-RSC isolated from MEFs and human HeLa cell lines indicated similar recruitment kinetics for TANK and NEMO, while AZI2 recruitment was delayed and resembled that of A20 (Supplementary Fig. 7a–d).

a–d Immunoblot analysis of TNF-RSC isolated from WT ST2 cells (a, b) or WT and NEMOKO ST2 cells (c, d). Cells were stimulated with SF-TNF (500 ng/ml) for the indicated time points, and lysates were subjected to anti-Flag immunoprecipitation. As a control, SF-TNF was added post-lysis to unstimulated samples. A representative experiment (a, c) and quantification of recruitment of indicated proteins to TNF-RSC from three independent experiments normalized to WT cells, mean + SEM (b, d). e, f Isolation of SF-TANK or SF-AZI2 via anti-Flag immunoprecipitation from HEK293FT cells transiently overexpressing these proteins with NEMO. A representative experiment (e) and quantification of three experiments normalized to WT cells, mean + SEM (f). g–j Immunoblot analysis of TNF-RSC isolated from ST2 WT or A20KO cells (g, h) or NEMO-deficient HeLa cells reconstituted with NEMOWT or NEMOL227P (i, j), performed as in a-d. Representative experiments (g, i) and quantification of recruitment of indicated proteins to TNF-RSC from three (h) or four (j) independent experiments normalized to the WT cells, mean + SEM. #, nonspecific band.
It was previously reported that NEMO recruits both TANK and AZI2 to TNF-RSC22. We confirmed these findings by showing that NEMO-deficient ST2 cells had a severe defect in the TANK, AZI2, and TBK1 recruitment (Fig. 7c, d). Interestingly, our data revealed that NEMO was also required for A20 recruitment (Fig. 7c, d), consistent with similar observations made in the context of the IL-17 receptor signaling complex49 and Lymphotoxin β (LTβ) receptor signaling complex50. Furthermore, TNF-RSC isolated from the NEMOKO HeLa cell line did not contain TANK, AZI2, or A20 and reintroducing WT NEMO, but not NEMO lacking amino acids (AA) 211-240 required for TANK and AZI2 interaction22,45, restored the recruitment of these proteins (Supplementary Fig. 7e, f). Therefore, NEMO is required to recruit TANK, AZI2, and also A20 to TNF-RSC.
To elucidate whether TANK and AZI2 bind directly to NEMO, we overexpressed NEMO with SF-tagged TANK or AZI2 in HEK293FT cells. Upon cell lysis, we isolated SF-TANK and SF-AZI2 via Flag immunoprecipitation. While both adapters were associated with TBK1, only SF-TANK interacted directly with NEMO and induced TBK1 phosphorylation upon overexpression (Fig. 7e, f). This suggested that AZI2 is recruited to TNF-RSC in a NEMO-dependent manner but not through direct interaction. Since AZI2 and A20 required NEMO for their association with TNF-RSC, and their recruitment kinetics were closely correlated, we aimed to explore whether A20 enables AZI2 recruitment and retention within the complex. Indeed, A20KO ST2 cells failed to recruit AZI2 to the TNF-RSC (Fig. 7g, h), which was rescued by the reconstitution of these cells with A20 (Supplementary Fig. 7g, h). Similarly, AZI2 recruitment was nearly abolished in HeLa cells deficient in A20 (Supplementary Fig. 7i, j).
Gene coding NEMO is located on the X chromosome, and its deficiency in males leads to prenatal lethality. However, specific mutations in this gene are tolerated, but male patients develop ectodermal dysplasia with immunodeficiency (EDA-ID). A previous study reported a patient diagnosed with EDA-ID due to L227P mutation in NEMO51,52, which is localized in the region AA211-240 required for A20, AZI2, and TANK recruitment to TNF-RSC (Supplementary Fig. 7e, f). To test whether this mutation impacts TNF-induced TBK1 phosphorylation, we expressed NEMO WT or NEMO L227P mutant in NEMOKO HeLa cells. Analysis of TNF-RSC revealed that TANK, AZI2, A20, and TBK1 are absent from the complex in cells expressing NEMO L227P (Fig. 7i, j). Interestingly, NEMO L227P association with TNF-RSC was enhanced compared to NEMO WT (Fig. 7i, j), which is in accord with our observation that ablation of TANK and AZI2 promotes NEMO recruitment (Supplementary Fig. 4e, f).
Altogether, our results indicate that during the initial stage of TNF-RSC assembly, TANK directly links TBK1 with NEMO. Subsequently, NEMO facilitates A20 association with M1-ubiquitin linkages within the complex19, which promotes the recruitment of TBK1 through AZI2 (Fig. 8a). While NEMO is released at later time points to terminate the activation of NF-κB signaling, A20 and AZI2 remain associated with the complex, ensuring the presence of TBK1 in the late stages of complex formation (Fig. 8b). In this manner, TBK1 can remain associated with the TNF-RSC to control RIPK1 activation both at the early and late stages of complex assembly.

a, b Schematic representation of the differential recruitment of TANK and AZI2 to TNF-RSC. NEMO is recruited to K63- and M1-ubiquitin linkages formed by cIAP1/2 and LUBAC, respectively. NEMO is associated with TANK-TBK1 and promotes recruitment of A20, which enables recruitment of AZI2-TBK1 (a). At a later time, NEMO and TANK-TBK1 are released from the TNF-RSC. However, A20 remains associated with the TNF-RSC due to M1-binding and keeps recruiting AZI2-TBK1 (b).
Discussion
In the present work, we demonstrate that the proteins TANK and AZI2 play a crucial yet redundant role in recruiting TBK1 to TNF-RSC. Cell lines deficient in TBK1 show slightly augmented activation of NF-κB and MAPK signaling pathways and are sensitive to TNF-induced cell death20,22. We observed similar effects in various TANK and AZI2-deficient cells, which have severe defects in TBK1 activation. Notably, although activation of the NF-κB pathway and production of antiapoptotic proteins is enhanced in Tank/Azi2DKO cells, it is not sufficient to overcome the enhanced formation of cell death-inducing complex II. This is in accord with the notion that activation of NF-κB transcriptional responses and regulation of RIPK1 activity are two separate cell death checkpoints23. Although our data indicate that the major role of these two adapters is to enable the activation of TBK1 upon TNF stimulation, they may have additional functions in regulating immune homeostasis. For example, both adapters were described to interact with the kinase IKKε53,54,55. Ablation of IKKε in mice does not lead to an overt phenotype, suggesting it is not required for protection against cell death56. However, IKKε can partially compensate for TBK1 deficiency in specific settings, including TNF signaling22,25.
The absence of either TANK or AZI2 in mice is well tolerated, indicating that these adapters can compensate for each other to preserve immune homeostasis in unchallenged mice. However, deficiency in both proteins triggers a severe autoinflammatory disease characterized by multiorgan inflammation, including glomerulonephritis, liver granulomas, dermatitis, and disruption of splenic architecture. Additionally, Tank/Azi2DKO animals have delayed hair growth and the absence of subdermal fat, but the precise cause of these features remains unclear. Even though the phenotype of Tank/Azi2DKO mice is very severe, it does not result in complete embryonic lethality, as observed in TBK1 knockout mice on the C57BL/6 background24,56. This fits with our observation that TBK1 phosphorylation was strongly decreased, but not completely blocked, in the cytoplasm of Tank/Azi2DKO cells. TBK1 is activated via autophosphorylation or phosphorylation by IKKα/β21. It is possible that in Tank/Azi2DKO cells, a portion of cytoplasmic TBK1 might be phosphorylated by IKKα/β and partially inhibit TNF-induced cell death.
The deletion of TNFR1 rescued Tank/Azi2DKO animals, while the ablation of RIPK1 kinase activity provided substantial, but not complete, rescue. Tank/Azi2DKO/Ripk1KD/KD animals suffered from liver damage, which was accompanied by an increased proportion of isotype-switched B cells in these mice. Bone marrow transfer experiments demonstrated that Tank/Azi2DKO B cells are intrinsically prone to undergo isotype switching, which is in accordance with a previous study showing that TBK1 is a negative regulator of IgA switching in B cells57. Since B cells isolated from Tank/Azi2/Tnfrsf1aTKO do not show an isotype-switched phenotype, it is likely that this process relies on TNFR1 signaling but does not require RIPK1 activity.
One of our unexpected discoveries was that the absence of TANK and AZI2 leads to male sterility, which was rescued by the ablation of TNFR1 or the inactivation of RIPK1 kinase activity. This is intriguing since it shows that TNF-mediated cell death can be responsible for decreased sperm count and motility, indicating that aberrant TNF signaling might underlie fertility problems more broadly. Interestingly, TNF-induced necroptosis was suggested to promote the aging of the testes in WT mice58. These data open the possibility that some cases of male infertility might be caused by aberrant TNF-induced cell death and could be treated by inhibition of this pathway.
Although there is a significant functional compensation between TANK and AZI2 in vivo, they have different amino acid sequences and structures, suggesting that the existence of these two adapters is not simply due to the duplication of ancestral genes. Instead, they likely have distinct roles in controlling inflammation. Both proteins are recruited via NEMO but markedly differ in their recruitment kinetics. TANK binds directly to NEMO, which is associated with TNF-RSC during the initial stage of complex assembly. In contrast, AZI2 does not interact with NEMO but requires deubiquitinase A20, which is recruited to TNF-RSC in a NEMO-dependent manner. However, once part of the complex, A20 and AZI2 remain associated with M1-linked polyubiquitin chains even after NEMO is released19. Although the role of NEMO in recruiting A20 is surprising, it is in accordance with the previous study showing that short C-terminal NEMO deletion abolished recruitment of A20 to TNF-RSC and caused inflammatory disease in a human patient59. Our data suggest that TANK and AZI2 cooperate to enable TBK1 activation during the whole duration of TNF-RSC assembly.
The role of TBK1 within TNF-RSC is to suppress RIPK1-mediated induction of cell death20,22. RIPK1 interacts directly with TNFR1 and TRADD dead domains and is recruited to TNF-RSC independently on NEMO60,61,62. The existence of two TBK1 binding adapters, one recruited early via NEMO and the second recruited later via A20, ensures that TBK1 can regulate RIPK1 activity during different stages of TNF-RSC assembly. In accord with the critical role of NEMO in recruiting TANK, A20, AZI2, and TBK1 to the TNF-RSC, we identified that human NEMO mutation L227P prevents the recruitment of all these proteins. AlphaFold prediction of NEMO structure63 indicates that this mutation is localized in the alpha helix segment and might cause broader protein assembly disruption. Therefore, other pathogenic NEMO mutations reported in human patients might also drive the disease due to the block in TBK1 activation and enhanced TNF-induced cell death.
Combined, our results show that either TANK or AZI2 expression is sufficient to prevent the development of autoinflammatory disease. However, in the case of immune challenges, such as pathogen infection, where TNF is produced in substantial quantity for a long-term period, both adapters might be required to limit TNF-induced cell death and tissue damage. Albeit we focused on TNF signaling, it is likely that TANK and AZI2 recruit TBK1 to regulate downstream signaling from any receptor that employs NEMO and A20, such as NOD1/2, CD40, IL-1 receptor, or Toll-like receptor (TLR) family members64. In accord, TBK1 and TANK were reported to suppress the NF-κB signaling pathways upon stimulation of various TLRs33,65,66. However, whether TANK and AZI2 play a redundant role in regulating signaling from different immune receptors remains to be addressed. Collectively, our work establishes TANK and AZI2 as core components of TNF-RSC that recruit TBK1 to prevent aberrant cell death induction and subsequent development of autoinflammatory disease.
Methods
Cell cultures and reagents
All cell lines were cultured at 37 °C in a humidified atmosphere with 5% CO2 in complete Dulbecco’s Modified Eagle’s Medium (DMEM) (Merck), which was supplemented with 10% fetal bovine serum (FBS) (Biosera) and 1% penicillin-streptomycin antibiotics (ATB) (Biosera). ST2 cells were kindly provided by J. Balounova, and HeLa, HEK293FT, ØNX-Eco, and ØNX-Ampho cells were kindly provided by T. Brdicka (both from the Institute of Molecular Genetics, Prague, Czech Republic). MEFs were derived from E11.5 mouse embryos and immortalized by transduction with SV40 large T antigen. To prepare BMDMs, bone marrow cells isolated from the tibia and femur of 8-9-week-old WT or Tank/Azi2DKO mice were cultured in DMEM/10% FBS/ATB supplemented with 1% non-essential amino acids (Merck) and 25 ng/ml M-CSF (Peprotech). The medium was changed after 2, 5, and 7 days. After 7 days, more than 95% of cells were positive for murine macrophage marker F4/80 as measured by flow cytometry. Subsequently, BMDMs were either directly used for experiments or cultivated with routine changing of the fully supplemented media every two days for up to 18 days. Cell lines were regularly tested for the presence of Mycoplasma using the Mycoplasmacheck services (Eurofins Genomics).
The antibodies and reagents used in this study are listed as part of Supplementary Dataset 2.
Recombinant proteins and protein isolation
All recombinant ligands contained the CD33 leader at the N-terminus, followed by a 6xHis Tag, which enabled the secretion and purification of proteins, respectively. In the case of SF-tagged ligands, 2xStrep and 1xFlag sequences were included. The ligands contained murine TNF (AA80-235) or human TNF (AA77-233) sequences. Mouse TNF was used to stimulate ST2 cells, MEFs, and BMDMs, while human TNF was used to stimulate HeLa cells.
The coding sequences for individual ligands were prepared using the GeneArt Gene Synthesis service (Thermo Fisher Scientific) and inserted into the pcDNA3.1 vector. Plasmids were transfected into HEK293FT cells using polyethylenimine (PEI) transfection. After three days, cell supernatants containing the produced proteins were collected. Samples were loaded on a His GraviTrap TALON column (Cytiva), previously equilibrated with a purification buffer (50 mM sodium phosphate, pH 7.4, 300 mM NaCl). The columns were washed with a purification buffer containing 20 mM imidazole and eluted with a purification buffer containing 350 mM imidazole. Subsequently, the samples were loaded on a centrifugal filter (10 kDa molecular weight cut-off, Millipore), washed several times with purification buffer to remove imidazole, and concentrated. The concentration of purified proteins was determined using NanoDrop One (Thermo Fisher Scientific). Finally, 50% glycerol was added, and for long-term storage, ligands were kept at −80 °C.
Preparation of knockout cell lines and exogenous protein expression
ST2 and HeLa cell lines deficient in specific proteins were prepared using the CRISPR-Cas9 approach. Sequences of target sites for particular genes were selected using the CHOPCHOP web tool67 and inserted into pSpCas9(BB)-2A-GFP (PX458) (Addgene plasmid number 48138)68. The target sites are listed as part of Supplementary Dataset 2. Obtained plasmids were transfected into cells using Lipofectamine 2000 (Invitrogen), and EGFP-positive cells were sorted using FACSAria™ Fusion (BD Biosciences). Single-cell clones were prepared, and the deficiency of a specific protein was verified by immunoblotting or sequencing of the DNA surrounding the target sites.
To reintroduce the proteins of interest, the coding sequences for the specified proteins were inserted into a retroviral pBabe vector, which expresses the GFP marker under the SV40 promoter. All the constructs were sequenced. Subsequently, ØNX-Eco cells (used for ST2 cells reconstitution) or ØNX-Ampho cells (used for HeLa cells reconstitution) were transfected with the pBabe-GFP vector containing coding sequences for the indicated proteins or an empty vector, using Lipofectamine 2000. Supernatants were collected, filtered through a 0.2 μl filter, and then incubated with target cells in the presence of 6 μg/ml polybrene. The cells were subsequently centrifugated (1 230 g, 30 °C, 45 min). GFP-positive cells were sorted using BD FACSAria™ Fusion.
To express proteins in HEK293FT cells, cells were transfected with NEMO and SF-TANK (SF-tag added at C-terminus) or SF-AZI2 (SF-tag at N-terminus) coding vectors using PEI.
Analysis of signaling pathways and immunoprecipitation
ST2, MEF, BMDM, or HeLa cell lines were washed and incubated in serum-free DMEM for at least 30 minutes. Subsequently, the cells were stimulated with mouse or human TNF (100 ng/ml) for the specified time points. Next, the cells were lysed on ice in lysis buffer (30 mM Tris, pH 7.4, 120 mM NaCl, 2 mM KCl, 2 mM EDTA, 10% glycerol, 10 mM chloroacetamide, Complete protease inhibitor cocktail, and PhosSTOP tablets (Roche)) containing 1% n-Dodecyl-β-D-Maltoside (DDM) (ThermoFisher Scientific). The lysates were incubated on ice for 30 minutes and cleared by centrifugation (21 130 g, 30 minutes, 2 °C). The cleared lysates were mixed with sodium dodecyl sulfate (SDS)-containing sample buffer, reduced by 50 mM dithiothreitol (DTT), and heated at 92 °C for 3 minutes.
To isolate TNF-RSC, the cells were stimulated with SF-TNF as indicated. After solubilizing the samples in lysis buffer containing 1% DDM, the cleared lysates were subjected to immunoprecipitation with anti-Flag M2 affinity agarose gel (Merck) overnight at 4 °C. Alternatively, to isolate proteins overexpressed in HEK293FT cells, cleared lysates from cells overexpressing indicated proteins were incubated with M2 beads overnight at 4 °C. The following day, M2 beads were washed with 0.1% DDM-containing lysis buffer four times. Beads were resuspended in an SDS sample buffer containing 50 mM DTT to release immunoprecipitated proteins, and the samples were heated at 92 °C for 3 minutes.
For the immunoprecipitation of RIPK1, cells were stimulated with TNF (250 ng/ml) for 24 h in the presence or absence of the caspase inhibitor zVAD (20 µM). RIPK1 antibody was added to pre-cleared lysates for 2 h. Subsequently, Protein A/G PLUS-Agarose (Santa Cruz Biotechnology) was added, and samples were incubated overnight at 4 °C. Upon washing the beads in lysis buffer containing 0.1% DDM, beads were resuspended in SDS sample buffer containing 50 mM DTT and heated at 92 °C for 3 minutes to elute the proteins.
The obtained samples were analyzed by immunoblotting. The chemiluminescence from HRP-fused secondary antibodies was detected using ChemidocTM MP Imaging System and analyzed using ImageLab software v6.1 (both Bio-Rad Laboratories).
Analysis of cell death
WT or TANK/AZI2-deficient ST2 cells, HeLa cells, or BMDMs were seeded on 96-well plates. The following day, cells were stimulated with mouse or human TNF (100 ng/ml) in the presence of 125 nM Incucyte Cytotox Red Dye (Sartorius) or 200 nM YOYO3 dye (ThermoFisher Scientific) in DMEM/10% FBS/ATB. In some experiments, IKKα/β inhibitor TPCA-1 (5 µM), caspase inhibitor zVAD (20 µM), and/or RIPK1 inhibitor Nec-1s (20 µM) were added. The induction of cell death was assessed at two-hour intervals using Incucyte Live-Cell Analysis Systems (Sartorius). Samples were analyzed in triplicates, and at least two images per well were captured.
To determine the percentage of cell death of high-throughput live cell imaging data, custom-made software was developed69,70. In short, brightfield- and fluorescence images (for cell death marker acquisition) of different cell lines and stages of apoptosis, necroptosis, pyroptosis, and ferroptosis induction (healthy, 30-, 60- or 90 % cell death) were acquired using Incucyte Live-Cell Analysis Systems. For necroptosis induction, HT29, HeLa cells expressing RIP3, MEFs, SCLC, and U2OS cells were treated with TNF (100 ng/ml), MCL1 inhibitor S63845 (5 μM), and zVAD (10 µM). For ferroptosis induction, HeLa, NIH-3T3, and HEK293 cells were treated with ferroptosis-inducing compound RSL3 (2 µM for NIH-3T3 and HEK293 or 4 µM for HeLa). For apoptosis induction, HeLa and SCLC cells were treated with Ccl-2 inhibitor ABT‐737 (5 μM) and S63845 (5 μM). For pyroptosis induction, BMDMs were preincubated for two hours with LPS (200 ng/ml) and then treated with inflammasome inducer Nigericin (2.5 µM). To validate the specificity of cell death induction, the respective inhibitors were used in control conditions: zVAD (10 µM) for apoptosis, NSA (2 µM) or Nec-1 (10 µM) for necroptosis, Fer-1 (5 µM) for ferroptosis and DSF (5 µM) for pyroptosis. Various brightfield images were manually annotated and assembled into a data set for the training data generation. For this, the area of living and dead cells was labeled, whereby the live/dead classification was based on the presence or absence of a cell death marker. Additionally, the center of cells was labeled. This dataset was used to train a U-Net-based artificial intelligence network. This custom-made software allows the precise segmentation of cells and the prediction of whether a single cell is alive or dead using only a brightfield image as input. As output, masks of living and dead cells and their position are obtained, allowing the numerical assessment of living and dead cells. Based on those numbers, the % of cell death was calculated. Cell death prediction was validated by cell death marker.
For analysis of cell death using flow cytometry, WT or Tank/Azi2DKO ST2 cells in DMEM/10% FBS/ATB were pretreated with zVAD (20 µM) and/or Nec-1s (20 µM) for 15 minutes at 37 °C. Subsequently, TNF (250 ng/ml) was added as indicated. Cells were incubated for 24 h. After incubation, the supernatants containing dead cells were collected. The attached cells were detached with Trypsin/EDTA solution (Biosera) for 10 minutes. Subsequently, they were collected, mixed together with the supernatant, centrifugated, and resuspended in PBS containing PI (Thermo Fisher Scientific). The proportion of dead cells was determined by detecting the percentage of PI-positive cells using the BriCyte E3 flow cytometer (Mindray).
RNA sequencing
WT and Tank/Azi2DKO BMDMs were seeded on six-well plates and stimulated with mouse TNF (100 ng/ml) for 2 h. The BMDMs were independently obtained from 3 pairs of animals. Subsequently, cells were collected, and RNA was isolated in five separate experiments using the Zymo Research Kit (R1055). The cDNA libraries were prepared using the KAPA mRNA Hyperprep Kit (Roche KK8580). Single-end sequencing was performed on Illumina NextSeq 2000 using the NextSeq 2000 P3 Reagents (50 cycles) kit (Illumina 20046810). The quality of reads was verified using FastQC v0.11.7 and MultiQC v1.17 tools71. The reads were aligned to mouse genome GRCm39 using salmon v1.10.072. The quantification of results was done using R packages tximport v1.26.173. The downstream analysis, including normalization, raw count modeling, and differential expression analyzes on complete count matrices, as well as subsets of specific samples, was done using the R package DESeq2 v1.38.374. The p-value was calculated using the Wald test in the DESeq2 R package and corrected for multiple comparisons with the Benjamini–Hochberg procedure. The changes in gene expression are listed in Supplementary Dataset 1. Visualization and gene set enrichment analysis were performed with the packages EnhancedVolcano v1.16.0, pheatmap v1.0.12, and fgsea v1.24.0. The analysis was done on R v4.2.1.
Mice
The mice were housed in a specific-pathogen-free facility at the Institute of Molecular Genetics of the Czech Academy of Sciences (IMG) in compliance with the laws of the Czech Republic. All animal protocols for the mice experiments were approved by the Resort Professional Commission for Approval of Projects of Experiments on Animals of the Czech Academy of Sciences, Czech Republic. The mice were provided with a standard rodent breeding diet and water ad libitum. The animal facility maintained a 12-h light and 12-h dark cycle, with a temperature of 22 ± 1 °C and a relative humidity of 55 ± 5%. Experimental and control animals were co-housed, with both males and females included in the study. Experimental mice were euthanized by cervical dislocation or by isofluorane inhalation at overdose.
The murine strains utilized in this study were generated at the Czech Center for Phenogenomics at IMG. To introduce targeted indel mutations into the Tank and Azi2 genes, a mixture containing Cas9 mRNA (100 ng/ml) and target-specific sgRNA (50 ng/ml) was microinjected into zygotes derived from C57BL/6 N mice. Zygote electroporation was employed to deliver sgRNAs targeting Tnfrsf1a and Ripk1 genes, as described elsewhere75. In the case of producing the RIPK1KD allele harboring the inactivating D138N mutation, a single-stranded DNA template (10 μM) was included. The resulting embryos were then implanted into foster mothers.
Genomic DNA was isolated from tail biopsies of newborn mice, and the DNA sequence surrounding the CRISPR/Cas9 recognition site was amplified by PCR using Phusion polymerase (Thermo Fisher Scientific). Subsequently, the amplified DNA was sequenced. The selected founders were backcrossed onto the C57BL/6 J background for at least three generations. Genotyping primers were designed to distinguish between the WT and modified alleles using Taq DNA polymerase (Top-Bio). The sequences of the oligonucleotides used for generating the mice and their genotyping can be found in Supplementary Dataset 2.
Clinical biochemistry and histology
To collect blood samples, mice were placed under terminal isoflurane anesthesia. Retro-orbital sinus bleeding was performed using glass capillaries, and the blood was collected in lithium heparin-coated tubes. After collection, each tube was gently inverted to ensure proper mixing and then kept at room temperature. The samples were centrifuged within one hour of collection (5 000 g, 10 minutes, 8 °C). The resulting plasma samples were frozen at −80 °C until further analysis. Biochemical parameters were analyzed using the AU480 clinical chemistry analyzer (Beckman Coulter). The analysis included measuring the concentrations of urea, phosphate, potassium, magnesium, total bilirubin, AST, cholesterol, triglycerides, HDL, LDL, albumin, and total protein. The concentrations of globulins were calculated as the difference between the total protein and albumin concentrations. The concentration of cytokines and antibody isotypes in the plasma was measured using the Bio-Plex 200 system (Bio-Rad). The Bio-Plex Pro Mouse Cytokine Th17 Panel A (Bio-Rad) was utilized to detect cytokines. Antibody Isotyping 7-Plex Mouse ProcartaPlex (Invitrogen) assay was employed to evaluate antibody concentrations.
Tissue samples from the back skin, kidney, liver, spleen, pancreas, skeletal muscle, white adipose tissue, and testes of 20-24-week-old mice were fixed in 4% formaldehyde solution in PBS overnight and transferred to 70% ethanol. The samples were processed by the Leica ASP6025 Vacuum Tissue Processor according to the program Standard processing overnight, embedded in Paraplast X-tra (Leica Biosystems), using the Tissue Embedding Station Leica EG1150. The five μm thick sections were prepared using the Leica Fully Motorized Rotary Microtome RM2255-FU. The obtained slides were stained by Hematoxylin H (Biognost) and Eosin Y (Carl Roth) using the Leica ST5010-CV5030 Stainer Integrated Workstation. Congo red staining was done by using the Congo red Staining Kit (Roche) in Ventana Staining Machine for Special Stains (Roche) according to producer manuals. Picrosirius red staining was done manually using Weigert’s Iron Hematoxylin Set and Direct Red 80 (Merck) according to producer protocol. To detect apoptotic cells, tissue samples were stained using TUNEL Assay Kit HRP-DAB (Abcam) according to the manufacturer’s instructions. Apoptotic cells with exposed 3’-OH ends of DNA fragments were marked by terminal deoxynucleotidyl transferase-mediated addition of biotinylated deoxynucleotides and detected by staining with streptavidin conjugated to horseradish peroxidase (HRP), followed by the addition of diaminobenzidine (DAB). Samples were counterstained with methyl green. Images of histological slides were obtained using the Axio Scan Z1 (Zeiss) scanner and evaluated in the software ZEN v3.4 (Zeiss). The average thickness of the subdermal fat layer was calculated by measuring 5 different locations in the skin of each analyzed animal. The proportion of swollen glomeruli was calculated from 1 mm × 1 mm areas. The number of liver granulomas was calculated from 2 mm × 2 mm areas.
Analysis of immune cell populations
Spleen and peripheral lymph nodes were removed from 8–12-week-old mice and minced to obtain single-cell suspensions. In the case of the spleen, red blood cells were lysed in ACK buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA- Na2, pH 7.4). Cells were resuspended in FACS buffer and stained on ice with LIVE/DEAD near-IR dye (Invitrogen).
To analyze B cells, the samples were stained with the following antibody panel: CD19-PE, IgM-BV421, IgD-PerCP_Cy5.5, CD23-APC, and CD1d-PE-Cy7. B cells (CD19+) were gated to identify isotype-switched cells (IgM−, IgD−).
For the analysis of myeloid cells, the samples were stained with the following antibody panel: CD3-FITC, CD19-FITC, NK1.1-FITC, CD11b-BV421, CD11c-AF700, Ly6C-PE-Cy7, and Ly6G-AF647. Myeloid cells (CD3−, CD19−, NK1.1−, CD11b+) were separated into three subsets: monocytes/macrophages (CD11c−, Ly6G−), neutrophils (CD11c−, Ly6G+), and dendritic cells (CD11c+). All the fluorescently labeled antibodies are listed as part of Supplementary Dataset 2.
Samples were measured on Cytek Aurora flow cytometer, and data were analyzed using FlowJo software v10.10.0 (BD Biosciences). The gating strategy is shown in Supplementary Fig. 8.
Bone marrow transfer
Bone marrow was isolated from 4–5-week-old Tank/Azi2DKO mice harboring Ly5.2 or WT mice expressing Ly5.1/5.2. Cells were mixed in a 1:1 ratio, and 2 × 106 cells in 200 µl of PBS were intravenously injected into lethally irradiated (6 Gy) 6-week-old recipient Ly5.1 mice. After 12–14 weeks, the spleen and lymph nodes were harvested and analyzed by flow cytometry.
Male fertility and sperm count
To evaluate male fertility, males of the specified genotype were paired with WT females for a maximum duration of 40 days or until pups were born, after which the males were separated. The number of newborn pups was recorded.
Sperm count was obtained after thorough mincing of epididymides in PBS at room temperature using a Burker chamber. Motility was determined as previously described76 on sperm isolated by puncturing the epididymides and capacitation in Toyoda-Yokoyama-Hosi (TYH) medium at 37 °C and 5% CO2 for one hour. The Computer-assisted sperm analysis system (Hamilton-Thorne, version 12.3, standard settings for mouse sperm) with Leja slides (double chamber, 0.1 mm deep) was employed to assess the percentage of motile and progressively motile spermatozoa via video recordings of over one thousand cells per male.
Statistics and reproducibility
The used statistical tests are indicated. In the case of one-way ANOVA, two-way ANOVA, and unpaired two-tailed t-test, the normality was assessed using the Kolmogorov-Smirnov test or Shapiro-Wilk test. Statistical analysis was performed and graphs were plotted using Prism v10.2.3 (GraphPad).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The bulk RNA sequencing data have been deposited in Sequence Read Archive (SRA) under the accession code PRJNA1095284. Source data are provided with this paper.
Code availability
The code for the custom-made software CellLocator v0.9.2 used to determine the percentage of cell death in high-throughput live cell imaging data can be accessed in the public repository Zenodo70 [https://zenodo.org/records/13774183]. Source data are provided with this publication.
Change history
27 February 2025
This article was originally published under a CC BY-NC-ND 4.0 license, but has now been made available under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. license. The PDF and HTML versions of the paper have been modified accordingly.
References
Kupka, S., Reichert, M., Draber, P. & Walczak, H. Formation and removal of poly-ubiquitin chains in the regulation of tumor necrosis factor-induced gene activation and cell death. FEBS J 283, 2626–2639 (2016).
Micheau, O. & Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003).
Degterev, A. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4, 313–321 (2008).
Cho, Y. S. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
He, S. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111 (2009).
Zhang, D. W. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 (2009).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Cai, Z. et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 16, 55–65 (2014).
Varfolomeev, E. & Vucic, D. RIP1 post-translational modifications. Biochem J 479, 929–951 (2022).
Dondelinger, Y. et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell 60, 63–76 (2015).
Geng, J. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun 8, 359 (2017).
Jaco, I. et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell 66, 698–710.e695 (2017).
Menon, M. B. et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat Cell Biol 19, 1248–1259 (2017).
Dondelinger, Y. et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat Cell Biol 19, 1237–1247 (2017).
Kirisako, T. et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J 25, 4877–4887 (2006).
Haas, T. L. et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell 36, 831–844 (2009).
Kanayama, A. et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell 15, 535–548 (2004).
Rahighi, S. et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 136, 1098–1109 (2009).
Draber, P. et al. LUBAC-Recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep 13, 2258–2272 (2015).
Xu, D. et al. TBK1 suppresses RIPK1-Driven apoptosis and inflammation during development and in aging. Cell 174, 1477–1491.e19 (2018).
Clark, K. et al. Novel cross-talk within the IKK family controls innate immunity. Biochem J 434, 93–104 (2011).
Lafont, E. et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol 20, 1389–1399 (2018).
van Loo, G. & Bertrand, M. J. M. Death by TNF: a road to inflammation. Nat Rev Immunol 23, 289–303 (2023).
Bonnard, M. et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J 19, 4976–4985 (2000).
Eren, R. O., Kaya, G. G., Schwarzer, R. & Pasparakis, M. IKKepsilon and TBK1 prevent RIPK1 dependent and independent inflammation. Nat Commun 15, 130 (2024).
Marchlik, E. et al. Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J Leukoc Biol 88, 1171–1180 (2010).
Taft, J. et al. Human TBK1 deficiency leads to autoinflammation driven by TNF-induced cell death. Cell 184, 4447–4463.e20 (2021).
Sun, Y. et al. Targeting TBK1 to overcome resistance to cancer immunotherapy. Nature 615, 158–167 (2023).
Runde, A. P., Mack, R., S, J. P. & Zhang, J. The role of TBK1 in cancer pathogenesis and anticancer immunity. J Exp Clin Cancer Res 41, 135 (2022).
Wild, P. et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233 (2011).
Fang, R. et al. MAVS activates TBK1 and IKKepsilon through TRAFs in NEMO dependent and independent manner. PLoS Pathog 13, e1006720 (2017).
Zhang, C. et al. Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398 (2019).
Kawagoe, T. et al. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat Immunol 10, 965–972 (2009).
Fukasaka, M. et al. Critical role of AZI2 in GM-CSF-induced dendritic cell differentiation. J Immunol 190, 5702–5711 (2013).
Ma, X. et al. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci USA 109, 9378–9383 (2012).
Kishore, N. et al. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J Biol Chem 277, 13840–13847 (2002).
Liu, T., Zhang, L., Joo, D. & Sun, S. C. NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2, 17023 (2017).
Ngo, K. A. et al. Dissecting the regulatory strategies of NF-kappaB RelA target genes in the inflammatory response reveals differential transactivation logics. Cell Rep 30, 2758–2775.e2756 (2020).
Karin, M. & Lin, A. NF-kappaB at the crossroads of life and death. Nat Immunol 3, 221–227 (2002).
Le Hir, M. et al. Differentiation of follicular dendritic cells and full antibody responses require tumor necrosis factor receptor-1 signaling. J Exp Med 183, 2367–2372 (1996).
Polykratis, A. et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol 193, 1539–1543 (2014).
Cicognani, C. et al. Serum lipid and lipoprotein patterns in patients with liver cirrhosis and chronic active hepatitis. Arch Intern Med 157, 792–796 (1997).
Moustafa, T. et al. Alterations in lipid metabolism mediate inflammation, fibrosis, and proliferation in a mouse model of chronic cholestatic liver injury. Gastroenterology 142, 140–151.e112 (2012).
Ryzhakov, G. & Randow, F. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J 26, 3180–3190 (2007).
Chariot, A. et al. Association of the adaptor TANK with the I kappa B kinase (IKK) regulator NEMO connects IKK complexes with IKK epsilon and TBK1 kinases. J Biol Chem 277, 37029–37036 (2002).
Clark, K., Takeuchi, O., Akira, S. & Cohen, P. The TRAF-associated protein TANK facilitates cross-talk within the IkappaB kinase family during Toll-like receptor signaling. Proc Natl Acad Sci USA 108, 17093–17098 (2011).
Bosanac, I. et al. Ubiquitin binding to A20 ZnF4 is required for modulation of NF-kappaB signaling. Mol Cell 40, 548–557 (2010).
Tokunaga, F. et al. Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-kappaB regulation. EMBO J 31, 3856–3870 (2012).
Draberova, H. et al. Systematic analysis of the IL-17 receptor signalosome reveals a robust regulatory feedback loop. EMBO J 39, e104202 (2020).
Chen, Y. G. et al. LUBAC enables tumor-promoting LTbeta receptor signaling by activating canonical NF-kappaB. Cell Death Differ https://doi.org/10.1038/s41418-024-01355-w (2024).
Schweizer, P., Kalhoff, H., Horneff, G., Wahn, V. & Diekmann, L. [Polysaccharide specific humoral immunodeficiency in ectodermal dysplasia. Case report of a boy with two affected brothers]. Klin Padiatr 211, 459–461 (1999).
Doffinger, R. et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet 27, 277–285 (2001).
Fujita, F. et al. Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol Cell Biol 23, 7780–7793 (2003).
Nomura, F., Kawai, T., Nakanishi, K. & Akira, S. NF-kappaB activation through IKK-i-dependent I-TRAF/TANK phosphorylation. Genes Cells 5, 191–202 (2000).
Goncalves, A. et al. Functional dissection of the TBK1 molecular network. PLoS One 6, e23971 (2011).
Hemmi, H. et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med 199, 1641–1650 (2004).
Jin, J. et al. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-kappaB signaling. Nat Immunol 13, 1101–1109 (2012).
Li, D. et al. RIPK1-RIPK3-MLKL-dependent necrosis promotes the aging of mouse male reproductive system. Elife 6, e27692 (2017).
Zilberman-Rudenko, J. et al. Recruitment of A20 by the C-terminal domain of NEMO suppresses NF-kappaB activation and autoinflammatory disease. Proc Natl Acad Sci USA 113, 1612–1617 (2016).
Hsu, H., Xiong, J. & Goeddel, D. V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 81, 495–504 (1995).
Pobezinskaya, Y. L. et al. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat Immunol 9, 1047–1054 (2008).
Ermolaeva, M. A. et al. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat Immunol 9, 1037–1046 (2008).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Shimizu, Y., Taraborrelli, L. & Walczak, H. Linear ubiquitination in immunity. Immunol Rev 266, 190–207 (2015).
Gitlin, A. D. et al. N4BP1 coordinates ubiquitin-dependent crosstalk within the IkappaB kinase family to limit Toll-like receptor signaling and inflammation. Immunity 57, 973–986.e977 (2024).
Gao, T. et al. Myeloid cell TBK1 restricts inflammatory responses. Proc Natl Acad Sci USA 119, e2107742119 (2022).
Labun, K. et al. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res 47, W171–W174 (2019).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308 (2013).
Vorndran, M. R. H. & Roeck, B. F. Inconsistency Masks: Removing the Uncertainty from Input-Pseudo-Label Pairs. arXiv:2401.14387 https://ui.adsabs.harvard.edu/abs/2024arXiv240114387V (2024).
Vorndran, M. R. H. & Roeck, B. F. MichaelVorndran/CellLocator: CellLocator v0.9.2 (v0.9.2). Zenodo. https://doi.org/10.5281/zenodo.13774183 (2024).
Ewels, P., Magnusson, M., Lundin, S. & Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419 (2017).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014).
Jenickova, I. et al. Efficient allele conversion in mouse zygotes and primary cells based on electroporation of Cre protein. Methods 191, 87–94 (2021).
Kusari, F., Mihola, O., Schimenti, J. C. & Trachtulec, Z. Meiotic epigenetic factor PRDM9 impacts sperm quality of hybrid mice. Reproduction 160, 53–64 (2020).
Acknowledgements
We thank J. Stefanovic, L. Cupak, and H. Freislebenova for their technical assistance, D. Zudova for histopathology analysis, and Michael R. H. Vorndran for help with cell death experiments analysis. This project was supported by a Czech Science Foundation grant (21-25251S), EMBO Installation grant (4420), and Charles University grant (PRIMUS/20/MED/003) awarded to P.D., European Union’s Horizon 2020 research and innovation program under grant agreement No 802878 (ERC Starting Grant FunDiT) awarded to O.S., and Charles University grant (406322) awarded to M.P. We acknowledge Charles University institutional funding (Cooperation and UNCE/MED/016), core funding of the Institute of Molecular Genetics of the Czech Academy of Sciences, Czech Republic (IMG ASCR) (RVO 68378050), the project National Institute for Cancer Research (Program EXCELES, LX22NPO5102) and the project National Institute of virology and bacteriology (Program EXCELES, LX22NPO5103) - both funded by the European Union - Next Generation EU, and grant SVV 260637 provided by the Ministry of Education, Youth and Sports of the Czech Republic (MEYS). The project was further supported by grant LM2023036 from the MEYS provided to the Czech Center for Phenogenomics at IMG, ASCR and by the German Research Foundation (DFG) project SFB1403 (414786233) and by the German Federal Ministry of Education and Research (BMBF) project 16LW0213. A.U., M.P., and T.T. were students at the First Faculty of Medicine, and D.K., A.S., and V.N. were students at the Faculty of Science at Charles University in Prague.
Author information
Authors and Affiliations
Contributions
A.U., D.K., A.S., M.P., T.T., A.D., A.D., D.P., R.D.G., P.K., O.F. and O.M. planned, performed, and analyzed the experiments. V.N. performed RNAseq analysis. B.F.R. analyzed cell death experiments. J.P., J.L., Z.T., R.S. and O.S. supervised the experiments. P.D. conceived the study and wrote the manuscript. All authors commented on the manuscript draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ujevic, A., Knizkova, D., Synackova, A. et al. TBK1-associated adapters TANK and AZI2 protect mice against TNF-induced cell death and severe autoinflammatory diseases. Nat Commun 15, 10013 (2024). https://doi.org/10.1038/s41467-024-54399-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54399-4
