
Abstract
Type VI secretion systems (T6SSs) are macromolecular assemblies that deliver toxic effector proteins between adjacent bacteria. These effectors span a wide range of protein families that all lack canonical signal sequences that would target them for export. Consequently, it remains incompletely understood how conserved structural components of the T6SS apparatus recognize a diverse repertoire of effectors. Here, we characterize a widespread family of adaptor proteins, containing the domain of unknown function DUF4123, that enable the recognition and export of evolutionarily unrelated effectors. By examining two nearly identical paralogs of the conserved T6SS spike protein, VgrG, we demonstrate that each spike protein exports a structurally unique effector. We further show that the recruitment of each effector to its respective spike protein requires a cognate adaptor protein. Protein–protein interaction experiments demonstrate that these adaptor proteins specifically tether an effector to a structurally conserved but sequence divergent helix-turn-helix motif found at the C-terminus of its cognate VgrG. Using structural predictions and mutagenesis analyses, we elucidate the molecular contacts required for these interactions and discover that these adaptor proteins contain a structurally conserved N-terminal lobe that has evolved to bind VgrG helix-turn-helix motifs and a structurally variable C-terminal lobe that recognizes diverse effector families. Overall, our work provides molecular insight into a mechanism by which conserved T6SS components recognize structurally diverse effectors.
Similar content being viewed by others
Introduction
Bacteria secrete toxic proteins to inhibit the growth of competitor microorganisms and to perturb the physiology of host cells during infection1. In Gram-negative bacteria, the presence of an outer membrane has driven the evolution of sophisticated protein secretion apparatuses that span the cell envelope and export toxic effectors into nearby cells. The type VI secretion system (T6SS) is one such apparatus harbored by a wide range of Gram-negative species that deliver toxic effectors to both bacterial and eukaryotic cells, thus contributing to microbial competition and virulence2,3.
The T6SS comprises two main molecular assemblies: a membrane complex that spans both cell membranes and enables protein secretion across the cell envelope and a secreted tail-tube complex that binds to effector proteins and delivers them into target cells3. The tail-tube complex consists of stacked hemolysin co-regulated protein (Hcp) hexamers and a spike composed of a trimer of valine-glycine repeat protein G (VgrG)4,5. VgrG spikes are often capped by a single copy of a proline-alanine-alanine-arginine (PAAR) protein, which is proposed to sharpen the tail-tube complex and thus enable its penetration of the competitor cell membrane6,7. Upon secretion by a T6SS, the tail-tube complex and its bound effectors are delivered into the recipient cell where they target conserved physiological processes to inhibit cell growth8,9,10,11,12,13,14.
Unlike proteins exported by other secretory pathways, T6SS effectors do not contain a linear signal sequence or universal trafficking domain that facilitates their recognition by conserved structural components of the T6SS apparatus15. Instead, T6SS effectors are exported by physically associating with the tail-tube complex by one of several different mechanisms. Some effectors exist as C-terminal domains of VgrG, PAAR, or Hcp proteins and are therefore fused to these exported tail-tube proteins16,17,18. However, most described T6SS effectors are not C-terminal extensions of VgrG or PAAR proteins and must, therefore, be associated with the tail-tube complex by other mechanisms. Several accessory proteins have been reported to contribute to the formation and stability of the tail-tube complex prior to secretion19,20,21. However, in most cases, the molecular role of these protein families remains poorly understood21.
The best-characterized accessory proteins involved in T6SS effector secretion are chaperones belonging to the Eag family12,20,22,23,24,25. These proteins are associated with a subset of T6SS effectors that contain N-terminal transmembrane domains, which facilitate effector translocation across the target cell cytoplasmic membrane following delivery22. Prior to secretion, Eag chaperones bind to these transmembrane domains and shield them from the aqueous environment, thus maintaining their cognate effectors in a soluble and secretion-competent state20. However, it is important to note that Eag chaperones are not themselves directly involved in effector recruitment to the tail-tube complex because Eag-associated effectors contain N-terminal PAAR domains.
Several reports implicate domain of unknown function (DUF) 4123 proteins in the recruitment of diverse T6SS effectors for secretion26,27,28,29,30. In contrast to Eag chaperones, these proteins bind to a soluble effector and a VgrG or PAAR protein and thus serve as adaptors that tether the effector to the tail-tube complex26. Although several DUF4123 homologs have been found to mediate interactions between effector C-terminal extensions of VgrG or PAAR proteins, the molecular contacts that underly these interactions have not been elucidated, and thus the mechanism of effector recognition by DUF4123 proteins remains unknown28,30. Interestingly, effectors from multiple evolutionarily unrelated protein families have been reported to require a DUF4123 protein for interaction with VgrG or PAAR and subsequent export through the T6SS26,27,28,31. This observation suggests that DUF4123 proteins may serve as a link between conserved T6SS tail-tube complex proteins and structurally diverse effectors, thus broadening the T6SS effector repertoire. However, to date, no comprehensive analysis has defined the complete range of T6SS effectors that rely on a DUF4123 protein for secretion. Furthermore, how a single adaptor family can interact with effectors that share no conserved features remains poorly understood.
In this work, we report the discovery that DUF4123 is not a single protein family as previously defined, but rather, it contains multiple sub-families that have divergently evolved to each recognize a unique effector family. Using a combination of predictive structural analyses and targeted mutagenesis, we demonstrate that DUF4123 proteins contain a structurally conserved N-terminal lobe that binds to a short helix-turn-helix motif at the C-terminus of their cognate VgrG spike protein. We further show that each DUF4123 sub-family is defined by the presence of a unique C-terminal lobe that each recognizes a cognate co-evolving effector family. Collectively, our data elucidate the molecular mechanisms by which the DUF4123 family of adaptor proteins diversify the effector repertoire of the T6SS spike.
Results
DUF4123 proteins co-occur with diverse T6SS toxin families
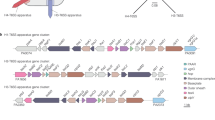
DUF4123 proteins are required for the export of T6SS effectors in several different Gram-negative bacteria26,27,28,29,30. However, the abundance and diversity of DUF4123-dependent T6SS effectors have yet to be explored in a systematic manner. All characterized DUF4123 proteins are encoded in close genomic proximity to the effectors that they export26,27,28,29,31. Therefore, we reasoned that gene co-occurrence analysis of T6SS-containing bacteria would identify a diverse repertoire of T6SS effectors and their corresponding DUF4123 proteins. We first searched for all DUF4123-annotated genes present in Pseudomonas, Burkholderia, Salmonella, Shigella, Escherichia, Enterobacter, Yersinia, Serratia, and Vibrio because species belonging to these genera harbor well-characterized T6SSs and therefore likely also encode T6SS-associated DUF4123 proteins. This initial search identified 1869 DUF4123 encoding genes found in 713 unique genomes. We next examined genes found 10 kb upstream or downstream of each DUF4123 gene to identify co-occurring genes that encode candidate T6SS effectors. This approach identified five distinct DUF4123 gene neighborhoods, with each neighborhood defined by the presence of a conserved effector family (Fig. 1A). These effector families include recombination hotspot (Rhs) proteins, Tle family phospholipases, α/β hydrolases, and marker for type six (MIX) domain-containing toxins, all of which are established T6SS effectors with previously described antibacterial and/or anti-host activities9,11,32,33. Additionally, our analysis identified DUF4123 proteins encoded adjacent to multi-antimicrobial extrusion (MATE) proteins, which a recent study suggests may constitute a previously overlooked family of T6SS effectors (Fig. 1A)34. Interestingly, each of these neighborhoods contains a vgrG gene upstream of the gene encoding DUF4123, which suggests that these DUF4123 proteins may serve as a general mechanism of effector recruitment to VgrG. In sum, our informatic analysis reveals that DUF4123 genes co-occur with five distinct families of unrelated T6SS effectors and that each of these effectors likely requires a cognate DUF4123 protein for its T6SS-dependent export from the cell.

A Genomic context of representative DUF4123 proteins belonging to each identified gene neighborhood cluster. Neighboring genes are colored according to the effector family, indicated by the legend. DUF4123 proteins are colored in blue, and VgrG proteins are colored in gray. B Phylogenetic distribution of 1869 DUF4123 proteins encoded by the genera Burkholderia, Enterobacter, Escherichia, Pseudomonas, Salmonella, Serratia, Shigella, Vibrio, and Yersinia. Colors correspond to gene neighborhood clusters as indicated by the legend.
The co-occurrence of DUF4123 genes with unrelated effector families suggests that DUF4123 is not a single homogenous protein family but instead likely exists as multiple sub-families that have each evolved to recognize a distinct group of effectors. Prior informatic examination of DUF4123 proteins did not examine sequence diversity within this family and thus may have overlooked the sequence features that would differentiate such sub-families from one another29. We reasoned that a more in-depth phylogenetic analysis of these proteins may reveal molecular signatures within DUF4123 sub-families that specifically recognize and export distinct families of effectors. To explore this possibility, we first constructed a phylogenetic tree of the DUF4123 sequences used for our gene co-occurrence analysis (Fig. 1B). In line with our hypothesis, these sequences formed several evolutionarily divergent clades and within each clade are DUF4123 sequences encoded by multiple bacterial genera, indicating that the diversity within the DUF4123 family does not mirror the evolutionary relatedness of the bacteria encoding them (Fig. S1A). Upon mapping the five distinct DUF4123 gene neighborhoods onto our phylogenetic tree, it instead became apparent that the relatedness of DUF4123 proteins strongly correlates with the family of co-occurring effectors, bolstering our hypothesis that sub-families of DUF4123 proteins likely harbor distinct sequences that facilitate the specific recruitment of a given effector family to its cognate VgrG protein (Fig. 1B). Based on these findings, we conclude that divergent evolution within the DUF4123 protein family likely underlies the ability of these proteins to facilitate the export of unrelated effector families.
Pseudomonas aeruginosa encodes two nearly identical VgrG paralogs that export distinct toxins
The co-evolution between DUF4123 sub-families and their adjacently encoded toxins suggests that DUF4123 proteins exhibit a high degree of specificity towards their cognate toxin. If this is the case, multiple DUF4123 proteins belonging to different sub-families should be found encoded alongside distinct effector types within the same bacterial genome. Indeed, our informatic search identified three such DUF4123 proteins encoded by the model T6SS bacterium Pseudomonas aeruginosa PA14 (Fig. 2A). These three DUF4123 proteins are each encoded downstream of a VgrG protein, and these VgrGs are closely related at the amino acid sequence level. VgrG4a and VgrG6 are 72% identical, whereas VgrG6 and VgrG14 share 96% sequence identity. The striking sequence similarity between VgrG6 and VgrG14 led us to focus on the gene clusters encoding these proteins because they presumably represent an instance in which two nearly identical T6SS apparatus components export unrelated effectors. Despite co-occurring with nearly identical VgrG paralogs, the DUF4123 proteins associated with VgrG6 and VgrG14 are only 19% identical at the amino acid level. This observation suggests that these DUF4123 proteins are involved in the recruitment of distinct effectors and would, therefore, be an ideal system to test our hypothesis that DUF4123 sub-families have evolved to recruit unrelated effectors to conserved T6SS spike proteins. We therefore focused our experimental efforts on characterizing the adaptor-effector pairs associated with VgrG6 and VgrG14 to better understand the molecular basis for effector recruitment to the T6SS apparatus. To reflect the co-occurrence of these genes with vgrG6, and vgrG14, respectively, and to maintain consistency with existing DUF4123 gene nomenclature, we named these DUF4123 genes type VI adaptor protein (tap) 6 and tap14.

A Schematic representation of three DUF4123 gene clusters in Pseudomonas aeruginosa strain PA14. B, C Outcome of intraspecific growth competitions between the indicated P. aeruginosa PA14 donor and recipient strains. The competitive index is calculated as a change (final/initial) in the donor-to-recipient ratio. Error bars represent mean values +/− SEM, n = 3 biological replicates. Asterisks indicate statistically significant differences in the competitive indices between indicated donor and recipient strain (p < 0.05). Differences between groups were calculated using an ordinary one-way ANOVA with multiple comparisons against the parent strain. p-value in panel B is 0.03. In panel C, the p-value for the mean difference between the parent and ∆vgrG6 competed against the ∆ptx2∆pti2 strain is 0.04. In panel C, the p-value for the mean difference between the parent and the ∆vgrG14 strain competed against the ∆rhsP2∆rhsI2 recipient is 0.0059. All other p-values are not significant.
In order for P. aeruginosa PA14 to serve as a suitable system to investigate DUF4123 function, we first needed to identify the cognate toxins exported by VgrG6 and VgrG14. P. aeruginosa harbors three distinct T6SSs, referred to as the H1-, H2-, and H3-T6SS, and each of these systems exports a unique repertoire of effectors35. Recently published genetic data implicate VgrG14 as the spike protein required for the export of the antibacterial toxin RhsP2 through the H2-T6SS11. Given its sequence similarity to VgrG14, we reasoned that VgrG6 likely also transits the H2-T6SS. However, a toxin that relies on VgrG6 for its delivery into target cells has not yet been identified. Our gene co-occurrence analysis found that tap6 co-occurs with a MIX-domain-containing protein encoded by the PA14_69520 locus (Fig. 2A). Given that MIX-domain-containing proteins are known to function as antibacterial T6SS effectors, we hypothesized that this gene encodes an antibacterial toxin that relies on VgrG6 for its H2-T6SS-dependent export33. We also identified an additional open reading frame downstream of this putative effector, PA14_69510, that encodes a protein of unknown function. Given that antibacterial T6SS toxins are invariably encoded adjacent to cognate immunity proteins that protect toxin-producing cells from the activities of their own toxins or those of kin cells, we predict that this open reading frame encodes the immunity factor that confers resistance to the putative effector (Fig. 2A).
If PA14_69520 encodes an H2-T6SS exported antibacterial toxin, deletion of PA14_69520 and its putative immunity gene PA14_69510 should render P. aeruginosa susceptible to toxin injected by a strain with a constitutively active H2-T6SS. To test this assertion, we conducted a series of bacterial competition experiments between a parental donor strain and a recipient lacking both PA14_69520 and PA14_69510. Because the molecular cues that trigger H2-T6SS expression and activation remain incompletely understood, the parent strain used in these experiments lacks both rsmA and amrZ, two genes encoding negative regulators of the H2-T6SS36. Using this genetic background, we found that the parental donor strain displayed a nearly 1000-fold fitness advantage against the recipient lacking PA14_69520 and PA14_69510 (Fig. 2B). Moreover, this fitness advantage was abrogated in donor strains that lack either the putative toxin gene or tssM2, a gene encoding a critical H2-T6SS apparatus component (Fig. 2B)11. Since the genetic background we employed constitutively expresses all three P. aeruginosa T6SSs, the finding that tssM2 is required for the observed PA14_69520-dependent co-culture fitness advantage confirms our prediction that this toxin transits the H2-T6SS. Additionally, we found that plasmid-borne expression of PA14_69510, the gene downstream of PA14_69520, significantly recovered growth of the PA14_69520 sensitive recipient strain, indicating that PA14_69510 functions as an immunity gene that confers protection against PA14_69520-mediated killing (Fig. S2A). From these data, we conclude that PA14_69520 and PA14_69510 encode an H2-T6SS antibacterial toxin-immunity pair and we henceforth refer to these proteins as protein toxin exported by the H2-T6SS (Ptx2) and protein toxin immunity of the H2-T6SS (Pti2), respectively.
The existence of vgrG6 and ptx2 within the same gene cluster suggests that the gene product of the former is likely responsible for the export of the latter. However, because VgrG6 and VgrG14 are 96% identical at the amino acid level, we considered the possibility that VgrG14 may also be capable of exporting Ptx2. To address this possibility, we constructed donor strains lacking either vgrG6 or vgrG14 and tested their competitive fitness against our Ptx2-susceptible recipient (∆ptx2∆pti2). In these experiments, the donor strain lacking vgrG14 displayed a competitive advantage that was comparable to the parent strain, whereas the donor strain lacking vgrG6 was unable to outcompete the Ptx2-susceptible recipient (Fig. 2C). This finding indicates that Ptx2 is specifically exported by VgrG6. We next asked if the same specificity exists between VgrG14 and its associated toxin, RhsP2, using the same donor strains as above co-cultured with a recipient lacking rhsP2 and its cognate immunity gene, rhsI211. As reported previously, deletion of vgrG14 abrogates the rhsP2-dependent fitness advantage and we found that deletion of vgrG6 has no effect on this phenotype, indicating that VgrG14 is specifically required for RhsP2 delivery into target cells (Fig. 2C). Taken together, these data demonstrate that although they are nearly identical proteins, VgrG6 and VgrG14 specifically export the unrelated toxins Ptx2 and RhsP2, respectively. Thus, PA14 represents an ideal model system to study how two nearly identical VgrG paralogs specifically export cognate toxins that bear no evolutionary relatedness to one another.
Tap proteins simultaneously interact with toxins and VgrG proteins
Having established that VgrG6 and VgrG14 specifically export Ptx2 and RhsP2, respectively, we next set out to determine the role of tap6 and tap14 in the delivery of these effectors. Our bioinformatic data suggests that divergent evolution within the DUF4123 family has led to the emergence of multiple sub-families that each facilitate the export of a distinct family of effectors (Fig. 1). Given that Tap6 and Tap14 belong to different clades of our phylogenetic tree, we hypothesized that these proteins are specifically required for the delivery of their cognate effectors Ptx2 and RhsP2, respectively. To test this hypothesis, we constructed strains lacking either tap6 or tap14 and tested their fitness against our Ptx2-susceptible (Δptx2 Δpti2) and RhsP2-susceptible (ΔrhsP2 ΔrhsI2) recipients (Fig. 3A). In line with our prediction, deletion of tap6 in donor cells abrogates their fitness advantage against Ptx2-susceptible recipients whereas deletion of tap14 has no effect (Fig. 3A). Likewise, deletion of tap14 abrogates the competitive advantage of donor cells against RhsP2-susceptible recipients but deletion of tap6 does not (Fig. 3A). When taken together with our prior findings, these data demonstrate that tap6 and tap14 are specifically required for the export of their cognate effectors by closely related VgrG spike proteins.

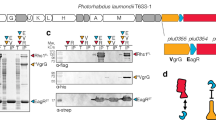
A Outcome of intraspecific growth competitions between the indicated P. aeruginosa PA14 donor and recipient strains. The competitive index is calculated as a change (final/initial) in the donor-to-recipient ratio. Error bars represent mean values +/− SEM, n = 3 biological replicates. Asterisks indicate statistically significant differences in the competitive indices between indicated donor and recipient strain (p < 0.05). Differences between groups were calculated using an ordinary one-way ANOVA with multiple comparisons against the parent strain. Against the ∆ptx2∆pti2 recipient, the p-value for the mean difference between parent and ∆tap6 is <0.0001. Against the ∆rhsP2∆rhsI2 recipient, the p-value for the mean difference between parent and ∆tap14 is 0.0004. All other p-values are not significant. B Schematic representation of VgrG6 and VgrG14. The inset represents pairwise alignment from residues 644-680. C AlphaFold3 models of VgrG6 and VgrG14 trimers. Colored regions represent residues 653–680. D Western blot analysis of co-purification experiments between the indicated heterologously expressed tagged (hexahistidine (H) or FLAG (F)) proteins. E Size exclusion chromatograms of indicated combinations of recombinant proteins. F Western blot analysis of immunoprecipitation experiments between the indicated heterologously expressed tagged (VSV-G or V5) proteins. G Western blot analysis of co-purification experiments between the indicated heterologously expressed tagged (hexahistidine (H) or FLAG (F)) proteins. RhsP2 was detected using a polyclonal antibody raised against a truncation of this protein lacking its C-terminal toxin domain. Control lanes represent co-expression of VgrG14CT-His and RhsP2 without Tap14-FLAG. The asterisk represents a non-specific band detected by the RhsP2 polyclonal antibody. Western blots are representative of three independent experiments.
Several reports implicate other DUF4123 proteins as molecular tethers that interact with both VgrG and effector proteins, thus linking them together26,28,29. However, the molecular determinants underlying the specificity of these interactions are unknown. We therefore sought to characterize the protein–protein interactions that are likely responsible for the Tap6- and Tap14-mediated effector export that we observe in our bacterial competition experiments. Closer inspection of the amino acid sequences of VgrG6 and VgrG14 reveals that these proteins are in fact 100% identical across 652 of their 680 residues and differ by only 28 residues at their C-terminus (Fig. 3B). These C-terminal regions are predicted by AlphaFold3 to form short helix-turn-helix (HTH) motifs that protrude outwards from the trimeric VgrG spike via an unstructured linker (Fig. 3C)37. Given that these HTH motifs are the only regions of sequence divergence between VgrG6 and VgrG14, we reasoned that they must be responsible for the specificity of these VgrG proteins towards their cognate effectors. We therefore hypothesized that each VgrG C-terminal HTH motif specifically interacts with its cognate DUF4123 protein, as has been shown for other DUF4123 homologs30. We further hypothesized that Tap6 and Tap14 additionally interact with their cognate effectors via a second protein–protein interaction site. To test this idea, we first performed co-purification experiments in which we co-expressed in Escherichia coli His6-tagged versions of the isolated HTH motifs, hereafter referred to as VgrG6CT or VgrG14CT, with FLAG-tagged Tap6 or Tap14. Nickel affinity purification of these overexpressed VgrG HTH motifs revealed that VgrG6CT co-purifies with Tap6 but not Tap14, whereas VgrG14CT co-purifies with Tap14 but not Tap6 (Figs. 3D and S3B). This finding indicates that the divergent HTH motifs of VgrG6 and VgrG14 specifically interact with Tap6 and Tap14, respectively. Additionally, our finding that the HTH motifs of these VgrG proteins in isolation are sufficient for interaction with their cognate DUF4123 proteins suggests that these motifs are directly responsible for the specificity observed in our bacterial competition experiments.
If Tap6 and Tap14 do indeed tether Ptx2 and RhsP2 to VgrG6 and VgrG14, respectively, then we would expect Tap6 and Tap14 to also specifically interact with their cognate effectors. To test this hypothesis, we expressed and purified both effectors and DUF4123 proteins to homogeneity and subjected pairwise mixtures of these proteins to size exclusion chromatography (SEC) (Figs. 3E and S3C, D). In doing so, we found that purified Ptx2 co-elutes as a high molecular weight complex with purified Tap6, indicating that these proteins form a stable complex in vitro. Tap14, however, does not form such a complex with Ptx2 because these proteins elute from the column at distinct volumes that are consistent with their monomeric molecular weights (Fig. S3C). By contrast, purified Tap14 elutes as a complex with RhsP2, whereas Tap6 does not (Fig. 3E). Taken together, these results indicate that Tap6 and Tap14 specifically interact with their cognate effectors Ptx2 and RhsP2 in vitro.
We next constructed a P. aeruginosa PA14 strain bearing epitope-tagged alleles of vgrG6 and ptx2 at their native chromosomal loci and confirmed that these epitope tags do not disrupt Ptx2 function (Fig. S3E). This strain enabled us to perform co-immunoprecipitation experiments to investigate interactions between VgrG6 and Ptx2 in vivo. In line with our genetic and biochemical data, we found that Ptx2 co-precipitates with VgrG6 and thus forms a complex with this spike protein in vivo (Fig. 3F). Moreover, deletion of tap6 in this epitope-tagged background abrogates Ptx2 co-precipitation with VgrG6, demonstrating that Tap6 mediates the interaction between this effector and its cognate VgrG. In a parallel line of investigation, we were unable to detect RhsP2 using either a custom polyclonal antibody raised against purified protein or an epitope-tagged version of the toxin when expressed from its native chromosomal locus. Therefore, we instead relied on our heterologous co-expression system in E. coli to investigate complex formation between RhsP2 and VgrG14. In co-expressing RhsP2 with His6-tagged VgrG14CT in the presence or absence of Tap14, we found that RhsP2 only co-purifies with VgrG14CT when Tap14 is present, confirming that Tap14 tethers RhsP2 to VgrG14 (Fig. 3G). Together, these biochemical analyses demonstrate that DUF4123 proteins form ternary complexes with their cognate VgrG and effector proteins, thus physically connecting them.
Tap proteins possess a structurally conserved N-terminal VgrG-binding lobe
Having experimentally determined the network of protein–protein interactions formed by Tap6 and Tap14 and their cognate VgrG and effector proteins, we next turned to AlphaFold3 to model each of the individual proteins and the biochemically validated complexes in which they participate. We started by modeling Tap6 and Tap14 because their predicted structures could provide insight into how they connect VgrG proteins to diverse effector families. AlphaFold3 generated high-confidence predictions for both proteins and revealed that they both adopt a bilobed domain architecture. The N-terminal lobe is comprised of a mixed α/β secondary structure, whereas the C-terminal lobe consists of α-helices (Figs. 4A and S4A). Interestingly, the structural overlay of these predictions shows that the N-terminal lobes of Tap6 and Tap14 are structurally quite similar (Cα RMSD 2.1 Å over 177 aligned residues), whereas their C-terminal lobes are not (Cα RMSD 5.2 Å over 63 aligned residues) (Fig. 4A). To probe the generality of this observation, we analyzed the predicted structures of additional DUF4123 homologs from each of the different sub-families identified by our bioinformatics analyses and found that the N-terminal lobe is a conserved structural feature of these proteins despite the low level of sequence conservation across the DUF4123 protein family (Fig. S4B). Given our data showing that both Tap6 and Tap14 interact with C-terminal HTH motifs of VgrG proteins, we speculated that this structurally conserved lobe serves as a platform for VgrG binding. Consistent with this idea, AlphaFold3 multimer predicts that both Tap6 and Tap14 interact with the C-terminal HTH motifs of their cognate VgrG proteins through their N-terminal lobe (Figs. 4B, C and S4A).

A AlphaFold3 models of the indicated proteins. Gray shapes represent the N- and C-terminal domains as indicated. B, C AlphaFold3 model of the complexes formed between the indicated proteins. The C-terminal HTH motifs of VgrG6 and VgrG14 are colored yellow and green, respectively. D, E Molecular contacts formed at the predicted interface between the indicated proteins and western blot analysis of co-purification experiments between the indicated heterologously expressed tagged (hexahistidine (H) or FLAG (F)) proteins. Western blots are representative of three independent experiments.
To experimentally validate the AlphaFold3 multimer models, we introduced mutations into Tap6 and Tap14 at sites of predicted interaction and examined whether the mutated proteins co-purify with their cognate VgrG HTH motifs when co-expressed in E. coli. In doing so, we found that the substitution of hydrophobic residues in Tap6 at the predicted Tap6/VgrG6 binding interface with glutamine substantially reduced its ability to co-purify with VgrG6CT (Fig. 4D). Similarly, swapping the charge of both acidic and basic residues at this interface also disrupted co-purification. For both types of mutations, not all sites were equally disruptive, with the L188Q and R120E variants still retaining the capacity to maintain an interaction, albeit more weakly than wild-type Tap6. In a similar line of experimentation, site-specific mutagenesis of residues in the N-terminal lobe of Tap14 at the predicted Tap14/VgrG14 binding interface disrupted co-purification with VgrG14CT (Fig. 4E). Based on these findings, we conclude that DUF4123 proteins specifically interact with an HTH motif found at the C-terminus of their cognate VgrG via a structurally conserved N-terminal lobe.
The C-terminal lobe of Tap14 recognizes the cage domain of RhsP2
Having identified the molecular contacts involved in DUF4123/VgrG interactions, we next set out to characterize the interactions between DUF4123 proteins and their cognate effectors. The diversity of predicted structures for the C-terminal lobe of DUF4123 proteins suggests that effector recognition is mediated by this region of the protein. Therefore, we next attempted to model these complexes using AlphaFold3. However, because it has few close sequence or structural homologs, AlphaFold3 was unable to confidently predict the structure of Ptx2 and, therefore, could not confidently predict the complex formed between Ptx2 and Tap6. Therefore, rather than basing our functional studies on a potentially inaccurate structural model, we instead focused our characterization of the DUF4123 C-terminal lobe on the complex formed between Tap14 and RhsP2.
As its name suggests, RhsP2 belongs to the well-characterized Rhs protein family, which includes several antibacterial toxins associated with the T6SS as well as host-targeting toxins that rely on other protein delivery systems38,39,40. Characterized Rhs proteins contain three distinct domains: an N-terminal domain involved in Rhs protein export and/or delivery into target cells, a middle β-cage domain that encapsulates the Rhs toxin domain, and a C-terminal domain that harbors toxin activity38,40. These domains are autoproteolytically cleaved prior to Rhs protein export, which is thought to allow the C-terminal toxin domain to exit the Rhs cage upon toxin delivery, although the precise molecular events underlying this process remain incompletely understood41. In line with this precedent, we find that purified RhsP2 exists as three distinct species when analyzed by SDS-PAGE and that the molecular weights of these species correspond to the predicted sizes of each of its three domains (Fig S3D).
To understand how RhsP2 is recruited to the H2-T6SS by Tap14, we first predicted the structure of the complex formed by these proteins using AlphaFold3 (Figs. 5A and S5A, B). Consistent with our hypothesis that the C-terminal lobe of DUF4123 proteins mediates the physical interaction with cognate effectors, the C-terminal lobe of Tap14 was confidently predicted to interact with the β-cage domain of RhsP2 (RhsP2cage). To experimentally validate this prediction, we next co-expressed a His6-tagged version of the isolated β-cage domain of RhsP2 and tested its ability to co-purify with Tap14. In line with the AlphaFold3 prediction, Tap14 co-purified with RhsP2cage but did not co-purify with RhsP2’s N-terminal domain (RhsP2N-term), which was included to assess the specificity of the interaction (Fig. 5B). This finding demonstrates that Tap14 recognizes RhsP2 through the β-cage domain of the effector.

A AlphaFold3 prediction of the complex formed between RhsP2Cage and Tap14. B Western blot analysis of co-purification experiments between the indicated heterologously expressed tagged (hexahistidine (H) or FLAG (F)) proteins. RhsP2 was detected using a polyclonal antibody raised against a truncation of this protein lacking its C-terminal toxin domain. C Molecular contacts present at the predicted interface between RhsP2Cage and Tap14. D Western blot analysis of co-purification experiments between the indicated heterologously expressed tagged (hexahistidine (H) or FLAG (F)) proteins. E Electrostatic surface representation of the indicated proteins. Dashed line indicates surface contacting RhsP2Cage in the AlphaFold3 model depicted in B. Color corresponds to charge as indicated by the scale bar. Western blots are representative of three independent experiments.
We next examined the predicted binding interface between Tap14 and RhsP2 to gain insight into the molecular contacts that mediate their interaction. Alphafold3 predicts that the interaction between Tap14 and RhsP2cage involves numerous electrostatic contacts between a positively charged surface on Tap14 and a negatively charged surface near the C-terminal end of RhsP2’s β-cage domain (Fig. 5A, C). Therefore, we introduced charge-reversing mutations into tap14 and rhsP2 at residues that form the predicted binding interface between these proteins and assessed co-purification of these variants when co-expressed in E. coli (Figs. 5D and S5C). Reversing the charge of residues in Tap14 or RhsP2cage at the predicted binding interface between these proteins disrupts their co-purification and provides experimental validation of our AlphaFold3 prediction. Interestingly, the positively charged RhsP2-binding site found on the surface of Tap14 is not present in Tap6, suggesting that electrostatic interactions may be a feature of Rhs-associated DUF4123 proteins but not for DUF4123 proteins associated with other effector families (Fig. 5E). Indeed, upon predicting the complexes formed between several Rhs-associated DUF4123 proteins and the β-cage domain of their cognate effectors, we found that in every instance AlphaFold3 confidently predicts the formation of a binding interface between a positively charged DUF4123 C-terminal lobe and a negatively charged patch on the cage domain of its associated Rhs effector (Fig. S5D). Importantly, this electrostatic interface is consistently predicted across DUF4123–Rhs pairs that share low sequence identity with each other (DUF4123 mean sequence identity 37.2%, Rhscage mean sequence identity 51.5%), suggesting that this binding mechanism is a conserved feature of DUF4123-Rhs interactions.
Taken together with our prior analysis of the molecular contacts involved in Tap14 binding to VgrG14CT, these data indicate that Tap14 is a bilobed protein with an N-terminal lobe that mediates interaction with the HTH motif of VgrG14 and a C-terminal lobe that binds the cage domain of RhsP2. Furthermore, we identified a positively charged surface that is a unique feature of Rhs-associated DUF4123 proteins, indicating that an electrostatic mechanism of Rhs recognition is likely widely conserved across this sub-family of DUF4123 proteins.
Discussion
Protein secretion systems endow bacteria with the ability to compete with other organisms and perturb the physiology of host cells during infection1. Although the T6SS has previously been shown to export a broad repertoire of effectors belonging to multiple protein families, the molecular mechanisms by which these effectors are specifically recognized among the diverse pool of proteins in the bacterial cytoplasm have remained elusive21. In this work, we discover previously overlooked heterogeneity within the DUF4123 protein family that facilitates the interaction of these proteins with evolutionarily unrelated T6SS effectors, thus diversifying the effector repertoire of the T6SS tail-tube complex. Furthermore, we provide the first molecular characterization of the ternary complexes formed between VgrG, DUF4123, and effector proteins, and we identify structural features of DUF4123 proteins that enable specific recognition of their cognate effectors. Given that DUF4123 proteins are encoded by a wide range of bacteria and co-occur with several distinct families of effectors, we expect that our findings in P. aeruginosa represent a widely conserved mechanism of effector recruitment to the T6SS.
Several families of accessory proteins have been reported to participate in the recruitment of T6SS effectors to the tail-tube complex20,22,26. Proteins belonging to the Eag (DUF1795) family interact with transmembrane domains (TMDs) present in PAAR effectors, thus serving as chaperones that stabilize these effectors prior to secretion20,22. Proteins belonging to the DUF2169 family have also been shown to co-occur with PAAR effectors, but their role in effector recruitment and/or stability remains unknown30,42. Unlike Eag chaperones, DUF4123 proteins do not play a role in effector stability or solubility because our work and that of others demonstrate that DUF4123-associated effectors are stable in the absence of their cognate adaptor26. Therefore, Eag proteins and DUF4123 proteins play distinct but complementary roles in effector recruitment: the former is a molecular chaperone that stabilizes TMD-containing PAAR effectors prior to their secretion, whereas the latter is a molecular tether that links soluble effectors to VgrG spike proteins.
Although our work offers molecular insight into the protein–protein interactions underlying DUF4123-mediated effector recognition, several questions remain regarding the biological significance of these proteins. It remains unclear why some T6SS effectors exist as C-terminal domains of VgrG or PAAR proteins while others, such as those recruited by DUF4123 proteins, are distinct polypeptides. One possibility is that the formation of a VgrG-DUF4123-effector complex enables the disassembly of effectors from the VgrG protein following delivery to the recipient cell. This may allow a single T6SS tail-tube complex to export effectors that act in different cellular compartments since a subset of these effectors must dissociate from the complex to reach their targets. However, whether DUF4123 proteins enable effector dissociation and how these effectors would dissociate only after delivery to the target cell remains unknown. One possibility is that the VgrG-DUF4123-effector complex may be secreted in a meta-stable state that is triggered to dissociate by the physical conditions of the recipient cell periplasm. Interestingly, the VgrG HTH motifs we examined in our bioinformatic analysis contain a conserved pair of cysteine residues (data not shown). The oxidizing environment of the recipient cell periplasm may trigger the formation of a disulfide bond between these residues, which could subsequently cause dissociation of the VgrG-DUF4123-effector complex. Alternatively, it is possible that other physical differences between the producing cell cytoplasm and the recipient cell periplasm, such as pH or the concentration of potassium or sodium ions, could serve as a signal that stimulates complex dissociation.
Our bioinformatic analyses found that DUF4123 proteins contain two discrete lobes: a structurally conserved N-terminal VgrG-binding lobe and a structurally diverse C-terminal toxin-binding domain. Interestingly, in some instances, these two domains appear to be encoded by separate open reading frames, as is the case for the split DUF4123 proteins TecT and co-TecT28. TecT tethers the DNAse effector TseT to its cognate PAAR protein in a manner that requires the activity of co-TecT. TecT, which only consists of the structurally conserved region of DUF4123 proteins, interacts with a C-terminal extension in its associated PAAR protein and is, therefore, functionally analogous to the N-terminal lobe of the DUF4123 proteins described in this work. Likewise, co-TecT interacts with the TseT effector and thus performs the same function as the C-terminal lobe of Tap14. The observation that the two lobes of DUF4123 adaptors are sometimes encoded by two open reading frames further strengthens our conclusion that they function independently.
In summary, our molecular characterization of DUF4123 proteins reveals that this heterogenous family of adaptors diversifies the effector repertoire of the T6SS spike by binding to evolutionarily distinct effectors and tethering them to closely related VgrG proteins. Thus, DUF4123 proteins enable T6SSs to recognize and secrete a diverse range of effectors, arming bacteria with a broad arsenal of toxins with which to kill or perturb nearby cells.
Methods
Bacterial strains and growth conditions
Pseudomonas aeruginosa strains were derived from the sequenced strain PA1443. Cultures were grown in lysogeny broth (LB) medium (10 g/L tryptone, 10 g/L NaCl, 5 g/L yeast extract) at 37 °C shaking at 220 revolutions per minute (RPM). Solid media contained either 1.5% or 3% agar (w/v), as indicated.
E. coli strain XL1 blue (Novagen) was used for plasmid maintenance. Strain SM10 was used for conjugative transfer, and strain BL21 (DE3) pLysS (Novagen) was used for protein expression. E. coli strains were grown in LB at 37 °C in shaking at 220 RPM. Cultures were supplemented with 50 µg/mL kanamycin, 100 µg/mL ampicillin, 15 µg/mL gentamicin (E. coli), 30 µg/mL gentamicin (P. aeruginosa), 25 µg/mL irgasan (P. aeruginosa), 1 mM b-D-1-thigalatopyranoside (IPTG), or 40 µg/mL 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal), as indicated. A detailed list of strains and plasmids used in this study can be found in Tables S1 and S2.
Bioinformatic analyses
AnnoTree (v1.2)44 and AnnoView (v2.0)45 and their associated databases were used to explore the phylogenomic distribution and gene neighborhoods surrounding genes encoding DUF4123 domains. All protein-coding sequences from GTDB version r214 were downloaded and then annotated by mapping sequences to the Uniref100 database using DIAMOND46, with an E-value threshold of 1e-5, and with both query and subject alignment coverage thresholds of 70%. This was followed by extracting the protein domain functional annotation from the Uniref100 database. All sequences annotated as DUF4123 were collected and filtered by length―only sequences with lengths greater than 220 and less than 350 were selected to reduce spurious matches. The sequences were then further filtered to those within the genera Pseudomonas, Burkholderia, Salmonella, Shigella, Escherichia, Enterobacter, Yersinia, Serratia, and Vibrio. The sequences were aligned using MUSCLE v5, and a phylogenetic tree was built using FastTree, using default parameters. iTOL was used for tree visualization47. Each sequence has a unique gene ID, and the gene IDs were used as center genes in gene neighborhood analysis45. Other annotations around the center gene within +/− 10 kb ranges were collected for co-occurrence analysis. FP-Growth, a data mining algorithm used for finding frequent item sets, was applied to find the most frequent combinations of functional annotations associated with the chosen center gene.
DNA manipulation, plasmid construction, and mutant strain generation
All expression and allelic exchange plasmids were constructed using standard restriction enzyme-based cloning procedures48. Primers were synthesized by Integrated DNA Technologies. Oligonucleotide sequences are provided in supplementary dataset S3. Phusion polymerase, restriction enzymes, and T4 ligase were obtained from New England Biolabs. Sanger sequencing was performed by The Center for Applied Genomics at the Hospital for Sick Children in Toronto, Ontario. Plasmids used for heterologous expression include pETDuet-1, pET29b, and pET28b (Novagen).
Chromosomal mutants were introduced to P. aeruginosa by double allelic exchange as previously described49. Approximately 500 bp flanks upstream and downstream of the region to be deleted were amplified by PCR and spliced together by overlap-extension PCR. The resulting amplicon was ligated into the allelic exchange vector pEXG2, which was subsequently transformed into SM10 and introduced into P. aeruginosa by conjugation. Merodiploids were selected on media containing 30 µg/mL gentamicin and 25 µg/mL irgasan. Counterselection using the sacB marker was performed by streaking merodiploids onto LB agar lacking NaCl that contained 5% (w/v) sucrose. Strains that grow on sucrose and are gentamicin sensitive were screened by colony PCR for the deleted gene. Chromosomal mutants were validated by PCR amplification of the appropriate region and Sanger sequencing of the resulting amplicon.
Chromosomal fusion mutants (V5, VSV-G) were generated by the same method as deletion mutants, with the appropriate tag sequence spliced between the 500 bp flanking regions by overlap-extension PCR.
Bacterial competition assays
Competition assays were performed in co-culture as previously described50. Cultures of the indicated donor and recipient strains were grown overnight in LB at 37 °C and were diluted to an OD600 of 1.0. Donor and recipient strains were combined to a ratio of 5:1 (donor:recipient) and 10 µL of the resulting mixture was spotted onto nitrocellulose membranes overlaid on 3% LB agar. To enumerate the initial donor:recipient ratio, 10-fold serial dilutions of the initial mixture were plated on 1.5% agar LB containing 40 µg/mL X-gal. Recipient strains, which harbor a constitutively expressed lacZ allele at a neutral chromosomal site, were differentiated from donor strains by their blue phenotype on media containing X-gal. Co-culture competitions were incubated at 25 °C for 20 h before being resuspended in 1 mL LB and serially diluted to enumerate the final donor:recipient ratio. Competitive index was calculated as (final donor:recipient ratio)/(initial donor:recipient ratio).
Protein expression and purification
E. coli BL21 (DE3) pLysS (Novagen) harboring expression vectors were grown shaking at 220 RPM at 37 °C in LB containing the appropriate antibiotics. Upon reaching an OD600 of 0.6, protein expression was induced by the addition of IPTG to a final concentration of 1 mM. Protein were expressed at 18 °C for 18–24 h. Cells were pelleted by centrifugation at 9000 × g for 20 min and resuspended in buffer containing 50 mM HEPES NaOH pH 7.5, 300 mM NaCl before lysis by sonication. Lysates were clarified by centrifugation at 36,000 × g for 45 min. Lysates were loaded onto a 2 mL gravity flow Ni-NTA column pre-equilibrated with buffer containing 50 mM HEPES NaOH pH 7.5, 300 mM NaCl, and 10 mM imidazole pH 7.5 (wash buffer). Columns were washed three times with 20 mL of wash buffer before His6-tagged proteins were eluted by the addition of 4 mL of buffer containing 50 mM HEPES NaOH pH 7.5, 300 mM NaCl, 400 mM imidazole pH 7.5. Eluted proteins were further purified by gel filtration on a HiLoad 16/600 superdex 200 size exclusion chromatography column (GE Healthcare) equilibrated with buffer containing 20 mM HEPES NaOH pH 7.5, 150 mM NaCl. The purification of each protein was determined by SDS-PAGE, followed by staining with Coomassie Brilliant Blue R250. Proteins were concentrated to 5 mg/mL using a 10 kDa molecular weight cutoff (Tap6, Tap14) or a 100 kDa molecular weight cutoff (Ptx2, RhsP2) centrifugal filter device (MilliporeSigma). Protein concentration was measured using a NanoDrop instrument (ThermoFisher). Proteins were used immediately or snap-frozen in liquid nitrogen before being stored at −80 C.
To protect against its toxic effect, a catalytically inactive mutant of RhsP2 (E1576Q) was purified instead of wild-type11.
Co-purification and co-immunoprecipitation experiments
For co-purification experiments, E. coli BL21 (DE3) pLysS strains harboring the indicated vectors were grown in 100 mL LB containing appropriate antibiotics shaking (220 RPM) at 37 °C. Cells were harvested by centrifugation at 9000 × g, resuspended in buffer containing 50 mM HEPES NaOH pH 7.5, 500 mM NaCl, and lysed by sonication. Lysates were clarified as above. Lysates were loaded onto a 500 µL gravity flow Ni-NTA column pre-equilibrated with 10 mL buffer containing 50 mM HEPES NaOH pH 7.5, 500 mM NaCl, and 10 mM imidazole pH 7.5 (wash buffer). Columns were washed four times with 10 mL wash buffer. His6-tagged proteins/protein complexes were eluted in 500 µL buffer containing 50 mM HEPES NaOH pH 7.5, 300 mM NaCl, and 400 mM imidazole. Proteins present in the input and eluted fractions were analyzed by Western blotting.
For co-immunoprecipitation experiments, P. aeruginosa strains encoding the indicated epitope-tagged proteins at their native chromosomal loci were diluted to an OD600 of 0.1 and grown in 100 mL LB at 25 °C shaking (220 RPM). When an OD600 of 0.8 was reached (7–8 h of growth), cultures were harvested by centrifugation at 9000 × g for 20 min. Cells were resuspended in 4 mL buffer containing 50 mM HEPES NaOH pH 7.5, 300 mM NaCl, 2% (w/v) glycerol, and lysed by sonication (IP buffer). Lysates were clarified by centrifugation at 36,000 × g for 20 min, and input protein fractions were collected. Lysates were combined with 20 µL anti-VSV-G resin (Sigma) pre-equilibrated with 3 mL of IP buffer and incubated for 1 h, rotating at 4 °C. VSV-G-beads were pelleted by centrifugation at 40 × g for 5 min and washed three times with 15 mL IP buffer. Bound proteins were eluted by the addition of 100 µL 2x Laemelli sample buffer. Input and immunoprecipitated proteins were analyzed by western blotting.
Size exclusion chromatography
Analytical size exclusion chromatography was performed using a 10/300 GL Superdex 200 size exclusion column (GE Healthcare) on an AKTA system (GE Healthcare). 1 mg of each of the indicated purified proteins was combined, incubated for 10 min at room temperature, and loaded onto the size exclusion column pre-equilibrated with buffer containing 20 mM HEPES NaOH pH 7.5, 150 mM NaCl. To confirm complex stability, proteins that co-eluted as a peak were pooled, concentrated, and loaded onto the size exclusion column a second time. Eluted proteins were analyzed by SDS-PAGE, followed by staining with Coomassie Brilliant Blue R250.
Antibody generation
A custom polyclonal antibody for RhsP2 was generated for this study. Because full-length RhsP2 is toxic to E. coli, overexpression of a RhsP2 variant lacking the C-terminal toxin domain was performed for antibody production. This protein was purified as described above, except that PBS buffer was used for all stages instead of HEPES NaOH pH 7.5 NaCl. 5 mg of purified protein was sent to GenScript for polyclonal antisera production. The specificity of our custom RhsP2 antibody was determined by Western blotting of purified RhsP2 and comparison to blotting of a different protein purified from the same E. coli protein expression cell line.
Western blotting
Western blotting was performed as previously described for rabbit anti-VSV-G (MilliporeSigma V4888, 1:5000), rabbit anti-FLAG (MilliporeSigma F7425, 1:5000), mouse anti-His (GenScript A00186-100, 1:5000) and detected using anti-rabbit (New England Biolabs 7074 V) or anti-mouse New England Biolabs 7076S) horseradish peroxidase-conjugated secondary antibodies (1:5000) as appropriate51. Custom polyclonal rabbit anti-RhsP2 (this study) was used as a titer of 1:5000. Briefly, proteins were separated using 12% acrylamide SDS-PAGE run at 95 V for 15 min, followed by 195 V for 40 min. Proteins were subsequently wet transferred to a nitrocellulose membrane using the Mini Trans-Blot electrophoretic transfer system (Bio-Rad). The transfer was run at 103 V for 33 min, after which the blot was blocked in Tris-buffered saline containing 0.05% (v/v) Tween-20 (TBS-T) and 5% (w/v) non-fat milk (Bio-Rad) for 30 min with gentle agitation at room temperature. Primary antibody was added at the titer indicated above and incubated for 1 h at room temperature with gentle agitation. The blot was subsequently washed 3 times for 5 min each with 10 mL TBS-T. The secondary antibody was added at 1:5000 titer and incubated at room temperature for 45 min with gentle agitation. Blots were washed 3 times with 10 mL TBS-T before developing Clarity Max ECL substrate (Bio-Rad).
Statistics and reproducibility
No statistical method was used to predetermine sample size, and no data were excluded from the analyses. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The bioinformatic data generated in this study are available in supplemental dataset S1. Raw data for plotted figures are available in the source data file. Raw data for blots and gels are provided in Supplemental Fig. S6. AlphaFold3 models and confidence metrics are provided in Supplemental Dataset S2. Source data are provided in this paper.
References
Klein, T. A., Ahmad, S. & Whitney, J. C. Contact-dependent interbacterial antagonism mediated by protein secretion machines. Trends Microbiol. 28, 387–400 (2020).
Russell, A. B., Peterson, S. B. & Mougous, J. D. Type VI secretion system effectors: poisons with a purpose. Nat. Rev. Microbiol. 12, 137–148 (2014).
Cherrak, Y., Flaugnatti, N., Durand, E., Journet, L. & Cascales, E. Structure and activity of the type VI secretion system. Microbiol. Spectr. 7, https://doi.org/10.1128/microbiolspec.PSIB-0031-2019 (2019).
Ballister, E. R., Lai, A. H., Zuckermann, R. N., Cheng, Y. & Mougous, J. D. In vitro self-assembly of tailorable nanotubes from a simple protein building block. Proc. Natl Acad. Sci. USA 105, 3733–3738 (2008).
Leiman, P. G. et al. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc. Natl Acad. Sci. USA 106, 4154–4159 (2009).
Shneider, M. M. et al. PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature 500, 350–353 (2013).
Wood, T. E., Howard, S. A., Wettstadt, S. & Filloux, A. PAAR proteins act as the ‘sorting hat’ of the type VI secretion system. Microbiology 165, 1203–1218 (2019).
Russell, A. B. et al. Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475, 343–347 (2011).
Russell, A. B. et al. Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature 496, 508–512 (2013).
Ahmad, S. et al. An interbacterial toxin inhibits target cell growth by synthesizing (p)ppApp. Nature 575, 674–678 (2019).
Bullen, N. P. et al. An ADP-ribosyltransferase toxin kills bacterial cells by modifying structured non-coding RNAs. Mol. Cell 82, 3484–3498 e3411 (2022).
Whitney, J. C. et al. An interbacterial NAD(P)(+) glycohydrolase toxin requires elongation factor Tu for delivery to target cells. Cell 163, 607–619 (2015).
Jurenas, D. et al. Photorhabdus antibacterial Rhs polymorphic toxin inhibits translation through ADP-ribosylation of 23S ribosomal RNA. Nucleic Acids Res. 49, 8384–8395 (2021).
Ting, S. Y. et al. Bifunctional immunity proteins protect bacteria against FtsZ-targeting ADP-ribosylating toxins. Cell 175, 1380–1392 e1314 (2018).
Silverman, J. M., Brunet, Y. R., Cascales, E. & Mougous, J. D. Structure and regulation of the type VI secretion system. Annu. Rev. Microbiol. 66, 453–472 (2012).
Whitney, J. C. et al. Genetically distinct pathways guide effector export through the type VI secretion system. Mol. Microbiol. 92, 529–542 (2014).
Pukatzki, S., Ma, A. T., Revel, A. T., Sturtevant, D. & Mekalanos, J. J. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc. Natl Acad. Sci. USA 104, 15508–15513 (2007).
Ma, J. et al. The Hcp proteins fused with diverse extended-toxin domains represent a novel pattern of antibacterial effectors in type VI secretion systems. Virulence 8, 1189–1202 (2017).
Silverman, J. M. et al. Haemolysin coregulated protein is an exported receptor and chaperone of type VI secretion substrates. Mol. Cell 51, 584–593 (2013).
Ahmad, S. et al. Structural basis for effector transmembrane domain recognition by type VI secretion system chaperones. Elife 9, e62816 (2020).
Unterweger, D., Kostiuk, B. & Pukatzki, S. Adaptor proteins of type VI secretion system effectors. Trends Microbiol. 25, 8–10 (2017).
Quentin, D. et al. Mechanism of loading and translocation of type VI secretion system effector Tse6. Nat. Microbiol. 3, 1142–1152 (2018).
Alcoforado Diniz, J. & Coulthurst, S. J. Intraspecies competition in Serratia marcescens is mediated by type VI-secreted Rhs effectors and a conserved effector-associated accessory protein. J. Bacteriol. 197, 2350–2360 (2015).
Ma, J. et al. PAAR-Rhs proteins harbor various C-terminal toxins to diversify the antibacterial pathways of type VI secretion systems. Environ. Microbiol. 19, 345–360 (2017).
Jurenas, D. et al. Mounting, structure and autocleavage of a type VI secretion-associated Rhs polymorphic toxin. Nat. Commun. 12, 6998 (2021).
Unterweger, D. et al. Chimeric adaptor proteins translocate diverse type VI secretion system effectors in Vibrio cholerae. EMBO J. 34, 2198–2210 (2015).
Pei, T. T. et al. Intramolecular chaperone-mediated secretion of an Rhs effector toxin by a type VI secretion system. Nat. Commun. 11, 1865 (2020).
Burkinshaw, B. J. et al. A type VI secretion system effector delivery mechanism dependent on PAAR and a chaperone-co-chaperone complex. Nat. Microbiol. 3, 632–640 (2018).
Liang, X. et al. Identification of divergent type VI secretion effectors using a conserved chaperone domain. Proc. Natl Acad. Sci. USA 112, 9106–9111 (2015).
Bondage, D. D., Lin, J. S., Ma, L. S., Kuo, C. H. & Lai, E. M. VgrG C terminus confers the type VI effector transport specificity and is required for binding with PAAR and adaptor-effector complex. Proc. Natl Acad. Sci. USA 113, E3931–E3940 (2016).
Miyata, S. T., Unterweger, D., Rudko, S. P. & Pukatzki, S. Dual expression profile of type VI secretion system immunity genes protects pandemic Vibrio cholerae. PLoS Pathog. 9, e1003752 (2013).
Gunther, P. et al. Structure of a bacterial Rhs effector exported by the type VI secretion system. PLoS Pathog. 18, e1010182 (2022).
Salomon, D. et al. Marker for type VI secretion system effectors. Proc. Natl Acad. Sci. USA 111, 9271–9276 (2014).
Duran, D. et al. An orphan VrgG auxiliary module related to the type VI secretion systems from Pseudomonas ogarae F113 mediates bacterial killing. Genes 14, 1979 (2023).
Barret, M., Egan, F., Fargier, E., Morrissey, J. P. & O’Gara, F. Genomic analysis of the type VI secretion systems in Pseudomonas spp.: novel clusters and putative effectors uncovered. Microbiology 157, 1726–1739 (2011).
Allsopp, L. P. et al. RsmA and AmrZ orchestrate the assembly of all three type VI secretion systems in Pseudomonas aeruginosa. Proc. Natl Acad. Sci. USA 114, 7707–7712 (2017).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Jackson, A. P., Thomas, G. H., Parkhill, J. & Thomson, N. R. Evolutionary diversification of an ancient gene family (Rhs) through C-terminal displacement. BMC Genom. 10, 584 (2009).
Jones, C., Hachani, A., Manoli, E. & Filloux, A. An Rhs gene linked to the second type VI secretion cluster is a feature of the Pseudomonas aeruginosa strain PA14. J. Bacteriol. 196, 800–810 (2014).
Meusch, D. et al. Mechanism of Tc toxin action revealed in molecular detail. Nature 508, 61–65 (2014).
Busby, J. N., Panjikar, S., Landsberg, M. J., Hurst, M. R. & Lott, J. S. The BC component of ABC toxins is an RHS-repeat-containing protein encapsulation device. Nature 501, 547–550 (2013).
Santos, M. N. M. et al. Redundancy and specificity of type VI secretion vgrG loci in antibacterial activity of Agrobacterium tumefaciens 1D1609 strain. Front. Microbiol. 10, 3004 (2019).
Lee, D. G. et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 7, R90 (2006).
Mendler, K. et al. AnnoTree: visualization and exploration of a functionally annotated microbial tree of life. Nucleic Acids Res. 47, 4442–4448 (2019).
Wei, X. et al. AnnoView enables large-scale analysis, comparison, and visualization of microbial gene neighborhoods. Brief. Bioinform 25, bbae229 (2024).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82 (2024).
Green, M. R., Sambrook, J. & Sambrook, J. Molecular Cloning: A Laboratory Manual. 4th edn (Cold Spring Harbor Laboratory Press, 2012).
Hmelo, L. R. et al. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat. Protoc. 10, 1820–1841 (2015).
Hood, R. D. et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7, 25–37 (2010).
Colautti, J., Bullen, N. P. & Whitney, J. C. Lack of evidence that Pseudomonas aeruginosa AmpDh3-PA0808 constitute a type VI secretion system effector-immunity pair. Mol. Microbiol. 119, 262–274 (2023).
Acknowledgements
We thank Andrew McArthur for access to computational resources, Lindsey Marmont for critical feedback on the manuscript, and members of the Whitney lab for helpful discussions. J.C. is supported by an MD-PhD Canada Graduate Scholarship from the Canadian Institutes of Health Research (CIHR). This work was supported by an NSERC Discovery Grant (RGPIN-2019-04266) to A.C.D. and a CIHR project grant to J.C.W. (PJT-175011). J.C.W. is the Canada Research Chair in Molecular Microbiology and holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Author information
Authors and Affiliations
Contributions
J.C. and J.C.W. conceived the study. J.C., S.S.T., N.P.B., and J.C.W. contributed to the experimental design. J.C., S.S.T., and N.P.B. conducted experiments. H.T., D.H., and A.C.D. conceived and conducted bioinformatic analyses. J.C. and J.C.W. wrote the manuscript. All authors provided feedback on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Colautti, J., Tan, H., Bullen, N.P. et al. A widespread accessory protein family diversifies the effector repertoire of the type VI secretion system spike. Nat Commun 15, 10108 (2024). https://doi.org/10.1038/s41467-024-54509-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54509-2
This article is cited by
-
Structural insights into the antibacterial function of the Pseudomonas putida effector Tke5
The EMBO Journal (2026)
-
Biophysical characterization of Eag chaperones suggests the mechanism of effector transmembrane domain release
Nature Communications (2026)
-
The type VI secretion system and associated effector proteins
Nature Reviews Microbiology (2025)
