
Abstract
Post-operative recurrence rates of stage I non-small cell lung cancer (NSCLC) range from 20% to 40%. Nonetheless, the molecular mechanisms underlying recurrence hitherto remain largely elusive. Here, we generate genomic, epigenomic and transcriptomic profiles of paired tumors and adjacent tissues from 122 stage I NSCLC patients, among which 57 patients develop recurrence after surgery during follow-up. Integrated analyses illustrate that the presence of predominantly solid or micropapillary histological subtypes, increased genomic instability, and APOBEC-related signature are associated with recurrence. Furthermore, TP53 missense mutation in DNA-binding domain could contribute to shorter time to recurrence. DNA hypomethylation is pronounced in recurrent NSCLC, and PRAME is the significantly hypomethylated and overexpressed gene in recurrent lung adenocarcinoma (LUAD). Mechanistically, hypomethylation at TEAD1 binding site facilitates the transcriptional activation of PRAME. Inhibition of PRAME restrains the tumor metastasis via downregulation of epithelial–mesenchymal transition-related genes. We also identify that enrichment of AT2 cells with higher copy number variation burden, exhausted CD8 + T cells and Macro_SPP1, along with the reduced interaction between AT2 and immune cells, is essential for the formation of ecosystem in recurrent LUAD. Finally, multi-omics clustering could stratify the NSCLC patients into 4 subclusters with varying recurrence risk and subcluster-specific therapeutic vulnerabilities. Collectively, this study constitutes a promising resource enabling insights into the biological mechanisms and clinical management for post-operative recurrence of stage I NSCLC.
Similar content being viewed by others

Introduction
Lung cancer is deemed to be the most frequently diagnosed malignant tumor and leading cause of cancer-related mortality worldwide1. Approximately 85% of lung cancer cases are non-small cell lung cancer (NSCLC), while lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) serve as the two major subtypes of NSCLC2,3. In current clinical practice, surgery is the standard of care for early-stage NSCLC2. However, despite surgical resections, recurrence remains a significant challenge. As estimated, 20–40% of patients with stage I NSCLC experience tumor recurrence4,5,6. Approximately 80% recurrences occur within 2 years after surgery and result in treatment failure7. The 5-year post-recurrence survival is dismal, ranging from 15% to 16.6%5,8. Therefore, there is an urgent need to decipher the biological mechanisms underlying post-operative recurrence among stage I NSCLC.
To understand the events that facilitate tumorigenesis and related processes including recurrence and metastasis, established studies have demonstrated the complex interplay of molecular abnormalities and interactions within the tumor microenvironment9,10. For instance, genomic instability, arising from molecular events like TP53 mutations and intratumoral heterogeneity, contributes to lung cancer recurrence and metastasis6,11,12,13,14. There also exists a crucial association between DNA differential methylation in early-stage NSCLC and subsequent recurrence15. Importantly, tumors are complicated ecosystems composed of a diverse array of cell types whose interactions are integral to tumor evolution. In lung cancer, the enrichment of alveolar type 2 (AT2) cells and reduced proportion of alveolar type 1 (AT1) cells represent higher malignancy16. Furthermore, a pronounced enrichment of CD163+ macrophages is correlated with more aggressive cancer subtype and poorer prognosis17, and metastatic lung tumor is characterized by an immunosuppressive state featuring increased M2-type macrophages18,19. However, comprehensive molecular characterization for post-operative recurrence of stage I NSCLC is lacking. Additionally, single platform profiling fails to capture the complexity of the tumor ecosystem, with research on epigenomic patterns remaining limited. Therefore, the integration of multi-omics analyses emerges as a crucial step to unravel the comprehensive molecular characteristics contributing to post-operative recurrence, ultimately informing personalized clinical decision-making.
In this study, we conduct genomic, transcriptomic and epigenomic analyses on samples from 122 stage I NSCLC patients based on whole-exome sequencing (WES), nanopore sequencing, RNA sequencing (RNA-seq) and single-cell RNA sequencing (scRNA-seq) techniques. Our multi-omics study has explored the molecular aberrations and depicted the tumor ecosystem associated with early recurrence after surgical resections. This study provides a promising resource enabling future research on stage I NSCLC recurrence to investigate the biological mechanisms and uncover potential therapeutic strategies.
Results
Overview of the stage I NSCLC cohort
To comprehensively illustrate the multi-omics characteristics of post-operative recurrence in early-stage NSCLC, samples from 122 stage I NSCLC patients without prior therapy were enrolled. Frozen fresh (FF) tumors and paired adjacent normal tissues were collected from 47 patients. DNA and RNA were extracted and profiled by WES, nanopore sequencing and RNA-seq. Formalin-fixed and paraffin-embedded (FFPE) tumors and matched normal adjacent tissues from 61 patients were profiled using WES. Furthermore, 14 fresh resected (FR) tumors and 11 adjacent normal samples from remaining 14 patients were involved in scRNA-seq. According to at least 3 years of follow-up, patients were subordinated into recurrent (Rec, n = 57) and non-recurrent (NonRec, n = 65) groups (Fig. 1a). Clinicopathological characteristics are provided in Supplementary Table 1.

a Schematic representation of the study design. b Clinicopathological features and somatic gene mutations between Rec and NonRec NSCLC groups in both FF and FFPE cohorts. Clinicopathological features and somatic gene mutations of cases with extrathoracic recurrence (c) and intrathoracic recurrence (d). e Violin plot depicting contributions of mutational signatures in Rec (n = 25) and NonRec (n = 22) groups. f Violin plot depicting HRD score in Rec (n = 53) and NonRec (n = 55) groups. P-values are obtained from unpaired two-sided Wilcoxon ranked-sum test. The boxes indicate the median value, interquartile range, with whiskers extending from the box boundaries to upper/lower quartile ± 1.5 interquartile range. LASC, lung adenosquamous carcinoma. The schematic in (a) was created using BioRender (https://www.biorender.com). Source data are provided as a Source Data file.
Primary NSCLC tumors from patients who develop recurrence exhibit distinct genomic features
To reveal the genomic characteristics associated with the post-operative recurrence, we performed a comprehensive analysis covering 108 NSCLC patients, including 47 patients with FF tissues and 61 patients with FFPE tissues, which unveiled a spectrum of somatic mutations, gene signatures, clonal architectures, and structural variations (SVs) across Rec and NonRec groups.
Consistent with previous studies20,21, we found that known somatic mutations in oncogenes and tumor suppressor genes including TP53 (47%), EGFR (30%), APC (11%) were prevalent in our cohort (Fig. 1b, Supplementary Data 1). We also applied a finer subtype classification to LUAD samples, assigning them to low-grade (lepidic), mid-grade (acinar, papillary) and high-grade (micropapillary, solid) predominant groups22. The recurrence rate was highest in patients with tumors in high-grade predominant group. TP53 mutation was the most prevalent mutation among all groups, yet its occurrence was significantly higher in the predominantly high-grade group (75%) than in the mid-/low-grade predominant groups (46% and 29%, respectively; Supplementary Fig. 1a). Moreover, TP53 mutation was more frequent in Rec group of LUAD (Supplementary Fig. 1b), suggesting a potentially higher malignant nature. Then, we further analyzed the mutations related to site-specific differences and timing of recurrence. Compared to the intrathoracic recurrence, the patients developed extrathoracic recurrence had higher mutation proportion of known driver genes including TP53 and EGFR (Fig. 1c-d). TP53 missense mutation in DNA-binding domain has been reported to influence the function of p5323, thus we also investigated its role in timing of recurrence and revealed that it was significantly associated with poor recurrence-free survival (RFS) (Supplementary Fig. 1c).
According to mutation profiles, we identified 4 mutational signatures (Sig1, Sig2, Sig3 and Sig4) (Supplementary Fig. 2a, b). Sig 1 is associated with the APOBEC family, while Sig 2 related to defective DNA mismatch repair (dMMR) is defined by C > T transitions24, both of which were increased in Rec group (Fig. 1e). In addition, despite no difference on copy number variation (CNV) burden and tumor mutation burden (TMB) between Rec and NonRec groups, the homologous recombination deficiency (HRD) score was significantly higher in LUAD Rec group (Fig. 1f, Supplementary Fig. 2c-d).
To shed light on the clonal architectures associated with NSCLC recurrence after surgery, we performed the phylogenetic analysis using PyClone-VI. Among all patients, multiple clones were profiled in most cases (Supplementary Fig. 3a). Then we examined whether the patterns of phylogeny were associated with the lung cancer recurrence. Survival analysis indicated LUAD with multiple clones was associated with a significantly worse RFS (Supplementary Fig. 3b). In addition, among LUAD cases, the inferred phylogeny demonstrated that driver mutations including EGFR, MET and ALK occurred in clones with high cellular prevalence, which could be designated as early mutations triggering tumorigenesis. Tumor suppressor TP53 mutation rarely occurred in clones with maximum cellular prevalence in LUAD NonRec group, while its frequency in major clones of LUAD Rec group was significantly increased (Supplementary Fig. 3c, d), indicating a potential contributor for LUAD recurrence. For LUSC, the tumor suppressors were mostly detected in major clones among both Rec and NonRec groups (Supplementary Fig. 3a, e). Long-read sequencing could enhance the characterization of SVs that was possibly associated with NSCLC recurrence25,26, thus we also detected SVs between Rec and NonRec groups. Among the somatic SV landscape, duplications emerged as the predominant type with a total of 891 in our cohort, followed by 687 deletions (Supplementary Fig. 3f). Based on reported NSCLC-related genes20,21,26,27, we identified SV statuses in selected oncogenes and tumor suppressors (Supplementary Fig. 3g). Among these SVs, in the LUSC Rec group, patient FF_33 had a deletion in PTEN, whose expression was significantly decreased compared with normal sample (FPKM: 975.294 vs 2648.635) (Supplementary Fig. 3h). Moreover, another case FF_41 in LUAD Rec group exhibited a significant duplication in EGFR, and the RNA-seq indicated a sharply increased expression level compared with paired normal sample (FPKM: 7777.238 vs 2102.873) (Supplementary Fig. 3i). Particularly, both of them had no somatic mutation for corresponding genes, suggesting that SVs have the potential to regulate downstream transcriptomic alterations and trigger the NSCLC recurrence after surgery.
To summarize, we provided a comprehensive genomic landscape and analyzed distinct genomic factors linked with NSCLC recurrence. We delineated that increased genomic instability characterized by a TP53 mutation-dominant phylogenetic pattern, higher HRD score, signatures of dMMR and APOBEC cytidine deaminases could contribute to lung cancer recurrence after surgery.
DNA methylation associated with post-operative recurrence
Alterations to the epigenetic landscape, exemplified by DNA methylation, have contributed to tumorigenesis, recurrence and metastasis in various cancers, including lung cancer15,28. Here, we applied nanopore sequencing data to evaluate DNA methylation profiles between Rec and NonRec NSCLC groups. We identified differentially methylated regions (DMRs) with Wald test (P < 0.05 and difference > 0.1). The differential methylation landscapes of LUAD and LUSC between Rec and NonRec groups were presented in Fig. 2a and Fig. 2b respectively. In general, 11,412 DMRs were found in LUAD, while 28,671 DMRs were identified in LUSC (Supplementary Fig. 4a). In both LUAD and LUSC, we observed similar trends in distributions of DMRs across different regulatory areas. Most of these changes happened in the intron regions, followed by the distal intergenic areas and promoter regions.

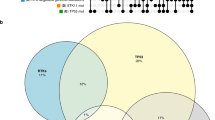
Distribution of DMRs in LUAD (a) and LUSC (b). c The comparison of methylation level between Rec (LUAD, n = 18 and LUSC, n = 5) and NonRec (LUAD, n = 10 and LUSC, n = 12) groups. P-values are obtained from unpaired two-sided Wilcoxon ranked-sum test. The boxes indicate the median value, interquartile range, with whiskers extending from the box boundaries to upper/lower quartile ± 1.5 interquartile range. d Venn diagrams depicting the hypomethylated and upregulated genes between Rec and NonRec groups in LUAD (top) and LUSC (bottom). Activity map integrating the association between methylation of TF binding site and expression of target genes in LUAD (e) and LUSC (f). g Correlation between methylation level of TEAD1 binding motif and PRAME expression. h Correlation of TEAD1 and PRAME expression. P-values are obtained from the two-sided t-test, and “R” represents the Pearson correlation coefficient. Source data are provided as a Source Data file.
Analysis of CpG methylation levels indicated that both LUAD and LUSC displayed the significant hypomethylation in Rec group compared to NonRec group (Fig. 2c). To identify the epigenetically dysregulated pathways in NSCLC recurrence, we conducted pathway enrichment analyses of hypomethylated genes in the Rec group (Supplementary Fig. 4b-c), which demonstrated that EMT in LUAD and inflammatory response in LUSC were activated. EMT is a well-known cellular program participating in malignant progression through reshaping intercellular and cell-extracellular interactions29. In addition, KEGG analysis demonstrated that Rap1 signaling pathway and ECM-receptor interaction were enriched in both LUAD and LUSC Rec groups.
DNA methylation could regulate the gene expression, and the interplay of differential methylation and gene expression in primary lung cancer lesions has been utilized to predict cancer progression30. In total, we identified 1642 and 3613 significantly hypomethylated genes in Rec groups of LUAD and LUSC, respectively. Additionally, we examined expression levels of hypomethylated genes in Rec group. It was intriguing that we discovered 6 genes in LUAD and 8 genes in LUSC that were hypomethylated and upregulated simultaneously in Rec group (Fig. 2d). Transcription factors (TFs) binding could be diminished by methylation on their DNA binding sites31. Thus, to investigate how methylation of TF binding sites impacts expression of target genes, we identified sets of TFs driving the overexpression of hypomethylated genes for further analyses. Subsequently, we utilized the activity scores to evaluate the association between expression of specific genes and the methylation level of TF binding sites (Fig. 2e, f). Here, we observed that the most strongly activated gene was PRAME located on chromosome 22. Among the relative TFs of PRAME, TEAD1 plays a pivotal role in regulating tissue homeostasis and tumorigenesis. Aberrant activation of TEAD1 and its coactivator YAP/TAZ have been implicated in progression of various cancers32,33,34. Our results also revealed that hypomethylation at the TEAD1 binding site was correlated with PRAME overexpression (Fig. 2g), and the TEAD1 expression was positively related to PRAME expression (Fig. 2h).
In summary, significant hypomethylation in Rec group was observed compared to NonRec group, which might affect the gene expression levels and lead to post-operative recurrence.
Transcriptomic signatures related to stage I NSCLC recurrence after surgery
Differential expression analysis was utilized to identify transcriptomic features associated with post-operative recurrence in stage I NSCLC. There were a total of 85 differentially expressed genes (DEGs) in LUAD and 206 DEGs in LUSC between Rec and NonRec groups (adjusted P < 0.05 and |log2FoldChange| > 1) (Fig. 3a, b, Supplementary Fig. 5a). Highly expressed genes in recurrent LUAD included PRAME, DRAIC, and DUXAP8 (Fig. 3a), while COL22A1, TIMD4, and PLA2G2D were significantly upregulated in Rec group of LUSC (Fig. 3b). Then we integrated clinical data to evaluate the prognostic value of DEGs. DRAIC and DUXAP8 in LUAD, as well as TIMD4 and COL22A1 in LUSC were demonstrated as potential “High-Risk Genes” correlated with poorer RFS (Supplementary Fig. 5b). Interestingly, DRAIC and DUXAP8 were considered as oncogenes promoting the progression of breast cancer and pancreatic cancer, respectively35,36. Moreover, the cavity-resident macrophages with high expression of TIMD4 could impair anti-tumor activity of CD8 + T cells to facilitate the cancer progression37.

a Volcano plot showing DEGs in Rec and NonRec LUAD groups (DESeq2 method). b Volcano plot showing DEGs in Rec and NonRec LUSC groups (DESeq2 method). c EMT and angiogenesis scores between Rec and NonRec LUAD groups. d EMT and angiogenesis scores between Rec and NonRec LUSC groups. e KEGG and GO pathway enrichment between Rec and NonRec LUAD groups (hypergeometric distribution). f KEGG and GO pathway enrichment between Rec and NonRec LUSC groups (hypergeometric distribution). g The naïve, exhausted, and cytotoxic states assessment derived from ssGSEA scores between Rec (n = 23) and NonRec (n = 22) groups. P-values are obtained from GSEA (c, d) and unpaired two-sided Wilcoxon ranked-sum test (g). The boxes indicate the median value, interquartile range, with whiskers extending from the box boundaries to upper/lower quartile ± 1.5 interquartile range. Source data are provided as a Source Data file.
To explore the biological processes driving the lung cancer recurrence, we then conducted gene set enrichment analysis (GSEA) on the MSigDB Hallmarks gene sets and revealed enriched pathways through comparison of Rec and NonRec groups (Fig. 3c, d, Supplementary Fig. 5c). Activation of the pathways such as EMT and angiogenesis, known to trigger cancer cell migration and dissemination38,39, was observed in Rec groups of LUAD and LUSC (Fig. 3c, d). In addition, GO and KEGG analyses also indicated extracellular matrix and cell adhesion molecules-related pathways were upregulated in Rec group (Fig. 3e, f ). Notably, extracellular matrix remodeling could contribute to detachment of epithelial cells from adjacent cells and basement membrane, which is essential for EMT40. Cell adhesion molecules such as integrin might lead to tumor angiogenesis41. Therefore, EMT and angiogenesis are common biological behaviors associated with the stage I NSCLC recurrence.
The tumor immune microenvironment (TIME) plays the crucial role in lung cancer progression. To characterize the TIME correlated to NSCLC recurrence, the single sample gene set enrichment analysis (ssGSEA) scores were calculated with defined gene sets (Naïve: TCF7, SELL, LEF1, CCR7; Exhausted: LAG3, TIGIT, PDCD1, HAVCR2, CTLA4; Cytotoxic: TNFSF10, CST7, GZMA, GZMB, NKG7, GNLY, IFNG, PRF1). And we observed that the naïve and exhausted signatures were evidently higher in the Rec group (P = 0.03179, 0.01580, respectively), while the cytotoxic score was not significantly different between Rec and NonRec groups, suggesting an immunosuppressive environment conducive to recurrence (Fig. 3g).
Above all, our investigation revealed the key DEGs associated with NSCLC recurrence and highlighted that the activated EMT and angiogenesis signatures might contribute to the post-operative recurrence of stage I NSCLC. Moreover, the higher exhausted score was observed in Rec group than NonRec group, suggesting a suppressive TIME.
PRAME as a critical gene for stage I LUAD recurrence after surgery
As aforementioned, we observed that PRAME was highly expressed and hypomethylated in LUAD Rec group. In function, PRAME regulates the cell death and retinoic acid receptor signaling42, which could contribute to the tumor progression and worse prognosis.
To achieve an integrated perspective, we created a coordinate axis integrating the dimensions of transcriptomic and epigenomic results in LUAD Rec group compared to NonRec group, where the PRAME gene was identified to be the most significant gene considering both differential expression and differential methylation levels (Fig. 4a, Supplementary Data 2). In correlation analysis, an inverse correlation was evident between methylation and expression of PRAME in LUAD samples (Fig. 4b). Further, based on the expression level of PRAME, LUAD samples were classified into PRAME-high and PRAME-low groups. Hallmark analysis showed that multiple proliferation and metastasis-related pathways such as EMT, MYC targets v1, E2F targets and MTORC1 signaling were significantly positively enriched in PRAME-high group, compared to PRAME-low group (Fig. 4c). Moreover, survival analysis showed high expression of PRAME was associated with poor RFS with a P-value of 0.0065 (Fig. 4d).

a Scatter plot correlating the expression fold change with the differential methylation level of overlap genes. b Scatter plot demonstrating hypomethylation of PRAME was associated with its upregulated expression between Rec and NonRec groups. P-values are obtained from the two-sided t-test. “R” represents the Pearson correlation coefficient (a, b). c Biological processes involved in PRAME high/low expression. P-values are obtained from GSEA. d Kaplan-Meier analysis showing the RFS characterized by low (blue) and high (red) expression of PRAME. The P-value is obtained from the two-sided log rank test. e The cell migration rates of PRAMEOE cells compared to PRAMENC A549 cells (n = 3 biological replicates). GSEA revealing the enrichment of EMT (f) and E2F targets (g) in the PRAMEOE cells compared to PRAMENC cells. P-values are obtained from GSEA. h KEGG pathway enrichment of PRAMEOE cells compared to PRAMENC cells (hypergeometric distribution). i The relative expression levels of EMT-related genes in PRAMENC and PRAMEOE A549 cells (n = 3 technical replicates). j Representative western blotting pictures of three independent experiments for PRAME, mTOR, Rap1, and PCNA in PRAMENC and PRAMEOE A549 cells. k Relative cell viability of sgPRAME and sgScr A549 cells (n = 3 biological replicates). l The cell migration rates of sgPRAME and sgScr A549 cells (n = 3 biological replicates). m The relative expression levels of EMT-related genes in sgPRAME and sgScr A549 cells (n = 3 technical replicates). n Representative western blotting pictures of three independent experiments for PRAME, mTOR, Rap1, and PCNA in sgPRAME and sgScr A549 cells. o Representative brightfield (up) and fluorescent (down) images showing metastatic lesions in the lungs of recipient mice with sgPRAME and sgScr A549 cells. p Representative images of H&E staining of the lungs of recipient mice with sgPRAME and sgScr A549 cells. q. Statistical graphs showing the lesion number of metastases in the lungs of recipient mice (n = 5 mice). Data are shown as mean ± SD (e, i, l, m) and mean ± SEM (q) (Two-tailed Student’s t test). Data (k) are shown as mean ± SD (Two-way anova). Source data are provided as a Source Data file.
To investigate the role of PRAME in LUAD recurrence, we firstly amplified PRAME cDNA into A549, PC9, and H1299 cells (PRAME-OE cells). Wound healing assay revealed a significantly enhanced cell migration rate in PRAME-OE cells, compared to control cells (Fig. 4e and Supplementary Fig. 6a, d). Transcriptome analysis indicated that PRAME overexpression upregulated the EMT gene signature, the E2F targets gene signature, and multiple pathways involved in cell proliferation and migration (Fig. 4f–h). The results of RT-qPCR validated that the relative expression levels of the EMT-related genes ACTA2, COL1A1, DAB2, MMP2, and TAGLN were upregulated after PRAME overexpression (Fig. 4i and Supplementary Fig. 6b, e). Consistent with transcriptome analysis, western blotting showed that proliferation and migration-associated proteins mTOR, PCNA, and Rap1 were upregulated in PRAME-OE cells compared to control cells (Fig. 4j and Supplementary Fig. 6c, f). Additionally, we introduced small interfering RNA (siRNA) targeting PRAME gene to repress its expression in A549, H1299, and PC9 cells. siPRAME cells displayed significantly suppressed cell proliferation and migration compared to siNC cells. Western blotting showed that PRAME gene silencing significantly inhibited the expression of mTOR, PCNA, and Rap1 proteins (Supplementary Fig. 7).
To further validate the in vivo role of PRAME, we designed two single guide RNAs (sgRNAs) with the CRISPR Design Tool and transduced sgRNAs into A549 cells with Cas9. PRAME disruption was confirmed by western blotting. PRAME disruption significantly inhibited cell proliferation and migration (Fig. 4k, l). RT-qPCR showed that the relative expression levels of the EMT-related genes ACTA2, COL1A1, MMP2, and TAGLN were significantly repressed in sgPRAME A549 cells compared to those with sgScr (Fig. 4m). Consistently, western blotting showed that PRAME disruption reduced the expression levels of cell proliferation and migration-related proteins mTOR, Rap1, and PCNA (Fig. 4n). Then sgPRAME cells were transplanted into the recipient mice through tail vein injection. 24 days after transplantation, mice were harvested. Biopsy showed some lesions with specific mcherry fluorescent protein expression in lungs, which indicated that the lesions were derived from the transplanted cells. Decreased lesions were observed in the lungs of recipient mice with sgPRAME cells than in the lungs of control mice (Fig. 4o). Consistently, pathological analyses showed that there were reduced lesions in lungs of recipient mice with sgPRAME cells (Fig. 4p, q). Taken together, these data strongly suggested that PRAME deficiency inhibited LUAD metastasis in mice.
To summarize, our integrated analysis identified the PRAME gene as a key mediator of LUAD recurrence due to its high expression and hypomethylation. We comprehensively validated the biological function of PRAME through a series of in vitro and in vivo experiments. These findings elucidated the crucial function of PRAME activation in enabling LUAD progression, highlighting its potential as therapeutic target for preventing recurrence.
Loss of AT2 features and gain of malignancy are related to LUAD recurrence after surgery
To unveil the role of tumor ecosystem in post-operative recurrence of LUAD, we collected fresh tumor specimens and adjacent non-tumor samples from 14 patients for scRNA-seq. Following quality control procedures, high-quality transcriptomes from a total of 118,860 cells were obtained (Supplementary Data 3). Annotated by known markers, these cells were assigned into 15 major clusters including epithelial cells (AT1, AT2, basal, ciliated, and club cells), fibroblasts, endothelial cells as well as immunocytes (lymphocytes, and myeloid cells) and visualized by t-distributed stochastic neighbor embedding (tSNE) (Fig. 5a and Supplementary Fig. 8a, b). Although all cell clusters were presented in both Rec and NonRec groups, AT2 cells were more abundant in the Rec group, while AT1 cells and CD8 + T cells were more enriched in NonRec group (Fig. 5b).

a tSNE depicting the composition of 15 different phenotypes from LUAD patients including 10 patients in NonRec group and 4 patients in Rec group. b Cell percent ratio in NonRec and Rec groups. c The significantly enriched hallmarks of highly expressed genes in AT2 cells between Rec and NonRec groups (hypergeometric distribution). d CNV inferred by scRNA-seq. e Ridge plot and heatmap showing the cell density and dynamic changes in gene expression of AT2 cells with the pseudotime. Source data are provided as a Source Data file.
Since AT2 cells were considered as the origin of LUAD43,44,45, we first investigated the transcriptomic heterogeneity of AT2 cells between Rec and NonRec groups. According to gene expression profile, AT2 cells were assigned into AT2-SFTPC cells characterized by the high expression of typical AT2 markers including SFTPC, and AT2-like cells defined by decreased expression of SFTPC as well as upregulated cell proliferation genes46,47. We further subgrouped AT2-like cells into 5 clusters (AT2-like 1-5), among which AT2-like 2 took a higher proportion in Rec group (Supplementary Fig. 8c). The pathway enrichment indicated the hallmarks of tumor progression including hypoxia, EMT, and angiogenesis were enriched in AT2 cells of Rec group (Fig. 5c), which were mainly reflected in AT2-like 1 and AT2-like 2 clusters (Supplementary Fig. 8d, e). To further investigate the malignant state of AT2 cell subpopulations, inferCNV was applied with adjacent normal tissues as the reference. As expected, almost no CNV events were detected in normal tissues. AT2 cells from the Rec group exhibited relatively higher heterogeneity of CNV than NonRec group (Fig. 5d). For instance, CNV amplifications were more enriched in the chromosomes 7 and 8 in AT2 cells from the Rec group, and deletions on chromosomes 18, 19 and 20 were also evident. Given the relationship between cancer and large-scale CNV, AT2-like cells were termed LUAD cells (Supplementary Fig. 8f). Specifically, AT2-like 2 subpopulation obtained significantly larger scale of chromosomal CNVs than other clusters, demonstrating a more malignant phenotype. In addition, to better understand the evolutionary dynamics of AT2 cell clusters during LUAD recurrence, we performed pseudotime analysis using Monocle 2. We detected two different differentiation trajectories. The route 1 trajectory exhibited the increased abundance of AT2-SFTPC cells, which was mainly consisted of cells from NonRec group. However, throughout the route 2 developmental trajectory with high expression of MDK, SAA1, and ITGB8 genes, the proportion of AT2-SFTPC cells was reduced, while AT2-like 2 maintained a high proportion. Moreover, route 2 ended with cells from Rec group, and the CNV burden exclusively accumulated along the route 2 trajectory (Fig. 5e, Supplementary Fig. 8g). Therefore, route 2 resembled the cellular trajectory of post-operative recurrence. Overall, we have provided insights into the heterogeneity of AT2 cells and identified AT2-like 2-cell subpopulation, which might contribute to LUAD recurrence.
Transcriptional reprogramming of immunocytes towards a recurrent microenvironment in LUAD
To resolve the distinctive TIME contributing to the lung cancer recurrence, we next conducted unsupervised clustering for macrophages/monocytes and CD8 + T cells. Macrophages/monocytes held the highest abundance in myeloid cells (Fig. 5a), which are reported to be pivotal in reshaping tumor microenvironment (TME)48. Here, they were subclustered into 5 clusters, namely Macro_KLRB1, Macro_FABP4, Macro_PPARG, Macro_SPP1 and monocytes (Fig. 6a). The pathway enrichment exhibited an activated involvement in inflammatory pathways in macrophages/monocytes of Rec group compared to NonRec group (Fig. 6b). The Macro_SPP1 highly expressing CD14 was defined as monocyte-derived macrophages (MDMs) playing crucial pro-tumorigenic role, while Macro_PPARG was considered as the tissue-resident macrophages (TRM)49,50. Despite the similar proportions of macrophages/monocytes among groups (Fig. 5b), the abundance of specific subpopulations, including Macro_SPP1 and monocytes, was higher in Rec group than NonRec group (Fig. 6c). Furthermore, pseudotime analysis revealed that Macro_SPP1 and monocytes were predominantly at the end phase of differentiation path characterized by upregulation of VEGFA, a crucial modulator of angiogenesis51, and activation of inflammation-related pathways via KEGG enrichment (Fig. 6d). Consistently, hallmark pathway analysis also indicated that inflammatory response was enriched in these two clusters (Fig. 6e, f ). Taken together, our analyses revealed that enrichment of Macro_SPP1 and monocytes contributed to the lung cancer recurrence via triggering angiogenesis and tumor-promoting inflammation.

a tSNE plots of macrophages/monocytes colored by subtypes. b The significantly enriched hallmarks of highly expressed genes in macrophages/monocytes between Rec group and NonRec group (hypergeometric distribution). c Cell percent ratio of macrophage/monocyte subclusters between Rec and NonRec groups. d Ridge plot and heatmap showing the cell density and dynamic changes in gene expression of macrophages/monocytes with the pseudotime. The significantly enriched hallmarks of highly expressed genes in Macro_SPP1 (e) and monocytes (f) (hypergeometric distribution). Source data are provided as a Source Data file.
CD8 + T cells were assigned into 5 subclusters (CD8-CCR6, CD8-GZMH, CD8-GZMK, CD8-LAG3, and CD8-XCL1). Compared with the NonRec group, the higher proportions of CD8-CCR6, CD8-LAG3, and CD8-GZMK were observed in Rec group (Supplementary Fig. 9a). To illustrate the transcriptional heterogeneity of CD8 + T cells between Rec and NonRec groups, we assessed the expression states of cytotoxic, exhausted and naïve phenotypes. Among these subpopulations, CD8-CCR6 overexpressed both exhausted markers (CTLA4 and TIGHT) and naïve genes (SELL and CCR7) and thus designated as early-exhausted CD8 + T cells. CD8-LAG3 were defined as exhausted CD8 + T cells, CD8-GZMH showed the cytotoxic feature, while CD8-XCL1 and CD8-GZMK exhibited the naïve characteristics (Supplementary Fig. 9b). We next investigated the cell transitions of CD8 + T cells. In pseudotime analysis, the transition initiated with CD8-GZMH, the cytotoxic subtype with higher proportion in the NonRec group, and eventually reached an exhausted state (Supplementary Fig. 9c). Additionally, the pathway analysis also revealed the immunodeficiency-related pathway was enriched, and cytotoxicity-associated pathway was repressed in CD8 + T cells from Rec group compared to NonRec group (Supplementary Fig. 9d, e). Thus, the immune-suppressed TME featured by low cytotoxic and high exhausted state was associated with post-operative recurrence of lung cancer.
Cell-cell interactions play a crucial role in cancer progression52. We used CellPhoneDB to decipher the potential crosstalk contributing to lung cancer recurrence based on ligand-receptor (L-R) interactions. Generally, the cellular interactions were much fewer in Rec group than NonRec group (Supplementary Fig. 10a, b). Specifically, the reduced crosstalk between AT2 cells and myeloid cells in Rec group was evident, indicative of the less contact between tumor cells and corresponding environment. In spite of the decreased interactions, the cellular interactions in regard to CD8-GZMK and CD8-CCR6 were stronger in Rec group than NonRec group (Supplementary Fig. 10c, d). Therefore, the general diminished cell-cell communications, along with the enhanced regulations of exhausted T cells, might be the essential biological behavior contributing to the colonization of lung cancer cells in the recurrent sites.
To better support our findings, we applied immune infiltration and survival analyses based on bulk RNA-seq data for major subphenotypes. Consistent with findings of scRNA-seq analysis, the abundance of CD8-CCR6, CD8-GZMK, CD8-LAG3, Macro_SPP1, and monocytes were significantly higher in Rec group than NonRec group (Supplementary Fig. 10e, f ). The AT2 like 2 and Macro_SPP1 were indeed related to poorer prognosis (Supplementary Fig. 10g, h), further validating their role in recurrence.
In conclusion, the enrichment of Macro_SPP1 with highly inflammatory signature, immunosuppressive state, and reduced cell-cell communications in TME might be crucial for stage I LUAD recurrence.
Multi-omics integration yields insights into the recurrence-risk stratification and precise therapy of NSCLC
Given that we have characterized the genomic, epigenomic, and transcriptomic landscape between Rec and NonRec groups, unsupervised non-negative matrix factorization (NMF) clustering was adopted to divide the tumors into 4 subclusters (nmf1, nmf2, nmf3, nmf4) (Fig. 7a, Supplementary Fig. 11a,b). Survival analyses demonstrated integrated subtypes could help stratify the recurrence risk for patients, among which nmf1 and nmf2 exhibited a high risk of recurrence, nmf3 represented a moderate risk of recurrence, while cases in nmf4 showed a low risk of recurrence (Supplementary Fig. 11c). We further delved into the multi-omics characteristics of each subtype. Despite of high frequency of EGFR mutation in LUAD, tumors in both nmf1 and nmf2 groups lacked EGFR mutation (Fig. 7a, b). The nmf1 subtype was mainly composed of tumors from both LUAD and LUSC Rec groups, which was featured by enrichment of EMT, angiogenesis, and hypoxia pathways, as well as highest exhausted score (Fig. 7c–f, Supplementary Fig. 11d), suggesting the strong invasiveness and potential of immune escape. For nmf2 subcluster primarily involving tumors from LUAD Rec group, MYC targets, G2M checkpoint, E2F targets pathways were activated, and dMMR signature was also enriched (Fig. 7f, Supplementary Fig. 11e). The nmf3 subcluster consisted of LUAD tumors, and half of them were from Rec group, characterized by high proportion of EGFR mutations (Fig. 7a, b). Lastly, the nmf4 including an overwhelming majority of tumors from LUSC NonRec group was distinguished by a remarkable smoking signature, as well as comparatively low level of angiogenesis score (Fig. 7d, Supplementary Fig. 11e).

a Heatmap showing the multi-omics integration of NSCLC tumors into 4 NMF-derived clusters. Panels exhibiting clinical features, mutations of selected genes, methylation level, mutational signatures, and angiogenesis score. b The proportions of gene mutation for each subcluster. The enrichment of EMT (c), angiogenesis (d), and hypoxia (e) assessed via ssGSEA in each subcluster. f Heatmap of pathway enrichments in each subcluster detected by ssGSEA. P-values are obtained from unpaired two-sided Wilcoxon ranked-sum test (c–e) and GSEA (f). The boxes indicate the median value, interquartile range, with whiskers extending from the box boundaries to upper/lower quartile ± 1.5 interquartile range. *P value < 0.05. Source data are provided as a Source Data file.
Based on the molecular characteristics above, we further focused on therapeutic vulnerabilities of certain subcluster. The nmf1 subcluster exhibited the highest level of angiogenesis activation and exhausted score calculated by the expression of genes including LAG3, TIGIT, PDCD1, and CTLA4 (Fig. 7d, Supplementary Fig. 11d). The previous studies have highlighted that the anti-angiogenic agents such as bevacizumab could improve the prognosis of NSCLC with angiogenesis features, and T cells with high expression of exhausted genes are closely related to immunotherapy efficacy53,54. Interestingly, VEGFR inhibitor-immune-checkpoint inhibitor (ICI) combinations were reported to enhance the efficiency of immunotherapy55. Therefore, combination therapy of ICIs and anti-angiogenic agents might be an optimal treatment for individuals in nmf1. In addition, Aurora kinase inhibition has been proved to be a novel therapeutic strategy for cancers with MYC amplification, and targeting WEE1 could inhibit the G2M pathway56,57, indicative of the potential options for nmf2 subtype. The recurrence rate in nmf3 group, exhibiting high EGFR mutation rate, was as high as 50% among our cohort (Fig. 7a). Thus, EGFR-TKI might be benefit for cases with EGFR mutation categorized into nmf3 cluster to decrease the risk of recurrence after surgery. Additionally, the patients in nmf4 subcluster were mainly from NonRec group, hence the adjuvant therapy might be unnecessary. These results underscored the potential of multi-omics subcluster-based therapy in NSCLC to guide the personalized treatment after surgery and reduce the risk of recurrence.
Discussion
Cancer recurrence is a complex process orchestrated by distinct molecular features of tumor cells and the dynamic variations within the tumor microenvironment58,59,60,61. To comprehensively elucidate the biological behavior of post-operative recurrence in NSCLC, we performed multi-omics profilings on a large cohort with NSCLC to analyze the crucial molecular characteristics promoting the post-operative recurrence. We found that NSCLC samples in the Rec group exhibited an increased genomic instability and DNA hypomethylation. Transcriptomic analyses also revealed the activation of EMT and angiogenesis pathways in the Rec group. Specifically, we identified and validated PRAME as a critical gene promoting the LUAD recurrence. Additionally, our study presented a high-resolution landscape of the cellular heterogeneity and intercellular crosstalk in Rec and NonRec groups of NSCLC. Finally, we conducted multi-omics clustering to stratify the recurrence risk of NSCLC patients and guide precise post-operative treatment.
The genomic instability featured by dysregulation of oncogenes and tumor suppressor genes, clonal evolution, and SVs, has been documented to promote tumor progression and recurrence62,63. In our study, the mutation frequency of TP53 was significantly increased in the Rec group of LUAD. TP53 mutation is a frequent event in multiple cancers, which might be associated with cancer recurrence64,65. We also underscored that the presence of predominantly solid or micropapillary pattern, highly invasive LUAD histological subtypes66, was related to LUAD recurrence. In addition, our results revealed APOBEC and dMMR signatures were enriched in Rec group compared with NonRec group, both of which have been reported to drive the DNA damage and tumor evolution67,68, and higher HRD score was found in LUAD Rec group. The tumor clonal heterogeneity is highly represented by genomic complexity and offers soil for tumor evolution and recurrence14,69. Thus, we further investigated the clonal architectures and revealed that a TP53 mutation-dominant phylogenetic pattern might promote the LUAD recurrence. In terms of large-scale chromosomal alterations, we found PTEN deletion and EGFR duplication could facilitate the recurrence via gene expression regulation. In summary, our analyses emphasized the importance of enhanced genomic instability in lung cancer recurrence after surgery.
Combination of the epigenetic and transcriptomic profiles could provide deeper insights into biological mechanism of NSCLC recurrence. In general, the recurrent cases exhibited a hypomethylation pattern. We conducted pathway enrichment analyses of hypomethylated genes in the Rec group and found the activation of crucial pathways associated with cancer recurrence such as EMT, Rap1 signaling, and ECM-receptor interaction. The hypomethylation could be linked to gene expression, therefore transcriptome analyses also showed that the same pathways including EMT were significantly positively enriched in the Rec group. Consistently, previous studies showed that DNA hypomethylation was an adverse factor for tumor recurrence and metastasis70,71,72. Binding sites for stemness- and proliferation-associated genes OCT4, SOX2, NANOG, and SIN3A are specifically hypomethylated in breast cancer71. In this study, we explored the methylation of TF binding sites in regulating targeted gene expression. In Rec group, we found hypomethylation of TEAD1 binding site associated with higher expression of PRAME. TEAD1 directly binds the YAP/TAZ complex, acting as gene expression regulators and harmonizing cellular growth, differentiation and development, and Hippo-YAP pathway is frequently dysregulated in human cancers73,74,75. Our findings further support TEAD1 as a potential therapeutic target to prevent NSCLC recurrence, with promise in precision oncology76. Correlative analyses between the DNA methylation and transcriptomic profiles showed enrichment of multiple proliferation and recurrence-related pathways and genes in the Rec group. Further, these high-risk transcriptomic and epigenetic traits specific to the Rec group may serve as predictors of post-operative recurrence risk and inform clinical treatment77.
Additionally, we identified and validated PRAME as a potential biomarker and therapeutic target, which has been defined as a cancer-testis antigen78,79. High levels of PRAME expression have been reported to correlate with unfavorable OS in medulloblastoma and acute myeloid leukemia80,81. And in acute lymphoblastic leukemia, targeting PRAME could prevent recurrence after hematopoietic stem cell transplant82. In our study, PRAME exhibited significant hypomethylation and overexpression in the Rec group of LUAD, associated with poor RFS. To validate its function, we conducted both in vitro and in vivo assays. The results demonstrated that PRAME overexpression significantly enhanced the proliferative and migratory abilities of LUAD cells, while PRAME silencing inhibited these capabilities. Furthermore, compared to the control group, mice with sgPRAME had fewer lung metastatic lesions, indicating deficiency of PRAME could significantly suppress lung cancer metastasis. Therefore, these results indicate that PRAME may play an important role in promoting LUAD recurrence and could serve as potential therapeutic target.
The TME plays a pivotal role in tumor initiation and progression83. AT2 cells act as stem cells in lung regeneration after damage84, and are often considered a major origin of LUAD13,85. In this study, we found that the significantly enriched AT2-like 2 subpopulation in the LUAD Rec group had a higher CNV burden and was involved in activated pathways such as EMT and angiogenesis, collectively indicating the increased malignancy. Notably, we discovered MDK and SAA1, the malignant genes promoting cancer progression86,87, may be responsible for LUAD recurrence. Additionally, we observed that CD8 + T cells in the Rec group were in a low cytotoxic and high exhausted state, which has been also observed in hepatocellular carcinoma recurrence88. Regarding myeloid cells, the Macro_SPP1 marked by inflammatory signature was enriched in the Rec group. Indeed, monocyte-derived Macro_SPP1 represents a shared cell state in lung cancer, pulmonary fibrosis, and COVID-1989, and is predominant in liver metastasis with a pro-metastasis role90. Consistently, highly angiogenic, inflammatory, and immune-suppressed TME profiled in our study were the distinguished features for highly proliferative and invasive LUAD91. Moreover, the lack of intercellular interactions and enhanced regulations of exhausted T cells were evident in Rec group, which could contribute to the formation of suppressive TIME. Interestingly, a recent study also revealed that reduced interactions would reshape the metastatic microenvironment of pancreatic cancer92. Overall, scRNA-seq analyses revealed the enrichment of highly aggressive AT2-derived malignant cells, exhausted CD8 + T cells, and Macro_SPP1 might be an important mechanism responsible for lung cancer recurrence.
The principal innovation of this study lies in the application of multi-omics technology to comprehensively analyze the characteristics in early-stage NSCLC associated with recurrence. Unsupervised clustering stratified patients into high-risk (nmf1, nmf2), moderate-risk (nmf3), and low-risk (nmf4) recurrence groups, and each subcluster showed the distinct biological features and therapeutic vulnerabilities. The nmf1 subtype displayed enrichment of angiogenesis and the highest exhausted score, indicating the potential response to the combination therapy of anti-angiogenic drugs and immunotherapy93. The nmf2 subtype showed a significant activation of MYC targets, G2M checkpoint pathways and thus may benefit from aurora kinase inhibitors and WEE1 inhibitors56,94. Furthermore, the nmf3 subtype characterized by a high frequency of EGFR mutations may be sensitive to EGFR-TKI. Lastly, we propose that adjuvant therapy is not required for nmf4 subtype exhibiting the lowest risk of recurrence. Therefore, the multi-omics subclusters might guide the precise treatment and reduce the post-operative recurrences of stage I NSCLC.
Nevertheless, further efforts should be applied to validate outcomes of this study. Although the multi-omics profilings could inform the stratification of recurrence risk and precise post-operative management, experimental evidence and prospective multicenter studies are required for clinical application of our findings. Additionally, the cohort in our study involves resectable samples from primary lung cancers, which may limit the investigation of recurrent tumors. Therefore, incorporation of primary tumors and matched specimens from recurrent sites could better elucidate the developmental trajectory of lung cancer recurrence. Despite the existing limitations, our study provides a valuable insight into the mechanisms underlying NSCLC recurrence.
Taken together, this study has systematically provided a landscape of stage I NSCLC recurrence through multi-omics comparisons conducted from genomic, epigenomic, and transcriptomic dimensions. We hope that these findings will contribute to more effective recurrence risk stratification and precise therapy after surgery. This dataset could also represent a valuable resource to facilitate future exploration on basic and clinical research of NSCLC post-operative recurrence.
Methods
Patients and ethics statement
A total of 122 patients diagnosed as having NSCLC at West China Hospital, Sichuan University in China between 2014 and 2020 were enrolled in this study, among which 57 patients had recurrence during the follow-up until August 2024. All patients were treated surgically, and those received neoadjuvant therapy before surgery were excluded. Tumors and matched distal normal lung tissues were obtained during surgery. Normal tissues were obtained from a location 5 cm away from the tumor margin. All samples were evaluated by two expert pathologists independently to determine the pathological status. The tumor staging was determined according to the TNM system of the International Association for the Study of Lung Cancer (9th edition)95. This study protocol was approved by the Institutional Review Board of West China Hospital, Sichuan University (Ethics: Project identification code: 2020.232). As the information on sex or gender was not relevant in this study, no related analysis was carried out. The clinical characteristics of patients were shown in Supplementary Table 1, and all patients provided written informed consent for the collection of samples and the publication of relevant clinical information before enrollment.
Sample collection and preparation
Immediately after surgery, the resected tumors along with matched normal tissues were promptly processed. For the FF samples, tissues were snap-frozen in liquid nitrogen at −80 °C until further processing. The FFPE samples were fixed in 10% neutral buffered formalin for 24–48 h, ensuring thorough penetration and fixation, followed by embedding in paraffin to provide structural preservation suitable for long-term storage. The FR samples were put in Hank’s Balanced Salt Solution (HBSS, Life Technologies), and finely minced into cubes smaller than 0.5 mm3 using scalpels and transferred to a 15 mL conical tube (BD Falcon) with 8 mL pre-warmed HBSS, 1 mg/mL collagenase I as well as 0.5 mg/mL collagenase IV.
Library preparation for whole exome sequencing and data preprocessing
For WES, FF samples were processed using AllPrep DNA/RNA Mini Kit following the manufacturer’s protocol to efficiently extract high-quality DNA. In contrast, FFPE samples underwent deparaffinization and then DNA extraction. The concentration of DNA was assessed by Qubit® DNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, USA). DNA quantity and quality were monitored with 1% agarose gel. For DNA sample preparation, a total of 0.6 μg of genomic DNA per sample was utilized as input. Both FF and FFPE DNA samples were fragmented to an average size of 180–280 bp and subjected to DNA library creation following established Illumina paired-end protocols. To capture the exome, the Agilent SureSelect Human All ExonV6 Kit (Agilent Technologies, Santa Clara, CA, USA) was employed in accordance with the manufacturer’s instructions. Subsequently, the libraries were sequenced on the Illumina Novaseq platform, generating 150 bp paired-end reads.
Fastp (v.0.23.4) was used for quality control of the sequencing data with default parameters. MultiQC was then used to aggregate the quality control results across all samples. The sequencing reads demonstrated high base quality scores across the length of the reads. Next, reads were aligned to the reference genome GRCh38.p13 from GENCODE using BWA (v.0.7.17) mem with default parameters. Gene annotations were obtained from the GENCODE (v.42) gtf file. Samtools (v.1.6) was then utilized to sort the alignments and merge bam files across multiple lanes for the same sample. PCR duplicates were marked and removed using Sambamba (v.0.6.6-2)96. And the bam files were sorted again and indexed using samtools. Base recalibration was performed with GATK BaseRecalibrator, using known variants from dbsnp_146.hg38.vcf.gz, Mills_and_1000G_gold_standard.indels.hg38.vcf.gz, and 1000G_phase1.snps.high_confidence.hg38.vcf.gz. Finally, GATK ApplyBQSR was used to recalibrate the base quality scores across all reads.
Somatic variant calling
Single-nucleotide variants (SNVs) were called using GATK Mutect2 in tumor-normal mode, where a tumor sample was matched with a normal sample from the same patient. To filter out likely false positive calls due to deamination artifacts, Mutect2 was used to enable filtering on read orientation. GATK GetPileupSummaries, CalculateContamination, and LearnReadOrientationModel tools were subsequently utilized to calculate sample contamination. GATK FilterMutectCalls was used to further filter the initial Mutect2 callsets based on the previously calculated contamination and orientation bias metrics. Filtering criteria were: (1) removal of variants with base quality <18 and depth <10; (2) removal of variants with depth <5 in the tumor sample. Indels called by both Strelka2 and MuTect2 were considered true positives. Finally, called SNVs were annotated using GATK Funcotator and the somatic annotation database funcotator_dataSources.v1.7.20200521 s. Maftools package was utilized for variant visualization. Driver gene analysis was performed using Oncodrive and MutSigCV (v.1.4) algorithms, and important mutated genes included TP53, EGFR, and KRAS, etc.
Mutation signature analysis
We employed the NMF to explore mutation signatures, in which trinucleotideMatrix, estimateSignatures, and extractSignatures were used to detect signature profiles. Subsequently, Catalog of Somatic Mutations in Cancer (COSMIC) database was adopted as reference to define mutation patterns97. Cosine similarity (ranging from 0 to 1) was utilized for matching.
Copy number variation analysis
To identify copy number states from WES data, Sequenza (v.3.0.0) was used for read count normalization and segmentation to identify genomic regions with potential CNVs98.
Clone architecture analysis
Clone numbers were calculated based on variant allele frequency (VAFs) of somatic mutations employing PyClone-VI (v.0.1.1), and the parameters -c 40 -d beta-binomial -r 10 were used99. The input included read count of somatic mutation, copy numbers, and cellularity estimated from tumor and paired normal WES data using Sequenza (v.3.0.0).
HRD quantification
ScarHRD (v.0.1.1) was adopted to detect the HRD level from WES data using CNV as input100. HRD score was calculated via summarization of large-scale transitions (LST), telomeric allelic imbalance (TAI), and loss of heterozygosity (LOH).
Library preparation for transcriptome sequencing and data processing
RNA was extracted from FF tissues using AllPrep DNA/RNA Mini Kit following the manufacturer’s protocol. The quantity and quality of the extracted RNA were determined by Qubit® RNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, USA), 1% agarose gels, and Bioanalyzer 2100 system. The RNA sample preparations employed a total amount of 2 μg RNA per sample as input material. Following the manufacturer’s recommendations, sequencing libraries were created using the NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA), when index codes were added to assign sequences to each sample. The libraries were then sequenced on the Illumina Novaseq technology, yielding 150 bp paired-end reads. Illumina short reads were aligned with BWA (v.0.7.17).
Quantification of gene expression level
Fastp (v.0.23.4) was used for quality control of the raw sequencing reads with default parameters. RSEM (v.1.2.28) was utilized for gene-level quantification, using the GENCODE GRCh38.p13 genome assembly and GENCODE (v.42) annotation. Then we filtered genes with low expression levels (fragments per kilobase of transcript per million fragments mapped, FPKM < 1) in all groups.
Definition of DEGs
The R package DESeq2 (v.1.38.3) was applied for differential expression analysis based on the gene count matrix from RSEM quantification. DEGs were identified using thresholds of adjusted P-value (padj) <0.05 and absolute log2 fold change (|log2FC|) > 1.
Gene set enrichment analysis
In pathway enrichment analysis, with 0.05 as a cutoff for FDR value, hallmark gene sets from Molecular Signatures Database of R package msigdbr (v.7.5.1) were evaluated by ssGSEA using the R package “GSVA” (v.1.46.0). Each enrichment score represented the degree of which the genes in a particular gene set were coordinately up- or down-regulated within a sample. Gene Ontology (GO) enrichment analyses were performed using the R package clusterProfiler (v.4.6.2).
Library preparation for nanopore sequencing and data processing
For nanopore sequencing, the genomic DNA was extracted using QIAGEN Genomic-tip 100/G (QIAGEN, Germany) according to the manufacturer’s instructions. The quality of the DNA was assessed by monitoring DNA degradation and RNA contamination through pulsed field gel electrophoresis. DNA concentration and purity were analyzed by Qubit® DNA Assay Kit in Qubit® 4.0 Fluorometer (Invitrogen, USA) and Nanodrop 2000 (Thermo, USA), respectively.
For library preparation, a total amount of 8 μg DNA per sample was used as input. The 1D library was generated using SQK-LSK109 (Oxford Nanopore Technologies, UK) following manufacturer’s recommendations. Briefly, the genomic DNA was fragmented to 30 kb by sonication and purified by AMPure XP beads (Oxford Nanopore Technologies, UK). The DNA fragments were then subjected to end polishing, A-tailing, and ligation with sequencing adapters and motor proteins via NEBNext Quick T4 DNA Ligase (NEB, USA) for nanopore sequencing. Nanopore libraries were loaded onto the flow cell with sequencing buffer and loading beads, subsequently undergoing sequencing on the PromethION platform. Nanopore sequencing data were processed by NanoFilt with default parameters (v.2.5.0). Read alignments were performed against the latest human genome reference (hg38) using minimap2 (v.2.1.7)101.
DNA methylation analyses
To call methylation on nanopore sequencing data, we used the nanopolish call-methylation module102. Nanopolish was used to preprocess result from the nanopore sequencer, and minimap2 aligned nanopore data to the reference genome to enable methylome characterization101. The output contained information including the genomic location of CG dinucleotides, supporting read IDs, and log likelihood ratios comparing the probability of methylation versus non-methylation at each CpG site. Positive log likelihood ratios provided evidence for methylation. We calculated per-site methylation frequencies across all samples at all covered CG sites in the genome and filtered out low coverage sites (depth 5x). Then differential analysis was conducted between tumor and normal groups using DMLtest from the DSS package, subsetting sites with absolute methylation differences > 0.1 between tumors and normals as tumor-specific. DMLtest and callDMR were utilized to identify DMRs in Rec tumors compared with NonRec tumors in LUAD and LUSC. Finally, we performed gene set enrichment analysis (GSEA) on the DMRs using clusterProfiler and org.Hs.eg.db to uncover pathways associated with methylation changes in Rec group. The representative sequence statistics of the long reads have been presented in Supplementary Data 4.
Methylation driver genes and associated transcription factors
We found DMRs linked to hypomethylated genes with higher expression in the Rec group. Using HOMER103, we predicted transcription factors for these DMRs. Activity plots showed a correlation between motif hypermethylation and downregulated gene expression, defining motifs with stronger downregulation as having higher “inactivation strength”.
Further, we leveraged HOMER “find motifs” function to map transcription factor binding motifs to specific genomic regions corresponding to the repertoire of identified DMRs in promoter region. This enabled us to link motifs of interest to putative methylation-driven target genes. Then, we filtered motifs to those corresponding to protein-coding transcription factor genes. Specifically, we required Pearson correlation coefficients below −0.3 between transcription factor binding motif methylation and target gene expression across our sample cohort. To ensure a robust dataset, we filtered out duplicate and low-scoring motifs, keeping the top 5 motifs by score for each target gene.
SV analyses based on long-read data
To explore somatic SVs from long read WGS data, the sequences were mapped to GRCh38 and detected via SAVANA (v.1.0.4). Then, AnnotSV (v.3.3.8) was used to annotate the SVs104.
Single-cell library preparation and sequencing
Single-cell suspensions were converted to barcoded scRNA-seq libraries by using the Chromium Single Cell 3’ Library, Gel Bead & Multiplex Kit, and Chip Kit (10x Genomics). The libraries were designed to have approximately 7,000 cells per library. Samples were processed using kits pertaining to either the V2 or V3 barcoding chemistry of 10x Genomics. Libraries were sequenced on an Illumina NovaSeq 6000.
Processing scRNA-seq raw data
Single cell RNA-seq reads were processed using the 10x Genomics Cell Ranger pipeline (v.7.1.0) with default parameters. We deployed the Cell Ranger “mkgtf” to filter genes and “mkref” to construct the reference genome alignment index with the reference genome GRCh38.p13 from Gencode (v.42)105. Quantification analysis of each sample was performed with “count” utility. To remove doublets, the DoubletFinder (v.2.0.3) was employed106. Specifically, cells with less than 10 expressed genes and cells with fewer than 200 detected genes were excluded. Subsequently, for each sample, a Seurat object (v.4) was created, predicted doublets were filtered out, and the objects were merged. Cells whose mitochondrial gene expression exceeded 10% of total expression were filtered out based on the aggregated data from all samples. Cells with fewer than 200 or more than 7500 detected genes were also filtered out. The Harmony algorithm (v.0.1.1) was then utilized for batch integration, dimensionality reduction, and unsupervised clustering of the integrated dataset107.
Clustering and cell type annotation
To reduce dimensionality, the differentially expressed genes were calculated via principle component analysis (PCA), and tSNE was further used with default settings. Using the first 20 principal components and a resolution value of 0.3, we applied the FindClusters function to generate distinct cell clusters. We identified the gene expression markers for each cluster using FindAllMarkers function with the Wilcoxon Rank Sum test. We focused on genes detected in at least 10% cells in either group with an average log (fold change) of at least 0.25 between the two groups. Cell clusters were annotated as known cell types utilizing canonical marker genes.
Cell-type composition analysis
To investigate potential differences in cell-type compositions between Rec and NonRec groups, we calculated the percentages of cell types in each sample, which subsequently were visualized as boxplots by scRNAtoolVis R package.
Pseudotime analysis
To speculate the pseudotime trajectory of cells, we applied Monocle (v.2.26.0)108. Firstly, newCellDataSet was used to create an object with the parameter negbinomial size and gene-cell matrix of UMI counts. Next, DDRTree was used for reducing dimensionality and constructing tree-like trajectories. Finally, we visualized the cell density along the pseudotime by R packages ggplot2 and ggridges.
InferCNV analysis
InferCNV package (v.1.14.2) was utilized to infer CNVs in AT2 cells to explore the malignant degree of each subcluster. The cells from adjacent normal sample were considered as reference, and genes with average read counts less than 0.1 were filtered out. Further, the signal was also denoised.
Cell–cell interaction
Cell–cell interactions of cell types were estimated through Cell-PhoneDB (v.4.0.0) with default parameters, which inferred potential interactions between two cell types according to the gene expression of receptor-ligand pairs. Subsequently, the adjacency matrices were created for cell–cell interactions and visualized via heatmaps. After excluding interactions among identical cellular lineages, the receptor-ligand pairs within different cell types were visualized. Interactions with P-value < 0.05 were considered statistically significant.
Survival analysis
Survival analyses were performed using the R package “survminer” (v.0.4.9). Recurrence-free survival (RFS) and overall survival (OS) rate were estimated with Kaplan-Meier method using the survfit function.
Specifically, we obtained the top 50 highly expressed genes representing each cell subgroup based on scRNA-seq data. Subsequently, the ssGSEA algorithm from the GSVA R package was utilized to compute ssGSEA scores for each sample using the TCGA RNA-seq gene expression data. Samples were divided into high and low groups based on the median ssGSEA score. Finally, survival analysis was conducted between the high and low scoring groups.
Multi-omics data integration
In order to further elucidate the clinical relevance of genes identified through multi-omics analyses in sample classification, an unsupervised machine learning approach was employed using NMF based on mRNA TPM expression from FF samples. The implementation of this algorithm was achieved through the R package NMF (v.0.26). Different rank values from 2-8 were tested, with 100 random runs for each rank value. The optimal rank of 4 was selected for further analysis based on model performance. Heatmaps were generated to visualize the classification of samples by NMF-derived clusters. Then, the data from genomic, epigenomic, and transcriptomic analyses were integrated in each subcluster. The ssGSEA scores were used to evaluate the enrichment of pathways among clusters, and RFS was estimated with Kaplan-Meier for every NMF-based subgroup.
Mice
Mice were kept in a specific pathogen-free animal facility at Sichuan University with autoclaved food, water, and bedding. All mouse experiments were approved by the Animal Care and Use Committee of Sichuan University and were performed in compliance with the Guide for the Care and Use of Laboratory Animals of Sichuan University. BALB/c-nu mice (male, 8 weeks old, approximately 20 g weight) used in the experiments were purchased from Jiangsu Gempharmatech Co. Ltd. In the mouse experiments, the tumor volumes did not exceed the maximal permitted tumor volume of 1,000 mm3.
Cell culture
Human lung carcinoma (A549, PC9, and H1299) cells were purchased from the American Type Culture Collection (ATCC) and were cultured in medium containing 10% (vol/vol) fetal bovine serum and penicillin (100 U ml−1)/streptomycin (0.1 mg ml−1), placed at 37 °C, 5% CO2 cell incubator.
Gene editing and efficiency testing
Gene-specific sgRNA oligos targeting PRAME were cloned into the lentiviral vector V2TC, which bicistronically expresses sgRNAs and mCherry. We designed sgRNAs (Supplementary Data 5) with the CRISPR Design Tool (http://crispr.mit.edu/). The V2TC-sgRNA plasmids were transfected into HEK293T cells (ATCC, Cat# CRL-1573) along with the helper plasmids psPAX2 (RRID: Addgene_12260) and pMD2.G (RRID: Addgene_12259) using the calcium phosphate transfection method. The lentivirus-containing supernatant was harvested 36 and 48 h after transfection and used for infection experiments. Genomic DNA was isolated from infected cells, and mutation validation was performed by the T7E1 (Vazyme, Cat# EN303-01) assay.
Western blotting
Cell lysates were extracted in RIPA buffer (Beyotime, Cat# P0013) supplemented with protease inhibitors (Beyotime, Cat# P1045). Protein concentration was determined using Bicinchoninic Acid Assay (Thermo Scientific, 23227). SDS–PAGE gel electrophoresis and blotting onto PVDF membranes were performed. The following antibodies were used: Anti-PRAME (Abcam, Ab219650), GAPDH (CST, 2118), Rap1 (CST,2399 s), mTOR (CST,2983 T), and PCNA (CST, 13110 s). Images were developed by NcmECL Ultra Reagent (NCM biotech).
RNA extraction and RT–qPCR
Total RNA was extracted from cells using TRIzol (Applied Biosystems, 15596026). RT SuperMix (TSINGKE, TSK314S) was used for reverse transcription according to the manufacturer’s protocol. Quantitative PCR was performed in triplicate on CFX96 Touch Real-Time PCR Detection System (Bio-Rad) with ArtiCanCEO SYBR qPCR Mix (TSINGKE, TSE401). GAPDH was used as normalization control. The relative expression of genes was calculated using the 2-ΔΔCt method. Primer sequences are reported in Supplementary Data 6.
Wound healing assay
Cell migration ability was determined using wound healing assay. PC9, A549 and H1299 cells were seeded into 24 well plates. Cells grew until 90% confluency. The cell layer was then gently scratched through the central axis using a P200 pipette tip. Floating cells were washed away. Images were captured at the same position at 0, 24, and 48 h. Cell migration was analyzed using imageJ software.
Mouse tail vein injection and lung metastasis
A total of ten BALB/c-nu mice (male, approximately 20 g weight) at 8-week of age were randomly separated into two groups (sgPRAME group and sgScr group). For tail vein injection, each mouse was injected with 2 × 106 cells in a 100 µL volume within 10 s. 24 days later, mice were sacrificed, and metastatic lesions of fresh lungs were detected with fluorescence stereo microscopy. Further, lungs were fixed in 4% paraformaldehyde, and H&E staining was performed according to the standard protocol.
Statistical analyses
No statistical method was used to predetermine sample size. Standard statistical tests were utilized to depict data distribution. For ordered categorical and continuous variables, Student’s t-test, ANOVA, and Wilcoxon ranked-sum test were used. Pearson’s correlation coefficients were used to describe linear dependence. In addition, survival analyses were performed using Kaplan-Meier curves (log rank test). For in vitro experiments, wound healing assay, western blot, qPCR, and cell proliferation assay were repeated three times independently. For in vivo experiment, the measurements of tumor lesion number were performed blindly. The numbers of independent experiments, samples, or events were indicated in the figure legends. Data were presented as mean ± SD, mean ± SEM or otherwise illustrated in figure legends. R package and GraphPad Prism were used for statistical analyses. P < 0.05 indicated statistical significance.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequence data in this study have been deposited to Genome Sequence Archive (GSA) in BIG Data Center, Beijing Institute of Genomics (BIG) with accession numbers of HRA003362 for WES, nanopore sequencing, bulk RNA-seq data [https://ngdc.cncb.ac.cn/gsa-human/browse/HRA003362] and HRA007834 for scRNA-seq data [https://ngdc.cncb.ac.cn/gsa-human/browse/HRA007834]. The sequencing data are available under restricted access which could be provided for scientific research complying with the provisions of law due to concerns about patient privacy. Readers could make an access request through GSA for Human and send email to corresponding author with a detailed proposal. Once access is granted, the data will be available within 4 weeks. Source data are provided with this paper.
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Herbst, R. S., Morgensztern, D. & Boshoff, C. The biology and management of non-small cell lung cancer. Nature 553, 446–454 (2018).
Riely, G. J. et al. Non-small cell lung cancer, version 4.2024, NCCN clinical practice guidelines in oncology. J. Natl Compr. Canc Netw. 22, 249–274 (2024).
Altorki, N. et al. Lobar or sublobar resection for peripheral stage IA non-small-cell lung cancer. N. Engl. J. Med. 388, 489–498 (2023).
Mahvi, D. A., Liu, R., Grinstaff, M. W., Colson, Y. L. & Raut, C. P. Local cancer recurrence: the realities, challenges, and opportunities for new therapies. CA Cancer J. Clin. 68, 488–505 (2018).
Chen, K. et al. Spatiotemporal genomic analysis reveals distinct molecular features in recurrent stage I non-small cell lung cancers. Cell Rep. 40, 111047 (2022).
Jones, G. D. et al. A genomic-pathologic annotated risk model to predict recurrence in early-stage lung adenocarcinoma. JAMA Surg. 156, e205601 (2021).
Wang, C., Wu, Y., Shao, J., Liu, D. & Li, W. Clinicopathological variables influencing overall survival, recurrence and post-recurrence survival in resected stage I non-small-cell lung cancer. BMC Cancer 20, 150 (2020).
Ginsburg, O., Ashton-Prolla, P., Cantor, A., Mariosa, D. & Brennan, P. The role of genomics in global cancer prevention. Nat. Rev. Clin. Oncol. 18, 116–128 (2021).
Phan, T. G. & Croucher, P. I. The dormant cancer cell life cycle. Nat. Rev. Cancer 20, 398–411 (2020).
Martínez-Ruiz, C. et al. Genomic–transcriptomic evolution in lung cancer and metastasis. Nature 616, 543–552 (2023).
Al Bakir, M. et al. The evolution of non-small cell lung cancer metastases in TRACERx. Nature 616, 534–542 (2023).
Jamal-Hanjani, M. et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 376, 2109–2121 (2017).
Wang, X. et al. Genetic intratumor heterogeneity remodels the immune microenvironment and induces immune evasion in brain metastasis of lung cancer. J. Thorac. Oncol. 19, 252–272 (2024).
Brock, M. V. et al. DNA methylation markers and early recurrence in stage I lung cancer. N. Engl. J. Med. 358, 1118–1128 (2008).
Kaiser, A. M. et al. p53 governs an AT1 differentiation programme in lung cancer suppression. Nature 619, 851–859 (2023).
Sorin, M. et al. Single-cell spatial landscapes of the lung tumour immune microenvironment. Nature 614, 548–554 (2023).
Zhang, Q. et al. The spatial transcriptomic landscape of non-small cell lung cancer brain metastasis. Nat. Commun. 13, 5983 (2022).
Maynard, A. et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell 182, 1232–1251.e22 (2020).
The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Moreira, A. L. et al. A grading system for invasive pulmonary adenocarcinoma: a proposal from the International Association for the Study of Lung Cancer pathology committee. J. Thorac. Oncol. 15, 1599–1610 (2020).
Boettcher, S. et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599–604 (2019).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Beyter, D. et al. Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat. Genet. 53, 779–786 (2021).
Sakamoto, Y. et al. Phasing analysis of lung cancer genomes using a long read sequencer. Nat. Commun. 13, 3464 (2022).
Haga, Y. et al. Whole-genome sequencing reveals the molecular implications of the stepwise progression of lung adenocarcinoma. Nat. Commun. 14, 8375 (2023).
Davalos, V. & Esteller, M. Cancer epigenetics in clinical practice. CA Cancer J. Clin. 73, 376–424 (2023).
Haerinck, J., Goossens, S. & Berx, G. The epithelial-mesenchymal plasticity landscape: principles of design and mechanisms of regulation. Nat. Rev. Genet. 24, 590–609 (2023).
Teixeira, V. H. et al. Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. Nat. Med 25, 517–525 (2019).
Yin, Y. et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356, eaaj2239 (2017).
Dey, A., Varelas, X. & Guan, K. L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 19, 480–494 (2020).
Wu, B. K., Mei, S. C., Chen, E. H., Zheng, Y. & Pan, D. YAP induces an oncogenic transcriptional program through TET1-mediated epigenetic remodeling in liver growth and tumorigenesis. Nat. Genet 54, 1202–1213 (2022).
Pearson, J. D. et al. Binary pan-cancer classes with distinct vulnerabilities defined by pro- or anti-cancer YAP/TEAD activity. Cancer Cell 39, 1115–1134.e12 (2021).
Li, S., Jia, H., Zhang, Z. & Wu, D. DRAIC promotes growth of breast cancer by sponging miR-432-5p to upregulate SLBP. Cancer Gene Ther. 29, 951–960 (2022).
Lian, Y. et al. DUXAP8, a pseudogene derived lncRNA, promotes growth of pancreatic carcinoma cells by epigenetically silencing CDKN1A and KLF2. Cancer Commun. (Lond.) 38, 64 (2018).
Chow, A. et al. Tim-4(+) cavity-resident macrophages impair anti-tumor CD8(+) T cell immunity. Cancer Cell 39, 973–988.e9 (2021).
Nieto, M. A., Huang, R. Y., Jackson, R. A. & Thiery, J. P. EMT: 2016. Cell 166, 21–45 (2016).
Altorki, N. K. et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat. Rev. Cancer 19, 9–31 (2019).
Dongre, A. & Weinberg, R. A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 20, 69–84 (2019).
Aman, J. & Margadant, C. Integrin-dependent cell-matrix adhesion in endothelial health and disease. Circ. Res 132, 355–378 (2023).
Pujol, J. L. et al. Safety and immunogenicity of the PRAME cancer immunotherapeutic in patients with resected non-small cell lung cancer: a phase I dose escalation study. J. Thorac. Oncol. 11, 2208–2217 (2016).
Mainardi, S. et al. Identification of cancer initiating cells in K-Ras driven lung adenocarcinoma. Proc. Natl Acad. Sci. USA 111, 255–260 (2014).
Travaglini, K. J. et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625 (2020).
Han, G. et al. An atlas of epithelial cell states and plasticity in lung adenocarcinoma. Nature 627, 656–663 (2024).
Wang, Z. et al. Deciphering cell lineage specification of human lung adenocarcinoma with single-cell RNA sequencing. Nat. Commun. 12, 6500 (2021).
Yang, D. et al. Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution. Cell 185, 1905–1923.e25 (2022).
Barry, S. T., Gabrilovich, D. I., Sansom, O. J., Campbell, A. D. & Morton, J. P. Therapeutic targeting of tumour myeloid cells. Nat. Rev. Cancer 23, 216–237 (2023).
Cheng, S. et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809.e23 (2021).
Casanova-Acebes, M. et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 595, 578–584 (2021).
Pérez-Gutiérrez, L. & Ferrara, N. Biology and therapeutic targeting of vascular endothelial growth factor A. Nat. Rev. Mol. Cell Biol. 24, 816–834 (2023).
Armingol, E., Officer, A., Harismendy, O. & Lewis, N. E. Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 22, 71–88 (2021).
Liu, Z. L., Chen, H. H., Zheng, L. L., Sun, L. P. & Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target Ther. 8, 198 (2023).
Chen, Y. et al. Spatiotemporal single-cell analysis decodes cellular dynamics underlying different responses to immunotherapy in colorectal cancer. Cancer Cell 42, 1268–1285.e7 (2024).
Wang, M., Herbst, R. S. & Boshoff, C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat. Med 27, 1345–1356 (2021).
Stewart, E. et al. Identification of therapeutic targets in rhabdomyosarcoma through integrated genomic, epigenomic, and proteomic analyses. Cancer Cell 34, 411–426.e19 (2018).
Dauch, D. et al. A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med 22, 744–753 (2016).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Marusyk, A., Janiszewska, M. & Polyak, K. Intratumor heterogeneity: the Rosetta Stone of therapy resistance. Cancer Cell 37, 471–484 (2020).
Zhang, J., Späth, S. S., Marjani, S. L., Zhang, W. & Pan, X. Characterization of cancer genomic heterogeneity by next-generation sequencing advances precision medicine in cancer treatment. Precis Clin. Med. 1, 29–48 (2018).
Wang, L., Jia, Q., Chu, Q. & Zhu, B. Targeting tumor microenvironment for non-small cell lung cancer immunotherapy. Chin. Med J. Pulm. Crit. Care Med. 1, 18–29 (2023).
McGranahan, N. & Swanton, C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168, 613–628 (2017).
Al-Rawi, D. H., Lettera, E., Li, J., DiBona, M. & Bakhoum, S. F. Targeting chromosomal instability in patients with cancer. Nat. Rev. Clin. Oncol. 21, 645–659 (2024).
George, J. et al. Evolutionary trajectories of small cell lung cancer under therapy. Nature 627, 880–889 (2024).
Yang, F. et al. Chemotherapy and mismatch repair deficiency cooperate to fuel TP53 mutagenesis and ALL relapse. Nat. Cancer 2, 819–834 (2021).
Karasaki, T. et al. Evolutionary characterization of lung adenocarcinoma morphology in TRACERx. Nat. Med 29, 833–845 (2023).
Isozaki, H. et al. Therapy-induced APOBEC3A drives evolution of persistent cancer cells. Nature 620, 393–401 (2023).
Supek, F. & Lehner, B. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 521, 81–84 (2015).
Lengel, H. B. et al. Genomic mapping of metastatic organotropism in lung adenocarcinoma. Cancer Cell 41, 970–985.e3 (2023).
Guo, H. et al. DNA hypomethylation silences anti-tumor immune genes in early prostate cancer and CTCs. Cell 186, 2765–2782.e28 (2023).
Gkountela, S. et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 176, 98–112.e14 (2019).
Na, F. et al. KMT2C deficiency promotes small cell lung cancer metastasis through DNMT3A-mediated epigenetic reprogramming. Nat. Cancer 3, 753–767 (2022).
Kim, J. et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 538, 114–117 (2016).
Tang, Y. et al. Selective inhibition of STRN3-containing PP2A phosphatase restores Hippo tumor-suppressor activity in gastric cancer. Cancer Cell 38, 115–128.e9 (2020).
Park, H. W. et al. Alternative Wnt signaling activates YAP/TAZ. Cell 162, 780–794 (2015).
Murciano-Goroff, Y. R., Suehnholz, S. P., Drilon, A. & Chakravarty, D. Precision oncology: 2023 in review. Cancer Discov. 13, 2525–2531 (2023).
Pradat, Y. et al. Integrative pan-cancer genomic and transcriptomic analyses of refractory metastatic cancer. Cancer Discov. 13, 1116–1143 (2023).
Wadelin, F. et al. Leucine-rich repeat protein PRAME: expression, potential functions and clinical implications for leukaemia. Mol. Cancer 9, 226 (2010).
Epping, M. T. et al. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell 122, 835–847 (2005).
Orlando, D. et al. Adoptive immunotherapy using PRAME-specific T cells in medulloblastoma. Cancer Res 78, 3337–3349 (2018).
Mumme, H. et al. Single-cell analysis reveals altered tumor microenvironments of relapse- and remission-associated pediatric acute myeloid leukemia. Nat. Commun. 14, 6209 (2023).
Naik, S. et al. Donor-derived multiple leukemia antigen-specific T-cell therapy to prevent relapse after transplant in patients with ALL. Blood 139, 2706–2711 (2022).
Wen, L. et al. Single-cell technologies: from research to application. Innovation 3, 100342 (2022).
Nabhan, A. N., Brownfield, D. G., Harbury, P. B., Krasnow, M. A. & Desai, T. J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359, 1118–1123 (2018).
Gardner, E. E. et al. Lineage-specific intolerance to oncogenic drivers restricts histological transformation. Science 383, eadj1415 (2024).
Ren, X. et al. Single-cell transcriptomic analysis highlights origin and pathological process of human endometrioid endometrial carcinoma. Nat. Commun. 13, 6300 (2022).
Cerezo-Wallis, D. et al. Midkine rewires the melanoma microenvironment toward a tolerogenic and immune-resistant state. Nat. Med. 26, 1865–1877 (2020).
Sun, Y. et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 184, 404–421.e16 (2021).
Sikkema, L. et al. An integrated cell atlas of the lung in health and disease. Nat. Med. 29, 1563–1577 (2023).
Liu, Y. et al. Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell 40, 424–437.e5 (2022).
Kortlever, R. M. et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 171, 1301–1315.e14 (2017).
Zhang, S. et al. Single cell transcriptomic analyses implicate an immunosuppressive tumor microenvironment in pancreatic cancer liver metastasis. Nat. Commun. 14, 5123 (2023).
Kuo, H. Y., Khan, K. A. & Kerbel, R. S. Antiangiogenic-immune-checkpoint inhibitor combinations: lessons from phase III clinical trials. Nat. Rev. Clin. Oncol. 21, 468–482 (2024).
Liu, Q. et al. Proteogenomic characterization of small cell lung cancer identifies biological insights and subtype-specific therapeutic strategies. Cell 187, 184–203.e28 (2024).
Rami-Porta, R. et al. The International Association for the Study of Lung Cancer lung cancer staging project: proposals for revision of the TNM stage groups in the forthcoming (ninth) edition of the TNM classification for lung cancer. J. Thorac. Oncol. 19, 1007–1027 (2024).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Favero, F. et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 26, 64–70 (2015).
Roth, A. et al. PyClone: statistical inference of clonal population structure in cancer. Nat. Methods 11, 396–398 (2014).
Sztupinszki, Z. et al. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. npj Breast Cancer 4, 16 (2018).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Simpson, J. T. et al. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 14, 407–410 (2017).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Geoffroy, V. et al. AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 34, 3572–3574 (2018).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e4 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Acknowledgements
We are thankful to all the patients for their voluntary participation in the study. This work was supported by grants from the National Natural Science Foundation of China (82470109, 92159302, 32170592), the Science and Technology Project of Sichuan (2022ZDZX0018), the Science and Technology Project of Chengdu (2023-YF09-00007-SN), the 1.3.5 Project of State Key Laboratory of Respiratory Health and Multimorbidity (RHM24208), the 1.3.5 Project of Disciplines Excellence (ZYYC23027), the 1·3·5 project of Artificial Intelligence (ZYAI24016), West China Hospital, Sichuan University, and Postdoctoral Program of West China Hospital, Sichuan University (2020HXBH084).
Author information
Authors and Affiliations
Contributions
W.L., C.W., and L.Z. conceived and designed the study. C.W., J.L., J.C., and Z.W. performed the experiments and analyzed the data. L.S. and X.C. contributed reagents, materials, and analysis tools. G.Z., J.W., C.L., and R.Q. assisted with methodology. C.W., J.L., J.C., and Z.W. wrote the original draft. All authors reviewed and edited the manuscript. W.L., C.W., and L.Z. supervised the study and acquired funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yutaka Suzuki, and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, C., Li, J., Chen, J. et al. Multi-omics analyses reveal biological and clinical insights in recurrent stage I non-small cell lung cancer. Nat Commun 16, 1477 (2025). https://doi.org/10.1038/s41467-024-55068-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55068-2
This article is cited by
-
Transcription factor ZNF266 suppresses cancer progression by modulating CA9-mediated intracellular pH alteration in lung adenocarcinoma
Respiratory Research (2025)
-
Collaborative assessment of the risk of postoperative progression in early-stage non-small cell lung cancer: a robust federated learning model
Cancer Imaging (2025)
-
China Protocol for early screening, precise diagnosis, and individualized treatment of lung cancer
Signal Transduction and Targeted Therapy (2025)
