
Abstract
The fallopian tube undergoes extensive molecular changes during the menstrual cycle and menopause. We use single-cell RNA and ATAC sequencing to construct a comprehensive cell atlas of healthy human fallopian tubes during the menstrual cycle and menopause. Our scRNA-seq comparison of 85,107 pre- and 46,111 post-menopausal fallopian tube cells reveals substantial shifts in cell type frequencies, gene expression, transcription factor activity, and cell-to-cell communications during menopause and menstrual cycle. Menstrual cycle dependent hormonal changes regulate distinct molecular states in fallopian tube secretory epithelial cells. Postmenopausal fallopian tubes show high chromatin accessibility in transcription factors associated with aging such as Jun, Fos, and BACH1/2, while hormone receptors were generally downregulated, a small proportion of secretory epithelial cells had high expression of ESR2, IGF1R, and LEPR. While a pre-menopausal secretory epithelial gene cluster is enriched in the immunoreactive molecular subtype, a subset of genes expressed in post-menopausal secretory epithelial cells show enrichment in the mesenchymal molecular type of high-grade serous ovarian cancer.
Similar content being viewed by others


Introduction
The fallopian tubes (FT) play a vital role in women’s reproductive function. After ovulation, the FT fimbriae pick up the egg from the ovary and muscular contractions and the ciliated epithelial surface cells transport the egg toward the uterus through the ampullary part of the FT, where fertilization takes place. The zygote is then transported toward the narrow intrauterine segment of the FT, from which the blastocyst descends and implants into the uterus1. The FT serves as an anatomical and immunological barrier against colonization of the abdominal cavity by pathogens. An infection of the tubal lumen (salpingitis) from an ascending infection through the vagina may lead to occlusion of the fimbriated tubal end. This can result in hydrosalpinx, a high risk of ectopic pregnancy, and tubal factor infertility2. We know that during menopause, bereft of the protective hormonal environment, the FT undergoes atrophy3. However, little is known about cellular changes in the tubes associated with the transition to menopause and if any of these changes predispose women to disease. The FT is now recognized as a potential site of origin of high-grade serous cancer (HGSC) since regions of dysplasia with a high proliferation rate, activation of the PI3 kinase pathway, and p53 overexpression are detected in the mucosa of the FT of women suffering from invasive HGSC4,5. Indeed, a mutational analysis comparing p53 mutations in serous tubal intraepithelial carcinomas (STIC) and HGSC showed identical mutations in paired STICs and concurrent HGSC6.
To better understand the biology of tubal diseases and HGSC, it is crucial that we characterize the cellular makeup of the FT throughout the menstrual cycle and menopause. Single-cell technologies have revolutionized our understanding of healthy organs7 and their associated diseases8 by allowing the characterization of individual cells, revealing their unique gene expression and regulation patterns. Prior single-cell research has led to the recognition of diverse subtypes of FT cells and identified cellular changes that occur with common FT disease9,10,11,12,13. However, this research has not systematically compared cellular diversity, gene expression, and the regulation of these cells in different phases of the menstrual cycle or by menopausal status. By leveraging single-cell gene expression and chromatin accessibility assays, we deciphered the molecular transformations occurring in specific cell types during the menstrual cycle and menopause. We then generated a high-quality FT cell atlas that allowed us to examine the physiological changes and cellular functions in the human FT during aging. This atlas provides insights that will substantially improve our understanding of diseases affecting the fallopian tubes.
Results
Cellular composition of pre-menopausal human fallopian tubes
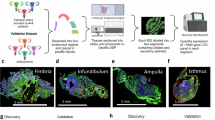
After removing doublets and cells with high mitochondrial content, 85,107 scRNA-seq cells from pre-menopausal donors (ampulla and fimbriae only) were integrated with 46,111 scRNA-seq cells from post-menopausal donors (before further analysis (Supplementary Table 1). After scRNA-seq and bioinformatics analysis (Fig. 1A, Supplementary Data 1), unsupervised clustering identified 19 clusters, representing cell types typically identified microscopically in hematoxylin and eosin-stained pre-menopausal FT (Fig. 1B, Fig. 2A, Supplementary Data 2). Based on the expression of canonical marker genes (Supplementary Data 3)10,11, the 19 FT clusters were classified into 12 major cell types (Supplementary Fig. 1A, B, Supplementary Data 4): Ciliated epithelial (CE), secretory epithelial (SE) 1–3, smooth muscle (SM), pericyte/vascular (P/V) 1-2, endothelial (EN) 1-2, lymphatic endothelial (LE), stromal (ST) 1-3, mast (MA), T and natural killer (T/NK) cells, macrophages (MP), monocytes (MN), B and plasma cells (BP) 1-2. A comparison of the three different FT anatomical sites showed similar cell distributions for almost all cell types. The isthmus had the highest percentage of SM cells and lowest percentages of CE and SE cells, consistent with the isthmus’s function of transporting the ovum towards the uterine cavity. In the fimbriae, the prevalent cells are CE and SE cells, consistent with the large surface area required to increase the chances of the egg being picked up by the FT (Supplementary Fig. 1B)14.

A Pre-menopausal fallopian tube (FT) fimbria (F; n = 10), ampulla (A; n = 10) and isthmus (I; n = 5) were characterized using scATAC-seq and scRNA-seq. To study the cellular and molecular changes driven by the menstrual cycle and by menopause in the normal human fallopian tube both datasets were integrated. B Representative hematoxylin and eosin (H&E) staining of normal pre-menopausal FT fimbria. Dashed lines and roman numerals indicate principal cell compartments defined by H&E in the FT. Black lines indicate stretches of ciliated (1) or secretory (2) epithelial cells, and (3) stromal cells. Different cell types identified in the FT by H&E are at higher magnification. Specific cell types are marked with circles or by an arrowhead and annotated by numbers. Created in BioRender. Weigert, M. (2024) BioRender.com/s53z837.

A Canonical cell types and subtypes found in the ampulla (n = 10) and fimbria (n = 10) of the normal pre-menopausal fallopian tube. The UMAP plot visualizes the 19 cell clusters identified using scRNA-seq. B Dot plot showing normalized gene expression levels of canonical marker genes associated with common cell types identified in the dataset (scRNA-seq). C The UMAP plot visualizing the 16 cell clusters identified in pre-menopausal ampulla (n = 5) and fimbria (n = 5) using scATAC-seq. Cell type labels were transferred from scRNA-seq data and match abbreviations described in (A). Cell types SE3, EN2 and B/P2 were not identified in this analysis. D Heatmap showing transcription factor (TF) activity enrichment by cell clusters based on scATAC-seq data. Abbreviations: SE1-3 = 3 subtypes of secretory epithelial cells; CE = ciliated epithelial cells; T/NK = T and natural killer cells; MP = macrophages; MN = monocytes; MA = mast cells; ST1-3 = 3 subtypes of stromal cells; SM = smooth muscle cells; LE = lymphatic endothelial cells; EN1-2 = 2 subtypes of endothelial cells; P/V1-2 = 2 subtypes of pericytes and vascular smooth muscle cells; B/P1-2 = 2 subtypes of B cells and plasma B cells.
Several cell types contained distinct subtypes. Within the SE cell type, we found three subtypes. SE1 and SE2/3 are distinguishable by different levels of OVGP1 expression, and SE3 cells expressed several cell cycle genes (Fig. 2B). Though rare, we observed SE3 in all pre-menopausal samples (58 cells detected, Supplementary Data 5). All ST cell clusters expressed the mesenchymal progenitor marker PDGFRA15. ST1 cells strongly express myofibroblast markers POSTN and NR2F2. ST2 cells feature high expression of CD34, a stem cell marker and adhesion molecule and a constitutive stromal component in normal tissue secreting collagen and facilitating cell migration16. ST3 cells express canonical fibroblast and myofibroblast markers (COL1A1, POSTN). We further subtyped EN cells into EN1 and EN2 based on the expression patterns of KDR and CLDN5, respectively. The two subtypes of P/V differ in the expression levels of canonical pericyte markers MCAM, ACTA2, MYH11, and TAGLN, which are considerably lower in P/V2 than in P/V1. However, P/V2 shows strong NR2F2 expression. Lastly, B/P cells comprise two subtypes: B/P1 expresses B and plasma cell markers, while B/P2 only expresses B cell marker CD79A (Fig. 2B).
We performed scATAC-seq (10X Genomics) on a subset of pre-menopausal samples and matched them to their corresponding scRNA-seq data (Supplementary Table 1). The cells from the ampulla and fimbriae from 10 donors were processed separately and integrated in silico. We annotated the cluster identity of each sample by transferring labels from its corresponding scRNA-seq. Specifically, using the scRNA-seq cluster information as a reference, each cell in scATAC-seq was annotated by searching for the best-matched cluster from the same sample. We identified 68,150 cells from the pre-menopausal FT (Fig. 2C and Supplementary Fig. 1C) from scATAC-seq with matched scRNA-seq compartments (Supplementary Data 6). The majority of scRNA-seq and scATAC-seq clusters matched well, confirming the high quality of both datasets. However, SE3, EN2, and B/P2 cell subtypes were not detected in scATAC-seq.
To characterize the regulatory landscape in pre-menopausal FT, we assessed the activities of 870 transcription factors (TF) listed in the cisBP database17 across all major cell types (Supplementary Fig. 1D and Data 7). Our analysis revealed notable cell-type–specific TF activities, such as strong TCF family activity in B/P1; STAT activity in EN1. CE cells showed increased activity in FOX family members, responsible for ciliogenesis, cell growth, and differentiation (16); T/NK cells in the ETV family, which are involved in monocyte differentiation (17); and GATA activity in MA. Our TF enrichment analyses identified differences in TF activities in cell subtypes, further corroborating our decision to subtype these major cell types. For instance, SE1 shows higher accessibility to GATA, TCF, and HOX TF, while SE2 was characterized by HOX, which determines cell and tissue identities (18), SOX, which regulates cell fate (19), and the accessibility of the transcriptional repressor, BACH. ST1 and ST2 show differences in the activities of both the TCF and CEBP families. P/V1 and P/V2 vary in the activities of EBF1, which is involved in the cell lineage commitment in pericytes (20) and the MEF family, which modulates blood vessel maturation (21) (Fig. 2D).
Menopause changes gene expression in fallopian tube cells
We compared the cellular composition of our pre-menopausal FT samples with the post-menopausal FT samples analyzed previously11. The results indicate that the cellular composition and gene expression of the FT undergoes dramatic changes during menopause (Fig. 3A). Among all cell types, the abundance of CE, SE, and T/NK cells decreased significantly in post-menopausal women, while the abundance of EN1, EN2, P/V1, and P/V2 cells increased. There was a trend of more stromal cells (ST2, ST3) in postmenopausal women, but the observed variability was high between samples. While we observed dramatic changes in cell composition, canonical cell marker expression did not change significantly during menopause (Supplementary Fig. 2A).

A Differences in cellular composition between pre- (n = 10) and post-menopausal (n = 7) women in percentages for combined anatomical sites (ampulla and fimbria). Data are represented using a boxplot showing the median and first and third quantile. Two-sided t-test. SE2 p-value < 0.045, CE p-value < 0.00059, T/NK p-value < 0.000049, EN1 p-value < 0.0059, EN2 p-value < 0.0029, P/V1 p-value < 0.0058, P/V2 p-value < 0.014. B Number of differentially expressed genes in pre- and post-menopausal women by cell clusters using scRNA-seq. Number of DEGs is for ampulla and fimbria combined. C Dot plot showing normalized gene expression levels of selected, differentially expressed genes specific to menopausal status by individual cell clusters (scRNA-seq). D Heatmap showing absolute differences in selected transcription factor (TF) activities by cell clusters using scATAC-seq. The z-score difference is calculated by directly subtracting the average pre-menopausal TF activity from the post-menopausal activity (Post - Pre). E Heatmap showing relative changes in selected transcription factor (TF) activities by cell clusters using scATAC-seq data. The relative changes are expressed as a percentage change from the pre-menopausal levels ((Post–Pre) / Pre * 100). F Dot plot showing normalized gene expression levels of selected genes in pre- and postmenopausal ST subtypes (scRNA-seq). G Dot plot showing the enrichment of selected gene ontology (GO) terms for each ST subtype in pre- and post-menopausal women based on scRNA-seq data. FDR-corrected p-value are shown.
To understand the changes in gene expression profiles between pre- and post-menopausal FT, we conducted cell-type–specific differential gene expression analysis using pseudo-bulk donor-level data. Among all cell types, SE cells exhibited the most differentially expressed genes (DEGs) (Fig. 3B, Supplementary Data 8), followed by MP, ST1, T/NK, ST2, and CE. Several genes were specific to menopausal status: Post-menopausal donors expressed senescence and aging markers such as ICAM1 (MP, MN, MA, and EN1-2), CXCL2 (SE, MP, MN, LE and EN1-2), ZNF31 (P/V1-2, all immune and stromal cells), ELL2 (B/P1, P/V1-2 and immune cells), and BAG3 (P/V1-2, EN1-2, SM, and ST1-3) (Fig. 3C and Supplementary Fig. 2B). PCSK1N (SM and P/V1-2), OVGP1 (SE cells), CRISP3, and CYBA (SE cells, B/P1-2, and immune cells), were higher expressed in pre-menopausal cell types (Fig. 3C). Pre-menopausal women exhibited expression in IFITM1/2 (ST1-3, EN1-2, P/V2, and T/NK) and PLCG2 (EN1-2 and LE), which are critical for viral defense and age-related inflammatory responses18,19,20.
After menopause, there was a decrease in stromal and immune cell interactions in both the ampulla and fimbria (CellPhoneDB, Supplementary Fig. 2C). When cell-type–specific activity scores of TFs in post-menopausal FT were compared to pre-menopausal levels, the JUN and FOS families of TFs showed significantly higher activity after menopause in all cell types except ST3 (Fig. 3D) consistent with their role in cellular aging21. Similarly, the BACH1 and BACH2 TFs, markers of aging and senescence22, displayed better chromatin accessibility in post-menopausal samples for all cell types except ST3 and P/V2 (Fig. 3D, Supplementary Data 9). To avoid overlooking TFs with high relative changes but low absolute changes, we assessed the percentage of change in activities relative to their pre-menopausal levels (Fig. 3E). In post-menopausal donors, the androgen receptor (AR) and the glucocorticoid receptor (NR3C1) exhibited an increase in ST3 and SM; the early growth response (EGR) family activities increased in SE1 and MA; and TWIST1, an EMT marker, increased in ST1 and MP (Fig. 3E).
During menopause, stromal cells in the fallopian tubes undergo significant changes that lead to histologic thinning of the FT wall and a reduction in the fold complexity of epithelial cells on the luminal side of the FT23. Our analysis revealed a molecular upheaval associated with these physiological changes (Fig. 3F). Before menopause, ST1 cells showed high expression levels of genes such as IGF1, TMSB4X, COX5B, COX4I1, NDUFB2, NDUFS5, and ribosomal protein-coding genes, which all drastically decreased in post-menopausal samples indicating a decrease in mitochondrial and ribosomal functions with age24. Similarly, NUPR1 and PIH1D1 expression were high in pre-menopausal ST2 cells but decreased in the post-menopausal ST2 cells. Conversely, the expression of NCL, HNRNPK, SELENOK, and GADD45B surged in ST1 after menopause. Likewise, the chromatin binding protein PSIP1 and MYC increased in ST2, and FUS, MALAT1, SON, RBM39, HNRNPH1, and HNRN2B1 were highly expressed in ST3. Additionally, the expression of EGR1 increased drastically in both ST2 and ST3 after menopause (Fig. 3F). The GO enrichment analysis (Fig. 3G, Supplementary Data 11) of the top expressed genes in each ST group revealed that pre-menopausal STs are enriched in genes required for metabolic activity (e.g. ribosome biogenesis, ATP metabolism). In contrast, post-menopausal STs are enriched in genes involved in RNA destabilization, cell aging, and apoptosis (Supplementary Data 11).
Menopause significantly changes secretory epithelial cells
We had previously studied the cellular makeup of FT from post-menopausal donors. Our analysis of the differences between pre- and post-menopausal samples uncovered a distinct cluster of cells unique to the post-menopausal cohort. We named this cluster SE post-specific to differentiate it from the remaining post-menopausal SE. We also found clusters of cells, which we designated SE1/2/3-pre in pre-menopausal samples. SE3-pre was not detected in any post-menopausal samples (Fig. 4A and Supplementary Fig. 3A).

A Pre- and post-menopausal SE cells identified using UMAP profiling. The pre-menopausal fallopian tube (n = 10) is characterized by three SE subtypes (SE1 pre, SE2 pre, and SE3 pre), while the post-menopausal fallopian tube (n = 7) is characterized by two SE subtypes (SE post and SE post-specific). The number of cells identified in each subtype is shown in the panel (scRNA-seq). B Dot plot showing normalized gene expression levels of selected markers associated with proliferation, stemness, and PEG-cells in pre- and postmenopausal SE subtypes based on scRNA-seq data. C Dot plot showing normalized gene expression levels of hormone receptors in pre- and postmenopausal SE subtypes based on scRNA-seq data. D Heatmap showing the average expression of target genes in SE pre- and post-menopausal subtypes at the individual sample level based on scRNA-seq data (ampulla and fimbria). E Heatmap showing the absolute transcription factor activities in SE subtypes using the scATAC-seq data. F Heatmap showing relative transcription factor activities in SE subtypes using scATAC-seq data. The Z-score is obtained by centering and re-scaling across SE subtypes for each TF. G Dot-plot showing the enrichment of selected gene ontology (GO) terms for each SE subtype based on scRNA-seq. FDR-corrected p-values are shown. H Spatial transcriptomics profiling of normal pre-menopausal fallopian tube fimbria (n = 1) using 10x Genomics Visium. Pre-menopausal fallopian tube fimbria were analyzed for spatial expression of KRT7, KRT15, TUBB, OVGP1 and INSR1.
Premenopausal SE1 is characterized by increased expression of MHC class II proteins (HLA-DPA1, HLA-DQA1) and PAX8 (Fig. 4B and Supplementary Fig. 3B), a known lineage marker for high-grade serous ovarian cancer used as a clinical biomarker25. In contrast, SE2-pre is defined by high expression of PEG cell markers26(LGR5, PGR, TNFRSF19, CD24). Lastly, SE3-pre is distinguished by chromatin remodeling markers (RUVBL1, HMGB2, SMC1A), DNA repair markers (FANCD2, MSH2, and MSH6), and TUBB, a major constituent of microtubules important for mitosis and an indication that these cells are cycling. In contrast, post-menopausal SE cell clusters exhibited decreased expression levels of PEG, MHC II, DNA repair, and chromatin remodeling markers (Fig. 4B and Supplementary Fig. 3B). This further indicates that SE cells undergo significant changes during menopause.
Pre- and post-menopausal SE cells also differ in hormone receptor expression (Fig. 4C and Supplementary Fig. 3C). SE1-pre cells strongly express AR, INSR, and NPR1, while SE2-pre is characterized by its strong expression of progesterone receptor (PGR), and SE3-pre displays moderate expression of PGR and PGR membrane component 1 (PGRMC1). In post-menopausal SE cells, the overall expression levels of hormone receptors are decreased. Still, there is a small proportion of cells represented by the SE-post cluster with a higher average expression of ESR2, IGF1R, and LEPR. Keratins (KRT) are intermediate filament proteins that provide mechanical support and organization to epithelial tissues27. We found that KRT 13, 15, and 17 are associated with menopause and are only expressed in SE post-specific cells (Fig. 4D).
The TF enrichment analysis indicated major differences in TF activity among the different SE groups (Fig. 4E-F). The SOX TF family, especially SOX9/13, involved in tissue repair and regeneration28, showed activity in all pre-menopausal SE groups but showed the highest activity in SE2-pre, indicating a higher regenerative potential29. PAX-1/5/9 were active in SE1-pre and SE-post, consistent with their role in regulating SE gene expression30. In contrast, TP-53/63/73, involved in regulating apoptotic processes31, were only active in post-menopausal samples. The SE post-specific cell cluster had the strongest activity of TP53 (Fig. 4E, F and Supplementary Fig. 3D) as well as the highest activity in TFCP2L1, FOX-A1/A2/B2, and RUN-X2/X3, indicating p53-mediated DNA damage signaling. AR activities decreased while proto-oncogenes from the MAF TF family showed strong chromatin accessibility after menopause (Fig. 4E, F).
Gene ontology (GO) enrichment analysis of the top expressed genes in each SE group suggests a specific function for each cell cluster (Fig. 4G, Supplementary Data 10). SE1-pre is hormone-responsive and interacts with immune cells. SE2-pre expresses genes activated by fibroblast growth factor and microtubule function. SE3-pre expresses many genes involved in DNA replication and immune effector processes. Post-menopausal SE cells are enriched in genes necessary for migration and hormone response32, while SE post-specific exhibits enrichment in genes required for the p53-mediated response to cellular stress.
To validate the scRNA-seq findings, we performed full spatial transcriptomics (Visium) on independent pre-menopausal samples. Using KRT7 as a marker for the SE pre-menopausal cells, we found that TUBB, OVGP1, and INSR co-localized with KRT7-expressing areas, providing additional evidence for their role in the pre-menopausal functions of SE cells. As expected, we did not detect expression of KRT15 in any of the analyzed cells (Fig. 4H).
Menstrual cycle hormones regulate cellular states in secretory cells
To examine gene expression changes during the proliferative and secretory phases of the menstrual cycle33, we isolated cells from donors with normal menstrual cycles (25–32 days). Our initial observation revealed that SE2-pre cells were predominantly identified in samples from the proliferative phase and marked by high OVGP1 expression, whereas SE1-pre cells were mostly found in the secretory phase and had considerably lower OVGP1 expression (Fig. 5A–D and Supplementary Fig. 4A). To validate the menstrual-cycle–specific expression of OVGP1 in SE cells, we performed IHC on patient-matched samples. Strong OVGP1 protein staining emerged in the FT epithelium during the proliferative phase but was decreased during the secretory phase (Fig. 5B). Estrogen signaling and OVGP1 expression reaches its peak during the late proliferative phase and ovulation, where it supports sperm decapacitation, sperm-ovum binding and embryo development34,35. We validated the post-menopausal expression of OVGP1 (Supplementary Fig. 4B). We confirmed that SE1-pre cells are the dominant SE cell cluster during progesterone treatment, indicating that progesterone may be important for SE1-pre cells (Supplementary Fig. 4A, B) while SE2-pre cells and OVGP1 expression are associated with estrogen levels (Fig. 5C, D and Supplementary Fig. 4A), consistent with our scRNA-seq and ATAC-seq results (Fig. 3D–F). This suggests that they represent distinct cellular states rather than separate cell subtypes, with each state being associated with a specific phase/ hormone of the menstrual cycle.

A Differences in cellular composition between the proliferative- (n = 4) and secretory phases (n = 3) of the menstrual cycle based on scRNA-seq data (two-sided t-test). Data are represented using a boxplot showing the median and first and third quantile. N describes the number of patients/ samples for each respective menstrual cycle phase. SE1 p-value < 0.0018, SE2 p-value < 0.0035. B Representative OVGP1 immunohistochemistry in the fallopian tube of patients in the proliferative phase (n = 2) and in the secretory phase (n = 2). C Scatter plot comparing gene expression levels of pre-menopausal SE1 (secretory phase) and SE2 cells (proliferative phase) using pseudo-bulk RNA analysis. D Volcano plot derived from pseudo bulk analysis. The volcano plot shows the differential gene expression analysis of genes expressed in SE cells (SE 1/2/3-pre) based on the proliferative and secretory phases. E Number of differentially expressed genes between the proliferative- and secretory phases of the menstrual cycle by cell clusters based on scRNA-seq data. F Dot plot showing normalized scRNA-seq derived gene expression levels of selected genes that differ in the proliferative and secretory phase in ST subtypes. G Representative immunofluorescence staining of IGF1 and IGF2 in primary human stromal and epithelial fallopian cell co-culture of one patient (n = 3). The co-culture was stimulated for 8 h with estrogen (E4) or progesterone (P4). IGF1 protein in green, IGF2 protein in red and nuclei/ DAPI in blue. White dotted lines mark epithelial cells while white stars mark stromal cells. H Ligand-receptor interactions, detected by CellPhoneDB, between SE and ST subtypes (left) and between SE cells (right) separated by the menstrual phase using scRNA-seq data. I RNA velocity analysis of SE1- and SE2-pre cells. Arrows indicate the location of the estimated future cell state. Long vectors mark rapid transition events (i.e., large changes in gene expression), while short arrows indicate homeostasis. J Median latent time for pre-menopausal SE cells during the proliferative- and secretory phase of the menstrual cycle, highlighting temporal positions for SE1- and SE2-pre.
SE cells displayed the greatest number of DEGs in both menstrual phases (Fig. 5E), followed by ST1 cells. Through this analysis, we discovered additional markers for SE1-pre and SE2-pre cells (Fig. 5D, Supplementary Data 12), besides the markers identified using cell-level analysis (Fig. 5C). During the proliferative phase, DEGs in SE2-pre are predominantly associated with COPI-coated vesicle functions transporting cargo between the Golgi and the endoplasmatic reticulum36. Conversely, in the secretory phase, SE1-pre shows DEGs enriched in clathrin-coated vesicle functions (Supplementary Fig. 4C). Similarly, in ST1 cells, elevated energy consumption and protein folding are linked to the proliferative phase, whereas integrin binding and cell-matrix adhesion are associated with the secretory phase (Fig. 5F). There was an inverse expression of IGF1 and IGF2 in ST1 cells during the menstrual phases (Supplementary Fig. 4D).
A co-culture of primary human stromal and epithelial cells from patients operated on for benign conditions was stimulated with progesterone, which induced IGF2 protein expression, while estrogen treatment (E2) induced IGF1 (Fig. 5G).
We further investigated the alterations in receptor:ligand interactions during the menstrual cycle phases using the CellPhoneDB database. In pre-menopausal SE cells, the proliferative phase was marked by increased communication between collagen molecules and the integrin α2β1 complex. At the same time, we observed a significant increase in BMP7 and EFNB1 interactions during the secretory phase (Fig. 5H). Overall, the proliferative phase had fewer receptor:ligand interactions than the secretory phase (Fig. 5H and Supplementary Fig. 4F).
RNA velocity analysis utilizes both un-spliced (nascent) and spliced (mature) mRNA molecules to predict potential directionality and the time it takes to transition between different cell states37,38. We overlaid the velocity vectors in UMAP space to reveal the direction of cellular transitions between SE1- and SE2-pre during the menstrual cycle. The RNA velocity vectors in pre-menopausal SE cells formed a circle, suggesting being part of a cyclic biological process (Fig. 5I). This is consistent with the menstrual cycle pattern, a repeating transition between SE cell states during the cycle. The median latent time was much shorter during the secretory phase, indicating that the export of mature mRNA to the cytoplasm occurs faster during the secretory phase (Fig. 5J and Supplementary Fig. 4E). SE cells were more transcriptionally active during the secretory phase, consistent with the CellPhoneDB analysis (Fig. 5H).
Secretory epithelial cell markers can subtype ovarian cancer
Given the mounting evidence suggesting that FT epithelial cells are the cell of origin for HGSC39, we sought to determine how different FT epithelial cell markers identified in our study are represented in The Cancer Genome Atlas (TCGA) dataset reporting bulk RNA-seq data of various cancers40. We focused on the top differentially expressed genes that we identified in SE cells from pre-menopausal (SE1/2/3-pre) and post-menopausal samples (SE-post, SE-post-specific) and performed a deconvolution analysis of the different TCGA molecular subtypes of ovarian, breast, head and neck, and lung squamous cell carcinoma (Fig. 6A).

A Heatmaps showing the top 740 up-regulated genes in our post-menopausal SE cells across the various molecular subtypes of high-grade serous ovarian cancer, breast cancer (BRCA), head and neck squamous cell carcinoma (HNSC), and lung squamous cell carcinoma (LUSC) as described by TCGA. B Heatmap showing the top 50 expressed markers in pre- and post-menopausal SE subtypes across the ovarian cancer molecular subtypes as identified by TCGA. C Principal component analysis plot for TCGA samples (n = 304) using 186 SE markers (SE3; n = 50 genes, and SE post-menopausal; n = 136 genes). D Pathway enrichment analysis of selected SE marker sets derived from post-menopausal SE (gene set 1) and SE3 (gene set 2). FDR-corrected p-values are shown.
The SE markers upregulated in post-menopausal women (740 genes, Supplementary Data 13) adeptly recapitulated the previously described molecular subtypes of HGSC: immunoreactive, proliferative, mesenchymal, and differentiated40. A subset of 136 genes (Supplementary Data 13) are highly expressed in the mesenchymal subtype of HGSC (Fig. 6A). Upon further scrutiny of the top expressed markers found in the different pre- and post-menopausal SE cell types, we discerned that the top 50 SE3-pre gene markers (Supplementary Data 13) were prominently expressed in the immunoreactive TCGA HGSC subtype (Fig. 6B). By juxtaposing these patterns with those from other cell types and TCGA cancer types, we were able to confirm that these associations are uniquely tethered to the SE cells in ovarian cancer. We combined the top 50 SE3-pre gene markers associated with immunoreactive HGSC (Fig. 6B), and the 136 SE-post markers associated with mesenchymal HGSC in the post-menopausal FT and performed principal component analysis. Strikingly, the 186 markers effectively separated immunoreactive, proliferative, and mesenchymal TCGA HGSC subtypes, but not the differentiated subtype (Fig. 6C, Supplementary Fig. 5A). The 136 SE-post genes associated with the mesenchymal subtypes showed enrichment in pathways associated with cell migration and cell junction assembly (Fig. 6A). The 50 SE3-pre gene markers, associated with the immunoreactive subtype (Fig. 6B) exhibited enrichment in apoptosis, chemotaxis, response to tumor necrosis factor, and cell adhesion (Fig. 6D). The immunoreactive subtype, as identified by the 50 SE3-pre genes, demonstrated notably better survival compared to proliferative and mesenchymal subtypes combined (Supplementary Fig. 5B).
Discussion
Our study presents compelling evidence that menopause leads to substantial changes in the cellular composition and gene expression profiles of the FT. Uterine and ovarian atrophy during menopause has been extensively described41, and our findings indicate that the FT also undergoes previously undescribed atrophic changes marked by age-associated loss of ciliated, secretory and T/NK cells and an increase in vasculature (EN1-2 and P/V1-2). These cellular changes were accompanied by significant molecular alterations indicative of aging and cellular stress responses, especially in stromal and epithelial cells. Almost all cell types, with the notable exception of ST3 cells, showed increased accessibility of TF from the AP-1 and Bach families, which are both involved in regulating cellular senescence42.
Using scRNA- and scATAC-seq, our study delineates the molecular and transcriptional alterations that occur within FT cells, especially SE and ST cells, throughout the menstrual cycle. In SE cells, we found that hormones regulate gene expression and transcription. During the proliferative phase of the menstrual cycle, OVGP1 expressing SE2-pre cells dominate the FT epithelium. These highly proliferative cells are characterized by the expression/activity of epithelial progenitor genes (SOX9/LGR5/PGR/Bach1-2) similarly as the previously described PEG cells26,43 and the NCS2-6 cell cluster10. During the secretory phase of the menstrual cycle, SE1-pre cells, characterized by markers of mature secretory cells (PAX8/ KRT7), are the predominant cell type in the FT epithelium. We show that SE1-pre cells are enriched in pathways responsive to steroid hormone regulation and MHC class II assembly and processing, which fosters immune cell tolerance and ensures successful fertilization44,45. Progesterone alters stromal cell transition and changes epithelial-stromal crosstalk during embryo attachment and implantation44. Our data showed increased and strong epithelial-stromal crosstalk/ communication during the secretory phase. SE2-pre cells were enriched in pathways involved in growth factor stimulation, which in the uterus are involved in the proliferation/ thickening of the uterine lining29,33. The stromal cells of the FT show similar menstrual cycle dependent changes in cell number, functions, and epithelial-stromal communication (8). It is probable that the gene expression in the different cell types of the uterus and FT, which share a common embryological origin (mesoderm) and are connected through the uterotubal junction, are similarly regulated throughout the menstrual cycle.
It is becoming increasingly clear that reproductive hormones coordinate gene expression and transcription in the uterus, ovary, and fallopian tube to optimize and synchronize ovulation, sperm and ovum movement, fertilization in the FT, and embryo implantation in the uterus. The FT epithelium is currently regarded as the site of origin for most HGSC39,46. The earliest event of malignant epithelium transformation is marked by the outgrowth of SE cells and mutations or loss of TP534. The ovarian TCGA data categorized HGSC into four molecular subtypes: proliferative, immunoreactive, differentiated and mesenchymal40. We showed that genes upregulated in SE-post and SE3-pre cell clusters can be used successfully to classify the proliferative, immunoreactive and mesenchymal subtypes of HGSC. This supports the theory that SE cells may be the progenitors of HGSC, particularly for these three subtypes, suggesting a different cell of origin for the differentiated subtype47,48. Previous work by Dinh et al., 2021, uncovered six epithelial signatures and applied them to the TCGA RNA-seq profiles of 394 HGSC using deconvolution analysis12. Strikingly, their transitional cluster (‘unclassified 2 + 3’) showed enrichment in the immunoreactive molecular subtype. While comparing genes expressed in each cluster, we found that the majority of our top 100 SE3-pre DEGs overlapped with their transitional cluster (66 out of 100).
The post-menopausal SE cell cluster (‘SE-post specific’) has increased TF activities of p53 family members, RUNX3 activity, and enrichment in DNA damage repair and p53 signal transduction pathways. Notably, RUNX3’s role as a tumor suppressor or oncogene is contingent upon p53 status49. Given that p53 mutations are ubiquitous in HGSOC50, it remains undetermined whether the ‘SE-post specific’ cell cluster is a putative precursor that may lead to malignant cell transformation or if this cell cluster is destined for apoptotic cell death.
In summary, we have systematically characterized gene expression and TF regulation in the FT before and after menopause and during different menstrual cycle phases. These datasets serve as a valuable resource for investigating the biological functions of the female reproductive tract (FT) and shed light on the underlying reasons for the differential prevalence of certain diseases among pre-menopausal and post-menopausal women.
Limitations of our study include that we did not measure blood hormone levels to determine menstrual cycle phases but determined them based on patient history and endometrial pathology. We reported three patients in the secretory phase and four patients in the proliferative phase of the menstrual cycle. For three patients, we did not know the cycle phase due to the use of contraception. Analyses were performed on combined cells from ampulla and fimbria for scRNA- and scATAC-seq samples unless indicated otherwise. For similar reasons, and due to the limited number of pre-menopausal isthmus (I) samples collected, we did not include them in the menstrual cycle analysis. Ethnicity might be a confounding factor in our study since six out of seven patients in the postmenopausal cohort were Caucasian, and eight out of ten were African American in the premenopausal cohort.
scATAC-seq was only performed on a subset of donors used for scRNA seq (D14– 18), resulting in fewer cell numbers sampled. As a result, we could not identify all the cell types identified in scRNA-seq in our scATAC-seq analysis.
Methods
All research complies with all relevant ethical regulations and procedures described herein were conducted under the approval of the Institutional Review Board at the University of Chicago. We obtained informed consent, including consent to data from this study being used in medical publications or presentations from all study participants prior to their surgery.
Experimental model and subject details
We collected fallopian tube fimbria (F), isthmus (I), and ampulla (A) samples from 10 pre-menopausal women. Healthy women under the age of 50 undergoing surgery for fibroids, prolapse or elective sterilization were approached for study participation. All eligible women were approached regardless of socioeconomic factors, race or ethnicity. Participants were not compensated for partaking in this study. A gynecologic pathologist (R.L., A.B.) determined the menstrual cycle phases for each patient. Phases were determined based on the microscopic appearance of the normal endometrium or abstracted from the pathology report (Supplementary Table 1). We obtained informed consent from all study participants prior to their surgery.
Cohort description and fallopian tube collection
The cohort for this study included 17 healthy female patients (designated as D1-7, and D9-D19; Supplementary Table 1) who donated FT collected at the time of surgery for benign gynecological conditions at the University of Chicago. The FT collected did not show any major macroscopic or histologic abnormalities (Fig. 1A). We analyzed the different anatomic regions, including the isthmus, ampulla, and fimbriae (Fig. 1A), using scRNA-seq and scATAC-seq.
The single cell results of the seven post-menopausal samples (D1-D7) were previously reported11. For this study we collected the same FT anatomic regions from an additional ten pre-menopausal women (D9-D18) for scRNA- or scATAC-seq, and Visium by 10x Genomics (D19). We expeditiously transported fresh tissues from the operating room to the laboratory for cell dissociation, followed by scRNA-seq and scATAC-seq assays.
Immunohistochemistry
We used formalin-fixed and paraffin-embedded tissue blocks from samples collected as part of this study. Briefly, five µm sections were stained with hematoxylin and eosin or with commercially available antibodies using the Leica Bond RX automated stainer (Leica Biosystems) using the following antibody: OVGP1 at 1:500 (Thermofisher Scientific, PA5-64007).
Tissue dissociation, primary cell isolation and hormone treatments
For tissue dissociations, 2-3 mm (100 mg) cross-sections of FT segments (Ampulla, Fimbria and Isthmus) were digested independently using a two-stage protocol to separate epithelial and stromal cells. In the first stage epithelial cells were digested using pronase (7 U/mg, Sigma-Aldrich, Cat#10165921001) in Opti-MEM (Thermo Fisher Scientific, Cat# 31985-070) at 37 °C for 15 min followed by a filtration step to separate epithelial cells from the remaining stromal portion. The stroma-fibroblast supernatant underwent a second digest with DNAse I (20,000 U/mL, Millipore Sigma, Cat#4536282001), collagenase IV (120 U/mg, Sigma-Aldrich, Cat#5138), Hyaluronidase (2,000 U/mg, Sigma-Aldrich, Cat#H3884) in HBSS (Corning, Cat#21-022-CV) at 37 °C for 15 min. The epithelial and stromal-fibroblast suspensions were then combined and passed through a 70 µm filter prior to centrifugation at 400 rcf for 7 min and resuspension in DMEM (Thermofisher Scientific, A1443101) with FBS (FBS, Applied biological materials, TM997-100). Red blood cells were removed using red blood cell lysis solution, EasySep RBC Depletion Reagent (Stemcell Tech, Cat#18170).
For in vitro studies, fallopian tube epithelial cells (FTECs) were isolated and cultured as previously described51. Following epithelial cell removal, we used the remaining stromal and fibroblast fraction for further processing. The stroma and fibroblast fraction was rinsed in PBS and digested (12–18 h) in collagenase type III (1000 U, Worthington Biochemical, LS004206) in 10% fetal bovine serum (FBS, Applied biological materials, TM997-100) in DMEM. Following digestion, cells were centrifuged, and the resulting pellet washed in PBS. Fallopian tube stromal cells (FTSCs) were plated in DMEM and 10% FBS.
The FTECs were plated at 250,000 cells on primaria coated 6-well tissue culture plates (Corning, 353846) and the FTSCs were plated at 200,000 cells on 6-well tissue culture plates. Both FTECs and FTSCs were cultured at 5% CO2, 37 °C for 24–48 hrs or until 70% confluence was achieved. Cells were then primed with either 500 nM β-estradiol (E2, Sigma-Aldrich, E2758), medroxyprogesterone 17-acetate (P4, Sigma-Aldrich, M1629) or DMSO (Control) for 8 h before cell were harvested and RNA isolated.
Immunofluorescence
Chamber slides were coated with fibronectin (5 µg/mL, BD biosciences, 356008). Human FTEC and FTSC cells were plated at 70,000 and 30,000 cells/ well respectively. Cells were treated with 500 nM medroxyprogesterone 17-acetate or 500 nM β-estradiol for 8 hrs, fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 and blocked with goat serum. Slides were stained overnight at 4 °C in goat serum (Thermo Fisher Scientific, Cat#16210064) using the following primary antibodies: IGF1 (1:200, OriGene Technologies, TA805748S) and IGF2 (1:200, ThermoFisher scientific, MA532485), followed by fluorescently labeled secondary antibodies (1:200, Alexa Fluor 488 and 568, Invitrogen) and Hoechst 33258 (1:200, Molecular probes, H-3569) for 1 h. We imaged slides on a Nikon Eclipse Ti2 microscope and processed images with NIS-Elements (Nikon).
10X Genomics 3’ Single-cell RNA sequencing
Samples from 10 pre-menopausal women were used for 10x Genomics 3’ scRNA-seq. Single-cell suspensions were washed in PBS + 0.4% BSA and resuspnded in PBS + 0.4% BSA to achieve a target cell count of 700-1200 cells/µl. 3’ scRNA-seq libraries were generated targeting 8000 cells per sample. Sequencing libraries were constructed using 15 amplification cycles following 10X Genomics protocols. The resulting libraries were sequenced using the Illumina NextSeq 500, (using 75 cycle kits) or Novaseq 6000 (using 100 cycle kits) at 30,000–50,000 reads per cell.
Single-cell RNA sequencing data analysis
We converted and demultiplexed raw binary base call (BCL) sequencing data into FASTQ files using the wrapper function, cellranger mkfastq from the Cell Ranger software52. In brief, raw sequencing reads were aligned to human genome reference hg38, filtered, and quantified as unique molecular identifier (UMI) using cellranger count. We refined the UMI count matrix by applying a systematic analysis workflow (scRICA)53 with the following quality control (QC) criteria: (1) cells must express at least 200 gene features, and each gene feature must be present in a at least 3 cells; (2) removal of doublets and triplets as identified by DoubletDecon54; and (3) filtering of low-quality cells with ≥20% mitochondrial content. Next, UMI count matrices were log-normalized and cells from different samples were integrated via Seurat v4.0 RPCA method. Unwanted variations were eliminated prior to dimensionality reduction using PCA and construction of a shared nearest-neighbor (SNN) graph. The SNN graph was constructed for graph-based clustering via Seurat function FindNeighbors and FindClusters correspondingly, at resolution of 0.5.
We then compared the cell clusters obtained from unsupervised learning to our post-menopausal study, and manually annotated them using literature curated canonical cell type markers (Supplementary Data 2). We used HIPPO to refine SE subtypes. Premature termination was prevented by applying a z-score threshold of 1 and maintaining the default outlier proportion at 0.001% in HIPPO. The resulting SE sub-clusters were further characterized using additional gene marker lists.
Ligand–receptor analysis with CellPhoneDB
We utilized normalized gene expression data from seven pre-menopausal donors with clearly identified menstrual cycle phases as input for ligand-receptor analysis. The CellPhoneDB database v2.0.0 (https://www.cellphonedb.org/) was employed to detect ligand-receptor interactions55. For each of the two distinct menstrual cycle phases, we determined significant ligand–receptor interactions between any pair of cell types, using a statistically significant level of 0.05. To examine and compare the differences between the menstrual cycle phases, we chose the ligand–receptor interactions that were most closely related to hormone response.
Functional enrichment analysis
For functional enrichment analysis, we used various identified gene sets as input files, including top genes obtained from SE cells from different menstrual cycle phases, SE cells from pre- or post-menopausal patients, and the top 200 over-expressed genes in each sub-cluster of SE and ST cells. We analyzed these gene sets with the Gene Ontology (GOs) database, concentrating on three functional categories: biological processes (BP), cellular components (CC), and molecular functions (MF). The entire analysis was conducted in R using the bioconductor package clusterProfiler. Enriched functions were identified at FDR-corrected p-values of 0.1, and we presented selected GO terms in dot-plots for visualization.
Velocity analysis
We used scVelo for scRNA velocity analysis56 to derive spliced and unspliced count matrices for each cell in our scRNA-seq dataset37.
These matrices enabled us to estimate the rates of splicing and transcription for individual genes within each cell. Using BAM files containing aligned reads for each sample, we provided input to scVelo, which merged the unspliced and spliced matrices of each sample into an AnnData object for efficient single-cell data storage and manipulation.
To facilitate downstream analyses, we categorized cells into subsets based on their assigned cell types and phenotypes. Subsequently, using a dynamical model, we computed both velocity stream and latent time for each cell in the dataset. The velocity stream describes both the direction and magnitude of RNA changes over time, while latent time represents the elapsed duration since the initiation of the transcriptional program.
Single-cell ATAC sequencing
Single-cell suspensions were lysed on ice for 4 min to obtain intact nuclei, followed by nuclei tagmentation at 37 °C for 1 hr to generate scATAC-seq libraries. For scATAC-seq we targeted 8000 cells with 6000–8000 nuclei per sample. We used Agilent 2100 Bioanalyzer traces to evaluate library quality and performed sequencing with the University of Chicago Core Facility on the Illumina NextSeq 500 or NovaSeq 6000 platforms. Libraries were sequenced at 25,000–30,000 (at PE75 and PE100, respectively) per nucleus.
Pre-processing and quality control of scATAC-seq data
We performed scATAC-seq data analysis following the scATAC-pro workflow57. In brief, raw sequencing data were aligned to human hg38 reference genome and R Seurat v4.0 and Signac v1.4.058 were used for further analysis. Quality control metrics for samples to be included in term-frequency inverse-document-frequency (TFIDF) normalization were: (1) cells with peak region fragments between 1000 and 20,000; (2) fraction of fragments in peaks >15%; (3) blacklist ratio <0.05; (4) nucleosome signal <10; and (5) TSS enrichment >2. Next, we performed label transfer (from scRNA-seq) and merged Seurat objects from each sample based on the common peak set created by merging peaks from all the datasets. Dimensional reduction was performed using singular value decomposition of the TFIDF matrix and UMAP. Batch effects were corrected on the first 25 latent semantic indexing (LSI) components using Harmony59. The first LSI was excluded because it was highly correlated with sequencing depth. Gene activity scores were estimated using Seurat’s GeneActivity60 and corrected data were used for unsupervised clustering analysis using FindNeighbor and FindClusters functions60.
Integration of scRNA-seq and scATAC-seq datasets
We used cells from matched scRNA-seq as a reference to predict and obtain cell types in our scATAC-seq data11. The variable features of the scRNA-seq data were used as a reference, while the gene activity matrix generated using Seurat’s GeneActivity60 from scATAC-Seq data was used as the query data. Transfer anchors were learned in Seurat using FindTransferAnchors60, and cell type labels were predicted with TransferData60 with the scATAC-seq LSI reduction as weight.reduction input. We assigned each cell in the scATAC-seq with a cell type (subcluster) identity from the matching scRNA-seq data based on the first 30 LSI components corrected by Harmony, excluding the first one. We only retained cells with a prediction score > 0.5 (denoted by prediction.score.max) for further analysis. We performed label transfer for each patient separately.
Transcription factor motif analysis
Transcription factor (TF) activities were estimated from Harmony Harmony integrated scATAC-seq data using chromVAR v3.1461. Transcription factors and their binding motifs listed in human_pwms_v2 (cisBP)17 were used as input to chromVAR v3.1461 for positional weight matrix calculation. We applied RunChromVAR in Signac v1.4.0 to calculate cell type-specific TF activities. Differential TF activities for each cell type by menopausal status were computed with FindMarkers using Bonferroni-adjusted p-values < 0.05. We performed TF enrichment analysis of all motifs for each cell separately. We then compared and displayed the results using absolute differences using a z-score that subtracts the average pre-menopausal TF activity from the post-menopausal activity (Post - Pre), or by displaying relative changes, expressed as a percentage change from the pre-menopausal levels ((Post - Pre) / Pre * 100).
Tissue preparation and image acquisition for spatial transcriptomics (Visium)
Fresh fallopian tube fimbriae were embedded in cryomolds with ice-cold Tissue Tek O.C.T and the blocks stored at -80°. Prior to the placement of 10 µm frozen sections onto the gene expression and tissue optimization slides (10x Genomics), we analyzed RNA Integrity Number values to determine RNA quality. The fimbria of one patient was used in quadruplicate. Tissues were optimized according to the manufacturers’ user guide (Visium spatial gene expression reagents kits- tissue optimization; document number: CG000238 Rev D). The slides and samples were processed as described by the manufacturer user guide for frozen samples (Visium spatial gene expression reagent kit, document number: CG000239 Rev E) and libraries generated. Samples were sequenced on the Illumina NovaSeq using 75 bp PE. Raw sequencing data was pre-processed using spaceranger v1.3 from 10X Genomics. The data was then loaded using Load10X Spatial function in R Seurat and normalized using sctransform. The SpatialFeaturePlot function was applied to visualize the gene expression in the histology image.
Analysis of TCGA data
TCGA gene expression data were retrieved from GDAC Firehose. R survival and survminer packages were used for survival analysis on the TCGA OV cohort. We used the Kaplan Meier method to generate survival curves and used log rank test to compare the survival curves regarding gene expression levels.
Resource availability
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Mengjie Chen (mengjiechen@uchicago.edu). Requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mengjie Chen (mengjiechen@uchicago.edu).
Statistics and reproducibility
No statistical methods were used to predetermine sample sizes, but our cohort sizes are similar or larger to those reported in previous publications. Investigators were not blinded to conditions during clinical data collection and analysis. Bioinformatic analyses were performed blind to molecular characteristics. The clinical samples used in this study were non-randomized with no intervention, and all samples were interrogated equally. Thus, controlling for covariates among clinical samples is not relevant. Unless specified otherwise, lines represent means, and error bars represent standard error of the means. We indicated sample sizes, statistical tests, and p-values in the figure legends. All sample sizes noted represent biological replicates unless stated otherwise.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw scRNA-seq and ATAC-seq data generated in this study have been deposited in the European Genome-Phenome Archive (EGA) database under accession code EGAS50000000628. The data contains sensitive human genomic and phenotypic information, necessitating controlled access to protect participant confidentiality and comply with ethical standards. Access is restricted to authorized researchers and clinicians who have obtained approval from the relevant Data Access Committee (DAC). To request access, you must contact the DAC associated with this dataset through EGA. Contact details are provided on the dataset’s EGA page. The response time for access requests varies depending on the DAC’s review process, up to 3 months. Once access is granted, the duration for which you can use the data is specified in the Data Access Agreement. This period is determined by the DAC and may vary based on the nature of the research and ethical considerations. The processed scRNA-seq data are available at CELLxGENE under the following link: https://cellxgene.cziscience.com/collections/380ade76-e561-49a8-afb2-0f10b39c2c72. The scRNA-seq and ATAC-seq data used in this study are available in the EGA database under accession code EGAS00001006780. Source data are provided with this paper.
References
Holdsworth-Carson, S. J., Menkhorst, E., Maybin, J. A., King, A., Girling, J. E. Cyclic processes in the uterine tubes, endometrium, myometrium, and cervix: pathways and perturbations. Mol. Hum. Reprod. 29, gaad012 (2023).
Rigby, C. H. et al. The immune cell profile of human fallopian tubes in health and benign pathology: a systematic review. J. Reprod. Immunol. 152, 103646 (2022).
Crow, J., Amso, N. N., Lewin, J. & Shaw, R. W. Morphology and ultrastructure of fallopian tube epithelium at different stages of the menstrual cycle and menopause. Hum. Reprod. 9, 2224–2233 (1994).
Karst, A. M. et al. Stathmin 1, a marker of PI3K pathway activation and regulator of microtubule dynamics, is expressed in early pelvic serous carcinomas. Gynecol. Oncol. 123, 5–12 (2011).
Labidi-Galy, S. I. et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 8, 1093 (2017).
Kuhn, E. et al. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma-evidence supporting the clonal relationship of the two lesions. J. Pathol. 226, 421–426 (2012).
Gray, G. K. et al. A human breast atlas integrating single-cell proteomics and transcriptomics. Dev Cell. 57, 1400–1420 (2022).
Travaglini, K. J. et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625 (2020).
Wang, Y. et al. Spatial transcriptomic analysis of ovarian cancer precursors reveals reactivation of IGFBP2 during pathogenesis. Cancer Res. 82, 4528–4541 (2022).
Ulrich, N. D. et al. Cellular heterogeneity of human fallopian tubes in normal and hydrosalpinx disease states identified using scRNA-seq. Dev. Cell. 57, 914–29.e7 (2022).
Lengyel, E. et al. A molecular atlas of the human postmenopausal fallopian tube and ovary from single-cell RNA and ATAC sequencing. Cell Rep. 41, 111838 (2022).
Dinh, H. Q. et al. Single-cell transcriptomics identifies gene expression networks driving differentiation and tumorigenesis in the human fallopian tube. Cell Rep. 35, 108978 (2021).
Hu, Z. et al. The repertoire of serous ovarian cancer non-genetic heterogeneity revealed by single-cell sequencing of normal fallopian tube epithelial cells. Cancer Cell. 37, 226–42.e7 (2020).
Eddy, C. A. & Pauerstein, C. J. Anatomy and physiology of the fallopian tube. Clin. Obstet. Gynecol. 23, 1177–1193 (1980).
Muhl, L. et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 11, 3953 (2020).
Diaz-Flores, L. et al. CD34+ stromal cells/fibroblasts/fibrocytes/telocytes as a tissue reserve and a principal source of mesenchymal cells. Location, morphology, function and role in pathology. Histol. Histopathol. 29, 831–870 (2014).
Weirauch, M. T. et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014).
Gomez-Herranz, M., Taylor, J. & Sloan, R. D. IFITM proteins: Understanding their diverse roles in viral infection, cancer, and immunity. J. Biol. Chem. 299, 102741 (2023).
Chan, J. M. et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell. 39, 1479–96.e18 (2021).
Basisty, N. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599 (2020).
Karakaslar, E. O. et al. Transcriptional activation of Jun and Fos members of the AP-1 complex is a conserved signature of immune aging that contributes to inflammaging. Aging Cell. 22, e13792 (2023).
Uittenboogaard, L. M. et al. BACH2: a marker of DNA damage and ageing. DNA Repair 12, 982–992 (2013).
Hwang, T. S. & Song, J. Morphometrical changes of the human uterine tubes according to aging and menstrual cycle. Ann. Anat. 186, 263–269 (2004).
Ehinger, J. K. et al. Mitochondrial function in peripheral blood cells across the human lifespan. NPJ Aging 10, 10 (2024).
Reddy, J. et al. Predicting master transcription factors from pan-cancer expression data. Sci. Adv. 7, eabf6123 (2021).
Paik, D. Y. et al. Stem-like epithelial cells are concentrated in the distal end of the fallopian tube: a site for injury and serous cancer initiation. Stem Cells 30, 2487–2497 (2012).
Wu, Y. J. et al. The mesothelial keratins: a new family of cytoskeletal proteins identified in cultured mesothelial cells and nonkeratinizing epithelia. Cell 31, 693–703 (1982).
Yao, Y., Yao, J. & Bostrom, K. I. SOX transcription factors in endothelial differentiation and endothelial-mesenchymal transitions. Front Cardiovasc Med. 6, 30 (2019).
Garcia-Alonso, L. et al. Mapping the temporal and spatial dynamics of the human endometrium in vivo and in vitro. Nat. Genet. 53, 1698–1711 (2021).
Nameki, R., Chang, H., Reddy, J., Corona, R. I. & Lawrenson, K. Transcription factors in epithelial ovarian cancer: histotype-specific drivers and novel therapeutic targets. Pharm. Ther. 220, 107722 (2021).
Flores, E. R. et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416, 560–564 (2002).
Brodowska, A. et al. Estrogen and progesterone receptor immunoexpression in fallopian tubes among postmenopausal women based on time since the last menstrual period. Int. J. Environ. Res. Public Health. 18, 9195 (2021).
Holt, E. H., Lupsa, B., Lee, G. S., Bassyouni, H., Peery, H. E. Hormonal control of reproduction in the female: the menstrual cycle. In Goodman’s Basic Medical Endocrinology (eds. Holt, E. H, Lupsa, B., Lee, G. S., Bassyouni, H., Peery H. E.) 433–472 (Elsevier, 2021).
Zhao, Y., Vanderkooi, S. & Kan, F. W. K. The role of oviduct-specific glycoprotein (OVGP1) in modulating biological functions of gametes and embryos. Histochem Cell Biol. 157, 371–388 (2022).
Punyadeera, C. et al. Oestrogen-modulated gene expression in the human endometrium. Cell Mol. Life Sci. 62, 239–250 (2005).
Taylor, R. J., Tagiltsev, G. & Briggs, J. A. G. The structure of COPI vesicles and regulation of vesicle turnover. FEBS Lett. 597, 819–835 (2023).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Gorin, G., Fang, M., Chari, T. & Pachter, L. RNA velocity unraveled. PLoS Comput Biol. 18, e1010492 (2022).
Piek, J. M. et al. Dysplastic changes in prophylactically removed fallopian tubes of women predisposed to developing ovarian cancer. J. Pathol. 195, 451–456 (2001).
The Cancer Genome Atlas N. Integrated genomic analyses of ovarian carcinoma. Nature. 474, 609–615 (2011).
Laszczynska, M., Brodowska, A., Starczewski, A., Masiuk, M. & Brodowski, J. Human postmenopausal ovary–hormonally inactive fibrous connective tissue or more? Histol. Histopathol. 23, 219–226 (2008).
Zhou, Y., Wu, H., Zhao, M., Chang, C. & Lu, Q. The bach family of transcription factors: a comprehensive review. Clin. Rev. Allergy Immunol. 50, 345–356 (2016).
Ng, A. et al. Lgr5 marks stem/progenitor cells in ovary and tubal epithelia. Nat. Cell Biol. 16, 745–757 (2014).
Robertson, S. A., Care, A. S. & Moldenhauer, L. M. Regulatory T cells in embryo implantation and the immune response to pregnancy. J. Clin. Invest. 128, 4224–4235 (2018).
Green, E. S., et al. Regulatory T cells are paramount effectors in progesterone regulation of embryo implantation and fetal growth. JCI Insight. 8, e162995 (2023).
Karst, A. M., Levanon K., Drapkin, R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc. Natl Acad. Sci. USA. 108, 7547–7552 (2011).
Perets, R. et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca; Tp53; Pten models. Cancer Cell. 24, 751–765 (2013).
Karst, A. M. et al. Cyclin E1 deregulation occurs early in secretory cell transformation to promote formation of fallopian tube-derived high-grade serous ovarian cancers. Cancer Res. 74, 1141–1152 (2014).
Heinze, K. et al. RUNX3 transcript variants have distinct roles in ovarian carcinoma and differently influence platinum sensitivity and angiogenesis. Cancers (Basel). 13, 476 (2021).
Ahmet, A. A. et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 221, 49–56 (2010).
Karst, A. M. & Drapkin, R. Primary culture and immortalization of human fallopian tube secretory epithelial cells. Nat. Protoc. 7, 1755–1764 (2012).
Zheng, G. X. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
scRICA: single-cell RNA-Seq integrative comparative analysis. https://github.com/yan-cri/scRICA/?tab=readme-ov-file#readme (2021).
DePasquale, E. A. K. et al. DoubletDecon: Deconvoluting doublets from single-cell RNA-sequencing data. Cell Rep. 29, 1718–27.e8 (2019).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 15, 1484–1506 (2020).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Yu, W., Uzun, Y., Zhu, Q., Chen, C. & Tan, K. scATAC-pro: a comprehensive workbench for single-cell chromatin accessibility sequencing data. Genome Biol. 21, 94 (2020).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–87.e29 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–902.e21 (2019).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. ChromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Acknowledgements
We thank the women who kindly donated the tissues and the surgeons that collected tissue (Dr. Laura Douglass, Dr. Perpetua T. Goodall, and Dr. Obianuju Sandra S. Madueke-Laveaux). We also thank the Human Tissue Resource Center and the Genomics Facility. Grant funding: This work is supported by the Chan Zuckerberg Initiative (to E.L., O.B., M.W., M.C.). M.C. is supported by the NIH (R01 GM126553 and R01 HG011883), the National Science Foundation (NSF 2016307), and the Sloan Research Fellowship Program. A.B. is supported by NIH DP2AI158157. E.L. is supported by R35CA264619, RO1CA211916, RO1CA237029, the Ovarian Cancer Research Alliance (OCRA), and Bears Care, the charitable arm of the Chicago Bears Football Club.
Author information
Authors and Affiliations
Contributions
E.L., A.B., and M.C. jointly oversaw project design and analysis. Computational analyses were performed by Y.L., L.Z., and M.C. Tissue dissociations and 10X Genomics single-cell RNA and ATAC-seq assays were performed by H.E., P.B., S.A., R.K. and M.W, under A.B.’s guidance. M.W. performed Visium (by 10x Genomics) experiments, immunofluorescence, and immunohistochemistry. M.W. designed all images made with Biorender.com. P.B. and M.W. performed all tissue isolations and cultures for in vitro studies. R.L. and A.J.B. read histology and immunohistochemistry. S.A., P.B. and R.K. consented all women donating tissues. E.L., M.W., Y.L., and M.C. wrote the manuscript. All authors reviewed and agreed on the final version of the submitted manuscript.
Corresponding authors
Ethics declarations
Competing interests
E.L. receives research funding to study ovarian cancer from Arsenal Bioscience and AbbVie through the University of Chicago unrelated to this work. All other authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Ahmed Ahmed, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Weigert, M., Li, Y., Zhu, L. et al. A cell atlas of the human fallopian tube throughout the menstrual cycle and menopause. Nat Commun 16, 372 (2025). https://doi.org/10.1038/s41467-024-55440-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55440-2
This article is cited by
-
Spatial transcriptomic profiling of the human fallopian tube epithelium reveals region-specific gene expression patterns
Communications Biology (2025)
-
NNMT inhibition in cancer-associated fibroblasts restores antitumour immunity
Nature (2025)
-
Impact of BRCA mutations, age, surgical indication, and hormone status on the molecular phenotype of the human Fallopian tube
Nature Communications (2025)
