
Abstract
Fullerene derivatives are widely employed as efficient electron-transporting layers (ETLs) in p-i-n perovskite photovoltaics but face challenges in mitigating interfacial recombination losses and ensuring stable film morphology. Non-fullerene acceptors (NFAs), commonly utilized in organic photovoltaics, present a promising alternative to fullerene-based ETLs. Nevertheless, the suboptimal performance of NFA-based devices underscores the need for molecular engineering to tailor their properties. Herein, we develop two Y-type NFAs, Y-Phen and Y-CE, by substituting the benzothiadiazole core of Y6 with higher-polarity phenanthroline and crown ether. These modifications effectively enhance carrier kinetics by (1) promoting ordered molecular assembly on the perovskite surface through supramolecular interactions, thereby optimizing interfacial energetic alignment, and (2) improving the molecular packing to facilitate efficient charge transport. Using Y-CE as the ETL, the device achieves a certified power conversion efficiency (PCE) of 25.59%. Furthermore, the optimized device exhibits less than 10% degradation in PCE after 1440 hours of thermal aging. This work offers valuable insights into designing NFA-based ETLs for high-performance perovskite photovoltaics.
Similar content being viewed by others

Introduction
Metal halide perovskite solar cells (PSCs) have attracted significant interest as a promising photovoltaic technology for solar energy conversion1,2,3. This rapid progress is attributed to various strategies for regulating perovskite crystallization and passivating defects to improve perovskite quality, as well as the application of self-assembled monolayers (SAMs) to optimize hole extraction4,5,6,7,8,9. As a result, the certified power conversion efficiency (PCE) of planar-structured inverted PSCs has rapidly surpassed 26%. However, despite the equally critical role of the electron-transporting layer (ETL) in determining the device efficiency and stability10,11, it has not received sufficient attention. For inverted PSCs, an effective ETL must satisfy three key criteria: (1) high electrical conductivity and electron mobility for effective charge transport, (2) suitable energy levels to ensure optimal interfacial energetic alignment for electron extraction and hole blocking, and (3) physical compatibility with perovskite layer to establish an intimate molecular contact that facilitates charge transfer across the interface while preventing ion migration and moisture penetration. Due to their suitable lowest unoccupied molecular orbital levels (LUMO) and high isotropic conductivity, fullerene, and its derivatives fulfill most of these requirements, making them effective ETLs for inverted PSCs12,13,14.
Nonetheless, fullerene-based ETLs present notable limitations. Their band-tail-induced energy disorder extends into the bandgap, introducing additional electronic states that lower quasi-Fermi level splitting, which in turn reduces open-circuit voltages (VOC)15,16. Additionally, the symmetrical, low-polarity, high-specific-surface-area structure of fullerenes primarily assemble through weak van der Waals forces and π–π interactions17. These interactions cause the easy aggregation of fullerene molecules to lower the surface free energy of derived films18. Such morphological changes adversely impact interfacial contact, compromising device stability under thermal and operational stress. To address this, structural modifications aimed at increasing intermolecular interactions are essential to raise the energy barrier for molecular motion19. Recent alternatives, including rylene imide-based organic semiconductors and inorganic metal oxides (e.g., SnO2), have been employed20. However, PSCs utilizing these materials as ETLs often exhibit lower PCEs compared to those using fullerenes, likely due to mismatched energy levels, rough film morphology, and interface incompatibility. Hence, the development of more effective and stable ETL materials remains crucial for achieving highly efficient PSCs.
Non-fullerene acceptors (NFAs), represented by Y6, were initially developed for high-performance bulk heterojunction organic solar cells and are promising candidates for ETLs due to their structural modification feasibility and excellent electronic properties. These NFAs form ordered aggregates through strong intermolecular interactions, which arise from their planar structures, multiple functional end-groups, and π–π interactions within core groups. Since the introduction of Y6 in 2019, various NFAs have been designed by modifying heteroatoms on the A-D-A backbone, distinct alkyl chains, 2-(3-oxo-2,3-dihydroinden-1-ylidene) (IC) group terminations, and exploring a range of fused cores such as benzothiadiazole, benzotriazole or quinoxaline derivatives21,22,23,24. These developments offer significant advantages for Y-type NFAs as ETL candidates in inverted PSCs for the following reasons: (1) compatibility with solution processing to form dense and homogeneous films; (2) suitable electron mobility and tunable energy levels that align well with the perovskite layer25,26; (3) effective defect passivation through the presence of heteroatoms; and (4) a balance between robust aggregation and smooth morphology achieved through appropriate structural modifications27. In a comparative study, C60, PC61BM, and Y6 were deposited on perovskite films and subjected to thermal aging conditions to assess morphological stability. Both C60 and PC61BM films showed an alteration in surface roughness after thermal aging (Supplementary Fig. 1 and Note 1), due to self-aggregation induced by molecular motion. This morphological change was independent of the perovskite substrate, as thermal treatment had minimal impact on the perovskite film itself (Supplementary Fig. 2). In contrast, the perovskite/Y6 film maintained its original morphology well, demonstrating the robust morphology stability of the NFA film. These merits make NFAs promising candidates to replace fullerenes as efficient ETLs in inverted PSCs.
However, the highest PCE for PSCs using Y6-based NFAs as ETL has only reached approximately 21%. When used as an interlayer between the perovskite and fullerene ETL, a moderate PCE of 24.5% was achieved. Beyond the issues of film morphology, energy level alignment, and interface compatibility previously discussed, the limited application of Y-type NFAs in high-performance PSCs may be attributed to energy disorder-related carrier behaviors at both the perovskite/NFA interface and within the bulk phase (Supplementary Fig. 3). First, carrier transport in Y-type NFAs films is strongly influenced by molecular packing through π–π interactions, with ordered molecular assembly potentially achieved through modifying targeted functional group28,29,30,31. Second, the orientation and adhesion of dipolar molecules on the perovskite surface significantly impact directional carrier movement across the interface32. A dipolar molecule with varying orientations can yield distinct electronic properties on perovskite surfaces with different compositions33,34. For Y6, while heteroatoms (e.g., S, F, O, N) provide binding sites to passivate defects on the perovskite35, the lack of controlled interaction between dipolar groups and the perovskite surface may lead to unfavorable interface energy level alignment, therefore impacting charge extraction efficiency. Therefore, it would be ideal to functionalize NFAs with specific groups that promote dipolar supramolecular interactions with perovskite, enabling desirable energy level alignment for electron extraction while improving molecular stacking to enhance electron transport.
To realize these hypotheses, two Y-type NFAs, Y-Phen and Y-CE, were designed and synthesized by substituting the benzothiadiazole core with phenanthroline and crown ether (15-crown-5, 15C5), respectively, through a fused quinoxaline structure. This rational molecular design increased the molecular dipole, promoting both an ordered arrangement of interfacial NFA molecules and enhancing π–π interactions within the bulk film, particularly for Y-CE, as supported by DFT calculation, surface electronic analysis, and X-ray crystallography. The optimized energetics of Y-CE-based PSCs achieved a certified performance exceeding 25%, one of the highest values reported for PSCs with NFA-based ETLs. Furthermore, reduced charge accumulation at the perovskite/NFA interface significantly enhanced device stability.
Results
Tailored dipole NFAs as ETLs for high-performance inverted PSCs
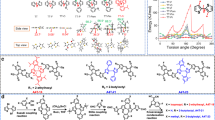
To address energy disorder in Y6-based NFAs for their application as efficient ETLs in high-performing inverted PSCs, a structural design focusing on inducing an optimal dipole moment was considered. This approach aims to improve both the interfacial contact between the NFA and the perovskite surface, as well as the molecular packing. Electron-rich phenanthroline and crown ether 15C5 groups, which contain multiple N and O sites (Fig. 1a), were employed for this purpose. The introduction of phenanthroline and 15C5 into Y-Phen and Y-CE was realized through successive reactions of reductive ring-opening with in-situ oxidation, followed by condensation with 1,2-diamine or o-quinone, as detailed in Supplementary Fig. 4 and Fig. 5. The corresponding structural characterizations are presented in Supplementary Figs. 6–19. Surface electrostatic potential analysis revealed increased negative potentials at the cores of both Y-Phen and Y-CE (Fig. 1b), resulting in elevated dipole moment from 1.07 D for Y6 to 2.30 D for Y-Phen, and 7.12 D for Y-CE. These NFAs demonstrated excellent solubility in aprotic polar solvents, enabling the solution processing of dense and uniform films on the perovskite film (Supplementary Fig. 20). Moreover, both Y-Phen and Y-CE displayed excellent thermal stability (Supplementary Fig. 21a), blue-shifted absorption compared to Y6 (Supplementary Fig. 21b), and slightly higher mobility than Y6 (Supplementary Fig. 21c). The LUMO energy levels of Y6, Y-Phen and Y-CE were determined to be −4.10, −3.86 and −3.90 eV, respectively (Supplementary Fig. 22)21, which match well with the conduction band minimum (CBM) of the perovskite layer.

a, b Molecular structures a and their electrostatic potential maps b of the modified NFAs. c, d J–V curves c and performance statistics d of NFA-based PSCs. e Certified J–V curves of champion PSC. f Performance comparison of inverted PSCs incorporating NFAs as ETLs, either individually or in combination with fullerene.
To investigate the potential of these modified NFAs as ETLs in inverted PSCs, fullerene-free devices were fabricated with the configuration of ITO/CbzNaph/Perovskite/NFA/BCP/Ag, where CbzNaph represents a p-type self-assembled monolayer (SAM)36, BCP represents bathocuproine and formamidine-cesium perovskite composition (FA0.95Cs0.05PbI3) was used. After optimizing the thickness of NFA film (Supplementary Fig. 23 and Note 2), the PCE was significantly enhanced to 23.72% and 24.11% for PSCs with Y-Phen and Y-CE as ETLs, respectively, compared to 21.96% for devices using Y6 as the ETL. The enhancement was clearly reflected in both the VOC and fill factor (FF) parameters, with VOC and FF increasing from 1.127 V and 79.57% for Y6 to 1.163 V and 83.03% for Y-Phen, and further to 1.168 V and 84.08% for Y-CE (Fig. 1c, Supplementary Fig. 24 and Table 1). The performance statistics of the fabricated devices are shown in Fig. 1d. Notably, the enhancement in device performance correlates with the increase in the dipole moment of the modified NFAs37,38, particularly in the FF parameter. This performance is comparable to results obtained using fullerene (Supplementary Fig. 25). To further enhance device performance, NFA materials were incorporated into a modified device structure with a ternary perovskite composition (FA0.85MA0.1Cs0.05PbI3) as the active layer (details in Experimental Section). The optimized Y-CE-based device exhibited a PCE of 25.61% with an FF parameter exceeding 85% (Supplementary Fig. 26). One of the best-performing devices was sent to the National PV Industry Measurement and Testing Center, where it received a certified PCE of 25.59% (Fig. 1e and Supplementary Fig. 27). This result ranks among the highest reported, surpassing those using single NFAs or NFA/fullerene combinations (Fig. 1f and Supplementary Table 2). This achievement underscores the importance of rational molecular design of NFAs as efficient ETLs for high-performance PSCs.
The interfacial orientation of NFAs correlates with their energetic alignment
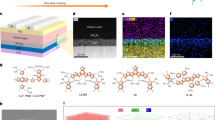
The interfacial contact between the perovskite layer and NFAs is crucial for efficient carrier extraction, necessitating optimal molecular orientation at the interface for proper energetic alignment. To investigate the molecular contact of NFAs on the perovskite (PVSK) layer, density functional theory (DFT) calculations were first performed to optimize the adsorption modes of various structural segments of Y6, Y-Phen, and Y-CE on perovskite structure. Two primary surface terminations — PbI2-rich and FAI-rich — were examined. Initially, we compared the binding energy between the Y6 backbone (Y6 structure without the IC group) and a single IC group with the perovskite structure. The IC group exhibited two binding modes with the PbI₂-rich surface through Pb-N and Pb-F interactions, while it adsorbed onto the FAI-rich surface in a flat-lying mode (Supplementary Fig. 28). The stronger binding interaction between the Y6 backbone and both PbI₂-rich and FAI-rich perovskite surfaces, compared to the IC group alone (Fig. 2a), suggests that Y6 preferentially binds to perovskite through its A-D-A backbone. The optimized adsorption structure of the Y6 backbone on both perovskite terminations resulted in dihedral angles of 24.78° and 16.49° for the PbI₂-rich and FAI-rich surfaces, respectively (Fig. 2b, Supplementary Fig. 29 and Fig. 30). These binding modes likely arise from the strong interactions between the perovskite and the heteroatoms in both the core and A-D-A backbone of Y6.

a Calculated binding energies for NFA-bound FAPbI3 (100) surface. b, c Optimized structures for FAPbI3 (100) terminations adsorbed with NFA units. d Single-crystal structure of PbI2-Phen and FAI-15C5. e XPS spectra of PVSK/NFA films showing the comparison of Pb-S and C-S-C signals. f Schematic diagram of the band diagram of NFA-coated PVSK film.
In contrast, the Y-Phen and Y-CE backbones adopt a more tilted configuration on the perovskite surface, with their chelating phenanthroline and crown ether groups anchoring to the perovskite. This results in larger dihedral angles of 50.55° and 36.68° for Y-Phen, and 33.12° and 29.48° for Y-CE on the PbI₂-rich and FAI-rich surfaces, respectively. Calculations indicate that substituting the core of the Y6 backbone with higher-polarity phenanthroline and crown ether groups can significantly enhance interactions with the perovskite, promoting directional molecular assembly with a more upright orientation. Specifically, Y-Phen and Y-CE show the lowest adsorption energies of −2.03 and −0.87 eV on the PbI₂-rich and FAI-rich surfaces, respectively. To further verify the anchoring effect of phenanthroline on PbI₂ and crown ether on FAI, single crystals of PbI₂-Phen and FAI-15C5 were grown from mixed FAPbI3/Phen and FAPbI3/15C5 solutions due to their strong interactions (Fig. 2d and Supplementary Table 3). In the [PbI2(Phen)]2 dimers, each Pb center is chelated by two phenanthroline ligands through four N–Pb bonds, with distance ranging from 2.64 to 2.86 Å (Supplementary Fig. 31). In the FAI-15C5 crystal, multiple hydrogen bonds with distances of 3.12–3.49 Å for C–H⋅⋅⋅I and 2.36–2.79 Å for C–H⋅⋅⋅N were observed, linking hydrogen interaction from 15C5 donors to I/N atoms in the FAI acceptors (Supplementary Fig. 32). These findings suggest that the modified phenanthroline and crown ether groups serve as dominant anchoring sites on the perovskite surface39,40, potentially impacting NFA molecule orientation and binding modes at the perovskite/NFA interface.
High-resolution X-ray photoelectronic spectroscopy (XPS) was further employed to investigate the interfacial orientation of NFAs. NFA/PVSK films were rinsed with chlorobenzene and compared to control samples of pure NFA and pure perovskite films deposited on ITO substrates. Significant shifts in the Pb 4f and I 3d peaks were observed for Y6/PVSK, Y-Phen/PVSK, and Y-CE/PVSK (Supplementary Fig. 33), indicating clear interfacial interactions between the NFAs with perovskite. For Y6/PVSK, the O 1s (C=O bond) and F 1s (C–F bond) peaks shifted to higher energies relative to pure Y6 (Supplementary Fig. 34a, d), suggesting an increased oxidation state resulting from Coulombic interactions between the photoemitted electrons and the ion core. The N 1s spectrum did not distinguish due to the complex C–N bonds from FA+ cation and Y6, although the CN-PVSK interaction was confirmed41,42. Notably, a characterized signal of the Pb–S bond emerged in Y6/PVSK (Fig. 2e)42,43, supporting a relatively flat orientation of the Y6 molecule on the perovskite surface to enable the potential interactions through various heteroatoms from the A-D-A backbone, core and IC groups.
In contrast, no Pb-S signals were detected for Y-Phen/PVSK and Y-CE/PVSK, indicating suppressed interfacial interaction between the S atoms (from the A-D-A backbone) and the perovskite. Additionally, no shifts in the F 1s (C–F) and O 1s (C=O) peaks were observed (Supplementary Fig. 34). These findings align with DFT-optimized structures, where the skeletons of Y-Phen and Y-CE are lifted away from the perovskite surface by their phenanthroline and crown ether cores. This supramolecular force-driven molecule arrangement helps reduce energy disorder at the interface and promotes a uniform dipole direction, with the negative core directed toward the perovskite layer and the positive end toward the NFA bulk. This configuration holds promise to raise the work function of the perovskite surface and enhance charge collection efficiency37,38.
Ultraviolet photoelectron spectroscopy (UPS) was further employed to assess the impact of interfacial dipoles on the energy levels of perovskite films. To probe the electronic effect of NFAs at the PVSK/NFAs interface, freshly deposited NFAs were rinsed with chlorobenzene, leaving only the bound NFAs on the perovskite surface. The results are presented in the energy level scheme (Fig. 2f and Supplementary Fig. 35). The work function (WF) of the pure perovskite film was measured at 4.50 eV, which decreased to 4.45 eV, 4.38 eV and 4.301 eV for Y6/PVSK, Y-Phen/PVSK and Y-CE/PVSK samples, respectively. These values indicate interfacial dipoles (∆Ф) of −0.05 eV, −0.12 eV, and −0.20 eV for Y6, Y-Phen, and Y-CE, respectively, which can induce an additional downward shift in the local vacuum level of the perovskite surface37,44. The larger WF reductions in PVSK/Y-Phen and PVSK/Y-CE can be attributed to the ordered molecular orientations with higher dipole moments (Fig. 1b), which enhance majority carrier transport while repelling minority carriers. Therefore, dipole-tailored NFAs like Y-Phen and Y-CE can effectively optimize energetic alignment by constructing an ordered interfacial molecular bridge between the perovskite and NFA ETLs, enabling efficient electron extraction at the interface and enhancing device performance through increased Voc and FF values.
Enhanced molecular packing of NFAs for bulk charge transport
To further unveil the effects of core structure modification on molecular packing, which is crucial for charge transport in the bulk, single-crystal X-ray diffraction measurements of Y-Phen and Y-CE were performed (Supplementary Fig. 36, Fig. 37, and Note 3). Details of the crystal growth method are provided in the Experimental Section, and crystallographic parameters are summarized in Supplementary Table 4. Both Y-Phen and Y-CE molecules feature banana-shaped units akin to the Y6 molecule but with enhanced coplanarity. The dihedral angles of the terminated IC groups are 2.1° for Y-Phen and 2.0° for Y-CE, significantly smaller than the 25.1° angle in Y6 (Supplementary Fig. 38). This increased coplanarity is consistent with other quinoxaline-derivatized NFAs, where improved coplanarity within one molecule is favorable for closer intermolecular packing45,46,47. Regarding the 3D packed frameworks, both Y-Phen and Y-CE form rectangle nets, but with distinct packing modes. Y-Phen exhibits three packing modes: end-to-end (mode 1), dual end-to-central (mode 2), and end-to-central (mode 3), with π–π stacking distances of 3.404(4), 3.395(4) and 3.304(4) Å, respectively (Fig. 3a). Y-CE also shows three packing modes: end-to-end (mode 1) and two types of dual end-to-central units (modes 2 and 3), with slightly longer π–π distances of 3.428(7), 3.425(8) and 3.499(6) Å, respectively (Fig. 3b). These distinct packing modes likely stem from difference between the aromatic phenanthroline and the bulky non-aromatic crown ether groups, potentially influencing the molecular assembly from the solution phase to the solid film.

a, b 3D packing frameworks of a Y-Phen and b Y-CE. c Time-dependent UV-vis absorption spectra during the solution processing of NFA films. d In-situ monitoring of the absorption peak intensity and position of NFAs as a function of film forming time during spin coating. e–g 2D GIWAXS patterns of e Y-Phen, f Y-CE films, and g the corresponding 1D profiles.
In-situ UV-vis absorption measurements were conducted to clarify the differences in molecular aggregation processes. The 2D absorption patterns are shown in Supplementary Fig. 39, with the evolution of peak intensity and position in the corresponding films depicted in Fig. 3c, d. Film formation occurs in three successive stages: (1) pre-aggregation, driven by solvent evaporation and increasing solution concentration; (2) molecular assembly, transitioning from the solution phase to the solid phase, as indicated by a red-shift in NFA absorption; and (3) stabilization of peak intensity and position48. Compared to Y6, the elongated process 2 for Y-Phen and Y-CE could allow sufficient time for molecular assembly, which is helpful for forming an ordered packing structure in the resultant films.
Grazing incidence wide-angle X-ray scattering (GIWAXS) was performed to investigate the crystallinity of the NFA films. As shown in Fig. 3e–g and Supplementary Fig. 40, all NFAs exhibited a characteristic π-π stacking diffraction peak at q~1.8 Å−1 in the out-of-plane direction, indicating a face-on crystallite orientation. Notably, Y-CE displayed higher diffraction intensity and evident spots compared to Y-Phen, with both showing stronger diffraction than Y6. Moreover, the prominent peaks of Y-Phen and Y-CE in the low q region (around 0.5 Å−1) can be originated from long-range π-π stacked ordering, which was also observed in other Y-type NFAs49. The calculated d-spacing and crystalline coherence length values were summarized in Supplementary Table 5. Additionally, the deposition of NFAs onto the perovskite surface effectively reduced the roughness of the perovskite film44. Among these, Y-Phen and Y-CE demonstrated more ordered molecular stacking both in the NFA bulk and at the PVSK/NFA interface, achieving a greater reduction in roughness compared to Y6 (Supplementary Fig. 41). This conformal film morphology is beneficial for the interfacial contact between adjacent layers, potentially contributing to better charge extraction and transport properties in Y-Phen and Y-CE NFAs.
Charge carrier dynamics between perovskite and NFA
The significant benefits of modified NFAs on carrier behaviors were further investigated. Ultrafast transient absorption (TA) spectroscopy measurements were utilized to compare the interfacial carrier dynamic. The pump light was directed through the NFA and the perovskite layer, with a 450 nm laser weakly exciting the NFAs, resulting in short exciton lifetimes due to minimal charge separation (Supplementary Fig. 42). Compared to pure perovskite, the PVSK/NFA samples showed a characteristic ground-state bleaching (GSB) peak for NFAs around 850 nm and charge characteristics near 900 nm, with longer lifetimes than those of pure NFAs (Supplementary Fig. 43). This suggests that electron transfer occurs at the PVSK/NFA, resulting in long-lived charges. By analyzing the GSB dynamics of perovskite, electron transfer rates in the different PVSK/NFA systems could be determined (Fig. 4a–d). Both perovskite and perovskite/NFA samples displayed a prominent GSB peak at 780 nm with a high-energy tail near the bandgap, attributed to state-filling effects. Additionally, the higher-energy tails of the GSB peak at 0.1 ps correspond to hot carrier cooling dynamics, arising from the rapid distribution of initial non-equilibrium carriers into a Fermi-Dirac distribution via elastic scattering50,51. Two key observations were made: (1) perovskite/NFA films showed a faster contraction in the higher-energy tail compared to perovskite films, likely due to the formation of a charge transfer state between the perovskite and NFA, which facilitates efficient hot-electron extraction; (2) the GSBs at 780 nm in PVSK/NFA decayed significantly within a few nanoseconds, demonstrating electron transfer from PVSK to NFA. Notably, Y-CE demonstrated the highest electron extraction rate (Fig. 4e), which should be ascribed to optimized energetics from the interfacial dipole, emphasizing the crucial role of the interfacial dipole in facilitating carrier extraction.

a–d TA spectra of a PVSK, b PVSK/Y6, c PVSK/Y-Phen, and d PVK/Y-CE films at different time delays. e Corresponding TA decay kinetics. f Confocal PL mapping images for PVSK and PVSK/NFA films. The collection range was set from 700–800 nm, with a scale bar of 10 μm.
Confocal PL mapping measurement was conducted to evaluate carrier transport within the NFA ETLs. The NFA coating markedly reduced the PL intensity of the perovskite, with the PVSK/Y-CE showing notably lower brightness, indicating efficient carrier extraction and transport from the perovskite layer to the dipole-tailored NFA layer (Fig. 4f). Time-resolved PL spectroscopy further proved the strong PL quenching effect of the Y-CE layer (Supplementary Fig. 44), with the lifetime decreasing from 32.4 ns for PVSK/Y6 to 21.9 ns for PVSK/Y-Phen and 9.6 ns for PVSK/Y-CE. These results elucidate the mechanism behind the enhanced performance of NFA-based PSCs, underscoring the importance of dipole-tailored NFAs in molecular design. Improved carrier dynamics can significantly boost device performance, reduce carrier accumulation, and potentially enhance device stability.
Improved stability of NFA-based inverted PSCs
The stability of PSCs using NFAs as ETL was further evaluated. Unencapsulated devices were aged under continuous heating at 65 °C in a nitrogen-filled glovebox. Devices with Y-Phen and Y-CE being the ETLs exhibited minimal PCE decay (<10%) after 1440 h, whereas the Y6-based device showed a decline of over 20% during the same period (Fig. 5a). This illustrates that the modified NFAs, with strong supramolecular interactions, can effectively bind to the perovskite surface while preserving their original carrier transport properties under thermal stress. Additionally, the devices were encapsulated with UV-cured adhesive and placed in a nitrogen-filled test box. Their performance was monitored at the maximum power point (MPP) under continuous illumination in ambient air with 30–40% relative humidity (RH) at approximately 45 °C. As plotted in Fig. 5b, the Y6-based device displayed a rapid decline in PCE within the initial 200 h, while both Y-Phen and Y-CE-based devices could retain over 90% of their initial PCEs after being operated for 1200 h, performing comparably to devices using C60 as ETL (Supplementary Fig. 45). To understand the stability improvement in NFA-based PSCs under continuous operation condition, particularly with Y-CE as the ETL, real-time charge carrier distributions in complete devices were analyzed using cross-sectional Kelvin probe force microscopy (KPFM) under open-circuit illumination conditions52,53. In this setup, the quasi-Fermi levels are split, charge carriers become unextractable, and may accumulate at selective heterointerfaces of opposite polarity54. As shown in Fig. 5c, d, the device with Y6 as the ETL exhibits significant charge accumulation at the perovskite/ETL interface, indicating inefficient carrier extraction and bulk charge transport. In contrast, the Y-CE device shows reduced charge accumulation at the perovskite/ETL interface, aligning with predictions that a larger dipole moment improves interfacial energetics and facilitates directional carrier movement. Since excessive charge accumulation at the interface is a critical factor in device degradation, the Y-CE ETL, with its enhanced charge extraction and transport capabilities, contributes to improved operational stability. Advances in interface energetics regulation, achieved through the design of ammonium cations and counter-anions or double-side 2D/3D heterojunctions53,55, have been developed to prevent potential charge trapping and accumulation at the perovskite-charge transport layer interface, further improving device stability.

a Intrinsic stability of unencapsulated devices aged at 65 °C in a N2-filled glovebox. b MPP tracking of encapsulated PSCs measured at approximately 45 °C under LED illumination. Electric field distribution in (c) Y6- and d Y-CE-based PSCs under illumination and open-circuit conditions, acquired through cross-sectional KPFM. Scale bars: 200 nm.
Device stability under atmospheric conditions was also monitored, showing a continuous performance decay across all NFA-based devices (Supplementary Fig. 46). While the hydrophobicity of the perovskite films was enhanced (Supplementary Fig. 47) through the deposition of functional NFA molecules56, this did not prevent performance decline. Studies have reported that both oxygen and water impact the stability of NFA-based devices by altering the NFAs’ electronic structure57,58. Although NFAs demonstrate considerable resistance to water vapor due to weak hydrogen bonding interactions, they are more susceptible to oxygen. Oxygen can attach at multiple positions on the NFA structure via non-covalent interactions, shifting both the highest occupied molecular orbital and the lowest unoccupied molecular orbital deeper. This significant alteration can increase pinning energy, ultimately degrading the performance of NFA-based devices. To enhance ambient stability, robust encapsulation technologies and the development of rigid NFA molecular designs that mitigate oxygen’s impact on energy levels are essential.
Discussion
Two functional NFAs, Y-Phen and Y-CE, were designed and synthesized by modifying the benzothiadiazole core of a normal Y-based structure with higher-polarity phenanthroline and crown ether groups. These modifications facilitate strong supramolecular interactions with the perovskite, thereby promoting ordered assembly and orientation of the NFAs at the perovskite surface. This molecular design simultaneously optimizes the PVSK/NFA interfacial energetic alignment, improves the conjugated planarity of the NFAs, and boosts charge extraction and transport efficiency. Collectively, these improvements enable efficient carrier dynamics in inverted PSCs, yielding a certified PCE of 25.59% with device stability comparable to fullerene-based ETLs. This work paves a pathway for developing NFAs as efficient ETLs in high-performance, fullerene-free perovskite photovoltaics.
Methods
Materials
All materials were used as received without further purification. These include dimethylformamide (DMF, 99.99%, J&K), dimethyl sulfoxide (DMSO, 99.70%, J&K), isopropanol (IPA, 99.50%, J&K) and chlorobenzene (CB, 99.90%, J&K). MAI, CsI, MACl, phenmethylammonium iodide (PMAI), and 4-fluoro-phenethylammonium iodide (pF-PEAI) were bought from Xi’an Polymer Light Technology. PbI2 (99.9985%) and PbCl2 were bought from TCI, while FAI was sourced from Dysol. Piperazinium iodide (PI) and CbzNaph were synthesized according to previous reports34,36.
Growth of single-crystal
Single crystals of Y-Phen and Y-CE were grown by layering pure methanol over their chloroform solutions (1 mg/mL) and allowing them to stand undisturbed at room temperature. PbI2-Phen crystals were obtained by mixing a PbI2 solution in DMF (5 mg/mL, 1 mL) with a phenanthroline solution in dichloromethane (3 mg/mL, 1 mL), which was left undisturbed at room temperature. FAI-15C5 crystals were obtained by mixing an FAI solution in water (12 mg/mL, 1 mL) with a 15C5 solution in methanol (20 mg/mL, 1 mL), which was evaporated under ambient conditions.
Device fabrication
FACsPbI3-based PSCs were fabricated as follows. The pre-patterned ITO glass substrates were cleaned by sonication in detergent, deionized water, acetone, and IPA successively, each for 15 min. Subsequently, the cleaned ITO glass substrates were dried in an oven at 80 °C for 24 h and treated with O2 plasma for 30 min before use. A CbzNaph SAM (1.5 mg/mL) was then spin-coated onto the ITO glass substrates at 3000 r.p.m. for 30 s and annealed at 100 °C for 10 min. A 1.4 M perovskite precursor solution with the formula FA0.95Cs0.05PbI3 was prepared by dissolving FAI, PbI2, and CsI in DMF:DMSO solvent (4:1 volume ratio). Thirty minutes before film deposition, 32.27 mg of PbI2, 11.67 mg of PbCl2, 9.38 mg of MACl, and 1 mg of pF-PEAI were added to 1 mL of the solution. 40 uL of the perovskite precursor was spin-coated at 5000 r.p.m. for 50 s, with 180 uL of CB antisolvent added to the center of the wetted film 5 s before the end of the process. The film was then annealed at 100 °C for 30 min. Subsequently, a PI solution (0.3 mg/mL in IPA) was spin-coated onto the prepared perovskite at 3000 r.p.m. for 30 s and annealed at 100 °C for 10 min. The NFAs were weighted under ambient conditions, dissolved in CB solvent within a N2-filled glovebox, and stirred at 60 °C for 30 min. The resulting solution was cooled to room temperature and spin-coated onto the perovskite film at 3000 r.p.m. for 30 s. All the spin-coating processes were conducted in an N2-filled glovebox with O2 and H2O levels below 5 ppm and a controlled temperature of approximately 20 °C. Finally, a 6-nm BCP and 100-nm Ag were thermally evaporated in a high-vacuum chamber (<2 × 10−6 torr) through a metal shadow mask.
FA0.85MA0.1Cs0.05PbI3-based PSCs were fabricated as follows. The cleaned FTO glass substrate was treated with UV ozone for 30 min before being transferred into an N2-filled glovebox for device fabrication. A CbzNaph (1.5 mg/mL) solution was spin-coated onto the substrates at 3000 r.p.m. for 30 s, followed by annealing at 100 °C for 10 min. A 1.65 M perovskite precursor solution was prepared by dissolving the components in a DMF:DMSO mixed solvent (4:1 volume ratio). Additionally, 10 mol% MACl, 5 mol % PbI2, and 0.2 mol% PMAI were added, and the precursor solution was stirred at room temperature for 3 h before deposition. For film processing, 40 uL of the precursor solution was applied to the substrate and spin-coated at 1000 r.p.m. for 5 s, followed by 5000 r.p.m. for 30 s. During the final 12 s, 180 uL of CB was dripped as an antisolvent. The resulting perovskite films were annealed at 100 °C for 30 min. For surface passivation, a solution of PI (0.3 mg/mL) and choline chloride (0.2 mg/mL) in an IPA:DMF mixed solvent (100:1 volume ratio) was dynamically spin-coated onto perovskite films at 5000 r.p.m. for 30 s, followed by annealing at 100 °C for 5 min. The NFAs were weighted under ambient conditions, dissolved in CB solvent within a N2-filled glovebox, and stirred at 60 °C for 30 min. The resulting solution was spin-coated onto the perovskite film at 3000 r.p.m. for 30 s. Finally, a 6-nm BCP and 100-nm Cu were thermally evaporated to complete device fabrication, followed by thermal evaporation of 100 nm of MgF2 onto the glass side of the devices as an antireflection layer.
Device characterizations
The J–V characteristics of the devices were measured in a N2-filled glovebox using a Keithley 2400 Source Meter under simulated sunlight from a solar simulator (SS-F5, EnliTech). To achieve an AM 1.5 G (100 mW/cm2) solar simulator light intensity, a National Renewable Energy Laboratory (NREL)-calibrated silicon solar cell (with a KG-2 filter) was employed. During testing, the perovskite solar cells were covered with a shading mask with an aperture area of 0.04 cm2 to ensure accurate current density measurements from the J–V curves. The J–V measurements were conducted in sweep mode with both reverse and forward scans at a scan rate of 10 mV/s and a voltage step of 0.02 V. External quantum efficiency (EQE) curves were obtained using an EQE measurement system (QE-R, EnliTech). Cyclic voltammetry measurements were conducted using a CHI1020D electrochemical workstation. TRPL measurement was conducted using an FLS1000 photoluminescence spectrometer system (Edinburgh), with a 375 nm pulsed excitation laser. XPS and UPS analysis were conducted using a Thermo Fisher ESCALAB XI + X-ray photoelectron spectrometer. UV light from a non-monochromatic He I source with an energy of 21.21 eV was utilized for the measurement. In-situ UV-vis spectra were gathered using a custom-built facility within a temperature-controlled nitrogen-filled glovebox. The optical probe is positioned 2–5 mm from the sample, with a halogen lamp serving as the light source. The optical fiber was arranged perpendicular to the sample to capture light reflected from the thin silver film on the substrate’s back. AFM and KPFM images were obtained using an Oxford Instruments Asylum Research Cypher ES. GIWAXS was carried out at the BL14B1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF). X-ray with a wavelength of 0.6887 A˚ and energy of 18.00264 keV was used, and 2D GIWAXS patterns were captured with a MarCCD 225 detector at a sample-to-detector distance of 522.052 mm. Subsequently, the 2D GIWAXS patterns were analyzed using FIT2D software.
Single-crystal analysis
The black, lustrous prism or needle samples, measuring 0.30 × 0.11 × 0.05 mm3 for Y-Phen and 0.45 × 0.06 × 0.02 mm3 for Y-CE, were mounted on a loop with oil. Data were collected at −173 °C using a Bruker APEX II single-crystal X-ray diffractometer with Mo-Kα radiation, equipped with a Miracol X-ray optical collimator. The data were integrated and scaled using SAINT and SADABS within the APEX2 software package by Bruker. Solution by direct methods (SHELXT or SIR97) provided a complete heavy-atom phasing model consistent with the proposed structure. The structure was refined using difference Fourier synthesis with SHELXL. Additional details are provided in the Supporting Information.
Density functional theory (DFT) calculations
DFT calculations were performed using the projector-augmented wave (PAW) method in the Vienna Ab initio Simulation Package (VASP). The generalized gradient approximation (GGA) with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional was employed. The van der Waals (vdW) interactions were included in the calculations using the zero-damping DFT-D3 method of Grimme. A uniform grid of 6 × 6 × 6 k-mesh in the Brillouin zone was used to optimize the crystal structures of cubic-phase FAPbI3 in bulk, 4 × 4 × 1 k-mesh for FAPbI3 slabs, and 2 × 2 × 1 k-mesh for NFAs-passivated FAPbI3. The 4 × 3 FAPbI3 supercells with three or four octahedral layers were exposed (100) surface terminated with both FAI-rich and PbI2-rich surfaces. The slab replicas were separated by ∼20 Å of vacuum. The energy cutoffs of the wavefunctions were set at 500 eV for the bulk and slabs and 450 eV for the supercells and interfaces. Each crystal structure was optimized until forces on single atoms were less than 0.015 eV/Å. The binding energies between NFAs and FAPbI3 were calculated as ENFAs/FAPbI3 (binding) = E (NFAs/FAPbI3) – E (NFAs) – E (FAPbI3), where E (NFAs/FAPbI3) is the total energy for NFAs-passivated FAPbI3; E (NFA) and E (FAPbI3) are the energies of isolated NFA molecule and FAPbI3 supercell, respectively. For the molecular NFAs calculations, the geometries were optimized using B3LYP functional with 6-31+G(d,p) basis set as implemented in the Gaussian16 program. The electrostatic potential maps for NFAs were obtained at the same theoretical level.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2361362 (PbI2-Phen), 2361363 (FAI-15C5), 2361364 (Y-CE) and 2361365 (Y-Phen). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Source data are provided with this paper.
References
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Lee, M. M., Teuscher, J., Miyasaka, T., Murakami, T. N. & Snaith, H. J. Efficient hybrid solar cells based on meso-superstructured organometal halide perovskites. Science 338, 643–647 (2012).
Wu, S. et al. Redox mediator-stabilized wide-bandgap perovskites for monolithic perovskite-organic tandem solar cells. Nat. Energy 9, 411–421 (2024).
Liu, C. et al. Bimolecularly passivated interface enables efficient and stable inverted perovskite solar cells. Science 382, 810–815 (2023).
Li, M., Liu, M., Qi, F., Lin, F. R. & Jen, A. K. Self-assembled monolayers for interfacial engineering in solution-processed thin-film electronic devices: design, fabrication, and applications. Chem. Rev. 124, 2138–2204 (2024).
Qu, G. et al. Conjugated linker-boosted self-assembled monolayer molecule for inverted perovskite solar cells. Joule 8, 2123–2134 (2024).
Huang, X. et al. Solvent racing crystallization: Low-solvation dispersion cosolvents for high-quality halide perovskites in photovoltaics. Joule 7, 1556–1573 (2023).
Zhou, Y. Y., Herz, L. M., Jen, A. K. Y. & Saliba, M. Advances and challenges in understanding the microscopic structure-property-performance relationship in perovskite solar cells. Nat. Energy 7, 794–807 (2022).
Zhang, X. et al. Advances in inverted perovskite solar cells. Nat. Photon. 18, 1243–1253 (2024).
Jiang, Q. & Zhu, K. Rapid advances enabling high-performance inverted perovskite solar cells. Nat. Rev. Mater. 9, 399–419 (2024).
Yang, B. P., Peng, S. M. & Choy, W. C. H. Inorganic top electron transport layer for high performance inverted perovskite solar cells. Ecomat 3, e12127 (2021).
Said, A. A. et al. Sublimed C60 for efficient and repeatable perovskite-based solar cells. Nat. Commun. 15, 708 (2024).
Shui, Q. J. et al. Evaporable fullerene indanones with controlled amorphous morphology as electron transport layers for inverted perovskite solar cells. J. Am. Chem. Soc. 145, 27307–27315 (2023).
Yin, J., Shi, X., Wang, L., Yan, H. & Chen, S. High-performance inverted perovskite solar devices enabled by a polyfullerene electron transporting material. Angew. Chem. Int. Ed. 61, e202210610 (2022).
Shao, Y. C., Yuan, Y. B. & Huang, J. S. Correlation of energy disorder and open-circuit voltage in hybrid perovskite solar cells. Nat. Energy 1, 15001 (2016).
Lian, J., Lu, B., Niu, F., Zeng, P. & Zhan, X. Electron‐transport materials in perovskite solar cells. Small Methods 2, 1800082 (2018).
Perez, E. M. & Martin, N. pi-pi interactions in carbon nanostructures. Chem. Soc. Rev. 44, 6425–6433 (2015).
Pont, S., Foglia, F., Higgins, A. M., Durrant, J. R. & Cabral, J. T. Stability of polymer:PCBM thin films under competitive illumination and thermal stress. Adv. Funct. Mater. 28, 1802520 (2018).
Tian, C. et al. Efficient and stable inverted perovskite solar cells enabled by inhibition of self-aggregation of fullerene electron-transporting compounds. Sci. Bull. 66, 339–346 (2021).
Johnson, S. A. et al. Improving the barrier properties of tin oxide in metal halide perovskite solar cells using ozone to enhance nucleation. Joule 7, 2873–2893 (2023).
Yuan, J. et al. Single-junction organic solar cell with over 15% efficiency using fused-ring acceptor with electron-deficient core. Joule 3, 1140–1151 (2019).
Xie, M., Wei, Z. & Lu, K. Quinoxaline-based Y-type acceptors for organic solar cells. Chem. Sci. 15, 8265–8279 (2024).
Yang, Y. The original design principles of the Y-series nonfullerene acceptors, from Y1 to Y6. ACS Nano 15, 18679–18682 (2021).
Zhang, Y. et al. Recent progress of Y6‐derived asymmetric fused ring electron acceptors. Adv. Funct. Mater. 32, 202205115 (2022).
Li, S., Li, C.-Z., Shi, M. & Chen, H. New phase for organic solar cell research: emergence of Y-series electron acceptors and their perspectives. ACS Energy Lett. 5, 1554–1567 (2020).
Qi, F., Fan, B., Fan, Q. & Jen, A. K. Y. Non‐fullerene acceptors with heteroatom substitution on the core moiety for efficient organic photovoltaics. InfoMat 6, e12595 (2024).
Ma, L., Zhang, S. & Hou, J. Crystal structures in state-of-the-art non-fullerene electron acceptors. J. Mater. Chem. A 11, 481–494 (2023).
An, K. et al. Mastering morphology of non-fullerene acceptors towards long-term stable organic solar cells. Nat. Commun. 14, 2688 (2023).
Shi, Y. et al. Small energetic disorder enables ultralow energy losses in non‐fullerene organic solar cells. Adv. Energy Mater. 13, 202300458 (2023).
Kupgan, G., Chen, X.-K. & Brédas, J.-L. Low energetic disorder in small-molecule non-fullerene electron acceptors. ACS Mater. Lett. 1, 350–353 (2019).
Liu, S. et al. High-efficiency organic solar cells with low non-radiative recombination loss and low energetic disorder. Nat. Photon. 14, 300–305 (2020).
Jiang, Y. et al. Reducing energy disorder in perovskite solar cells by chelation. J. Am. Chem. Soc. 144, 5400–5410 (2022).
Mariotti, S. et al. Interface engineering for high-performance, triple-halide perovskite-silicon tandem solar cells. Science 381, 63–69 (2023).
Li, F. et al. Regulating surface termination for efficient inverted perovskite solar cells with greater than 23% efficiency. J. Am. Chem. Soc. 142, 20134–20142 (2020).
Hong, J. A. et al. Efficient and moisture-stable inverted perovskite solar cells via n-type small-molecule-assisted surface treatment. Adv. Sci. 10, e2205127 (2023).
Jiang, W. et al. π-expanded carbazoles as hole-selective self-assembled monolayers for high-performance perovskite solar cells. Angew. Chem. Int. Ed. 61, e202213560 (2022).
Chen, Q., Wang, C., Li, Y. & Chen, L. Interfacial dipole in organic and perovskite solar cells. J. Am. Chem. Soc. 142, 18281–18292 (2020).
Zhang, C. et al. Reduction of nonradiative loss in inverted perovskite solar cells by donor-pi-acceptor dipoles. ACS Appl. Mater. Interfaces 13, 44321–44328 (2021).
Huang, X. et al. Solvent gaming chemistry to control the quality of halide perovskite thin films for photovoltaics. Acs. Cent. Sci. 8, 1008–1016 (2022).
Huang, X., Cheng, F., Wu, B. & Zheng, N. Intermediate chemistry of halide perovskites: origin, evolution, and application. J. Phys. Chem. Lett. 13, 1765–1776 (2022).
Wang, J. et al. Highly efficient all-inorganic perovskite solar cells with suppressed non-radiative recombination by a Lewis base. Nat. Commun. 11, 177 (2020).
Hu, Q. et al. Improving efficiency and stability of perovskite solar cells enabled by a near-infrared-absorbing moisture barrier. Joule 4, 1575–1593 (2020).
Liu, J., Ejtemaei, M., Nguyen, A. V., Wen, S. & Zeng, Y. Surface chemistry of Pb-activated sphalerite. Miner. Eng. 145, 106058 (2020).
Wang, H. et al. Interface dipole management of D-A-type molecules for efficient perovskite solar cells. Angew. Chem. Int. Ed. 63, e202404289 (2024).
Liu, K. et al. Organic solar cells with over 19% efficiency enabled by a 2D-conjugated non-fullerene acceptor featuring favorable electronic and aggregation structures. Adv. Mater. 35, e2300363 (2023).
Liang, H. et al. A rare case of brominated small molecule acceptors for high-efficiency organic solar cells. Nat. Commun. 14, 4707 (2023).
Qi, F. et al. Regiospecific N-alkyl substitution tunes the molecular packing of high-performance non-fullerene acceptors. Mater. Horiz. 9, 403–410 (2022).
Wang, Y. et al. Control of the crystallization and phase separation kinetics in sequential blade‐coated organic solar cells by optimizing the upper layer processing solvent. Adv. Energy Mater. 13, 2203496 (2023).
Zhang, G. et al. Delocalization of exciton and electron wavefunction in non-fullerene acceptor molecules enables efficient organic solar cells. Nat. Commun. 11, 3943 (2020).
Li, M. et al. Slow cooling and highly efficient extraction of hot carriers in colloidal perovskite nanocrystals. Nat. Commun. 8, 14350 (2017).
Wang, P. et al. Tetraphenylethene-based hole transporting material for highly efficient and stable perovskite solar cells. Sci. China Chem. 67, 2009–2015 (2024).
Cao, F. et al. Redox-sensitive NiOx stabilizing perovskite films for high-performance photovoltaics. J. Am. Chem. Soc. 146, 11782–11791 (2024).
Tan, S. et al. Stability-limiting heterointerfaces of perovskite photovoltaics. Nature 605, 268–273 (2022).
Cai, M. et al. Control of electrical potential distribution for high-performance perovskite solar cells. Joule 2, 296–306 (2018).
Azmi, R. et al. Double-side 2D/3D heterojunctions for inverted perovskite solar cells. Nature 628, 93–98 (2024).
Sui, Y. et al. Understanding the role of crown ether functionalization in inverted perovskite solar cells. ACS Energy Lett. 9, 1518–1526 (2024).
Wang, C., Ni, S., Braun, S., Fahlman, M. & Liu, X. Effects of water vapor and oxygen on non-fullerene small molecule acceptors. J. Mater. Chem. C 7, 879–886 (2019).
Li, W., Liu, D. & Wang, T. Stability of non‐fullerene electron acceptors and their photovoltaic devices. Adv. Funct. Mater. 31, 202104552 (2021).
Acknowledgements
A.K.Y.J. thanks the sponsorship of the Lee Shau-Kee Chair Professor (Materials Science), and the support from the APRC Grants (9380086, 9610419, 9610440, 9610492, 9610508) of the City University of Hong Kong, the MHKJFS Grant (MHP/054/23) and MRP Grant (MRP/040/21X) from the Innovation and Technology Commission of Hong Kong, the Green Tech Fund (202020164) from the Environment and Ecology Bureau of Hong Kong, the GRF grants (11304424, 11307621, 11316422) and CRS grants (CRS_CityU104/23, CRS_HKUST203/23) from the Research Grants Council of Hong Kong, and the Guangzhou Huangpu Technology Bureau (2022GH02). C. Z. acknowledges the support of the National Natural Science Foundation of China (52163018) and the Hong Kong scholar program (XJ2022019). J.Y. acknowledges financial support from Hong Kong Polytechnic University (Grant no. P0042930) and a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. PolyU 25300823). W.L. acknowledges financial support from the Beijing Natural Science Foundation (JQ21006). D.L. acknowledges the financial support from the Research Grants Council of Hong Kong through a Collaborative Research Equipment Grant (C1015-21EF).
Author information
Authors and Affiliations
Contributions
X.H., C.Z., W.L., and A.K.-Y.J. conceived the idea. X.H. and C.Z. designed the project. C.Z. and W.L. supervised the molecular design. D.X. synthesized Y-CE. Q.X. synthesized Y-Phen. X.H. fabricated the devices and conducted relevant characterizations. D.W. and Q.F. helped optimize device performance. Q.L. and D.L. conducted the TA test. J.Y. performed the DFT calculations. Z.S. conducted the GIWAXS characterization. F.C. and B.W. performed the cross-sectional KPFM characterization. Z.Z. and S.-W.T. helped collect the in-situ absorption data. W.J., K.L., and F.R.L. offered helpful discussions. C.Z. grew and analyzed the single crystals. W.K. solved the single-crystal structures. X.H., C.Z., and A.K.-Y.J. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zeguo Tang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, X., Xia, D., Xie, Q. et al. Supramolecular force-driven non-fullerene acceptors as an electron-transporting layer for efficient inverted perovskite solar cells. Nat Commun 16, 1626 (2025). https://doi.org/10.1038/s41467-025-56060-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56060-0
This article is cited by
-
Perylene-based Electron Transport Materials in Perovskite Solar Cells To Improve Power Conversion Efficiency
Journal of Fluorescence (2025)
