
Abstract
The literature documenting the value of drug-like molecules found in natural products is vast. Although many dietary and herbal remedies have been found to be effective for treating intestinal inflammation, the identification of their active components has lagged behind. In this study, we find that a major ginger component, furanodienone (FDN), is a selective pregnane X receptor (PXR) ligand with agonistic transcriptional outcomes. We show that FDN binds within a sub-pocket of the PXR ligand binding domain (LBD), with subsequent alterations in LBD structure. Using male mice, we show that orally provided FDN has potent PXR-dependant anti-inflammatory outcomes that are colon-specific. Increased affinity and target gene activation in the presence of synergistically acting agonists indicates further opportunities for augmenting FDN activity, efficacy and safety. Collectively, these results support the translational potential of FDN as a therapeutic agent for the treatment and prevention of colonic diseases.
Similar content being viewed by others

Introduction
Crohn’s disease and ulcerative colitis afflict millions of patients globally, with China and the United States currently having the highest case numbers. These numbers are still rising due to changes in industrialization and diet1, and are expected to increase far more rapidly in less developed countries as their economies catch up and more Westernized lifestyles are adopted2,3. Particularly noteworthy is the expected exponential increase in the number of individuals with inflammatory bowel disease (IBD) in populous regions of Asia, Latin America, and Africa4.
Until 2021, the direct costs of managing Crohn’s disease and ulcerative colitis were approximately 12,000 and 9000 US dollars per patient per year, respectively5. Additionally, IBD patients often suffer from extraintestinal complications, severely affecting their quality of life6, with incalculable additional expenses. Consequently, a substantial and growing global burden of IBD is expected over the next 30 years.
Numerous studies have established connections between IBD pathogenesis, environmental factors, and gut microbiota. IBD onset and prevalence are correlated with aberrant and persistent T cell-mediated immune responses7. Not surprisingly, IBD risk genes are primarily associated with a diversity of immune functions. These include innate immune processes such as intestinal physical barrier maintenance and autophagy8,9. Consequently, most available therapeutic interventions for IBD primarily aim to suppress inflammation, frequently by targeting specific inflammatory molecules10. Nonetheless, the efficacies of current therapies are limited to subsets of patients11. Furthermore, chronic immunosuppressive treatments are associated with infectious and neoplastic complications11. Given that IBD is also associated with epithelial damage and dysbiosis, leading to a dysregulated gut mucosal barrier12, promoting mucosal healing without relying solely on immunosuppression is a key strategy for developing new and safer IBD treatments.
While there is a wealth of knowledge on IBD pathogenesis and the effectiveness and side effects of pharmacological treatments, research focused on the effects of complementary and integrative medicine to help treat or cure IBD has been limited13. Ginger (Zingiberaceae) family species are one of the most consumed dietary condiments in the world. They have a medicinal history spanning over 5000 years with applications for diseases such as flu, nausea, arthritis, migraines, and hypertension. Clinical trials and in vivo studies have also investigated the therapeutic effects of ginger supplementation, ginger extracts, and ginger-derived nanoparticles for the treatment of patients with ulcerative colitis to alleviate diarrhea and associated pain14,15, and in alleviating the severity of ulcerative colitis induced by substances such as dextran sulfate sodium (DSS)16,17,18. Pharmaceutical studies have also focused on specific ginger components, showing that gingerols exhibit suppressive effects on colon carcinomas19. Despite these findings, current evidence suggests that the consumption of ginger extract alone has relatively weak effects as compared to its potential19. Our lack of a complete understanding of its components and mechanistic actions limits its potential usefulness.
Nuclear receptors (NRs) serve as general sensors for many dietary components and nutrients20. Thus, it is not surprising that NRs play critical roles in maintaining intestinal homeostasis, doing so by responding to appropriate cues and modulating intestinal barrier integrity, transporters, and local inflammatory responses21. Analyses of IBD patient biopsies, as well as findings from animal studies, have shown a close correlation between NR biology and IBD pathogenesis11. In particular, the NRs farnesoid X receptor (FXR), pregnane X receptor (PXR), peroxisome proliferator-activated receptor α and γ (PPARα and γ), liver receptor homolog 1 (LRH-1), and V-erbA-related protein 2 (EAR-2) have all been shown to regulate epithelial intestinal cell integrity and to modulate intestinal immune cell numbers and activities21,22,23,24,25,26,27.
Here, using NR-wide affinity purification-untargeted mass spectrometry (APUMS)28,29, along with mass spectrometry-guided natural product discovery, we show that the ginger compound 5E, 9E-furanodienone (FDN)30 is a highly selective PXR agonist. Crystallographic analysis shows that FDN occupies the ligand-binding pocket (LBP) of PXR but leaves a previously identified sub-pocket site used by some larger ligands (delimited by helices H3, H11, and H1231) vacant. Further analyses show that FDN, and steroids binding in the adjacent pocket, bind PXR synergistically, thereby enhancing its efficacy and potency. That said, oral treatment with FDN alone was able to significantly mitigate systemic inflammation and colitis in mice with no deleterious effects in other tissues, suggesting potential therapeutic applications for the treatment of IBD.
Results
Affinity pulldowns identify FDN as a potential hPXR or hCAR ligand
To examine the extent by which NRs are expressed in human colon tissues, we analyzed transcriptomic data from 373 sigmoid colon samples and 406 transverse colon samples sourced from GTEx32. The data indicate that most NRs are expressed in these regions of the colon, with the exception of those typically expressed in neuronal and retinal tissues (Nr1f2, Nr2e1, and Nr2e3), and those primarily present in hormone-producing tissues (Nr0b1 and Nr5a1) (Fig. 1a, b). While some of the detected NRs show low levels of expression, their roles in the pathogenesis and treatment of IBD have been well documented and are significant21. Hence, to identify ligands for all potential NR targets, we utilized an activity metabolomics approach using His-tagged NR ligand-binding domains (LBDs) for all NRs except NR0B1 and NR0B2, which exhibited poor solubility and stability when expressed in E.coli.

a, b Relative abundance of NR expression in GTEx colon-sigmoid (n = 373) and colon-transverse (n = 406) specimens obtained from the GTEx Portal. The center line of the boxplot indicates the median value. Whiskers are drawn to 10% and 90% values with points below and above indicated as individual points. c–f Volcano plots showing the distribution of mass features differentially enriched by PXR, CAR, RXRβ, and RXRγ from ginger in comparison to vehicle (DMSO). p-values were determined by a two-tailed unpaired Student’s t-test. Dashed lines represent significance cut-offs (p-value ≤ 0.05, fold change ≥ 1000). Masses of enriched metabolites are highlighted. g Mass features identified in (c–f) are selectively enriched by the corresponding receptors. Pulldowns were performed in triplicate in at least three independent experiments, and data are expressed as mean ± SD. h Structure of FDN. E labels indicate double bond geometry. TPM transcript per million, FDN furanodienone. Source data are provided in the Source Data file.
Due to challenges with additional NR LBD solubilities, we employed a strategy to mimic structural stabilization by coactivators. This involved tethering an 18-mer Steroid receptor coactivator-1 (SRC1) LxxLL peptide motif to problematic LBD C-terminal ends. Another option used was co-expression with the retinoid X receptor (RXR) LBD, which forms heterodimers with a subset of NRs and has previously been used to facilitate crystallization studies33. LBD sequences were selected as reported in the protein data bank (PDB). The subsequent 46 LBD fusion constructs (described in Supplementary Table 1), were expressed, affinity purified, and confirmed by mass spectrometry or Western blots.
Since many known NR ligands are commercially available, we tested a subset of corresponding LBD fusion constructs for activity using available ligands and APUMS28,29. Next, the fusion proteins were used to test for potential interactions with the secondary metabolites present in our ginger (Zingiber officinale) extract. The extract mixture was spiked into an E. coli lysate containing a His-tagged NR LBD fusion protein. Following incubation and Nickel affinity purification, the samples were analyzed by untargeted LC-MS to identify masses enriched by the NR pulldown. Control pulldowns were conducted using NR LBD fusion proteins without the addition of extract. Non-specific and low-affinity interactions were further minimized by disregarding masses pulled down by most other NRs.
Among the 46 NRs tested, only PXR (NR1I2), CAR (NR1I3), RXRβ (NR2B2), and RXRγ (NR2B3) yielded clearly and specifically enriched mass features from the tested ginger extract (Fig. 1c–f). Using electrospray ionization in positive (ESI+) mode LC-MS, four mass features (m/z = 231.1372 and 232.1406; 412.2678 and 413.2712) were pulled down by both PXR (p = 0.022; 25,772-fold enrichment, p = 0.013; 4219-fold enrichment, p = 0.002; 5096-fold enrichment, and p = 0.0002; 1358-fold enrichment, respectively, Fig. 1c) and CAR (p = 0.009; 25,267-fold enrichment, p = 0.011; 4432-fold enrichment, p = 0.0004; 97,755-fold enrichment, and p = 0.0007; 22,864-fold enrichment, respectively, Fig. 1d). The first two masses were subsequently assigned as isotopic peaks of ion [C15H19O2]+ (∆m = −5.6 ppm). The other two were assigned as isotopic peaks of ion [C22H34O6 + NH4]+ (∆m = −5.6 ppm). Two mass features (m/z = 345.2052 and 346.2086) were pulled down by both RXRβ (p = 1.48e-07; 18,505-fold enrichment, and p = 1.72821e-05; 4750-fold enrichment, respectively, Fig. 1e) and RXRγ (p = 0.02268; 16,545-fold enrichment, and p = 0.036739; 3009-fold enrichment, respectively, Fig. 1f). Both masses were subsequently assigned as isotopic peaks of ion [C19H30O4Na]+ (∆m = 3.0 ppm). Enriched features selected by each NR are summarized in Fig. 1g.
To elucidate the structures of the identified masses, 15 kg of dried ginger root, powdered and extracted with 95% ethanol, was fractionated using normal and reverse-phase liquid chromatography. Using NMR and X-ray diffraction (Supplementary Fig. 1), compound 1 with m/z of 231.1372 was identified as (5E, 9E)-3,6,10-trimethyl-8,11-dihydro-7H-cyclodeca[b]furan-4-one (FDN, Fig. 1h). During the isolation, compound 2 with m/z of 412.2678 became decarboxylated (−44 Dalton), with the byproduct elucidated as 10-gingerol. Compound 3 with m/z of 345.2052 was identified as 8-gingerol.
Selective effects of FDN on NR transcriptional activity
To further assess FDN specificity, we conducted an examination of FDN binding to all 48 human NRs using the comprehensive FACTORIAL NR assay system34. When used at a concentration of 1 µM, PXR was the only responsive NR (Fig. 2a). At higher doses, agonistic effects were also observed with estrogen receptor α and β (ERα and ERβ), and PPARγ (Fig. 2a). Despite its presence in hCAR pulldowns, no agonistic response was detected for hCAR (Fig. 2a). To further evaluate how FDN affects the relative transcriptional activities of PXR and CAR, we utilized a stable HeLa-based cell line (HG5LN) that expresses the GAL4 DBD fused to either the hPXR or hCAR LBDs, along with a UAS-luciferase reporter. The results with hPXR were in line with our pulldown assay, with an EC50 value of 2.8 µM and approximately 80% of SR12813 full agonist activity (Fig. 2b). For hCAR, FDN exhibited only weak agonistic activity and an EC50 value that could not be determined due to inability to reach saturation, presumably due to cell toxicity at the higher concentrations (Fig. 2c). Taken together, FDN interactions with CAR appear to be weak and ineffective.

a Trans-FACTORIAL assay showing the effect of FDN at different concentrations on the transcriptional activities of all 48 human NRs in HepG2 cells. Rings with different diameters indicate “1” no effect; “10,” a 10-fold increase; and “0.1,” a 10-fold decrease. Each profile is an average of three independent Trans-FACTORIAL NR assays. Rifampicin was used as a positive PXR control (Supplementary Fig. 3). b Transcriptional responses of PXR in GAL4-hPXR-LBD-transfected HG5LN cells exposed to different concentrations of FDN and SR12813 (n = 3). c HG5LN cells expressing GAL4-hCAR-LBD were treated with increasing amounts of FDN or CITCO in the presence of inverse agonist, PK11195 (agonism mode), or FDN in the absence of PK11195 (antagonism mode) (n = 3). d HELN-hERα, HELN-hERβ, and HG5LN-PPARγ cells were exposed to different concentrations of FDN (n = 3). Data are mean ± SD. e Isothermal titration calorimetry (ITC) characterization of PXR LBD interaction with FDN. Representative thermograms of heat are shown in the upper panel and corresponding binding isotherms are in the lower panel. The Kd value is the mean of two independent experiments. f Respective thermodynamic signatures of binding of FDN to the PXR LBD (n = 3 replicates) and represent the mean ± SD. g FDN placed in its 2Fo-Fc electron density map. Carbon atoms discussed in the text are labeled. h Close-up view of the ligand-binding pocket of PXR bound to FDN. Key PXR residues in contact with the compound (cut-off 4.2 Å) are depicted as orange sticks. Other residues are displayed as lines. Residues and secondary structural elements discussed in the text are labeled. The dashed line depicts a hydrogen bond. i Superposition of FDN- and dabrafenib (DAB)-bound PXR (orange, PDB code 6HJ2) structures. FDN furanodienone. Source data are provided in the Source Data file.
To assess the potential off-target effects of FDN on ERα, ERβ, and PPARγ in subsequent animal studies, we utilized HELN-hERα, HELN-hERβ, and HG5LN-PPARγ luciferase reporter cell lines to determine their efficacies. Consistent with the FACTORIAL NR assay, FDN required relatively high concentrations to activate these receptors, and with only weak activation (approximately 15%, 30%, and 18% respectively for ERα, ERβ, and PPARγ, versus control agonist activity: Fig. 2d). This may explain previous findings of FDN suppressing 17β-estradiol (E2)-stimulated ERα signaling35, acting as a competing partial agonist when present at high concentration.
Further characterization of ligand-receptor interactions
As a first approach to ascertain the biochemical nature of the interactions between FDN, hPXR, and hCAR, we used isothermal titration calorimetry (ITC). Titration of a solution of FDN using the His-tagged hPXR-LBD-SRC1 fusion protein resulted in an exothermic binding event (Fig. 2e, upper panel). In contrast, titration of FDN with the hCAR LBD did not reveal any significant interactions (Supplementary Fig. 2). Analysis of the total heat changes plotted against increasing concentrations of the PXR-LBD-SRC1 protein indicated a binding stoichiometry of approximately 1:1 (ligand: protein, mol/mol, Fig. 2e, bottom panel). Curve fitting analysis yielded a dissociation constant of 4.5 µM. A positive Gibbs free energy (ΔG) value (−7.28 ± 0.40 kcal/mol, Fig. 2f) indicates that ligand binding is spontaneous. FDN binding also yielded a favorable enthalpy (ΔH) value (−2.40 ± 2.45 kcal/mol: Fig. 2f), accompanied by a significant loss of entropy (ΔS) (16.38 ± 8.60 cal/mol/deg: Fig. 2f), indicative of a reduction in LBD conformational freedom36.
Structural analyses of the FDN-PXR interaction
To gain further structure-based insights into the mode of action of FDN, we solved the crystal structure of the bound PXR LBD (hereafter PXR) at a resolution of 1.95 Å (Supplementary Table 2). The structure reveals a canonical PXR active conformation with the C-terminal helix, H12 (also termed AF-2 or activation helix-2), capping the LBP, with FDN placed by electron density within the LBP (Fig. 2g). Note that the electron density of FDN is quite diffuse compared to surrounding residues, indicating a dynamic binding mode. Despite this, the polder omit-map, calculated with Phenix (Adams, Acta Cryst D, 2010) shows an electron density that is not biased by the ligand model, as this has been removed from the structure when calculating the map (Supplementary Fig. 4). This map represents a volume in which the FDN model can be inserted convincingly with no alternative orientations.
Consistent with its moderate affinity for PXR (EC50 = 2.77 µM), FDN is a rather small compound that interacts with a small set of eight PXR LBP residues (4.2 Å distance cut-off, Fig. 2h and Supplementary Fig. 4). For comparison, the potent PXR agonist and anti-cancer drug dabrafenib (DAB)37 occupies a much larger volume and is involved in many more contacts with LBP residues (Fig. 2i), in line with its higher affinity for the receptor (EC50 = 87 nM). The two main anchoring points for FDN involve (1) multiple contacts between its ten-membered ring and the so-called PXR π-trap, composed of amino acids F288, W299, and Y30631, and (2) a hydrogen bond that is likely formed between the carbonyl moiety of FDN and Q285 (Fig. 2h). Additionally, the FDN furan ring is engaged in several contacts with M243, M246, S247, and F288 (Fig. 2h).
Alleviation of DSS-induced colitis by FDN
Given the extensive traditional use of ginger species to treat intestinal inflammation conditions, and the known roles of PXR in IBD27, we next tested the potential of FDN for alleviating intestinal inflammation using a mouse model of colitis induced by DSS. DSS administration disrupts the integrity of the colon epithelial cell barrier, allowing increased bacterial penetration and subsequent inflammation and damage. Prior to initiating this study, we tested whether FDN can activate mouse PXR (mPXR), as many hPXR agonists do not27. Using a HEK293 cell line expressing a mPXR LBD-GAL4 fusion protein, along with a UAS-luciferase reporter, we found that FDN does indeed activate mPXR with a similar efficacy to that found with hPXR (EC50 = 4.9 µM, Supplementary Fig. 5).
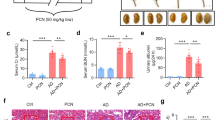
For the DSS study, mice were treated with FDN or a previously validated and tested mPXR agonist, pregnenolone 16α-carbonitrile (PCN)38, both at a relatively low concentration of 10 mg/kg. To reduce experimental costs and complexity, male mice were used exclusively for these studies. Both compounds provided similar protective activities, including reduced disease index scores (Fig. 3a), attenuation of weight loss (Fig. 3b), and reduced colon shortening (Fig. 3c, d). Histological analysis of colon sections stained with hematoxylin and eosin (H&E) also revealed reduced tissue damage in the agonist-treated mice (Fig. 3e, f).

Colitis was induced by administration of 2.8% DSS in water (w/v) in C57BL/6 (6–8-week age old, n = 6/group). Mice were pre-treated for 3 days prior to DSS treatment with either vehicle, PCN (10 mg/kg), or FDN (10 mg/kg), and then exposed to DSS for an additional 7 days. Representative data from one of two independent experiments are shown. a Disease activity index. b Body weight changes during DSS. Data were analyzed by one-way ANOVA and are expressed as mean ± SEM. p-values were determined by comparing them to the DSS group. c Images of colons. d Colon length. e Microphotographs of H&E stained sections of colons. Scale bar: 100 μm. n = 3 biological samples per group, the representative example shown. f Histological score. a, b, d, f Data were analyzed by one-way ANOVA with multiple comparisons and are expressed as mean ± SD. p-values are in comparison to the DSS group. FDN furanodienone, PCN pregnenolone-16a carbonitrile, DSS dextran sodium sulfate. Source data are provided in the Source Data file.
Mechanisms of PXR-mediated colon protection
To validate that the protective intestinal effects observed with FDN are PXR-dependent, we conducted additional experiments to address mechanisms of FDN action and PXR dependence. We started by looking at three known PXR target genes. First, we noted that FDN, and the positive control PCN, both upregulate expression of the prototypic PXR target genes Cyp3a11, which facilitates the metabolism and elimination of xenobiotics39, and Mdr1a, a PXR-responsive transporter that exports toxic bile acids, metabolites and xenobiotics40 (Fig. 4a). Similar responses were observed when monitoring expression of both gene products by Western blotting (Supplementary Fig. 6a). Strong induction was also observed with Cyp2b10 (Fig. 4a), a detoxification gene that is cooperatively regulated by both PXR and CAR41.

a, b Colonic mRNA expression of cyp3a11 (n = 5 for No DSS, n = 3 for DSS, n = 4 for DSS + PCN, n = 4 for DSS + FDN), mdr1a (n = 6 for No DSS, n = 6 for DSS, n = 4 for DSS + PCN, n = 5 for DSS + FDN), and cyp2b10 (n = 4 for No DSS, n = 5 for DSS, n = 5 for DSS + PCN, n = 3 for DSS + FDN) (a), and gsta (n = 6 except n = 5 for No DSS), and gstm (n = 6) (b) mRNA expression levels in wild-type mice. c Protein expression levels of p-p65, p65, p-IκBα, and IκBα in the colon measured by immunoblotting. α-Tubulin was used as the loading control. Experiments were performed in triplicate and repeated twice with similar results. d Colonic IL-6 (n = 6 mice), IL-1β (n = 6 mice except n = 5 for DSS and DSS + FDN), and TNF-α (n = 6 mice) levels. e Representative immunofluorescence images of Occludin (green) and ZO-1 (red) distributions in colon sections. n = 3 biological samples per group, the representative example shown. f Relative fluorescent intensity of Occludin and ZO-1 (n = 3 mice). a, b Data were analyzed by two-way ANOVA with multiple comparisons and are expressed as mean ± SD. d, f Data were analyzed by one-way ANOVA with multiple comparisons and are expressed as mean ± SD. p-values are in comparison to the DSS group. FDN furanodienone, PCN pregnenolone-16a carbonitrile, DSS dextran sodium sulfate. Source data are provided in the Source Data file.
We also analyzed the expression of glutathione S-transferase (GST) genes, which are crucial for cellular detoxification42. The genes Gsta3, Gstm1, Gstm2, Gstm3, Gstm6 and Gstm7 all showed approximately two-fold increases in expression in FDN-treated groups compared to mice treated with DSS alone (Fig. 4b). Together, these results suggest that FDN mediates much of its activity by promoting the expression of detoxification pathway genes in the colon.
The activation of NF-κB signaling pathways is another established mechanism by which PXR has been shown to mediate anti-inflammatory effects43. To test for this, we looked at levels of phosphorylated NF-κB p65 and IκBα in the colon. DSS treatment alone led to the pronounced phosphorylation of both proteins in the colons of DSS-treated mice, while cotreatment with either FDN or PCN reversed these levels back to normal (Fig. 4c). Consistent with our observed results with NF-κB components, levels of key NF-κB-regulated markers of ulcerative colitis including IL-6, IL-1β, and TNF-α44, were also decreased by the addition of either PXR agonist (Fig. 4d).
Activation of PXR or inhibition of NF-κB has also been shown to improve intestinal barrier function22,45. Accordingly, we found that DSS-mediated downregulation of occludin and ZO-1, important components of tight junctions46 and mediators of mucosal repair47, were reverted by both PCN and FDN treatment (Fig. 4e, f).
To further test whether the effects seen in DSS-treated mice are due to selective activation of PXR, FDN treatment of DSS-induced colitis mice was also assessed in PXR knockout (Nr1i2−/−) animals (Fig. 5). Treatment of Nr1i2−/− mice with FDN failed to restore any of the protective measures noted above in wild-type mice (Fig. 5a–k). Together, these results indicate that FDN exerts its protective activities via PXR. These results are also consistent with the lack of effects seen by FDN on all other tested NR target genes within the intestines (Supplementary Fig. 6b, c).

a–d Colitis induced using DSS in wild-type C57BL/6 (6–8-week age old, n = 6 mice/group), and Nr1i2−/− (6–8-week age old, n = 6 mice/group) mice. Mice were pre-treated for 3 days by intragastric administration with either vehicle, FDN (10 mg/kg), or PCN (10 mg/kg) and then for another 7 days during DSS treatment. a, c Percentage body weight loss. b, d Disease activity index. e, f Representative colon images. g Colon lengths. h Microphotographs of H&E stained sections. Scale bar indicates 100 μm. n = 3 mice per group, a representative example is shown. i–k colonic IL-6, IL-1β and TNF-α levels (n = 6 mice). a–d Data were analyzed by one-way ANOVA and are expressed as mean ± SEM. g, i–k Data were analyzed by two-way ANOVA with multiple comparisons and are expressed as mean ± SD. p-values were determined by comparison to the DSS group. FDN furanodienone, PCN pregnenolone-16a carbonitrile, DSS dextran sodium sulfate. Source data are provided in the Source Data file.
FDN tissue-specificity
While previous studies have shown that PXR agonists can exert protective properties in the colon, these have been offset by deleterious effects in the liver. The liver is a major site of PXR expression and activity, with roles in lipid metabolism and xenobiotic inactivation48,49. However, PXR overexpression and hyperactivity can lead to liver enlargement, hepatotoxicity, and drug inactivation48,49. Importantly, treatment with either PCN or FDN at therapeutic concentrations did not elicit any signs of liver toxicity (Fig. 6a, b) or in any other internal organs (Supplementary Fig. 7a).

a H&E staining of the liver from acute UC mice. Scale bar = 100 μm. n = 3 mice, biological samples per group, a representative example is shown. b Liver-to-body weight ratios (liver index) in treated mice. Serum levels of alanine transaminase (ALT), aspartate aminotransferase (AST), malondialdehyde (MDA), superoxide dismutase (SOD), and glutathione (GSH) were measured (n = 6 mice). c RT-qPCR analysis of PXR target gene expression cyp3a11 and cyp2b10 levels in liver (n = 6 mice). d RT-qPCR analysis of cyp3a4, cyp2b6, and ugt1a1 mRNA expression in PHH (from three donors) treated for 48 h with vehicle (Veh, 0.1% DMSO) or indicated concentrations of ligands (RIF and FDN at 10 μM; CITCO at 3 μM). Results were obtained from experiments performed in triplicate. b Data are shown as means ± SD, one-way ANOVA test, p-values compared to the DSS group. c, d Data were analyzed by two-way ANOVA with multiple comparisons and are expressed as mean ± SD. p-values compared to the DSS group (c) or the Veh (DMSO) group (d). RIF, rifampicin is a human PXR agonist. CITCO is a human CAR agonist. Veh vehicle, FDN furanodienone, PCN pregnenolone-16a carbonitrile, DSS dextran sodium sulfate. Source data are provided in the Source Data file.
In terms of target gene expression, FDN showed little to no effects on liver PXR target genes (Fig. 6b). Although PCN and FDN both weakly upregulated Cyp2b10 compared to DSS treatment alone (Fig. 6b), this is likely due to decreased levels of PXR and CAR protein caused by DSS treatment (Supplementary Fig. 7b). These DSS-induced decreases were reverted to normal when mice were co-dosed with FDN or PCN. Interestingly, while FDN restored expression of the PXR target gene Cyp3a11 back to baseline, PCN induced significantly higher levels of Cyp3a11 expression (Fig. 6c). FDN showed no effects on other tested NR target genes (Supplementary Fig. 7c)
Similar results were observed using primary human hepatocytes. FDN treatment elicited weak or no effects on the human PXR target genes Cyp3a4, Cyp2b6, and Ugt1a1 (Fig. 6d). Given that FDN applied by oral gavage induces strong PXR-dependent effects in the colon with no effects on target gene expression in liver or primary hepatocytes, it appears to have excellent potential for the safe treatment of IBD.
Synergistic interaction of PXR with FDN and steroids
As a xenosensor, human PXR has a large, flexible, and hydrophobic LBP made up of four distinct sub-pockets, to which small compounds can bind in unique combinations and orientations according to size, structure, and chemical composition31. Our structure reveals that FDN occupies the same binding site, and approximately the same volume, as organochlorine pesticides such as endosulfan (END, Fig. 7a). This observation is consistent with their very similar EC50 values (EC50 = 7.0 µM for END and 2.8 µM for FDN)31, the slightly higher potency of FDN being most likely due to the additional hydrogen bond with Q285 (Fig. 2h). In this binding mode, a significant portion of the LBP, delineated by helices H3, H11, and H12, is left vacant, with the potential to accommodate a second substance such as the natural hormone E2, as shown in Fig. 5a. Our previous work revealed that END and E2 can bind to PXR simultaneously in a cooperative manner, activating the receptor synergistically31. These cooperative interactions with endogenous metabolites or xenobiotics may lead to alternative physiological outcomes.

a Superposition of the structures of FDN-bound PXR and PXR bound to E2 and END (orange, PDB code 7AXK). b FDN docking pose obtained in the presence of E2. The docking pose (in gray) matches the PXR: FDN crystallographic structure (in violet, RMSD = 1.67 Å), and correctly reproduces the H-bond with residue Q285. c FDN docking pose obtained in the presence of EE2. The docking pose (in gray) matches the crystallographic structure (in violet, RMSD = 1.52 Å), and correctly reproduces the H-bond with residue Q285. d Crystal structure overlaying differential HDX indicates alternations in the structural conformation of PXR-SRC1 due to SR12813, FDN, FDN/E2, or FDN/EE2 (PDB ID: 3HVL) binding. Structures are color-coded according to the color bar at the bottom of the figure where colors represent differences in deuterium uptake (%D). Regions highlighted in black are areas not covered in HDX analysis. e Fractional uptake difference heatmaps showing variations of deuterium uptake between ligand-bound PXR-SRC1. f, g HG5LN GAL4-PXR-LBD cells were exposed to different concentrations of FDN in combination with E2 or EE2 (n = 4). Dashed lines represent the theoretical activation curves obtained for the additive combination of individual compound activities calculated using the Bliss independence model86. Assays were performed in at least three independent experiments, and data are expressed as mean ± SEM. h RT-qPCR analysis of Mdr1, Cyp3a4, and Cyp1a1 mRNA expression in hPXR-overexpressing LS174T cells treated for 48 h with solvent (0.1% DMSO) or the indicated ligands (RIF and FDN at 10 μM; E2 and EE2 at 3 μM). Results were obtained from three separate experiments performed in triplicate. i–k RT-qPCR analysis of Cyp3a4, Ugt1a1, and Mdr1 mRNA expression in human ileum organoids treated for 24 h with solvent (0.1% DMSO) or the indicated ligands (RIF and FDN at 10 μM; EE2 at 3 μM). Results were obtained from three separate experiments performed in quadruplicate. h–k Data were analyzed by two-way ANOVA with multiple comparisons and are expressed as mean ± SD. E2 17β-estradiol, EE2 17α-ethinylestradiol, END endosulfan, FDN furanodienone, PCN pregnenolone-16a carbonitrile, RIF rifampicin. Source data are provided in the Source Data file.
To gain further insight into potential combinatorial interactions, we first used in-silico molecular docking to investigate differences in predicted FDN binding affinities in the presence or absence of E2 or EE2. Starting with the PXR structure 4X1G, which already contains EE2, the docking of FDN yielded a binding geometry similar to that observed in the PXR: FDN crystallographic structure (Fig. 7b), with a more favorable docking score (XP score = −9.223 kcal/mol) compared to that of FDN alone (XP score = −8.723 kcal/mol). Similarly, docking FDN in the PXR structure 7AXK, which contains E2, also results in a structure similar to the PXR: FDN crystal structure (Fig. 7c), also with a more favorable docking score (XP score = −8.895 kcal/mol) compared to that of FDN alone (XP score = −8.725 kcal/mol). These results predict a significant impact on FDN binding affinity in the presence of other ligands. The induced-fit effects of these ligands ensure enough space for the simultaneous placement of both ligands, and the overall increase in hydrophobicity within the cavity is likely to enhance LBD stability and activity significantly. Notably, the synergistic effect predicted with EE2 is stronger than with E2, as reflected by the greater improvement of the binding energy in the former case.
To gain further insight into the conformational perturbations of PXR structure upon FDN binding with either E2 or EE2, hydrogen-deuterium exchange (HDX) coupled with mass spectrometry (HDX-MS) was utilized. HDX experiments were conducted in the presence or absence of ligands, with an average sequence coverage of 75.6 and an average peptide redundancy of 2.83. The HDX-MS analyses revealed significant structural changes in PXR in the presence of FDN, FDN/E2, and FDN/EE2 (Fig. 7d). PXR-E2 and PXR-EE2 complexes were tested as well, with an average sequence coverage of 78.5% and peptide redundancy of 2.76, but for these complexes, the HDX-MS analysis showed no statistical significance in deuterium exchange when bound with E2 or EE2. It is important to note that for the PXR-E2 complex, due to solubility issues, the concentration of ligand used would have resulted in a ‘fraction bound’ of 75% given the known EC50 of 10.0 µM (all other complexes were measured at 90% occupancy or higher). However, if structural changes occur in the presence of E2, differential deuterium uptake should still be statistically significant (Fig. 7d).
Our results also indicate that in the presence of either SR12813 (full agonist), FDN, FDN/E2, or FDN/EE2, PXR undergoes structural compacting within the LBD, including helices H3 (246–257), H6/H7 (319–325), and H9/H10 (400–411). Furthermore, the data indicates a substantial conformational increase in helical integrity in the C-terminal helix H12 (412–428), which is a key player in recruiting coactivator partners (Fig. 7d). In the combined presence of FDN/E2 or FDN/EE2, there was a further decrease in helix H12 deuterium uptake from 15% with FDN alone to 18.9% and 19.4% with FDN/EE2 and FDN/E2, respectively (Fig. 7e), consistent with additional stabilization of the active AF-2 conformation. Indeed, H12 exchange in the presence of FDN/E2 and FDN/EE2 is more like the effects of the full agonist SR12813 than the partial agonist FDN alone (Supplementary Fig. 8).
Previous work on other NRs has also stressed the importance of helix H3 in ligand and coactivator binding50. Interestingly, the deuterium uptake profile for FDN, FDN/E2, and FDN/EE2 in the H3 region all showed higher H3 stabilization than SR12813 (Supplementary Fig. 8 and Table 3), with FDN/E2 and FDN/EE2 providing higher H3 stability than FDN alone, with the deuterium exchange protection rising from 25.9% with FDN to 29.8% and 30.2% with FDN/EE2 and FDN/E2, respectively (Fig. 7d, e). Thus, E2 and EE2 both augment the structural effects of FDN on PXR in ways that suggest a transition to full agonist activity.
Synergistic transcriptional activation of PXR by FDN and steroids
To elucidate the transcriptional effects of this synergy, we queried whether FDN can synergistically activate PXR transcription with a second molecule using the reporter cell line HG5LN GAL4-PXR-LBD. When co-treated with FDN and either E2 or EE2, both the potency and efficacy of FDN on PXR activation were clearly enhanced (Fig. 7f, g). In agreement with the docking experiments, the less favorable FDN/E2 mixture decreased the EC50 2-fold (Fig. 6f), while the more favorable FDN/EE2 mixture decreased the EC50 down to the nanomolar range (Fig. 7g). For the latter, Emax agonist activity also rose to match that of the full agonist SR12813 (Fig. 7f, g).
Next, we compared the abilities of E2, EE2, and FDN, alone or in combination, to increase known PXR target gene expression in the human colon adenocarcinoma-derived cell line LS174T (Fig. 7h). Rifampicin (RIF) was chosen as the positive control here as its affinity for hPXR (2.7 μM)51 is comparable to FDN. Consistent with our reporter assay and docking results, we found that the relative effectiveness of FDN in activating the PXR target genes Mdr1 and Cyp3a4 was modest but significantly increased when used in combination with EE2 (Fig. 7h). As expected, neither RIF nor FDN/EE2 mixture affected the expression of the aryl hydrocarbon receptor (AhR) target gene Cyp1a1 (Fig. 7h).
To gain physiological insight into the potential transcriptional impacts of FDN, along and in combination with EE2, we compared their activities in activating hPXR in human ileum organoids, which differentiate to model natural intestinal tissue structures. Remarkably, treatment with FDN alone for 24 h resulted in robust induction of Cyp3a4, comparable to the effects of RIF, and cotreatment with EE2 further induced Cyp3a4 expression (Fig. 7i). The effects on ugt1a1 were somewhat different, with FDN alone weakly inducing expression compared to RIF, but with cotreatment with EE2 again surpassing the effects of RIF (Fig. 7j). In the case of Mdr1, FDN alone had no effect on expression, but was once again more effective than RIF when used in combination with EE2 (Fig. 7k). Similar results were also observed with colonic organoids (Supplementary Fig. 9). As expected, depletion of PXR (Nr1i2) in ileum organoids resulted in no response to PXR ligand treatment (Fig. 7i–k). Collectively, these results demonstrate that the induction of hPXR target genes depends on specific ligand combinations and target cell types. Differences may also be seen with more acute treatments due to the absence of feedback and secondary target gene responses.
Discussion
Ginger extracts have been administered widely to treat numerous ailments, with intestinal afflictions featuring prominently. However, recent studies have shown that, in some contexts, ginger consumption can also aggravate intestinal inflammation52. These contradictory effects may be due to the complexity of ingredients in ginger and their means of extraction and administration. This combination of potential and complexity motivated us to identify active components in ginger and their mechanisms of action.
Our analysis of 46 human NRs for interactions with ginger-derived small molecules identified specifically binding compounds for three NRs, PXR, CAR, and RXR. Further analysis of a compound that was pulled down by both PXR and CAR revealed its identity as FDN. Although FDN was first isolated and identified in 196953, and a variety of biological activities have been reported54, its mediators have never been determined. Here, using multiple approaches, we found that FDN binds most strongly and consistently with the nuclear receptor PXR, prompting us to focus further analyses on the nature and potential of this FDN-PXR interaction.
PXR holds excellent potential as a mediator for the treatment of IBD, as shown previously by the actions of rifaximin, a known PXR agonist55. However, rifaximin, also being an antibiotic, also influences the gut bacterial community through a PXR-independent pathway55. Selectively targeting PXR in the intestine is a challenge due to its unusually promiscuous LBP, which is capable of interacting with a variety of natural products, toxins, and drugs56. Examples of the latter, such as the chemotherapeutic agent paclitaxel57, the antibiotic rifampicin51, and the anti-depressant hyperforin58, are clearly not PXR-specific. In contrast, our work shows that the natural compound FDN functions as a highly selective PXR agonist with no observed side effects after acute oral treatment in mice.
Most existing PXR agonists also activate PXR systemically with consequential side effects. For example, PXR activation by RIF in the liver of humanized PXR transgenic mice causes lipid accumulation, resulting in hepatic steatosis and insulin resistance59. In contrast, selective activation of intestinal PXR has shown beneficial effects on diet-induced metabolic disorders60. Importantly, we found that FDN treatment, alone or in combination with E2 or EE2, does not activate PXR in primary human hepatocytes (Supplementary Fig. 10), whereas RIF treatment leads to strong PXR activation (Supplementary Fig. 10). The former is likely due, in part, to rapid clearance or non-uptake of FDN by hepatocytes, though other mechanisms are also possible. In summary, FDN on its own elicits a potent, PXR-dependent tissue-selective response in the colons of DSS-treated mice. Since these studies were performed only in male mice, follow-up studies will be required to see if there are any sex-specific variations.
Interestingly, while FDN is a relatively small PXR ligand (230 Da) compared to the aforementioned ligands (500–900 Da), its binding affinity and EC50 for PXR are comparable to some larger agonists such as RIF and its analogs51. Though smaller than RIF, our structural analyses show that FDN engages with the same aromatic F/W/Y π-trap site in the LBP51. As with another small ligand that binds this site31,51,61, it also appears to have strong interactions with H3 and weaker ones with H12. This H3-centric effect appears to be responsible, in part, for the more limited and target gene-selective responses of FDN that limit the potential side effects of stronger PXR modulators that act predominantly via H12 positioning62.
Modern medicine tends to rely on the use of single-drug therapies. Many natural remedies, however, benefit from the combinatorial actions of multiple components63. In the case of ginger, this may apply to another abundant ginger component 8-gingerol, which we have identified as an RXR-interacting metabolite in our NR pulldowns. As with many NRs, PXR performs much of its function as a heterodimer with RXR. Thus, FDN and 8-gingerol may also elicit synergistic outcomes via combinatorial actions on RXR/PXR heterodimer pairs. These combinations may be less toxic than those that strongly activate PXR alone.
Previous studies have shown that the PXR LBP can accommodate two separate molecules31. These cooperative interactions increase individual ligand-binding affinities and lead to differential transcriptional outcomes. Likewise, our study shows that FDN binds PXR synergistically with EE2 and elicits differential PXR target gene outcomes, both in vitro and in intestinal organoids. Although the synergy in this study is synthetic, it remains possible that endogenous steroid-like molecules, or structurally related microbial metabolites, may also bind cooperatively to PXR together with FDN. While potentially advantageous, the ability of FDN to activate PXR cooperatively also merits caution, given the role of PXR as a master regulator of xenobiotic metabolism. The PXR-regulated drug efflux pump MDR1, for instance, facilitates drug resistance to many anti-cancer drugs64. Conversely, MDR1 also governs the tolerance of bile acids in the ileum65. Notably, FDN only weakly upregulated Mdr1 in human intestinal cells and intestinal organoids, but this activation was greatly increased in combination with EE2. CYP3A4, which primarily contributes to the first-pass metabolism of clinical drugs66, and UGT1A1, which is critical for the detoxification of endogenous and exogenous substrates67, were also highly expressed by the combined presence of FDN and EE2. As noted earlier, this type of enhanced PXR activation in the liver would be highly problematic. However, the limited actions of orally provided FDN to the colon may also limit the synergistic actions of EE2 and related agonists to the colon, where they may be wholly beneficial.
In addition to the transition from partial agonism to full agonism, it is also possible that PXR synergy with some ligands may affect receptor activity negatively by reorienting H12 non-poductively62. Therefore, the implications of these combinatorial interactions on intestinal physiology and drug treatment outcomes require further investigation.
As noted in the introduction, the incidence of IBD is anticipated to increase substantially in developing countries in the coming years. Given the costs and side effects of current therapeutic options, the accessibility, affordability, and safety profile of FDN position it as an alternative candidate for addressing the escalating burden of IBD. This is particularly significant in regions where existing treatments may be economically restrictive or associated with adverse effects. In this regard, we note that FDN is much more abundant in other Curcuma species68 as well as in wild ginger (Siphonochilus aethiopicus)69, myrrhs (Commiphora)70, and laurels (Lauraceae)71, which are widespread in Africa, East, South and Southeast Asia as well as the Arabian Peninsula.
FDN may also have potential for the treatment of colorectal cancer. For example, FDN has been shown to inhibit the growth of human colorectal cancer cells such as RKO, SW480, HT-29, SW620, and LoVo cells (data accessible at https://www.proteinatlas.org/ENSG00000144852-NR1I2/cell+line) by inducing G0/G1 cell cycle arrest72. Although these actions were achieved using FDN concentrations exceeding those needed for PXR activation, these cell lines are known to express only low levels of PXR. Transfection of PXR into HT-29 cells also suppresses cell proliferation by arresting G0/G1, but these cells failed to respond to RIF73. In humanized PXR mice, rifaximin treatment prevents inflammation-induced intestinal tumor formation74. On the other hand, chronic PXR activity can also enhance the aggressiveness of metastatic colon cancer cells and resistance to chemotherapy or radiation75,76,77. These findings underscore the complex role of PXR in carcinogenesis. Its potential as an anti-cancer target is also complicated by its physical and genetic interactions with other NRs such as RXR and CAR78. Leveraging the PXR-selectivity of FDN may offer promising strategies that circumvent many of these issues. Strategic FDN delivery methods should also be considered for testing in other PXR-related diseases such as cholestatic liver diseases79, neurodegenerative diseases80, kidney injury81, and aging82.
Methods
All animal and cell-based experiments described and carried out comply with relevant ethical regulations and procedures. Animal protocols were approved by the Academic Ethics Committee of ZunYi Medical University Zhuhai Campus (ZHSC-2-(2023)017). Freshly isolated primary human hepatocytes were obtained from BioIVT (Baltimore, MD), with procurement and use not requiring further institutional board review. The human ileum enteroid cell line IL2003 and colonic enteroid cell line TC2003, received at passage 2, were obtained via a materials transfer agreement from Baylor College of Medicine. Cell lines were generated from patients undergoing bariatric surgery under an IRB-approved protocol at Baylor College of Medicine.
Reagents and antibodies
Methylcellulose (M112869) was purchased from Aladdin; Pregnenolone Carbonitrile (PCN, P911021) was purchased from Macklin; Dimethyl Sulfoxide (DMSO) was purchased from Sigma. ELISA kits for mice IL-6, IL-1β, and TNF-α were purchased from BioLegend. Dextran Sulfate Sodium Salt (DSS) was purchased from MP Biomedical. Occludin-1 antibody (27260-1-AP) and ZO-1 antibody (21773-1-AP) were obtained from Proteintech. FastPure Cell/Tissue Total RNA Isolation Kit V2 (RC112-01) was from Nanjing Vazyme Biotech. PrimeScriptTM RT reagent Kit with gDNA Eraser (RR047A) and TB Green® Premix Ex Taq™ II (RR820Q) were purchased from Takara. All antibodies used are described in Supplementary Table 5.
Plant material and extraction
The aerial parts of Zingiber officinale were purchased from Longxi, Gansu, China, in December 2020. The plant was identified by Dr. X. Wang (School of Pharmacy, Lanzhou University of China). Dried roots of Zingiber officinale (15 kg) were powdered and extracted with EtOH-H2O (3 × 20 L, 95:5, v/v, three times) at room temperature. The combined extracts were concentrated to give a crude extract, which was partitioned with EtOAc and H2O. The EtOAc fraction was then concentrated using a rotary evaporator and resuspended in DMSO.
Animal experiments
6–8 week-old C57BL/6J male mice were obtained from SeBiona Bio-Tech. 8–10 week-old global PXR knockout male mice (Nr1i2−/− mice) were obtained from the Shanghai Model Organisms Center, Inc. Mice were pathogen-free and housed at 20–22 °C, 45–65% humidity with a strict 12 h light/dark cycle and monitored daily. Mice were acclimated for 7 days prior to experiments. C57BL/6J male mice and PXR−/− male mice were randomly divided into 4 groups (n = 6 per group). FDN and PCN were solubilized in water containing 3% DMSO and 0.1% methylcellulose. Administration was intragastric at 10 mg/kg once daily for 10 consecutive days. All mice were administered 2.8% (w/v) DSS in drinking water for the DSS-induced acute UC model, with the control group given normal drinking water. Before drinking the 2.8% DSS water, FDN and PCN were pre-treated for 3 days and then continued for another 7 days. Body weights were recorded daily, and the disease activity index was scored based on body weight loss, stool consistency, and fecal bleeding, with each indicator scored from 0 to 4. On day 11, mice were sacrificed, and colon, heart, liver, spleen, lung, and kidney tissues were harvested for analysis.
The expression levels of cytokines in the colon tissues, including IL-1β, TNF-α, and IL-6, were detected via ELISA kits. For real-time quantitative polymerase chain reaction (qPCR) experiments, the total RNA was extracted from frozen colon, ileum, and liver tissues using Tissue Total RNA Isolation Kits according to the protocol. RNA was transcribed into cDNA by PrimeScriptTM RT reagent kits. Next, RT-qPCR was performed using TB Green® Premix Ex Taq™ II Kits. Relative mRNA expression was calculated using GAPDH as the endogenous control. Colon, heart, liver, spleen, lung, and kidney tissues were embedded in paraffin, cut into 3–5 μm sections, and stained with hematoxylin/eosin (H&E). To demonstrate the protective effect of FDN on intestinal barrier function, immunofluorescence assays were also carried out. Colon tissue sections were stained with occludin-1 antibody conjugated with AlexaFluor-488 and ZO-1 antibody conjugated with CY3, with nuclei marked using DAPI.
Serum and liver biochemical analyses
Serum was collected to determine alanine transaminase (ALT) and aspartate aminotransferase (AST) levels. After weighing, liver tissue was immediately frozen in liquid nitrogen. Malondialdehyde (MDA), superoxide dismutase (SOD), and glutathione (GSH) were quantified by spectrophotometry.
Western blotting
Total protein was extracted using RIPA lysis buffer containing PMSF and phosphorylation inhibitor for 30 min on ice. Protein was quantified by BCA, and then 5× loading buffer was added for SDS PAGE and Western analyses. After incubation with antibodies (listed in Supplementary Table 5), signals were detected using a ChemiDocTM Imaging System. Uncropped blots are available in the provided Source Data file.
Construction of expression plasmids
Human NR1A1 (147–370), NR1A2 (203–461), NR1B1 (176–421), NR1B2 (176–421), NR1B3 (183–417), NR1C1 (192–468), NR1C3 (238–503), NR1D1 (281–614), NR1D2 (381–579), NR1F2 (200–559), NR1F3 (261–507), NR1H4 (217–472), NR1I1 (118–427), NR2A1 (142–378), NR2A2 (103–408), NR2B1 (225–462), NR2B2 (293–528), NR2B3 (233–463), NR2F6 (165–393), NR3B1 (193–423), NR3B2 (204–432), NR3B3 (229–457), NR4A1 (351–598), NR4A2 (353–598), NR4A3 (394–626), NR5A1 (221–461), NR5A2 (300–541), and NR6A1 (249–480) LBD amplicons with 15-bp extensions complementary to each end of the BsERI digested pET28-MHL backbone were produced by PCR and inserted into pET28-MHL using an In-Fusion HD Cloning kit. Human LBD-SRC1 expression plasmids were made by amplifying the NR1C2 (169–440), NR1F1 (270–523), NR1H2 (216–461), NR1H3 (200–447), NR1I2 (130–434), NR1I3 (103–352), NR2E1 (180–383), and NR2E3 (217–410) with 15-bp extensions complementary to each end of a predigested BsERI digested pET28-MHL-SRC1 vector that has the SRC1 peptide already in place via In-Fusion HD cloning. For co-expression with RXR (NR2B1), human NR2C1 (348–590), NR2C2 (341–583), NR2F1 (184–410), NR2F2 (177–403), NR3C1 (521–777), NR3C2 (731–984), NR3C3 (678–933), and NR3C4 (671–920) LBDs were cloned into a pACYCDuet-1 plasmid containing a non-tagged NR2B1 LBD (225–462) via Gibson Assembly Cloning.
LBD expression and pulldowns
NR LBD plasmids were transformed into BL21(DE3) cells (NEB, C2527) and cultures grown in LB medium at 37 °C until an OD600 of 1.0 was reached. The temperature was then decreased to 17 °C, and cultures were induced with 0.1 mM isopropyl 1-thio-β-D-glucopyranoside (IPTG, BioShop IPT001) for 16 h. Harvested bacterial pellets were resuspended in a buffer containing 20 mM Tris/HCl (pH 8.2), 300 mM NaCl, 5 mM imidazole (BioShop, IMD508), and 0.5 mM DTT (BioShop, DTT001). After sonication (5 min at 40% amplitude using cycles of 10 s-on/10 s-off) and centrifugation at 18,000 × g at 4 °C for 20 min, supernatants were incubated with ginger extract (1:100, v:v) and 7 μL of HIS-select nickel magnetic agarose beads (H9914, Sigma–Aldrich) for 30 min at 4 °C, then for 15 min at room temperature. Supernatants and washes were removed using a magnetic stand. After washing 3 times with 100 μL of buffer containing 20 mM Tris/HCl (pH 8.2), 300 mM NaCl, and 30 mM imidazole, the His-tagged LBD-ligand complexes were released with 100 μL of buffer containing 20 mM Tris/HCl (pH 8.2), 300 mM NaCl and 300 mM imidazole28.
LC-MS analysis
LC-MS sample preparation and data analysis were performed as described in a previous publication28, with minor modifications. Elution samples (n = 3) were loaded onto a Zeba spin desalting plate (Thermo Fisher Scientific, 7 K MWCO (89807)), then centrifuged at 1500 × g at 4 °C for 2 min. The desalted samples were then evaporated using a SpeedVac and resuspended in HPLC-grade methanol (Fisher Chemical A4564). Samples were analyzed using a Q Exactive orbitrap mass spectrometer equipped with a Vanquish ultra-high performance liquid chromatography (UPLC) system (Thermo Fisher Scientific, USA). Samples were loaded onto a reverse-phase column (AccucoreTM Vanquish C18+, 1.5 µm 50 × 2.1 mm, Thermo Fisher), and eluted with a gradient starting with 90% mobile phase A (H2O (Fisher Chemical 022934-M6) containing 0.1% formic acid (Fisher Chemical A11750) and 10% mobile phase B (methanol containing 0.1% formic acid) at time zero, with a flow rate of 0.15 mL/min, then increasing to 50% mobile phase B at 2 min, 100% mobile phase B at 5 min, then kept isocratic for 3 min before returning to 90% mobile phase A over the next 0.5 min, and finally kept isocratic for 1.5 min. The temperatures of the column and sample compartments were maintained at 40 °C and 10 °C, respectively. Data were acquired in full MS mode. Spray voltage was 3.5 kv using a sheath gas flow rate of 30 L/h, auxiliary gas flow rate of 7 L/h, and capillary temperature of 300 °C. Parameters for full MS scans were recorded at resolution R = 70,000 with a maximum of 3 × 106 ions collected within 200 ms in both negative and positive modes.
Untargeted metabolomics data analysis
Untargeted metabolomics analyses were performed using an in-house R program. Specifically, raw MS files were converted to open format (.mzXML). Chemical features were detected using the XCMS package at 2.5 ppm mass accuracy. Features were matched across different samples with a 0.5 min retention time tolerance window. Putative metabolite features were selected by calculating the fold change of features from treatments relative to controls. Specifically, only those features with 1000-fold higher abundance (p < 0.05), using ginger extract versus vehicle controls, were considered true metabolite features.
LC-MS guided sample fractionation
The EtOAc extract (45 g) with the MS signal identified in the previous procedure was subjected to silica gel (200-300 mesh, zcx-II, Qingdao Yuminyuan Silicone Reagent Co. Ltd, PR China) column chromatography with hexane/ethyl acetate gradients (from 20:1 to 1:1, v/v) and ethyl acetate (100%) to yield compound 1 (FDN), add then further purified by semi-preparative HPLC (85% MeOH-H2O) to yield compound 2 (10-gingerol) and compound 3 (8-gingerol).
NMR analysis of isolated compounds
NMR spectra were recorded on a BRUKER (AVANCE NEO 600) 600 MHz spectrometer. Chemical shifts are expressed in δ (ppm) and referenced to the residual solvent signals.
Compound 1 (FDN)
1H-NMR (CDCl3, 600 MHz): 7.05 (1H, s, H12), 5.78 (1H, s, H-5), 5.15 (1H, dd, J = 5.16 Hz, H-1), 3.67 (2H, d, J = 5.20 Hz, H9), 2.44, (1H, dt J = 11.32, 3.64 Hz, H3a), 1.85, (1H, m, H-3b), 2.15–2.29 (1H, m, H-2), 2.10 (3H, s, H-13), 1.97 (3H, s, H-14), 1.27 (3H, s, H-15).
13C-NMR (CDCl3, 150 MHz): 130.4 (C-1), 26.3 (C-2), 40.5 (C-3), 145.5 (C-4), 132.3 (C-5), 189.6 (C-6), 122.0 (C-7), 156.3 (C-8), 41.5 (C-9), 135.2 (C-10), 123.6 (C-11), 137.9 (C-12), 9.48 (C-13), 18.5 (C-14), 15.6 (C-15).
Compound 2 (10-gingerol)
1H-NMR (CDCl3, 600 MHz) δ: 6.81 (1H, d, J = 8.0 Hz, H-5′), 6.67 (1H, d, J = 2.0 Hz, H-2′), 6.65 (1H, dd, J = 8.0, 2.0 Hz, H6′), 4.02 (1H, m, H-5), 3.85 (3H, s, 3′-OCH3), 2.83 (2H, m, H-1), 2.72 (2H, m, H-2), 2.53 (2H, m, H-4), 1.26 (16H, m, H6 ~ 13), 0.88 (3H, t, J = 7.1 Hz, H-14). 13C-NMR (CDCl3, 150 MHz) δ: 36.5 (C-1), 45.4 (C-2), 211.5 (C-3), 49.4 (C-4), 67.7 (C-5), 31.9 (C-6), 29.6 (C-7), 29.6 (C-8), 29.6 (C-9), 29.3 (C-10), 29.3 (C-11), 25.5 (C-12), 22.7 (C-13), 14.1 (C-14), 111.0 (C-1′), 132.6 (C-2′), 144.0 (C-3′), 146.5 (C-4′), 120.7 (C-5′), 114.5 (C-6′), 55.9 (3′-OCH3).
Compound 3 (8-gingerol)
1H-NMR (CDCl3, 600 MHz) δ: 6.83 (1H, d, J = 8.0 Hz, H-5′), 6.69 (1H, d, J = 2.0 Hz, H-2′), 6.67 (1H, dd, J = 8.0, 2.0 Hz, H6′), 4.04 (1H, m, H-5), 3.88 (3H, s, 3′-OCH3), 2.85 (2H, m, H-1), 2.75 (2H, m, H-2), 2.55 (2H, m, H-4), 1.29 (12H, m, H6~11), 0.89 (3H, t, J = 7.1 Hz, H12). 13C-NMR (CDCl3, 150 MHz) δ: 36.5 (C-1), 45.4 (C-2), 211.5 (C-3), 49.4 (C-4), 67.7 (C-5), 31.8 (C-6), 29.5 (C-7), 29.3 (C-8), 29.2 (C-9), 25.5 (C-10), 22.7 (C-11), 14.1 (C-12), 111.0 (C-1′), 132.6 (C-2′), 144.0 (C-3′), 146.5 (C-4′), 120.7 (C-5′), 114.5 (C-6′), 55.9 (3′-OCH3).
FDN crystallography
The X-ray crystal structure of FDN was determined at T = 301.37 K from data collected to μ = 0.622 mm–1 with Cu Kα radiation on a ROD, Synergy Custom system, and HyPix diffractometer. C15H18O2, M = 230.29, monoclinic, space group P21/n (no. 14), a = 7.49360 (10), b = 15.4242 (2), c = 11.02380 (10) Å, α = 90, β = 96.3640 (10), γ = 90°, V = 1266.31 (10) Å3, Z = 4, Dcalc = 1.208 g/cm3, F (000) = 496.0. A total of 14,380 reflections were collected in the range of 9.902° <2θ < 153.126°, including 2562 independent reflections (Rint = 0.0171, Rsigma = 0.0103). The final R1 was 0.0417 (I > 2σ(I), and wR2 was 0.1203 (all data). The goodness of fit on F2 was 1.056. The relative configuration was determined as (5E, 9E).
Isothermal titration calorimetry
Purified hPXR-SRC1 protein (25 µM) was dissolved in 400 μL of PBS and supplemented with 0.5% DMSO. All experiments were performed in a MicroCal Auto-iTC200 instrument (Malvern Panalytical, Marlvern, United Kingdom) at 25 °C. Ligands were used at a concentration of 200 μM. Protein was titrated into cells in a series of 19 injections (injection 1 = 0.4 µL, injection 2–19 = 2 µL) 180 s apart. Control experiments performed by injection of the protein into the ligand solution yielded insignificant heats of dilution, and heat responses were subtracted. Integrated heat effects were analyzed using Microcal Origin 7 software (Malvern Instruments).
PXR crystal structure
The human PXR LBD (130–434) containing a fragment of the steroid receptor coactivator-1 (SRC1, 623-710) to enhance PXR stability, was purified as previously described61. Crystals of the PXR-FDN complex were obtained by mixing 1 μL of protein (4 mg/mL) pre-incubated (1 h) with 3 molar equivalent of ligand and 1 μL of precipitant (50–100 mM imidazole pH 7.0–7.4, 8–14% (v/v) isopropanol), and equilibrated against a reservoir of 500 μL of precipitant. Crystals appeared in 24–48 h. Diffraction data were collected at the ID23-1 beamline at the European Synchrotron Radiation Facilities, Grenoble, France. Data were processed and scaled with XDS and XSCALE (Kabsch, Acta Cryst D, 2010). The structure was solved and refined using Phenix (Adams, Acta Cryst D, 2010) and COOT (Emsley and Cowtan, Acta Cryst D, 2004). Data collection and refinement statistics are summarized in Supplementary Table 2. Figures were prepared using PyMOL (http://pymol.org/).
Molecular docking
Structures of the FDN ligand were downloaded from PubChem and then prepared with the LigPrep utility in the Schrödinger suite. The ligand structure was then optimized using the Prime Macrocycle conformational search utility with the OPLS3e force field in implicit water. Docking was performed using Glide XP (extra precision)83 included in the Schrödinger suite 2019–3. Grids for PXR structures were set up using default parameters and the binding box was centered in the center of mass of the FDN crystallographic ligand. Redocking of FDN within its crystallographic structure resulted in a good binding pose (RMSD = 1.1 Å from the crystallographic structure; XP score = −8.962 kcal mol−1), thus confirming the ability of the approach to predict the interactions of this ligand within the binding site.
Bottom-up HDX-MS
PXR-SRC1 (20 mM Hepes, 150 mM NaCl pH 8.2) was incubated with SR12813, FDN, FDN/E2, FDN/EE2 at a 1:10 molar ratio (protein: ligand) with a final concentration of 6% DMSO. PXR-SRC1 was also incubated with a 1:10 molar ratio with EE2 and a 1:40 molar ratio with E2. A 3 μL protein sample was diluted into 57 μL equilibration buffer (10 mM phosphate buffer, pH 7.5) or deuterated reaction buffer (10 mM phosphate buffer, 150 mM NaCl, pH 7.5 in D2O). After labeling times of 1, 10, and 30 min at 25 °C, samples were mixed with equal volumes of quench buffer (100 mM phosphate buffer, pH 2.5) at 0 °C. Subsequently, the quenched sample was injected into the HDX platform, where the protein underwent digestion by passing through an enzymatic Nepenthesin-2/Pepsin column (2.1 × 20 mm, Affipro, AP-PC-006) at 15 °C. The resulting peptides were captured and desalted using a (size) ACQUITY UPLC BEH C18 VanGuard Pre-column (2.1 × 5 mm, Waters, 186003975), followed by separation on an ACQUITY UPLC BEH C18 column (2.1 × 50 mm, Waters, 186002350) with a linear gradient of 5–35% acetonitrile over 12 min at 4 °C. Mass spectrometer data was acquired using a Cyclic IMS Mass spectrometer (Waters, MA). HDX analyses were conducted in triplicates with a single preparation of each protein-ligand complex. Deuterium incorporation was converted to a percentage and averaged over all time points. A 3σ statistical significance of the differential HDX was calculated. Peptide identification and deuterium uptake were analyzed using ProteinLynx Global server (PLGS) and DynamX. PyMOL 2.5 software was utilized for protein visualization.
Reporter cell lines and stable gene expression assays
HG5LN, HG5LN hCAR, HG5LN hPXR, HGELN, HGELN-ERα, HGELN-ERβ, and HGELN-GAL-PPARγ cell lines were established as previously described31,84. Cell lines were seeded at a density of 40,000 cells per well in white opaque 96-well plates and treated with test compounds; 24 h later, the culture medium was replaced by DMEM containing test compounds. Assays were performed in the absence of serum in order to avoid ligand capture by serum proteins. Following incubation with test compounds, cells were incubated for 16 h and then the culture medium was replaced with a test medium containing 0.3 μM luciferin solution and then analyzed by fluorescence quantification.
PXR target gene responses in human colonic carcinoma cells
5 × 105 LS174T cells transfected with pSG5-hPXR (500 ng per well) were seeded in 6 well plates (Sarstedt, NC 28658). After 24 h of transfection, cells were treated with vehicle (0.1% DMSO), positive control (rifampicin; 10 µM), FDN (10 µM), E2 (3 µM) and EE2 (3 µM) or FDN + E2 and FDN + EE2, respectively for 24 h. Total RNA was isolated using an RNeasy Plus Mini Kit (Qiagen, 74134) and cDNAs were synthesized using a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher, 4368814) according to the manufacturer’s protocol. RT-PCR was conducted using PowerUp SYBR Green Master Mix (Thermo Fisher, A25742) to check the expression of the CYP1A1, CYP3A4, and MDR1 genes. Three independent experiments were performed, and each RT-PCR measurement was performed in tetraplicate. Expression values were normalized to the internal control gene GAPDH.
Intestinal organoid culture
Cells were passaged twice and maintained in WRNE (Advanced DMEM with 50% L-WRN cell line conditioned medium, 1× GlutaMAX, 1× Pen/Strep, 1× B27 supplement, 1× N2 supplement, 50 ng/mL epidermal growth factor, 10 mM nicotinamide, 10 nM gastrin I, 500 nM A-83-01, 10 μM SB202190, 1 mM N-acetylcysteine) in Matrigel beads with regular passage 1–2 times per week with 1:3 splits. Transwells (24-well, Corning) were coated with 40 µg/ml human collagen (Sigma–Aldrich) in 0.6% acetic acid. Enteroids were dissociated into single cells with TrypLE and 10 µM Rock Inhibitor for 20 mins at 37 °C. Cell concentration was adjusted to 1 × 106 cells/ml in WRNE, and 250 µL seeded to the apical compartment of Transwells, with 500 µL WRNE media in the basolateral compartment. Monolayers were grown in WRNE until confluence (2–3 days) then media was changed to Differentiation media (Differentiation media: WRNE without L-WRN cell line conditioned medium, SB202190 and nicotinamide). Monolayer integrity was verified by Lucifer Yellow exclusion before experiments. For PXR target gene expression, PXR agonists were added to the apical compartment for 24 h, followed by RNA extraction for RT-qPCR analysis.
CRISPR-CAS9 knockdown of PXR
Normal tissue culture plates (24-well) were coated with 40 µg/ml human collagen for 2 h at 37 °C. Enteroids were dissociated into small clumps of 3–6 cells with TrypLE supplemented with 10 µM Rock Inhibitor for 10 mins at 37 °C, followed by manual pipetting with a P-1000 micropipette. Cells were seeded onto Collagen in WRNE without antibiotics. After an 8 h attachment period, Enteroids were incubated with Edit-R All-in-one Lentiviral vectors (pAIOsgRNA hEF1a, from Horizon Bioscience), which encode both Cas9 protein and the PXR target gene gRNA and puromycin resistance. The gRNA targeted PXR exon 4 with the following sequence: CTCCCATTTCAAGAATTTCC. Positive control vectors were Edit-R Human All-in-one DNMT3B hEF1a-EGFP Lentiviral sgRNA and negative control vectors were Edit-R All-in-one Lentiviral sgRNA Non-targeting Controls, both expressing GFP. Lentivirus was seeded onto cells in WRNE overnight at an MOI of 10, supplemented with 10 µM polybrene and 10 µM Rock Inhibitor. Then, a layer of Matrigel (150 µL) was overlaid onto the cells to reform the 3-D structure. After three passages in WRNE supplemented with puromycin (2–4 µg/ml), knockout clones were dissociated into single cells and serial diluted in 96-well plates to form single colonies. Single colonies were then selected for Sanger sequencing (Cornell genomics core) and mismatch cleavage assay (GeneArt Genomic Detection kit, Thermo Fisher) to verify INDEL formation. Sanger Sequencing data was analyzed using Geneious Prime software.
Primary human hepatocyte (PHH) assay
Freshly isolated PHHs with over 90% viability were obtained from BioIVT (Baltimore, MD) and seeded at 0.75 × 106 cells/well in a 12-well biocoat plate in INVITROGRO CP Medium (BioIVT). On the second day after cells attached, PHHs were overlaid with Matrigel (0.25 mg/mL) for sandwich culture in complete Williams’ E medium before treatment with 0.1% DMSO, CITCO (1 µM), RIF (10 µM), FDN (10 µM), E2 (3 µM) and EE2 (3 µM) or FDN + E2 and FDN + EE2, respectively for another 24 h. Cells were harvested using TRIzol reagent (Thermo Fisher, Rockford, IL) and total RNA was isolated and reverse transcribed to cDNA using a High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA) following the manufacturer’s instructions. RT-PCR assays were performed on an ABI StepOnePlus Real-Time PCR System (Applied Biosystems) using SYBR Green PCR Mastermix (Qiagen). Primer sequences for cyp3a4, cyp2b6, ugt1a1, and glyceraldehyde-3-phosphate dehydrogenase (gapdh) are shown in Supplementary Table 4. Expression values were quantified using the 2ΔΔCt method, normalizing to gapdh as the housekeeping gene and fold change compared to control values (n = 3).
Statistical analyses
One-way ANOVA tests with multiple comparisons tests (for >3 groups) and two-tailed unpaired t-tests (for 2 groups) were performed as specified in figure legends, using Prism 9 (GraphPad Software, Inc., La Jolla, CA). All data were shown as means ± SEM or means ± SD as specified in the figure legends. Data were considered to be significant if p < 0.05. At least three independent experiments were performed in pulldowns, ITC, and luciferase reporter assays. Two independent experiments were performed in HDX-MS and human intestinal organoid assays. Human intestinal organoid assays were randomly allocated into experimental groups. Images were unbiasedly collected and analyses were performed blind. All attempts at replication were successful. For mouse experiments, the sample size was determined based on previous studies22. All biological replicates supporting reproducibility are described in each figure legend. No data were excluded in other analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Metabolomic data that support the findings of this study have been deposited in MassIVE with ID: MSV000093761. FDN Crystal structure has been deposited in Cambridge Structural Database (CSD/CCDC) under Deposition Number 2376591. Protein structures used in this study have been deposited in PDB under code 1XVP (https://doi.org/10.2210/pdb1XVP/pdb), 7AXK (https://doi.org/10.2210/pdb7AXK/pdb), 4X1G (https://doi.org/10.2210/pdb4X1G/pdb), and 3HVL (https://doi.org/10.2210/pdb3HVL/pdb). Source data are provided with this paper.
Code availability
The code used in the pulldown analysis is available from https://github.com/huiUofT/humanCAR85.
Change history
03 March 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-57509-y
References
Wang, R., Li, Z., Liu, S. & Zhang, D. Global, regional and national burden of inflammatory bowel disease in 204 countries and territories from 1990 to 2019: a systematic analysis based on the Global Burden of Disease Study 2019. BMJ Open 13, e065186 (2023).
Kaplan, G. G. & Windsor, J. W. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 18, 56–66 (2021).
Kuenzig, M. E. et al. Twenty-first century trends in the global epidemiology of pediatric-onset inflammatory bowel disease: systematic review. Gastroenterology 162, 1147–1159.e1144 (2022).
Collaborators, G. B. D. I. B. D. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 5, 17–30 (2020).
Burisch, J. et al. The cost of inflammatory bowel disease in high-income settings: a Lancet Gastroenterology & Hepatology Commission. Lancet Gastroenterol. Hepatol. 8, 458–492 (2023).
Ott, C. & Scholmerich, J. Extraintestinal manifestations and complications in IBD. Nat. Rev. Gastroenterol. Hepatol. 10, 585–595 (2013).
Powrie, F. T cells in inflammatory bowel disease: protective and pathogenic roles. Immunity 3, 171–174 (1995).
Xavier, R. J. & Podolsky, D. K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434 (2007).
Khor, B., Gardet, A. & Xavier, R. J. Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317 (2011).
Kobayashi, T. & Hibi, T. Improving IBD outcomes in the era of many treatment options. Nat. Rev. Gastroenterol. Hepatol. 20, 79–80 (2023).
Villablanca, E. J., Selin, K. & Hedin, C. R. H. Mechanisms of mucosal healing: treating inflammatory bowel disease without immunosuppression? Nat. Rev. Gastroenterol. Hepatol. 19, 493–507 (2022).
Kotla, N. G. & Rochev, Y. IBD disease-modifying therapies: insights from emerging therapeutics. Trends Mol. Med. 29, 241–253 (2023).
Sigall-Boneh, R. et al. Research gaps in diet and nutrition in inflammatory bowel disease. a topical review by D-ECCO Working Group [Dietitians of ECCO]. J. Crohns Colitis 11, 1407–1419 (2017).
Chen, J. C. et al. Ginger and its bioactive component inhibit enterotoxigenic Escherichia coli heat-labile enterotoxin-induced diarrhea in mice. J. Agric. Food Chem. 55, 8390–8397 (2007).
Zhang, C. et al. Ginger relieves intestinal hypersensitivity of diarrhea predominant irritable bowel syndrome by inhibiting proinflammatory reaction. BMC Complement. Med. Ther. 20, 279 (2020).
Nikkhah-Bodaghi, M., Maleki, I., Agah, S. & Hekmatdoost, A. Zingiber officinale and oxidative stress in patients with ulcerative colitis: a randomized, placebo-controlled, clinical trial. Complement. Ther. Med. 43, 1–6 (2019).
Guo, S. et al. Ginger alleviates DSS-induced ulcerative colitis severity by improving the diversity and function of gut microbiota. Front. Pharmacol. 12, 632569 (2021).
Zhang, M. et al. Edible ginger-derived nanoparticles: a novel therapeutic approach for the prevention and treatment of inflammatory bowel disease and colitis-associated cancer. Biomaterials 101, 321–340 (2016).
Nachvak, S. M. et al. Ginger as an anticolorectal cancer spice: a systematic review of in vitro to clinical evidence. Food Sci. Nutr. 11, 651–660 (2023).
Scholtes, C. & Giguere, V. Transcriptional control of energy metabolism by nuclear receptors. Nat. Rev. Mol. Cell Biol. 23, 750–770 (2022).
Klepsch, V., Moschen, A. R., Tilg, H., Baier, G. & Hermann-Kleiter, N. Nuclear receptors regulate intestinal inflammation in the context of IBD. Front. Immunol. 10, 1070 (2019).
Venkatesh, M. et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 41, 296–310 (2014).
Bouguen, G. et al. Intestinal steroidogenesis controls PPARgamma expression in the colon and is impaired during ulcerative colitis. Gut 64, 901–910 (2015).
Bayrer, J. R. et al. LRH-1 mitigates intestinal inflammatory disease by maintaining epithelial homeostasis and cell survival. Nat. Commun. 9, 4055 (2018).
Klepsch, V. et al. Nuclear orphan receptor NR2F6 as a safeguard against experimental murine colitis. Gut 67, 1434–1444 (2018).
Fu, T. et al. FXR mediates ILC-intrinsic responses to intestinal inflammation. Proc. Natl Acad. Sci. USA 119, e2213041119 (2022).
Cheng, J., Shah, Y. M. & Gonzalez, F. J. Pregnane X receptor as a target for treatment of inflammatory bowel disorders. Trends Pharmacol. Sci. 33, 323–330 (2012).
Liu, J. et al. The omega-3 hydroxy fatty acid 7(S)-HDHA is a high-affinity PPARalpha ligand that regulates brain neuronal morphology. Sci. Signal 15, eabo1857 (2022).
Liu, J. et al. Diindoles produced from commensal microbiota metabolites function as endogenous CAR/Nr1i3 ligands. Nat. Commun. 15, 2563 (2024).
Hikino, H. et al. Sesquiterpenoids. Part XLVII. Structure, configuration, conformation, and thermal rearrangement of furanodienone, isofuranodienone, curzerenone, epicurzerenone, and pyrocurzerenone, sesquiterpenoids of Curcuma zedoaria. J. Chem. Soc. Perkin Trans. 1, 478–484 (1975).
Delfosse, V. et al. Mechanistic insights into the synergistic activation of the RXR-PXR heterodimer by endocrine disruptor mixtures. Proc. Natl Acad. Sci. USA 118, e2020551118 (2021).
Consortium, G. T. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Xu, R. X. et al. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol. Cell 16, 919–928 (2004).
Medvedev, A. et al. Comprehensive assessment of NR ligand polypharmacology by a multiplex reporter NR assay. Sci. Rep. 12, 3115 (2022).
Li, Y. W. et al. Furanodienone inhibits cell proliferation and survival by suppressing ERalpha signaling in human breast cancer MCF-7 cells. J. Cell Biochem. 112, 217–224 (2011).
Leavitt, S. & Freire, E. Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 11, 560–566 (2001).
Creusot, N. et al. The anti-cancer drug dabrafenib is a potent activator of the human pregnane X receptor. Cells 9, 1641 (2020).
Xie, W. et al. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl Acad. Sci. USA 98, 3375–3380 (2001).
Anzenbacher, P. & Anzenbacherova, E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol. Life Sci. 58, 737–747 (2001).
Cao, W. et al. The xenobiotic transporter Mdr1 enforces T cell homeostasis in the presence of intestinal bile acids. Immunity 47, 1182–1196.e1110 (2017).
Choi, S., Neequaye, P., French, S. W., Gonzalez, F. J. & Gyamfi, M. A. Pregnane X receptor promotes ethanol-induced hepatosteatosis in mice. J. Biol. Chem. 293, 1–17 (2018).
Langmann, T. et al. Loss of detoxification in inflammatory bowel disease: dysregulation of pregnane X receptor target genes. Gastroenterology 127, 26–40 (2004).
Zhou, C. et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J. Clin. Invest. 116, 2280–2289 (2006).
Neurath, M. F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 14, 329–342 (2014).
Chen, S. et al. Epithelial PBLD attenuates intestinal inflammatory response and improves intestinal barrier function by inhibiting NF-kappaB signaling. Cell Death Dis. 12, 563 (2021).
Al-Sadi, R. et al. Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G1054–G1064 (2011).
Kuo, W. T. et al. The tight junction protein ZO-1 is dispensable for barrier function but critical for effective mucosal repair. Gastroenterology 161, 1924–1939 (2021).
Jiang, Y. et al. Pregnane X receptor regulates liver size and liver cell fate by yes-associated protein activation in mice. Hepatology 69, 343–358 (2019).
Teng, S. & Piquette-Miller, M. Hepatoprotective role of PXR activation and MRP3 in cholic acid-induced cholestasis. Br. J. Pharmacol. 151, 367–376 (2007).
Heidari, Z. et al. Definition of functionally and structurally distinct repressive states in the nuclear receptor PPARgamma. Nat. Commun. 10, 5825 (2019).
Lin, W. et al. Structure-guided approach to modulate small molecule binding to a promiscuous ligand-activated protein. Proc. Natl Acad. Sci. USA 120, e2217804120 (2023).
Zhao, B. et al. 6-Shogaol derived from ginger inhibits intestinal crypt stem cell differentiation and contributes to irritable bowel syndrome risk. Research 7, 0524 (2024).
Hikino, H. et al. Structure and conformation of the sesquiterpenoids furanodienone and isofuranodienone. J. Chem. Soc. D Chem. Commun. https://doi.org/10.1039/C29690000662 (1969).
Messina, F., Gigliarelli, G., Palmier, A. & Marcotullio, M. C. Furanodienone: an emerging bioactive furanosesquiterpenoid. Curr. Org. Chem. 21, 305–310 (2017).
Ma, X. et al. Rifaximin is a gut-specific human pregnane X receptor activator. J. Pharmacol. Exp. Ther. 322, 391–398 (2007).
Staudinger, J. L., Ding, X. & Lichti, K. Pregnane X receptor and natural products: beyond drug-drug interactions. Expert Opin. Drug Metab. Toxicol. 2, 847–857 (2006).
Synold, T. W., Dussault, I. & Forman, B. M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 7, 584–590 (2001).
Moore, L. B. et al. St. John’s wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc. Natl Acad. Sci. USA 97, 7500–7502 (2000).
Cai, X., Young, G. M. & Xie, W. The xenobiotic receptors PXR and CAR in liver physiology, an update. Biochim. Biophys. Acta Mol. Basis Dis. 1867, 166101 (2021).
Zhang, J. et al. B3galt5 functions as a PXR target gene and regulates obesity and insulin resistance by maintaining intestinal integrity. Nat. Commun. 15, 5919 (2024).
Delfosse, V. et al. Synergistic activation of human pregnane X receptor by binary cocktails of pharmaceutical and environmental compounds. Nat. Commun. 6, 8089 (2015).
Garcia-Maldonado, E. et al. Chemical manipulation of an activation/inhibition switch in the nuclear receptor PXR. Nat. Commun. 15, 4054 (2024).
Atanasov, A. G., Zotchev, S. B., Dirsch, V. M., & Supuran, C. T., International Natural Product Sciences Taskforce Natural products in drug discovery: advances and opportunities. Nat. Rev. Drug Discov. 20, 200–216 (2021).
Kurimchak, A. M. et al. The drug efflux pump MDR1 promotes intrinsic and acquired resistance to PROTACs in cancer cells. Sci. Signal 15, eabn2707 (2022).
Chen, M. L. et al. CAR directs T cell adaptation to bile acids in the small intestine. Nature 593, 147–151 (2021).
Thummel, K. E. Gut instincts: CYP3A4 and intestinal drug metabolism. J. Clin. Invest. 117, 3173–3176 (2007).
Chen, S. et al. Intestinal glucuronidation protects against chemotherapy-induced toxicity by irinotecan (CPT-11). Proc. Natl Acad. Sci. USA 110, 19143–19148 (2013).
Chen, M. et al. Analysis of genetic and chemical variability of five Curcuma species based on DNA barcoding and HPLC fingerprints. Front. Plant Sci. 14, 1229041 (2023).
Igoli, N. P., Obanu, Z. A., Gray, A. I. & Clements, C. Bioactive diterpenes and sesquiterpenes from the rhizomes of wild ginger (Siphonochilus aethiopicus (Schweinf) B.L Burtt). Afr. J. Tradit. Complement Altern. Med. 9, 88–93 (2012).
Dekebo, A., Dagne, E. & Sterner, O. Furanosesquiterpenes from Commiphora sphaerocarpa and related adulterants of true myrrh. Fitoterapia 73, 48–55 (2002).
Joshi, S. C. & Mathela, C. S. Antioxidant and antibacterial activities of the leaf essential oil and its constituents furanodienone and curzerenone from Lindera pulcherrima (Nees.) Benth. ex hook. f. Pharmacognosy Res. 4, 80–84 (2012).
Jiang, Y., Wang, X. & Hu, D. Furanodienone induces G0/G1 arrest and causes apoptosis via the ROS/MAPKs-mediated caspase-dependent pathway in human colorectal cancer cells: a study in vitro and in vivo. Cell Death Dis. 8, e2815 (2017).
Ouyang, N. et al. Pregnane X receptor suppresses proliferation and tumourigenicity of colon cancer cells. Br. J. Cancer 102, 1753–1761 (2010).
Cheng, J. et al. Activation of intestinal human pregnane X receptor protects against azoxymethane/dextran sulfate sodium-induced colon cancer. J. Pharmacol. Exp. Ther. 351, 559–567 (2014).
Bansard, L. et al. Niclosamide induces miR-148a to inhibit PXR and sensitize colon cancer stem cells to chemotherapy. Stem Cell Rep. 17, 835–848 (2022).
Dong, Y. et al. Pregnane X receptor is associated with unfavorable survival and induces chemotherapeutic resistance by transcriptional activating multidrug resistance-related protein 3 in colorectal cancer. Mol. Cancer 16, 71 (2017).
Niu, X. et al. Nuclear receptor PXR confers irradiation resistance by promoting DNA damage response through stabilization of ATF3. Front. Oncol. 12, 837980 (2022).
Bwayi, M. N. et al. Molecular basis of crosstalk in nuclear receptors: heterodimerization between PXR and CAR and the implication in gene regulation. Nucleic Acids Res. 50, 3254–3275 (2022).
Sonoda, J. et al. Pregnane X receptor prevents hepatorenal toxicity from cholesterol metabolites. Proc. Natl Acad. Sci. USA 102, 2198–2203 (2005).
Langmade, S. J. et al. Pregnane X receptor (PXR) activation: a mechanism for neuroprotection in a mouse model of Niemann-Pick C disease. Proc. Natl Acad. Sci. USA 103, 13807–13812 (2006).
Yu, X. et al. Nuclear receptor PXR targets AKR1B7 to protect mitochondrial metabolism and renal function in AKI. Sci. Transl. Med. 12, eaay7591 (2020).
Fan, S. et al. Pregnane X receptor agonist nomilin extends lifespan and healthspan in preclinical models through detoxification functions. Nat. Commun. 14, 3368 (2023).
Friesner, R. A. et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196 (2006).
Riu, A. et al. Characterization of novel ligands of ERα, Erβ, and PPARγ: the case of halogenated bisphenol A and their conjugated metabolites. Toxicol. Sci. 122, 372–382 (2011).
Peng, H. Diindoles produced from commensal microbiota metabolites function as endogenous CAR/Nr1i3 ligands. Zenodo https://doi.org/10.5281/zenodo.10684290 (2024).
Zhao, W. et al. A new bliss independence model to analyze drug combination data. J. Biomol. Screen 19, 817–821 (2014).
Acknowledgements
J.L. was supported by a Charles H. Best Postdoctoral Fellowship and Precision Medicine Initiative (PRiME) Fellowship. We acknowledge experimental assistance from the staff of the European Synchrotron Radiation Facility (Grenoble, France) during data collection. L.S. was supported by the Czech Science Foundation (22-00355S). Work performed was supported by a Canadian Institutes of Health Research (PJT-186117) to H.M.K., a New Frontiers in Research Fund grant (NRFRE-2019-00901) to H.M.K. and H.P., National Institutes of Health grants (R01CA222469) to S.Ma., a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grants (RGPIN480432, and CRDPJ597036) to D.J.W., a National Natural Science Foundation of China grant (82204227) to X.W., a Key-Area Research and Development Program of Guangdong Province, China grant (2020B1111110003) to S.W., and an Agence Nationale de la Recherche SYNERGY grant (ANR-23-CE34-0006), to P.B. and W.B. Infrastructure support was also provided by the Canada Foundation for Innovation, Ontario Research Fund and NSERC Research Tools and Instrument grants. The Centre de Biologie Structurale is a member of France-BioImaging and the French Infrastructure for Integrated Structural Biology, two national infrastructures supported by the French National Research Agency (ANR-10-INBS-04-01 and ANR-10-INBS-05).
Author information
Authors and Affiliations
Contributions
J.L., D.W., P.B., W.B., S.Mani, L.B., H.P., J.M., H.W., S.W., and H.M.K. designed the experiments. J.L. and Y.G. performed the protein expression, characterization, pulldown, and ligand identification experiments shown in Fig. 1. J.L. also performed the data collection shown in Fig. 1a, b, and characterizations depicted in Fig. 2 and Supplementary Fig. 2. X.W., G.Z., Z.B., and M.Guo performed the experiments in Figs. 3–6, and Supplementary Figs. 6 and 7. X.W., Z.B., and S.H. also performed the extraction, isolation, and structural elucidation for compounds 1–3. V.C. performed the HDX experiments in Fig. 7d, e, and Supplementary Fig. 8. M.Grimaldi performed the experiments in Fig. 2b–d, and Fig. 7f, g. C.Carivenc and S.S. performed experiments depicted in Fig. 2g–i, Fig. 6a, Supplementary Fig. 4, and Supplementary Table. 2. H.L. and L.S. performed the experiments in Fig. 7h and Supplementary Fig. 5. S.Motta and Y.L. performed the experiments in Fig. 7b,c. C.Costello performed the experiments in Fig. 7i–k and Supplementary Fig. 9. L.L. performed the experiments in Fig. 6d and Supplementary Fig. 10. M.J. performed the data analysis. J.L. wrote the initial manuscript. S.Mani and H.M.K. revised and edited the manuscript. All authors reviewed the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Huichang Bi, Emre Turer, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, X., Zhang, G., Bian, Z. et al. An abundant ginger compound furanodienone alleviates gut inflammation via the xenobiotic nuclear receptor PXR in mice. Nat Commun 16, 1280 (2025). https://doi.org/10.1038/s41467-025-56624-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56624-0
