
Abstract
Zika virus (ZIKV) is a member of the Flaviviridae family and causes congenital microcephaly and Guillain–Barré syndrome. Currently, there is a lack of approved vaccines or therapies against ZIKV infection. In this study, we profile vRNA‒host protein interactomes at ZIKV stem‒loop B (SLB) and reveal that interleukin enhancer binding factor 3 (ILF3) and DEAH-box helicase 9 (DHX9) form positive regulators of antiviral RNA inference in undifferentiated human neuroblastoma cells and induced pluripotent stem cell-derived human neural stem cells (iPSC–NSCs). Functionally, ablation of ILF3 in brain organoids and Nestin-Cre ILF3 cKO foetal mice significantly enhance ZIKV replication and aggravated ZIKV-induced microcephalic phenotypes. Mechanistically, ILF3/DHX9 enhance DICER processing of ZIKV vRNA-derived siRNAs (vsiR-1 and vsiR-2) to exert anti-flavivirus activity. VsiR-1 strongly inhibits ZIKV NS5 polymerase activity and RNA translation. Treatment with the vsiR-1 mimic inhibits ZIKV replication in vitro and in vivo and protected mice from ZIKV-induced microcephaly. Overall, we propose a novel therapeutic strategy to combat flavivirus infection.
Similar content being viewed by others
Introduction
ZIKV has a single-stranded, positive-sense RNA genome ~10.8 kb in length that contains two flanking noncoding regions (UTRs) and a single long open reading frame (ORF) encoding a polyprotein: 5′-C-prM-E-NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5-3′. ZIKV circulates as two genetic lineages, African and Asian. The ancestral African lineage (MR766) is maintained in a sylvatic transmission cycle and infects few human hosts1,2, whereas a recent African strain caused increased transmissibility in neural progenitor cells (NPCs) and increased fetal pathogenicity3,4,5. The epidemic outbreak of the Asian lineage in Micronesia and French Polynesia has caused increasing public health concerns and is associated with a higher rate of congenital ZIKV syndrome (CZVS), manifesting as microcephaly and Guillain–Barré syndrome6,7. As Zika transmission has persisted in certain countries since 20188,9, the World Health Organization has cautioned that ZIKV remains a significant and enduring health challenge10. Currently, there is still a lack of approved vaccines or specific therapies to prevent or treat ZIKV infection. Thus, it is critical to address ZIKV infection, understand its pathogenesis, and develop therapeutic and prevention strategies.
The 5′UTR of the ZIKV genome, ~100 nucleotides in length, contains two conserved stem-loop structures, stem-loop A (SLA) and stem-loop B (SLB), which play essential roles in ZIKV infectivity and pathogenicity11,12,13. It is unclear whether and how these RNA elements contribute to diverse infectivity, transmissibility, and neurovirulence between geographic lineages of ZIKV12,14. SLB has been reported to serve as a critical platform for RNA‒RNA or RNA‒protein interactions in the ZIKV life cycle. SLB is indispensable for the recruitment of NS5, an RNA-dependent RNA polymerase, which initiates viral RNA (vRNA) synthesis15,16. In addition, SLB and capsid hairpin element (cHP), a stem-loop located downstream of the AUG start codon, form base pairs with the 3′ end for genome cyclization12. The region 5′ upstream of the AUG start codon region (5′ UAR) flanking the stem (UFS), located within SLB, is the key region for long-range RNA‒RNA interactions and the switch from genome cyclization to NS5 binding12,13,17,18. It remains obscure which, if any, host proteins contribute to this process. As ZIKV infection is more efficient in primary human neural stem cells19, it is important to uncover how SLB contributes to ZIKV infection and neuropathogenicity in human undifferentiated neuronal cells.
The RNA interference (RNAi) pathway, an evolutionarily conserved posttranscriptional gene-silencing mechanism, mediates essential antiviral immunity in eukaryotes20,21,22,23,24. In the context of viral infection in hosts, two main types of small RNAs generated via the RNAi pathway, namely, host-derived microRNAs (miRNAs) and virus-derived small interfering RNAs (vsiRNAs), have been revealed to exert antiviral activity20. DICER, a host endoribonuclease in the RNAi pathway, recognizes and cleaves the stem-loop structure of precursor miRNAs and vsiRNAs into 21–22 nt double-stranded RNAs (dsRNAs), generating miRNAs and vsiRNAs, respectively25,26. After DICER processing, one strand of dsRNAs is unwound and loaded into the AGO2/RNA-induced silencing complex (RISC)26. Although miRNAs and vsiRNAs share DICER/AGO2 processing machinery in RNAi, these two small RNAs exert antiviral activity through distinct molecular mechanisms25,26. Mature miRNAs target host mRNAs or vRNAs through base-pair complementarity, leading to the inhibition of mRNA stability or translation, whereas vsiRNAs direct the cleavage of cognate vRNAs26. Recently, genome-wide screening identified RDO5 and VIR1 as positive and negative regulators of antiviral RNAi in Arabidopsis thaliana27. It remains an open question whether certain host proteins may regulate antiviral RNAi in mammals. To date, the host regulators of antiviral RNAi in undifferentiated neuronal cells with stem-like properties remain obscure and require further investigation.
In this work, we profile ZIKV vRNA‒host protein interactomes at the ZIKV SLB element in an undifferentiated human neuroblastoma cell line, SK-N-BE(2). We identify four general host-interacting proteins and multiple strain-specific interacting proteins. Interleukin enhancer binding factor 3 (ILF3) and DEAH-box helicase 9 (DHX9) are characterized as positive regulators of antiviral RNA inference in SK-N-BE(2) cells and iPSC–NSCs. Functionally, ablation of ILF3 in brain organoids and Nestin-Cre ILF3 cKO fetal mice significantly enhanced ZIKV replication and aggravated ZIKV-induced microcephalic phenotypes. Mechanistically, ILF3/DHX9 enhances DICER processing of ZIKV SLB and cHP-derived vsiRNAs (vsiR-1 and vsiR-2, respectively). We further reveal that vsiR-1 exerts anti-flavivirus activity by inhibiting ZIKV NS5 polymerase activity and RNA translation. Treatment with the vsiR-1 mimic inhibits ZIKV replication in vitro and in vivo and protects mice from ZIKV-induced microcephaly. Overall, our study proposes a novel therapeutic strategy to combat flavivirus infection.
Results
RNA-pull-down coupled with mass spectrometry (MS) identified ZIKV vRNA‒host protein interactomes at the ZIKV SLB element in human neuroblastoma cells
We applied RNA-pull-down coupled with MS to identify host proteins that bind to the ZIKV SLB element in undifferentiated human neuroblastoma SK-N-BE(2) cells. Mock cells without virus infection were used as the control group. SK-N-BE(2) cells were infected with African lineage MR766 strains and Asian lineage PRVABC.59 strains (Multiplicity of infection, MOI = 1) for 48 h. To purify host proteins that are specifically recruited at the 5′SLB of ZIKV vRNA, we designed a biotin-labeled RNA oligo that hybridizes with the 5′SLB of ZIKV vRNA through sequence complementarity. A negative control was generated using a biotin-labeled oligo with scrambled sequences28. We performed RT-qPCR assays to assess the enrichment region of the pulled-down RNAs across the entire length of the ZIKV vRNA (including the SLB, prM, NS2A, and NS5 regions). The results revealed that SLB oligo pulled-down RNAs were specifically enriched in the ZIKV SLB region in virus-infected samples (Fig. 1A). Both SLB and scramble oligo pulled-down RNAs, such as 18S RNA, were depleted for highly abundant host RNAs (Fig. 1A). The SLB oligo pull-down specificity was further confirmed by Northern blotting29 (Supplementary Fig. 1A). The SLB element of ZIKV vRNA derived from ZIKV-infected cells could be efficiently pulled down by the biotin-labeled SLB oligo but not by biotin-labeled scramble oligo (Fig. 1A and Supplementary Fig. 1A).

A RNA-pull-down efficiency at ZIKV genome in mock, ZIKV MR766 or PRVABC.59-infected SK-N-BE(2) cells (Multiplicity of infection, MOI = 1, 48 hpi) were assessed by RT-qPCR. Target viral RNA was pull-down by biotin-labeled scramble oligo or SLB oligo. Four primer pairs were used to assess the enrichment across ZIKV vRNA. The position of each PCR amplicon is presented as below diagram. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. *p < 0.05, **p < 0.01. B Venn diagram illustrates the identification of human proteins interacting with ZIKV SLB vRNA in mock, ZIKV MR766, or PRVABC.59-infected cells. C Gene Ontology (GO) analysis shows the enriched signal pathways among common SLB-interacting proteins for MR766 and PRVABC.59 strains. The size of the dot represents enriched gene numbers. And dots are colored according to p values calculated by hypergeometric test. GO terms with p value adjust <0.05 are plotted. D Venn diagram illustrates the intersecting results of ZIKV SLB-interacting proteins in SK-N-BE(2) and Huh7 cells. E Western blot analysis of ILF3 and DHX9 protein pull-downed by biotin-labeled scramble oligo or SLB oligo in mock, ZIKV MR766, or PRVABC.59-infected SK-N-BE(2) cells (MOI = 1, 48 hpi). GAPDH was applied as a loading control. n = 4 biological replicates/group. F Venn diagram illustrates ILF3-interacting proteins in mock or ZIKV MR766-infected cells. G Western blot detection of ILF3 and DHX9, NS5, E protein in ILF3 endogenous IP samples from mock, ZIKV MR766 or PRVABC.59-infected SK-N-BE(2) cells (MOI = 1, 48 hpi). GAPDH was applied as a loading control. n = 3 biological replicates/group. H Western blot detection of ILF3 and DHX9 interactions in ILF3 endogenous IP samples from mock or PRVABC.59-infected SK-N-BE(2) cells (MOI = 1, 48 hpi) with or without RNase A (20 μg/ml) treatment. n = 3 biological replicates/group. I Western blot analysis of ILF3, NS5, E, and GAPDH protein expressions in ZIKV-infected SK-N-BE(2) cells at indicated time points (MOI = 1). n = 3 biological replicates/group. J Western blot showing the distribution of ILF3, NS5, E, and Histone H3 in the nuclear or cytoplasmic fraction from ZIKV-infected SK-N-BE(2) cells at indicated time points (MOI = 1). n = 3 biological replicates/group. K Immunofluorescence showing the localization of ILF3 among mock, ZIKV MR766 or PRVABC.59-infected SK-N-BE(2) cells (MOI = 1) at 48 hpi (scale bar, 7.5 μm). Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01, ***p < 0.001. Source data are provided as a Source Data file.
The RNA‒protein complexes pulled down by scrambled oligo or the SLB probe were subsequently identified by MS. The proteins associated with SLB pull-down RNA oligo compared to proteins with scrambled oligo in each group were identified as the enriched proteins. The enriched proteins in the virus-infected samples compared with the mock control samples were defined as the specific interactors. The interactome profiling identified 61 host proteins in common between the ZIKV MR766 and PRVABC.59 strains, whereas 25 proteins and 74 proteins exclusively bound to the MR766 and PRVABC.59 strains, respectively (coverage > 0.12) (Fig. 1B, Supplementary Data 1). Gene Ontology (GO) enrichment analyses revealed that the 61 common proteins were enriched in the replicative life cycle of ZIKV, such as transcription initiation factor binding (Fig. 1C). Moreover, MR766-specific recruited host factors were enriched in the RNA splicing, processing and modification pathways (Supplementary Fig. 1B), whereas PRVABC.59-specific recruited host factors were enriched in the transcription machinery binding and siRNA binding pathways (Supplementary Fig. 1C).
We further integrated two studies of ZIKV vRNA–host protein interactomes30,31 in a well-differentiated cell line, Huh7, to identify common host factors recruited at SLB of ZIKV vRNA. This integrative analysis identified four common ZIKV SLB-interacting host proteins, ILF3, DHX9, DDX17, and RBM14, among Huh7 and SK-N-BE(2) cells (Fig. 1D, Supplementary Fig. 1D, E, Supplementary Data 2). In ZIKV MR766 and PRVABC.59-infected SK-N-BE(2) cells, ZIKV SLB-interacting factors were enriched in mRNA metabolic process, RNA splicing, processing and transcription (Supplementary Fig. 1D), whereas in ZIKV-infected Huh7 cells, ZIKV SLB-interacting proteins were enriched in amide biosynthetic process, translation and mRNA metabolic process (Supplementary Fig. 1E).
RNA-dependent interactions between ILF3 and DHX9 recruit SLB of ZIKV vRNA
We confirmed that ILF3, a dsRNA protein, and DHX9, an RNA helicase, are SLB-interacting factors for the ZIKV MR766 and PRVABC.59 strains in SK-N-BE(2) cells (Fig. 1A, E). By analyzing ILF3 complexes in uninfected mock cells and MR766-infected SK-N-BE(2) cells (MOI = 1), we identified eight common ILF3-interacting proteins between mock-infected cells and ZIKV-infected cells, whereas 18 proteins and 30 proteins specifically bound to ILF3 in mock-infected and ZIKV-infected cells, respectively (Fig. 1F, Supplementary Table 1). GO analysis suggested that ILF3 interactors in ZIKV-infected cells were enriched in double or single RNA-binding pathways (Supplementary Fig. 1G). Intriguingly, we found that ILF3 and DHX9 formed a protein complex in mock-, MR766- and PRVABC.59-infected SK-N-BE(2) cells (Fig. 1G). Under RNase A treatment, the interactions between ILF3 and DHX9 were diminished, suggesting that the interaction was dependent on RNA (Fig. 1H). ILF3 was also found to interact with the ZIKV NS5 protein, which was indispensable for recruiting the 5′UTR promoter and initiating vRNA synthesis in ZIKV-infected cells (Fig. 1G). In contrast, we failed to observe an interaction between ILF3 and the ZIKV E protein (Fig. 1G). Together, these results demonstrated that RNA-dependent interactions between ILF3 and DHX9 are recruited to the SLB of ZIKV vRNA.
ILF3 is upregulated and transported into the cytoplasm upon ZIKV infection
We next examined the expression and localization of ILF3 and DHX9 in ZIKV-infected SK-N-BE(2) cells. ZIKV infection induced a time-dependent upregulation of ILF3 in SK-N-BE(2) cells (Fig. 1I). Moreover, the cytoplasmic fraction of ILF3 was significantly increased in MR766- and PRVABC.59-infected SK-N-BE(2) cells (Fig. 1J). Immunofluorescence confirmed that in mock-infected cells, ILF3 was located in the nucleus, whereas in ZIKV-infected cells, ILF3 was exported to the cytoplasm (Fig. 1K).
ILF3 inhibits ZIKV replication and propagation in human neuroblastoma cells and iPSC–NSCs
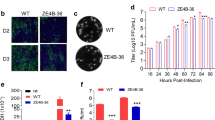
In MR766- and PRVABC.59-infected SK-N-BE(2) cells, knockdown of ILF3 did not alter viral copy numbers at 2 or 4 h post-infection (hpi) (Supplementary Fig. 2A–C), suggesting that ILF3 does not affect the virus entry or attachment steps. Notably, ZIKV viral copy numbers were significantly increased in ILF3-knockdown cells at 24 or 48 hpi (Supplementary Fig. 2B, C, Fig. 2B, C), as were NS5 and E protein expression levels (Fig. 2A). Furthermore, ZIKV viral titers were strongly increased in ILF3-knockdown cells at 48 hpi (Fig. 2D). Immunofluorescence revealed that ZIKV particle and dsRNA production levels were strongly induced in ILF3-knockdown cells upon ZIKV infection (Fig. 2E, F).

A Western blot analysis of ILF3, ZIKV-E, NS5 and GAPDH protein expression in ZIKV-infected control or ILF3-knockdown SK-N-BE(2) cells (MOI = 1) at 48 hpi. n = 3 biological replicates/group. RT-qPCR analysis of ILF3 RNA expression (B) and ZIKV viral RNA copies (C) in ZIKV-infected control or ILF3-knockdown SK-N-BE(2) cells (MOI = 1) at 48 hpi. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. ***p < 0.001. D Viral titers in ZIKV-infected control or ILF3-knockdown SK-N-BE(2) cells (MOI = 1) were quantified by plaque assay at 48 hpi. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. ***p < 0.001. Immunofluorescence showing Flavivirus (E) and dsRNA (F) expression in ZIKV-infected control or ILF3-knockdown SK-N-BE(2) cells (MOI = 1) at 48 hpi (scale bar, 50 μm). Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. ***p < 0.001. G RT-qPCR analysis of ILF3 expression and viral RNA copies in ZIKV-infected control or ILF3-knockdown iPSC–NSC cells (MOI = 1) at 48 hpi. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01, ***p < 0.001. Immunofluorescence showing Flavivirus (H) and dsRNA (I) expression in ZIKV-infected control or ILF3-knockdown iPSC–NSC cell (MOI = 1) at 48 hpi (scale bar, 50 μm). Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01. J Western blot analysis of ILF3, NS5, E, and GAPDH protein expression in ZIKV-infected control or ILF3 overexpressed SK-N-BE(2) cell (MOI = 1) at 48 hpi. n = 3 biological replicates/group. K RT-qPCR analysis of viral RNA copies in ZIKV-infected control or ILF3 overexpressed SK-N-BE(2) cell (MOI = 1) at 48 hpi. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. *p < 0.05, **p < 0.01. L Western blot analysis of ILF3, NS5, E, and GAPDH protein expression in ZIKV-infected control or ILF3, NF90, NF110 WT or dsRNA-binding motif (dsRBM) mutant overexpressed SK-N-BE(2) cells (MOI = 1) at 48 hpi. n = 3 biological replicates/group. Source data are provided as a Source Data file.
ZIKV infection is more efficient in primary human neural stem cells than in mature cortical neurons19. Therefore, we further investigated the role of ILF3 upon ZIKV infection in iPSC–NSCs, which were positive for the NSC markers Nestin and SOX2 and negative for the differentiated cell type marker GFAP (Supplementary Fig. 2D, E). Additionally, we tested the viability of control or ILF3-knockdown iPSC–NSCs. A CCK-8 assay revealed that the knockdown of ILF3 expression did not affect cell viability (Supplementary Fig. 2F); however, the knockdown led to an increase in ZIKV viral copy numbers in iPSC–NSCs at 48 h post-infection (Fig. 2G). Immunofluorescence revealed that ZIKV particle and dsRNA production were strongly induced in ILF3-knockdown NSCs upon ZIKV infection (Fig. 2H, I). Furthermore, ZIKV viral titers were strongly increased in ILF3-knockdown NSCs (Supplementary Fig. 2G). Together, these results indicate that ILF3 inhibits ZIKV replication and propagation in human neuroblastoma cells and NSCs.
ILF3 antiviral activity is dependent on the dsRNA-binding motif (dsRBM)
The ILF3 gene encodes diverse isoforms, including nuclear factors 90 and 110 (NF90 and NF110), due to alternative splicing and alternative polyadenylation32,33. These two isoforms both contain two dsRBMs34. We examined whether the dsRBM of ILF3 is involved in antiviral activity against ZIKV infection. In MR766- and PRVABC.59-infected SK-N-BE(2) cells, overexpression of WT ILF3, WT NF110 or WT NF90 significantly inhibited ZIKV NS5 and E protein expression and viral copy numbers (Fig. 2J–L). However, overexpression of an NF110 or NF90 dsRBM mutant attenuated the antiviral activity of ILF3 against ZIKV infection (Fig. 2L).
DHX9 inhibits ZIKV replication and propagation in human neuroblastoma cells and iPSC–NSCs
In MR766- and PRVABC.59-infected SK-N-BE(2) cells, knockdown of DHX9 did not alter viral copy numbers from 2 h to 4 hpi (Supplementary Fig. 3A, B), suggesting that DHX9 does not affect virus entry or attachment. Notably, ZIKV viral copy numbers, ZIKV E protein expression, and viral titers were significantly increased in DHX9-knockdown cells at 48 hpi (Fig. 3A–D). Immunofluorescence further revealed that viral particles were strongly induced in DHX9-knockdown iPSC–NSCs upon ZIKV infection (Fig. 3E). These results suggest that DHX9 inhibits ZIKV replication and propagation in human neuroblastoma cells and iPSC–NSCs.

A Western blot analysis of DHX9, ZIKV-E, and GAPDH protein expression in ZIKV-infected control or DHX9 knockdown SK-N-BE(2) cells (MOI = 1) at 48 hpi. n = 3 biological replicates/group. RT-qPCR analysis of DHX9 RNA expression (B) and ZIKV viral RNA copies (C) in ZIKV-infected control or DHX9 knockdown SK-N-BE(2) cells (MOI = 1) at 48 hpi. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01, ***p < 0.001. D Viral titers in ZIKV-infected control or DHX9 knockdown SK-N-BE(2) cells (MOI = 1) were quantified by plaque assay at 48 hpi. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01. E Immunofluorescence showing Flavivirus expression in ZIKV-infected control or DXH9 knockdown SK-N-BE(2) cells (MOI = 1) at 48 hpi (scale bar, 50 μm). Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01. F WB analysis of ILF3, DHX9, DENV-NS3, DENV-E, and GAPDH protein expression in DENV-infected control or ILF3 or DHX9 knockdown SK-N-BE(2) cells (MOI = 1) at 48 hpi. n = 2 biological replicates/group. G RT-qPCR analysis of viral RNA copies in DENV-infected control, ILF3 or DHX9 knockdown SK-N-BE(2) cells (MOI = 1) at 48 hpi. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01. H Viral titers in DENV-infected control, ILF3 or DHX9 knockdown SK-N-BE(2) cells (MOI = 1) were quantified by plaque assay at 48 hpi. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01. I, J Immunofluorescence showing Flavivirus expression in DENV-infected control, ILF3 or DHX9 knockdown iPSC–NSC cells (MOI = 1) (scale bar, 20 μm) at 48 hpi. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01, ***p ≤ 0.001, two-tailed Student’s t-test. Source data are provided as a Source Data file.
ILF3 and DHX9 inhibit dengue virus (DENV) replication and propagation
DENV and ZIKV are structurally similar flaviviruses that coexist in tropical regions in Asia35. We therefore explored the role of ILF3 and DHX9 in the life cycle of DENV. In DENV-infected SK-N-BE(2) cells, knockdown of ILF3 or DHX9 led to increased DENV-NS3 and DENV-E protein expression levels, as well as increased viral copy numbers, viral titers, and viral particles (Fig. 3F–J). These results suggest that ILF3 or DHX9 inhibits DENV replication and propagation in human neuroblastoma cells and iPSC–NSCs.
ILF3 knockout enhances ZIKV-associated microcephaly in iPSC–NSC-derived brain organoids and a mouse model
Here, we used iPSC–NSC-derived brain organoids to model ZIKV infection during human brain development. Studies have demonstrated that iPSC-derived human brain organoid is a well-established model to explore the effect of ZIKV infection in the developing brain tissue36,37,38. One-to-five-week-old brain organoids closely mimic the fetus period which is most vulnerable to ZIKV infection. Based on these findings, therefore in our study, the brain organoids on day 7 were used for viral infection. After ZIKV MR766 and PRVABC.59 infection, control lentivirus-treated cerebral organoids showed a ~40% reduction in size compared with mock cells on Day 15 (Fig. 4A), which mimics ZIKV-associated microcephaly and neurovirulence3,7,39,40,41. The organoid sections were subsequently stained for the progenitor marker SOX2 and the neuron marker Tuj1 (Supplementary Fig. 4A). The microphotographs revealed that the control organoid (D15) predominantly comprised NPCs (~70% of the cells), as indicated by positive SOX2 staining. The control organoids also comprised a small proportion of differentiated neurons (~30% of the cells), which was represented by Tuj1-positive staining. ZIKV PRVABC.59 infection caused substantial reduction of SOX2 fluorescence intensity and Tuj1+ differentiated neurons (Supplementary Fig. 4A). We also found that ILF3-knockdown did not affect the size of brain organoids or the proportion of hNPCs or differentiated neurons under mock conditions (Fig. 4A, B and Supplementary Fig. 4A, B). Under ZIKV infection, while compared with control organoids, ILF3 shRNA lentivirus-treated brain organoids were significantly smaller in size (Fig. 4A, B and Supplementary Fig. 4A). Moreover, knockdown of ILF3 led to a small reduction in the number of Tuj1+ differentiated neurons (Supplementary Fig. 4A). RT-PCR confirmed that the number of vRNA copies was increased in ILF3-depleted brain organoids during ZIKV infection (Fig. 4B).

A Demonstrative Brain organoids and size measurement in mock or ZIKV-infected control or ILF3-depleted brain organoids (MOI = 1) at day 15 (scale bar, 200 μm). Upper panel: schematic diagram of brain organoid protocol. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. n.s., non-significance, *p ≤ 0.05, **p < 0.01. B RT-qPCR showing ILF3 mRNA expression and viral RNA copies in mock or ZIKV-infected control or ILF3-depleted brain organoids (MOI = 1) at day 15. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01, ***p ≤ 0.001. C Flowchart demonstrated the experiment procedure of ZIKV-associated microcephaly control or Nestin-Cre ILF3 cKO mice model. D Survival rate. Lethality was monitored for 10 days. Kaplan–Meier survival curves were analyzed by the log-rank test, **p < 0.01, n = 8 mice/group. Representative image of mock or ZIKV-infected WT or Nestin-Cre ILF3 conditional knockout mice (cKO) mice (2 × 105 PFU/mice), brain size (E), and brain weight (F) captured at 7 dpi. Bars, mean ± SD, n = 8 mice/group, two-tailed Student’s t-test. n.s., non-significance, *p ≤ 0.05, **p < 0.01. G Clinical score measured in mock or ZIKV-infected control or ILF3 cKO mice (2 × 105 PFU/mice) for 7 days. Bars, mean ± SD, n = 8 mice/group, one-way ANOVA analysis. *p ≤ 0.05, **p < 0.01. H Immunofluorescence showed the relative expression of GFP, SOX2, and ZIKV-E in mock or ZIKV-infected control or ILF3 cKO mice (scale bar, 100 μm). Bars, mean ± SD, n = 3 mice, two-tailed Student’s t-test. **p < 0.01. I RT-qPCR showing viral RNA copies in ZIKV-infected control or ILF3-cKO mice brain at 7 dpi. Bars, mean ± SD, n =6 mice/group, two-tailed Student’s t-test, **p < 0.01. J Virus titers were quantified by virus plaque assay from ZIKV-infected WT or ILF3-cKO mice brain at 7 dpi. Bars, mean ± SD, n = 6 mice/group, two-tailed Student’s t-test, **p < 0.01. K Hematoxylin and eosin stain (HE) staining of the hippocampus of mock and ZIKV-infected mice brain (Scale bar, 100 μm). Red arrow: infiltrated mononuclear cells. n = 3 mice. Source data are provided as a Source Data file. C was created with BioRender.com.
We further used newborn ILF3 Nestin-cKO conditional knockout mice as a model to investigate the role of ILF3 in ZIKV infection (Fig. 4C). Upon ZIKV infection, half of the control mice had survived at Day 7, whereas half of the Nestin-Cre ILF3 cKO mice had survived at Day 5 (Fig. 4D). Moreover, ZIKV infection led to strong reductions in body and brain size/weight in control mice, whereas an even smaller body and a smaller brain size/weight were observed in ZIKV-infected Nestin-Cre ILF3 cKO mice (Fig. 4E, F, Supplementary Fig. 4C, D). Notably, Nestin-Cre ILF3 cKO mice presented more severe clinical symptoms than control mice did upon ZIKV infection (Fig. 4G). RT-qPCR confirmed the loss of ILF3 in the brains of Nestin-Cre-GFP adenovirus-injected mice (Nestin-Cre ILF3 cKO) (Supplementary Fig. 4C). We also found that ZIKV E protein expression was strongly upregulated in ZIKV-infected Nestin-Cre ILF3 cKO mice, whereas SOX2 protein expression was strongly reduced compared with that in ZIKV-infected control mice (Fig. 4H). RT-PCR and virus plaque assays revealed that the number of vRNA copies and the viral titers were greater in Nestin-Cre ILF3 cKO mice than in control mice during ZIKV infection (Fig. 4I, J). Moreover, haematoxylin‒eosin staining demonstrated that ZIKV-infected WT mouse brains presented massive neutrophilic infiltration, frequently visible perivascular cuffs of mononuclear cells, and disruption of normal cytoarchitecture in the hippocampus, whereas ZIKV-infected ILF3 cKO mice presented more severe inflammation and cytoarchitecture disruption in the hippocampus (Fig. 4K and Supplementary Fig. 4E). In contrast, mock infection caused no pathological changes in WT or ILF3 cKO mice (Fig. 4J and Supplementary Fig. 4E). These results suggest that Nestin-Cre ILF3 cKO mice are more prone to develop lethal disease and accelerated ZIKV-associated microcephaly upon challenge with ZIKV PRVABC.59 strains.
ILF3 is recruited with DICER at the ZIKV SLB and enhances DICER processing of SLB-derived siRNA
We further investigated the mechanism by which ILF3 affects ZIKV replication. Small RNA sequencing (Seq) and AGO-associated RNA-seq were conducted using ZIKV MR766-infected SK-N-BE(2) cells (Fig. 5A, B and Supplementary Fig. 5A–C). The western blot showed that AGO2 protein was specifically pulled-down by AGO2 antibody (Supplementary Fig. 5A). A known miRNA, let-7a, was detected by northern blot in Ago2 IP as positive control (Supplementary Fig. 5B). ZIKV vsiRNA-1 were also detected in AGO2 IP of ZIKV-infected cell lysates, but not with Mouse IgG negative control (Supplementary Fig. 5B). In both analyses, abundant vRNA reads (Supplementary Fig. 5C), ranging from 18–28 nt in length, were readily detected in MR766-infected mock SK-N-BE(2) cells. After mapping with the ZIKV genome, 20–23 nt vRNA reads were unequally generated from the positive and negative strands across the ZIKV genome (Fig. 5A, B). A total of 5291 vsiRNAs were identified across the ZIKV genome from the small RNA-seq data, 19.07% of which were mapped to the 5′UTR. In addition, 257 vsiRNAs were identified across the ZIKV genome from the AGO-associated RNA-seq data, 19.77% of which were mapped to the 5′UTR (Supplementary Fig. 5D). The vsiRNAs produced from the SLB (nts 89–111) and cHP regions (nts 132–157) were annotated as vsiR-1 and vsiR-2, respectively (Supplementary Fig. 5E). Northern blotting confirmed the production of vsiR-1 and vsiR-2, which are ~21 nt in length, in MR766- and PRVABC.59-infected SK-N-BE(2) cells (Fig. 5C). RT-qPCR analysis revealed that the knockdown of ILF3 led to strong reductions in vsiR-1 and vsiR-2 in ZIKV MR766- and PRVABC.59-infected SK-N-BE(2) cells (Fig. 5D, Supplementary Fig. 5F). In ZIKV-infected iPSC–NSC-derived brain organoids and Nestin-Cre ILF3 cKO mice, vsiR-1 and vsiR-2 expression levels were strongly repressed compared with those in the control (Fig. 5E, F). Moreover, knockdown of DHX9 also led to downregulation of vsiR-1 and vsiR-2 in ZIKV MR766- and PRVABC.59-infected SK-N-BE(2) cells (Supplementary Fig. 5G), suggesting the common regulatory role of ILF3 and DHX9 in vsiRNA production.

Distribution of vsRNA reads (20–23 nt in size) in the positive- and negative-stranded ZIKV 5′UTR genome from total small RNA seq (A) or AGO-IP small RNA seq (B), respectively. The relative abundances of positive- and negative-stranded vsRNAs are indicated. C Northern Blot validated the expression of vsiR-1 and vsiR-2. n = 2 biological replicates/group. D RT-qPCR analysis of ZIKV vsiR-1 and vsiR-2 expression in ZIKV-infected control or ILF3-knockdown SK-N-BE(2) cells (MOI = 1) at 48 hpi. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. ***p ≤ 0.001. RT-qPCR analysis of ZIKV vsiR-1 and vsiR-2 expression in ZIKV-infected control or ILF3-knockdown brain organoid (E) or Nestin-Cre ILF3-cKO mice (F). Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. *p < 0.05, **p < 0.01. G Western blot showing the interactions with ILF3, DICER, and AGO proteins in ILF3 endogenous IP from mock, ZIKV MR766, or PRVABC.59-infected cells (MOI = 1) at 48 hpi. n = 3 biological replicates/group. H PrismNET illustrates the enrichment of ILF3 at 5′UTR of ZIKV vRNA. Upper panel, two heatmap tracks display sequence response and structure response respectively; red color showing highly attention region. Bottom panel red color illustrates the enrichment position of ILF3 at ZIKV 5′UTR vRNA. I RIP-qPCR shows the enrichment of ILF3, DICER, and AGO2 at SLB and cHP region of ZIKV vRNA in ZIKV MR766-infected SK-N-BE(2) cells. Five primer pairs were used to assess the enrichment across ZIKV vRNA. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. *p < 0.05, **p < 0.01, ***p ≤ 0.001. J RNA EMSA was performed using purified DICER and ILF3 protein as indicated and probed with biotin-labeled ZIKV 5′UTR RNA or ZIKV 5′UTR mutant RNA (M1B). n = 2 biological replicates/group. K DICER processing assay shows vsiRNA cleavage ability with DICER, ILF3 along or DICER and ILF3 together to biotin-labeled ZIKV 5′UTR RNA or ZIKV 5′UTR mutant RNA (M1B) respectively. n = 2 biological replicates/group. Source data are provided as a Source Data file.
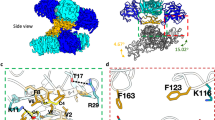
We next investigated how ILF3 could modulate the abundance of vsiRNAs. We purified the ILF3 complex in mock-, MR766- and PRVABC.59-infected SK-N-BE(2) cells and found that ILF3 interacted with DICER in these cells (Fig. 5G). In contrast, we failed to observe an interaction between ILF3 and AGO2 or ZIKV-E among these cells (Fig. 5G). We further applied PrismNET42, a deep learning tool that integrates RNA structure and CLIP-Seq data, to predict cellular protein‒RNA interactions. The saliency map derived from PrismNET analysis was applied to show the sequence and structural signatures of the ILF3-binding site in the ZIKV 5′UTR. The high-response sites in red in the top region represent the putative ILF3 recognition sites in the stem structures of SLA and SLB (nts 63–71; nts 85–92; nts 108–120, respectively) (Fig. 5H). By RNA-binding protein immunoprecipitation (RIP) assay, we validated the enrichment of ILF3, DICER, and AGO2 at the stem structure of SLB but not at the ILF3-negative region (458 and 3354 nt) (Fig. 5I).
We next performed an RNA EMSA experiment to assess the binding sites of ILF3 and DICER at the ZIKV SLB element in vitro. Biotin-labeled ZIKV 5′UTR RNA was used as a WT probe. By targeting three putative ILF3-binding sites in the ZIKV SLB element, we designed three biotin-labeled mutant probes as negative controls. Two mutations, M1A (AAAC(90–93)GGGG) and M1B (UU(99–100)GG), labeled in green-colored font (Supplementary Fig. 5H), disrupted the base pairing of SLB. One mutation, M2A (AAAAA(111–115)GGGGG), labeled in green-colored font (Supplementary Fig. 5H), disrupted the base pairing of the 5′-UAR-flanking stem (UFS). The results revealed that ILF3 enhanced the recruitment of the DICER protein to ZIKV 5′UTR WT RNA (Fig. 5J, Supplementary Fig. 5I). Recruitment was strongly reduced on the mutant probes M1A and M1B, which disrupted putative ILF3 binding sites (Fig. 5J, Supplementary Fig. 5I). The data suggest that the interaction at the SLB element revealed by EMSA is specific. Two ILF3-binding sites, AAAC(90–93) and UU(99–100) in the SLB stem structure, were important for the interaction. Moreover, ILF3 enhanced vsiRNA processing by DICER at the ZIKV SLB element, whereas DICER processing ability was attenuated in the SLB element, which had mutated UU(99–100) ILF3-binding sites (Fig. 5K). Together, these findings suggest that ILF3 interacts with DICER at the ZIKV SLB for vsiRNA cleavage.
vsiR-1 and vsiR-2 inhibited ZIKV replication in vitro
After 48 h of transfection of vsiR-1 and vsiR-2 mimics into ZIKV MR766- and PRVABC.59-infected SK-N-BE(2) cells, we found that ZIKV vRNA copies, protein expression levels, and viral titers were strongly repressed (Fig. 6A–C, Supplementary Fig. 6A).

In ZIKV-infected SK-N-BE(2) cells (MOI = 1, 48 hpi), RT-qPCR, western blot, and virus plaque assay analysis of viral copies (A), ZIKV-E, GAPDH expressions (B) and viral titer (C) by scramble or indicated vsiRNA mimic transfection (MOI = 1, 48 hpi). Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01, ***p ≤ 0.001. D Western blot showing ZIKV-E protein expression by depletion of DICER or AGO2 in ZIKV-infected control or ILF3- overexpressed cells (MOI = 1, 48 hpi). n = 2 biological replicates/group. E RT-PCR showing viral RNA copies in ZIKV-infected control or ILF3 overexpressed cells with or without DICER or AGO2 presence. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. n.s., non-significance, *p < 0.05. In ZIKV-infected control or ILF3-knockdown cells (MOI = 1, 48 hpi), RT-qPCR (F) and WB (G) analysis of viral RNA copies and relative protein expressions by scramble or indicated vsiRNA mimic transfection. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. n.s., non-significance, ***p ≤ 0.001. H, I In ZIKV-infected control or DHX9 knockdown cells (MOI = 1, 48 hpi), RT-qPCR analysis (G) and WB (H) analysis of viral RNA copies and relative protein expressions by scramble or indicated vsiRNA mimic transfection. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. n.s., non-significance, **p < 0.01, ***p ≤ 0.001. J In vitro NS5 polymerase activity assay measured by control or vsiR-1 mimic treatment. Relative intensity of newly synthesized biotin-labeled RNAs was quantified by Image J software. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01. In vitro (K) and in vivo (L) Luciferase activity of ZIKV 5R3 RNA by control or vsiR-1 mimic transfection. Bars, mean ± SD, n = 3 biological replicates/group, two-tailed Student’s t-test. **p < 0.01. Source data are provided as a Source Data file.
To test whether vsiR-1 and vsiR-2 had potential off-target effects, we synthesized two vsiRNA mutant mimics, which mutated two “seed” positions for vsiR-1 and vsiR-2, respectively (Supplementary Fig. 6B). It has been reported that off-target events require matches between the mRNA and sequences that occur at “seed” positions in the targeting oligonucleotide43,44. Thus, off-target effects can occur when the siRNA is partially complementary to one or more cellular mRNAs (in addition to the target). After transfecting WT and mutant mimics into ZIKV-infected SK-N-BE(2) cells, we found that the number of viral proteins, the number of vRNA copies, and the viral titers were reduced in only two vsiRNA WT mimic-transfected cells (Supplementary Fig. 6C, D). The results indicated that there were no detectable off-target effects with vsiR-1 or vsiR-2.
ILF3 antiviral activity is dependent on the DICER/AGO2 pathway
We next asked whether the antiviral function of ILF3 occurs through a DICER- or AGO2-dependent pathway. Compared with that in control cells, ZIKV E protein expression in ILF3-overexpressing 293T cells was strongly inhibited upon ZIKV infection (Fig. 6D, E). However, ablation of DICER and AGO2 attenuated the antiviral activity of ILF3 (Fig. 6D, E).
We next determined whether the antiviral function of the ILF3/DHX9 complex occurs through vsiR-1 or vsiR-2 upon ZIKV infection. We transfected two vsiRNA mimics into MR766- and PRVABC.59-infected control or ILF3- or DHX9-knockdown SK-N-BE(2) cells (Supplementary Fig. 6F–K). We found that increased ZIKV-E protein expression and viral copy numbers in ILF3- (Fig. 6F, G) or DHX9-knockdown cells (Fig. 6H, I) were strongly inhibited in vsiRNA mimic-transfected cells. These results indicate that ILF3 is involved in DICER processing and AGO-mediated vsiRNA maturation, which are important against viral infection.
vsiR-1 inhibits ZIKV replication in vitro through inhibiting ZIKV NS5 polymerase activity and RNA translation
vsiR-1 attracted our attention because of its localization at the 5′UTR SLB, which is upstream of the AUG start codon and critical for genome cyclization12,18. We then investigated the mechanism by which vsiR-1 regulates ZIKV replication. We applied a ZIKV RNA reporter system consisting of a Renilla luciferase ORF with a ZIKV RNA 5′UTR and 3′UTR (5R3). The vsiR-1 mimic greatly inhibited the abundance of newly synthesized RNAs (Fig. 6J), suggesting that vsiR-1 could inhibit NS5 polymerase activity. In cell-free translation systems (Fig. 6K) and upon the transfection of 5R3 RNA (Fig. 6L), we found that the vsiR-1 mimic strongly repressed Renilla luciferase activity, whereas firefly luciferase activity remained unaffected, suggesting that vsiR-1 inhibits translation of the ZIKV RNA reporter.
vsiR-1 inhibits ZIKV replication in vivo and protects mice from ZIKV-induced microcephaly
In scramble mimic-treated control mice, ZIKV infection led to strong reductions in body and brain size/weight, whereas even smaller bodies and smaller brain size/weight were observed in scramble mimic-treated Nestin-Cre ILF3 cKO mice (Fig. 7A, B). In contrast, vsiR-1 mimic treatment in ZIKV-infected control or Nestin-Cre ILF3 cKO mice reversed the reductions in body and brain size/weight (Fig. 7A, B). Moreover, vsiR-1 mimic treatment in ZIKV-infected control or Nestin-Cre ILF3 cKO mice protected the mice from ZIKV-induced clinical symptoms (Fig. 7C). HE staining revealed that after treatment with the vsiR-1 mimic, ZIKV-infected mice displayed normal cytoarchitecture in the hippocampus (Fig. 7D, Supplementary Fig. 7A). Immunofluorescence and viral plaque assays further revealed that after vsiR-1 mimic treatment, ZIKV E protein expression and the ZIKV titer were strongly repressed in control and Nestin-Cre ILF3 cKO mice (Fig. 7E, F), whereas SOX2 protein expression was strongly upregulated compared with that in scramble mimic-treated mice (Fig. 7E). We next analyzed histological sections of the brain, heart, kidney, lung and liver in toxicity test. The HE results revealed no toxicity in vsiR-1-treated animals (Supplementary Fig. 7B). Additionally, we tested cell viability following vsiR-1 or vsiR-2 mimic transfection. A CCK-8 assay revealed that vsiR-1 and vsiR-2 mimics did not affect cell viability (Supplementary Fig. 7C). Taken together, our data indicated that vsiR-1 and vsiR-2 mimics do not have toxic effects.

A Representative image of mock or ZIKV-infected WT or Nestin-Cre ILF3 cKO mice under scramble or vsiR-1 mimics treatment. Brain size (Middle) and brain weight (bottom) were captured at 7 dpi. Bars, mean ± SD, Mock group (n = 3 mice/group), ZIKV infection group (n = 5 mice/group), two-tailed Student’s t-test. *p < 0.05, **p < 0.01. Body weight change (B) and clinical score (C) were measured in mock or ZIKV-infected control or ILF3 cKO mice under scramble or vsiR-1 mimics treatment for 7 days. Bars, mean ± SD, Mock group (n = 3 mice/group), ZIKV infection group (n = 5 mice/group), one-way ANOVA analysis. *p < 0.05, **p < 0.01, ***p < 0.001. D HE staining of the hippocampus brain of indicated mice group (Scale bar, 100 μm). n = 3 mice. E Immunofluorescence showing the relative expression of GFP, SOX2, and ZIKV-E in the indicated mice treatment group. The immunofluorescence intensity was quantified by Image J software. Bars, mean ± SD, n = 3 mice, two-tailed Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001. F Virus titers were quantified by virus plaque assay analysis from ZIKV-infected WT or ILF3-cKO mice under scramble or vsiR-1 mimics treatment. Bars, mean ± SD, n = 5 mice, two-tailed Student’s t-test. n.s., non-significance, **p < 0.01, ***p < 0.001. G Working model. Source data are provided as a Source Data file. G was created with BioRender.com.
Discussion
RNAi is a highly conserved gene-silencing mechanism that exerts antiviral effects on several RNA viruses in mammals22,23,24,45. In well-differentiated cells, the core cellular RNAi pathway components, including DICER and the AGO complex, are frequently involved in the processing of virus-derived vsiRNAs, which leads to the cleavage of cognate vRNAs26. However, host regulators of antiviral RNAi remain poorly understood in undifferentiated neuronal cells with stem-like properties. Recently, a few positive or negative regulators of antiviral RNAi, such as RDO5 and VIR1, have been identified in plants27. However, plants and vertebrates have very different RNAi systems with very divergent functions, though studies in plants provide important hints in the field of RNAi46. These findings suggest that in addition to DICER and AGO, several potential host factors act as essential antiviral RNAi regulators in plants and vertebrates. ADAR1 has been identified as a negative regulator of antiviral RNAi against recombinant Sendai virus in mammalian cells47. Therefore, exploring novel host regulators of antiviral RNAi in undifferentiated neuronal cells is interesting. In this study, we identified a dsRNA-binding protein, ILF3, and an RNA helicase, DHX9, from ZIKV SLB vRNA–host protein profiling, which comprises a positive regulatory complex of antiviral RNA inference in undifferentiated human neuroblastoma cells and iPSC–NSCs (Fig. 7G).
Genomic variation in protein-coding regions is one of the key mechanisms affecting ZIKV properties48,49. However, mutations in protein-coding regions, which make up 95% of the ZIKV genome, are not efficient in elucidating the distinct neuropathogenicity caused by Asian or recent African strains of ZIKV. Multiple studies in well-differentiated cells have proposed critical roles of the ZIKV 5′ and 3′UTRs in viral infection and pathogenesis through host interactions12,30,31. As ZIKV infection is more efficient in primary human neural stem cells than in mature neurons19, how these conserved RNA elements contribute to ZIKV infection and neuropathogenicity in undifferentiated neuronal cells with stem-like properties remains to be determined. Therefore, we profiled vRNA–host protein interactomes at the 5′UTR SLB element of the ZIKV African MR766 and Asian PRVABC.59 strains in human neuroblastoma cells. The results revealed that 61 common host proteins were enriched in RNA-binding and neurodegenerative disorder pathways, indicating essential roles in the replicative life cycle and the neuropathogenicity of ZIKV. Moreover, MR766-specific recruited host factors were enriched in the RNA splicing, processing, and modification pathways, whereas PRVABC.59-specific recruited host factors were enriched in the transcription machinery binding and siRNA binding pathways, which is consistent with findings that RNAi may be crucial in ZIKV-related microcephaly50.
The ILF3 gene encodes diverse isoforms, including nuclear factors 90 and 110 (NF90 and NF110), due to alternative splicing and alternative polyadenylation. NF90 has been reported to modulate HIV, HBV, HCV, and DENV replication33. However, the underlying molecular mechanism of this modulation remains obscure. In this study, we demonstrated that ILF3 exerts an anti-flavivirus function by enhancing DICER processing of vsiRNAs. After ZIKV infection, ILF3 is exported to the cytoplasm. Although nuclear NF90 has been shown to inhibit pri-miRNA cropping through competition with the DROSHA/DGCR8 complex51,52, the function of ILF3 in the cytoplasm, especially in the context of viral infection, remains unknown. We identified a strong interaction between ILF3 and DICER in the ZIKV SLB stem structure. However, we failed to detect the interaction between ILF3 and AGO2 in neuroblastoma cells, though this interaction has been characterized in HeLa, HEK293, and HEK293T cells53,54,55, suggesting a distinct role of ILF3 in undifferentiated neuronal cells. Moreover, the antiviral activity of ILF3 is dependent on the DICER/AGO2 pathway. Together, our findings indicate a central role of ILF3 in the host antiviral RNAi pathway, which may play an essential role in ZIKV-related microcephaly (Fig. 7G).
DHX9, also known as nuclear DNA helicase II (NDH II) and RNA helicase A (RHA), belongs to the SF2 superfamily of nucleic acid unwinding enzymes56. Recently, DHX9 was identified as a human RISC-associated factor via its dsRNA-binding domains (dsRBDs)57,58. DHX9 depletion reduced the association of siRNA with AGO257. During viral infection, the DHX9 protein was enriched with DICER in virus-infected cells. However, the regulatory mechanism and biological significance of this phenomenon under viral infection remain to be further explored. Based on the integrated proteomics analysis from GeneCards (https://www.genecards.org/), it demonstrates that in fetal brain, the protein expression of ILF3 and DHX9 is higher than that in brain, frontal cortex, cerebral cortex, cerebrospinal fluid, and spinal cord. Overall, ILF3 and DHX9 are regularly expressed across various cell types. In this study, we demonstrated the RNA-dependent interactions of ILF3 and DHX9 with the SLB element of ZIKV vRNA. In ZIKV-infected cells, DHX9 may unwind the RNA structure of the SLB element and function as a RISC-loading factor facilitating vsiRNA–RISC complex assembly.
RNA virus infection in mammals leads to robust production of abundant vsiRNAs, whereas the viral suppressor of RNAi (VSR) protein remains defective24. Recent studies have demonstrated genome-wide profiling of ZIKV-derived vsiRNAs and PIWI-interacting ZIKV vsiRNAs from infected mosquito59 to cultured mammal cells, newborn mice, and adult mammals60,61. We understand that small RNA deep sequencing provided sequence annotation of ZIKV vsiRNAs, therefore experimental validation and the regulatory mechanisms still require to be determined. In this study, we experimentally validated two vsiRNAs, vsiR-1, and vsiR-2, which were processed by the ILF3/DHX9/DICER complex from SLB and cHP of ZIKV vRNA in human neuroblastoma cells, respectively. Mechanistically, we demonstrated that vsiR-1 exerts anti-flavivirus activity by inhibiting ZIKV NS5 polymerase activity and RNA translation (Fig. 7G). VsiR-2 is derived from the cHP stem-loop, which encompasses the translational AUG start codon. Similar to how miR-122 functions as an RNA chaperone that guides the formation of a functional internal ribosome entry site (IRES), vsiR-2 may stimulate ZIKV IRES-dependent translation, probably by stabilizing a certain structure of the IRES that is required for initiation62,63,64. Treatment with the vsiR-1 mimic inhibited ZIKV replication in vitro and in vivo and protected mice from ZIKV-induced microcephaly. Together, our findings suggest a novel therapeutic target to combat flavivirus infection.
Methods
Inclusion and ethics statement
The study conforms to the guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised in 1996). All procedures of mice experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Jinan University, number IACUC-20200930-02 and 20210712-07. C57BL/6 WT and ILF3flox/flox mice used in this study were supplied by the Gempharmatech Co., Ltd. (Jiangsu, China) and housed under specific-pathogen-free conditions.
Cell lines
Vero, HEK293T, and SK-N-BE(2) cells were maintained in an incubator at 37 °C with 5% CO2. Vero and HEK293T cells cultured in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO, Grand Island, NY, USA) supplemented with 10% Fetal Bovine Serum (FBS), 100 U/ml penicillin, and 100 mg/ml streptomycin sulfate. SK-N-BE(2) cells were maintained in DMEM/F12 (DMEM/F12) (GIBCO, Grand Island, NY, USA) supplemented with 10% FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin sulfate. C6/36 cells were maintained at 30 °C in MEM (GIBCO, Grand Island, NY, USA) supplemented with 10% heat-inactivated FBS with 1% penicillin and streptomycin (Grand Island, NY, USA) and 1% tryptose phosphate broth (Sigma) (St Louis, MO, USA). Induced pluripotent stem cells (iPSC)-derived Human Neural Stem Cells (iPSC–NSC) were purchased from CIB (Shenzhen, China) and supplemented with NSC culture media (CIB, Shenzhen, China). All cultured cells were regularly tested and found to be mycoplasma-free.
Viruses
ZIKV MR766 (GeneBank: MW143022.1) and PRVABC.59 (GeneBank: KU501215), were gifts from Prof. Jianfeng Dai (Soochow University, China). Virus stocks were propagated in Vero cells after inoculating at a MOI of 0.02 and harvesting supernatants at 4 days post-infection (dpi). The titers of ZIKV stocks were determined by standard plaque assay on Vero cells.
Animal model
To construct neural stem cell-specific knockout ILF3 mice, Control or Nestin-Cre-GFP adenovirus (OBiO Tech, Shanghai, China) were stereotactically injected into subventricular zone (SVZ) of C57BL/6 ILF3flox/flox mice, which contains both NSC and NPCs, at postnatal days 1–265. Animals of similar sex (littermates where possible) were used to control for covariates. At day 8 post-infection, we confirmed conditional loss of ILF3 in Nestin-Cre-GFP adenovirus-injected mice (ILF3-cKO) brain. Subsequently, control or ILF3-cKO mice were stereotactically injected with 2 mg of IFNR-1 blocking antibody (Cat.1021L220, Leinco Technologies, St. Louis, USA) in SVZ and then exposed to PBS or 2 × 105 PFU of ZIKV PRVABC.59 strain, which can cause neurodegeneration in CNS of mice66. For scr or vsiR-1 mimics treatment, 2 nM siRNA mimics (RiboBio, Guangzhou, China) were injected in SVZ of mice brain every 2 days. The body weight, health scores, and mortality of mice were measured daily67,68. The death of Suckling mice was taken as the experimental endpoint.
Plasmids, shRNA, and vsiRNA mimics transfection
The plasmid overexpressing ILF3 and ILF3 shRNA plasmid were both ordered from GeneCopoeia (Guangzhou, China). The NF90 and NF110 WT and Mut plasmids were gifts from Dr. Chen-LingLing’s lab34. DHX9 shRNA plasmid was ordered from Beijing Qingke Biotechnology. ZIKV 5R3 plasmid was constructed with 5R3 RNA template30. After reverse transcription, ZIKV 5R3 fragment was inserted into backbone of pGL3 basic plasmid. ShRNA and vsiRNA mimics were ordered from Ribobio and Genepharma. Lipo 3000 (Invitrogen, USA) or Jet-polyethylenimine (Polyplus, France) were used for transfection. The sequence information is listed in Supplementary Table 2.
Lentivirus package
HEK293T cells were seeded in 10 cm dishes and transfected with sh-ILF3 or negative control empty vector along with the packing plasmids pLVX and pMD2.G using Jet-polyethylenimine (PEI) transfection reagent. After 36 h post-transfection, cell supernatants containing lentivirus were collected and filtrated with 0.45 μm filter. Target cells were infected with filtered viral supernatant with 8 μg/ml polybrene (Biosharp, China).
Lentivirus transduction of brain organoids
The lentiviral particles were first concentrated by the Lenti-PacTM Lentivirus Concentration Solution (GeneCopoeia, China). To transduce organoids, 1 μl of lentivirus (titer was 10–12 genome copies per ml) and 8 μg/ml polybrene were added into each well of a six-well plate containing 2 ml medium and 3-5 organoids. Culture media was changed every 2 days after lentivirus infection69.
RNA-pull-down
SK-N-BE(2) cells were infected with two subtypes of ZIKV, MR766, and PRVABC.59, respectively, for 48 h (MOI = 1). Uninfected cells were used as control group. After crosslinking with formaldehyde, control cells, and infected cells were then harvested. Biotin-labeled RNA oligo that specifically hybridized with 5′SLB of ZIKV vRNAs were used to capture vRNA‒protein complex in the cell lysates. A biotin-labeled scramble RNA oligo was applied as a negative control28.
After overnight incubation, Streptavidin beads were added into protein extracts for purifying vRNA‒protein complex. The purified RNA was collected and analyzed by qRT-PCR to analyze the enrichment of ZIKV vRNA. Protein samples were loaded onto an SDS-PAGE gel and excised for MS analysis. GAPDH blot was added as a loading control in the input lanes to show equal starting material. The proteins associated with SLB pull-down RNA oligo compared to proteins with scramble oligo in each group were identified as the enriched proteins. The enriched proteins in the “virus infection” samples compared to the “mock” control samples were considered as specific SLB-interacting proteins. RNA oligos were synthesized from Beijing Tsingke Biotech Co., Ltd (Beijing, China). The sequence information for RNA-pull-down oligos and RT-PCR primers is listed in Supplementary Table 2.
Western blot
Cells were washed with PBS and dissolved in lysis buffer (50 mM Tris-HCl pH7.4, 300 mM NaCl, 1% TritonX-100, 5 mM EDTA, and 10%glycerol, Protease inhibitor, Roche, cat.04693116001). Protein concentration was measured by Bradford assay (Bio-Rad, Richmond, CA). The samples were then boiled and separated on 10% or 12% SDS-PAGE gels and transferred to polyvinylidene difluoride membrane (Millipore, MA, USA). The membranes were blocked for 1 h in blocking buffer (5% skimmed milk and 0.1% Tween 20 in TBS) at room temperature. After incubation with primary and secondary antibodies, ECL blotting reagents (Millipore) were used for immunoblot detection.
Gene Ontology (GO) enrichment analysis
GO enrichment analysis was performed using a hypergeometric test to assess the over-representation of GO terms in the input gene set relative to the background gene set. Multiple testing correction was performed using the Benjamini-Hochberg method to calculate the false discovery rate (FDR) and the p value adjust. GO terms with p value adjust of less than 0.05 were considered statistically significant.
Confocal microscopy
Briefly, cells cultured on glass-bottomed dishes were fixed with 4% paraformaldehyde for 15 min at room temperature. After washing three times with PBS, cells were permeabilized for 15 min in PBS containing 0.2% Triton X-100, and then blocked with PBS containing 3% BSA. Cells were incubated with primary antibodies (in PBS with 0.1% Triton X-100 and 3% BSA) overnight at 4 °C and then with secondary antibodies (in PBS with 0.1% Triton X-100 and 3% BSA) for 1 h at room temperature, followed by DAPI (Sigma) staining for 10 min. Images were captured using a Leica TCS SP8 confocal microscope.
Nuclear and cytoplasmic fractionation experiment
The nuclear/plasma protein Extraction kit (Cat.PC306, Epizyme, China) was used for nuclear and cytoplasmic fractionation experiment. The fractionated proteins were loaded onto an SDS-PAGE gel for Western Blot analysis. Nuclear marker: Lamin A/C and cytoplasmic marker: GAPDH were used for control.
Co-immunoprecipitation
The cells were harvested by means of ice-cold 1X PBS and lysed with 1 ml of lysis buffer (20 mM Tris-HCl, pH7.4–7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40) containing phosphatases and proteases inhibitors, at 4 °C for 1 h. Flag and IgG antibodies were added into cell lysate as a positive and negative control, respectively, and incubated with protein G magnetic beads for 4 h. After wash, the precipitates were loaded onto an SDS-PAGE gel for Western Blot analysis.
qRT-PCR
Total RNA was extracted with TRIzol reagent (Invitrogen) and reverse transcribed by oligo dT or random primer (Promega). For quantitate miRNA and vsiRNA, small RNAs were reverse transcribed by miRNA First Strand cDNA Synthesis kit (Tailing Reaction, Sangon Biotech). The following Real-time PCR was performed using the Roche LC480 and SYBR RT-PCR Kits (DBI Bio-science, Ludwigshafen, Germany) in a reaction mixture of 20 μl SYBR Green PCR master mix, 1 μl cDNA diluted template, and RNase-free water. qRT-PCR was performed as follows32,70,71. Briefly, Sequence-specific primers for ILF3, DHX9, and GAPDH mRNA were used for reverse transcription. Targeting ZIKV E gene was used to detect ZIKV RNA. Targeting the DENV E gene was used to detect DENV RNA. qRT-PCR was performed on a LightCycler 2.0 (Roche) system with the One Step PrimeScript RT-PCR kit (TaKaRa). Purified RNA transcript was 10-fold serial diluted and used as standards for calculation of RNA copy number32,70,71. The value of GAPDH was used as internal control. And the values in the gene-knockdown cells were quantified relative to the control samples, either in mock group or each ZIKV strain-infected group, respectively. Data are mean ± SD, n = 3 independent biological samples. qRT-PCR primers are listed in Supplementary Table 2.
Small RNA-seq
Small RNAs (<200 bp) were Trizol extracted and excised from the gel. Libraries of small RNAs were prepared according to the manufacturer’s instruction with TruSeq Small RNA Library Preparation Kits (Illumina) and sequenced at Illumina HiSeq 2000. The sequencing reads were mapped to human genome version hg19 (build GRCh37) using Bowtie 2.2.5. All miRNAs were annotated by miRbase (version 22.1)52. The small RNA reads that could not be mapped to the human genome were then mapped to ZIKV genome52. The sequencing reads only allow −2 or +2 nt to be templated by the corresponding genomic sequence at the 3′ ends. The length and location of ZIKV-derived vsiRNAs were calculated using in-house Perl scripts. The counts of complementary pairs of 18–28 nt vsRNAs in each library in different distance categories were calculated using in-house Perl scripts following basic principle23,60.
AGO-associated RNA sequencing
To discriminate functional ZIKV-derived viRNAs from ZIKV genome, we performed AGO-associated RNA sequencing with minor modifications as follows61. ZIKV-infected cells (MOI = 1) were harvested after 48 h infection and lysed by RIPA buffer. Cell lysates were incubated overnight with magnet beads which were conjugated with IgG or AGO2 antibody (CST, AGO2 C34C6, Cat #2897). The antibody has been validated in the IP experiment37,72,73,74. The beads were washed with RIP buffer: 20 mM HEPES, pH 7.5, 150 mM NaCl, 2.5 mM MgCl2•6H2O, 250 mM sucrose, 0.05% (v/v) NP-40 and 0.5% (v/v) Triton X-100 containing 10 U ml−1 of RNasin (Promega), 1 mM DTT, 0.1 mM PMSF and EDTA-free protease inhibitor cocktail. The beads were washed five times each for 5 min at 4 °C. After IP with AGO2 or IgG antibody, the cellular lysates were loaded onto SDS-PAGE for western blot analysis. Immunoprecipitated AGO2 protein complex was validated by western blot. And AGO2-associated RNAs or mouse IgG control-associated RNAs were validated by northern blot. After validation, AGO2-associated RNAs and mouse IgG control-associated RNAs were sent to construct small RNA libraries and sequenced on HiSeq3000. The sequencing reads were mapped to human genome version hg19 (build GRCh37) using Bowtie 2.2.5. All miRNAs were annotated by miRBase52. The small RNA reads that were specifically enriched in AGO2 IP and could not be mapped to the human genome were then mapped to ZIKV genome52,61. The sequencing reads only allow −2 or +2 nt to be templated by the corresponding genomic sequence at the 3′ ends. The length and location of ZIKV-derived vsiRNAs were calculated using in-house Perl scripts. The counts of complementary pairs of 18-28nt vsRNAs in each library in different distance categories were calculated using in-house Perl scripts following basic principles23,60.
Northern blot
Total RNA was extracted by Trizol (Life Technologies). Ten to thirty micrograms of total RNA or pull-downed RNA were run on 20% (w/v) acrylamide/8 M urea gels and then transferred onto Hybond-N membranes. After transfer and UV crosslink, Biotin-labeled RNA probes that reverse complement to the positive strand of targeted vsiRNAs were hybridized with membrane in Hybridization Buffer at 37 °C overnight29. Biotin-labled RNA Probes that target ZIKV SLB and cHP-derived-vsiRNA are named RNA probes vsiR-1 and vsiR-2, respectively. U6 snRNA detection served as an internal control in Northern blot.
Pulled-down RNAs by biotinylated oligonucleotides were separated by 15% (w/v) acrylamide/8 M urea gels. After being transferred and crosslinked onto Hybond-N membranes, the pulled-down RNAs were hybridized with a digoxin (DIG)-labeled RNA probe, which was specifically hybridized with 5′SLB of ZIKV vRNAs. After wash steps, the membranes were visualized by DIG nucleic acid detection kit (Roche).
AGO2-associated RNAs or a mouse IgG control-associated RNAs were separated by 15% (w/v) acrylamide/8 M urea gels, and hybridized with biotin-labeled RNA probe. Detection of let-7a miRNA in AGO2 IP was demonstrated as a positive control75. After washing with 2× SSC buffer five times, the membrane was visualized by LightShift Chemiluminescent RNA EMSA Kit (Thermo). RNA probes were synthesized from Beijing Tsingke Biotech Co., Ltd (Beijing, China). The sequence information is listed in Supplementary Table 2.
PrismNET analysis
PrismNET is a deep learning tool integrating RNA structure data and CLIP‒Seq, predicting cellular protein‒RNA interactions42. Briefly, we followed the workflow of the PrismNet web server to analyze ILF3 putative binding sites at ZIKV vRNAs76. Sequence and Structure modes were selected as input types. After processing the web server, the saliency maps, including binding sites and sequence-structure integrative motifs, can be obtained. The saliency map includes two portions: the top sequence region presents all the nucleotides; whereas the bottom structure region displays the icSHAPE score.
RNA EMSA
RNA EMSA was performed using indicated recombinant proteins51. Biotin-labeled ZIKV 5′UTR WT or mutant RNA were synthesized from Beijing Tsingke Biotech Co., Ltd (Beijing, China) or in vitro transcribed by T7 biotin-labeled transcription kit (RIBOBIO, China) from ZIKV 5R3 T7-promoter plasmids or synthesized from Beijing Tsingke Biotech Co., Ltd (Beijing, China). RNA/protein complexes were run on 20% (w/v) acrylamide/8 M urea gels. After hybridized on Hybond-N membranes, RNA/protein complexes were visualized by LightShift Chemiluminescent RNA EMSA Kit (Thermo). The sequence information of biotin-labeled ZIKV WT or mutant RNA is listed in Supplementary Table 2.
NS5 RNA polymerase activity assay
The NS5 RNA polymerase activity assay was modified as follows77,78,79. The 5′UTR RNA templates were in vitro transcribed from ZIKV 5R3 plasmids containing T7 promoter. One microgram of ZIKV RNA template and 500 ng of NS5 full-length protein were added to a reaction buffer containing 50 mM HEPES (pH 7.3), 0.4 mM dithiothreitol (DTT), 3 mM MgCl2 and 0.4 mM of ATP, GTP, CTP and biotinylated-UTP ribonucleotides. The ribonucleotide UTP was labeled in an equimolar ratio of biotinylated-UTP for quantification of newly replicated RNA. The reaction mixtures were incubated for 1 h at 30 °C with control mimics or viR-1 mimics, respectively. Then the reactions were stopped with addition of EDTA at a final concentration of 10 mM. The newly synthesized RNA products were biotin-labeled, and separated on 20% (w/v) acrylamide/8 M urea gels. After hybridization on Hybond-N membranes, RNAs were visualized by LightShift Chemiluminescent RNA EMSA Kit (Thermo Fisher, USA). The image was quantified using ImageQuant software.
In vitro translation assay
The capped 5R3 and Fluc RNAs were generated using an in vitro T7 RNA Synthesis Kit (Ribobio). Capped RNA (5R3) combined with Fluc RNA (molar ratio 4:1) were subjected to the retic lysate IVT system (Invitrogen, AM1200) together with control mimics or viR-1 mimics, respectively. Then an in vitro translation reaction was performed as per the manual instructions. Luciferase activities were detected using a Dual-Luciferase reporter assay kit (Promega, E1910).
hiPSC-NSC-derived brain organoids
hiPSC-derived human brain organoid is a well-established model to explore the effect of ZIKV infection in the developing brain tissue36,37,38. hiPSC-NSC-derived brain organoids were grown according to protocol adapted from Lancaster and Knoblich80,81. For organoid culture media, we used commercial kit (STEMdiffTM Cerebral Organoid kit, Cat. 08570) and followed its protocol. Briefly, the embryoid bodies (EBs) were formed from human iPSCs. On day 5, the EBs were transferred to a 24-well plate for neural ectoderm induction. On day 7, neuroectodermal tissues are then transferred to Matrigel droplets, containing 4 ng/ml FGF, 4 ng/ml EGF, and 50 mM Rho-associated protein kinase (ROCK) inhibitor. After 3 days, neuroepithelial bud expands and grows in a 6 cm dish. On day 10, Matrigel droplets are transferred to the spinning bioreactor.
One-to-five-week-old brain organoids closely mimic the fetus period which is most vulnerable to ZIKV infection36,37,82. Human brain organoids loss typical structure by 6 days after viral infection and undergo an overt shrinkage at day 15 after viral infection36,37. Herein, the brain organoids on day 7 were used for ZIKA viral infection, and harvested at day 15. The organoids were initially infected with sh-NC or sh-ILF3 lentivirus at day 5 (titer was 10–12 genome copies per ml). From day 7, control or ILF3-knockdown brain organoids were infected with ZIKV (MOI = 1) every 3 days. The brain organoids were harvested on day 15. The organoid sections were stained for neural progenitor’s marker SOX2, Neurons’ marker TuJ1, and ZIKV E protein.
Behavioral assessment
The health score of mice was measured as follows67,68: (1) Complete health score was 0; (2) 1 point for no vitality or loss of appetite; (3) 2 points for slight weakness and weakness in the limbs; (4) paralysis of one leg and one foot, claudication score 3 points; (5) Total paralysis of hind or forelimb 4 points; (6) Dying or dying is 5 points. The status of mice was detected daily, and the score was recorded.
Quantification and statistical analysis
All results are presented as the mean ± SEM or SD. Statistical analysis was carried out using Student’s t-test (two-tailed unpaired) for two groups, ANOVA for multi-group comparison, and the Kaplan–Meier method for mouse survival as indicated, all using GraphPad Prism 10 unless otherwise noted. The statistical analysis of viral titers and vRNA copies has been performed a log10-transformation, and plotted in logarithmic scale. For all tests, p < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001). All experiments were repeated independently at least two times. The experimental groups were randomly assigned. Experimental sample size and replicate numbers (n) are indicated in the figure legends. Data acquisition and analysis were performed randomly. No data were excluded from the analysis. Blinding was applicable in this study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data used to generate figures in this paper are provided in the Supplementary Information and Source Data file. Supplementary information is provided in this paper. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD050964, PXD048671, PXD054904. The Small RNA-seq, IgG, and AGO-associated small RNA seq data are available in NCBI’s SRA database (https://www.ncbi.nlm.nih.gov/sra) with accession numbers: SRR30869521, SRR30869522, SRR30869525, SRR30869526, SRR30010836, SRR30010837, SRR30010838, SRR30010839. Source data are provided with this paper.
References
Haddow, A. D. et al. Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage. PLoS Negl. Trop. Dis. 6, e1477 (2012).
Dick, G. W., Kitchen, S. F. & Haddow, A. J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 46, 509–520 (1952).
Gabriel, E. et al. Recent Zika virus isolates induce premature differentiation of neural progenitors in human brain organoids. Cell Stem Cell 20, 397–406.e5 (2017).
Aubry, F. et al. Recent African strains of Zika virus display higher transmissibility and fetal pathogenicity than Asian strains. Nat. Commun. 12, 916 (2021).
Bohm, E. K. et al. Zika virus infection of pregnant Ifnar1(-/-) mice triggers strain-specific differences in fetal outcomes. J. Virol. 95, e0081821 (2021).
Weaver, S. C. et al. Zika virus: history, emergence, biology, and prospects for control. Antivir. Res. 130, 69–80 (2016).
de Oliveira, W. K. et al. Infection-related microcephaly after the 2015 and 2016 Zika virus outbreaks in Brazil: a surveillance-based analysis. Lancet 390, 861–870 (2017).
Yadav, P. D. et al. Detection of Zika virus disease in Thiruvananthapuram, Kerala, India 2021 during the second wave of COVID-19 pandemic. J. Med. Virol. 94, 2346–2349 (2022).
Giron, S. et al. Vector-borne transmission of Zika virus in Europe, southern France, August 2019. Eur. Surveill. 24, 1900655 (2019).
WHO. WHO to identify pathogens that could cause future outbreaks and pandemics (2022).
Song, Y., Mugavero, J., Stauft, C. B. & Wimmer, E. Dengue and Zika virus 5’ untranslated regions harbor internal ribosomal entry site functions. mBio 10, e00459–19 (2019).
Li, P. et al. Integrative analysis of Zika virus genome RNA structure reveals critical determinants of viral infectivity. Cell Host Microbe 24, 875–886.e5 (2018).
Li, X. D., Deng, C. L., Yuan, Z. M., Ye, H. Q. & Zhang, B. Different degrees of 5’-to-3’ DAR interactions modulate Zika virus genome cyclization and host-specific replication. J. Virol. 94, e01602–e01619 (2020).
Bernardo-Menezes, L. C. et al. An overview of Zika virus genotypes and their infectivity. Rev. Soc. Bras. Med. Trop. 55, e02632022 (2022).
Lee, E. et al. Structures of flavivirus RNA promoters suggest two binding modes with NS5 polymerase. Nat. Commun. 12, 2530 (2021).
Choi, K. H. The role of the stem-loop A RNA promoter in flavivirus replication. Viruses 13, 1107 (2021).
Alvarez, D. E., Lodeiro, M. F., Luduena, S. J., Pietrasanta, L. I. & Gamarnik, A. V. Long-range RNA-RNA interactions circularize the dengue virus genome. J. Virol. 79, 6631–6643 (2005).
Liu, Z. Y. et al. Viral RNA switch mediates the dynamic control of flavivirus replicase recruitment by genome cyclization. eLife 5, e17636 (2016).
Miner, J. J. & Diamond, M. S. Understanding how Zika virus enters and infects neural target cells. Cell Stem Cell 18, 559–560 (2016).
Guo, Z., Li, Y. & Ding, S. W. Small RNA-based antimicrobial immunity. Nat. Rev. Immunol. 19, 31–44 (2019).
Samuel, G. H., Adelman, Z. N. & Myles, K. M. Antiviral immunity and virus-mediated antagonism in disease vector mosquitoes. Trends Microbiol. 26, 447–461 (2018).
Maillard, P. V. et al. Antiviral RNA interference in mammalian cells. Science 342, 235–238 (2013).
Li, Y., Lu, J., Han, Y., Fan, X. & Ding, S. W. RNA interference functions as an antiviral immunity mechanism in mammals. Science 342, 231–234 (2013).
Qiu, Y. et al. Human virus-derived small RNAs can confer antiviral immunity in mammals. Immunity 46, 992–1004.e1005 (2017).
Maillard, P. V., van der Veen, A. G., Poirier, E. Z. & Reis e Sousa, C. Slicing and dicing viruses: antiviral RNA interference in mammals. EMBO J. 38, e100941 (2019).
Wang, J. & Li, Y. Current advances in antiviral RNA interference in mammals. FEBS J. https://doi.org/10.1111/febs.16728 (2023).
Liu, S. et al. Identification of positive and negative regulators of antiviral RNA interference in Arabidopsis thaliana. Nat. Commun. 13, 2994 (2022).
Song, C. et al. Circular RNA Cwc27 contributes to Alzheimer’s disease pathogenesis by repressing Pur-alpha activity. Cell Death Differ. 29, 393–406 (2022).
Akiyama, Y., Kharel, P., Abe, T., Anderson, P. & Ivanov, P. Isolation and initial structure-functional characterization of endogenous tRNA-derived stress-induced RNAs. RNA Biol. 17, 1116–1124 (2020).
Zhang, S. et al. Comparison of viral RNA-host protein interactomes across pathogenic RNA viruses informs rapid antiviral drug discovery for SARS-CoV-2. Cell Res. 32, 9–23 (2022).
Ooi, Y. S. et al. An RNA-centric dissection of host complexes controlling flavivirus infection. Nat. Microbiol. 4, 2369–2382 (2019).
Barbier, J. et al. An NF90/NF110-mediated feedback amplification loop regulates dicer expression and controls ovarian carcinoma progression. Cell Res. 28, 556–571 (2018).
Castella, S., Bernard, R., Corno, M., Fradin, A. & Larcher, J. C. Ilf3 and NF90 functions in RNA biology. Wiley Interdiscip. Rev. RNA 6, 243–256 (2015).
Li, X. et al. Coordinated circRNA Biogenesis and Function with NF90/NF110 in Viral Infection. Mol. Cell 67, 214–227.e217 (2017).
York, A. Oiling the Flavivirus replication machinery. Nat. Rev. Microbiol. 16, 455 (2018).
Watanabe, M. et al. Self-organized cerebral organoids with human-specific features predict effective drugs to combat Zika virus infection. Cell Rep. 21, 517–532 (2017).
Slonchak, A. et al. Zika virus noncoding RNA cooperates with the viral protein NS5 to inhibit STAT1 phosphorylation and facilitate viral pathogenesis. Sci. Adv. 8, eadd8095 (2022).
Sutarjono, B. Can we better understand how Zika leads to microcephaly? A systematic review of the effects of the Zika virus on human brain organoids. J. Infect. Dis. 219, 734–745 (2019).
Anfasa, F. et al. Phenotypic differences between Asian and African lineage Zika viruses in human neural progenitor cells. mSphere 2, e00292-17 (2017).
Simonin, Y., van Riel, D., Van de Perre, P., Rockx, B. & Salinas, S. Differential virulence between Asian and African lineages of Zika virus. PLoS Negl. Trop. Dis. 11, e0005821 (2017).
Nakayama, E. et al. Neuroinvasiveness of the MR766 strain of Zika virus in IFNAR-/- mice maps to prM residues conserved amongst African genotype viruses. PLoS Pathog. 17, e1009788 (2021).
Sun, L. et al. Predicting dynamic cellular protein-RNA interactions by deep learning using in vivo RNA structures. Cell Res. 31, 495–516 (2021).
de Souza, N. Off-targets in RNAi screens. Nat. Methods 11, 480 (2014).
Franceschini, A. et al. Specific inhibition of diverse pathogens in human cells by synthetic microRNA-like oligonucleotides inferred from RNAi screens. Proc. Natl. Acad. Sci. USA 111, 4548–4553 (2014).
Ding, S. W., Han, Q., Wang, J. & Li, W. X. Antiviral RNA interference in mammals. Curr. Opin. Immunol. 54, 109–114 (2018).
Agrawal, N. et al. RNA interference: biology, mechanism, and applications. Microbiol. Mol. Biol. Rev. 67, 657–685 (2003).
Uhl, S. et al. ADAR1 biology can hinder effective antiviral RNA interference. J. Virol. 97, e0024523 (2023).
Yuan, L. et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 358, 933–936 (2017).
Agrelli, A., de Moura, R. R., Crovella, S. & Brandao, L. A. C. Mutational landscape of Zika virus strains worldwide and its structural impact on proteins. Gene 708, 57–62 (2019).
Zeng, J. et al. The Zika virus capsid disrupts corticogenesis by suppressing dicer activity and miRNA biogenesis. Cell Stem Cell 27, 618–632.e9 (2020).
Sakamoto, S. et al. The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol. Cell. Biol. 29, 3754–3769 (2009).
Grasso, G. et al. NF90 modulates processing of a subset of human pri-miRNAs. Nucleic Acids Res. 48, 6874–6888 (2020).
Meister, G. et al. Identification of novel argonaute-associated proteins. Curr. Biol. 15, 2149–2155 (2005).
Hock, J. et al. Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells. EMBO Rep. 8, 1052–1060 (2007).
Grasso, G. et al. NF90 interacts with components of RISC and modulates association of Ago2 with mRNA. BMC Biol. 20, 194 (2022).
Fuller-Pace, F. V. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 34, 4206–4215 (2006).
Robb, G. B. & Rana, T. M. RNA helicase A interacts with RISC in human cells and functions in RISC loading. Mol. Cell 26, 523–537 (2007).
Fu, Q. & Yuan, Y. A. Structural insights into RISC assembly facilitated by dsRNA-binding domains of human RNA helicase A (DHX9). Nucleic Acids Res. 41, 3457–3470 (2013).
Varjak, M. et al. Characterization of the Zika virus induced small RNA response in Aedes aegypti cells. PLoS Negl. Trop. Dis. 11, e0006010 (2017).
Xu, Y. P. et al. Zika virus infection induces RNAi-mediated antiviral immunity in human neural progenitors and brain organoids. Cell Res. 29, 265–273 (2019).
Zeng, J. et al. Functional mapping of AGO-associated Zika virus-derived small interfering RNAs in neural stem cells. Front. Cell. Infect. Microbiol. 11, 628887 (2021).
Goergen, D. & Niepmann, M. Stimulation of Hepatitis C Virus RNA translation by microRNA-122 occurs under different conditions in vivo and in vitro. Virus Res. 167, 343–352 (2012).
Conrad, K. D. et al. MicroRNA-122 dependent binding of Ago2 protein to hepatitis C virus RNA is associated with enhanced RNA stability and translation stimulation. PLoS ONE 8, e56272 (2013).
Schult, P. et al. microRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat. Commun. 9, 2613 (2018).
Ming, G. L. & Song, H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702 (2011).
Manangeeswaran, M., Ireland, D. D. & Verthelyi, D. Zika (PRVABC59) Infection Is Associated with T cell Infiltration and Neurodegeneration in CNS of Immunocompetent Neonatal C57Bl/6 Mice. PLoS Pathog. 12, e1006004 (2016).
Li, S. et al. Zika virus fatally infects wild type neonatal mice and replicates in central nervous system. Viruses 10, 49 (2018).
Hayashida, E. et al. Zika virus encephalitis in immunocompetent mice is dominated by innate immune cells and does not require T or B cells. J. Neuroinflamm. 16, 177 (2019).
Wang, M. et al. Morphological diversification and functional maturation of human astrocytes in glia-enriched cortical organoid transplanted in mouse brain. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02157-8 (2024).
Wang, W. et al. Zika virus infection induces host inflammatory responses by facilitating NLRP3 inflammasome assembly and interleukin-1beta secretion. Nat. Commun. 9, 106 (2018).
Liu, Z. Y. et al. Novel cis-acting element within the capsid-coding region enhances flavivirus viral-RNA replication by regulating genome cyclization. J. Virol. 87, 6804–6818 (2013).
Li, Q. et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 382, 1199–1207 (2020).
Luo, Y. Y. et al. Hsa_Circ_0098181 suppresses hepatocellular carcinoma by sponging miR-18a-3p and targeting PPARA. Front. Pharmacol. 13, 819735 (2022).
Miyazaki, Y., Du, X., Muramatsu, S. & Gomez, C. M. An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNA1A second cistron. Sci. Transl. Med. 8, 347ra394 (2016).
Boudreau, R. L. et al. Transcriptome-wide discovery of microRNA binding sites in human brain. Neuron 81, 294–305 (2014).
Xu, Y. et al. PrismNet: predicting protein-RNA interaction using in vivo RNA structural information. Nucleic Acids Res. 51, W468–W477 (2023).
Sacramento, C. Q. et al. The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci. Rep. 7, 40920 (2017).
Sariyer, I. K. et al. Suppression of Zika virus infection in the brain by the antiretroviral drug rilpivirine. Mol. Ther. 27, 2067–2079 (2019).
Zhao, B. et al. Structure and function of the Zika virus full-length NS5 protein. Nat. Commun. 8, 14762 (2017).
Lancaster, M. A. & Knoblich, J. A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 9, 2329–2340 (2014).
Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
Qian, X. et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165, 1238–1254 (2016).
Acknowledgements
We thank Dr. Jianfeng Dai for providing ZIKV MR766 and PRVABC.59 strains; Dr. Qiangfeng Cliff Zhang’s Lab for providing PrismNET analysis42 and ZIKV 5R3 RNA30; Dr. Wenbiao Wang for providing technique support; Dr. Chen-LingLing’s Lab for providing NF90 and NF110 WT and Mut plasmids34. We deeply thank Prof. Jianguo Wu for his effusive dedication and constructive guidance. This work was supported by the National Natural Science Foundation of China (82072834 to X.C., 32400132 to Z.L.), Guangdong Natural Science Foundation (2023A1515010318, 2021A1515011065 to X.C.).
Author information
Authors and Affiliations
Contributions
L. Zhiwei, Y. Yu, J. Dai, and X. Chen conceived the study. L. Zhiwei, Y. Yu, and X. Chen designed the main study and experiments. L. Zhiwei, Y. Gu, Y. Liu, W. Chen, P. Pan, Z. Yue, J. Ruan, L. Zhu, G. Li, and X. Xia performed cellular and animal experiments. H. Liu, L. Zhiwei, X. Lu, Z. Chen, and X. Chen performed Small RNA-seq and bioinformatic analysis. L. Zhiwei, L. Zhou, and Y. Liu constructed related plasmids. L. Zhiwei, Y. Gu, Y. Yu, and X. Chen interpreted data and drafted figures. L. Zhiwei, Y. Gu, J. Dai, Y. Yu, and X. Chen prepared the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sumana Sanyal, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lei, Z., Gu, Y., Liu, Y. et al. Identification of antiviral RNAi regulators, ILF3/DHX9, recruit at ZIKV stem loop B to protect against ZIKV induced microcephaly. Nat Commun 16, 1991 (2025). https://doi.org/10.1038/s41467-025-56859-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56859-x
