
Abstract
The Ebola virus, a member of the Filoviridae family, causes severe hemorrhagic fever in humans. Filamentous virions contain a helical nucleocapsid responsible for genome transcription, replication, and packaging into progeny virions. The nucleocapsid consists of a helical nucleoprotein (NP)–viral genomic RNA complex forming the core structure, to which VP24 and VP35 bind externally. Two NPs, each paired with a VP24 molecule, constitute a repeating unit. However, the detailed nucleocapsid structure remains unclear. Here, we determine the nucleocapsid-like structure within virus-like particles at 4.6 Å resolution using single-particle cryo-electron microscopy. Mutational analysis identifies specific interactions between the two NPs and two VP24s and demonstrates that each of the two VP24s in different orientations distinctively regulates nucleocapsid assembly, viral RNA synthesis, intracellular transport of the nucleocapsid, and infectious virion production. Our findings highlight the sophisticated mechanisms underlying the assembly and functional regulation of the nucleocapsid and provide insights into antiviral development.
Similar content being viewed by others


Introduction
The Ebola virus (EBOV), which belongs to the Filoviridae family in the Mononegavirales order, causes fatal hemorrhagic fever in both humans and non-human primates1. Although recent advancements in the availability of Food and Drug Administration-approved vaccines and antibody drugs exist2, their therapeutic targets are limited to glycoproteins (GP), which are responsible for virus entry into target cells. Therefore, exploring the structures and functions of other viral proteins and understanding the molecular mechanisms underlying EBOV replication are required for the development of antiviral therapeutics.
The EBOV nucleocapsid, responsible for the transcription and replication of non-segmented, single-stranded, negative-sense genomic viral RNA (vRNA), is composed of a nucleoprotein (NP), a viral protein (VP24), a VP30, a VP35, and polymerase L, in which a helical NP–vRNA complex serves as the core structure1. VP24 and VP35, essential for the formation of nucleocapsid-like structures (NCLSs)3,4, bind externally to the helical NP–vRNA helix5,6, forming protrusions on its surface7,8,9,10. VP35, which is analogous to the P protein of other mononegaviruses11, is a polymerase cofactor that bridges the NP–vRNA helix and L during vRNA synthesis12,13,14,15. VP35 also binds to nascent NPs to prevent oligomerization before forming the helical NP–vRNA complex16,17. Unlike VP35 that bridges between NP and L, the molecular properties of VP24 in NCLS assembly remain poorly understood due to the complexity of its functions and the limited structural information available. Currently, it is established that VP24 suppresses transcription and replication of the vRNA18,19. Additionally, it is essential for the intracellular transport of nucleocapsids and, thus, genome packaging into progeny virion20,21. This suggests that VP24 may switch nucleocapsid functions from RNA synthesis to vRNA transport for virion formation. However, the underlying structural basis for its regulatory functions remains elusive because of the lack of knowledge regarding the detailed nucleocapsid structure.
Previously, using single-particle cryo-electron microscopy (cryo-EM), we determined the high-resolution structure of the NP–RNA complexes from EBOV and its close relative Marburg virus (MARV), at 3.6 Å and 3.1 Å resolution, respectively, and identified essential interactions on NP for the formation of the helical NP–RNA complex and for the transcription and replication of the vRNA22,23. In addition, studies using cryo-electron tomography (cryo-ET) determined the nucleocapsid structure in authentic EBOV particles and the NCLS structure in virus-like particles (VLPs) at 9.1 Å and 7.3 Å resolution, respectively8. These studies generated a model of the nucleocapsid structure, in which a repeating unit consists of two NPs with two protruding VP24s in different orientations8. However, the absence of a high-resolution structure of the nucleocapsid hinders our understanding of the structural characteristics and functions of the EBOV nucleocapsid assembly.
This study reports the in situ structure of EBOV NCLS in VLPs at the 4.6 Å resolution. These results advance our understanding of the molecular mechanisms underlying EBOV replication and contribute to the development of antiviral therapeutics.
Results
Cryo-EM structure of the EBOV NCLS in VLPs
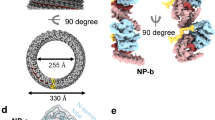
To better understand the molecular interactions among the components that regulate the assembly and function of the EBOV nucleocapsid, we attempted to determine the high-resolution structure of the EBOV NCLS within VLPs using single-particle cryo-EM, which differs from previous studies using cryo-ET7,8. Consequently, we co-expressed NP, VP24, VP35, VP40, and GP in a human cell line, HEK293T (human embryonic kidney) cells, and produced NCLS-containing VLPs that were morphologically indistinguishable from authentic EBOV particles24. Initially, we employed a single-particle analysis, imposing helical symmetry, and determined the helical NCLS structure inside the VLP at an overall resolution of 7.5 Å (Fig. 1a, Supplementary Fig. 1). Our cryo-EM map shows a good fit with the cryo-ET maps of the NCLS in the VLP and the nucleocapsid in authentic virion8 (Fig. 1b). It maintains a consistent helical parameter (Supplementary Table 1), suggesting that our cryo-EM structure accurately represents the native conformation of the EBOV nucleocapsids. The structure of the NP–RNA complex located within the inner layer of the NCLS is analogous to the high-resolution structure as reported previously22 (Supplementary Fig. 2). The external protrusions on the helical NP–RNA complex were presumed to be two VP24 molecules, as also described by previous cryo-ET analysis8. We also detected additional map densities outside the VP24 protrusions that remained uncharacterized, as reported in cryo-ET analysis8 (Fig. 1c, shown in dark gray). Next, to identify the molecular interactions within the nucleocapsid, we focused on its repeating unit, composed of two NPs and two VP24s. We achieved a higher-resolution structure by expanding the helical symmetry and further processing the repeating unit (Supplementary Fig. 1). The resulting map, with a 4.6 Å resolution, facilitated the identification of the NP–RNA complex that constitutes the inner core of the nucleocapsid and the positioning of VP24s on the NP–RNA complex (Fig. 1c, d). Unfortunately, we could not identify the outer map of the VP24 protrusions owing to the limited resolution (Fig. 1c, shown in dark gray). The repeating unit of the NCLS was composed of two adjacent copies of NP-associating RNA and two differently oriented VP24 molecules (Fig. 1c, d): NP-1 (shown in pink) featured a large protrusion of additional density on the outside of the core, which included one copy of VP24-1 (shown in lime green), and NP-2 (shown in beige) had a smaller protrusion that contained one copy of VP24-2 (shown in violet).

a Helical reconstruction of the NCLS complex at 7.5 Å resolution, with RNA colored in red, nucleoproteins (NP-1 and NP-2) in pink and beige, and viral proteins VP24-1 and VP24-2 in lime green and violet. b Overlay of the cryo-electron tomography (cryo-ET) map of the VLP (virus-like particle) (EMD-3871 [https://www.ebi.ac.uk/emdb/EMD-3871]) and the authentic virus (EMD-3873 [https://www.ebi.ac.uk/emdb/EMD-3873]) compared with the cryo-EM map obtained in this study, showing structural congruence. c Symmetry-expanded and focus-refined cryo-EM map at 4.6 Å resolution showing the arrangement of NPs and VP24 molecules, with VP24-1 in lime green and VP24-2 in violet alongside NPs (NP-1 in pink and NP-2 in beige). Adjacent repeating units are shown in light gray; areas, where the model could not be assigned, are shown in dark gray. d Atomic model of the NCLS. The color scheme for NPs and VP24s matches that of Fig. 1a. e Comparison of the NP structures derived from this study with the previously published purified NP structure (PDB ID: 5Z9W), shown in orange. The left image shows a side view, while the inset displays a 90° rotated top view with the cryo-EM map from this study. The arrow indicates a local structural difference between these NP structures.
The overall architecture of the NP core closely resembled that of the purified NP–RNA complex (Fig. 1e). The root-mean-square deviation (RMSD) of backbone atoms was 1.05 Å after superposition of both NP-1 and NP-2 to the NP protomer of purified NP–RNA complex (Protein Data Bank (PDB) ID: 5Z9W)22. Notably, a marginal difference was observed in the local structure of the N-terminal lobe domains of both NP-1 and NP-2 (Fig. 1e indicated by arrows). This observation suggests that the NPs adjust their position at the inter-rung interface after binding to VP24 and VP35, as also indicated in previous cryo-ET study8.
NP–VP24 interaction in the NCLS
The NP–VP24 interaction involves three distinct interfaces. One interface is formed via the interaction between the C-terminal helix of NP-1 and a loop on VP24-1 (Fig. 2a, b), whereas the other two interfaces primarily rely on electrostatic interactions between the N-terminal lobes of NPs and VP24s (Fig. 2c–e). The 169–173 loop region of VP24, in which the YxxL motif is included, is recognized as a crucial site for NP–VP24 interactions and intracellular transport of nucleocapsids21,25. Our structure revealed an interaction between the C-terminal α-helix 18 of NP-1 and the 169–173 loop of VP24-1 (Fig. 2b). Notably, the loop region exhibited considerable flexibility, and the local resolution of this region was limited (Supplementary Fig. 3A, B); hence, the precise positioning of each amino acid in this loop could not be determined.

a Atomic model of the NCLS showing the helical repeating unit containing two NPs (NP-1 in pink and NP-2 in beige) and two VP24 molecules (VP24-1 in lime green and VP24-2 in violet). b Close-up view of the interface between the 169–173 loop of VP24-1 and the α18 helix of NP-1, with the mesh representing the cryo-EM map. c Electrostatic surface potential maps of the NCLS from two orientations, 180° apart. The maps show the distribution of charges around NP-1 and NP-2, with VP24-1 and VP24-2, respectively. Electrostatic surface potential was calculated using the Delphi web-server57. Scale from −5 (red) to +5 kcal mol−1 e−1 (blue). d Close-up view of the NP-1 and VP24-1 interface, detailing electrostatic contacts. Residues possibly involved in interactions labeled and presented in stick representation. e Close-up view of the NP-2 and VP24-2 interface.
Examining the other interfaces mediated via the N-terminal lobe of the NPs and each VP24 subunit, electrostatic interactions emerge as the predominant driving force for their binding (Fig. 2c). In our structural model, R59 of VP24-1 is inserted into the negatively charged region of NP-1 as shown Fig. 2c, which is mainly composed of residues E61 and E141, forming a possible salt bridge (Fig. 2d). Additionally, H196 and R202 of NP-1 are close to VP24-1 within an interactable distance. Examining the adjacent pair of NP-2–VP24-2, it is observed that R132 of NP-2 is located where it could interact with the negatively charged region of VP24-2 (Fig. 2c, e). Notably, K148 of VP24-2 is inserted into the negatively charged region comprising residues E61 and E141 of NP-2, resembling the positioning of R59 in the adjacent VP24-1. This observation suggested that the negatively charged surface of the NP N-terminal lobe is important for binding both VP24s.
VP24–VP24 interactions in the NCLS
Compared to our two VP24 models, a previously reported crystal structure of the EBOV VP24 dimer26 showed substantial differences in arrangement, potentially owing to its crystal packing (Supplementary Fig. 3C). In addition, the crystal structure shows different conformations with RMSDs of 3.66 Å for VP24-1 and 2.35 Å for VP24-2 (Supplementary Fig. 3C). Thus, the VP24 dimer in the crystal packing does not represent the arrangement and conformation within the NCLS; however, it may instead depict an interferon antagonist form27. Notably, the two VP24s observed in our NCLS structure exhibit a substantial difference in their conformation at the main chain level, potentially owing to their association with each NP, with RMSD of 1.37 Å (Supplementary Fig. 3D).
In our model, no apparent inter-unit interaction, such as that between VP24-1 in a repeating unit and VP24-2 in a neighboring repeating unit, is observed. This is because of the distinct positioning of each VP24 unit, separated from those in neighboring units. Notably, an atomic model could not be constructed for the N- and C-termini (1–9 a.a. and 232–251 a.a. for VP24-1, and 1–27 a.a. and 232–251 a.a. for VP24-2) (Supplementary Fig. 3A, E), leaving the possibility of interactions via this region open. In contrast, there seems to be an intra-unit interaction between VP24-1 and VP24-2 (Fig. 3a), with a specific region showing a potential contact based on a cut-off distance (>7 Å). Within this interface, VP24-2 forms a hydrophobic pocket, in which a hydrophobic residue, I35 of VP24-1, is inserted (Fig. 3b, c). Given that VP24-VP24 interactions are limited to this hydrophobic interaction with I35 (Fig. 3a), the binding affinity between the two VP24s within the NCLS would be relatively weak.

a (Left) A contact map showing the interaction sites between VP24-1 and VP24-2. The dotted box indicates a key area where contacts are enriched. (Right) A magnified inset of the region indicated by the dotted box in the left panel. b The atomic model showing the spatial arrangement of VP24-1 (lime green) and VP24-2 (violet) in ribbon representation, with the molecular lipophilicity potential map of the VP24-2 structure (in surface representation) colored in blue (hydrophilic) to orange (hydrophobic). c Close-up view of the hydrophobic pocket formed at the interface between VP24-1 and VP24-2, corresponding to the dotted box in the contact map (a). This close-up demonstrates residue I35 of VP24-1 resides within the hydrophobic pocket of VP24-2. NCLS, nucleocapsid-like structure.
NP–VP24 interaction within cells
To identify the specific residues responsible for NCLS formation and function, we generated eight NP and VP24 mutants based on our structural model. We designed four NP mutants at the interface with VP24-1 (E61K, H196A, and R202A) and VP24-2 (E61K and R132A), three VP24 mutants at the interface with NP-1 (R59A and N171A) and NP-2 (K148A), and one VP24 mutant at the hydrophobic interface with another VP24 (I35E). The E61K of the NP is located within both interfaces. Initially, to investigate the interactions between NP and VP24, we performed an immunoprecipitation (IP) assay using these mutants (Fig. 4a). Among all mutants, the VP24 N171A mutant lost its ability to interact with NP, consistent with previous reports21. In contrast, the other mutants exhibited interactions between NP and VP24. This result suggested that the interaction of the 169–173 loop of VP24-1 with the C-terminal α-helix 18 of NP-1 is primarily responsible for the interaction between NP and VP24.

a. Immunoprecipitation assay using FLAG-tagged NP and VP24 mutants. Left panel, whole cell lysates were analyzed using western blotting with anti-VP24 and anti-FLAG antibodies. Right panel, VP24s, co-immunoprecipitated (IP) with FLAG-tagged NPs using an anti-FLAG antibody, were analyzed using western blotting with anti-VP24 and anti-FLAG antibodies. Beta-actin was detected as a loading control. Experiments were performed three times independently. b Intracellular localization of NP and VP24 mutants in cells co-expressing FLAG-tagged NP, VP24, and VP35.The cells were immunostained using anti-NP (red) and anti-VP24 (green) antibodies. Nuclei were stained with Hoechst 33342 (blue). The absence of NP or VP24 is indicated by (-), and the MOCK control demonstrates cells without plasmid transfection. Scale bars represent 10 µm. Experiments were performed three times independently. Source data are provided as a Source data file.
NP expression induces the formation of inclusion bodies (IBs), the sites for viral genome transcription and replication28, where VP24 is recruited during virus replication29. Therefore, to examine whether these mutants accumulate in IBs, we examined the intracellular localization of the NP and VP24 mutants using an immunofluorescence assay (IFA). When wild-type NP, VP24, and VP35, the minimal components of NCLS formation3,4, were co-expressed, VP24 was localized within the IBs formed by NP expression, as reported previously20,21,29 (Fig. 4b). VP24 N171A, which showed no interaction with NP as demonstrated by the IP assay, was not localized within IBs. Unexpectedly, VP24 R59A, located at the interface between VP24-1 and NP-1, showed minimal recruitment into IBs. The distribution of NP and VP24 R59A are highly interpenetrated, unlike VP24 N171A, although there is little co-localization between the two (Fig. 4a). All other mutants with mutations at the interfaces between VP24-2 and NP-2 and between VP24-1 and VP24-2, showed the localization of VP24 within the IBs, similar to the wild-type combination (Fig. 4b). These results suggest that VP24 N171 is crucial for the interaction between NP and VP24 and that the interaction between NP-1 and VP24-1 is important for the recruitment of VP24 into IBs. Unfortunately, the residues in the α-helix 18 of NP-1 that are critical for the interaction with the 169–173 loop of VP24-1 could not be identified, although several NP mutants were examined (Supplementary Fig. 4). A higher resolution structure of this region is needed to determine the specific interaction in the future.
Impact on polymerase activity and NCLS formation
To investigate the amino acid residues involved in nucleocapsid function, we performed a minigenome assay using the mutants and assessed their impact on the transcription and replication activity of the nucleocapsid. In the presence of wild-type VP24, we observed a significant reduction in the transcription and replication activity, as previously reported19,30 (Fig. 5a). The VP24 N171A and R59A mutants, which did not localize within the IBs, showed significantly reduced inhibitory activity compared to VP24 WT, whereas the other VP24 mutants showed similar reductions as observed with wild-type VP24 (Fig. 5a). We were unable to quantitatively evaluate the inhibitory activity of VP24 for most NP mutants because they showed reduced transcription and replication activities, even in the absence of VP24 (Fig. 5b). However, in the NP mutants, the transcription and replication activities of the nucleocapsid composed of NP H196A and R202A, which localize at the interface between NP-1 and VP24-1 and show sufficient transcription and replication activity, were mildly inhibited by wild-type VP24 (Fig. 5c). This suggested that the interaction between NP-1 and VP24-1 plays a role in the inhibition of transcription and replication by VP24.

a–c Transcription and replication activity evaluated using the minigenome assay with NP and VP24 mutants. The results are presented in box-and-whiskers plots. The plots detail the maxima, 75th percentile, median, 25th percentile, and minima, with outliers represented by dots. The activities are compared between a Wild-type VP24 and its mutants, with the activity of wild-type NP in the absence of VP24 normalized to 100%. b Wild-type NP and NP mutants, with the activity of wild-type NP normalized to 100%. (c) Wild-type NP and NP mutants, with the activity of each specific NP type in the absence of VP24 normalized to 100%. n = 3 for each technical replicate. All experiments were performed in three biological replicates (n = 9 in total). p values from ANOVA-Dunnett’s test are indicated as ***p <0.001, **p <0.01, *p < 0.05. d Ultrathin section electron microscopy of HEK293T cells co-expressing NP and VP24 mutants and VP35. Each lower panel shows a close-up view of the square area of the upper panel. Red arrows indicate the NCLS. Scale bars represent 100 nm. Source data are provided as a Source data file.
To investigate whether each mutant is assembled into NCLS, we performed ultrathin section electron microscopy of cells co-expressing NP, VP24, and VP35. Similar to wild-type NP, NP E61K, and NP R202A mutants formed tubular structures with dense walls approximately 50 nm in diameter, representing NCLS3,4 (Fig. 5d). As expected from the results obtained using IFA, VP24 N171A and R59A mutants did not form NCLS structures. However, other mutants, such as NP R132A and VP24 K148A (located at the interface between NP-2 and VP24-2) and VP24 I35E (located at the interface between VP24-1 and VP24-2), were also unable to form NCLS in cells (Fig. 5d). These results demonstrated that NCLS formation is not necessary for the inhibition of viral genome transcription and replication. Rather, it is likely that the interactions between VP24-1 and NP-1 via N171 and electrostatic interactions are important for its inhibitory activity. In addition, the interactions of VP24-2 with both NP-2 and VP24-1 are potentially important for NCLS formation, although the interaction between NP-1 and VP24-1 is probably a prerequisite.
NCLS formation is necessary for its intracellular transport
NP, VP24, and VP35 are essential for the release of the nucleocapsid from IBs and the subsequent intracellular transport of the nucleocapsid20,21,30, which is a prerequisite for the packaging of the nucleocapsid into progeny virions mediated by the matrix protein VP404,31. As NCLS formation was unrelated to the inhibition of viral RNA synthesis (Fig. 5a–c), we hypothesized that NCLS formation is required for its intracellular transport. Therefore, we co-expressed NP, VP35, and one of the VP24 mutants, along with VP30-GFP which was used to visualized nucleocapsid movement using immunofluorescence microscopy. Here, we focused on VP24 mutants that did not form the NCLS but instead formed incomplete NCLS, yet showed different regulatory activities for transcription and replication: VP24 N171A, which did not inhibit transcription and replication; VP24 R59A, which showed reduced inhibitory activity for transcription and replication; VP24 I35E and VP24 K148A, which inhibited transcription and replication. In the presence of wild-type VP24, the NCLSs exhibited long-distance intracellular movement (>5 µm) per cell, as reported previously20. In contrast, considerably fewer incomplete NCLSs exhibited long-distance movement in the presence of VP24, N171A, R59A, I35E, K148A (Fig. 6a, b). Furthermore, signal trajectory analysis demonstrated that the incomplete NCLSs containing VP24 mutants exhibited significantly shorter movements than the nucleocapsid containing wild-type VP24 (Fig. 6c). These results suggested that intracellular NCLS transport is independent of the inhibition of vRNA transcription and replication; however, it relies on the formation of NCLS.

a Maximum intensity projections of time-lapse images recorded for 3 min with 3 s intervals, showing the intracellular distribution of NCLS composed of wild-type VP24 and VP24 I35E, VP24 R59A, VP24 K148A, and VP24 N171A mutants. Scale bars represent 10 µm. The magnified image of the square area is displayed, with a scale of 2 µm. b Analysis of the number of NCLS showing long-range movement within the cell. Each point represents the number of moving NCLS per cell, with the average indicated by horizontal lines. Variants that are statistically significant compared to the wild type (wt) from one-sided Student’s t-test are marked with asterisks (*p < 0.05; ***p < 0.001, ****p < 0.0001). All experiments were performed in three biological replicates. c Analysis of the trajectory length of NCLS movement within cells. Data points represent the length of each NCLS movement, with the trend showing a significant reduction in movement distance for VP24 mutants compared to wild-type. Variants that are statistically significant compared to the wild type (wt) from one-sided Student’s t-test are marked with asterisks (*p < 0.05; ***p < 0.001, ****p < 0.0001). All experiments were performed in three biological replicates. Source data are provided as a Source data file.
NCLS formation is important for infectious virus production
To assess whether incomplete NCLS formation followed by its inefficient transport affects infectious virion formation, we generated infectious virus-like particles (iVLPs) using various VP24 mutants (I35E, R59A, K148A, and N171A). We then examined the amounts of viral proteins in the iVLPs using western blotting (Fig. 7a). VP40 was detected in all iVLPs at comparable levels, suggesting similar budding efficiency of all iVLPs. Similarly, a consistent amount of NP was detected in all iVLPs, suggesting that NP incorporation efficiency is independent of VP24. In contrast, the levels of VP24 and VP35 were decreased in all iVLPs comprising VP24 mutants. Notably, iVLPs comprising VP24 N171A mutant showed a failure to incorporate VP24, suggesting that interaction of VP24 with NP via N171 is critical for incorporating the NCLS into iVLPs. Other iVLPs with VP24 mutants showed a substantial decrease in VP24 incorporation, suggesting partial incorporation of VP24 into the iVLPs as incomplete NCLSs.

a iVLP production using VP24 mutants. Left panel, cell lysate of plasmid-transfected P0 cells were analyzed using western blotting using anti-NP, -VP24, -VP35, and -VP40 antibodies. Beta-actin was detected as a loading control. Right panel, purified iVLPs from the supernatants of P0 cells were analyzed using western blotting. b Luciferase activity on P1 cells infected with respective iVLPs. The results are presented in box-and-whiskers plots. The plots detail the maxima, 75th percentile, median, 25th percentile, and minima, with outliers represented by dots. The activities are compared between Wild-type VP24 and its mutants, with the activity of VP24 wild-type normalized to 100%. n = 3 for each technical replicate. All experiments were performed in three biological replicates (n = 9 in total). p values from ANOVA-Dunnett’s test are indicated as ***p <0.001, **p <0.01, *p < 0.05. ***p <0.001. iVLP infectious virus-like particle, NP nucleoprotein. Source data are provided as a Source data file.
Finally, we assessed the infectivity of the iVLPs containing VP24 mutants. iVLPs collected from the supernatant of plasmid-transfected cells (P0 cells) were used to infect naïve target cells without any pre-transfection (P1 cells). Luciferase activity in P1 cells was measured to evaluate the levels of infectious iVLP production in P0 cells. While iVLP generated without VP24, referred to as VP24(-), was produced at levels similar to other iVLPs (Fig. 7a), it showed a significant reduction in luciferase activity in P1 cells (Fig. 7b), suggesting that the iVLPs lacking VP24 were less infectious, probably due to reduced primary transcription activity of the incomplete nucleocapsids18. Notably, all iVLPs containing VP24 mutants, which either failed to incorporate or only partially incorporated VP24, showed a significant reduction in luciferase activities, indicating that the incorporation of NCLS, rather than incomplete NCLS, is important for infectious iVLP formation. Collectively, these findings suggest that nucleocapsid formation is important for infectious virions production.
Discussion
The EBOV nucleocapsid is essential for viral RNA transcription and replication, and genome packaging into progeny virions. Despite advances in understanding the respective structures of its components8,14,15,16,17,22,26,27,32,33,34,35 and the accumulated knowledge of nucleocapsid functions12,13,16,19,21,23,25,33,36,37, the structural basis of nucleocapsid functions remains poorly understood. Here, using cryo-EM single-particle analysis, we reported a 4.6 Å resolution structure of the EBOV NCLS within VLPs. Structure-based mutagenesis revealed specific interactions between the two NPs and two VP24s that govern the regulation of viral RNA synthesis, nucleocapsid assembly, intracellular nucleocapsid transport, and infectious VLP production. This study deciphered the structure-function relationship of the nucleocapsid and revealed the distinct roles of each of the two VP24 molecules on the nucleocapsid during the viral life cycle, suggesting a more sophisticated regulatory mechanism than previously believed.
Based on our structural and functional analyses, we proposed a mechanistic model for the role of VP24s in regulating viral RNA synthesis and nucleocapsid formation (Fig. 8). Among the mutations located within the NP–VP24 interfaces, only the N171A mutation on VP24 eliminated the binding affinity of VP24 with NP (Fig. 4a), suggesting that the interaction between the 169–173 loop of VP24-1 and the C-terminal α-helix 18 of NP-1 is indispensable for NP–VP24 interactions. Thus, VP24 initially binds to NP-1 on the NP–RNA helix as VP24-1 via its 169–173 loop (Fig. 8a). Notably, VP24 R59A, similar to VP24 N171A (Fig. 4b), was not recruited into IBs, suggesting that VP24 R59 is likely important for stabilizing the NP-1–VP24-1 interaction following the initial NP-1–VP24-1 interaction via the 169–173 loop (Fig. 8a).

Our proposed model describes the two-phase strategy of NCLS formation and function. a The first phase depicts the role of VP24-1 in initial nucleocapsid assembly, highlighting that the interface with NP-1 is critical for the recruitment of VP24 into inclusion bodies (IBs) and termination of genome transcription and replication. This diagram highlights the importance of residue N171 in VP24-1, as a critical binding site for NP. Moreover, VP24-1 R59 and potentially NP-1 H196 are involved in recruiting VP24 to IBs and inhibiting transcription and replication. b The second phase illustrates the involvement of VP24-2 in the structural maturation of the nucleocapsid. Mutations in VP24 K148 and NP R132 at the NP-2–VP24-2 interface and VP24 I35 at the VP24-1–VP24-2 interface are crucial for nucleocapsid assembly. This stage highlights the necessity of these interactions for nucleocapsid formation and the subsequent transport of mature NCLS for the formation of progeny virions.
Using the minigenome assay, we found that VP24 (N171A and R59A) and NP (H196A and R202A), which have a mutation within the NP-1–VP24-1 interface, exhibited complete or partial failure to inhibit vRNA transcription and replication, whereas others that have a mutation within the NP-2–VP24-2 and the VP24-1–VP24-2 interface did not (Fig. 5a–c). These results strongly suggest that the binding of VP24-1 to NP-1 on the NP–RNA helix plays a critical role in inhibiting vRNA transcription and replication, rather than the previously believed mechanism involving NCLS formation (Fig. 8a). Although speculative, the proximity of VP24-1 to the template vRNA, closer than VP24-2 on the NP–RNA helix, suggested a potential steric hinderance on the movement of the L–VP35 polymerase complex for viral RNA synthesis. Further structural analyses of the nucleocapsid, including the L–VP35 complex, are required to understand the mechanisms underlying the inhibition of vRNA transcription and replication by VP24-1.
Our structural model demonstrates that VP24 N171, crucial for the NP–VP24 interaction, is not located within the NP-2–VP24-2 interface, suggesting that the interaction between VP24-2 and NP-2 is weaker than that between VP24-1 and NP-1. The initial binding of VP24-1 to NP-1 on the NP–RNA helix suggested that the binding of VP24-2 to NP-2 depends on the presence of VP24-1. It is anticipated to be stabilized by both electrostatic interaction with NP-2 and hydrophobic interaction with VP24-1. In our mutational analysis, VP24 K148A and NP R132A mutants, which have a mutation within the NP-2–VP24-2 interface, and the VP24 I35E mutant, which has a mutation within the VP24-1–VP24-2 interface, failed to form NCLS (Fig. 5d), suggesting the importance of VP24-2 for NCLS formation (Fig. 8b). Notably, VP24-1 is also crucial for NCLS formation, as mutants with VP24 and NP containing a mutation within the VP24-1dNP-1 interface were incapable of forming the NCLS. Such mutations (VP24 N171A, R59A, and potentially NP H196A) would hamper VP24 transport into IBs and the interaction between VP24-1 and VP24-2, leading to a failure in NCLS formation. Considering that the amino acids described in Fig. 8 are well conserved among filoviruses, especially between Ebola and Lloviu viruses (Supplementary Fig. 5A, B), the roles of each VP24 identified in this study are likely common among filoviruses.
Recently, Watanabe et al. reported the intracellular NCLS structure at 9 Å resolution using cryo-ET combined with sub-tomogram averaging, and suggested the existence of a C-terminal domain of VP35 on VP24-2 and an additional copy of the monomeric RNA-free NP on VP24-138. Although similar additional maps were observed for both the VP24 protrusions in our cryo-EM map (Fig. 1c), we were unable to annotate these molecules owing to the flexibility of this region. The flexibility of this layer in our model may be due to differences in the environment between intracellular IBs and VLPs. This additional outer layer, where an additional copy of NP is likely involved in the intra-rung interactions of nucleocapsids, suggests additional structural and functional regulation of the nucleocapsid, which was not addressed in our study. Analyzing the interactions between RNA-free NP and VP35 in the additional layer, presumed to be involved in progeny virion assembly, will be a notable focus for future research.
In conclusion, we determined the in situ NCLS structure within the EBOV VLP and revealed the structural basis of their distinct functions during the EBOV life cycle. The identification of specific residues required for the respective nucleocapsid functions revealed potential targets for therapeutic interventions. Although our findings will advance this field, they also highlight the complexity of nucleocapsid assembly and functional regulation. This study opens avenues for further research, particularly to understand the dynamic interplay between nucleocapsid components and elucidating the mechanisms governing their multiple functions.
Methods
Cell culture
HEK293T (human embryonic kidney) (CRL-3216, Lot 62296864), HEK293 (CRL-1573, Lot 61714301) cells were maintained in Dulbecco’s modified Eagle’s medium (D-MEM, D5796, Sigma-Aldrich, Burlington, MA, USA) supplemented with 10% fetal bovine serum (FCS, 10270-106, Thermo Fisher Scientific, Waltham, MA, USA) and 100 U/mL penicillin-streptomycin solution (P/S, 26253-84, Nacalai Tesque, Japan) at 37 °C and 5% CO2. Huh-7 (Human hepatoma, JTC-39, Lot 08282017) were maintained in D-MEM (low glucose) with L-glutamine (KSJ7029, Wako, Japan) supplemented with 10% FCS and 100 U/mL P/S. Vero (African green monkey kidney, Lot DSIU002) cells were maintained in Eagle’s minimum essential medium (E-MEM, 055-08975, Wako, Japan) supplemented with 10% FCS and 100 U/mL P/S.
Plasmids
Plasmids expressing EBOV proteins (pCAGGS-L, pCAGGS-GP, pCAGGS-VP24, pCAGGS-VP30, pCAGGS-VP35, pCAGGS-NP, and pCAGGS-NP-FLAG), the T7-driven EBOV minigenome encoding firefly luciferase (p3E5E-luc), T7 DNA-dependent RNA polymerase (pCAGGS-T7), and pTK Renilla luciferase were provided by Yoshihiro Kawaoka (University of Tokyo). pCAGGS-VP30-GFP for the VP30-GFP fusion protein was generated by cloning the EBOV VP30 open reading frame into the 5′ end of the gene encoding GFP, as previously described20. Mutant EBOV protein-expressing plasmids were generated via site-directed PCR mutagenesis, and their sequences were confirmed using Sanger sequencing (Supplementary Data 1).
Antibodies
The following antibodies were used for immunofluorescence microscopy: mouse Alexa Fluor 594-conjugated anti-FLAG-tag (M18-A59; MBL, Tokyo, Japan) at 1:1000 dilution, rabbit anti-VP24 (provided by Hideki Ebihara21 at the National Institute of Infectious Diseases) at 1:400 dilution, and Alexa Fluor 488-conjugated anti-rabbit (A-11008; Thermo Fisher Scientific) at 1:1000 dilution.
The following primary antibodies were used for western blot analysis (including samples from immunoprecipitation and iVLP assays): rabbit anti-EBOV NP (0301-012; IBT Bioservices, Rockville, MD, USA) at 1:10,000 dilution, rabbit anti-EBOV VP35 (0301-040; IBT Bioservices, Rockville, MD, USA) at 1:10,000 dilution, rabbit anti-EBOV VP40 (0301-010; IBT Bioservices, Rockville, MD, USA) at 1:10,000 dilution, rabbit anti-VP24 (provided by Yoshihiro Kawaoka19 at 1:10,000 dilution or provided by Hideki Ebihara21 at 1:10,000 dilution), and mouse anti-beta-actin (ab8226; Abcam, Cambridge, UK) at 1:10,000 dilution.
The following secondary antibodies were used for western blot analysis (including samples from immunoprecipitation and iVLP assays): horseradish peroxidase (HRP)-conjugated anti-rabbit (NA934; Cytiva, Marlborough, MA, USA) at 1:10,000 dilution and HRP-conjugated anti-mouse (NA931; Cytiva) at 1:10,000 dilution.
Expression and purification of EBOV VLPs
HEK293T cells (3.0 × 106 cells) seeded in a 10 cm dish were transfected using TransIT-293 (Mirus Bio, Madison, WI, USA) with 670 ng pCAGGS-GP, 4 µg pCAGGS-NP, 1 µg pCAGGS-VP24, 1 µg pCAGGS-VP35, 4 µg pCAGGS-VP40, 4 µg p3E5E-luc, and 1.5 µg pCAGGS-T7. Three days post-transfection, VLPs in supernatants were fixed with 1% paraformaldehyde (PFA). The supernatant containing VLPs was centrifugated at 1000 × g for 5 min at 4 °C to remove cell debris. VLPs in 7 ml of the supernatant were purified via ultracentrifugation using a Beckman SW41Ti rotor at 49,392 × g for 2 h at 4 °C, with 1 ml of 20% (w/v) sucrose cushion in Tris-HCl buffer (10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA) and the remaining volume filled with Tris-HCl buffer. The purified VLPs were resuspended in Tris-HCl buffer and diluted at 1.0 mg/ml. The VLP concentration was measured by absorbance at 280 nm.
Cryo-EM sample preparation and data collection
The total 2.5 µL sample was applied to both sides of glow-discharged 400-mesh R1.2/1.3 Cu grids (Quantifoil, Jena, Germany) at 10 mA 20 Pa for 30 s using a JEC-3000FC sputter coater (JEOL, Tokyo, Japan). The grids were blotted on both sides with filter papers, for 14 s at blotting force 0, at a humidity of 100% at 8 °C and rapidly frozen in liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific). Cryo-EM samples were initially screened using a Glacios cryo-EM operated at 200 kV and equipped with a Falcon4 camera installed at the Institute for Life and Medical Sciences, Kyoto University. Cryo-EM micrographs were acquired using a Titan Krios cryo-EM (Thermo Fisher Scientific) operated at 300 kV and equipped with a Gatan BioContinuum energy filter and Cs corrector (CEOS GmbH, Heidelberg, Germany) installed at the Institute for Protein Research, Osaka University. Data were collected using the Serial-EM software39 (v4.0) with a beam-image shift imaging scheme, with a K3 detector in counting and correlated double sampling imaging mode. The collection was performed at a magnification of 81,000× and a pixel size of 0.88 Å. The slit width of the energy filter was set to 20 eV. Each micrograph was exposed to a total dose of 60 e- per Ų, fractionated into 60 frames. The nominal defocus range for the dataset was −0.6 to −2 μm. The detailed imaging conditions are described in Supplementary Table 2.
Cryo-EM image processing
The detailed workflow is shown in Supplementary Fig. 2. The frames of each movie were aligned within RELION 4.0-beta40,41 on 5 × 5 patches, and contrast transfer function (CTF) estimations were performed using CTFFIND442. The coordinates of the NCLS were manually selected from 5897 micrographs. In total, 263,789 segments were extracted using a 720 × 720 pixel box size and an inter-box distance of 10%. The segments underwent two rounds of 2D classification in RELION 4.0-beta. The initial 3D model was generated from a previously reported cryo-ET map, EMD-3871, employing a 60 Å low-pass filter. Following a single round of 3D classification, without a local search for helical symmetry, 34,865 segments (binned at 1.2375 Å/pix) underwent further 3D refinement. Additional CTF refinement, Bayesian polishing, and additional 3D refinement, incorporating a local search for symmetry and SIDESPLITTER43, yielded a 7.5 Å cryo-EM map. Helical parameters were locally searched based on previously reported values8, resulting in a −28.1059° twist and a 5.8984 Å rise.
To enhance the structure of the NCLS repeating unit, we expanded the helical symmetry and subtracted signals outside the NCLS from the particles for subsequent classification and local refinement. Using the relion_particle_symmetry_expand command, symmetrized helical segments were expanded into each repeating unit (418,380 particles) according to their helical symmetry, ensuring an inter-box distance to prevent duplicates in the new particle sets. Subsequently, the signal outside the repeating unit (approximately 4-mer NPs + protrusions) was subtracted with a box size of 256 × 256 pixels. Thereafter, 3D classification without alignment was performed, focusing specifically on the repeating unit region with a mask. Finally. In total, 204,348 particles were imported into cryoSPARC44,45 (v3.3.1) for local refinement. The resulting cryo-EM map had an overall resolution of 4.6 Å.
Structural model building and refinement
For the initial model, we docked previously reported structures, PDB ID: 5Z9W for NP and PDB ID: 4M0Q for VP24, onto the main-chain scaffold of PDB ID: 6EHM. To precisely refine the atomic models in the intermolecular region, the two molecules surrounding the NPs were additionally fitted to our cryo-EM map. The initial refinement process was executed via Rosetta using Relax46,47,48. A total of 100 models were generated, and those with better energy scores and fit densities were manually inspected. Small modeling errors were manually corrected in COOT (v0.9.8.7)49,50, and in the refinement was reiterated in Rosetta using Relax. The final model was refined using Phenix51 with phenix.real_space_refine, followed by manual correction using COOT and ISOLDE (v1.6.0)52. After removing the unmodeled region, model validity was assessed using Phenix (v1.20) and MolProbity53 software. Detailed results of the atomic modeling and evaluation are shown in the Supplementary Table 2. The cryo-EM map and atomic models were displayed using UCSF Chimera X54 (v1.5) software. A contact map (Fig. 3a) was generated using the MAPIYA webserver55.
Protein immunoprecipitation
HEK293T cells (1.0 × 106 cells/well) were seeded in 6-well plates and transfected with FLAG-tagged NP and/or VP24 expression plasmids (1 µg/well each) using TransIT-293. Two days post-transfection, cells were washed with cold phosphate-buffered saline (PBS). Cells were resuspended in 400 μL of lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, cOmplete EDTA-free protease inhibitor tablets [Roche, Basel, Switzerland]), incubated for 1 h at 4 °C, and centrifuged at 12,000 × g for 10 min at 4 °C. The supernatant was incubated on a rotator overnight at 4 °C with an additional 16 μL of anti-FLAG M2 magnetic beads (Sigma-Aldrich. The beads were washed six times with wash buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, cOmplete EDTA-free protease inhibitor tablets) and subsequently eluted in 150 μL of wash buffer with 500 ng/μL FLAG peptide (Merck, Darmstadt, Germany) by rotation on a rotator for 2 h at 4 °C. The cell lysates and eluates were electrophoresed on a 4–15% sodium dodecyl sulfatepolyacrylamide gel (Bio-Rad Laboratories, Hercules, CA, USA) and subjected to western blot analysis.
Minigenome assay
HEK293T (1.25 × 105 cells/well) cells were cultured in 24-well plates. Each well was transfected with plasmids encoding EBOV nucleocapsid components, including 1,000 ng pCAGGS-L, 75 ng pCAGGS-VP30, 100 ng pCAGGS-VP35, 100 ng pCAGGS-NP, 200 ng firefly luciferase p3E5E-luc, 200 ng T7 DNA-dependent RNA polymerase (pCAGGS-T7), and 10 ng Renilla luciferase (pTK r.luc), with or without 100 ng pCAGGS-VP24. Transfection was performed using 4 µL TransIT 293 (Takara, Shiga, Japan) and 100 µL OPTI-MEM (Thermo Fisher Scientific). At 48 h post-transfection, cells were lysed using Passive Lysis buffer (Promega Madison, WI, USA). Luciferase activities were measured using a GloMax®-Multi+ Detection System (Promega), along with Luciferase Assay Reagent II and Stop & Glo® Reagent (Promega), following the manufacturer’s protocol.
Ultrathin section electron microscopy
HEK293T cells (5.0 × 105 cells/well) were seeded on a 6-well plate, transfected with 5 µg NP or NP mutant-encoding plasmids, 1 µg VP24 or VP24 mutant-encoding plasmids, and 1 µg VP35-encoding plasmids. At 24 h post-transfection, cells were fixed with aldehydes, postfixed with 1 % osmium tetroxide, dehydrated in a graded ethanol series, and embedded in an EPON 812 (TAAB, Berks, UK). Ultrathin sections were stained with uranyl acetate and lead citrate and observed using a Hitachi HT-7700 microscope operated at 80 kV (Hitachi Hi-Tech, Tokyo, Japan).
Immunofluorescence assay
HEK293 cells (2.5 × 105 cells) were plated in a 35 mm glass-bottom dish (Matsunami, Osaka, Japan) coated with collagen I (Corning, Corning, NY, USA). Cells were transfected with 1 µg/each of FLAG-tagged NP, VP24, and VP35 expressing plasmids. At 48 h post-transfect, transfected cells were fixed in 4% PFA in PBS for 15 min and subsequently permeabilized with 0.1% Triton X-100 (MP Biomedicals, Santa Ana, CA, USA) in PBS for 15 min. Cells were blocked with Blocking One solution (Nacalai Tesque, Kyoto, Japan) for 30 min, followed by overnight incubation at 4 °C with primary antibodies [anti-FLAG-tag-Alexa Fluor 594 (MBL, Tokyo, Japan) 1:1000; rabbit anti-EBOV-VP2421 1:400]. Thereafter, cells were incubated with secondary antibodies [Alexa Fluor 488 anti-rabbit (Thermo Fisher Scientific) 1:1000] for 1 h at room temperature. Cells were treated with Hoechst 33342 (1:1000; Thermo Fisher Scientific) for nuclei staining following the application of secondary antibodies. Section images were recorded using DeltaVision Elite (Cytiva, Marlborough, MA, USA) with a 60× objective and subsequently deconvolved and projected using SoftWoRx Version 6.5.2.
Live-cell microscopy
Huh-7 cells (1 × 104 cells) were seeded onto an 8-well slide (Ibidi, Gräfelfing, Germany). Transfection with plasmids expressing VP30-GFP, NP, VP24 or VP24 mutant, and VP35 was performed in 50 μL Opti-MEM (Thermo Fisher Scientific). The inoculum was removed at 1 h post-transfection, and a volume of 250 μL CO2-independent Leibovitz’s medium (Thermo Fisher Scientific) was added. Live-cell imaging was performed using a Keyence BZ-X 810 (KEYENCE, Osaka, Japan) with a 100× oil objective lens at 20 h post-transfection. Acquired images and movie sequences were processed using “Imaris tracking” software (Bitplane, Oxford Instruments, Abingdon, UK). Signals with a diameter greater than 0.5 μm for NCLSs were selected, and an autoregressive motion algorithm was applied for signal tracking. The detected trajectories that exceeded 5 μm in length, lasted over 15 s, and had a track straightness greater than 0.2 were included in the analyses. Obvious nonspecific trajectories observed in VP24 mutants, which were characterized by a series of linear lines, were removed from the analyses (Takamatsu, JVI 2022)20,56.
iVLP generation and infectivity measurement
HEK293T (1.0 × 106 cells/well) cells cultured in six-well plates were transfected using TransIT-293 (Mirus Bio) with 250 ng pCAGGS-GP, 125 ng pCAGGS-NP, 100 ng pCAGGS-VP24, 125 ng pCAGGS-VP35, 250 ng pCAGGS-VP40, 1 µg pCAGGS-L, 100 ng p3E5E-luc, and 250 ng pCAGGS-T7. At 24 h post-transfection, the cells were collected and subjected to western blot analysis. Simultaneously, the supernatant was collected and centrifuged at 1500 × g for 10 min at 4 °C to remove cell debris. Then, the collected supernatants were purified via ultracentrifugation using a Beckman SW55Ti rotor at 74,329 × g for 1 h at 4 °C, with 1 ml of 20% (w/v) sucrose cushion in Tris-HCl buffer (10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA) and the remaining volume filled with Tris-HCl buffer. The pellet of iVLP was resuspended with 50 µl of Tris-HCl buffer. The cell lysates and iVLPs were electrophoresed on a sodium dodecyl sulfatepolyacrylamide gel and subjected to western blotting. For the luciferase assay to evaluate iVLPs infectivity, producer HEK 293T cells (1 × 106 cells/well) were seeded in a six-well plate. Each well was transfected using 8 µl TransIT-293 (Mirus Bio) and 250 µl OPTI-MEM (Thermo Fisher Scientific) with 250 ng pCAGGS-NP, 250 ng pCAGGS-VP35, 100 ng pCAGGS-VP30, 3 µg pCAGGS-L, 250 ng pCAGGS-GP, 250 ng pCAGGS-VP40, 250 ng pCAGGS-T7, and 250 ng firefly luciferase p3E5E-luc, with or without 100 ng pCAGGS-VP24 (wt or mutant). At 72 h post-transfection, cells and iVLPs of three wells were collected and purified as described above. The target cells (Vero cells, 5 × 105 cells/well) were seeded in a six-well plate and infected with the purified iVLPs. The infected cells were lysed using Passive Lysis buffer (Promega Madison) at 48 h post-infection, and the cell lysates were subjected to luciferase assays. Luciferase activities were measured using a GloMax®-Multi+ Detection System (Promega), along with Luciferase Assay Reagent II (Promega), following the manufacturer’s protocol.
Statistical analyses
To assess statistically significant differences in the minigenome assay, we employed ANOVA-Dunnett’s test to correct for multiple hypotheses using the RStudio software. The results are reported as the mean ± s.d. with p <0.01 considered statistically significant. For the NCLS trajectory analysis of live-cell imaging, statistical analysis was performed using the Student’s t-test with GraphPad Prism version 10.0.0 for MacOS (GraphPad Software, Boston, MA, USA, www.graphpad.com), and p <0.0001 was considered statistically significant (****).
All experiments were performed in triplicate.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM maps generated in this study have been deposited in the Electron Microscopy Data Bank under accession code EMD-39081. The atomic coordinates reported in this paper have been deposited in the Protein Data Bank (PDB) under accession code 8Y9J. Raw cryo-EM movies were deposited in the Electron Microscopy Public Image Archive under the accession code EMPIAR-11932. PDB codes of previously published structures used in this study are 5Z9W, 4M0Q and 6EHM. EMDB codes of previously published structures used in this study are EMD-3871 and EMD-3873. Source data are provided as a Source data file. Source data are provided with this paper.
References
Sanchez, A., Geisbert, T. W. & Feldmann, H. In Fields Virology 1–82 (Lippincott Williams & Wilkins, 2011).
Liu, C.-H., Hu, Y.-T., Wong, S. H. & Lin, L.-T. Therapeutic strategies against Ebola virus infection. Viruses 14, 579 (2022).
Huang, Y., Xu, L., Sun, Y. & Nabel, G. J. The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and posttranslational modification of nucleoprotein. Mol. Cell 10, 307–316 (2002).
Noda, T. et al. Assembly and budding of Ebolavirus. PLOS Pathog 2, e99 (2006).
Watanabe, S., Noda, T. & Kawaoka, Y. Functional mapping of the nucleoprotein of Ebola virus. J. Virol. 80, 3743–3751 (2006).
Noda, T., Hagiwara, K., Sagara, H. & Kawaoka, Y. Characterization of the Ebola virus nucleoprotein–RNA complex. J. Gen. Virol. 91, 1478–1483 (2010).
Bharat, T. A. M. et al. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc. Natl. Acad. Sci. USA 109, 4275–4280 (2012).
Wan, W. et al. Structure and assembly of the Ebola virus nucleocapsid. Nature 551, 394–397 (2017).
Bharat, T. A. M. et al. Cryo-electron tomography of Marburg virus particles and their morphogenesis within infected cells. PLOS Biol. 9, e1001196 (2011).
Beniac, D. R. et al. The organisation of Ebola virus reveals a capacity for extensive, modular polyploidy. PLoS ONE 7, e29608 (2012).
Jamin, M. & Yabukarski, F. Chapter Four - Nonsegmented negative-sense rna viruses—structural data bring new insights into nucleocapsid assembly. In Advances in Virus Research (eds. Kielian, M., Mettenleiter, T. C. & Roossinck, M. J.) Vol. 97, 143–185 (Academic Press, 2017).
Miyake, T. et al. Ebola virus inclusion body formation and RNA synthesis are controlled by a novel domain of NP interacting with VP35. J. Virol. https://doi.org/10.1128/JVI.02100-19 (2020).
Groseth, A. et al. The Ebola virus ribonucleoprotein complex: a novel VP30-L interaction identified. Virus Res 140, 8–14 (2009).
Yuan, B. et al. Structure of the Ebola virus polymerase complex. Nature https://doi.org/10.1038/s41586-022-05271-2 (2022).
Peng, Q. et al. Molecular mechanism of de novo replication by the Ebola virus polymerase. Nature 622, 603–610 (2023).
Kirchdoerfer, R. N., Abelson, D. M., Li, S., Wood, M. R. & Saphire, E. O. Assembly of the Ebola virus nucleoprotein from a chaperoned VP35 complex. Cell Rep 12, 140–149 (2015).
Landeras-Bueno, S. et al. Sudan Ebolavirus VP35-NP crystal structure reveals a potential target for pan-filovirus treatment. mBio 10, e00734–19 (2019).
Hoenen, T. et al. Infection of naïve target cells with virus-like particles: implications for the function of Ebola virus VP24. J. Virol. 80, 7260–7264 (2006).
Watanabe, S., Noda, T., Halfmann, P., Jasenosky, L. & Kawaoka, Y. Ebola virus (EBOV) VP24 inhibits transcription and replication of the EBOV genome. J. Infect. Dis. 196, S284–S290 (2007).
Takamatsu, Y., Kolesnikova, L. & Becker, S. Ebola virus proteins NP, VP35, and VP24 are essential and sufficient to mediate nucleocapsid transport. Proc. Natl. Acad. Sci. USA 115, 1075–1080 (2018).
Banadyga, L. et al. Ebola virus VP24 interacts with NP to facilitate nucleocapsid assembly and genome packaging. Sci. Rep. 7, 7698 (2017).
Sugita, Y., Matsunami, H., Kawaoka, Y., Noda, T. & Wolf, M. Cryo-EM structure of the Ebola virus nucleoprotein–RNA complex at 3.6 Å resolution. Nature 563, 137–140 (2018).
Fujita-Fujiharu, Y. et al. Structural insight into Marburg virus nucleoprotein–RNA complex formation. Nat. Commun. 13, 1191 (2022).
Watanabe, S. et al. Production of novel ebola virus-like particles from cDNAs: an alternative to ebola virus generation by reverse genetics. J. Virol. 78, 999–1005 (2004).
Takamatsu, Y., Kolesnikova, L., Schauflinger, M., Noda, T. & Becker, S. The integrity of the YxxL motif of Ebola virus VP24 is important for the transport of nucleocapsid-like structures and for the regulation of viral RNA synthesis. J. Virol. 94, e02170–19 (2020).
Edwards, M. R. et al. The Marburg virus VP24 protein interacts with Keap1 to activate the cytoprotective antioxidant response pathway. Cell Rep 6, 1017–1025 (2014).
Zhang, A. P. P. et al. The Ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLOS Pathog 8, e1002550 (2012).
Hoenen, T. et al. Inclusion bodies are a site of ebolavirus replication. J. Virol. 86, 11779–11788 (2012).
Nanbo, A., Watanabe, S., Halfmann, P. & Kawaoka, Y. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci. Rep. 3, 1206 (2013).
Hoenen, T., Jung, S., Herwig, A., Groseth, A. & Becker, S. Both matrix proteins of Ebola virus contribute to the regulation of viral genome replication and transcription. Virology 403, 56–66 (2010).
Noda, T., Watanabe, S., Sagara, H. & Kawaoka, Y. Mapping of the VP40-binding regions of the nucleoprotein of Ebola virus. J. Virol. 81, 3554–3562 (2007).
Dong, S. et al. Insight into the Ebola virus nucleocapsid assembly mechanism: crystal structure of Ebola virus nucleoprotein core domain at 1.8 Å resolution. Protein Cell 6, 351–362 (2015).
Kirchdoerfer, R. N., Moyer, C. L., Abelson, D. M. & Saphire, E. O. The Ebola virus VP30-NP interaction is a regulator of viral RNA synthesis. PLOS Pathog. 12, e1005937 (2016).
Kirchdoerfer, R. N., Saphire, E. O. & Ward, A. B. Cryo-EM structure of the Ebola virus nucleoprotein–RNA complex. Acta Crystallogr. Sect. F Struct. Biol. Commun. 75, 340–347 (2019).
Leung, D. W. et al. Structure of the Ebola VP35 interferon inhibitory domain. Proc. Natl. Acad. Sci. USA 106, 411–416 (2009).
Biedenkopf, N., Lier, C. & Becker, S. Dynamic phosphorylation of VP30 is essential for Ebola virus life cycle. J. Virol. 90, 4914–4925 (2016).
Schudt, G. et al. Transport of Ebolavirus nucleocapsids is dependent on actin polymerization: live-cell imaging analysis of Ebolavirus-infected cells. J. Infect. Dis. 212, S160–S166 (2015).
Watanabe, R. et al. Intracellular Ebola virus nucleocapsid assembly revealed by in situ cryo-electron tomography. Cell. 187, 5587–5603 (2024).
Mastronarde, D. N. SerialEM: a program for automated tilt series acquisition on tecnai microscopes using prediction of specimen position. Microsc. Microanal. 9, 1182–1183 (2003).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
He, S. & Scheres, S. H. W. Helical reconstruction in RELION. J. Struct. Biol. 198, 163–176 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Ramlaul, K., Palmer, C. M., Nakane, T. & Aylett, C. H. S. Mitigating local over-fitting during single particle reconstruction with SIDESPLITTER. J. Struct. Biol. 211, 107545 (2020).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Fleishman, S. J. et al. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE 6, e20161 (2011).
Khatib, F. et al. Algorithm discovery by protein folding game players. Proc. Natl. Acad. Sci. USA 108, 18949–18953 (2011).
Maguire, J. B. et al. Perturbing the energy landscape for improved packing during computational protein design. Proteins 89, 436–449 (2021).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. Sect. Struct. Biol. 74, 519–530 (2018).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Badaczewska-Dawid, A. E., Nithin, C., Wroblewski, K., Kurcinski, M. & Kmiecik, S. MAPIYA contact map server for identification and visualization of molecular interactions in proteins and biological complexes. Nucleic Acids Res. 50, W474–W482 (2022).
Schudt, G., Kolesnikova, L., Dolnik, O., Sodeik, B. & Becker, S. Live-cell imaging of Marburg virus-infected cells uncovers actin-dependent transport of nucleocapsids over long distances. Proc. Natl. Acad. Sci. USA 110, 14402–14407 (2013).
Sarkar, S. et al. DelPhi Web Server: a comprehensive online suite for electrostatic calculations of biological macromolecules and their complexes. Commun. Comput. Phys. 13, 269–284 (2013).
Acknowledgements
We thank Hideki Ebihara (National Institute of Infectious Diseases) and Satoko Yamaoka (Mayo Clinic) for providing the VP24 antibody and Yoshihiro Kawaoka (University of Tokyo) for providing plasmids expressing EBOV viral proteins and the minigenome. This work was supported by the Cooperative Research Program (Joint Usage/Research Center program) of Institute for Life and Medical Sciences, Kyoto University, a Grant-in-Aid from the Japan Society for the Promotion of Science (JSPS) Fellows (21J12207) and an ANRI fellowship (to Y.F-F.), the Otsuka Toshimi Scholarship (to S.H.); Grant-in-Aid for the Ministry of Education, Culture, Sports, Science and Technology (MEXT) Leading Initiative for Excellent Young Researchers, JSPS Scientific Research (C) (21K07052), Japan Science and Technology Agency (JST) FOREST Program (JPMJFR214S) (to Y.S.); Japan Agency of Medical Research and Development (AMED: JP23fm0208101, JP22fm0208101, 22wm0325023), JSPS (22KK0115, 21K07059), MSD Life Science Foundation, Naito Foundation, Takeda Science Foundation (to Y.T.); and JSPS Core-to-Core Program A, Advanced Research Networks (JPJSCCA20190008), JSPS Grant-in-Aid for Challenging Exploratory Research (22K19431), JSPS Grants-in-Aid for Scientific Research, Fund for the promotion of Joint International Research (22KK0115), JST Core Research for Evolutional Science and Technology (JPMJCR20HA), AMED (24fm0208101j0008, JP23fm0208101 and 22fk0108552h0001), the cooperative research project program of the National Research Center for the Control and Prevention of Infectious Diseases, Nagasaki University (to T.N.); Grant for Joint Research Project of the Institute of Medical Science at the University of Tokyo, Joint Usage/Research Center Program of the Institute for Life and Medical Sciences at Kyoto University, the Takeda Science Foundation (to Y.S. and T.N.). This study was also supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under grant number JP23ama121001 (support number 5889).
Author information
Authors and Affiliations
Contributions
Y.F.-F., Y.S., and T.N. designed the study. Y.F.-F., S.H., A.H., Y.T., Y.N.N., K.H., Y.M., M.N., and Y.S. performed the experiments. Y.F.-F., S.H., Y.T., and Y.S. analyzed the data. Y.F.-F., S.H., Y.S., and T.N. wrote the manuscript. All the authors have reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Luca Zinzula and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fujita-Fujiharu, Y., Hu, S., Hirabayashi, A. et al. Structural basis for Ebola virus nucleocapsid assembly and function regulated by VP24. Nat Commun 16, 2171 (2025). https://doi.org/10.1038/s41467-025-57236-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57236-4
