
Abstract
Elevated fasting insulin levels (FI), indicative of altered insulin secretion and sensitivity, may precede type 2 diabetes (T2D) and cardiovascular disease onset. In this study, we group FI-associated genetic variants based on their genetic and phenotypic similarities and identify seven clusters with distinct mechanisms contributing to elevated FI levels. Clusters fall into two types: “non-diabetogenic hyperinsulinemia,” where clusters are not associated with increased T2D risk, and “diabetogenic hyperinsulinemia,” where T2D associations are driven by body fat distribution, liver function, circulating lipids, or inflammation. In over 1.1 million multi-ancestry individuals, we demonstrated that diabetogenic hyperinsulinemia cluster-specific polygenic scores exhibit varying risks for cardiovascular conditions, including coronary artery disease, myocardial infarction (MI), and stroke. Notably, the visceral adiposity cluster shows sex-specific effects for MI risk in males without T2D. This study underscores processes that decouple elevated FI levels from T2D and cardiovascular risk, offering new avenues for investigating process-specific pathways of disease.
Similar content being viewed by others
Introduction
Alterations in insulin secretion and sensitivity arise many years before the development of type 2 diabetes (T2D)1,2,3. Fasting insulin (FI) is an accessible trait to characterize molecular alterations that precede the development of T2D. FI is a biomarker with prognostic variability between individuals and populations4. Elevated FI levels can herald defects in mechanisms of insulin secretion and sensitivity and thus inform T2D pathophysiology and coronary artery disease (CAD) risk5,6. The specific molecular alterations associated with FI that are linked to disease risk remain largely unexplored.
The dynamic, overlapping, and molecular complexity underlying insulin sensitivity and resistance limits our knowledge about the impact of these processes in the development of T2D6,7. A better understanding of the physiological mechanisms could advance our pathophysiological understanding of the early stages of T2D and could contribute to more targeted therapeutic and preventive strategies. While previous studies have aimed at clustering T2D genetic variants8,9,10,11, these approaches often provide limited insights into the molecular mechanisms underlying glycemic regulation as people with T2D often present with other metabolic alterations due to disease or treatment. To date, genome-wide association studies (GWAS) have identified hundreds of genetic alleles associated with increased fasting serum insulin levels12,13,14,15,16. Very few of these genetic loci have been functionally characterized because it is clinically complex to separate functional mechanisms of insulin production and insulin action in experimental studies. Therefore, one approach to identifying molecular consequences of identified FI-associated GWAS variants is in silico bioinformatic analyses.
By implementing techniques previously applied to the clustering of T2D loci, we undertook a genetic approach to cluster FI-related variants based on their association with body composition, inflammatory, and lipid traits in individuals without diabetes from five ancestry groups to elucidate the mechanisms that underlie the variation in insulin resistance and insulin secretion and their impact on cardiometabolic outcomes.
Results
Using extant association summary statistics, we identified seven distinctive genetic clusters with defined genetic and trait similarities in the bNMF model. The clusters are labeled “FI-Lipodystrophy Cluster” (Supplementary Table 1), “Adiposity-driven Hyperinsulinemia Cluster” (Supplementary Table 2), “Insulin Resistance Mediated by Visceral Adiposity Cluster” (Supplementary Table 3), Preserved Insulin Secretion Cluster (Supplementary Table 4), “Elevated Insulin Secretion Cluster” (Supplementary Table 5), “Proinsulin Cluster” (Supplementary Table 6), and “FI-Liver/Lipid Cluster” (Supplementary Table 7), and are described in detail below.
We constructed partitioned polygenic scores (pPS) denoting genetic susceptibility to seven distinct mechanisms underlying FI levels among 1,104,258 individuals from five studies. The pPS showed similar distributions across cohorts and population groups (Supplementary Table 8). We classified the seven genetic clusters based on their association with T2D (Fig. 1; Source Data 1; Supplementary Data 1), which was not an input in the clustering. While all genetic clusters were defined by alleles increasing FI levels, three clusters were associated with non-elevated T2D risk (non-diabetogenic hyperinsulinemia), and four with increased T2D risk as well as strong effects on body fat distribution, liver, lipid, and inflammatory processes (diabetogenic hyperinsulinemia), which show mechanisms underlying impaired insulin sensitivity.

Panel A Forest plot displaying meta-analysis results from five cohorts (UKBB, MVP, ASPREE, MGBB, and FHS) for cluster associations with type 2 diabetes. Data points represent the mean effect size (odds ratio). Error bars indicate 95% confidence intervals (CIs) calculated using logistic regression models. All statistical tests were two-sided, with a significance threshold set at α = 0.0008, based on a Bonferroni adjustment for multiple comparisons. The dotted vertical line at 1 represents the null effect (no association). Panel B Each cluster is represented by a set of loci and a set of traits; The loci and traits represented in the plots are the top-weighted for better visualization; clusters might have more variants and traits. Loci with a number after the period “.xx” represents that there was more than one SNP at that locus. The green bars represent the weights with which each variant contributes to each cluster. In the case of traits, the color of the bars represents the direction of the effect that was found in each cluster, red for positive association and blue for negative association. Purple lines distinguished diabetogenic hyperinsulinemia clusters; increased fasting insulin, reduced glycemia, and preserved insulin secretion. Yellow lines distinguish clusters of non-diabetogenic hyperinsulinemia; increased fasting insulin due to a strong effect on body fat distribution, liver, lipid, and inflammatory processes. VAT visceral adipose tissue, SAT subcutaneous adipose tissue, 2hrG glucose at 2 hours after an oral glucose tolerance test, LEP leptin, HDL high-density lipoprotein, LVEDV left ventricular end-diastolic volume, WHR waist–hip ratio, PATadjHtWt pericardial adipose tissue adjusted by height to weight ratio, HOMAB Homeostasis Model Assessment of β-cell function, HIPC hip circumference, BFP body fat percentage; VATSAT VAT:SAT ratio, PI proinsulin, CIR corrected insulin response, FG fasting glucose, HBA1C hemoglobin A1C, LDL low-density lipoprotein. Source data are provided as a Source Data file.
Among the three clusters characterized by non-diabetogenic hyperinsulinemia, the first is a set of FI-increasing alleles with concomitant effects to lower glycemic trait levels (fasting glucose, 2h glucose, or A1c). The genetic variants within this cluster indicate processes linked to elevated levels of FI while maintaining beta-cell function. Notably, CELF1 and TCF7L2 were among the eight top-weighted loci. Interestingly, in the case of TCF7L2, it is in the opposite direction to the one described for T2D17. We refer to this cluster as “Preserved Insulin Secretion” (PIS) cluster. The second non-diabetogenic hyperinsulinemia cluster was a set of FI-increasing alleles with overlapping effects on increased corrected insulin response and decreased 2 h glucose, denoting a putative pathway of increased insulin secretion; referred to as “Elevated Insulin Secretion” cluster (EIS). This cluster included GRB10 and REEP3 within the 28 top-weighted loci. The third non-diabetogenic hyperinsulinemia cluster was a set of FI-increasing alleles with subtle effects on glucose homeostasis: increased levels of proinsulin, C-reactive protein, and gamma glutamine transferase (GGT), with HNF1A and ARAP1 among the six top-weighted loci. This cluster recapitulates a beta cell under stress, which we call “Proinsulin cluster”.
Among the four diabetogenic hyperinsulinemia genetic clusters, we first identified a set of FI-increasing alleles that were associated with increased waist circumference, body fat percentage, and subcutaneous adipose tissue (SAT). This suggests evidence of a cluster characterized by increased FI driven by generalized adiposity with FTO among the top-weighted 19 loci, we call it “adiposity-driven hyperinsulinemia cluster”. Second, we identified a set of FI-increasing alleles characterized by visceral adiposity, lower corrected insulin response, and sex-specific association with waist-adjusted BMI in males. In this cluster labeled as “Visceral Adiposity cluster”, the 25 top-weighted loci included MYO1A, BMP2, and ARL15 genes previously described for their association with lower BMI and energy storage into visceral and subcutaneous adipose tissue depots18,19. The third cluster was a set of FI-increasing alleles characterized by increased fat distribution in central compartments and a detrimental circulating and hepatic lipid deposition, which we called the “FI-lipodystrophy cluster”. The top 37 highly weighted loci included variants that lie in or near genes implicated in monogenic forms of lipodystrophy (PPARG, IRS1, LYPLAL1, and DNAH10)20. Finally, we identified a set of FI-increasing alleles with a concomitant effect on insulin sensitivity and alterations in liver metabolism and inflammation, which could correspond to hepatic insulin-resistance processes, which we called the “FI-liver/lipid”. Here, FI-increasing alleles in this cluster were associated with lower circulating triglycerides, albumin, C-reactive protein, and the glucokinase regulator GCKR as the top-weighted locus. Moreover, we noticed that the majority of variants in the FI-lipodystrophy cluster and Visceral Adiposity cluster originate from the FI-BMI adjusted GWAS, contrasting with the Adiposity-Driven Hyperinsulinemia cluster, where the majority of variants stem from non-adjusted sources (see Supplementary Data 2 for details on all variants used as input to the bNMF model).
Associations between FI-partitioned polygenic scores, T2D, and cardiometabolic outcomes
Our meta-analysis analyses showed that the non-diabetogenic hyperinsulinemia clusters were associated with differing risks of CAD (Fig. 2; Source Data 2; Supplementary Data 1). For example, in all individuals (with and without T2D), each 10-unit increase in the pPS of the PIS genetic cluster was associated with lower odds of T2D (OR 0.72, P < 10−300), CAD (OR 0.96, P < 10−6), MI (OR 0.95, P < 10−5). The lower T2D odds were also observed for the EIS cluster (OR 0.93, P < 10−16), but this cluster, unlike the previous one, was associated with increased odds of CAD (OR 1.04, P < 10−7), and HTN (OR 1.03, P < 10−8). No associations with metabolic outcomes were observed for the Proinsulin cluster.

Data points represent the mean effect size (odds ratio or beta estimate). Error bars indicate 95% confidence intervals (CIs). All statistical tests were two-sided, with a significance threshold set at p = 0.0008, based on a Bonferroni adjustment for multiple comparisons. The dotted vertical line at 1 or 0 represents the null effect (no association). Subgroup results are differentiated by colors, blue (non-T2D), pink (T2D). PIS preserved insulin secretion, EIS elevated insulin secretion, and VAT visceral adipose tissue. D: Diabetogenic; ND: Non-Diabetogenic. Panel A shows associations of fasting insulin clusters with cardiometabolic outcomes; HTN hypertension, CAD coronary artery disease, MI myocardial infarction, ISTR ischemic stroke, CKD chronic kidney disease. Panel B shows associations of fasting insulin genetic clusters with T2D-exclusive outcomes; DR diabetic retinopathy, diabetic neuropathy, INS insulin use. Panel C shows cluster associations with estimated Glomerular Filtration Rate (eGFR). Source data are provided as a Source Data file.
The pPS for the four genetic clusters underlying processes of diabetogenic hyperinsulinemia were all generally associated with an increased risk of T2D, with estimated effect sizes ranging from 1.22 (P < 10−300) for the FI-lipodystrophy cluster to 1.09 (P < 10−40) for the FI-Liver/Lipid cluster. However, directions of associations differed for metabolic outcomes, including CAD, MI, stroke, and eGFR. For example, the FI-lipodystrophy cluster was also associated with lower eGFR, increased odds for CKD, and most cardiovascular outcomes considered in this study, including CAD, MI, and stroke, while the FI-Liver/Lipid cluster was associated with lower odds for CAD, MI, and lower eGFR. Furthermore, in the sex-specific analysis, the Visceral Adiposity cluster showed an increased risk of MI in males without T2D (OR = 1.008, P = 9.8 × 10−3, p-value for interaction 0.018) (Fig. 3; Source Data 3; Supplementary Data 3). Notably, this effect remained consistent after adjusting for T2D status (P = 0.03), and it was not observed in males with T2D (OR = 0.99, P = 0.22). In general, our results did not display any significant interaction between this cluster and T2D status concerning cardiovascular risk (MI, stroke, or CAD) (P > 0.05).

Panel A shows the increased risk of MI in male individuals without type 2 diabetes (T2D) (N = 155,271; 8038 cases) in the visceral adiposity cluster. Data points represent the effect size (odds ratio) for the cluster’s association with MI. Error bars indicate 95% confidence intervals (CIs) calculated using logistic regression models. All statistical tests were two-sided. The vertical line at 1 represents the null effect (no association). Panel B shows 39 proteins that displayed sex interaction; higher levels in males compared with females. The values in the heatmap are the beta estimates from regression analysis of the visceral adiposity cluster polygenic score with Myocardial Infarction (MI) in the UK Biobank. Panel C displays the proteins that showed mediation effects with P < 10−280. p values were adjusted for multiple comparisons of 0.05/39 proteins tested (p = 0.0012). Source data are provided as a Source Data file.
Outcome associations with extreme pPS
Further, we investigated the extent to which the cluster associations were consistent when individuals were categorized as having extreme genetic risk (>90th percentile of a pPS). The analyses were generally consistent with our primary findings (Supplementary Data 4; Supplementary Fig. 1). However, the effect sizes notably increased in the 90th percentile group, in some cases up to six-fold in the case of the Visceral Adiposity and the PIS clusters for high and low T2D risk, respectively. This underscores the incremental risk associated with higher genetic risk.
FI-genetic clusters associations in ancestry diverse populations
To test the generalizability of our FI-cluster results, we performed ancestry-specific analyses (Fig. 4; Source Data 4; Supplementary Table 9; Supplementary Data 5). We found that the associations between FI-pPS and T2D susceptibility had the same direction of effect among all populations although with considerable differences in effect sizes. For example, the Visceral Adiposity cluster showed higher T2D risk in Hispanic than non-Hispanic Black and non-Hispanic White individuals. Similarly, the PIS-D cluster showed lower T2D risk in non-Hispanic Black participants than the rest of the populations. Only two out of the seven FI-pPS associations were replicated among South Asian ancestry individuals: PIS, and FI-lipodystrophy cluster.

Data points represent the mean effect size (odds ratio) for the combined effect across cohorts. Error bars indicate 95% confidence intervals (CIs) calculated using logistic regression models. All statistical tests were two-sided, with a significance based on a Bonferroni adjustment for multiple comparisons. The dotted vertical line at 1 represents the null effect (no association). SA South Asian, HS Hispanic, AF African, EU European. PIS preserved insulin secretion, EIS elevated insulin secretion, VAT visceral adipose tissue, D diabetogenic, ND non-diabetogenic. Panel A shows type 2 diabetes associations (T2D), hypertension (HTN), and myocardial Infarction (MI) with fasting insulin genetic clusters. Panel B shows associations of fasting insulin clusters with diabetes complications: diabetic retinopathy (DR), and insulin use across populations (INS). Source data are provided as a Source Data file.
FI-genetic clusters associations with diabetes complications
In a subgroup analysis to evaluate complications in individuals with T2D (n = 208,268), we observed that the PIS cluster was associated with lower odds of diabetic neuropathy (OR, 0.70, P < 10−5) diabetic retinopathy (OR 0.86, P < 10−18), and insulin use (OR 0.93, P < 10−7) (Fig. 2, Supplementary Data 1). Among the genetic clusters defined by elevated FI levels with deeper metabolic alterations, we found that FI-lipodystrophy and Adiposity-Driven Hyperinsulinemia clusters exhibited the largest number of associations with T2D complications including MI, diabetic retinopathy, CKD, ISTR, and insulin use. Interestingly, in individuals without T2D (n = 895,990), we observed that FI-lipodystrophy (P < 10−17), adiposity-driven hyperinsulinemia (P < 10−8), and EIS (P < 10−9) clusters maintained their associations with increased CAD and MI risk.
Molecular profiling of FI-partitioned genetic scores
We identified 447 significant proteins in at least one of the FI-pPS (P < 2.50e−06) (Supplementary Data 6). The Hepatic-Insulin Resistance cluster captured a large number of associations (N = 335), followed by the FI-lipodystrophy cluster (N = 128) (Supplementary Fig. 2). Adiponectin (ADIPOQ) and Leptin (LEP) were among the significant proteins in the FI-lipodystrophy cluster. Both proteins showed negative associations, consistent with the directional traits in the cluster input. Notably, associations with several proteins, including IGFBP2, LPL, FGF21, TNFRSF19, and TNFRSF21, showed effects in the opposite direction from the FI-Liver/Lipid cluster. Subclusters of significant proteins exhibited distinct T2D risk profiles among the FI clusters. For instance, the subcluster consisting of SHBG, CKB, IGFBP2, ADIPOQ, CD300LG, and LPL displayed positive associations within the FI-lipodystrophy cluster, aligning with the anticipated T2D profile. In contrast, the FI-Liver/Lipid cluster showed negative correlations, indicating an opposite T2D risk profile.
Next, to detect sex-specific associations with protein abundances, we identified that the Visceral Adiposity cluster revealed 39 significant protein associations with sex interaction to a nominal level (p < 0.01). There was a clear pattern where males with higher pPS had increased protein levels, a distinct pattern from females (Fig. 3; Supplementary Data 7). These sex-specific proteins were enriched in the lysosome (FDR = 0.01) and complement and coagulation cascades (FDR = 0.02) pathways (Supplementary Data 8). Given that the Visceral Adiposity cluster was associated with MI, we tested whether these proteins mediated the association with MI. Notably, three of the sex-specific proteins (CEACAM5, GALNT10, and SFRP1) mediated the risk between the Visceral Adiposity cluster and MI in males without T2D (P < 10−280) (Supplementary Data 9). These results support the role of this cluster in influencing cardiovascular risk specifically in males.
Comparison with T2D genetic clusters reveals FI-distinct domain
We also explored the contribution of FI-specific genetic variants and traits that differ from the T2D genetic domains, our results showed that Visceral Adiposity cluster does not resemble any of the T2D clusters. In terms of cluster-defining phenotypes, this cluster displays a maximum correlation of 0.36 (Supplementary Fig. 3) with T2D multi-ancestry clusters10. In the LD analysis between FI and T2D cluster variants, we found that of the 25 variants top-weighted in the visceral adiposity cluster, only three variants were in LD > 0.60 with T2D cluster loci10; rs1861882 (0.75), rs3775380 (0.94), and rs1011731 (0.96) (Supplementary Table 10). Furthermore, in the sex-specific protein associations, out of the 39 nominally significant proteins identified, 33 (84.6%) were distinct and did not show significant associations within any of the T2D genetic clusters, including the three proteins mediating the effect of this cluster with MI (Fig. 3C Supplementary Data 7). The rest of the clusters, particularly the FI-lipodystrophy, adiposity-driven hyperinsulinemia, and FI-Liver/Lipid clusters appear to capture some traits or genetic variants akin to their counterparts within T2D clusters, although with differences in direction or effect sizes for outcome associations (Supplementary Table 11).
Discussion
Twelve T2D genetic clusters have been identified related to mechanistic processes. We hypothesized that clustering using genetic loci associated with FI rather than with T2D would provide insight into the relationship between FI and T2D and identify new genetic clusters relevant to T2D, but not yet discovered because the genetic variants were not significantly associated with T2D at the genome-wide threshold. We identified seven distinctive FI genetic clusters representing different molecular processes for increasing FI levels, one of them with notable male-specific effects on protein levels and cardiovascular risk not previously captured by T2D genetic clusters. Our results ranged from clusters of variants with effects on increased FI, and a lack of increased risk of T2D (non-diabetogenic hyperinsulinemia), to clusters of variants that increased FI and T2D risk with demonstrated strong effects on body fat distribution, liver, lipid, and inflammatory processes (diabetogenic hyperinsulinemia). In addition, our findings provide important insights into granular metabolic processes that decouple increasing FI levels from cardiovascular conditions. The non-diabetogenic hyperinsulinemia clusters were surprisingly associated with both increased and decreased risks of cardiovascular conditions.
This work expands upon previous studies clustering T2D and FI-related loci8,9,10,21. Yaghootkar et al. studied 11 FI loci and eight traits, identifying a lipodystrophy-like genetic cluster, which was subsequently also recaptured in T2D genetic clustering efforts8,9,10. Our analysis advances prior research by including 230 FI loci and related traits and adds a more granular analysis of the mechanisms underlying T2D and cardiometabolic risk. Our results support the notion that insulin resistance associated with increased fat distribution in central compartments is a key contributing factor to T2D risk. Both the traits and genetic loci comprising the diabetogenic hyperinsulinemia clusters reflect this adverse metabolic physiology. The top loci in the visceral adiposity cluster MYO1A, BMP2, and ARL5 increased systolic and diastolic blood pressure, as well as hypertension22,23. Furthermore, rs1552245 in MYO1A has shown evidence of increased cardiovascular risk in non-T2D individuals24. The FI-lipodystrophy cluster with reduced subcutaneous adipose tissue and genetic risk driven by mostly BMI-adjusted variants relates to a “lipodystrophy-like” phenotype previously described by Yaghootkar21, Lotta25, Udler8, Kim9, and Smith10. Moreover, the adiposity-driven hyperinsulinemia cluster appears to represent a mechanism of insulin resistance concomitant with increments in body fat. (rs4280233 at FTO, rs10938397 at GNPDA2, and rs7138803 at BCDIN3D26,27,28. In addition, our findings support the evidence that excessive visceral adiposity strongly contributes to cardiovascular risk29. Consistent with an adverse impact of excessive visceral adiposity, the diabetogenic hyperinsulinemia clusters accurately captured the heightened risk of cardiovascular outcomes such as HTN, CAD, MI, ISTR, stroke, and CKD, associations that remained consistent regardless of the presence of T2D.
Our findings unveiled a diabetogenic cluster with notable sex-specific associations that influence cardiovascular risk in males, an aspect not previously captured by T2D genetic clusters. The primary reason why this cluster remained unidentified within the T2D clustering is that the genetic variants it comprises were not significantly associated with T2D at the genome-wide threshold. For instance, three of the top-weighted variants with low LD with the T2D multi-ancestry clusters did not reach genome-wide significance for T2D: rs1552245 at MYOLA (P = 0.078)30, rs979012 at BMP2 (P = 0.088)31, as well as rs4773173 at COL4A2 (P = 0.015)30. Moreover, the sex-specific effects observed within the visceral adiposity cluster arise from its impact on central fat distribution, as indicated by the higher waist adjusted by BMI in males. Furthermore, our study successfully captured the sex-specific effect of this cluster through proteomic associations. Biological sex has been shown to play a significant role in shaping the pathophysiology of cardiovascular diseases32. Recently, proteomic variances between males and females and their correlation with distinct rates of cardiovascular risk outcomes have been elucidated33,34,35,36, revealing sex-specific protein mechanisms in cardiovascular health. Particularly, disparities between sexes have been observed in MI, with men often experiencing earlier onset and greater severity compared to age-matched women32. In our study, we identified 39 proteins exhibiting higher abundances in males compared to females. Three of them mediate the association between Visceral Adiposity cluster and MI. One of them, SFRP1 stimulate adipogenesis in both human and murine in vitro models37,38, it also positively correlated with insulin sensitivity, possibly via effects on adipokine secretion39. Previous research has suggested a close association between SFRP1 and myocardial fibrosis, as well as cardiac remodeling40,41. The higher protein abundances in males observed in our study correlate to the effects of a loss of function observed in animal studies42 suggesting a compensatory response within the cellular system. The protein quantification downstream of the visceral adipose tissue genetic cluster, coupled with their interaction with sex, served as a novel avenue for identifying translatable therapeutic targets related to cardiovascular risk in males prior to the onset of T2D.
The non-diabetogenic clusters, characterized by alleles associated with increased FI and decreased risk of T2D, suggest mechanisms involving increased insulin resistance coupled with compensatory insulin production. In particular, the PIS cluster may be influenced by the gene expression of ADCY5, which is known to regulate insulin secretion from human islets43. This cluster features the opposite allele (rs11708067-G) compared to the T2D beta-cell cluster identified by Udler et al. 8, which contains the rs11708067-A allele. The rs11708067-A allele is linked to the reduced function of an islet enhancer, leading to decreased ADCY5 expression and impaired insulin secretion44. Similarly, the rs7903146-T allele in TCF7L2 is associated with compromised beta-cell function45. The alleles in the preserved beta-insulin secretion cluster (rs11708067-G, rs7903146-C, and rs11039290-A) contribute to incremental increases in insulin production. This increased production may enhance or preserve beta-cell function, potentially leading to lower blood glucose levels and reduced cardiometabolic risk. Alternatively, these findings could reflect an early compensatory response when glycemia is normal or only slightly elevated2,46,47. Furthermore, it is crucial to acknowledge that the alleles within the PIS cluster were selected for this analysis due to their association with increased FI levels, and they exhibit a decreased risk for T2D; as noted above, the alternative alleles increase T2D risk and are associated with reduced insulin secretion. The observed effects of the T2D-reducing alleles may be attributed to collider bias, particularly since the FI GWAS was conducted in individuals without T2D.
We identified two clusters where the association with T2D was decoupled from CAD risk, one diabetogenic and one non-diabetogenic cluster. The hepatic insulin resistance cluster was the only diabetogenic cluster that exhibited evidence of decreased cardiovascular risk. Top weighted loci in this cluster have been connected to fatty liver disease (GCKR)48 and hepatic glycogen storage (RP11-10A14.4/PPP1R3B)49,50,51, and the top-weighted trait was decreased serum triglyceride levels, suggesting that the cluster association with reduced CAD risk might be explained by loci impacting hepatic storage of triglycerides and/or glycogen. This cluster correlates with the liver lipid cluster previously identified by Udler et al. 8 and is consistent with the finding by DiCorpo et al. 52 that adjusting for serum triglycerides in a regression model partially attenuated the associations with CAD. The second instance of decoupled associations between T2D and CAD involved the non-diabetogenic EIS cluster which was significantly associated with reduced T2D risk but increased CAD risk.
Our study reveals several noteworthy associations that merit further examination. Notably, the FI-Liver/Lipid cluster demonstrates a significant association with a lower risk of MI. This finding is consistent with recent research by Ahmed et al. 53, which identified 13 genetic variants associated with impaired hepatic triglyceride export. Their study showed that liver fat-increasing alleles were correlated with a reduced risk of coronary artery disease and MI but an elevated risk of type 2 diabetes, underscoring the heterogeneous effects of liver fat on health outcomes. Additionally, the FI-Liver/Lipid cluster is associated with a reduced eGFR, a marker of kidney function. The connection between lower MI risk and reduced eGFR within the FI-Liver/Lipid cluster suggests that mechanisms implicated in diabetogenic pathways related to hepatic function may influence multiple disease outcomes through complex and interrelated mechanisms. This emphasizes the need for further research to elucidate the pathways linking insulin resistance with cardiovascular and renal diseases and to explore how these associations might inform personalized intervention strategies.
This study exhibits several strengths; first, the large-scale GWAS provided the necessary data to establish FI clusters for various pertinent outcomes. It utilized a substantial dataset (comprising over a million individuals) from diverse ancestries to assess both the correlation between these clusters and prevalent disease states and their ability to distinguish between diabetes and non-diabetes. The study also has some limitations; we acknowledge the potential bias stemming from the hyperbolic association, where elevated insulin secretion might be driven by insulin resistance as a compensatory mechanism rather than by genetic factors alone. Insulin responses, which reflect basal beta cell function, are inversely related to insulin action across varying degrees of glucose intolerance46,47,54. This physiological complexity complicates the task of isolating these two mechanisms, making it difficult to disentangle their individual genetic contributions. We also acknowledge some overlap in the GWAS discovery studies and the multi-ancestry cohort outcome studies. This inclusion may introduce overfitting in our models; however, we assessed the meta-analysis association analysis with and without the FHS cohort and did not observe significant differences in our results. Another limitation of our study is the inclusion of variants with subthreshold GWAS significance levels, with only 20.43% reaching genome-wide significance for FI (N = 47). However, to address this concern, we conducted a sensitivity analysis focusing on variants meeting the stringent criterion of p < 5 × 10−8. This stricter selection led to the identification of four clusters—PIS, Adiposity, VAT, and FI-Liver/Lipid—from a subset of 47 GWAS-significant variants, which corresponds closely to four of the original seven clusters, thereby reinforcing the robustness of our findings (Supplementary Fig. 4). Moreover, 88% of the full set of 101 variants highly weighted in at least one cluster were associated with at least one metabolic trait at genome-wide significance. This supports their relevance in metabolic regulation and provides clarity on how these alleles potentially influence FI, demonstrating that broader metabolic associations beyond FI are integral to understanding the allelic impact on metabolic traits. Another limitation is that SNPs and traits GWAS summary statistics utilized in the bNMF algorithm represent studies conducted in individuals of European ancestry only. To further enhance our understanding of the behavior of these clusters across diverse populations, future research should encompass multi-ancestry GWAS results. Finally, the sample size of South-Asian ancestry individuals might limit our capability to detect significant associations of FI genetic clusters with cardiovascular outcomes in this population group.
Our clustering approach offers valuable insights to decouple hyperinsulinemia from T2D and cardiovascular conditions, presenting a novel avenue for patient’s risk stratification for precision medicine approaches. The outcomes of this study can aid in identifying groups that require targeted therapeutic approaches, or exploring pathways aimed at preventing disease onset. This represents a significant stride towards genetically informed patient prevention and management of T2D, which is crucial given the limitations of current therapeutic approaches in addressing disease heterogeneity and predicting future disease trajectories.
In summary, through the examination of a key intermediate trait, FI, we gained detailed insights into the heterogeneity of T2D and cardiovascular conditions. By clustering FI-related loci and traits, we identified seven genetic clusters of FI that have both increasing and decreasing effects on CAD risk which are not always aligned with T2D risk. Our findings challenge the assumption that increasing FI levels always correlates with unfavorable outcomes. Moreover, the study of proteins downstream of process-specific genetic variation served as a novel avenue for identifying translatable therapeutic targets related to cardiovascular risk in males without T2D.
Methods
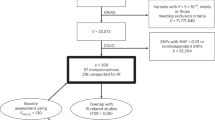
Figure 5 summarizes our study approach and the implemented analyses. To generate FI clusters, we gathered association summary statistics for FI and related metabolic traits to identify clusters of genetic variants and traits sharing similar physiological characteristics. Each cluster is defined by a set of FI-increasing alleles with weights derived from the clustering procedure. We used this information to generate partitioned polygenic scores (pPS) for the different clusters among 1,104,258 multi-ancestry individuals from five studies; Million Veterans Program (MVP), UK Biobank (UKBB), Mass General Brigham Biobank (MGBB), ASPirin in Reducing Events in the Elderly Study (ASPREE), Framingham Heart Study (FHS). A brief description of each contributing cohort is provided in Supplementary Data 10. We meta-analyzed the associations between each pPS and several cardiometabolic outcomes. Finally, we tested our FI-pPS against 2923 proteins available in UK Biobank55 to explore molecular mechanisms underlying each pPS, and to contrast these associations with previous T2D clusters. Analysis of the UK Biobank was conducted under application #42614. Data analysis was approved by the Mass General Brigham Institutional Review Board (Boston, MA).

This figure was created in BioRender. Sevilla, M. (2024) https://BioRender.com/u86n272.
Identification of genetic clusters
First, we leveraged association summary statistics to identify genetic variants previously associated with increased FI. These FI association summary statistics are listed in Supplementary Table 12. These GWAS were performed in population groups described as European ancestry and in individuals who did not have diabetes12,13,14,15,16. Then we obtained association summary statistics for the identified FI-associated genetic variants for additional traits that span cardio-metabolic domains (glycemic, anthropometric, body composition, inflammation, lipids, hormones, and liver function) available in the Accelerating Medicines Partnership Common Metabolic Diseases Knowledge Portal56 or the UK Biobank57. These traits were further pruned using a correlation threshold |r|<0.85 between the Z-scores of each trait in the SNP-trait matrix. This selection of non-completely overlapping traits is critical to ensure that each phenotype provides additional information and contributes to the generation of the matrix.
We used a Bayesian non-negative matrix factorization (bNMF) clustering approach to group FI-associated variants into clusters of genetic variants with molecular and clinical similarities. The bNMF algorithm factorizes the inputs into two matrices with an optimal rank K, corresponding to the association matrix of variants and traits to the number of clusters9. The maximum posterior solution at the most probable number of clusters was selected for downstream analysis. The input for the bNMF approach initially comprised 230 distinct genetic variants associated with fasting insulin (FI) at a significance level of p < 5 × 10−5 (Supplementary Data 2) and 43 traits (Supplementary Table 13). Additionally, we conducted a sensitivity analysis using a more stringent threshold, including only variants with p < 5 × 10−8 for FI in European (N = 39) or Multi-ancestry (N = 8) GWAS. The trait effect sizes were aligned to the FI risk-increasing allele, which was determined using the Lagou12 (FI) and Chen (FIadjBMI) GWAS13. The cut-off value to define top weighted variants and traits that characterize each cluster was 0.84. This was determined by the optimal threshold to define the beginning of the long tail of the distribution of clusters’ weights across all clusters. The most highly associated traits determined the main features of each cluster9. The bNMF algorithm was implemented in R with open-source code available at our GitHub repository (see the section “Code availability”).
Generation of FI-partitioned polygenic scores
pPS were calculated for each study participant in each cohort by summing the weights of the gene variants derived from the bNMF clustering algorithm, as has been described before8,9,10,52. Within each cohort, we used directly genotyped variants or imputed variants. If a genetic variant was unavailable or ambiguous, we used available proxies based on r2 ≥ 0.8. To prevent unintentional allele swapping, the frequencies of the alleles used in developing the clusters for hyperinsulinemia were compared with European ancestry individuals from the Mass General Brigham Biobank (MGBB). The genotyping information and imputation methods from the different studies included in our study are described in Supplementary Data 10. The median and distribution of the pPS were comparable across the included studies.
Biobank and cohort analyses
For individual-level data, informed consent was obtained from all participants in all datasets. We complied with all relevant ethical regulations when analyzing genetic data from human research participants. Individuals were not compensated for participation in this study.
Associations between FI-pPS, and cardiometabolic outcomes
The primary outcomes were T2D, CAD, myocardial infarction (MI), stroke, ischemic stroke (ISTR), hypertension (HTN), chronic kidney disease (CKD), and estimated glomerular filtration rate (eGFR). The rationale for investigating these outcomes was because of the association of FI levels with metabolic, renal, and cardiovascular conditions. Outcome definitions were generally consistent across studies, and the study-specific definitions can be found in Supplementary Data 10. These outcomes were based on the most recent information available from observational longitudinal studies and at baseline in the study with intervention; ASPREE.
Secondary outcomes included diabetes complications defined as neuropathy, retinopathy, kidney disease occurring in individuals with diabetes, and insulin use. Secondary outcome definitions can also be found in Supplementary Data 10.
Each cohort received a harmonized statistical analysis plan with a standardized definition of pPS, statistical models, covariates, and outcome definitions. We used multivariable regression models to estimate effect sizes and the odds of primary outcomes for each pPS. We computed pPS on a continuous scale and presented estimates for a 10-unit increase in polygenic scores. Models were adjusted for age, sex, and the 10 principal components (PCs) of genetic ancestry. For analysis in ASPREE, we also included the country of recruitment as a covariate. For analyses conducted in the UK Biobank, only PCs that reached the nominal significance threshold for association in the null model were included. Sex-stratified analyses were conducted in the UK Biobank across all outcomes to investigate potential sex-specific effects of the FI-pPSs. In a secondary analysis, in all cohorts, we categorized individuals according to the distribution of pPS and conducted association analyses comparing individuals in the top 10th distribution of each pPS against the rest. Because the median and distribution of the polygenic scores across the cohorts were consistent, we used the MGBB cohort to standardize the cutoff point to define high genetic risk among the five cohorts.
For multi-ancestry cohorts, we conducted subgroup analyses stratified by race/ethnicity population groups. To ensure accurate race/ethnic classification of participants, we employed a combination of self-reported race/ethnicity and genetic-derived continental ancestry approaches. We first classified individuals with their self-reported race/ethnicity and then cross-referenced this information with their genetic PCs to verify the accuracy of the classification. Individuals who did not match their self-reported race/ethnicity with their genetic PCs were excluded from the specific ancestry group. The MVP study used the Harmonized Ancestry and Race Ethnicity (HARE) algorithm58. We also conducted subgroup analyses stratified by T2D status.
Outcome-specific study-level regression coefficients were combined by inverse-variance weighted fixed effects meta-analysis. The R package “meta” version 4.18-2 was used to combine estimates of effect and produce an overall association test. We used the \({I}^{2}\) statistics to assess between-study heterogeneity. We considered a two-sided α level of 0.0008 based on a Bonferroni adjustment for performing 56 tests, including eight outcomes and seven genetic clusters. Statistical analyses were carried out using R software, versions 3.5.1 and 4.1.1.
Associations between FI-pPS and protein abundances
To explore molecular mechanisms underlying pPS and contrast these associations with previous T2D clusters we tested our FI-pPS against 2923 proteins available in UKBB55. Details regarding the assay have been described in detail59. Normalized protein expression (NPX) units are generated by normalization to the extension control and further normalization to the plate control and reported on a log2 scale. We used linear models to estimate the effect of the association of pPS and NPX, correcting for age, age2, sex, and 10 genetic principal components. Additionally, we performed models testing the interaction between sex and pPS to find sex-specific protein associations adjusting for the same covariates. All models were adjusted for multiple comparisons, setting a p-value of 2.50 × 10−6 as significant (0.05/2923 proteins/7 clusters). We also tested the association of these proteins with prevalent T2D adjusting for the same set of covariates. Using all proteins with at least one significant association with an FI-pPS, we performed hierarchical clustering of the effect estimates for each FI-pPS. Finally, we conducted a pathway analysis with the proteins identified in the sex-interaction analysis using the KEGG library (Kyoto Encyclopedia of Genes and Genomes) through the Enrichr tool60,61,62.
We employed a mediation analysis to evaluate the extent to which the association between genetic cluster and disease risk is mediated by the plasma proteins, adjusting for the covariates described above. Individual mediation analysis was performed using the R package mediation (v4.5.0; Tingley 2014). P values were adjusted for multiple comparisons of 0.05/39 proteins tested (p = 0.0012).
Comparison against existing T2D clusters
The comparison with existing T2D genetic clusters8,9,10 was performed across three levels of data: phenotypic, genetic, and protein associations. First, to explore phenotypic differences driving the formation of the clusters, we assessed the correlation between trait weights in FI and T2D clusters using Spearman correlation. Next, we tested the linkage disequilibrium (LD) of each top-weighted variant in the FI-clusters with the genetic variants included in the T2D genetic clusters10. Finally, we tested the extent to which the FI clusters captured different protein associations from T2D clusters10.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The UK Biobank (UKB) whole-genome sequence data can be accessed through UKB Research Analysis Platform (RAP), through the UKB approval system (https://www.ukbiobank.ac.uk). The Mass General Brigham Biobank (MGBB) individual-level data are available from https://personalizedmedicine.partners.org/Biobank/Default.aspx, where the data is available through Institutional Review Board (IRB) approval and therefore not publicly available. ASPirin in Reducing Events in the Elderly Study data is available through internal approval https://ams.aspree.org/public/ The Framingham Heart Study genomic data analyzed in the current study are available through restricted access via the Genotypes and Phenotypes (dbGaP) Exchange area in the database of repositories phs000974 and phs000007. The Million Veterans Program data that support the findings of this study are not openly available due to reasons of sensitivity and are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Code availability
Code for variant pre-processing, bNMF clustering, is available at https://github.com/gwas-partitioning/bnmf-clustering. https://doi.org/10.5281/zenodo.14218909 Code for polygenic risk scores is available at https://github.com/manning-lab/polygenic-risk-scores-pipeline. https://doi.org/10.5281/zenodo.14217974.
References
Taylor, R. Insulin resistance and type 2 diabetes. Diabetes 61, 778 (2012).
Stumvoll, M., Goldstein, B. J. & Van Haeften, T. W. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 365, 1333–1346 (2005).
Samuel, V. T. & Shulman, G. I. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J. Clin. Investig. 126, 12–22 (2016).
MO, G. et al. Fasting insulin reflects heterogeneous physiological processes: role of insulin clearance. Am. J. Physiol. Endocrinol. Metab. 301, 402–408 (2011).
Després, J.-P. et al. Hyperinsulinemia as an independent risk factor for ischemic heart disease. N. Engl. J. Med. 334, 952–958 (1996).
Wang, F., Han, L. & Hu, D. Fasting insulin, insulin resistance and risk of hypertension in the general population: a meta-analysis. Clin. Chim. Acta 464, 57–63 (2017).
Gutch, M., Kumar, S., Razi, S. M., Gupta, K. & Gupta, A. Assessment of insulin sensitivity/resistance. Indian J. Endocrinol. Metab. 19, 160 (2015).
Udler, M. S. et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med. 15, 1002654 (2018).
Kim, H. et al. High-throughput genetic clustering of type 2 diabetes loci reveals heterogeneous mechanistic pathways of metabolic disease. Diabetologia 66, 495–507 (2022).
Smith, K. et al. Multi-ancestry polygenic mechanisms of type 2 diabetes. Nat. Med. https://doi.org/10.1038/S41591-024-02865-3 (2024).
Suzuki, K. et al. Genetic drivers of heterogeneity in type 2 diabetes pathophysiology. Nature 627, 347–357 (2024).
Lagou, V. et al. Sex-dimorphic genetic effects and novel loci for fasting glucose and insulin variability. Nat. Commun. 12, 24 (2021).
Chen, J. et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet. 53, 840 (2021).
Laakso, M. et al. The metabolic syndrome in men study: a resource for studies of metabolic and cardiovascular diseases. J. Lipid Res. 58, 481–493 (2017).
Locke, A. E. et al. Exome sequencing of Finnish isolates enhances rare-variant association power. Nature 572, 323 (2019).
Dupuis, J. et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 42, 105 (2010).
Xue, A. et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 9, 1–14 (2018).
Klimentidis, Y. C. & Arora, A. Interaction of insulin resistance and related genetic variants with triglyceride-associated genetic variants. Circ. Cardiovasc. Genet. 9, 154–161 (2016).
Guiu-Jurado, E. et al. Bone morphogenetic protein 2 (BMP2) may contribute to partition of energy storage into visceral and subcutaneous fat depots. Obesity (Silver Spring) 24, 2092–2100 (2016).
Semple, R. K., Savage, D. B., Cochran, E. K., Gorden, P. & O’Rahilly, S. Genetic syndromes of severe insulin resistance. Endocr. Rev. 32, 498–514 (2011).
Yaghootkar, H. et al. Genetic evidence for a normal-weight ‘metabolically obese’ phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes 63, 4369–4377 (2014).
Evangelou, E. et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 50, 1412–1425 (2018).
Barton, A. R., Sherman, M. A., Mukamel, R. E. & Loh, P. R. Whole-exome imputation within UK Biobank powers rare coding variant association and fine-mapping analyses. Nat. Genet. 53, 1260–1269 (2021).
Zhao, W. et al. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat. Genet. 49, 1450–1457 (2017).
Lotta, L. A. et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet. 49, 17–26 (2016).
Peng, S. et al. FTO gene polymorphisms and obesity risk: a meta-analysis. BMC Med. 9, 1–15 (2011).
Cheung, C. Y. Y. et al. Genetic variants associated with persistent central obesity and the metabolic syndrome in a 12-year longitudinal study. Eur. J. Endocrinol. 164, 381–388 (2011).
Tang, L. et al. Meta-analyses between 18 candidate genetic markers and overweight/obesity. Diagn. Pathol. 9, 1–12 (2014).
Després, J. P. Body fat distribution and risk of cardiovascular disease. Circulation 126, 1301–1313 (2012).
Mahajan, A. et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 54, 560–572 (2022).
Mahajan, A. et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet. 46, 234–244 (2014).
Regitz-Zagrosek, V. & Kararigas, G. Mechanistic pathways of sex differences in cardiovascular disease. Physiol. Rev. 97, 1–37 (2017).
Barta, B. A. et al. Sex-related differences of early cardiac functional and proteomic alterations in a rat model of myocardial ischemia. J. Transl. Med. 19, 507–522 (2021).
Chandramouli, C. et al. Sex differences in proteomic correlates of coronary microvascular dysfunction among patients with heart failure and preserved ejection fraction. Eur. J. Heart Fail. 24, 681–684 (2022).
de Bakker, M. et al. Sex-based differences in cardiovascular proteomic profiles and their associations with adverse outcomes in patients with chronic heart failure. Biol. Sex Differ. 14, 29 (2023).
Li, M. et al. Core functional nodes and sex-specific pathways in human ischaemic and dilated cardiomyopathy. Nat. Commun. 11, 2843 (2020).
Lagathu, C. et al. Secreted frizzled-related protein 1 regulates adipose tissue expansion and is dysregulated in severe obesity. Int J Obes (Lond) 34, 1695–1705 (2010).
Gauger, K. J. et al. Mice deficient in Sfrp1 exhibit increased adiposity, dysregulated glucose metabolism, and enhanced macrophage infiltration. PLoS ONE 8, 78320 (2013).
Ehrlund, A. et al. Characterization of the Wnt inhibitors secreted frizzled-related proteins (SFRPs) in human adipose tissue. J. Clin. Endocrinol. Metab. 98, 503–508 (2013).
Gibb, N., Lavery, D. L. & Hoppler, S. Sfrp1 promotes cardiomyocyte differentiation in Xenopus via negative-feedback regulation of Wnt signalling. Development (Cambridge) 140, 1537–1549 (2013).
Huang, A. & Huang, Y. Role of Sfrps in cardiovascular disease. Ther. Adv. Chronic Dis. 11, https://doi.org/10.1177/2040622320901990 (2020).
Sklepkiewicz, P. et al. Loss of secreted frizzled-related protein-1 leads to deterioration of cardiac function in mice and plays a role in human cardiomyopathy. Circ. Heart Fail. 8, 362–372 (2015).
Hodson, D. J. et al. ADCY5 couples glucose to insulin secretion in human islets. Diabetes 63, 3009–3021 (2014).
Roman, T. S. et al. A type 2 diabetes-associated functional regulatory variant in a pancreatic islet enhancer at the ADCY5 locus. Diabetes 66, 2521–2530 (2017).
Cropano, C. et al. The rs7903146 variant in the tcf7l2 gene increases the risk of prediabetes/type 2 diabetes in obese adolescents by impairing b-cell function and hepatic insulin sensitivity. Diabetes Care 40, 1082–1089 (2017).
Efrat, S. Beta-cell expansion for therapeutic compensation of insulin resistance in type 2 diabetes. Int. J. Exp. Diabesity Res. 4, 1–5 (2003).
Cerf, M. E. Beta cell dynamics: beta cell replenishment, beta cell compensation and diabetes. Endocrine 44, 303–311 (2013).
Li, J. et al. Contribution of Rs780094 and Rs1260326 Polymorphisms in GCKR gene to non-alcoholic fatty liver disease: a meta-analysis Involving 26,552 Participants. Endocr. Metab. Immune Disord. Drug Targets 21, 1696–1708 (2020).
Kahali, B. et al. A noncoding variant near PPP1R3B promotes liver glycogen storage and MetS, but protects against myocardial infarction. J. Clin. Endocrinol. Metabolism 106, 372–387 (2021).
Stender, S. et al. Relationship between genetic variation at PPP1R3B and levels of liver glycogen and triglyceride. Hepatology 67, 2182–2195 (2018).
Manning, A. K. et al. A long non-coding RNA, LOC157273, is an effector transcript at the chromosome 8p23.1-PPP1R3B metabolic traits and type 2 diabetes risk locus. Front. Genet. 11, 521982 (2020).
DiCorpo, D. et al. Type 2 diabetes partitioned polygenic scores associate with disease outcomes in 454,193 individuals across 13 cohorts. Diabetes Care 45, 674–683 (2022).
Ahmed, A., Cule, M., Bell, J. D., Sattar, N. & Yaghootkar, H. Differing genetic variants associated with liver fat and their contrasting relationships with cardiovascular diseases and cancer. J. Hepatol. https://doi.org/10.1016/J.JHEP.2024.06.030 (2024).
Ferrannini, E. & Mari, A. Beta cell function and its relation to insulin actions in humans: z critical appraisal. Diabetologia 47, 943–956 (2004).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 622, 329–338 (2023).
Common Metabolic Disease Knowledge Portal - Home. https://hugeamp.org/.
UK Biobank — Neale lab. http://www.nealelab.is/uk-biobank.
Fang, H. et al. Harmonizing genetic ancestry and self-identified race/ethnicity in genome-wide association studies. Am. J. Hum. Genet. 105, 763–772 (2019).
Zhong, W. et al. Next generation plasma proteome profiling to monitor health and disease. Nat. Commun. 12, 1–12 (2021).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 14, 128 (2013).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Xie, Z. et al. Gene set knowledge discovery with enrichr. Curr. Protoc. 1, e90 (2021).
Acknowledgements
FHS: Supported by the National Heart, Lung and Blood Institute (NHLBI) contracts 75N92019D00031, HHSN268201500001I, and N01-HC-25195, and its contract with Affymetrix, Inc for genotyping services (contract number N02‐HL‐6‐4278). The analyses reflect intellectual input and resource development from the Framingham Heart Study investigators participating in the SNP Health Association Resource (SHARe) project. F.H.S. and J.B.M. are also supported by the National Institute for Diabetes and Digestive and Kidney Diseases (NIDDK) R01 DK078616, U01 DK078616, and UM1 DK078616, National Heart, Lung, and Blood Institute (NHLBI) R01 HL151855. ASPREE: We thank the ASPREE trial staff and participants, and the general practitioners and staff of the medical clinics who cared for the participants. The ASPREE Biobank is supported by a Flagship cluster grant (including the Commonwealth Scientific and Industrial Research Organization, Monash University, Menzies Research Institute, Australian National University, University of Melbourne); and grants (U01AG029824 and U19AG062682) from the National Institute on Aging and the National Cancer Institute at the National Institutes of Health, by grants (334047 and 1127060) from the National Health and Medical Research Council of Australia, and by Monash University and the Victorian Cancer Agency. MVP: This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration, and was supported by awards MVP003/MVP028 I01-BX003362, MVP009/MVP037 I01-BX005831, and MVP001 I01-BX004821. This publication does not represent the views of the Department of Veteran Affairs or the United States Government. MVP participants went through a counseling process before they enrolled and provided consent to have their electronic health records reviewed. The VA Central Institutional Review Board gave approval for the study protocol in accordance with the principles of the Declaration of Helsinki. Additional Funding: Supported in part by the NHLBI contracts R01HL151855 and NIDDK contract M1DK078616. The CHARGE Consortium infrastructure is partly supported by the National Heart, Lung, and Blood Institute (NHLBI) grant R01HL105756. This study was also supported by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. Research reported in this publication was supported by the National Institutes of Health for the project “Polygenic Risk Methods in Diverse Populations (PRIMED) Consortium,” with grant funding for Study Sites D-PRISM (U01HG011723). M.S.-G. is supported by American Diabetes Association grant 9-22-PDFPM-04 J.M. and R.S. are supported by W81XWH-17-1-0347 and W81XWH-17-1-0348, from the Congressionally Directed Medical Research Programs of the Department of Defense. M.S.-G. is supported by the Eris M. Field Chair in Diabetes Research. P.L. is supported by a National Heart Foundation Future Leader Fellowship (102604). J.J.M. is supported by an NHMRC Leadership Fellowship (IG1173690). M.S-G., D.D., N.W., J.D., C.L., J.B.M., A.K.M.: are supported by NIDDK UM01 DK078616 W., D.D., J.D., C.L., J.B.M. are supported by NHLBI R01HL151855 J.M. is partially supported by funding from the ADA (7-21-JDFM-005) and the National Institutes of Health (P30 DK40561 and UG1 HD107691). J.C.F. is supported by NIH K24 DK110550 S.R. is supported by VA award IK2-CX001907 and a Webb-Waring Biomedical Research Award from the Boettcher Foundation E.M.L. is supported by National Heart, Lung, and Blood Institute (NHLBI) R01 HL151855 and a Webb-Waring Biomedical Research Award from the Boettcher Foundation L.S.P. is supported in part by VA awards CSP #2008, I01 CX001899, I01 CX001737, and I01 BX005831; NIH awards R01 DK127083, R03 AI133172, R21 AI156161, U01 DK098246, UL1 TR002378; and a Cystic Fibrosis Foundation award PHILLI12A0. J.J.M. is supported by an Australian National Health and Medical Research Council (NHMRC) Leadership Award IG1173690. C.Y. is supported by a Vanguard Grant (108071-2024_VG) from the National Heart Foundation of Australia.
Author information
Authors and Affiliations
Contributions
M.S.-G., N.W., K.S., H.K.., D.D, A.J., E.L., and J.C. performed the analysis. A.K.M., M.U., and M.S.-G. conceived the study. M.S.-G., J.M., and N.W. wrote the first draft of the manuscript. A.K.M. and M.S.-G. are the guarantors of this work. J.M., M.U., J.B.M., J.C.F., S.R., J.D., D.D., R.S., and K.E.W. contributed to editing the manuscript (comments and edits). S.H., C.-T.L., C.Y., P.L., K.M.C., P.S.T., L.S.P., M.O.G., J.I.R., J.D., J.J.Z., and J.J.M. provided data and aided in data interpretation and manuscript revision. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.C.F. gets consulting honoraria from Novo Nordisk and AstraZeneca; speaker honoraria from Novo Nordisk, AstraZeneca, and Merck for research lectures over which J.C.F. had full control on content. J.B.M. is an Academic Associate for Quest Diagnostics’ endocrine R&D. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Timothy Frayling and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sevilla-González, M., Smith, K., Wang, N. et al. Heterogeneous effects of genetic variants and traits associated with fasting insulin on cardiometabolic outcomes. Nat Commun 16, 2569 (2025). https://doi.org/10.1038/s41467-025-57452-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57452-y
