
Abstract
The study of the stereochemistry of organic sulfur compounds has been ongoing for over a century, with S-chirogenic pharmacophores playing an essential role in drug discovery within bioscience and medicinal chemistry. Traditionally, the synthesis of sulfinamides featuring stereogenic sulfur(IV) centers involves a complex, multistep process that often depends on chiral auxiliaries or kinetic resolution. Here, we introduce an effective and versatile method for synthesizing diverse classes of S-chirogenic sulfinamides through selective aryl and alkenyl addition to sulfinylamines. This process is catalysed by a chiral nickel or cobalt complex under reductive conditions, and eliminating the need for preformed organometallic reagents. The method facilitates the incorporation of a diverse array of aryl and alkenyl halides at the sulfur position, enabling their integration into various biologically significant sulfur pharmacophores. Our detailed mechanistic investigations and density functional theory calculations provide insights into the reaction pathway, particularly highlighting the enantiocontrol mode during addition process.
Similar content being viewed by others

Introduction
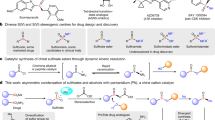
Enantiomerically enriched molecules with chiral sulfur centers, particularly in the oxidation states IV and VI, are pivotal across multiple fields such as catalysis, ligand design, materials science, agrochemistry, and pharmaceutical research1,2,3,4,5,6,7,8. Sulfinamides with S-chirogenic centers exemplify a unique balance between stability and chemical reactivity, making them essential for applications ranging from chiral auxiliaries9,10 to ligands in metal catalysis11 and as efficient organocatalysts12. Moreover, these compounds also function as valuable precursors for the stereoselective synthesis of a variety of S-chirogenic pharmacophores, including sulfinimidate esters, sulfinamides, sulfonimidoyl halides, and sulfoximines (Fig. 1a)13,14,15. These compounds have also been utilized as bifunctional chiral reagents through desulfinylation processes16,17. Conventional methods for producing these compounds typically involve complex, multistep procedures, often relying on chiral auxiliaries or kinetic resolutions18,19. Therefore, the development of a general and one-step approach to synthesize sulfinamides would represent a significant advancement, greatly enhancing the efficiency and simplicity of preparing chiral sulfur-containing molecules.

a Reaction diversity of S-chirogenic sulfinamides. b Asymmetric synthesis of chiral sulfinyl compounds by organocatalysts. c Metal-catalysed synthesis of S-chirogenic sulfinamides using arylboron compounds. d Reported results on asymmetric reductive addition for constructing chiral C − C bonds. e Asymmetric reductive addition of aryl and alkenyl halides to sulfinylamines to form C − S bonds.
There has been a surge in the development of asymmetric nucleophilic substitution reactions involving sulfur(IV) nucleophiles using organocatalysts, aimed at synthesizing S-chirogenic sulfinyl compounds (Fig. 1b). The research conducted by the Tan group marks a significant advancement in this domain, as they have successfully synthesized enantioenriched arylsulfinate esters through an asymmetric condensation of sulfinates with alcohols, employing a chiral pentanidium catalyst20. An asymmetric deoxygenation of sulfones was further developed to form chiral sulfinate esters by utilizing a catalyst that interacts with arylsulfonyl nitriles with alcohol through hydrogen bonding21. However, neither of these approaches is effective for the direct synthesis of sulfinamides. These challenges were later overcome, and chiral sulfinamides were synthesized through the nucleophilic substitution of arylsulfinates with amines, facilitated by bifunctional chiral 4-arylpyridine N-oxide catalysts22. Enantioenriched sulfinate esters and sulfinamides were also prepared via quinine-catalyzed reactions of activated sulfinates with alcohol and amine nucleophiles23. However, the limited commercial availability of S-containing substrates necessitates the de novo synthesis of the relevant substrates. Moving forward, there is a clear need for methods to prepare S-chirogenic sulfinamides that exploit commercially available reagents and substrates.
Sulfinylamines (R-NSO) have been pivotal as synthons in the synthesis of sulfinamides, which are typically generated through the nucleophilic addition of organometallic compounds such as Grignard and lithium reagents24,25. Building on this reactivity, the development of S-chirogenic sulfinamides using transition metal catalysts has become feasible (Fig. 1c)26. Recently, Zhang and Yang reported a copper-catalyzed asymmetric synthesis of sulfinamides via the addition of aryl boroxines to sulfinylamines27. In parallel, our group has discovered an asymmetric nickel-catalyzed arylation of sulfinylamines using more readily available arylboronic acids28. While many arylboron reagents are commercially available, specialized reagents like boroxines or arylboronic acids with complex frameworks still require pre-preparation. Moreover, alkenyl organometallic compounds are less readily available, thus rendering the synthesis of chiral alkenylsulfinamides particularly challenging and underexplored. Given that organohalides are frequently employed as precursors to the corresponding organometallic reagents, there is a pressing need to develop more efficient methodologies that permit the direct utilization of these abundant and structurally diverse feedstocks, thereby obviating the requirement for preformed organometallic reagents.
The Nozaki-Hiyama-Kishi (NHK) reaction is a classic example of reductive addition, typically used to produce allylic alcohols through the reaction of an aldehyde with an alkenyl halide in the presence of a terminal chromium reductant and a nickel catalyst29,30. Recent advancements in catalytic addition to aldehydes31,32,33,34, ketones35, imines36,37,38,39, enones40 with carbon electrophiles have significantly expanded the scope of this chemistry, enabling the formation of chiral C − C bonds under reductive conditions (Fig. 1d). Inspired by these developments, we hypothesized whether the direct addition of organohalides to sulfinylamines could lead to the formation of S-chirogenic centers41.
Here, we introduce an enantioselective nickel-catalyzed process that facilitates the addition of aryl halides to sulfinylamines, leading to the formation of S-chirogenic sulfinamides (Fig. 1e). Additionally, this reductive process can be extended to cobalt catalysis, which allows for asymmetric additions of vinyl halides to sulfinylamines, leading to the formation of chiral alkenylsulfinamides. By employing metal catalysts in conjunction with chiral bisoxazoline ligands and a metal reductant, sulfinylamines substituted with N-trityl (Tr) or N-TIPS groups undergo aryl and vinyl addition, demonstrating remarkable enantioselectivity.
Results
Preliminary studies and optimization
The feasibility of the enantioselective addition was explored by examining the reaction of reagent I with iodoarene 1a in the presence of a nickel catalyst (Fig. 2a). The optimum conditions for the reaction were achieved using 15 mol% Ni(cod)2 as a catalyst and 18 mol% dibenzofuran-4,6-bis(oxazoline) ligand L5 bearing two 3-methoxyphenyl substituents in a dry CH3CN solvent, with a reaction time of 5 days at 40 °C (entry 1). Under these carefully selected conditions, the desired product 1b was formed and isolated with a yield of 87%, and displaying 96/4 enantiomeric ratio (e.r.). When substituting the aryl group in the ligand with iPr (L1), tBu (L2) and Bn (L3), the formation of product 1b was significantly compromised (entries 2-4). The use of phenyl-substituted ligand L4 resulted in slightly lower enantioselectivity but still furnished compound 1b with a yield of 92% (entry 5). Using a more sterically bulky ligand L6, which contains two tert-butylphenyl groups led to a much lower outcome in both yield and enantioselectivity (entry 6). When a divalent nickel species such as Ni(dme)Br2 was employed as the catalyst, product 1b was obtained with a yield of 85%, but with only modest enantioselectivity (entry 7). Using other earth-abundant metals, such as Co(dme)Br2, only yielded trace amounts of the desired product 1b (entry 8). Interestingly, reducing the catalyst loading from 15 to 10 mol% had a profound effect on the reaction’s efficacy, resulting in a disappointing yield of 30%, while maintaining impeccable enantioselectivity (entry 9). Adjusting the reaction temperature to 60 °C under the established conditions led to a substantially poorer outcome (entry 10). The use of zinc powder as a reducing agent is essential for the reaction, as other reductants, such as manganese powder, resulted in much lower conversion (entry 11), and no conversion was observed in the absence of the reductant (entry 12). When using ordinary CH3CN that has not been dehydrated, the reaction yields are significantly reduced, and the ee is also greatly affected (entry 13). Additionally, substituting bromoarene 1a’ with substrate 1a in the system led to a 64% yield of 1b with an e.r. of 89/11 (entry 14). Furthermore, using chloroarene 1a” as the substrate produced only trace amounts of the desired product 1b (entry 15). With the optimized conditions in hand, we proceeded to investigate the effect of different S-substituents in sulfinylamine (Fig. 2b). Substrates bearing phenyl (II)42 and tert-octyl (III) groups exhibited only minimal formation of the desired products 1c and 1 d under the current reaction conditions. Encouragingly, when the sulfinylamine IV contained a TIPS group, the corresponding product 1e was obtained in good yield with high enantioselectivities using ligands L4 or L5. However, other silyl substitutes, such as TES (V) and SiPh3 (VI), displayed sluggish reactivity. Finally, the reaction of PhSO2–NSO (VII)43 did not yield any aryl addition product.

a Analysis of reaction parameters in the reaction of sulfinylamine I and iodoarene 1a. b Evaluation of diverse sulfinylamines. aReaction conditions: [Ni] (15 mol%), L* (18 mol%), 1a (3.0 equiv., 0.3 mmol), sulfinylamine I (1.0 equiv., 0.1 mmol), Zn dust (3.0 equiv., 0.3 mmol) in CH3CN (superdry, water ≤ 10 ppm) at 40 oC for 5 days under N2 atmosphere. The yields mentioned are isolated yields. E.r. values were determined via chiral HPLC bReact for 3 days with 1a (1.0 equiv., 0.1 mmol) and IV (3.0 equiv., 0.3 mmol).
We initially explored the versatility of the reaction by assessing the range of aromatic iodides combined with sulfinylamine I (Fig. 3a). Ligand L4 was also employed in some cases, exhibiting better enantioselectivity compared to ligand L5. Iodobenzene (2a) and its analogues, incorporating alkyl and aryl groups such as ethyl (3a), tert-butyl (4a), and phenyl (5a), yielded the corresponding products 2-5b in yields ranging from 62% to 85%, with good enantioselectivities. Furthermore, iodoarenes carrying electron-donating groups, such as benzyloxy (6a), methoxy (7-9a), methylthio (10a), and pyrazol (11a), at para, meta and ortho position exhibited excellent compatibility. Notably, the presence of one or more halogen groups including F (12-16a), Cl (17-18a), and Br (19a) did not influence the reaction. Of particular interest is the utilization of 1,4-diiodobenzene (20a), where one C–I bond could be retained under the reaction conditions, resulting in the formation of the corresponding product 20b with a yield of 74% and an e.r. of 95.5/4.5. Meanwhile, non-symmetric 1,4- diiodobenzene (21a) exhibited surprising regioselectivity and can obtain product 21b with a r.r. of 11/1 and an e.r. of 97/3. Aryl iodides with electron-withdrawing substituents, such as trifluoromethyl (22a) and trifluoromethoxy (23-24a), delivered products 22-24b in good yields and enantioselectivities. It is worth noting that carbonyl compounds, such as ester (25a) and amide (26a), which products 25-26b with delightful yields and enantioselectivitys. Among them, the absolute configuration of product 25b was determined to be R via X-ray crystallography. Expanding on this strategy, polycyclic arenes such as 2-iodonaphthalene (27a) and 2-iodo-9,9-dimethyl-9H-fluorene (28a) could be employed with satisfactory enantioselectivities. Heteroaryl iodides, such as thiophene 29a, readily underwent conversion into the corresponding product 29b with moderate outcomes. Unfortunately, other heteroaryl iodides, such as 4-iodopyridine, and 8-iodoquinoline, were not compatible and the corresponding products cannot be obtained (not shown in the Fig. 3). In addition, substrates derived from canagliflozin (30a) and empagliflozin (31a) proved to be compatible with this transformation. Building upon the success of asymmetric reductive addition, we further investigated the utilization of sulfinylamine IV bearing the TIPS group in these catalytic systems. As depicted in Fig. 3b, selected iodoarenes provided sulfinamides 2e, 14e, 18e, 20e, 23e and 31e with commendable enantioselectivities.

a Scope of iodoarenes with sulfinylamine I. b Selected examples of iodoarenes with sulfinylamine IV. aReaction conditions: Ni(cod)2 (15 mol%), L5 (18 mol%), 2-31a (3.0 equiv., 0.3 mmol), I (1.0 equiv., 0.1 mmol), Zn dust (3.0 equiv., 0.3 mmol) in CH3CN (superdry, water ≤10 ppm, 0.1 M) at 40 oC for 5 days under N2 atmosphere; Isolated yields; E.r. values determined by chiral HPLC. bUsing L4 instead of L5. cReact for 5 days at 10 oC. dShortened to 3 days with 2-31a (1.0 equiv., 0.1 mmol) and IV (3.0 equiv., 0.3 mmol).
Based on the above results, we further explored the use of alkenyl halides as substrates in our strategy for constructing S-chirogenic alkenylsulfinamides. Unfortunately, when aromatic iodides were replaced with alkenyl iodide 32a, the reaction with sulfinylamine I under the initial conditions produced only trace amounts of the desired product. Through systematic screening (see Supplementary information for details), we identified an optimized reaction protocol using Co(dme)Br2 as the catalyst, ligand L6, indium dust as the reductant, and nBu4NBF4 and 3 Å molecular sieves (MS) as additives in CH3CN solvent at 20 °C for 5 days (Fig. 4). Under these conditions, we isolated product 32b with a yield of 54% and an e.r. of 95.5/4.5. We then investigated the reactivity of sulfinylamine I with various alkenyl iodides. Substrates featuring the styrenyl motif with methyl (33a, 34a) and tert-butyl (35a) groups were successfully incorporated into the reaction, generating products 33b-35b with satisfactory yields and enantioselectivities. Styrenyl iodides containing methoxy groups (36a, 37a) were also compatible, yielding the desired products 36b and 37b with good enantioselectivities. Halide-containing substrates, including those with fluorine (38a, 39a), chlorine (40a), and bromine (41a, 42a), reacted smoothly with sulfinylamine I. Furthermore, substrates bearing electron-withdrawing groups, such as trifluoromethyl (43a) and trifluoromethoxy (44a), were equally compatible. The reaction of 2-vinylnaphthalene 45a under the current conditions resulted in satisfactory enantioselectivity. Additionally, a diene-substituted sulfinamide 46b was prepared in modest yield from alkenyl iodide 46a under the optimized reaction conditions. However, simple alkenyl iodides, such as iodoethene and 1-iodocyclohex-1-ene, produced only trace amounts of the desired products under the current reaction conditions (not shown in the Fig. 4).

Reaction conditions: Co(dme)Br2 (10 mol%), L6 (12 mol%), 32-46a (2.0 equiv, 0.2 mmol), I (1.0 equiv., 0.1 mmol), In dust (3.0 equiv., 0.3 mmol), nBu4NBF4 (2.0 equiv., 0.2 mmol) and 3 Å MS (50 mg) in CH3CN (superdry, water ≤10 ppm, 0.5 M) at 20 oC for 5 days under N2 atmosphere; Isolated yields; E.r. values determined by chiral HPLC.
To further demonstrate the synthetic utility of this approach, a series of experiments were then conducted (Fig. 5). Both type of products can undergo deprotection effectively (Fig. 5a). For example, anhydrous MsOH effectively served as a deprotecting agent, converting N-Tr sulfinamide 1b into primary sulfinamide 47 with a 70% yield while maintaining enantioselectivity. N-TIPS Sulfinamide 1e can also be deprotected to yield product 47 using TBAF under milder reaction conditions. Utilizing iodoarene 1a and sulfinylamine I as substrates, a 1.0 mmol-scale synthesis was performed smoothly under standard conditions, yielding product 1b with a 61% yield and retaining enantioselectivity (Fig. 5b). Sulfonimidoyl fluorides are valuable intermediates in chemical synthesis due to their potential for conversion into other significant compound classes through sulfur(VI) fluoride exchange (SuFEx) reactions44,45,46. The treatment of 1b with TBAF resulted in the formation of a sulfonimidoyl fluoride 48, containing a chiral sulfur(VI) center, achieved in a satisfactory yield, with only a slight decrease in enantioselectivity. Traditionally, sulfonimidamides are synthesized by reacting amines with sulfonimidoyl chlorides, which are obtained from the electrophilic chlorination of sulfinamides47. Building on our initial findings, treating compound sulfinamide 1b with tert-butyl hypochlorite led to a complete conversion to sulfonimidoyl chloride, which was then reacted with ammonium hydroxide to efficiently produce sulfonimidamide 49 with high enantioselectivity. Furthermore, compound 1b was efficiently transformed into tertiary sulfinamide 50 through a substitution reaction with allyl bromide. Enantioenriched sulfinamides are extremely versatile intermediates in organic synthesis, capable of being converted into other important compound families (Fig. 5c). Deprotection of 20e with TBAF, followed by reaction with pivalic anhydride, yielded sulfinamide 51 with an 84% yield and 96.5/3.5 e.r. Treatment of 51 with EtI using NaH resulted in the synthesis of chiral sulfoximine 52 through a stereospecific S-alkylation48. A stereospecific, oxygen-selective alkylation of 51 using isopropyl iodide, K2CO3, and DMPU formed chiral sulfimide 53 with a 75% yield and an e.r. of 96/4. In the presence of a copper catalyst, the asymmetric synthesis of chiral sulfoximine 54 with an aryl group was successful using diphenyliodonium salt as a coupling partner. To gain a more comprehensive understanding of the product structures, X-ray crystallographic analysis was performed on products 52-54, unequivocally confirming their absolute configurations. Finally, sulfinamide 51 was smoothly converted to the S-methylated product 55, a key precursor for the synthesis of proline-rich tyrosine kinase 2 (PYK2) inhibitor C, potentially offering a new anabolic treatment for osteoporosis49.

a Deprotection of compounds 1b and 1e. b Scale-up synthesis and follow-up transformations of chiral product 1b. c Subsequent transformations of chiral product 20e.
Discussion
Mechanistic investigation
To deepen our understanding of the reaction pathway, we conducted several mechanistic studies (Fig. 6). When equimolar amounts of ligand L4 were mixed with NiBr2(dme), the formation of a bivalent nickel complex 56 was confirmed via X-ray analysis (Fig. 6a). Nevertheless, further testing showed that compound 56 was an ineffective catalyst, delivering the product 1b with only a 16% yield and a 75/25 e.r. (Fig. 6b). This result suggests that the reduction of Ni(II) to Ni(I) to initiate the reaction is not the main pathway of the reaction. In contrast, reacting stoichiometric amounts of Ni(cod)2 with ligand L5 yielded the product 1b with a 78% yield and a 91/9 e.r., aligning with the outcomes observed in catalytic systems (Fig. 6c). These results highlight the critical role of the oxidative addition of Ni(0) species to iodoarenes, which form Ni(II) entities that subsequently undergo insertion into sulfinylamine. Moreover, arylzinc reagents 57a and 57b exhibited high reactivity in the nucleophilic addition to sulfinylamine I, yet both yielded the racemic product 1b (Fig. 6d). It rules out the in situ formation of arylzinc species, confirming zinc’s role as a reductant in the system. Lastly, the absence of nonlinear effects observed in the reaction between substrates I and 1a indicate that the active catalytic species is a monomeric nickel complex associated with a single ligand L4 (Fig. 6e).

a Synthesis of nickel complex 56. b Testing the catalytic reactivity of complex 56. c Evaluation of the reaction with stoichiometric amounts of nickel and ligand. d Reaction of sulfinylamine I with zinc reagents 57a-b. e Nonlinear effect experiments involving ligand L4, sulfinylamine I and 1a.
Based on the results described above, density functional theory (DFT) was utilized to investigate the detailed mechanism and the origin of enantioselectivity in the model reaction between aromatic iodide 1a and sulfinylamine I (Fig. 7). The chiral Ni(0) catalyst INT1A initially coordinates with 1a, followed by oxidative addition of the C–I bond through transition state TS3A to form the nickel(II) intermediate INT3A, with an activation free energy of 7.5 kcal·mol·1. Subsequently, this intermediate INT3A undergoes ligand exchange with reagent I to generate the cationic nickel intermediate INT4A. The migration insertion of the S = N bond of reagent I is crucial for determining enantioselectivity. Computational results indicate that the formation of the R configuration via transition state TS5A-R in the migration insertion process is kinetically favored, exhibiting significantly lower energy compared to the pathway through transition state TS5A-S (9.4 kcal·mol−1 vs 11.2 kcal·mol−1 relative to intermediate INT4A), consistent with our experimental observation. To gain deeper insights into the stereoselectivity, the non-covalent interactions of TS5A-R and TS5A-S have been systematically analyzed using the NCI (Non-Covalent Interaction) approach50. Structural analysis reveals the stereochemical preferences in transition state TS5A-R, where a significant S−O · · · H interaction is observed between the chiral skeleton of the ligand and the oxygen atom of the sulfinylamine. This interaction plays a crucial role in stabilizing the preferred transition state, thereby influencing the observed stereoselectivity. Subsequently, an excess amount of reducing agent facilitates the reduction of nickel(II) species INT5A-R to generate the zinc salt (R)-1b-Zn. This zinc salt undergoes protonolysis during the workup process to yield the final product (R)-1b51,52. In the presence of a substantial reducing agent, INT6A reduces to regenerate Ni(0) species, thereby completing the catalytic cycle53.

DFT-computed reaction pathways for the reaction of aromatic iodide 1a with sulfinylamine I (M06-D3/6-311 + G(d,p)-SDD(Ni, Zn, and I), SMD(MeCN)//B3LYP-D3/6-31 G(d,p)-SDD(Ni, Zn, and I)).
In summary, we have devised an efficient and selective process for reductive addition of aryl and alkenyl halides into sulfinylamines, generating chiral sulfinamides featuring S(IV) stereocenters, with the aid of a chiral nickel and cobalt catalysts. The resultant compounds facilitate the synthesis of assorted S-stereogenic derivatives through stereoselective manipulations. Insight into the reaction mechanism that delivers the selective aryl addition processes was gleaned through an integrated analysis of experimental and computational evidence. Given the burgeoning interest in chiral S-stereogenic derivatives for their potential applications in the pharmaceutical and agrochemical sectors, we anticipate a broad implementation of the methodologies delineated in this investigation.
Methods
General procedure for synthesizing S-chirogenic arylsulfinamides
In a nitrogen-filled glove box, to an oven-dried 8 mL screw-cap vial equipped with a magnetic stir bar was added Ni(cod)2 (15 mol%), L4 or L5 (18 mol%) and CH3CN (1 mL, superdry, water ≤ 10 ppm). The resulting solution was stirred for 30 min at room temperature. Then iodoarene 1-31a (0.3 mmol, 3.0 equiv.), sulfinylamine I (0.1 mmol, 1.0 equiv.) and Zn powder (0.3 mmol, 3.0 equiv) were added. The tube was sealed with a teflon-lined screw cap, removed from the glove box and the reaction was stirred at 40 °C for 5 days. Afterwards, the mixture was cooled to room temperature. Then, the work-up was performed by filtering through a short plug of silica gel, eluting with ethyl acetate (ca. 10 mL). The solvent was evaporated under reduced pressure. Purification via flash column chromatography afforded the desired products 1-31b.
Data availability
Crystallographic data for the structures of 25b, 52, 53, 54 and 56 reported in this paper have been deposited at the Cambridge Crystallographic Data Center under deposition numbers CCDC 2341522, 2348520, 2348517, 2348519 and 2351555. Copies of the data can be obtained free of charge via www.ccdc.cam.ac.uk/getstructures. All other data supporting the findings of the study, including experimental procedures and compound characterization, are available within the article and its Supplementary Information, or from the corresponding author upon request.
References
Johnson, C. R. The utilization of sulfoximines and derivatives as reagents for organic synthesis. Acc. Chem. Res. 6, 341–347 (1973).
Reggelin, M. & Zur, C. Sulfoximines: structures, properties and synthetic applications. Synthesis 2000, 1–64 (2000).
Okamura, H. & Bolm, C. Sulfoximines: Synthesis and catalytic appli-cations. Chem. Lett. 33, 482–487 (2004).
Drabowicz, J. Stereo-chemistry of organic sulfur compounds: more than 100 years of history, current state and further challenges, Phosphorus, Sulfur Silicon. Relat. Elem. 192, 145–148 (2017).
Lücking, U. Neglected sulfur(VI) pharmacophores in drug discovery: exploration of novel chemical space by the interplay of drug design and method devel-opment. Org. Chem. Front. 6, 1319–1324 (2019).
Wojaczyńska, E. & Wojaczyński, J. Modern stereoselective synthesis of chiral sulfinyl compounds. Chem. Rev. 120, 4578–4611 (2020).
Tilby, M. J. & Willis, M. C. How do we address neglected sulfur pharmacophores in drug discovery? Expert Opin. Drug Discov. 16, 1227–1231 (2021).
Zhang, X., Wang, F. & Tan, C.-H. Asymmetric synthesis of S(IV) and S(VI) stereogenic centers. JACS Au 3, 700–714 (2023).
Ellman, J. A., Owens, T. D. & Tang, T. P. N-tert-butanesulfinyl imines: versatile intermediates for the asymmetric synthesis of amines. Acc. Chem. Res. 35, 984–995 (2002).
Robak, M. T., Herbage, M. A. & Ellman, J. A. Synthesis and applications of tert-butanesulfinamide. Chem. Rev. 110, 3600–3740 (2010).
Otocka, S., Kwiatkowska, M., Madalińska, L. & Kiełbasiński, P. Chiral organosulfur ligands/catalysts with a stereogenic sulfur atom: applications in asymmetric synthesis. Chem. Rev. 117, 4147–4181 (2017).
Dinér, P., Sadhukhan, A. & Blomkvist, B. Chiral sulfinamides as highly enantioselective organocatalysts. ChemCatChem 6, 3063–3066 (2014).
Aota, Y., Kano, T. & Maruoka, K. Asymmetric synthesis of chiral sulfoximines via the S-arylation of sulfinamides. J. Am. Chem. Soc. 141, 19263–19268 (2019).
Zou, X., Wang, H. & Gao, B. Synthesis of sulfoximines by copper-catalysed oxidative coupling of sulfinamides and aryl boronic acids. Org. Lett. 25, 7656–7660 (2023).
Tsuzuki, S. & Kano, T. Asymmetric synthesis of chiral sulfimides through the O-alkylation of enantioenriched sulfinamides and addition of carbon nucleo-philes. Angew. Chem. Int. Ed. 62, e202300637 (2023).
Noten, E. A., Ng, C. H., Wolesensky, R. M. & Stephenson, C. R. J. A general alkene aminoarylation enabled by N-centred radical reactivity of sulfinamides. Nat. Chem. 16, 599–606 (2024).
Hervieu, C. et al. Chiral arylsulfinylamides as reagents for visible light-mediated asymmetric alkene aminoarylations. Nat. Chem. 16, 607–614 (2024).
Kagan, H. B. & Rebiere, F. Some routes to chiral sulfoxides with very high enantiomeric excesses. Synlett 1990, 643–650 (1990).
Zhu, R.-H. & Shi, X.-X. Practical and highly stereoselective method for the preparation of several chiral arylsulfinamides and aryl-sulfinates based on the spontaneous crystallization of diastereomeri-cally pure N-benzyl-N-(1-phenylethyl)-arylsulfinamides. Tetrahedron: Asymmetry 22, 387–393 (2011).
Zhang, X., Ang, E. C. X., Yang, Z., Kee, C. W. & Tan, C.-H. Synthesis of chiral sulfinate esters by asymmetric condensation. Nature 604, 298–303 (2022).
Huang, S. et al. Organo-catalytic asymmetric deoxygenation of sulfones to access chiral sulfinyl compounds. Nat. Chem. 15, 185–193 (2023).
Wei, T., Wang, H.-L., Tian, Y., Xie, M.-S. & Guo, H.-M. Enantioselective construction of stereogenic-at-sulfur(IV) centres via catalytic acyl transfer sulfinylation. Nat. Chem. 16, 1301–1311 (2024).
Liao, M. et al. Enantioselective sulfinylation of alcohols and amines by condensation with sulfinates. Chem 10, 1541–1552 (2024).
Davies, T. Q., Hall, A. & Willis, M. C. One-pot, three-component sulfonimidamide synthesis exploiting the sulfinylamine reagent N-sulfinyltritylamine, TrNSO. Angew. Chem. Int. Ed. 56, 14937–14941 (2017).
Ding, M., Zhang, Z.-X., Davies, T. Q. & Willis, M. C. A silyl sulfinylamine reagent enables the modular synthesis of sulfonimidamides via primary sulfinamides. Org. Lett. 24, 1711–1715 (2022).
Lo, P. K. T. & Willis, M. C. Nickel(II)-catalysed addition of aryl and heteroaryl boroxines to the sulfinylamine reagent TrNSO: the catalytic synthesis of sulfinamides, sulfonimidamides, and primary sulfonamides. J. Am. Chem. Soc. 143, 15576–15581 (2021).
Shi, Y., Yuan, Y., Li, J., Yang, J. & Zhang, J. Catalytic asymmetric synthesis of sulfinamides via Cu-catalyzed asymmetric addition of aryl boroxines to sulfinylamines. J. Am. Chem. Soc. 146, 17580–17586 (2024).
Xi, L., Fang, X., Wang, M. & Shi, Z. Asymmetric 2,3-addition of sulfinylamines with arylboronic acids enabled by nickel catalysis. J. Am. Chem. Soc. 146, 17587–17594 (2024).
Okude, Y., Hirano, S., Hiyama, T. & Nozaki, H. Grignard-type carbonyl addition of allyl halides by means of chromous salt. A chemospecific synthesis of homoallyl alcohols. J. Am. Chem. Soc. 99, 3179–3181 (1977).
Jin, H., Uenishi, J., Christ, W. J. & Kishi, Y. Catalytic effect of nickel(II) chloride and palladium(II) acetate on chromium(II)-mediated coupling reaction of iodo olefins with aldehydes. J. Am. Chem. Soc. 108, 5644–5646 (1986).
Zhu, Z., Xiao, J., Li, M. & Shi, Z. Nickel-catalysed intermolecular asymmetric addition of aryl iodides across aldehydes. Angew. Chem. Int. Ed. 61, e202201370 (2022).
Zhang, S. et al. Design and synthesis of tunable chiral 2,2’-bipyridine ligands: application to the enantioselective nickel-catalysed reductive arylation of aldehydes. Angew. Chem. Int. Ed. 61, e202117843 (2022).
Jiang, X. et al. Photoassisted cobalt-catalysed asymmetric reductive Grignard-type addition of aryl iodides. J. Am. Chem. Soc. 144, 8347–8354 (2022).
Jiang, H. et al. Photoinduced cobalt-catalysed desymmetrization of dialdehydes to access axial chirality. J. Am. Chem. Soc. 145, 6944–6952 (2023).
Huang, S. & Zhou, S. J. Nickel-catalysed enantioselective reductive arylation of common ketones. J. Am. Chem. Soc. 146, 12895–12900 (2024).
Xiao, J. et al. Enantioselective reductive (hetero)arylation of cyclic N-sulfonyl imines by cobalt catalysis. Angew. Chem. Int. Ed. 62, e202300743 (2023).
Zhang, L. et al. Nickel-catalysed enantioselective reductive arylation and heteroarylation of aldimines via an elementary 1,4-addition. J. Am. Chem. Soc. 145, 8498–8509 (2023).
Xia, T. et al. Cobalt-catalyzed asymmetric Aza-Nozaki–Hiyama–Kishi (NHK) reaction of α-imino esters with alkenyl halides. Angew. Chem. Int. Ed. 63, e202316012 (2024).
Wu, X. et al. Modular α-tertiary amino ester synthesis through cobalt-catalysed asymmetric aza-Barbier reaction. Nat. Chem. 16, 398–407 (2024).
Zhang, L. et al. Nickel-catalysed enantioselective reductive conjugate arylation and heteroarylation via an elementary mechanism of 1,4-addition. J. Am. Chem. Soc. 144, 20249–20257 (2022).
Wei, M.-K., Moseley, D., Bär, R., Sempere, Y. & Willis, M. C. Palladium-catalyzed addition of aryl halides to N-sulfinylamines for the synthesis of sulfinamides. J. Am. Chem. Soc. 146, 19690–19695 (2024).
Wang, B.-C. et al. Synthesis of S(IV)-stereogenic chiral thio-oxazolidinones via palladium-catalysed asymmetric [3+2] annulations. Angew. Chem. Int. Ed. 63, e202319728 (2024).
Bayeh, L., Le, P. Q. & Tambar, U. K. Catalytic allylic oxidation of internal alkenes to a multifunctional chiral building block. Nature 547, 196–200 (2017).
Teng, S., Shultz, Z. P., Shan, C., Wojtas, L. & Lopchuk, J. M. Asymmetric synthesis of sulfoximines, sulfonimidoyl fluorides and sulfonimidamides enabled by an enantiopure bifunctional S(VI) reagent. Nat. Chem. 16, 183–192 (2024).
Lou, T. S.-B. & Willis, M. C. Sulfonyl fluorides as targets and substrates in the development of new synthetic methods. Nat. Rev. Chem. 6, 146–162 (2022).
Greed, S. et al. Synthesis of highly enantioenriched sulfonimidoyl fluorides and sulfonimidamides by stereospecific Sulfur–Fluorine Exchange (SuFEx) reaction. Chem. Eur. J. 26, 12533–12538 (2020).
Zhang, Z.-X. & Willis, M. C. Sulfondiimidamides as new functional groups for synthetic and medicinal chemistry. Chem 8, 1137–1146 (2022).
Aota, Y., Kano, T. & Maruoka, K. Asymmetric synthesis of chiral sulfoximines through the S-alkylationof sulfinamides. Angew. Chem. Int. Ed. 58, 17661–17665 (2019).
Walker, D. P. et al. Sulfoximine-substituted trifluoromethylpyrimidine analogs as inhibitors of proline-rich tyrosine kinase 2 (PYK2) show reduced hERG activity. Bioorg. Med. Chem. Lett. 19, 3253–3258 (2009).
Johnson, E. R. et al. Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506 (2010).
Novotný, J., Bazzi, S., Marek, R. & Kozelka, J. Lone-pair–π interactions: analysis of the physical origin and biological implications. Phys. Chem. Chem. Phys. 18, 19472–19481 (2016).
Xu, Y. et al. Identifying π–π and π–lone pair interactions in a single-molecule junction. ACS Materials Lett 6, 1961–1967 (2024).
Day, C. S. et al. Elucidating electron-transfer events in polypyridine nickel complexes for reductive coupling reactions. Nat. Catal. 6, 244–253 (2023).
Acknowledgements
We would like to thank financial support from National Key R&D Program of China (2022YFA1503200), National Natural Science Foundation of China (Grants 22025104, 22171134, and 21972064), and the Fundamental Research Funds for the Central Universities (Grant 020514380254) for their financial support. The project is also supported by Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University. We are also grateful to the High-Performance Computing Center of Nanjing University for performing the numerical calculations in this paper on its blade cluster system.
Author information
Authors and Affiliations
Contributions
X.F., L.X., J.X. performed the experiments and analysed the data. M.W. performed the DFT calculation. Y. Z performed the crystallographic studies. M.C.W., Z.S. conceived and designed the study and wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fang, X., Xi, L., Wang, M. et al. Asymmetric reductive arylation and alkenylation to access S-chirogenic sulfinamides. Nat Commun 16, 2547 (2025). https://doi.org/10.1038/s41467-025-57471-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57471-9
This article is cited by
-
Organocatalytic kinetic resolution of sulfinamides by N/O exchange
Nature Communications (2025)
