
Abstract
Fluorescent π-conjugated polymers (FCPs) are known for their superior brightness but are still unavailable for highly multiplexed molecular imaging in single cells as they are hydrophobic and lack targeting capability toward biomolecules. Herein, we develop a π-conjugated polymer-based amplification (POSA) method to achieve highly multiplexed signal amplification. Optical amplification by virtue of the high brightness of FCPs makes POSA a simple and quick signal amplification technique that can spatially resolve the distribution of multiplexed proteins in single cells, with a 28- to 126-fold signal amplification effect. By this POSA method, we demonstrate that the high brightness of FCPs can be used to strengthen the images of subcellular biomolecules and showcase the phenomenon of optical amplification of FCPs at the cellular level. Additionally, with its sensitivity, ease of use, and quick imaging features, the POSA technique proves to be a valuable tool for advanced biological research.
Similar content being viewed by others

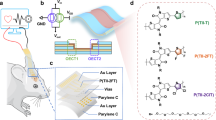

Introduction
Multiplexing immunohistochemistry (mIHC) techniques provide powerful tools to reveal single-cell spatial proteomics, which are essential for understanding normal cell physiology and disease pathogenesis1,2,3. Among the various methods, immunofluorescence (IF) has been most widely investigated because it relies on easily accessible fluorescence microscopy and offers exceptional imaging resolution4,5,6. However, IF still faces challenges in limited multiplexity due to the spectral crowding problem and inadequate sensitivity in imaging low-abundant proteins7,8. Recently, strategies such as immuno-SABER9 and CAD-HCR1 have been developed to address these challenges by employing multiplexed and cyclic DNA amplifications from the DNA-barcoded primary antibody. These strategies solve the problem by accumulating fluorescence dyes near the barcoded target (Fig. 1a). Although these state-of-the-art strategies are brilliant, they still suffer from the elaborate design and the high cost of a mass of DNA sequences for orthogonally executing multiple DNA amplification reactions10,11.

a Schematic of the DNA amplification and optical amplification using oligonucleotide-barcoded π-conjugated polymer nanoparticles (OCPNs). OCPNs are self-assembled from oligonucleotide grafted fluorescent π-conjugated polymers, showing much higher brightness than molecular dyes. b Workflow for spatial proteomics imaging analysis by POSA with five steps: (1) Various targets (T1 to Tn) are immunolabeled by orthogonal barcode sequences (BS) encoded primary antibodies (PAn-BSn); (2) Three distinctly colored OCPNs (PF OCPNs, PFBT OCPNs, and PFTBT OCPNs) with orthogonal imaging sequences (IS1-IS3) are hybridized to the corresponding BS sequences on the antibodies to image T1 to T3 simultaneously; (3) The fluorescent signals are erased by the introduction of erasing sequences (ES) to displace OCPNs from antibodies using toehold-mediated strand displacement reactions; (4) More targets are sequentially imaged in the next cycle; (5) The images are reconstructed and recolored to overlay all targets after imaging cycles.
Fluorescent nanoparticles can be a promising alternative to small molecular dyes for signal amplification, as they exhibit far greater brightness (Fig. 1a)12,13,14,15,16. However, the success of using nanoparticle imagers in spatial proteomics depends on several factors, including robust DNA modification, superior brightness, and negligible nonspecific intracellular retention during cyclic imaging17,18,19. So far, only DNA-modified quantum dots (QDs) have been used in subcellular multiplex protein imaging, and even then, they still need the DNA amplification technique to achieve a high signal amplification effect20. As a result, DNA-barcoded fluorescent nanoparticles that rely on brightness alone to achieve a signal amplification effect comparable to DNA technology are still unavailable.
π-Conjugated polymer nanoparticles have been recognized as the brightest fluorescent nanomaterials21,22,23. Their high brightness originated from the superior light-harvesting capability of fluorescence π-conjugated polymers (FCPs) comprised of overlapped π orbitals full of delocalized π electrons24,25,26. Backward-looking, the amplification effect of FCPs in vitro was first disclosed by Swager and co-workers and applied in developing chemo-/bio-sensors27. Chiu and Wu encapsulated π-conjugated polymer with a low weight content of amphiphilic polymer to develop ~10 nm polymer dots (Pdots), further which had been bio-conjugated with antibodies for subcellular protein imaging28. Recently, our group successfully synthesized oligonucleotide grafted π-conjugated polymer, which provides an accessible approach to fabricate small-size, colloidal stable, well water-dispersible, and oligonucleotide-barcoded π-conjugated polymer nanoparticles (OCPNs). Our previous work has shown that OCPNs are feasible for intracellular imaging applications. However, the signal amplification effect of OCPNs compared to small molecular dyes has yet to be demonstrated at the cellular level. Additionally, DNA-encoded OCPNs enable the use of highly bright FCP for multiplex protein spatial imaging, a capability that has not been achieved with any other FCP-based nanomaterials to date.
This work, based on OCPNs, has developed an optical signal amplification method called π-conjugated polymer-based optical signal amplification (POSA) for single-cell multiplexing and amplified protein imaging (Fig. 1b). The study shows that POSA could achieve a similar spatial resolution to the conventional IF technique but with significantly improved sensitivity due to the high brightness of OCPNs. Compared to small molecular dye imaging, POSA is able to gain 28- to 126-fold signal amplification effects (varied owing to the different proteins) at the cellular imaging level, making it equivalent to DNA amplification methods9. Moreover, the optical amplification principle makes POSA the most straightforward and quickest signal amplification technique. It shows a fast image rate within 10 minutes/cycle and becomes more accessible as only several short orthogonal sequences are needed in DNA design. In addition to the superior fluorescence brightness, OCPNs bear well-behaved subcellular penetration and minimal non-specific intracellular interactions. All these features determine that POSA could witness this progress of FCPs in bio-imaging applications. We apply POSA in imaging various kinds of proteins with different spatial distributions and abundances, such as cytoskeletal proteins, nucleoproteins, and organelle proteins. Based on three colored OCPNs fabricated from different fluorescent FCP cores, nine proteins in a single cell could be imaged through three cycles of three-color multiplexed imaging with a total time of less than 1 hour, which is faster than the DNA amplification methods1,9. This marks the successful application of FCPs for amplified imaging of the intracellular spatial distribution of proteins. Additionally, the POSA method, which offers rapid, sensitive, multiplexed, and precise imaging capabilities, has proven to be a valuable asset in biological and medical research.
Results
The establishment and principle of the POSA method
OCPNs with various fluorescent colors were synthesized based on different π-conjugated structures (Supplementary Figs. 1–8), i.e., polyfluorene (PF), poly(fluorene-benzothiadiazole) (PFBT), and poly(fluorene-thiophene-benzothiadiazole-thiophene) (PFTBT). For imaging acquisition, 20-nt oligonucleotides (the imaging sequences, ISn in abbreviation) were applied for OCPN synthesis, resulting in PF-g-IS OCPNs, PFBT-g-IS OCPNs, and PFTBT-g-IS OCPNs. All the DNA sequences used in this work are listed in Supplementary Table 1. Primary antibodies (PAn, with the specific type denoted by the subscript n, as shown in Supplementary Table 2) were combined with orthogonal 25-nt oligonucleotides (referred to as barcode sequences, BSn) to produce PAn-BSn conjugates (Supplementarys Figure 9,10)10. A pair of IS and BS contains a 15-nt complementary region for hybridization (Supplementary Fig. 11), allowing the staining of target proteins by OCPNs. Furthermore, a 10-nt spacer domain is included in the BSn sequence as the toehold overhang for the “erasing” step through DNA strand displacement reactions. These reactions are carried out using the fully complementary sequences of BSn, also known as the erasing sequences (abbreviated as ESn). The displacement reaction of ES to disassemble the hybridized PA-BS and OCPNs was verified by the agarose gel analysis (Supplementary Fig. 12). As a result, cyclic imaging can be performed, and in each cycle, three proteins can be simultaneously imaged by three distinctly colored OCPNs encoded with corresponding IS. Theoretically, unlimited multiplexity (3 × n) can be achieved (Fig. 1b).
To establish the POSA method, we selected PFBT-g-DNA (IS1 was used for these experiments) to optimize the particle size and optical properties of OCPNs, which play essential roles in intracellular fluorescence imaging. We adopted a mixed solvent of dichloromethane/dimethylformamide/dimethyl sulfoxide to dissolve the incompatible polymer and DNA simultaneously. The further purification process fully removed the unreacted polymers and DNA to avoid the influence of impurity on calculating DNA-graft efficiency (details in the Supplementary Information). Finally, the average DNA-graft efficiency was quantitatively analyzed by the absorptions of DNA and π-conjugated polymer in OCPNs (See the details in the Method part). Benefiting from the fact that PFBT-g-DNA is an amphiphilic polymer, the graft amount of DNA strands on each π-conjugated polymer chain could notably change the hydrophobic driving force for OCPN formation and, accordingly, the particle size29. The hydrodynamic size of OCPNs would be minimized down to 10 nm with the increase in average DNA-graft efficiency (Supplementary Fig. 13). The size of OCPNs can be adjusted between 10 to 90 nm (Supplementary Table 3), accompanied by a polydispersity index below 0.2, demonstrating relatively homogeneous size distributions. We noticed that OCPNs with large particle sizes showed increased standard deviation in size. It is assumed that as the PFBT-g-DNA with a lower average DNA-graft efficiency shows higher hydrophobicity, it needs more polymer chains to aggregate together to form sufficient DNA coverage, which results in a larger nanoparticle size. Also, the self-assembly processes of more polymer chains become more complicated, which results in increased size variations. In addition, PFBT OCPNs possessed a zeta potential of -42.2 mV, presenting high structural stability. The complexes formed by the hybridization of OCPNs with PA-BS still exhibited a high zeta potential value of -35.2 mV (Supplementary Fig. 11b). This demonstrated that the hybridization process would not affect the structural stability of OCPNs. Accordingly, we investigated the hybridization capability of the DNA strands on PFBT OCPNs of various sizes. The results revealed that the DNA on OCPNs exhibited quick kinetic rates of hybridization, which would not be affected by the particle size (Supplementary Fig. 14).
Using the optimized conditions, we synthesized PF OCPNs, PFBT OCPNs, and PFTBT OCPNs, respectively (Fig. 2a). We could observe that the characteristic peaks of DNA and the corresponding π-conjugated polymers co-existed in the UV-vis absorption spectra of the three OCPNs (Fig. 2b). All the average DNA-graft efficiencies of the three OCPNs were determined to be around 50% (Supplementary Fig. 15). All OCPNs used in the study showed high zeta potential values (~-40 mV) (Supplementary Table 3), which will promise high structural stability during the intracellular imaging application processes. Further characterized by dynamic light scattering (DLS) and transmission electron microscope (TEM), these OCPNs showed comparable hydrodynamic sizes of around 10 nm (Fig. 2d and Supplementary Figs. 16-17). In addition, these OCPNs showed separated absorption maximum, and their fluorescence spectra (Fig. 2c) revealed that each OCPN has its own non-interfering emission range, ensuring that they can be discriminated by confocal laser scanning microscope (CLSM) without crosstalk. Thus, the excitation wavelengths of PF OCPNs, PFBT OCPNs, and PFTBT OCPNs were set at 405 nm, 448 nm, and 488 nm, respectively, and their fluorescence signals were collected at 425-465 nm, 540-580 nm, and 650-700 nm, respectively. Moreover, all three OCPNs showed high molar absorptivity (ε) (Supplementary Table 4), and the absolute fluorescence quantum yields of PF OCPNs, PFBT OCPNs, and PFTBT OCPNs were determined to be 91 %, 55 %, and 11 % (Supplementary Fig. 18), respectively. We thoroughly analyzed the brightness (ε × QY) of OCPNs compared with commercial QDs and dyes and Pdots with similar spectral properties. Our findings, as reported in Supplementary Table 4, show that the brightness of OCPNs is almost two orders of magnitude higher than that of the dyes and an order of magnitude higher than QDs30,31,32,33. The good biocompatibility of OCPNs has also been proved by a cell viability assay (Supplementary Fig. 19).

a Schematic of FCP-g-DNA amphiphilic polymer synthesis via solid-phase “Click” reaction and the self-assembly of OCPN. The chemical structures of π-conjugated polymers are shown. b normalized UV-vis absorption spectra, (c) fluorescence spectra, (d) DLS data, and TEM images of OCPNs. e Fluorescent images of TOMM20 in HeLa cells employing AF488-labeled secondary antibody (AF488-SA). The anti-TOMM20 primary antibody (PA) was either adopted as is, modified with sulfhydryl group after the TCEP treatment, or modified with BS4. f Fluorescent images of TOMM20 in HeLa cells pre-immunolabelled by PA4-BS4 using PFBT-g-A20 OCPNs, PFBT-g-T20 OCPNs, and PFBT-g-IS4 OCPNs, respectively. g Orthogonal imaging of anti-Ki67 PA2-IS2, anti-Vimentin PA7-BS7, and anti-EGFR PA8-BS8 in HeLa cells using PFBT-g-IS2 OCPNs, PFBT-g-IS7 OCPNs, and PFBT-g-IS8 OCPNs. h Vimentin, Lamin A/C, and mitochondria were simultaneously imaged in HeLa cells using PFBT OCPNs (the green channel) and Cy5-labeled SA (the red channel). i Colocalization analysis between immunofluorescence (IF) and POSA for three proteins from (h) indicates a good correlation (Pearson’s R from 0.79 to 0.95). j Three-color multiplexed imaging of α-tubulin, mitochondria, and Lamin A/C in HeLa cells via POSA. Source data are provided as a Source Data file.
After optimizing OCPNs, we first conducted tests to validate the POSA method by examining the impact of DNA conjugation on the immunogenicity of the primary antibody. The conventional IF method stained by AF488-labeled secondary antibody (AF488-SA) was performed to detect TOMM20 with the unmodified anti-TOMM20 PA, the tris(2-carboxyethyl) phosphine (TCEP)-reduced anti-TOMM20 PA (PA-SH), and anti-TOMM20 antibody-BS4 conjugates (PA-BS4). As shown in Fig. 2e, the obtained images showed no significant difference. This verified that the PA maintained its specificity and affinity for protein recognition after chemical treatment and DNA conjugation. We further investigated the feasibility of POSA for fluorescent imaging. Three distinct IS strands, A20, T20, and IS4, were adopted to synthesize PFBT-g-A20 OCPNs, PFBT-g-T20 OCPNs, and PFBT-g-IS4 OCPNs. The HeLa cells pre-immunolabelled by anti-TOMM20 PA-BS4 were incubated with the above OCPNs, respectively. As shown in Fig. 2f, no fluorescence was detected from the cells incubated with PFBT-g-A20 OCPNs, suggesting the negligible non-specific interaction of OCPNs with the intracellular environment. Strong fluorescence in the whole cytoplasm was observed from cells incubated with PFBT-g-T20 OCPNs, as the T20 sequence is supposed to hybridize with the poly(A) tail of almost all mRNAs. Only the cells incubated with PFBT-g-IS4 OCPNs presented the morphology of TOMM20. These results indicated that OCPNs could freely diffuse and spread well in the fixed cells while interacting specifically with the barcoded antibodies34,35.
Additionally, we determined the imaging orthogonality of the POSA method. Three proteins, Vimentin, EGFR, and Ki67, were selected as the targets, and the corresponding PA were encoded with orthogonal IS strands to fabricate anti-Ki67 PA2-BS2, anti-Vimentin PA7-BS7, and anti-EGFR PA8-BS8. After incubation with a single antibody, cells were divided into three groups and imaged using PFBT-g-IS2 OCPNs, PFBT-g-IS7 OCPNs, and PFBT-g-IS8 OCPNs. Thus, a total of nine images were captured to visualize the three proteins. Figure 2g shows that only when we introduced OCPNs modified with the correct IS strand matching the corresponding antibody was an imaging signal produced. This result further showed that OCPNs cannot trigger unspecific signals without the encoded antibody for the protein targets.
Subsequently, we evaluated the accuracy of the POSA method for protein spatial location by comparing it with conventional IF images using colocalization analysis. Three proteins with distinct distribution and abundance, Vimentin (cytoskeleton), Lamin A/C (nuclear envelope), and mitochondria (organelle), were employed as targets and immunolabelled by corresponding PA-BS. The cells were first incubated with Cy5-labeled SAs and followed with PFBT OCPNs. The fluorescent images that were obtained from the green channel (OCPNs) and the red channel (Cy5) with identical z-positions of the objective lens were merged for colocalization analysis (Fig. 2h). The POSA method and the conventional IF method showed a high correlation with Pearson’s R = 0.86, 0.95, and 0.79 for Vimentin, Lamin A/C, and mitochondria imaging (Fig. 2i). This further confirmed that OCPNs could effectively access and target intracellular proteins.
Then, we employed the three distinctly colored OCPNs to simultaneously detect three independent proteins (Lamin A/C, α-tubulin, and mitochondria) in one imaging cycle and discriminated the proteins according to different excitation and emission properties of PF, PFBT, and PFTBT OCPNs. The cells were first immunolabeled with anti-α-tubulin PA1-BS1, anti-mitochondria PA3-BS3, and anti-Lamin A/C PA5-BS5, and further simultaneously stained by PFBT-g-IS1 OCPNs, PFTBT-g-IS3 OCPN, and PF-g-IS5 OCPNs. As shown in Fig. 2j, the fluorescent images detected by the PF OCPN, PFBT OCPN, and PFTBT OCPN channels presented the corresponding protein patterns, implying negligible crosstalk among different OCPNs.
The optical amplification effect of the POSA method
After evaluating the feasibility and specificity of the POSA method, we further assessed the signal amplification capability of the super-bright OCPNs. Notably, by increasing the size of the nanoparticles, the brightness of OCPNs can be adjusted as larger OCPNs contain more π-conjugated polymers and yield brighter fluorescence signals. Small OCPNs with a diameter of approximately 10 nm and large OCPNs with a diameter of around 26 nm were synthesized using the same IS strands with the aim of comparing their signal amplifying capability. It was observed that as the size of OCPNs was further increased to ~90 nm, the resolution of spatial imaging visibly declined (Supplementary Fig. 20). Therefore, we did not use larger OCPNs for the POSA method to ensure imaging specificity. To measure the amplification effect, we synthesized IS modified with AF488 as the unamplified control (AF488-IS). In addition, conventional IF was performed using AF488-SA as another control. Three independent proteins, mitochondria, Lamin A/C, and α-tubulin, were imaged by AF488-IS (unamplified control), AF488-SA (conventional IF), small OCPNs (~10 nm), and large OCPNs (~26 nm), respectively. The average signal intensity per unit area of the four groups was compared under the conditions that the fluorescent probes were kept identical in molar concentration for the staining step (Fig. 3a). Detailed determination process of the amplification effect was shown in Supplementary Fig. 21. 10 cells were selected as the region of interest (ROI), and the total signal intensity was divided by the total cellular area to count the average signal intensity per unit area, which was employed for the signal amplification effect estimation. The statistical data presented in Fig. 3b shows that OCPNs could achieve 28- to 126-fold amplification effects compared to the unamplified groups and elevate the fluorescence intensity by 6.5 to 35 folds compared to the group of conventional IF method. The signal distributions in protein images performed by various methods were analyzed in Supplementary Fig. 22, which further demonstrated the signal amplification effect of using OCPNs. Compared with the result of DNA-barcoded fluorescence QDs in similar multiplexed protein imaging, the amplification effect of POSA was found more effective20. In addition, the results confirmed that large OCPNs could further enhance the amplification effect for all three protein targets, and a relatively smaller difference was observed in the case of Lamin A/C between the large and small OCPNs. The reason might be that Lamin A/C proteins are located deep within the cells, away from the cell membrane. This phenomenon indicates that small OCPNs are more effective in navigating and reaching proteins in crowded intracellular environments.

a Fluorescent images of mitochondria, Lamin A/C, and α-tubulin in HeLa cells using various methods: the unamplified method using AF488-IS (the dye-modified sequence complementary to the barcode sequence on the primary antibody), the conventional IF based on AF488-SA, the POSA method via small PFBT OCPNs (~10 nm), and the POSA method via the large PFBT OCPNs (~26 nm). b Quantitative bar plots of average signal intensity per unit area were analyzed using the images in (a). The data represent the means ± standard deviation (error bar) of average signal intensity per unit area from ten cells (n = 10). c The CLSM images of HeLa cells before and after laser irradiation, where α-tubulin was stained using the POSA method, and Lamin A/C was stained using IF of AF488-SA. d The CLSM images of HeLa cells before and after laser irradiation, where α-tubulin was stained using IF of AF488-SA, and Lamin A/C was stained using the POSA method. The cross-sectional fluorescence intensity profiles are shown based on the yellow lines marked on the CLSM images. The histogram of the normalized fluorescence retention ratio of POSA and IF post-laser irradiation is shown, and the means ± standard deviations (error bar) of fluorescence retention ratios are presented from five cells pre- and post-laser irradiation (n = 5). Source data are provided as a Source Data file.
As a result of the signal amplifying effect, OCPNs could also shorten the staining time and enhance sensitivity for imaging. We shot a video (Supplementary Movie 1) to show a cyclic process of POSA. In the video, we successfully imaged mitochondria and erased this signal within 1 minute. Additionally, we were able to image the second protein target, Lamin A/C, directly without the need for a washing step. The entire process, which produced high-resolution fluorescent images of the two proteins, was completed in just 4 minutes. This quick rate can be attributed to the high brightness of OCPNs, which significantly enhanced imaging sensitivity. It is important to note that we stated in the general descriptions that the staining-erasing-staining cycle of POSA takes less than 10 minutes. This longer time frame accounts for the extended incubation period and washing step to ensure optimal imaging results. On the contrary, the conventional IF method generally requires a 30-minute incubation time for sufficient fluorescence staining, as one PA has to interact with several dye-labeled SA to generate an adequate signal5,36. Additionally, DNA amplification strategies often take several hours to complete the staining step due to the self-assembling process involved. On the other hand, the high brightness of OCPNs also improved the signal-to-noise ratio (SNR) of the POSA method. As shown in Supplementary Fig. 23, the mitochondrion protein in HeLa cells was imaged by the POSA and conventional IF methods, respectively. Since mitochondria are found throughout the cytoplasm, the signal-to-noise ratio (SNR) was determined by comparing the average fluorescence intensity of the cytoplasm (target signal) to that of the nucleus (background noise). Based on statistical data collected from 10 independent cells, the SNR of the POSA method was found to be over 50, which is significantly higher than the SNR of 11 obtained by the conventional IF method. We attribute this improved SNR level when using the POSA method to the high fluorescence brightness of OCPNs. Also, short oligonucleotides were used to fabricate OCPNs, which would help minimize non-specific DNA interactions.
Furthermore, when comparing the photostability of the POSA method by π-conjugated polymers and the conventional IF method with commercial dyes, the former was found to be more resistant to photobleaching37,38,39. We simultaneously imaged two independent proteins, Lamin A/C (located in the nucleus) and α-tubulin (located in the cytoplasm), by AF488-SA and OCPNs, respectively, to eliminate inconsistency among different cells and picture shot. The cells were divided into two groups and treated differently. In the first group, α-tubulin was stained using the POSA method, and Lamin A/C was stained with AF488-SA (Fig. 3c). In the second group, the protein staining methods were switched (Fig. 3d). Both groups were exposed to 488 nm laser irradiation for 3 minutes before imaging shots were taken. The fluorescent images of the cells pre- and post-laser irradiation were displayed. According to the cross-sectional fluorescence intensity profiles along the yellow lines, either α-tubulin or Lamin A/C stained by OCPNs showed significantly improved resistance to the photobleaching no matter where the proteins are located. The normalized retention ratio of fluorescence intensity for POSA and IF showed that the protein imaged with POSA maintained its 80% intensity after laser irradiation, demonstrating higher photostability compared to IF. This result has demonstrated the distinctive photostability of π-conjugated polymers in consideration of the critical role of probe photostability for IF imaging. It can avoid the risk of artifactual signals induced by excessive labels and tolerate extensive irradiation until reaching satisfactory imaging resolution.
Feasibility of cyclic in situ imaging by DNA strand displacement
In order to achieve highly multiplexed imaging, a rapid and gentle DNA strand displacement method is necessary to remove staining effectively40. For both the BS sequences modified on antibodies and the IS sequences modified on OCPNs, a single-strand domain is reserved as the toehold overhang for the “erasing” step of DNA strand displacement. Therefore, the Loc-displacement was designed to release OCPN signals by using ES to block the BS on antibody (Fig. 4a). To verify the feasibility of the Loc-displacement method, fixed HeLa cells were pre-immunolabelled with anti-Lamin A/C PA5-BS5 and following stained by PFBT-g-IS10 OCPNs. ES5, a 25-nt fully complementary strand with BS5, was adopted for the erasing step to displace OCPNs. As shown in Fig. 4b, the signals of OCPNs could be completely removed by the introduction of ES5 within 3 minutes. The following introduction of OCPNs could not re-stain the cells due to the full blocking of PA-BS. This indicated that the Loc-displacement method could realize a high-speed signal erasing but was only suitable for cyclic imaging of different protein targets. To verify the erasing effect for multiplexed imaging, the fluorescent signals from HeLa cells pre-stained by three colored OCPNs were successive erased following the introduction of corresponding ES (Supplementary Fig. 24), demonstrating the specificity of the strand displacement reaction.

a Schematic of fluorescence erasing step via Loc-displacement method. b The fluorescence images of Lamin A/C in HeLa cells were stained using OCPNs, then erased using the Loc-displacement reaction, and finally re-stained. The erasing process was completed within 3 minutes. c Cyclic imaging of Lamin A/C, mitochondria, α-tubulin, EGFR, GM130, and Vimentin by PFBT OCPNs during six cycles with the Loc-displacement reaction. d The merged image was reconstructed by false-colored fluorescent images from (c). e The zoom-in image of the marked region from (d). Source data are provided as a Source Data file.
Further, we designed six orthogonal BS sequences and barcoded six PAs to yield anti-Lamin A/C PA5-BS5, anti-mitochondria PA3-BS3, anti-α-tubulin PA1-BS1, anti-EGFR PA8-BS8, anti-GM130 PA6-BS6, and anti-Vimentin PA7-BS7. We tested the sequential cyclic imaging for the corresponding six proteins using the POSA method with the Loc-displacement method. To begin with, we marked fixed HeLa cells using the six PA-BS conjugates simultaneously. In the first cycle, we stained the cells with PFBT-g-IS5 OCPNs, imaged them, and then erased them by strand displacement with ES5. This whole process was completed in less than 10 minutes. We repeated this procedure six times using the orthogonally modified (IS5, IS3, IS1, IS8, IS6, and IS7) OCPNs for the successive imaging of six independent proteins (Fig. 4c). The results are shown in Fig. 4d with a zoom-in image in Fig. 4e. As all the proteins were imaged on the same single cells, the merged image could provide more information on the spatial distribution of multiple proteins.
Alternatively, NP-displacement was designed to release OCPN signals by using ES fully complementary to the IS sequence to block the activity of OCPNs (Fig. 5a). ES10 containing 25-nt fully complementary base pairs with IS10 was employed to displace OCPNs from PA5-BS5. As shown in Fig. 5b, the fluorescent signals could be erased in around 30 minutes. Compared with the Loc-displacement method, the NP-displacement method demanded a longer incubation time and a 40-fold higher amount of blocking DNA to fully erase the signal due to the densely packed DNA on the surface of OCPNs. Therefore, considering the imaging speed, Loc-displacement instead of NP-displacement method was selected for multiplexed protein imaging. However, it is important to note that using the NP-displacement method can preserve the activity of the antibody, allowing for further re-staining if needed for cyclic stains of the same proteins. This method can be used to image the same protein target repeatedly to evaluate whether cyclic imaging will induce signal loss and inconsistencies. In this case, fixed HeLa cells were labeled with anti-Lamin A/C PA5-BS5 and stained with PFBT-g-IS10 OCPNs. The cells were then treated with NP-displacement using ES10 to erase the fluorescence signal, re-stained with PFBT-g-IS10 OCPNs, and repeated for six cycles (Fig. 5c). The fluorescent images suggested that Lamin A/C could be imaged reversibly during six cycles. Additionally, we selected ten cells and analyzed their average fluorescent intensity for six cycles (Fig. 5d). The zoom-in fluorescent images of ten cells per cycle were shown in Fig. 5e with corresponding fluorescent intensity values determined by ImageJ software and marked at the top-left corners. The results confirmed no significant changes in cyclic imaging.

a Schematic of fluorescence erasing step via the NP-displacement method. b The fluorescence images of Lamin A/C in HeLa cells were stained using OCPNs, then erased using the NP-displacement reaction, and finally re-stained. The erasing step was completed in around 30 minutes. c Fluorescent images of Lamin A/C in HeLa cells for six staining and erasing cycles via NP-displacement method. The fluorescence intensity of ten cells was determined by ImageJ software for each cycle. d Statistical results of relative fluorescence intensity from (c). The means ± standard deviations (error bars) of relative fluorescence intensity are presented based on ten marked cells during six cycles (n = 6). e The zoom-in images of the marked cells from (c) in different cycles. The fluorescent intensity value of each cell was determined by ImageJ software and marked at the top-left corner of each zoom-in image. Source data are provided as a Source Data file.
Last, we conducted cyclic three-color multiplexed imaging using PF, PFBT, and PFTBT OCPNs to detect nine different proteins within a single cell. The nine proteins were divided into three groups and imaged in three cycles (Fig. 6a). The first group included EGFR (a plasma membrane marker), Lamin A/C (a nuclear envelope marker), and a cis-Golgi marker. The second group included mitochondria (a cytoplasmic protein), α-tubulin (a cytoskeleton marker), and Ki67 (a chromosome marker). The third group included TOMM20 (a transit peptide receptor at the surface of mitochondria), Lamin B1 (a nuclear envelope marker), and Vimentin (a cytoskeleton marker). In each imaging cycle, three out of the nine proteins were imaged using the properly modified PF, PFBT, and PFTBT OCPNs. The proteins were then erased by Loc-displacement for the next cycle of three-color POSA imaging. The entire process of one cycle was completed in less than 10 minutes. The results were displayed in Fig. 6b with a zoom-in image in Fig. 6c. We found that the distribution of the nine proteins mapped by the POSA method matched well with their locations. These results demonstrated that the POSA method could overcome the limit of spectral crowding and realize highly multiplex protein imaging in a very quick (less than one hour for nine-protein imaging) and accurate (for the proteins with different abundance) manner.

a Fluorescent images of the nine proteins during three cycles. 1: anti-EGFR PA8-BS8 (PF-g-IS8 OCPNs), anti-Lamin A/C PA5-BS5 (PFBT-g-IS5 OCPNs) and anti-GM130 PA6-BS6 (PFTBT-g-IS6 OCPNs); 2: anti-mitochondria PA3-BS3 (PF-g-IS3 OCPNs), anti-α-tubulin PA1-BS1 (PFBT-g-IS1 OCPNs) and anti-Ki67 PA2-BS2 (PFTBT-g-IS2 OCPNs); 3: anti-TOMM20 PA4-BS4 (PF-g-IS5 OCPNs), anti-Lamin B1 PA9-BS9 (PFBT-g-IS9 OCPNs), and anti-Vimentin PA7-BS7 (PFTBT-g-IS7 OCPNs). The erasing process was conducted via the Loc-displacement method. b A merged false-color image from (a). c The zoom-in image of the marked region of (b). Source data are provided as a Source Data file.
Discussion
In this work, we utilized the high brightness of FCPs to amplify the protein imaging in situ directly via the optical approach. We synthesized three-colored DNA amphiphilic polymers and optimized the sizes of their self-assembled products, OCPNs. The small size and densely packed DNA shell led OCPNs to access the intracellular environment freely without visible non-specific interactions. On this basis, the POSA method was developed by the coordination between OCPNs and DNA-barcoded antibodies. Using OCPNs, the POSA method has achieved a comparable imaging resolution to the conventional IF method. However, POSA has higher sensitivity and specificity and is less time-consuming (10 minutes/cycle). This is due to the 28- to 126-fold optical signal amplification effects of OCPNs. Additionally, this optical amplification strategy has greatly simplified DNA sequence design and cost. The toehold-mediated strand displacement reaction has enabled cyclic imaging for up to 6 cycles, proving the high fidelity of the POSA method in multiplexed imaging. The spatial distribution of nine independent proteins within single cells was mapped using three cycles of three-color POSA, taking less than an hour in total. Overall, by synthesizing π-conjugated polymer-DNA amphiphilic polymer and optimizing their self-assembly, we applied the optical amplification effect of FCPs in spatially resolved and multiplexed protein mapping in single cells. This innovative application simplified the signal amplification processes, making POSA advanced tools for clinical diagnostics and biomedical research.
Methods
The synthesis of OCPNs
The initial step in the synthesis process was to produce three fluorescent π-conjugated polymers with bromide substituents at the alkyl side chains, namely PF-Br, PFBT-Br, and PFTBT-Br. Next, the products were treated with NaN3 to convert the bromide modifications into azide groups, resulting in PF-N3, PFBT-N3, and PFTBT-N3. In parallel, a DNA synthesizer was adopted to yield the designed oligonucleotides on the solid support of CPG (controlled pore glass) through standard solid-phase phosphonamidite chemistry (Supplementary Fig. 7a). Then, the 5’-butyne phosphonamidite was employed as the last unit to introduce the alkyne group at the 5’ terminal of the oligonucleotide (Supplementary Fig. 7b). At last, the azide-modified π-conjugated polymers reacted with alkyne-modified oligonucleotides by solid-state copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction to yield FCP-g-DNA amphiphilic polymers (Supplementary Fig. 7c). Afterward, FCP-g-DNA were cleaved from the CPG support and transferred into the PBS medium (Supplementary Fig. 7d). We observed that the amphiphilic polymer directly self-assembled into OCPNs according to the TEM characterization (Supplementary Fig. 8a). Further analyzed by agarose gel electrophoresis (AGE), the products showed evenly distributed bands with much slower mobility than the free DNAs (Supplementary Fig. 8b), owing to the large self-assembled structure of OCPNs. Driven by the hydrophobic interaction of the π-conjugated part, these OCPNs bear π-conjugated cores and densely packed oligonucleotide shells.
Design of DNA sequences
Orthogonal DNA oligos used in this work were created by a sequence manipulation suite (Bioinformatics, version 2) and then designed by the NUPACK Design tool. The criteria of all sequences included stable bonding at RT, balanced nucleotide composition (GC content above 30%), less cross-hybridization, and fewer secondary structures in 1x PBS at RT. Each oligonucleotide selected for further experiments was examined by the IDT OligoAnalyzerTM tool to avoid non-specific interaction with mRNA. All DNA sequences employed are shown in Supplementary Table 1.
Estimating the average DNA-graft efficiency
First, the standard concentration curves of PF, PFBT, and PFTBT were plotted by their absorbance at 375, 450, and 515 nm versus concentrations (Supplementary Fig. 15). These standard curves were then used to determine the molar concentration of FCP repeat units, c(FCP), in OCPNs. The mass concentration of DNA in OCPNs, m(DNA), was measured at 260 nm using NanoDrop and then converted to molar concentration. The average DNA-graft efficiency of OCPNs was estimated using the following formula that took into account the presence of 2 azide functional groups in each FCP repeat unit.
\(\eta\): average DNA-graft efficiency of OCPNs;
\(m({DNA})\): the mass concentration of DNA in OCPNs;
\(c({FCP})\): the molar concentration of FCP repeat units in OCPNs;
\({M}_{w}({DNA})\): the molar mass of DNA;
\(V\): the volume of the tested solution of OCPNs;
\(n({DNA})\): the molar number of DNA in OCPNs;
\(n({FCP})\): the molar number of FCP repeat units in OCPNs;
\(n({reacted\; azide\; group})\): the molar number of the reacted azide groups on FCPs;
\(n({all\; azide\; group})\): the molar number of all available azide groups on FCPs.
Estimating the average amount of FCP molecular chains per OCPNs
Under some assumptions, we estimated the average number of FCP molecular chains per OCPN according to the diameter of a single nanoparticle measured by TEM. (1) Nanoparticle was regarded as a perfect sphere; (2) the volume of a nanoparticle core was accumulated by FCPs; (3) DNAs were distributed on the surface of nanoparticles. The following is the calculation method:
\(N\): the number of FCP molecular chains in each nanoparticle;
\(R\): the radius of a nanoparticle measured by TEM;
\(V\): the volume of a FCP molecular chain;
\(\rho\): the density of FCPs;
\(m\): the mass of a FCP molecular chain;
\({M}_{w}\): the molar mass of a FCP molecular chain;
\({N}_{A}\): Avogadro constant, \({N}_{A}=6.02\times {10}^{23}{{mol}}^{-1}\).
The ρ for three kinds of FCP is 1.2 g/cm3 41. Taking PFBT OCPNs with a size around 10 nm as an example, its diameter characterized by TEM is 8 nm, and the average molecular weight determined by GPC is 5.5 kDa. Thus, N, the number of FCP molecular chains forming an OCPN entity, is calculated to be 35. As the molecular weight of the PFBT repeat unit is 604 Da, the polymerization degree is determined to be 9. Thus, an OCPN entity comprises 320 polymer repeat units. According to the 54 % DNA-graft efficiency, 345 DNA strands are functionalized on each OCPN. Finally, according to the fitted curve of absorption value versus concentration shown in Supplementary Fig. 15, the absorption of PFBT in solution is determined by UV-vis. Then, the concentration is calculated by the fitted curve. So that the concentration of OCPNs is determined by the following formula:
\({C}_{{OCPN}}\): the concentration of OCPNs;
\({C}_{r{epeat\; unit}}\,\): the concentration of FCP repeat units;
\(N\): the number of FCP molecular chains composing an OCPN;
\(n\): the polymerization degree of FCPs.
General protocols of the POSA and IF methods
HeLa cell line (Cat # STCC10603P) purchased from Wuhan Servicebio Technology Co., Ltd. (China) was used for this study. The cells were tested negative for mycoplasma contamination by MycoBlueTM Mycoplasma Detector (Cat # D101-01). The cells were cultured in DMEM containing 10 % FBS and 1 % penicillin/streptomycin at 37 °C with 5 % CO2. HeLa cells were seeded in 96-well plates at a density of 104 cells per well and cultured for 24 hours. The cells were rinsed with 1x PBS for three sets of 3 minutes, then fixed with 4% (w/v) PFA for 20 minutes at RT. The fixation process was quenched with 100 μL fixation quencher buffer, 100 mM NH4Cl in 1x PBS, for 20 minutes. The cells were rinsed again with 1x PBS for three sets of 3 minutes. If needed, 100 μL storage buffer, 0.2 % (w/v) NaN3 in 1x PBS, could be added to some of the wells to store the cells at 4 °C. Then, the cells were treated with the permeabilization buffer, 0.1 % (v/v) Triton X-100 in 1x PBS, for 10 minutes at RT. The cells were rinsed with 1 x PBS for three sets of 5 minutes. Next, the cells were blocked with the blocking buffer, 0.2 mg/mL sheared salmon sperm DNA, 0.05 % (w/v) dextran sulfate, and 4 mM EDTA in BlockAidTM for 1 hour with gentle shaking. After removing the liquid, DNA-barcoded antibodies dissolved in blocking buffer were added into the wells and incubated overnight at 4 °C. The concentrations of each type of antibody used for immunolabelling are listed in Supplementary Table 2. The temperature of the cells was allowed to return to room temperature before rinsing them with 1 x PBS for three sets of 3 minutes. For the staining via AF488-labeled SA (Abcam, Cat # ab150077 and Cat # ab150113), SA dissolved in 1 x PBS with a final concentration of 26.6 nM was incubated with cells for 30 minutes at room temperature and then rinsed with 1x PBS for three sets of 3 minutes. For the staining via OCPNs, nanoparticles dissolved in hybridization buffer, 6 % (w/v) BSA, 0.05 % (w/v) dextran sulfate, 0.3 M glycine and 0.2 mg/mL sheared salmon sperm DNA in 2x SSC, with a final concentration of 15 nM were incubated with cells for 5 to 10 minutes and then rinsed with washing buffer, 2 % (w/v) BSA in 2x SSC, for three sets of 3 minutes.
CLSM setting
A confocal laser scanning microscope of Leica TCS-SP8 (Germany) with LAS X software (v4.4.0) was adopted for fluorescent imaging. The fluorescent images were captured at 2048 × 2048 resolution with a ×63 oil immersion objective. For general setting of POSA, PF OCPNs were excited at 405 nm with 5 % laser power, and the fluorescence was collected from 425 to 465 nm with 600 V smart gain; PFBT OCPNs were excited at 448 nm with 5 % laser power, and the fluorescence was collected from 540 nm to 580 with 600 V smart gain; PFTBT OCPNs were excited at 488 nm with 10 % laser power, and the fluorescence was collected from 650 to 700 nm with 900 V smart gain. For fluorescent imaging by IF alone, AF488-SA was excited at 488 nm with 10 % laser power, and the fluorescence was collected from 500 to 540 nm with 700 V smart gain; Cy5-SA (Abcam, Cat # ab6564 and ab6563) was excited at 638 nm with 15 % laser power, and the fluorescence was collected from 650 to 700 nm with 900 V smart gain. For the amplification effect evaluation, all samples were excited at 488 nm with 5 % laser power to collect the fluorescence from 500 to 580 nm with 750 V smart gain. For the photostability test, a setting of 2-fold filed amplification was employed for the irradiation region. The irradiation regions were exposed to a 488 nm laser with 100 % laser power for 3 minutes. Then, the whole region with a 1-fold filed amplification was captured by laser at 488 nm with 5 % laser power. The fluorescence was collected from 500 to 580 nm with 750 V smart gain.
Amplification effect evaluation
Three groups of cells were respectively immunolabelled by anti-α-tubulin PA1-BS1, anti-mitochondria PA3-BS3, and anti-Lamin A/C PA5-BS5 with an identical final concentration of 9.3 nM. All AF488-labeled oligos (AF488-IS1, AF488-IS3, and AF488-IS5), AF488-labeled SA, PFBT OCPNs (10 nm), and PFBT OCPNs (26 nm) were incubated with identical final concentrations of 15 nM. The AF488-labeled oligos and AF488-labeled SA were incubated with cells for 30 minutes and then rinsed with PBS-T, 0.1 % (v/v) Tween 20 in 1x PBS, for three sets of 3 minutes. The PFBT OCPNs (10 nm) and PFBT OCPNs (26 nm) were incubated with cells for 10 minutes and then rinsed with washing buffer for three sets of 3 minutes.
Photostability test
In the immunolabelling step, the concentration of the primary antibody used for IF was 18.6 nM and 9.3 nM for POSA. In the staining step, the concentration of AF488-SA for IF was 26.6 nM, while that of OCPNs for POSA was 6.6 nM. Setting 2-fold filed amplification to focus the laser on the irradiation regions for 3 minutes. The laser power was reduced to shoot the whole region with a 1-fold filed amplification setting.
Strand displacement reaction
For signal erasing during cyclic imaging by Loc-displacement reaction, IS dissolved in hybridization buffer with a final concentration of 100 nM were incubated with cells for 5 minutes (or until the disappearance of fluorescence signal under identical CLSM setting) and then rinsed with washing buffer for three sets of 1 minute. For NP-displacement reaction, corresponding IS dissolved in hybridization buffer with a final concentration of 4 to 5 μM were incubated with cells for 15 to 30 minutes and then rinsed with washing buffer for three sets of 3 minutes.
Statistics and reproducibility
Statistical tests were conducted using Origin software (version 2024b). Data were presented as the means ± standard deviation. Statistical comparison by analysis of variance was performed at a significance level of p ≤ 0.01 (**) and p ≤ 0.0001 (****) based on a Student’s t-test. No data were excluded from the analysis. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment. No statistical method was used to predetermine the sample size. Each experiment was performed in at least triplicate with consistent results. For the representative images shown in Fig. 2d, each type of OCPNs used in the study was characterized by TEM with at least five technical replicates as a routine quality control of OCPNs preparation. For representative images shown in Fig. 2e–j, three technical replicates were performed for each biological replicate, and a total of three biological replicates were produced with consistent results. For representative images shown in Figs. 4b and 5b, five biological replicates were performed with consistent results. For representative images shown in Figs. 4c and 6a, six biological replicates were performed with consistent results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The supporting data are available in the Supplementary Information. The sequence information of DNA and the concentration used information of antibodies are provided in the Supplementary Information (Supplementary Table 1-2). The raw images and quantification data of all figures in this study are provided in Source Data files. Source data are provided with this paper.
References
Liu, X. et al. Computer-aided design of reversible hybridization chain reaction (CAD-HCR) enables multiplexed single-cell spatial proteomics imaging. Sci. Adv. 8, eabk0133 (2022).
Lundberg, E. & Borner, G. H. H. Spatial proteomics: a powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 20, 285–302 (2019).
Thul, P. J. et al. A subcellular map of the human proteome. Science 356, eaal3321 (2017).
Lin, J.-R. et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 7, e31657 (2018).
Hickey, J. W. et al. Spatial mapping of protein composition and tissue organization: a primer for multiplexed antibody-based imaging. Nat. Methods 19, 284–295 (2022).
Gut, G. et al. Multiplexed protein maps link subcellular organization to cellular states. Science 361, eaar7042 (2018).
Zhao, Z. et al. Ultra-bright Raman dots for multiplexed optical imaging. Nat. Commun. 12, 1305 (2021).
Ijsselsteijn, M. E. & de Miranda, N. F. C. C. Advancing multiplexed imaging for enhanced tissue complexity analysis. Nat. Methods 20, 1280–1281 (2023).
Saka, S. K. et al. Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues. Nat. Biotechnol. 37, 1080–1090 (2019).
Black, S. et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat. Protoc. 16, 3802–3835 (2021).
Qing, Z. et al. In situ amplification-based imaging of RNA in living cells. Angew. Chem. Int. Ed. 58, 11574–11585 (2019).
Bai, X. et al. Semiconducting polymer dots as fluorescent probes for in vitro biosensing. J. Mater. Chem. B 10, 6248–6262 (2022).
Collot, M. et al. Stealth and bright monomolecular fluorescent organic nanoparticles based on folded amphiphilic polymer. ACS Nano 14, 13924–13937 (2020).
Algar, W. R. et al. Photoluminescent nanoparticles for chemical and biological analysis and imaging. Chem. Rev. 121, 9243–9358 (2021).
Luan, J. et al. Ultrabright fluorescent nanoscale labels for the femtomolar detection of analytes with standard bioassays. Nat. Biomed. Eng. 4, 518–530 (2020).
Trofymchuk, K. et al. Giant light-harvesting nanoantenna for single-molecule detection in ambient light. Nat. Photonics 11, 657–663 (2017).
Zrazhevskiy, P. et al. Cross‐platform DNA encoding for single‐cell imaging of gene expression. Angew. Chem. Int. Ed. 55, 8975–8978 (2016).
Zrazhevskiy, P. & Gao, X. Quantum dot imaging platform for single-cell molecular profiling. Nat. Commun. 4, 1619 (2013).
Egloff, S. et al. Amplified fluorescence in situ hybridization by small and bright dye-loaded polymeric nanoparticles. ACS Nano 16, 1381–1394 (2022).
Zhou, W. et al. Combining qdot nanotechnology and DNA nanotechnology for sensitive single‐cell imaging. Adv. Mater. 32, 1908410 (2020).
Feng, L. et al. Conjugated polymer nanoparticles: preparation, properties, functionalization and biological applications. Chem. Soc. Rev. 42, 6620–6633 (2013).
Tian, Z. et al. Amplified energy transfer in conjugated polymer nanoparticle tags and sensors. Nanoscale 2, 1999–2011 (2010).
Liu, Z. et al. Narrow-band polymer dots with pronounced fluorescence fluctuations for dual-color super-resolution imaging. Nanoscale 12, 7522–7526 (2020).
Qian, C.-g et al. Conjugated polymer nanomaterials for theranostics. Acta Pharmacol. Sin. 38, 764–781 (2017).
Xiao, F. et al. Hydrophobic interaction: a promising driving force for the biomedical applications of nucleic acids. Adv. Sci. 7, 2001048 (2020).
Guo, L. et al. Polymer dots as effective phototheranostic agents. Photochem. Photobiol. 94, 916–934 (2018).
Swager, T. M. The molecular wire approach to sensory signal amplification. Acc. Chem. Res. 31, 201–207 (1998).
Wu, C. et al. Bioconjugation of ultrabright semiconducting polymer dots for specific cellular targeting. J. Am. Chem. Soc. 132, 15410–15417 (2010).
Xiao, F. et al. Light-harvesting fluorescent spherical nucleic acids self-assembled from a DNA-grafted conjugated polymer for amplified detection of nucleic acids. Angew. Chem. Int. Ed. 61, e202115812 (2022).
Modi, S. et al. A DNA nanomachine that maps spatial and temporal pH changes inside living cells. Nat. Nanotechnol. 4, 325–330 (2009).
Zhang, J. et al. Ultrabright Pdots with a large absorbance cross section and high quantum yield. ACS Appl. Mater. Interfaces 14, 13631–13637 (2022).
Wu, I. C. et al. Squaraine-based polymer dots with narrow, bright near-infrared fluorescence for biological applications. J. Am. Chem. Soc. 137, 173–178 (2015).
Wu, C. et al. Ultrabright and bioorthogonal labeling of cellular targets using semiconducting polymer dots and click chemistry. Angew. Chem. Int. Ed. 49, 9436–9440 (2010).
Fong, L.-K. et al. The role of structural enthalpy in spherical nucleic acid hybridization. J. Am. Chem. Soc. 140, 6226–6230 (2018).
Randeria, P. S. et al. What controls the hybridization thermodynamics of spherical nucleic acids? J. Am. Chem. Soc. 137, 3486–3489 (2015).
Im, K. et al. An introduction to performing immunofluorescence staining. Methods Mol. Biol. 1897, 299–311 (2019).
Jiang, Y. et al. High‐precision mapping of membrane proteins on synaptic vesicles using spectrally encoded super‐resolution imaging. Angew. Chem. Int. Ed. 62, e202217889 (2023).
Biswas, D. S. et al. Long-range energy transfer between dye-loaded nanoparticles: observation and amplified detection of nucleic acids. Adv. Mater. 35, 2301402 (2023).
Wu, C. & Chiu, D. T. Highly fluorescent semiconducting polymer dots for biology and medicine. Angew. Chem. Int. Ed. 52, 3086–3109 (2013).
Halabi, E. A. & Weissleder, R. Light-deactivated fluorescent probes (FLASH-Off) for multiplexed imaging. J. Am. Chem. Soc. 145, 8455–8463 (2023).
Geng, J. et al. A general approach to prepare conjugated polymer dot embedded silica nanoparticles with a SiO2@CP@SiO2 structure for targeted HER2-positive cellular imaging. Nanoscale 5, 8593 (2013).
Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (22475094 to LL. T.), the Shenzhen Fundamental Research Programs (JCYJ20220530113612027 to LL. T.), Guangdong Basic and Applied Basic Research Foundation (2024A1515012055 to LL. T.), Shenzhen Science and Technology Program (SGDX20211123114002003 to J. L.), and the SUSTech high level of special funds (G03050K003 to LL. T.).
Author information
Authors and Affiliations
Contributions
LL.T. and WK.Z. conceived the project and designed the experiments. WK.Z., HF.J., and L.H conducted synthesis, characterizations, and assay development experiments. F.H., J.W., and J.L. provided technical support to the experiments and aided in interpreting the results. WK.Z. and LL.T. analyzed the data and co-wrote the manuscript with input from the other authors. LL.T. supervised the project. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
This work is related to a pending patent (2024105528370). The patent applicant is the institution Southern University of Science and Technology. LL. T., WK. Z., and F. H. are named as inventors. Status of the application: The patent application was filed at the China National Intellectual Property Administration. The specific aspect of the manuscript covered in the patent application is the protocols of the POSA method and its application in multiplexed protein imaging in cells and tissues. The remaining authors declare no competing interests.
Ethics
The HeLa cell line (purchased from Wuhan Servicebio Technology Co., Ltd. China, Cat # STCC10603P) used in this study is a commercially available standard cell line, which does not involve human genetic resources or individual privacy information. No animal or human experiments were included in this study.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, W., Jiang, H., Han, L. et al. POSA for fast, amplified and multiplexed protein imaging. Nat Commun 16, 2300 (2025). https://doi.org/10.1038/s41467-025-57589-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57589-w
