
Abstract
Despite the tremendous therapeutic promise of activating stimulators of interferon genes (STING) enable to prime robust de novo T-cell responses, biomechanics-mediated immune inhibitory pathways hinder the cytotoxicity of T cells against tumor cells. Blocking cancer cell biomechanics-mediated evasion provides a feasible strategy for augmenting STING activation-mediated anti-tumor therapeutic efficacy. Here, we fabricate a redox-responsive Methyl-β-cyclodextrin (MeβCD)-based supramolecular polyrotaxanes (MSPs), where the amphiphilic diselenide-bridged axle polymer loads MeβCD by the host-guest interaction and end-caping with two near-infrared (NIR) fluorescence probes IR783. The MSPs self-assemble with STING agonists diABZIs into nanoparticles (RDPNs@diABZIs), which enable simultaneous release of MeβCD and diABZIs in the redox tumor microenvironment. After the released diABZIs activate STING on antigen-presenting cells (APCs), de novo T-cell responses are initiated. Meanwhile, the released MeβCD depletes membrane cholesterol to overcome cancer-cell mechanical softness, which enhances the CTL-mediated killing of cancer cells. In the female tumor-bearing mouse model, we demonstrate that RDPNs@diABZIs lead to effective tumor regression and generate long-term immunological memory. Furthermore, RDPNs@diABZIs can achieve significant tumor eradication, with these mice remaining survival for at least 2 months.
Similar content being viewed by others


Introduction
Pharmacological activation of stimulator of interferon genes (STING) via STING agonists solves the inherent shortcomings of endogenous STING messenger 2’,3’-cyclic-GMP-AMP (cGAMP) and primes robust de novo T-cell responses, revolutionizing the cancer treatment1,2,3,4,5,6. T cells sense mechanical environments and exert forces at the immunological synapse to assist perforin drilling pores in the cell membrane, potentiating cytotoxicity against cancer cells7. Despite the tremendous therapeutic promise of STING agonists, cancer cells generally hamper T cells-mediated cytotoxicity by softening the cortical stiffness through membrane cholesterol enrichment and F-actin levels downregulation to achieve biomechanics-mediated immune inhibitory pathways8,9,10. Thus, manipulating cholesterol levels in the membrane lipid bilayer of cancer cells contributes to T-cell-mediated cancer treatments. Regulation of cholesterol levels often utilizes pharmacological or genetic interventions, such as statins and targeted silencing genes using short hairpin or small interfering RNA11,12. However, non-specific side effects and the cumbersome operation of these technologies limit the regulation of cancer cell stiffness at the in vivo level.
Macrocyclic methyl-β-cyclodextrin (MeβCD) has an intrinsic host-guest recognition with cholesterol, exhibiting special cholesterol-depleting capabilities in cell membranes13,14,15. Unfortunately, MeβCD is not specific to tumor cells and is rapidly cleared during circulation, thereby reducing their effectiveness in increasing the cancer-cell stiffness16,17,18. Thus, it is crucial to deliver plenty of MeβCD into the tumor microenvironment (TME) to turn the mechanical property of tumor cells and reinforce the T-cell-mediated killing of cancer cells. MeβCD-based supramolecular polyrotaxanes (MSPs) contain amphiphilic polymers that act as the axle, and plentiful macrocyclic MeβCD act as the wheels, driven by the host-guest interaction19,20,21,22. After sealing the MeβCD into MSPs, the MSPs enable self-assembling into nanoplatforms with high stability/biocompatibility for encapsulating therapeutic drugs for cancer theranostics23,24,25,26. However, to achieve the effective delivery and burst release of MeβCD into TME, the axle of MSPs is required to be cleavable in response to the acid or redox in TME27,28.
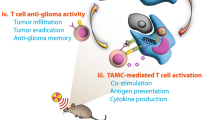
Herein, we fabricate redox-responsive MSPs with diselenide-bridged axle polymer by end-capping with two near-infrared (NIR) fluorescence probes IR78329,30,31,32. The MSPs further self-assembled with a STING agonist diABZIs into nanoparticles (RDPNs@diABZIs), which enabled simultaneous release of MeβCD and diABZIs in response to the redox TME33,34,35. The in vivo therapeutic process and the biodistribution of the RDPNs@diABZIs are visualized by fluorescence imaging (Fig. 1). After the accumulation of RDPNs@diABZIs in TME, the released diABZIs elicit STING activation on antigen-presenting cells (APCs) and then priming T-cell-mediated anti-tumor response. Meanwhile, the released MeβCD depletes membrane cholesterol to overcome cancer-cell mechanical softness, which enhances the cytotoxic T lymphocytes (CTLs)-mediated killing of cancer cells. In the tumor-bearing mouse model, we successfully demonstrate that RDPNs@diABZIs lead to effective tumor regression and generate long-term immunological memory. Furthermore, RDPNs@diABZIs can achieve significant tumor eradication, with these mice remaining survival for at least 2 months.

RDPNs@diABZIs nanoparticles, composed of the diselenide-bridged polyrotaxanes loaded with agonists diABZIs, generate robust anti-tumor immunity by synergistically enhancing T-cell intratumoral infiltration and T-cell force, thereby potentiating T-cell cytotoxicity. ROS reactive oxygen species, GSH Glutathione, CTL Cytotoxic T-Lymphocyte, DC Dendritic cell, STING stimulator of interferon genes, IFN Interferon.
Results
Synthesis and characterization of RDPNs@diABZIs
MeβCD-based supramolecular polyrotaxanes (MSPs) with diselenide-bridged axle polymer by end-capping with two NIR fluorescence probes IR783 (IR783-PEG-MeβCD-PDS) was synthesized, as detailed in the experimental section (Supplementary Fig. 1)36,37. As a control, MSPs with dicarbonyl-bridged axle polymer by end-capping with two NIR fluorescence probes IR783 (IR783-PEG-MeβCD-PTD) were also synthesized. H nuclear magnetic resonance (1H NMR) spectra were used to characterize the successful synthesis of polyrotaxane (Supplementary Figs. 2–6). By comparing the integration of the proton signals from MeβCD with those from the PEG segment, the average number of MeβCD units in the polyrotaxane was calculated to be 16.8 (Supplementary Fig. 6). Gel permeation chromatography (GPC) curves were shown that the average molecular weight (Mn) of IR783-PEG-MeβCD-PDS was determined to be 39.4 kDa, 22.6 kDa higher than PEG-PDS (Mn = 16.8 kDa), providing direct evidence for the formation of a polyrotaxane with approximately 17 MeβCD units (Fig. 2a). Additionally, thermogravimetric analysis confirmed that MeβCD is incorporated within the polymer PEG-PDS (Fig. 2b). The Fourier-transform infrared (FT-IR) spectrum of PEG-MeβCD-PDS displayed double characteristic peaks at 3433 cm-1 and at 3274 cm-1, corresponding to the N-H stretching of -NH2 groups (Supplementary Fig. 7). In contrast, the FT-IR spectrum of IR783-PEG-MeβCD-PDS showed the disappearance of the peak at 3274 cm-1, indicating the conversion of -NH2 to -NHR during the coupling reaction. These changes confirmed the successful coupling of IR783 to PEG-MeβCD-PDS. Moreover, the absorption spectrum of IR783 showed slightly red-shifting compared with that of IR783-PEG-MeβCD-PDS, indicating that IR783 is present on PEG-MeβCD-PDS (Fig. 2c). Under the same IR783 concentrations, the fluorescence intensity of IR783-PEG-MeβCD-PDS increased compared to that of free IR783 (Supplementary Fig. 8). These results suggested that IR783-PEG-MeβCD-PDS was successfully prepared, with IR783 capped into the polymer acting as a stopper. Eventually, polyrotaxane IR783-PEG-MeβCD-PDS was self-assembled into redox-responsive diselenide-bridged polyrotaxanes nanoformulations (RDPNs) in aqueous solution, with nonresponsive polyrotaxanes nanoparticles (NPNs) as control. STING agonists diABZIs were further encapsulated into the hydrophobic core of the nanoparticles (RDPNs@diABZIs) through simple ultrasonication38. The diABZIs-loaded RDPNs (RDPNs@diABZIs) were prepared with a diABZIs loading efficiency of 5.67% (Supplementary Fig. 13). Dynamic light scattering (DLS) results revealed that the hydrodynamic sizes of RDPNs, NPNs, and RDPNs@diABZIs were 94.09 ± 6.8 nm, 131.71 ± 9.4 nm, and 142.7 ± 8.5 nm, respectively (Fig. 2d). Additionally, the surface zeta potentials of these constructed NPs were found to be negatively charged, which were -1.05 ± 0.17 mV, -0.58 ± 0.12 mV, and -1.19 ± 0.35 mV, respectively (Fig. 2e). Moreover, RDPNs@diABZIs remained stable in various buffers for two weeks, demonstrating their stability in physiological systems, which provided assurance for subsequent experiments (Fig. 2e and Supplementary Figs. 9–11). The degradation behavior of RDPNs was investigated under oxidative conditions (H2O2), mimicking the in vivo TME condition39,40,41. As revealed by transmission electron microscopy (TEM) images, compared with the uniform spherical structure of RDPNs under PBS conditions, RPDNs aggregated and swelled after 12 h of exposure to H2O2 solution and fully collapsed into small fragments within 24 h. (Fig. 2g). Additionally, DLS results also revealed that as the concentration of hydrogen peroxide increased, the hydrodynamic sizes were gradually increased, indicating the RPDNs aggregated and swelled (Fig. 2h). On the contrary, the nonresponsive poly-MeβCD nanoparticles (NPNs) showed negligible changes in morphology and hydrodynamic sizes, and no degradation in the oxidative condition after 24 h of incubation (Supplementary Fig. 12). To investigate whether the degradation of RPDNs can induce the release of diABZIs, high-performance liquid chromatography (HPLC) was used to monitor the release behavior. RPDNs showed an increasing release of diABZIs as the concentration of hydrogen peroxide increased (Fig. 2i and Supplementary Fig. 13). They showed a burst release of approximately 78.5% of the cumulative total diABZIs in the oxidative condition after 6 h of incubation, fitting well with the degradation behavior.

a GPC curves of PEG-PDS and IR783-PEG-MeβCD-PDS. GPC was performed using N,N-dimethylformamide as the eluent. b TGA curves of MeβCD, PEG-PDS and IR783-PEG-MeβCD-PDS from 30 °C to 800 °C at a rate of 10 °C min-1 under nitrogen atmosphere. c Absorbance spectra of IR-783 and IR783-PEG-MeβCD-PDS. d, e The hydrodynamic diameters and zeta potential of NPNs, RDPNs, and RDPNs@diABZIs. n = 3 biologically independent samples per group, repeated three times. f The average hydrodynamic diameter of RDPNs@diABZIs were measured in PBS buffer over 2 weeks. n = 3 biologically independent samples per group, repeated three times. g, h TEM images and hydrodynamic diameters of RDPNs in different treatments with hydrogen peroxide (H2O2). Scale bar, 400 nm. i In vitro diABZIs release profile of RDPNs@diABZIs co-cultured with or without different concentrations of H2O2. n = 3 biologically independent samples per group, repeated three times. Data were expressed as means ± SD.
Cancer-cell stiffness is enhanced by depletion of cholesterol with RDPNs
Contributing to abnormal generation and cross-linking of extracellular matrix proteins, tumors are typically harder than their corresponding normal tissues42. Interestingly, membranes of cancer cells are often softer than non-malignant cells43,44,45. Cancer cells soften cell membranes via increasing cholesterol, which reduces the force exerted by T-cell synapses to help cancer cells evade T-cell killing. MeβCD, a cholesterol-depletion molecule, could increase cancer cell membrane stiffness (Fig. 3a). Compared to HC11 cells (murine breast epithelial cells), 4T1 cells (murine breast cancer cells) showed a significant increase in cholesterol levels (Fig. 3b and Supplementary Fig. 14). We further quantified the cholesterol levels in tumor tissues and normal tissues isolated from 4T1-bearing mice. The cholesterol levels in tumor tissues increased by 2.67 times compared to adjacent muscle tissues (Fig. 3c). Additionally, elevated cholesterol levels were analyzed in different types of tumor tissues (Supplementary Fig. 15). These results provide evidence that a common feature of cholesterol levels in various types of cancer cells was higher than those of normal cells.

a Illustrations of mechanical immuno-suppression induced by the softened cancer cells and mechanical immuno-activation induced by the stiffened cancer cells resulting from membrane cholesterol depletion. b Cholesterol levels in different cells. n = 3 biologically independent samples per group, repeated three times. c Cholesterol levels in 4T1 tumor tissues or the adjacent muscle tissues. n = 3 biologically independent samples per group, repeated three times. d, e Relative intracellular and plasma membrane levels of cholesterol in 4T1 cancer cells treated with PBS, MeβCD, NPNs, or RDPNs in vivo (d) and in vitro (e). n = 3 biologically independent samples per group, repeated three times. f, g The Filipin III staining of 4T1 tumor cells after different treatments. Experiments were performed three times with similar results. Scale bar, 100 nm. h Relative cortical stiffness of 4T1 cancer cells treated with different treatments. n = 6 biologically independent samples per group, repeated three times. i The schematic diagram of perforin forming pores in native cancer cells or stiffness cancer cells. j Viability of pre-treated 4T1 cancer cells after incubation with perforin at indicated concentrations at 37 °C for 20 min. n = 3 biologically independent samples per group, repeated three times. k Lysis percentage of 4T1 cancer cells pre-treated with activated CD8+ T cells at an E: T ratio of 10:1 for 5 h. n = 4 biologically independent samples per group. Data were expressed as means ± SD. G1: PBS, G2: MeβCD, G3: NPNs, G4: RDPNs. One-way ANOVA with Tukey’s post hoc test (b, d, e, h, k) or Two-tailed Student’s t-test (c). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To investigate the optimal therapeutic window for MeβCD in deleting membrane cholesterol, we quantified the cholesterol levels in 4T1 cells and T cells co-cultured with different concentrations of MeβCD in vitro. The half-maximal effective concentration (EC50) of MeβCD for cholesterol depletion in tumor cells was 3.85 mM, while no significant change was observed in the membrane cholesterol levels of T cells (Supplementary Fig. 16). The results indicated that T cells have a higher tolerance to MeβCD than 4T1 tumor cells. Additionally, the membrane cholesterol level of various cancer cells was reduced after co-culturing with MeβCD, while the intracellular cholesterol level showed negligible changes (Supplementary Fig. 17). Therefore, plasma membrane cholesterol levels could be controlled via MeβCD in various cancer cells.
Subsequently, we assessed the cell viability of 4T1 cells, T cell, or L929 (mouse fibroblast cell line) cells co-cultured with different drugs for 24 h. The changes in L929 cells and T cells treated with RDPNs were negligible (Supplementary Fig. 18). In contrast, the viability of 4T1 cells was significantly decreased. Moreover, no obvious change in the viability of 4T1 cells was observed after treatment with NPNs (Supplementary Fig. 19). Further, the analysis of live/dead staining and Annexin V-FITC/propidium iodide (PI) flow cytometry were consistent with the above results (Supplementary Figs. 20, 21). These results demonstrate that RDPNs exhibit selective cytotoxicity against cancer cells, while causing no damage to T cells and normal cells.
To verify whether RDPNs also can reduce membrane cholesterol levels, 4T1 tumor-bearing mice were used as models that were treated with MeβCD, RDPNs, or NPNs intratumorally (i.t). We found that the membrane cholesterol level of cancer cells treated with RDPNs dropped markedly to only 47.8% relative to PBS group (G1). In contrast, the membrane cholesterol level of 4T1 cancer cells treated with NPNs showed negligible changes (Fig. 3e). Additionally, similar results were obtained by intravenous injection (Supplementary Fig. 22). Moreover, the results in vitro were consistent with in vivo (Fig. 3d). Further, the membrane cholesterol level of 4T1 cancer cells were visualized or quantitatively analyzed via staining the Filipin III, a fluorescent dye that specifically binds to cholesterol (Fig. 3f, g and Supplementary Fig. 23). The fluorescence images and the flow cytometry analysis all revealed the outstanding cholesterol-depleting capability of RDPNs comparable to the same concentration of MeβCD. To further investigate whether RDPNs influence the membrane cholesterol levels of tumor-infiltrating T cells, we quantified the levels in cells in vitro and in vivo. The results showed that RDPNs had a negligible decrease on the membrane cholesterol levels of tumor-infiltrating T cells (Supplementary Fig. 24). We further investigated the functionality of tumor-infiltrating T cells after treating with RDPN. Compared to the PBS group, the population of tumor-infiltrating CD8+ T cells showed no significant change after RDPNs treatment (Supplementary Fig. 25). Additionally, the population of CD8+IFN-γ+ T cells and CD8+GzmB+ T cells also remained unchanged. These results demonstrate that RDPNs show a burst release of MeβCD in tumors, reducing membrane cholesterol levels of tumor cells while having negligible effects on membrane cholesterol levels and functionality of tumor-infiltrating T cells.
To investigate whether cancer-cell stiffness could be enhanced by depleting membrane cholesterol, we directly measured single-cell cortical stiffness using Atomic Force Microscopy (AFM)46 (Fig. 3h). We found that the cortical stiffness of 4T1 cancer cells treated with MeβCD and RDPNs decreased by 3.02 times and 2.89 times, respectively, relative to PBS group (G1). Moreover, similar results were observed in other cancer cells (Supplementary Fig. 26). These results demonstrated that cholesterol depletion in the cell membrane contributes to cancer cell stiffness.
Cancer-cell stiffness by RDPNs enhances T-cell-mediated cytotoxicity through T-cell forces
Cancer-cell softness impairs T-cell forces further preventing membrane pore formation by perforin47, which subsequently prevents the entry of granzyme B to initiate apoptosis. Cancer-cell stiffening via cholesterol depletion allowed for accelerating the speed of pore formation by synergistic mechanical force and perforin (Fig. 3i). To investigate whether cytotoxicity of different stiffness of cancer cells is related to perforin concentrations in the absence of activated T cells, differently pre-treated 4T1 cells were co-cultured with varying concentrations of perforin (Fig. 3j). Interestingly, in the absence of activated T cells, the cell viability of cancer cells of different stiffness was independent of the concentration of perforin. Next, we co-cultured activated CD8+ T cells with pre-treated 4T1 cells. Similar to pre-treated with MeβCD, RDPNs-pretreated 4T1 cells accelerated the speed of pore formation compared to native 4T1 cells (G1) (Fig. 3k). These findings indicated that cancer cell stiffening enhances T cell force-mediated cytotoxicity, mediated by increased membrane pore formation by perforin.
Activation of cGAS-STING Pathway by RDPNs@diABZIs in vitro
STING agonists diABZIs can activate the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway (Fig. 4b)48,49. To monitored the cytotoxicity of RDPNs@diABZIs, we co-cultured 4T1 cells and L929 cells with RDPNs@diABZIs. The result revealed that RDPNs@diABZIs exhibits selective toxicity towards cancer cells (Fig. 4a). To verify whether RDPNs@diABZIs enhance secretion of type-I Interferon (IFN-I) in APCs, we detected the levels of IFN-β secretion. cGAMP, as a second messenger to activate STING, rarely elicited the secretion of IFN-β with a half-maximum effective concentration (EC50) of 84.61 ± 3.11 µM in THP-1 (Fig. 4c). This can contribute to its acts as a negatively charged molecule. Interestingly, similar to EC50 of 5.15 ± 0.59 µM of STING agonists diABZIs, RDPNs@diABZIs also induced a concentration-dependent secretion of IFN-β in THP-1 cells with EC50 of 5.70 ± 0.71 µM (Fig. 4c). Compared with cGAMP, the delivery of diABZIs or RDPNs@diABZIs increased the IFN-β secretion levels by order of magnitude. Similarly, an enhancement in stimulating IFN-β secretion was observed in Bone Marrow Dendritic Cells (BMDCs), the EC50 of diABZIs, RDPNs@diABZIs, and cGAMP was 5.71 ± 0.12 µM, 6.42 ± 0.21 µM, 92.36 ± 6.90 µM (Fig. 4d). Compared to cells treated with free cGAMP, BMDCs stimulated by RDPNs@diABZIs exhibited significantly increasing expression of co-stimulatory molecules CD80 and CD86 (Fig. 4e–h). Western blot analysis revealed a noticeable phosphorylation of STING (p-STING) and IRF-3 (p-IRF-3) in response to diABZIs and RDPNs@diABZIs (Fig. 4i, j). The expression levels of pro-inflammatory cytokines tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and IL-1β also significantly increased (Fig. 4k-m). These results suggested that RDPNs@diABZIs can activate the cGAS-STING pathway and have the potential to stimulate T-cell activation.

a Viability of L929 and 4T1 co-cultured with RDPNs@diABZIs for 24 h. n = 5 biologically independent samples per group, repeated three times. b Schematic illustration of the activation of the GAS-STING pathway in dendritic cells (DCs). c, d Dose-dependent levels of secretion of IFN-β elicited by indicated formations in THP-1 and BMDCs. n = 3 biologically independent samples per group, repeated twice. e-h Representative flow cytometric plots and quantification of DC maturation surface marker expression (CD80+, CD86+) in BMDCs treated with indicated formulations for 24 h. n = 3 biologically independent samples per group, repeated twice. i, j Western blot image and semiquantitative analysis of p-STING and p-IRF-3 in THP−1 cells treated with various treatments. n = 3 biologically independent samples biologically independent animals per group. The samples derive from the same experiment and that blots were processed in parallel. k–m Cytokine secretion levels in the supernatants of BMDCs treated with indicated formulations for 24 h. n = 3 biologically independent samples per group, repeated twice. Data were expressed as means ± SD. G1: PBS, G2: cGAMP, G3: diABZIs, G4: RDPNs@diABZIs. One-way ANOVA with Tukey’s post hoc test (g, h, j, k, l). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Biodistribution and pharmacokinetic analysis
We evaluated the pharmacokinetics profiles by measuring the concentration of IR783. The elimination half-life (t1/2) of RDPNs and free IR783, calculated were 1.40 h and 0.31 h, respectively (Fig. 5a). Similarly, t1/2 of RDPNs@diABZIs and diABZIs were 1.5 h and 0.25 h, respectively (Fig. 5b). These provide evidence that nanomaterials can extend the blood circulation time of IR783 compared to free IR783. Additionally, the area under the curve (AUC) for RDPNs was about 10 times than IR783 (Fig. 5c). The concentration of IR783 in tumors sacrificed from injected with RDPNs was significantly enhanced compared with that injected with free IR783 (Fig. 5d). Therefore, we have reason to believe that RDPNs exhibit long-term retention characteristics in the tumor tissue.

a, c Plasma concentration-time profiles of IR783 and diABZIs post i.v. injection.n = 3 biologically independent animals per group, repeated twice. b The AUC0-t of IR783 and diABZIs analysis. n = 3 biologically independent animals per group, repeated twice. d Quantification of RDPNs in tumor and major organs by fluorescence spectrophotometer. n = 3 biologically independent animals per group. e, g The NIR fluorescence images and the relative fluorescence intensity of tumors in vivo of the 4T1-tumor bearing mice after intravenous injection of NPNs or RDPNs. n = 3 biologically independent animals per group, repeated twice. f, h Ex vivo NIR images and relative fluorescence intensity of different organs. n = 3 biologically independent animals per group, repeated twice. Data were expressed as means ± SD. One-way ANOVA with Tukey’s post hoc test (h) or two-tailed Student’s t-test (b, d). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Further, to investigate the biodistribution of RDPNs in the tumor-bearing mice model, we intravenously injected RDPNs or NPNs and monitored the fluorescence of IR783 using small animal in vivo imaging. In vivo fluorescence images and quantitative analysis all indicated enhanced tumor fluorescence signal with a peak at 12 h post-injection of RDPNs or NPNs (Fig. 5e, g). The fluorescence intensity gradually decreases after 24 h, but fluorescence is still visible at the tumor site at 48 h. The above results indicated a strong drug retention effect of RDPNs or NPNs. Furthermore, ex vivo images sacrificed at 48 h showed significant nanomedicines accumulation in the tumor site and little accumulation in the major organ (heart, liver, spleen, lung, Kidney), which indicated that nanomedicines were selectively accumulated to tumors (Fig. 5f, h).
RDPNs@diABZIs shift tumor immune microenvironment
The activation of the cGAS-STING pathway can prime innate and adaptive immune responses, which is beneficial for anti-tumor systemic immune responses50,51,52. An orthotropic 4T1 mouse breast tumor model was established. Surface expression of CD86 of dendritic cells (DCs, CD45+CD11c+CD86+) of the tumor-draining lymph node (TDLN) in RDPNs@diABZIs-treated mice was increased (Fig. 6a and Supplementary Fig. 27). Additionally, the total cell number and the fraction of activated DCs also were markedly increased, which exhibited approximately 2.23-fold activated DCs than those treated with PBS. (Fig. 6b and Supplementary Figs. 27, 28). The total cell number and the fraction of M1-like macrophages (CD11b+F4/80+CD86+) in the TME was significantly increased in mice-treated RDPNs@diABZIs than other groups (Fig. 6c, d and Supplementary Figs. 29, 30). On the contrary, RDPNs@diABZIs decreased the total cell number and the fraction of M2-like macrophages (CD11b+F4/80+CD206+) in TME compared with that received PBS only (Fig. 6e, h and Supplementary Figs. 31, 32). These results indicated repolarization or recruitment of macrophages with reduced immunosuppressive capacity. Notably, compared with those treated with others, the tumors in mice that received RDPNs@diABZIs increased the total number and the fraction of natural killer cells infiltrated (Fig. 6i and Supplementary Figs. 34-35). We further verified whether downregulating immune cells with immunosuppressive capacity and upregulating that with immune-activated capacity in TME can potentiate anti-tumor immune responses. RDPNs@diABZIs led to cytotoxic CD8+ T cells in TDLN increased compared with other groups (Fig. 6f, j and Supplementary Figs. 36-38). Immunofluorescence staining of the CD8 marker in tumor slices retrieved after mice treated with RDPNs@diABZIs showed enhanced infiltration of CD8+ T cells (Supplementary Fig. 40). The mice that received RDPNs@diABZIs elicited 2.43-fold more CD8+ T cells and 1.85-fold more CD4+ T cells than that treated PBS only. The expansion of CD8+IFN-γ+ T cells in mice receiving RDPNs did not exceed the baseline level, while diABZIs treatment resulted in a 2.67-fold increase in IFN-γ-secreting CTLs (Fig. 6g, k and Supplementary Fig. 39). Notably, RDPNs@diABZIs further elevated the frequency, with the expression level of CD8+IFN-γ+ T cells in TDLNs being 4.84 times higher than that of the PBS control group. We further measured a kinetics study over 7 days of pro-inflammatory cytokines in serum. The concentration of IFN-γ, TNF-α, and Granzyme B in serum increased rapidly, peaking at 72 h post-treatment with RDPNs@diABZIs (Fig. 6l and Supplementary Figs. 41-43). Subsequently, the concentration of pro-inflammatory cytokines decreased and returned close to benchmark level by 7 days after treatment. These findings indicate that RDPNs@diABZIs can initiate systemic T-cell immune responses, with the associated systemic inflammation being transient, which is essential to ensuring its safety.

a, b Representative flow cytometry histogram and relative quantification of CD86 expressions by DCs (CD45+CD11c+CD86+) in the TDLNs. n = 3 biologically independent animals per group. c, d Representative flow cytometric analysis and quantification of CD86 expressions in M1-like macrophages (CD45+CD11b+F4/80+CD86+) in tumors. n = 3 biologically independent animals per group. e, h Representative flow cytometry histogram and quantification of CD206 expressions in M2-like macrophages (CD45+CD11b+F4/80+CD206+) in tumors. n = 3 biologically independent animals per group. f, j, Representative flow cytometric analysis and relative quantification of CD8+ T cells (CD3+CD8+) in the TDLNs. n = 3 biologically independent animals per group. g, k Representative flow cytometric analysis and relative quantification of CD8+IFN-γ+ T cells (CD45+CD8+IFN-γ+) in the TDLNs. n = 3 biologically independent animals per group. i Quantification of NK cells (CD45+CD49b+) in spleen. n = 3 biologically independent animals per group. l Serum cytokine concentration of IFN-γ, TNF-α and Granzyme B. n = 3 biologically independent animals per group. Data were expressed as means ± SD. (G1: PBS, G2: NPNs, G3: RDPNs, G4: diABZIs, G5: RDPNs@diABZIs. DCs, Dendritic cells. NK cells, Natural killer cells. One-way ANOVA with Tukey’s post hoc test (b, c, h, i, j, k, l). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
RDPNs@diABZIs trigger robust anti-tumor therapeutic efficacy
Based on the above results, we next evaluated the anti-tumor effect of RDPNs@diABZIs in vivo. An orthotropic 4T1-luc mouse breast tumor model was established, and mice were randomly divided into five groups: PBS (G1), NPNs (G2), RDPNs (G3), diABZIs (G4), and RDPNs@diABZIs (G5). Mice were treated when the tumor volume reached approximately 100 mm3 and were intravenously injected with indicated formulations on days 0, 2, and 4. Mice treated with NPNs did not suppress tumor growth (Fig. 7a–d). On the contrary, the tumor volume of mice treated with RDPNs was inhibited. These demonstrated that MeβCD released from RDPNs reducing membrane cholesterol promotes cancer cell stiffness and induces its apoptosis, but it can’t be released from NPNs. Undoubtedly, tumor growth rates markedly were inhibited in mice that received diABZIs only, which contributed to enhanced anti-tumor immune responses induced by diABZIs-activated cGAS-STING pathway. Notably, mice treated with RDPNs@diAZBIs showed robust tumor regression and long-term survival of at least 2 months in about 83.3%. Consistent with the above results, mice treated with RDPNs@diAZBIs obtained more efficient therapeutic effects than other group according to the hematoxylin-eosin (H&E) and TUNEL staining results (Supplementary Figs. 44, 45). Next, blood chemistry analysis, including alanine aminotransferase (ALT), lactate dehydrogenase (LDH), aspartate transaminase (AST), urine acid (UA), creatine kinase (CK), and creatinine (CRE) were performed for long-term toxicity evaluation after treatments. The results showed all treatments did not induce significant side effects in the mice (Supplementary Fig. 46). Additionally, there were no significant pathological changes in major organs (heart, liver, spleen, lung, Kidney) H&E slices images (Supplementary Fig. 47).

a Respective bioluminescence images of tumors in mice on day 0, day 5, day 10, and day 15. b, c Individual and average tumor volume curves with indicated treatments. n = 6 biologically independent animals per group. d Survival curves after treatments. n = 6 biologically independent animals per group. e, f Representative flow cytometric analysis and quantification of memory T cells (CD3+CD8+CD44highCD62Llow, gated on CD3+CD8+ T cells) in the spleen. n = 4 biologically independent animals per group, repeated twice. g Survival curves after subcutaneous rechallenge. n = 5 biologically independent samples per group. h, i Bioluminescent images and fluorescence intensity of lung in BALB/C mice. n = 3 biologically independent animals per group. j Relative fluorescence intensity of ex vivo lungs. n = 3 biologically independent animals per group. G1: PBS, G2: NPNs, G3: RDPNs, G4: diABZIs, G5: RDPNs@diABZIs. Data are presented as mean ± s.d. One-way ANOVA with Tukey’s post hoc test (e) or two-tailed Student’s t-test (i, j). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
We next explored the mechanism of tumor regression and durable anti-tumor effect in mice-treated RDPNs@diAZBIs. To test the immunological memory, the memory T cells (CD8+CD44highCD62Llow) of the spleen retrieved from mice-treated indicated treatments were analyzed using flow cytometry. Compared to the PBS group (G1), mice treated with RDPNs@diABZIs exhibited a 2.33-fold increase in the fraction of memory T cells, a better result than other groups (Fig. 7e, f and Supplementary Fig. 48). This evidence suggested that mice established strong immune memory after RDPNs@diABZIs treatment. To further determine whether the anti-tumor immune responses induced by RDPNs@diABZIs were durable, the mice that survived following the first inoculation were re-challenged with 4T1-luc tumor cells by intravenous administration. Noticeable tumor nodules bioluminescence in lung in vivo and ex vivo was observed in mice of the control group (Fig. 7g–j and Supplementary Fig. 49). In contrast, mice treated with RDPNs@diABZIs showed no tumor nodules bioluminescence in lung, indicating the establishment of durable anti-tumor immune responses. These results provided evidence that RDPNs@diABZIs elicited robust anti-tumor efficacy and induced durable anti-tumor immunological memory.
Discussion
In summary, we have developed redox-responsive diselenide-bridged supramolecular polyrotaxanes nanoformulations to augment anti-tumor T-cell killing by overcoming cancer-cell mechanical softness. The redox-responsive supramolecular polyrotaxanes comprise the amphiphilic diselenide-bridged axle polymer loaded with MeβCD by the host-guest interaction and end-capped with two NIR fluorescence probes IR783. The supramolecular polyrotaxanes self-assemble with STING agonists diABZIs into nanoparticles. After RDPNs@diABZIs accumulate in the TME, MeβCD and diABZIs enable simultaneous release in response to the redox TME. The released diABZIs activate STING on APCs, and de novo T-cell responses are initiated. Meanwhile, the released MeβCD depletes membrane cholesterol to overcome cancer-cell mechanical softness, which enhances the T-cell-mediated killing of cancer cells. Overall, the strategy described here provides a feasible approach to initiate robust T-cell-mediated anti-tumor killing for cancer immunotherapy.
Methods
The research presented here complies with all relevant ethical regulations. The animal experiment procedures were conducted following an approved protocol (IACUC-20230035) by the Institutional Animal Care and Use Committee (IACUC) of Fujian Normal University. All the maximal tumor size/burden in our experiments did not exceed the maximal tumor size/burden permitted (2000 mm3). The all cells, including 4T1 cells, 4T1-Luc cells, B16-F10 cells, MC-38 (mouse colon cancer cell line, CBP60825), HC-11 cells, THP-1, and L929 cells were purchased from American Type Culture Collection (ATCC, Rockville MD). The materials and methods used in this study are described in detail in Supplementary.
Materials
Cell Counting Kit-8 was purchased from GLPBIO. Amplex Red cholesterol assay kit (Invitrogen) was purchased from Thermo Fisher Scientific. The Calcein-AM/PI staining kit was obtained from KeyGEN BioTECH Co., Ltd. (China). Annexin V-FITC/PI staining kit and D-Luciferin sodium salt were purchased from Yeasen Biotechnology (Shanghai) Co., Ltd. (China). All the antibody was purchased from BD Bioscience, Tonbo, Biolegend, Abcam, or Cell Signaling Technology. Dulbecco’s modified eagle medium was from Pricella Life Science & Technology Co., Ltd. Penicillin-Streptomycin Solution was from Beijing Solarbio Science & Technology Co., Ltd. Fetal Bovine Serum (FBS) was form ExCell Bio, China. Cell culture dishes/plates, round coverslips, 20-mmglass-bottom dishes, and centrifuge tubes were obtained from NEST Biotechnology Co. Ltd. (Wuxi, China). The ELISA kits were obtained from Elabscience® Biotechnology Co., Ltd. (China). Granulocyte–macrophage colony-stimulating factor (GM-CSF) was obtained from Beijing T&L Biological Technology Co., Ltd. Filipin III was obtained from MedChemExpress (Monmouth Junction, NJ, USA). All other reagents and solvents were purchased from Sigma-Aldrich. Ultrapure water from the Milli-Q® Pure water machine was used throughout the experiment.
Synthesis of IR783-PEG-PDS-MeβCD
The diselenide group was first introduced into a diol structure through the reaction of disodium diselenide (1.3 mmol, 0.265 g) and 6-Bromo-1-Hexanol (1.3 mmol, 0.235 g). The diselenide-contained blocks were synthesized via polymerization in a slight excess of toluene diisocyanate (TDI, 1.43 mmol, 0.2 mL) and diselenide-containing diols (1.3 mmol, 0.468 g). Then, the reactive TDI-terminated diselenide-containing polyurethane blocks (0.05 mmol, 0.086 g) were terminated by polyethylene glycol (PEG, 0.15 mmol, 0.75 g) denoted by PEG-PDS. PEG-PDS (0.05 mmol, 0.84 g) and IR783 (0.15 mmol, 0.012 g) were dissolved in DMF and allowed to stir for 2 h at room temperature. The above mixture was added dropwise into the saturated aqueous solution of MeβCD (50 mg/mL). The final mixture was allowed to stir for an additional 24 h at room temperature to obtain IR783-PEG-PDS-MeβCD. The solution was dialysed against deionized water for 2 d in a presoaked dialysis tubing ((MWCO) = 35,000 Da) to remove the residual DMF and free drug.
Preparation of RDPNs@diABZIs
The polymer RDPNs (100 mg) and agonists diABZIs (100 μg) were dissolved in DMF. The solution was added dropwise into water/DMF (10/1, v/v) solution in an ultrasonic vibrator (100 Hz). Then, the nanoparticles were dialyzed (molecular weight cut-off (MWCO) = 35,000 Da) against purified deionized water to remove the free drug for 48 h. The diABZIs content in RDPNs@diABZIs is quantitative analyzed by HPLC. The drug-loading content of diABZIs is calculated using the DLC formula. DLC were calculated as follows:
Cell lines and animals
Bone marrow cells were harvested from the femurs of male C57BL/6 mice and tibiae of mice and cultured in DMEM supplemented with fetal bovine serum and GM-CSF31. 4T1 cells (murine mammary carcinoma cell line, CBP60352), 4T1-Luc cells (murine mammary carcinoma cell line, CBP30102L), B16-F10 cells (mouse skin melanoma cell line, CBP60337), MC-38 (mouse colon cancer cell line, CBP60825), HC-11 cells (mouse mammary epithelium cell line, CBP61438), THP-1 (Human monocytic leukemia cells, CBP60518), and L929(mouse fibroblasts cell line, CBP60878) were purchased from American Type Culture Collection (ATCC, Rockville MD). The animal experiment procedures were conducted following an approved protocol (IACUC-20230035) by the Institutional Animal Care and Use Committee (IACUC) of Fujian Normal University. The mice are maintained under a 12-h light/12-h dark cycle at 18–23 °C and 50% humidity.
Quantification of the total cholesterol levels
Tumor tissues and muscle tissues near the tumor were collected from tumor-bearing mice and digested with collagenase IV for 1 h. Cells were extracted from the tissue fluid through a cell filter. Tumor cells were incubated with different treatments at 37 °C, and washed twice with PBS to consume cholesterol on the plasma membrane. Methanol/chloroform (1:2, v/v) was added for cholesterol extraction and shaken at room temperature for 2 h. Then, the organic phase containing cholesterol is collected and then is evaporated under a vacuum. Finally, the Amplex Red cholesterol assay kit was used to quantify the cholesterol in each sample.
Quantification of plasma membrane cholesterol levels
To quantify intracellular cholesterol, tumor cells were treated with cholesterol oxidase before extraction, oxidizing plasma membrane cholesterol. Plasma membrane cholesterol levels were calculated by subtracting intracellular cholesterol levels from total cellular cholesterol levels.
RDPNs@diABZIs promote the maturation of BMDCs through cGAS-STING pathway
BMDCs were generated by culturing bone marrow cells flushed from the femurs of C57BL/6 mice in BMDC medium: RPMI-1640 containing 10% FBS, penicillin/streptomycin, 20 ng/mL GM-CSF. The culture medium was half-replaced every 2 days, and the non-adherent and loosely adherent immature DCs were collected on day 8 and phenotyped by determining the expression of markers CD45 and CD11c. After being cultured with different treatments for 4-6 h, BMDCs were collected to analyze the surface markers CD86 and CD80 by flow cytometry.
Western blot analysis
After gel electrophoresis and protein transfer, anti-p-STING antibody (dilution ratio 1: 1000, cell signaling technology, 19781), anti-pIRF3 antibody (dilution ratio 1: 1000, cell signaling technology, 4947), anti-GADPH antibody (dilution ratio 1: 5000, cell signaling technology, 5174) at 4 °C overnight followed by incubation with anti-rabbit IGG-HRP secondary antibody (dilution ratio 1: 8000, cell signaling technology, 7076) Incubate at room temperature for 1 h. The blots were detected by Immobilon Western Chemiluminescent HRP Substrate (Millipore, WBKLS0100) and captured using ChemiDoc Imaging System (Bio-Rad).
Flow cytometry analysis of the immune cell population
Isolated tumor tissue, lymph node tissue, and spleen were digested into single-cell suspension and pre-incubated with anti-CD16/32 for 15 min at 4 °C. Monoclonal antibodies block non-specific binding and then stain the antibody with a diluted fluorescent dye. The antibodies involved in the experiment include anti-CD45-FITC、anti-CD3-APC、anti-CD4-APC-Cy7、anti-CD8-PE、anti-CD11b-PE-cy7、anti-CD11c-PE、 anti-F4/ 80-PE, anti-CD86-PE-cy7, anti-CD206-BV 421, anti-CD49b-APC, anti-CD62L-FITC and anti-CD44-PE. The stained cells were detected using flow cytometry.
In vivo anti-tumor activity
The animal experiment procedures were conducted following an approved protocol (IACUC-20230035) by the Institutional Animal Care and Use Committee (IACUC) of Fujian Normal University. The objective of the first tumor inoculation, 4T1-Luc cells (1 × 106) were subcutaneously injected into the right chest/flank of each female BALB/c mouse. Seven days later, the tumor-bearing mice were divided into 5 groups randomly: PBS (G1), NPNs (G2), RDPNs (G4), diABZIs (G4), RDPNs@diABZIs (G5). Mice in different groups were intravenously injected with indicated formulations on days 0, 2, and 4. The tumor volume was calculated according to the following formula: W2 × L / 2. Based on animal ethics, the mice with tumors exceeding 2000 mm3 would be euthanized in this study.
To establish the lung metastasis model, 4T1-Luc cells (1 × 105) were intravenously injected in naive mice or mice showing complete tumor regression after RDPNs@diABZIs treatment.
Statistical analysis
All quantitative data are expressed as mean ± standard deviation (SD) unless otherwise indicated. The significance between two groups was analyzed by two-tailed Student’s t-test. For multiple comparisons, one-way analysis of variance (ANOVA) with Tukey’s post hoc test was used. Statistical analysis was performed using Graph Pad Prism 9.0. P-values of <0.05 were considered significant. * P < 0.05, ** P < 0.01, ***P < 0.001, and ****P < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included in this article, its Supplementary information files, and source data files. All data underlying this study are available from the corresponding author upon request. Source data are provided with this paper.
References
Dane, E. L. et al. STING agonist delivery by tumour-penetrating PEG-lipid nanodiscs primes robust anticancer immunity. Nat. Mater. 21, 710–720 (2022).
Chen, X. et al. Chemically programmed STING-activating nano-liposomal vesicles improve anticancer immunity. Nat. Commun. 14, 4584 (2023).
Hopfner, K.-P. & Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Bio. 21, 501–521 (2020).
Decout, A., Katz, J. D., Venkatraman, S. & Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21, 548–569 (2021).
Garland, K. M., Sheehy, T. L. & Wilson, J. T. Chemical and biomolecular strategies for STING pathway activation in cancer immunotherapy. Chem. Rev. 122, 5977–6039 (2022).
Shae, D. et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat. Nanotechnol. 14, 269–278 (2019).
Basu, R. et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 165, 100–110 (2016).
Liu, Y. et al. Cell softness prevents cytolytic T-cell killing of tumor-repopulating cells. Cancer Res 81, 476–488 (2021).
Maja, M. et al. Surface cholesterol-enriched domains specifically promote invasion of breast cancer cell lines by controlling invadopodia and extracellular matrix degradation. Cell. Mol. Life Sci. 79, 417 (2022).
Lakk, M. et al. Membrane cholesterol regulates TRPV4 function, cytoskeletal expression, and the cellular response to tension. J. Lipid Res. 62, (2021).
Traber, G. M. & Yu, A.-M. RNAi-based therapeutics and novel RNA bioengineering technologies. J. Pharmacol. Exp. Ther. 384, 133–154 (2023).
Kurakula, H., Vaishnavi, S., Sharif, M. Y. & Ellipilli, S. Emergence of small interfering RNA-based gene drugs for various diseases. ACS Omega 8, 20234–20250 (2023).
Lei, K. et al. Cancer-cell stiffening via cholesterol depletion enhances adoptive T-cell immunotherapy. Nat. Biomed. Eng. 5, 1411–1425 (2021).
Murrell-Lagnado, R. D. Regulation of P2X purinergic receptor signaling by cholesterol. Curr. Top. Membr. 80, 211–232 (2017).
Butchbach, M. E., Tian, G., Guo, H. & Lin, C.-lG. Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J. Biol. Chem. 279, 34388–34396 (2004).
Xu, R. et al. Cholesterol Depletion and Membrane Deformation by MeβCD and the Resultant Enhanced T Cell Killing. ACS Appl. Mater. Interfaces 16, 6813–6824 (2024).
Blanchemain, N. et al. Methyl-β-cyclodextrin modified vascular prosthesis: Influence of the modification level on the drug delivery properties in different media. Acta Biomater. 7, 304–314 (2011).
Wei, D. et al. Nuclear-Targeting Lipid PtIV Prodrug Amphiphile Cooperates with siRNA for Enhanced Cancer Immunochemotherapy by Amplifying Pt-DNA Adducts and Reducing Phosphatidylserine Exposure. J. Am. Chem. Soc. 146, 1185–1195 (2023).
Takata, T. Polyrotaxane and polyrotaxane network: supramolecular architectures based on the concept of dynamic covalent bond chemistry. Polym. J. 38, 1–20 (2006).
Huang, F. & Gibson, H. W. Polypseudorotaxanes and polyrotaxanes. Prog. Polym. Sci. 30, 982–1018 (2005).
Mondjinou, Y. A. et al. Gd3+: DOTA-modified 2-hydroxypropyl-β-cyclodextrin/4-sulfobutyl ether-β-cyclodextrin-based polyrotaxanes as long circulating high relaxivity MRI contrast agents. Bioconjugate Chem. 29, 3550–3560 (2018).
Salústio, P. J. et al. Advanced technologies for oral controlled release: cyclodextrins for oral controlled release. Aaps Pharmscitech. 12, 1276–1292 (2011).
Yu, G. et al. Polyrotaxane-based supramolecular theranostics. Nat. Commun. 9, 766 (2018).
Wankar, J. et al. Recent advances in host–guest self‐assembled cyclodextrin carriers: Implications for responsive drug delivery and biomedical engineering. Adv. Funct. Mater. 30, 1909049 (2020).
Yin, H., Liu, F., Abdiryim, T. & Liu, X. Self-healing hydrogels: from synthesis to multiple applications. ACS Materials Lett. 5, 1787–1830 (2023).
Wei, D., Sun, Y., Zhu, H. & Fu, Q. Stimuli-Responsive Polymer-Based Nanosystems for Cancer Theranostics. ACS Nano 17, 23223–23261 (2023).
Shukla, T. et al. Novel supramolecular conjugated polyrotaxane as an acid-base controllable optical molecular switch. Sens. Actuators B: Chem. 243, 84–95 (2017).
Harada, A., Hashidzume, A., Yamaguchi, H. & Takashima, Y. Polymeric rotaxanes. Chem. Rew. 109, 5974–6023 (2009).
Li, P. et al. IR-783 inhibits breast cancer cell proliferation and migration by inducing mitochondrial fission. Int. J. Oncol. 55, 415–424 (2019).
Guan, Y. et al. Synthesis and biological evaluation of genistein-IR783 conjugate: cancer cell targeted delivery in MCF-7 for superior anti-cancer therapy. Molecules 24, 4120 (2019).
Ma, W. et al. Bioactivable STING Nanoagonists to Synergize NIR‐II Mild Photothermal Therapy Primed Robust and Long‐Term Anticancer Immunity. Adv. Mater. 35, 2303149 (2023).
Jin, D. et al. A Leaking-Proof Theranostic Nanoplatform for Tumor-Targeted and Dual-Modality Imaging-Guided Photodynamic Therapy. BME Front. 4, 0015 (2023).
Humphries, F. et al. A diamidobenzimidazole STING agonist protects against SARS-CoV-2 infection. Sci. Immunol. 6, eabi9002 (2021).
Wu, J. J., Zhao, L., Hu, H. G., Li, W. H. & Li, Y. M. Agonists and inhibitors of the STING pathway: Potential agents for immunotherapy. Med. Res. Rew. 40, 1117–1141 (2020).
Shi, J. et al. STING agonist and IDO inhibitor combination therapy inhibits tumor progression in murine models of colorectal cancer. Cell. Immunol. 366, 104384 (2021).
Ma, N., Li, Y., Xu, H., Wang, Z. & Zhang, X. Dual redox responsive assemblies formed from diselenide block copolymers. J. Am. Chem. Soc. 132, 442–443 (2010).
Xia, J., Li, T., Lu, C. & Xu, H. Selenium-containing polymers: perspectives toward diverse applications in both adaptive and biomedical materials. Macromolecules 51, 7435–7455 (2018).
Haaj, S. B., Magnin, A., Pétrier, C. & Boufi, S. Starch nanoparticles formation via high power ultrasonication. Carbohydr. Polym. 92, 1625–1632 (2013).
Lin, H. et al. Nanoparticle-triggered in situ catalytic chemical reactions for tumour-specific therapy. Chem. Soc. Rev. 47, 1938–1958 (2018).
Sun, Q. et al. H2O2/O2 self-supplementing and GSH-depleting Ca2+ nanogenerator with hyperthermia-triggered, TME-responsive capacities for combination cancer therapy. Chem. Eng. J. 425, 131485 (2021).
Li, L. et al. Burst release of encapsulated annexin A5 in tumours boosts cytotoxic T-cell responses by blocking the phagocytosis of apoptotic cells. Nat. Biomed. Eng. 4, 1102–1116 (2020).
Leight, J. L., Drain, A. P. & Weaver, V. M. Extracellular matrix remodeling and stiffening modulate tumor phenotype and treatment response. Annu. Rev. Cancer Biol. 1, 313–334 (2017).
Karges, J. Encapsulation of Ru (II) Polypyridine Complexes for Tumor-Targeted Anticancer Therapy. BME Front. 4, 0024 (2023).
Guo, X., Bonin, K., Scarpinato, K. & Guthold, M. The effect of neighboring cells on the stiffness of cancerous and non-cancerous human mammary epithelial cells. New J. Phys. 16, 105002 (2014).
Ramos, J. R., Pabijan, J., Garcia, R. & Lekka, M. The softening of human bladder cancer cells happens at an early stage of the malignancy process. Beilstein J. Nanotech. 5, 447–457 (2014).
Vargas-Pinto, R., Gong, H., Vahabikashi, A. & Johnson, M. The effect of the endothelial cell cortex on atomic force microscopy measurements. Biophys. J. 105, 300–309 (2013).
Voskoboinik, I., Whisstock, J. C. & Trapani, J. A. Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol. 15, 388–400 (2015).
Wang, X. et al. A protein-based cGAS-STING nanoagonist enhances T cell-mediated anti-tumor immune responses. Nat. Commun. 13, 5685 (2022).
Wang, K. et al. Gas therapy potentiates aggregation-induced emission luminogen-based photoimmunotherapy of poorly immunogenic tumors through cGAS-STING pathway activation. Nat. Commun. 14, 2950 (2023).
Pu, F., Chen, F., Liu, J., Zhang, Z. & Shao, Z. Immune regulation of the cGAS-STING signaling pathway in the tumor microenvironment and its clinical application. OncoTargets Ther. 1501-1516 (2021).
Samson, N. & Ablasser, A. The cGAS–STING pathway and cancer. Nat. Cancer 3, 1452–1463 (2022).
Jin, M. et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 12, 6565 (2021).
Acknowledgements
This work was supported by the National Nature Science Foundation of China (Z. Y., NSFC82302356), Innovation team of Photoelectric functional materials and devices for biomedical theranostics of Fujian Normal University (Z. Y., Y07204080K13), Postdoctoral Fellowship Program of China Postdoctoral Science Foundation (W. M., GZB20240152), the Natural Science Foundation of Fujian Province (Z. Y., 2023J01529).
Author information
Authors and Affiliations
Contributions
H.F. L., W.M., and Z.Y. conceived and designed the project. H.F.L., W.M., J.Q.L., S.Z., and P.Y.W. designed and performed all experiments. H.F.L., W.M., and Z.Y. performed data analysis. H.F.L. and W.M. wrote the manuscript. Z. Y. conducted proofreading of the manuscript. Z. Y. supervised the entire project. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhong Luo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luo, H., Lv, J., Wen, P. et al. Supramolecular polyrotaxane-based nano-theranostics enable cancer-cell stiffening for enhanced T-cell-mediated anticancer immunotherapy. Nat Commun 16, 2331 (2025). https://doi.org/10.1038/s41467-025-57718-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57718-5
