
Abstract
Obesity is associated with adverse effects on health and quality of life. Improved understanding of its underlying pathophysiology is essential for developing counteractive measures. To search for sequence variants with large effects on BMI, we perform a multi-ancestry meta-analysis of 13 genome-wide association studies on BMI, including data derived from 1,534,555 individuals of European ancestry, 339,657 of Asian ancestry, and 130,968 of African ancestry. We identify an intergenic 262,760 base pair deletion at the MC4R locus that associates with 4.11 kg/m2 higher BMI per allele, likely through downregulation of MC4R. Moreover, a rare FRS3 missense variant, p.Glu115Lys, only found in individuals from Finland, associates with 1.09 kg/m2 lower BMI per allele. We also detect three other low-frequency FRS3 missense variants that associate with BMI with smaller effects and are enriched in different ancestries. We characterize FRS3 as a BMI-associated gene, encoding an adaptor protein known to act downstream of BDNF and TrkB, which regulate appetite, food intake, and energy expenditure through unknown signaling pathways. The work presented here contributes to the biological foundation of obesity by providing a convincing downstream component of the BDNF-TrkB pathway, which could potentially be targeted for obesity treatment.
Similar content being viewed by others

Introduction
Obesity and its related disease burden is a major global health problem with rapidly rising worldwide prevalence1,2. Even though this development is driven by a complex interaction between environmental, cultural, and socioeconomic factors3,4, there is a strong genetic component to the vulnerability to these obesogenic conditions3,4. Genome-wide association studies (GWASs) of body mass index (BMI) have yielded sequence variants at over 1100 loci, most of which are common, non-coding variants with small effects4,5,6. A limitation of many GWASs is their inadequate representation of diverse populations, with most studies mainly including individuals of European ancestry6,7 and relatively small sample sizes of other ancestries5,7,8,9,10,11. This limits the ability to identify causative variants as their frequency can vary substantially between populations, e.g., because of environmental factors that interact with sequence variants and affect selection and genetic drift12,13. Recent whole-exome sequencing (WES) studies have provided important insights into the biology of obesity by yielding rare and low-frequency coding variants with large effects on BMI, implicating neuronal regulation of energy balance as a major contributor to obesity14,15,16,17.
In this study we conducted a multi-ancestry meta-analysis of GWASs with the aim of searching for sequence variants with large effects on BMI. We leveraged data derived from 2,005,180 individuals in 13 studies, thereof 1,534,555 of European ancestry, 339,657 of South and East Asian ancestry, and 130,968 of African ancestry (Table 1, Supplementary Data 1). Data from four of the 13 studies are not publicly available (Table 1). We focused on sequence variants with an absolute effect greater than 0.20 standard deviations (SD) of BMI, corresponding to 1 kg/m2. Here we report large-effect associations between four sequence variants at three loci and BMI. One of those variants is highly correlated with a large intergenic deletion at the MC4R locus that was identified among whole genome sequenced (WGS) individuals in the UK Biobank (UKB), thereby emphasizing the value of utilizing WGS data in such studies. Furthermore, we identified a Finnish missense variant in FRS3 that has a large BMI-lowering effect. FRS3 can therefore be characterized as a BMI-associated gene, further supported by three other FRS3 missense variants that associated with BMI in different ancestries in the study.
Results
We tested 189,440,750 sequence variants for association with BMI and used weighted Bonferroni significance thresholds to adjust for multiple testing (Supplementary Data 2)18. Out of 2635 variants previously reported to associate with BMI in two recent studies5,15 (of which some variants are not independent), we replicated 75% using data that has not been included in previous studies (P < 0.05 and a consistent effect direction; Supplementary Data 3–5).
Four variants at three loci associated with BMI with an effect greater than 1 kg/m2 (Table 2, Supplementary Data 6). All four variants associated with weight, but not with height (Supplementary Data 6), indicating that their effect on BMI is mediated through variation in weight rather than height.
Associations at the MC4R locus
A previously reported15 rare stop gained variant in MC4R, p.Tyr35Ter (rs13447324-T), associated with increased BMI (P = 2.5 × 10−13, effect = 0.60 SD or 3.12 kg/m2, 95% confidence interval [CI; 0.44, 0.76 SD]) in individuals of European ancestry in the Genetic Investigation of Anthropometric Traits (GIANT) consortium (allele frequency [AF] = 0.009%) and the UKB (AF = 0.005%; Supplementary Fig. 1). At the MC4R locus, we also found a 3’ untranslated region (3’ UTR) variant in PMAIP1, rs948848696-A, in the UKB (AF = 0.008%) that is close to but not correlated with p.Tyr35Ter (r2 = 0.00) and associated with a large increase in BMI (P = 2.1 × 10-10, effect = 0.85 SD or 4.42 kg/m2, 95% CI [0.59, 1.11 SD]; Supplementary Fig. 2). This variant is in high linkage disequilibrium (LD; r2 = 0.95) with a large 262,760 base pair (bp) deletion (chr18:59985753-60248513), found among 72 individuals of European ancestry in the UKB (AF = 0.008%; P = 4.5 × 10−9, effect = 0.79 SD or 4.11 kg/m2, 95% CI [0.53, 1.05 SD]). The association of rs948848696-A and the deletion with BMI were independent of more significant variants at the locus in the UKB dataset (Supplementary Data 7). The deletion is located 122,549 bp downstream of MC4R and overlaps common variants at the locus (Supplementary Fig. 3) that were among the first associations discovered in GWASs on BMI19. However, it does not overlap MC4R or other protein coding genes according to RefSeq20, and would thus not be detected with WES. MC4R encodes a G protein-coupled receptor involved in the leptin-melanocortin pathway, a potent appetite-regulating system in the hypothalamus4, and disruptive mutations in this gene are reported as the most common cause for severe monogenic obesity21,22,23. The deletion potentially leads to downregulation of MC4R through loss of regulatory elements, given its large size, its proximity to MC4R, and a BMI-increasing effect comparable to reported MC4R loss-of-function (LoF) variants14,15,16,17. However, functional studies are required to clarify the precise mechanism. Both sequence variants identified at this locus associated with self-reported larger comparative body size at age 10 in the UKB (Ncases = 67,722, Ncontrols = 140,977; P < 8.3 × 10−3; Supplementary Data 8).
Associations at the TMEM18 locus
An intergenic variant on chromosome 2p25, rs539478649-C, associated with lower BMI (P = 3.3 × 10−12, effect = −0.21 SD or −1.09 kg/m2, 95% CI [−0.27, −0.15 SD]; Supplementary Fig. 4). It was mainly found among individuals of European ancestry and was enriched in Iceland with an AF of 0.27%. This locus harbors common and low-frequency variants close to TMEM18 that are reported to associate with BMI24,25 (Supplementary Fig. 5), although the causal gene for these associations is still unknown26,27. Rs539478649-C was not significant (Padjusted = 0.057; Supplementary Data 7) after adjusting for two more significant variants that are in high LD (r2 > 0.8) with previously reported variants at the locus, rs939581-C24 (AFEur = 17.5%, r2 = 0.01 with rs539478649-C) and rs62106252-C25 (AFEur = 3.9%, r2 = 0.05 with rs539478649-C). The large effect of rs539478649-C is explained by 99% of its carriers also carrying both other variants.
Missense-variant associations in FRS3
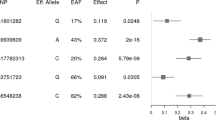
A rare missense variant in FRS3 on chromosome 6p21, p.Glu115Lys (rs773053137-T), associated with lower BMI (P = 6.6 × 10−12, effect = −0.21 SD or −1.09 kg/m2, 95% CI [−0.27, −0.15 SD]; Fig. 1a). The variant was only found in the FinnGen dataset28 (AF = 0.17%, Fig. 2a) where only two homozygotes have been identified among carriers. We further explored the BMI distribution among carriers and non-carriers of p.Glu115Lys by dividing BMI into five classes defined by the World Health Organization (WHO)29; underweight (BMI < 18.5 kg/m2), healthy weight (BMI 18.5–25 kg/m2), overweight (BMI 25–30 kg/m2), obesity (BMI 30–40 kg/m2), and severe obesity (BMI > 40 kg/m2). We observed a larger proportion of carriers with healthy weight (BMI 18.5–25 kg/m2; 40.8%) compared to the corresponding proportion of non-carriers (34.8%; P = 1.2×10-5; Fig. 3). In contrast, a smaller proportion of carriers was obese (BMI 30–40 kg/m2, 18.0%) compared to the corresponding proportion of non-carriers (23.0%; P = 4.2 × 10−5; Fig. 3). To determine if other coding variants in FRS3 associate with BMI without restricting to an effect larger than 1 kg/m2, we explored associations of the other 194 moderate- and high-impact FRS3 variants present in our datasets (Supplementary Data 9). Three missense variants associated with BMI in addition to p.Glu115Lys after accounting for multiple testing (P < 0.05/194 = 2.6 × 10−4; Supplementary Fig. 6, Supplementary Data 10). P.Pro137Arg (rs146730626-C) associated with lower BMI (P = 2.0 × 10-9, effect = −0.05 SD or −0.26 kg/m2, 95% CI [−0.07, −0.03 SD]; Fig. 1b) among individuals of South and East Asian ancestry, with higher frequency in East Asia (AF 1.80 to 3.97%; Fig. 2b). Its association was most significant among individuals of East Asian ancestry in the Taiwan Biobank (P = 6.0 × 10−7, effect = −0.14 SD or −0.73 kg/m2, 95% CI [−0.19, −0.08 SD]; Fig. 1b). P.Pro172Leu (rs74687105-A) also confers a BMI-lowering effect (P = 9.9 × 10−6, effect = −0.03 SD or −0.16 kg/m2, 95% CI [−0.04, −0.02 SD]; Fig. 1c, Supplementary Fig. 7) and is a common variant in African ancestry populations with an AF of 8.92 to 9.92%, and within Africa, the variant’s frequency increases from north to south (Fig. 2c). One FRS3 missense variant associated with higher BMI in our data, p.Arg316Gln (rs35744673-T; P = 1.3 × 10−11, effect = 0.03 SD or 0.16 kg/m2, 95% CI [0.02, 0.04 SD]; Fig. 1d, Supplementary Fig. 8), with the highest AF in European ancestry populations, ranging from 0.84 to 2.70% (Fig. 2d). As p.Arg316Gln is found in the Icelandic and Norwegian populations, we were able to test the variant for association with BMI at birth and in childhood, utilizing BMI data collected from Icelandic children over the years 1896 to 2017 and publicly available summary statistics from the Norwegian Mother, Father, and Child Cohort Study30 (MoBA; Supplementary Data 11). This variant associated with BMI collected at ages 3 months to 9 years (N = 65,767; P = 1.4 × 10−6, effect = 0.14 SD, 95% CI [0.08, 0.20 SD]) and 10 to 15 years (N = 68,172; P = 2.7 × 10−4, effect = 0.11 SD, 95% CI [0.05, 0.16 SD]), but not with BMI at birth (N = 84,818; P = 0.62, effect = −0.01 SD, 95% CI [−0.07, 0.04 SD]). P.Arg316Gln also associated with larger comparative body size at age 10 in the UKB (Ncases = 67,722, Ncontrols = 140,977; P = 6.8 × 10−4, OR = 1.10, 95% CI [1.04, 1.16]); Supplementary Data 8), thereby further validating its effect on childhood adiposity.

The plots illustrate the associations between the four FRS3 missense variants identified in the study and BMI: a p.Glu115Lys (rs773053137-T), b p.Pro137Arg (rs146730626-C), c p.Pro172Leu (rs74687105-A), and d p.Arg316Gln (rs35744673-T). Data are presented as effect sizes in standard deviation (SD) units with 95% confidence intervals, combined (from the meta-analysis) and for individual studies. Effect sizes were determined using a linear mixed model implemented in BOLT-LMM, assuming an additive genetic model, and two-sided P values were calculated. The P values presented in the figure have not been adjusted for multiple comparisons. The vertical dashed line indicates effect size equal to zero and horizontal dashed lines separate effect estimates by ancestry. Effects are not shown for allele frequencies below 0.1%. Source data are provided as a Source Data file. AF allele frequency, EUR European, SAS South Asian, EAS East Asian, AFR African, UKB UK Biobank, CHB/DBDS Copenhagen Hospital Biobank/Danish Blood Donor Study, GIANT Genetic Investigation of Anthropometric Traits, NashBio Nashville Biosciences, MVP Million Veteran Program, AAAGC African Ancestry Anthropometry Genetics Consortium.

Low allele frequencies are shown in light color, while higher frequencies are depicted in dark color. Gray indicates that zero participants reported being born in that country or region. Frequencies shown are for UK Biobank participants with origins in that region, except for Finland, for which the frequency in FinnGen was used. a p.Glu115Lys (rs773053137-T) is only found in Finland. b p.Pro137Arg (rs146730626-C) is predominantly found in East Asia but is present at lower frequencies in South Asia and the Indian Ocean. c p.Pro172Leu (rs74687105-A) is predominantly found in populations of African ancestry and is most common in Southern Africa. d p.Arg316Gln (rs35744673-T) is broadly distributed, but most common in populations of European ancestry. This figure was created with Natural Earth.

The BMI groups presented in the figure were defined according to the World Health Organization’s recommendations. Source data are provided as a Source Data file.
As it is unclear whether the missense variants in FRS3 act through loss or gain of function, we searched for insights into direction of effects by performing a FRS3 LoF burden analysis. We leveraged WGS data from the UKB (N = 426,912) and deCODE (N = 44,652), study populations with large available sets of WGS individuals. We found 44 carriers of 23 predicted LoF (pLoF) variants in the UKB and one carrier of a frameshift variant in deCODE (Supplementary Data 12). Carrying a LoF variant associated with higher BMI (PUK = 0.015, effectUK = 0.39 SD; PIceland = 0.08, effectIceland = 1.25 SD; PCombined = 0.0062, effectCombined = 0.43 SD or 2.24 kg/m2, 95% CI [0.12–0.74 SD]). This suggests that the three FRS3 missense variants that lower BMI are not acting through LoF, but rather gain-of-function. FRS3 is neither LoF intolerant (pLI = 0.06) nor constrained for missense variants (Z = 0.65; gnomAD, Broad Institute31), indicating that both LoF and missense variants in FRS3 are tolerable.
As high BMI is a risk factor for numerous conditions, including type 2 diabetes (T2D) and other cardiometabolic diseases1,2,32,33, we tested the FRS3 missense variants for association with 19 relevant cardiometabolic and adiposity-related traits (P value threshold = 0.05/61 = 8.2 × 10−4; Supplementary Data 13–17). Both p.Glu115Lys and p.Arg316Gln associated with obesity (BMI over 30 kg/m2) compared to normal or underweight (BMI under 25 kg/m2), with an effect consistent with the size and direction of their BMI effects (p.Glu115Lys: P = 1.2 × 10−7, OR = 0.66, 95% CI [0.57, 0.77]; p.Arg316Gln: P = 9.7 × 10−5, OR = 1.08, 95% CI [1.04, 1.13]; Supplementary Data 13 and 16, respectively). The effect sizes of all four missense variants on T2D were as expected when compared to an effect estimate of the causal relationship between BMI and T2D risk reported in a meta-analysis of Mendelian randomization studies34 (Supplementary Data 13-16). However, their associations with T2D and the rest of the phenotypes were not significant after adjusting for multiple testing (P > 8.2 × 10−4). We also explored if p.Arg316Gln, or correlated variants with r2 > 0.8, associated with mRNA expression (cis-expression quantitative trait loci [cis-eQTL]), mRNA splicing variations (splicing QTL [sQTL]), and plasma protein levels (protein QTL [pQTL]), but did not detect any QTL associations. We were not able to test the other missense variants for association with expression or protein levels because the required data were unavailable for FinnGen and ancestries other than European.
Discussion
FRS3 encodes the intracellular adaptor protein fibroblast growth factor receptor substrate 3 (FRS3), also known as FRS2β and SUC1-associated neurotrophic factor target 2 (SNT-2)35. It is most abundantly expressed in neuronal tissue36,37, and a study on mouse embryos suggests it is highly expressed in brain regions involved in energy balance regulation38 (i.e., the hypothalamus39, thalamus40, and lateral septal nucleus41). FRS3 has been shown to take part in the fibroblast growth factor (FGF) and neurotrophin signaling pathways35,38, which have key roles in both the developing and adult nervous system, including in neural cell proliferation, differentiation, and synaptic plasticity35,42,43,44. In addition to a role as an adaptor protein, FRS3 has been demonstrated to associate with microtubules and lysosomes within neurons, indicating a potential role in maintaining microtubule structure and stability, axonal transport, and intracellular lysosomal degradation45,46. The protein consists of 492 amino acids (AA) and has several motifs conserved across species, including a phosphotyrosine binding (PTB) domain (AA 13-115) that mediates the binding of FRS3 to its activating receptors35,47. According to UniProt47, the large-effect missense variant p.Glu115Lys is located within this functionally important PTB domain. The other missense variants, conferring smaller effects on BMI, are not located within established domains, but they are closer to binding sites of downstream proteins (located at AA 192, 287, 322, 417, and 455)35,47.
Several ligand-receptor pairs are known to activate FRS3, one of which has been associated with BMI and obesity; brain-derived neurotrophic factor (BDNF) and its tropomyosin kinase receptor B (TrkB, encoded by NTRK2)48,49. BDNF and TrkB have been recognized as major contributors to a complex regulation of appetite, food intake, and energy expenditure, and are believed to work downstream of MC4R and other proteins of the leptin-melanocortin pathway in the hypothalamus4,48,50,51,52,53. However, it is unknown which downstream pathways of BDNF and TrkB are activated to mediate their anorexigenic effect48,50. Studies on BDNF knockout mice have shown that impaired BDNF-TrkB signaling results in hyperphagia, obesity, and increased locomotor activity49,54. Furthermore, disruptive mutations and deletion syndromes of BDNF and NTRK2 in humans are reported to cause hyperphagia and obesity48,55,56,57,58, and in the case of NTRK2, also stereotyped behaviors, intellectual disability, and impaired nociception57. Consistent with this evidence, we found a frameshift variant in NTRK2, p.Gln247ArgfsTer4, that was carried by two individuals in the UKB, both with BMI > 55 kg/m2 (P < 0.05/36 pLoF variants in NTRK2 and BDNF = 1.4 × 10−3 for BMI in the UKB dataset). To explore whether neurologic traits caused by impaired BDNF-TrkB signaling associate with FRS3 function, we tested the FRS3 missense variants for association with attention deficit hyperactivity disorder (ADHD), autism, intellectual disability, and pain, but no association was detected (P > 0.05/8 = 6.3 × 10−3; Supplementary Data 13 and 16).
Disrupted BDNF-TrkB signaling has been proposed as a pathogenic mechanism in several neurological disorders other than obesity, including depression, amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), and Parkinson’s disease. Therefore a great interest has been taken in BDNF and other neurotrophic factors as therapeutic targets59,60,61. Trk inhibitors are already marketed as a treatment of Trk fusion-positive cancers, where side effects include substantial weight gain, dizziness, and withdrawal pain62, mimicking symptoms of BDNF knockout in mice49 and NTRK2 LoF in humans56,57. To treat obesity, the BDNF-TrkB signaling pathway would need to be activated, which is a challenge that has been difficult to overcome for other diseases of interest, mainly due to unfavorable pharmacokinetics of BDNF and other TrkB agonists59,60,61,63. A recently synthesized molecule induces Trk-signaling through positive allosteric modulation of the intracellular domain of Trk-receptors and is currently being explored as a treatment of AD and other cognitive disorders64. The results of a phase I trial suggest that this molecule overcomes many of the pharmacokinetic obstacles of TrkB agonists while maintaining favorable safety profiles in healthy subjects65. It effectively crosses the blood-brain-barrier and alters quantitative electroencephalography (qEEG) parameters65, indicating an effect on central Trk-signaling. This molecule therefore raises the prospect of targeting the BDNF-TrkB pathway in treatment of obesity.
By identifying a large-effect BMI-lowering missense variant in FRS3, a gene that encodes an adaptor protein known to act downstream of BDNF and TrkB, we postulate that FRS3 is a link in the undiscovered downstream pathways mediating the anorexigenic effects of BDNF-TrkB signaling. This speculation is supported by our burden analysis, which suggests that loss of FRS3 function has an increasing effect on BMI, consistent with BDNF knockout in mice49 and NTRK2 LoF in humans56,57. Additionally, the finding of three other FRS3 missense variants in populations of diverse ancestries, one of which also associated with BMI in childhood, supports that FRS3 has an effect on BMI. Since BDNF and TrkB are thought to be activated downstream of the leptin-melanocortin pathway52,53, FRS3 could be a downstream target of several highly potent proteins involved in the regulation of appetite, food intake, and energy expenditure4,39,66,67,68,69,70 (Fig. 4), and would therefore be an attractive therapeutic target for obesity treatment. The recent synthesis of an intracellular positive modulator of central Trk-signaling65,71 presents an opportunity for targeting TrkB or its downstream proteins, including FRS3. Further research on the specific role of FRS3 in the pathophysiology of obesity is needed to explore whether it could be a potent and safe therapeutic target.

Key anorexigenic components are illustrated in dark blue for the leptin-melanocortin pathway and lighter blue for the BDNF-TrkB-FRS3 pathway. Pointed arrows between the components represent activation of downstream proteins, where whole pointed arrows indicate that the linking mechanisms are well-established and dotted pointed arrows indicate that mechanisms remain to be clarified. The leptin-melanocortin pathway is activated in response to increased leptin levels and other hormones in the feeding state, resulting in decreased appetite and food intake, increased energy expenditure, and lower BMI. It also has an inhibiting mechanism, illustrated in light blue to the left, where AGRP acts as an antagonist of MC4R in response to low leptin levels with food deprivation, resulting in MC4R inhibition, increased appetite, decreased energy expenditure, and higher BMI. The BDNF-TrkB pathway is thought to be activated downstream of the leptin-melanocortin pathway to mediate its effect on energy balance. We postulate that FRS3 is a link in the undiscovered downstream signaling cascade of BDNF and TrkB. Components of these pathways are associated with both monogenic and polygenic forms of obesity, with the supporting evidence summarized to the right in the figure. Created in BioRender. Aegisdottir, H. (2025) https://BioRender.com/ a25a422. LEPR leptin receptor, AGRP agouti-related protein, POMC pro-opiomelanocortin, α-MSH α-melanocyte stimulating hormone, PCSK1 proprotein convertase subtilisin/kexin type 1, pLoF predicted loss-of-function, MC4R melanocortin 4 receptor, BDNF, brain derived neurotrophic factor, TrkB tropomyosin receptor kinase B, FRS3 fibroblast growth factor receptor substrate 3, EAF effect allele frequency.
Methods
Study design
We performed a meta-analysis of GWASs on BMI, utilizing data from 13 study populations of diverse ancestries. Individual level data on genotype and anthropometric measurements were utilized to conduct a GWAS on BMI for deCODE, UKB, Copenhagen Hospital Biobank/Danish Blood Donor Study (CHB/DBDS), Intermountain, and Nashville Biosciences (NashBio; Supplementary Data 18). Publicly available summary statistics were collected from other studies. Characteristics of study populations included in the meta-analysis, data sources, and references are summarized in Supplementary Data 1. We focused on associations with an effect larger than 1 kg/m2. To convert the effect sizes from SD to kg/m2, we multiplied the effect in SD with 5.2 kg/m2, which is the median of the standard deviations of BMI for the cohorts with available individual level data (Supplementary Data 18) and FinnGen. We noticed that a missense variant in LRRC45 on chromosome 17q25 associated with BMI with an effect close to this criterion (Supplementary Note 1; Supplementary Fig. 9). That variant has been reported5 but not specifically discussed in association with BMI. We also performed a replication analysis restricting to data that has not been included in previous BMI studies, i.e., data from deCODE, CHB/DBDS, Intermountain, and NashBio (Supplementary Data 3–5).
Ethics
We confirm that our research complies with all relevant regulations regarding the use of data from human participants and was conducted in accordance with the criteria set by the Declaration of Helsinki. Participants received no compensations. Study-specific ethics declarations are provided below (See Study populations and datasets).
Study populations and datasets
The Icelandic study population
The Icelandic deCODE genetics study has collected phenotypic and biological data from more than 170,000 volunteer participants through multiple research studies in Iceland. The study was approved by the National Bioethics Committe (NBC) of Iceland (VSN-17-076). The Icelandic childhood- and birth-BMI data were used under the NBC Licenses VSN-17-076 and VSN-15-169, respectively. The Icelandic ADHD and autism data were utilized under NBC Licenses VSN-15-047 and VSN-14-043, respectively, and data on intellectual disability were used as a part of those licenses. The Icelandic pain data were analyzed under NBC Licenses VSN-17-035 and VSN-12-162 (with amendments), issued following review by the Icelandic Data Protection Authority.
All participants who donated biological samples provided informed consent. The personal identities of the participants and biological samples were encrypted by a third-party system approved and monitored by the Icelandic Data Protection Authority. By using the well documented genealogy of the Icelandic population, genotypes of first and second degree relatives of those 170,000 participants can be predicted with familial imputation, which can further increase the power of GWASs involving the Icelandic population72,73. Information on height, weight, BMI, and other physical and biological measures used for association analyses was primarily obtained from Landspítali – The National University Hospital of Iceland, the Primary Health Care Clinics of the Capital Area, and from a range of research undertakings at deCODE genetics. Information on binary traits, for the most part based on ICD-coded diagnoses, was mainly obtained from Landspítali and registries kept by the Directorate of Health; the Causes of Death Register, the Register of Primary Health Care Contacts, and the Register of Contacts with Medical Specialists in Private Practice. The Icelandic childhood BMI phenotype data used in this study consists of 2,086,109 records of measured weight and height (or recumbent length) for 248,219 children and adolescents at ages from birth to 15 years, born in the years 1893–2017. The data originate from the Directorate of Health’s Birth Register and from routine data collection (health records) at school health services and well-child care in the greater Reykjavík area, for which data access was provided by the Primary Health Care of the Capital Area and the Reykjavik Municipal Archives.
BMI data used for genetic association testing in the GWAS was generated from weight and height measurements available for 127,837 individuals. In case of multiple height measurements, the mode of the measurements was selected to represent the individual’s height. BMI was calculated from the individual’s mode of height and each weight measurement. BMI values were then regressed in a generalized additive model with sex, year of birth (YOB), and age at measurement. In case of multiple residuals per individual, one final BMI value was obtained for each individual by averaging residuals. Averaged residuals were standardized before genetic association analysis, and only individuals 18 years of age or older were included. For the handling of the childhood BMI data, see Supplementary Methods.
The genotype data was based on 173,025 chip typed individuals, of whom 63,460 were also WGS72. WGS was performed using Illumina technology, including GAIIx, HiSeq, HiSeqX, and NovaSeq machines. Only samples with genome-wide average coverage of over 20x were included. The average genome-wide sequencing coverage was 39.8x (sd 14.2, min:20.0x, max:397.8x). Duplicated samples were discarded based on sequencing yield, and contaminated samples were detected using read_haps74 and removed. Chip-sequencing was performed using Illumina OmniExpress (N = 136,215) and HumanHap (N = 38,772) chips. Individual arrays were discarded if the total genotype yield was below 98%. Variants identified through WGS were imputed into the 173,025 chip genotyped participants using long-range phasing. Expected genotypes of first and second degree relatives of chip typed participants were incorporated in the analysis by integrating over possible genoypes75. This family imputation was performed without the genotypes being kept in storage. Single nucleotide polymorphisms (SNPs) and insertions/deletions (indels) were called using joint calling with GraphTyper (v.2.7.1)76.
The UK Biobank study population
The UKB is a large prospective cohort study that has collected phenotypic and biological information from approximately 500,000 individuals across the UK, aged between 40 and 69 years when recruited in 2006–201077. The North West Research Ethics Committee reviewed and approved the UKB protocol78 (ref.no.06/MRE08/65). All genotype and phenotype data were collected following an informed consent for all participants. This research was conducted using the UKB resource under application no. 56270.
BMI and other physical and biological measures were collected from participants with a baseline assessment, and data on ICD-coded diagnoses were collected from electronic health records (EHRs) and other registries77,78. BMI information was retrieved from data field 21001. In addition, we tested the large-effect variants reported in this study for association with the phenotype “Comparative body size at age 10”, retrieved from field 1687. We defined a case-control phenotype based on the question “When you were 10 years old, compared to average would you describe yourself as – Thinner/Plumper/About average/Do not know/Prefer not to answer”, asked on three separate occasions. Cases were defined as above average (plumper) and controls as below average (thinner), divided by genetic ancestries (British-Irish, South Asian, and African). Individuals who provided inconsistent answers to this question were excluded from the analysis.
Three cohorts of diverse ancestries were defined within the UKB dataset, based on genetic clustering of microarray genotypes informed by self-described ethnicity and supervised ancestry inference; British-Irish (XBI, N = 431,805), South Asian (XSA, N = 9252), and African (XAF, N = 9633)79. BMI values were regressed separately for each ancestry and sex using linear regression, with YOB, age, and principal components (PCs; 20 for XBI, 45 for XSA, and 20 for XAF) as covariates. In case of multiple residuals per individual, one final value was obtained by averaging residuals. Residuals for men and women were then combined and inverse-normally transformed, generating three separate standardized BMI lists for each ancestry.
The UKB genotype data used in the GWAS was based on 150,119 WGS participants79. Variants identified with WGS were imputed into other chip typed participants. All FRS3 pLoF variants included in the burden LoF analysis and the pLoF variants in NTRK2 and BDNF that were tested for association with BMI, including the NTRK2 frameshift variant p.Gln247ArgfsTer4, were identified in a dataset of 449,903 WGS participants with BMI measures80. The additional samples analyzed for this dataset were prepared and sequenced in the same way as for the preliminary one used in the GWAS meta-analysis79. The average genome-wide sequencing coverage for this dataset was 32.4x (sd 4.1, min:22.1x, max:162.1x). We ran GraphTyper v2.7.576 for genotyping SNPs and indels in all 490,549 individuals in the UKB genotyping set. We genotyped structural variants (SVs) by first running DRAGEN for discovering SVs in each individual. Then, we merged the SVs using svimmer81 and finally called the merged set of SVs using GraphTyper. In the original callset, the 263 kb deletion at chr18:59,985,753 was called by GraphTyper in 64 of the samples. Due to its high correlation with both the large-effect 3’ UTR variant rs948848696-A in PMAIP1 and a nearby 6.5 kb deletion at chr18:59,891,870, we manually inspected the sequence reads of all individuals called with at least one of those variants. Our inspection confirmed all genotyping calls of the SNP and the 6.5 kb. However, we found 8 individuals that had been misidentified as non-carriers for the 263 kb deletion. The 263 kb deletion has breakpoints on different GraphTyper graphs, causing variant calling to be less accurate. Therefore, the corrected genotyping of the 263 kb deletion has 72 carriers. Supplementary Fig. 3b, showing the chromosomal position of the deletion and whole-genome sequencing coverage for a single carrier, was created using Samplot82.
The Danish study population
The Danish data were obtained from the Copenhagen Hospital Biobank Oral Cardio-Metabolic health Study (CHB-OCMS)83 and the Danish Blood Donor Study (DBDS)84. The CHB-OCMS and DBDS have been approved by the Zealand Regional and National Committees on Health Research Ethics (SJ-989 and NVK-1900988) and the Danish Data Protection Agency (P-2022-913 and P-2019-99). CHB is a biobank based on residual blood samples from hospitalized and outpatient subjects in the capital region of Denmark. Instead of a written consent, the patients were informed about the opt-out possibility of having their biological specimens excluded from use in research in general. Since 2004, a national Register on Tissue Application (Vævsanvendelsesregistret) lists all individuals who have chosen to opt-out and whose samples cannot be used for research purposes. Before initiating this study, individuals who contacted the biobank to opt-out or were listed in the Register on Tissue Application were excluded. The CHB-OCMS was initiated in 2023 and involves a targeted selection of patients over 18 years of age with cardiometabolic diseases. BMI, blood lipid measurements, and ICD-coded diagnoses were collected from the BigTempHealth database at Novo Nordisk Foundation Center for Protein Research, Copenhagen University, and health registries. BMI data from the CHB were extracted from EHRs with natural language processing. The DBDS is an open prospective cohort study on Danish blood donors. All participants provide written informed consent and can withdraw from the study at any time. The eligibility criteria for blood donation are good physical health and body weight over 50 kg. Participants are aged 18 to 75 years. Information on BMI was obtained from questionnaires, and blood lipid measurements and ICD-coded diagnoses were collected from health registries.
The Danish genotype data used in this study consists of 375,216 chip typed individuals whose genotype was imputed based on a haplotype reference panel of WGS individuals from several cohorts85. Thereof, 224,540 had available BMI information. BMI values were regressed in a generalized additive model using sex, YOB, age at measurement, and 12 PCs as covariates. In case of multiple residuals per individual, one final BMI value was obtained by averaging residuals for each individual. Averaged residuals were standardized before genetic association analysis. DNA extraction from whole blood was performed at Copenhagen University Hospital or at deCODE genetics in Iceland. Genotyping was performed at deCODE genetics on behalf of CHB and DBDS while analysis was performed on a specialized, secure section of the Danish National Supercomputer for Life Sciences (Computerome). Analyses were quality tested by deCODE. The WGS protocol used for the CHB and DBDS samples was the same as for the deCODE samples72. A joint variant calling with GraphTyper (v.2.7.5)76 forms the basis of the imputation, which was done using the in-house work flow developed at deCODE genetics72. Phasing was carried out using SHAPEIT (version 4)86.
The Intermountain study population (USA)
Intermountain Healthcare is a healthcare system that consists of 33 hospitals and 385 clinics in Utah and surrounding states. The dataset was obtained from HerediGene, a general population study, and the INSPIRE Registry Study, which contains data on volunteer subjects, both healthy and diagnosed with a variety of medical conditions. The studies have been approved by the Intermountain Healthcare Institutional Review Board (IRB) and all participants have provided written informed consent. Eligibility criteria for both studies include being 18 years of age or older.
Measurements for height, weight, BMI, HbA1c, and blood lipids, as well as ICD-coded diagnoses, were retrieved from study records. Suspected data entry errors for weight, height, and BMI were removed by filtering out extreme values, and BMI was also calculated from individual’s mode of height and each weight measurement. BMI values were regressed separately by sex using linear regression, with YOB, age, and top four PCs as covariates. In case of multiple residuals per individual, one final value was obtained for each individual by averaging residuals, and residuals for men and women were finally combined and inverse-normally transformed.
The Intermountain genotype data consists of 138,006 chip typed individuals whose genotype was imputed based on the same haplotype reference panel as for the Danish dataset85. Thereof, 11,815 had available information on BMI and were eligible for inclusion in the GWAS. DNA extraction from whole blood and genotyping was performed at deCODE genetics in Iceland, and the WGS protocol was the same as described for the Icelanders72. Joint variant calling was performed using GraphTyper (v.2.7.5)76, and phasing was carried out using SHAPEIT (v4)86.
The Nashville Biosciences study population
Nashville Biosciences (NashBio) is a data and analytics provider wholly owned by Vanderbilt University Medical Center (VUMC, Tennessee, USA). NashBio harnesses VUMC’s extensive genomic and bioinformatics resources, including its biobank collection BioVU®87,88. BioVU® includes a collection of de-identified DNA samples that are linked to de-identified version of the VUMC’s EHRs, referred to as Synthetic Derivative (SD) database. The SD contains longitudinal clinical data from more than 3.6 million patients cared for at the VUMC’s hospitals and primary-care clinics since 2001. All patients of the VUMC consented to their residual samples from routine clinical testing and data being contributed to BioVU®. BioVU® extracts and banks germline DNA samples that are de-identified and only linked to the SD through a randomly assigned unique identifier, not back to the patient or their underlying medical record. The use of BioVU® is classified as non-human subject research by VUMC’s IRB, and NashBio is not required to seek study-specific consent for use of these datasets. The overall biobanking program is reviewed annually by the IRB to maintain this determination and make decisions about patient protections, privacy, and ethical issues. Each individual study seeking to use the SD database and BioVU® biobank is filed with VUMC’s IRB to validate its non-human subject classification and appropriate use of the data. NashBio takes steps to protect client confidentiality during this submission.
Germline DNA samples from more than 307,000 patients have been collected, and thereof, 250,000 samples were selected for WGS under the Alliance for Genomic Discovery, which is a collaboration between NashBio/VUMC and Illumina on one hand, and eight pharmaceutical companies (including Amgen/deCODE genetics) on the other hand89. The aim of the Alliance for Genomic Discovery is to accelerate development of therapeutics through large-scale genomics and to establish a preeminent clinical genomic resource for diverse ancestries. The Diverse Ancestry Cohort included in this study is a subset of the BioVU® cohort, consisting of 36,750 WGS individuals of diverse ancestries, primarily of African ancestry. We further restricted our sample set to 27,871 study participants that we assessed as carrying African genomic ancestry by analyzing their genotypes with supervised ADMIXTURE v1.2390, using the 1000 Genomes populations91 CEU (Utah Caucasian), CHB (Beijing Han), ITU (Indian Telugu), PEL (Peruvian), and YRI (Nigerian Yoruba) as training samples. We included individuals who were modeled by ADMIXTURE as either (1) carrying > 90% YRI-like ancestry, or (2) carrying > 30% YRI-like ancestry, > 2% CEU-like ancestry, and YRI-like plus CEU-like ancestry proportions summing to > 90%. Both in-house and previously published analyses of human diversity datasets such as Human Origins92 indicate that this filtering enriches for genomes similar to those carried by reference dataset samples labeled as African American (e.g., 1000 G “African Ancestry in Southwest US”) or Afro-Caribbean (e.g., 1000 G “African Caribbean in Barbados”), as well as those of West African origin (e.g., 1000 G “Yoruba in Ibadan, Nigeria”) whose genomic ancestry is related to the African genomic ancestry in African Americans and Afro-Caribbeans. We note that our subset was defined solely on the basis of genomic ancestry.
Measurements for height and weight were available from the SD database for 23,211 individuals of genetically determined African ancestry. Suspected data entry errors were removed by filtering out extreme values and BMI was calculated from individual’s mode of height and each weight measurement. Measurements for blood pressure, HbA1c, and total cholesterol, as well as information on ICD-coded diagnoses, were also retrieved from the SD database. For quantitative traits, including BMI, values were regressed separately by sex using linear regression and included age, YOB, and top seven PCs calculated by bigsnpr93 to adjust for population stratification as covariates. For PC calculation, we included variants with minor allele frequency (MAF) > 0.5% LD-pruned by plink –indep-pairwise 60000 6000 0.494, and samples without 3rd-degree or closer relatives according to king –ibdseg95, using bigsnpr’s OADP-projected coordinates for samples with relatives. To better account for ancestral diversity in the NashBio dataset, all samples regardless of assigned YRI ancestry proportion were used to calculate PCs. In case of multiple residuals per individual derived from the linear regression, one final value was obtained for each individual by averaging residuals. Residuals for men and women were then combined and inverse-normally transformed before genetic association analysis.
All samples were WGS using Illumina NovaSeq sequencing machines at deCODE genetics in Iceland. The average genome-wide sequencing coverage was 33.9x (sd 3.5, min:28.7x, max:66.4x), and joint variant calling was performed using GraphTyper (v.2.7.5)76. See Supplementary Methods for more detailed description on sample preparation and the WGS protocol.
The Finnish study population
The FinnGen study, controlled by the University of Helsinki, is a large-scale genomics initiative that has analyzed over 500,000 Finnish Biobank samples and correlated genetic variation with health data to understand disease mechanisms and predispositions28. The project is a collaboration between research organizations and biobanks within Finland and international industry partners. The FinnGen study has been approved by the Coordinating Ethics Committee of The Hospital District of Helsinki and Uusimaa (Nr HUS/990/2017). Participants in FinnGen provided informed consent for biobank research on basis of the Finnish Biobank Act. Alternatively, separate research cohorts, collected before the Finnish Biobank Act came into effect (in September 2013) and the start of FinnGen (August 2017), were collected on the basis of study-specific consent and later transferred to the Finnish biobanks after approval by Fimea, the National Supervisory Authority for Welfare and Health. Recruitment protocols followed the biobank protocols approved by Fimea. FinnGen has provided publicly available GWAS results for numerous phenotypes through their online FinnGen database. We utilized publicly available GWAS summary statistics for BMI and other relevant phenotypes from FinnGen Data Freeze 10.
The FinnGen samples were genotyped with Illumina (Illumina Inc., San Diego, CA, USA) and Affymetrix arrays (Thermo Fisher Scientific, Santa Clara, CA, USA). Genotype imputation in FinnGen Data Freeze 10 was performed with Beagle 4.1 using the Sequencing Initiative Suomi v4 population-specific reference panel developed from high-quality data for 8554 high-coverage (25×) whole-genome sequences in Finnish individuals. The variant call set for the reference panel was produced with the GATK HaplotypeCaller algorithm following GATK best practices for variant calling, and genotype, sample, and variant quality control were iteratively applied to the high-coverage whole-genome sequencing data using Hail framework v0.1.
Other study populations
Publicly available summary statistics were utilized from Genes & Health96, BioBank Japan9,97, the Korean Genome and Epidemiology Study (KoGES) Consortium10,98, Taiwan Biobank8,99, the Million Veteran Program5,100, the African Ancestry Anthropometry Genetics Consortium (AAAGC)11,101, and the GIANT consortium15 (see Data Availability). For GIANT, exome-array summary statistics derived from individuals of European, East Asian, and South Asian ancestry were utilized. Their summary statistics from individuals of African ancestry were excluded because of an overlap with the NashBio/BioVU® samples15.
Genetic association testing and meta-analysis
When testing for association between BMI and sequence variants in the deCODE, UKB, CHB/DBDS, Intermountain, and NashBio datasets, we used a linear mixed model implemented in BOLT-LMM102, used to test for association between sequence variants and quantitative traits, and assumed an additive genetic model. We used logistic regression to test for association between sequence variants and binary phenotypes, assuming an additive genetic model. For the Icelandic population, the logistic regression model included the following covariates: sex, county of birth, current age or age at death (first and second order terms), blood sample availability for the individual, whether the individual was whole genome sequenced, and an indicator function for the overlap of the lifetime of the individual with the time span of phenotype collection. For CHB/DBDS, the covariates were sex, age (first and second order terms), whether the individual was whole genome sequenced, and 12 PCs to adjust for population stratification. For UKB, PCs (20 for XBI, 45 for XSA, and 20 for XAF) were used to adjust for population stratification, and sex, age (first and second order terms) and whether the individual was whole genome sequenced were included as covariates in the logistic regression model. For Intermountain, the covariates were sex, age (first and second order terms), whether the individual was whole genome sequenced, top four PCs, and an indicator function for the overlap of the lifetime of the individual with the time span of phenotype collection. For NashBio, associations were adjusted for sex, age (first and second order terms), sequencing batch, and top seven PCs. All statistical tests performed were two-sided.
For the meta-analysis of summary-level statistics from different populations, we used a fixed-effects inverse variance method based on effect estimates and standard errors103. To account for inflation in test statistics due to cryptic relatedness and stratification, we applied the method of LD score regression104. For imputed variants in our data, those with an imputation info score < 0.95 were excluded from the meta-analysis. If variants were found in external datasets from which summary statistics were used in our study, the imputation info cutoff of 0.95 was used if information on the imputation info score was available. When information on the imputation info score was not available in external datasets, all variants were included.
For genome-wide significance (GWS), we accounted for multiple testing with a weighted Bonferroni adjustment using as weights the enrichment of variant classes with predicted functional impact among association signals estimated from the deCODE data (Supplementary Data 2)18. This yielded significance thresholds of 3.7 × 10−8 for variants with high impact (including stop-gained and loss, frameshift, splice acceptor or donor and initiator codon variants), 7.4 × 10−9 for variants with moderate impact (missense, splice-region variants and in-frame-indels), 6.8 × 10−10 for low-impact variants (including synonymous, 3’ and 5’ UTR, and upstream and downstream variants), 3.4 × 10−10 for other variants in DNase I hypersensitivity sites (DHS), and 1.1 × 10−10 for all other variants. We did not perform sex stratified analysis due to incomplete information or unavailable data for a large proportion of studies included in the meta-analysis.
To test for independent associations of large-effect GWS variants at the TMEM18 and MC4R loci, we performed a conditional analysis in the deCODE, UKB, CHB/DBDS and Intermountain datasets, adding the top variant for stronger signals at their locus as covariates. The top variants of stronger signals were identified in the datasets where associations of the large-effect BMI variants predominated, i.e., in the deCODE and UKB datasets, respectively. Conditional analysis was also performed for publicly available BMI summary statistics in FinnGen using conditional and joint association analysis (COJO)105, where the same top variants were used as covariates as for the other cohorts.
R v3.6.0 was used extensively to analyze data and create plots.
LoF burden analysis
For the burden analysis, carriers of a LoF were coded with genotype count 1 if they carry any LoF variants in FRS3 and their expected allele count was > 0.9, and 0 otherwise. For sequenced individuals, all identified LoF variants were used, and for imputed individuals (only applies to deCODE, Iceland), LoF genotypes were used if the LoF variant imputed had imputation information > 0.7. For deCODE, familial imputation was also used as in the GWAS. Only high-quality sequence variants were considered for selection. We used the following quality metrics from GraphTyper76 and considered variants where ABHet > 0.175, ABHom > 0.85, QD > 6, QUAL > 10, PASS_ratio < 0.05 and AAscore > 0.8. To further estimate the quality of the sequence variants, we regressed the alternative allele counts (AD) on the depth (DP) conditioned on the genotypes (GT). Deviations from the identity line on the regression indicate that the sequence variant is spurious or somatic. We filtered out variants with slope less than 0.5. For the gene burden analysis, we only used variants with MAF < 2%.
Secondary phenotypes
The four FRS3 missense variants identified in the study were tested for association with other relevant phenotypes in two separate analyses. The first analysis included 19 binary and quantitative phenotypes correlated with BMI, including several cardiometabolic disorders, whole body lean and fat mass measured by dual-energy X-ray absorptiometry (DXA), blood pressure, blood lipids, HbA1c, and grip strength. The second analysis included 4 binary neurologic phenotypes associated with impaired BDNF-TrkB signaling, i.e., autism, ADHD, intellectual disability, and pain.
The four missense variants were only tested for association with those phenotypes in datasets representing their predominant ancestry, yielding a Bonferroni adjusted P value thresholds of 8.2 × 10−4 (0.05/61) for the first analysis and 6.3 × 10−3 (0.05/8) for the second analysis. Several phenotypes were not available for ancestries other than European, including total fat and lean mass by DXA, and were thus not tested for all four missense variants. Phenotype definitions, data sources, and references are summarized in Supplementary Data 17.
Functional characterization of BMI-associated FRS3 missense variants
We sought to explore the functional effects of BMI-associated FRS3 missense variants by assessing their effects (and of correlated variants with r2 > 0.8) on mRNA expression (cis-eQTL), mRNA splicing variation (sQTL), and plasma protein levels (pQTL).
We could only test p.Arg316Gln (rs35744673-T) for association with expression or protein levels because the required data were unavailable for cohorts whose summary statistics were utilized in the study, including FinnGen, and for other ancestries than European. We tested whether p.Arg316Gln and correlated variants were sentinel cis-eQTLs in RNA-sequencing data at deCODE, derived from whole blood (N = 17,848) and adipose tissue (N = 700). The variants’ effect on the expression of genes within 1 megabase (Mb) window and with a sufficient expression (> 1 transcript per million) in the respective tissue was tested using a generalized linear regression, assuming an additive model for genetic effect. Expression association for p.Arg316Gln and correlated variants were also looked up in the GTEx online database V836. P.Arg316Gln and correlated variants were tested for association with levels of circulating proteins, measured with 4907 aptamers by SomaScan v4106 in 35,559 Icelanders at deCODE107, and levels of 2941 immunoassays using the Olink Explore 3072 in 46,218 participants of European ancestry in the UK Biobank108.
Variant frequency map
Data from the UK Biobank were utilized to create a frequency map for the four FRS3 missense variants identified in the study (Fig. 2a–d). UKB participants were first grouped by birth country. We then defined regional ancestry groupings with the aims that groups be representative of the region’s current population, be homogeneous by genetic ancestry, and have at least 200 individuals (for accurate estimation of variant frequencies).
We assessed the current genetic ancestry profiles of regions by: comparing our ancestry analyses79 to in-house and published results of human genome diversity datasets like Human Origins92 and HGDP109; comparing genetic ancestry results across neighboring countries; surveying country demographics through sources like The World Factbook110; and examining participants’ self-reported ethnicity information and UK census data111 to determine whether participants born in certain countries were representative of those countries’ current demographics.
In some cases, we split off ancestry-based groupings representing distinct populations or unrepresentative migrant communities (e.g., “South Asian ancestry born in Africa and West Asia”) to achieve homogeneous birthplace-based groupings. Groups depicted on map figures are those best representing the current demographic majority. For Finland, the FinnGen frequency is shown. If countries had fewer than 200 participant birthplaces, we merged them with neighboring countries with similar assessed ancestry profiles. Map geometries were obtained from Natural Earth via R package maps112 and manipulated with sf113.
Variant annotation
We used Variant Effect Predictor (VEP, release 100)114 to attribute predicted consequences to the variants sequenced in each dataset. We classified as high-impact variants those predicted as start-lost, stop-gain, stop-lost, splice donor, splice acceptor or frameshift, collectively called LoF variants.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The GWAS meta-analysis summary statistics are available at https://download.decode.com/form/2025-Jonsdottir-Sveinbjornsson. The sequence variants from the Icelandic population whole-genome sequence data have been deposited at the European Variant Archive under accession PRJEB15197. The Icelandic, Danish, Intermountain and NashBio individual-level data are not publicly available due to data privacy laws and policies. Those wishing to access individual-level data should contact the corresponding author, Kari Stefansson (kstefans@decode.is), to organize a visit to deCODE genetics’ facilities in Iceland, where data can be accessed and computation resources utilized to perform analyses that conform to the studies’ ethical approvals (see Methods). Individuals must provide a rationale for data access and work for a legitimate research organization with a track record of ethically conducted health-related research. Requests will be answered within one month or upon availability of the corresponding author. For the Danish studies, i.e., the CHB-OCMS and the DBDS, data access requires collaboration with principal investigators for the genetic cohorts (CHB-OCMS: Sisse Rye Ostrowski (Sisse.Rye.Ostrowski@regionh.dk), Erik Sørensen (Erik.Soerensen@regionh.dk) or Ole Birger Vesterager Pedersen (olbp@regionsjaelland.dk); DBDS: Sisse Rye Ostrowski (Sisse.Rye.Ostrowski@regionh.dk), Erik Sørensen (Erik.Soerensen@regionh.dk), Ole Birger Vesterager Pedersen (olbp@regionsjaelland.dk) or Christian Erikstrup (christian.erikstrup@skejby.rm.dk)). FinnGen data are publicly available and were downloaded from https://www.finngen.fi/en/access_results. In this study, FinnGen Data Freeze 10 was utilized. The individual-level genotype data from Finnish biobanks can be accessed through the Fingenious portal at https://site.fingenious.fi/en/ (for further information, contact contact@finbb.fi). The UK Biobank data were downloaded under application no. 56270. Individual-level genomic and phenotypic data from the UK Biobank are available to researchers upon application (https://ukbiobank.ac.uk). The BMI exome-array summary statistics from the GIANT consortium15 are publicly available at https://portals.broadinstitute.org/collaboration/giant/index.php/GIANT_consortium_data_files. The Genes & Health GWAS dataset, precomputed for all available phenotypes, is publicly available as a whole using gcloud CLI, installed from https://cloud.google.com/sdk/docs/install and then accessed at gs://genesandhealth_publicdatasets/ (see https://www.genesandhealth.org/researchers/data/ for more information). In this study, Genes & Health data version 5 was used for binary phenotypes and version 6 for quantitative phenotypes. GWAS summary statistics from the Taiwan Biobank are available as Supplementary Information in their published article8. BMI summary statistics from the BioBank Japan9 and the National Biobank of Korea10 are publicly available and can be accessed at https://pheweb.jp/pheno/BMI and https://koges.leelabsg.org/pheno/KoGES_BMI, respectively. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium summary results from genomic studies, including the AAAGC BMI GWAS summary level data utilized in this study11, are available from dbGaP under accession code phs000930.v10.p1. MVP BMI GWAS summary statistics used in this study5 are available from dbGaP under accession code phs001672.v12.p1. BMI summary level data at birth from the Norwegian Mother, Father and Child (MoBa) study30 were downloaded from https://www.fhi.no/en/ch/studies/moba/for-forskere-artikler/gwas-data-from-moba/. Other data supporting the findings of this study are available within the Article or its Supplementary Information. Source data are provided with this paper.
References
Chong, B. et al. Trends and predictions of malnutrition and obesity in 204 countries and territories: an analysis of the Global Burden of Disease Study 2019. eClinicalMedicine 57, 101850 (2023).
The GBD 2015 Obesity Collaborators. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 377, 13–27 (2017).
Albuquerque, D., Nóbrega, C., Manco, L. & Padez, C. The contribution of genetics and environment to obesity. Br. Med. Bull. 123, 159–173 (2017).
Loos, R. J. F. & Yeo, G. S. H. The genetics of obesity: from discovery to biology. Nat. Rev. Genet. 23, 120–133 (2022).
Huang, J. et al. Genomics and phenomics of body mass index reveals a complex disease network. Nat. Commun. 13, 7973 (2022).
Yengo, L. et al. Meta-analysis of genome-wide association studies for height and body mass index in ~700,000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649 (2018).
Young, K. L., Graff, M., Fernandez-Rhodes, L. & North, K. E. Genetics of obesity in diverse populations. Curr. Diab. Rep. 18, 1–10 (2018).
Wong, H. S. C. et al. Genome-wide association study identifies genetic risk loci for adiposity in a Taiwanese population. PLoS Genet. 18, 1–21 (2022).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Nam, K., Kim, J. & Lee, S. Genome-wide study on 72,298 individuals in Korean biobank data for 76 traits. Cell Genom. 2, 100189 (2022).
Ng, M. C. Y. et al. Discovery and fine-mapping of adiposity loci using high density imputation of genome-wide association studies in individuals of African ancestry: African ancestry anthropometry genetics consortium. PLoS Genet. 13, 1–25 (2017).
Klimentidis, Y. C., Abrams, M., Wang, J., Fernandez, J. R. & Allison, D. B. Natural selection at genomic regions associated with obesity and type-2 diabetes: East Asians and sub-Saharan Africans exhibit high levels of differentiation at type-2 diabetes regions. Hum. Genet. 129, 407–418 (2011).
Uusimaa, J. et al. The Finnish genetic heritage in 2022 – from diagnosis to translational research. Dis. Models Mech. 15, dmm049490 (2022).
Akbari, P. et al. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science 373, eabf8683 (2021).
Turcot, V. et al. Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat. Genet. 50, 26–35 (2018).
Curtis, D. Weighted burden analysis in 200,000 exome-sequenced subjects characterises rare variant effects on BMI. Int. J. Obes. 46, 782–792 (2022).
Zhao, Y. et al. Protein-truncating variants in BSN are associated with severe adult-onset obesity, type 2 diabetes and fatty liver disease. Nat. Genet. 56, 579–584 (2024).
Sveinbjornsson, G. et al. Weighting sequence variants based on their annotation increases power of whole-genome association studies. Nat. Genet. 48, 314–317 (2016).
Loos, R. J. F. et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat. Genet. 40, 768–775 (2008).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Vaisse, C. et al. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J. Clin. Investig. 106, 253–262 (2000).
Saeed, S. et al. Genetic variants in LEP, LEPR, and MC4R explain 30% of severe obesity in children from a consanguineous population. Obesity 23, 1687–1695 (2015).
Chami, N., Preuss, M., Walker, R. W., Moscati, A. & Loos, R. J. F. The role of polygenic susceptibility to obesity among carriers of pathogenic mutations in MC4R in the UK Biobank population. PLoS Med. 17, 1–20 (2020).
Thorleifsson, G. et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 41, 18–24 (2009).
Tachmazidou, I. et al. Whole-genome sequencing coupled to imputation discovers genetic signals for anthropometric traits. Am. J. Hum. Genet. 100, 865–884 (2017).
Larder, R. et al. Obesity-associated gene TMEM18 has a role in the central control of appetite and body weight regulation. Proc. Natl. Acad. Sci. USA 114, 9421–9426 (2017).
Landgraf, K. et al. The obesity-susceptibility gene TMEM18 Promotes Adipogenesis through Activation of PPARG. Cell Rep. 33, 108295 (2020).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
WHO Consultation on Obesity. Obesity: Preventing and managing the global epidemic: report of a WHO consultation. https://iris.who.int/handle/10665/42330 (2000).
Helgeland, Ø. et al. Genome-wide association study reveals dynamic role of genetic variation in infant and early childhood growth. Nat. Commun. 10, 4448 (2019).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Dai, H. et al. The global burden of disease attributable to high body mass index in 195 countries and territories, 1990–2017: an analysis of the Global Burden of Disease Study. PLoS Med. 17, 1–19 (2020).
Einarsson, G. et al. Sequence variants associated with BMI affect disease risk through BMI itself. Nat. Commun. 15, 9335 (2024).
Larsson, S. C. & Burgess, S. Causal role of high body mass index in multiple chronic diseases: a systematic review and meta-analysis of Mendelian randomization studies. BMC Med. 19, 1–10 (2021).
Dixon, S. J., MacDonald, J. I. S., Robinson, K. N., Kubu, C. J. & Meakin, S. O. Trk receptor binding and neurotrophin/fibroblast growth factor (FGF)-dependent activation of the FGF receptor substrate (FRS)-3. Biochim. Biophys. Acta 1763, 366–380 (2006).
Ardlie, K. G. et al. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Strober, B. J. et al. The GTEx consortium atlas of genetic regulatory effects across human tissues. Science 18, 1318–1330 (2020).
Gotoh, N., Laks, S., Nakashima, M., Lax, I. & Schlessinger, J. FRS2 family docking proteins with overlapping roles in activation of MAP kinase have distinct spatial-temporal patterns of expression of their transcripts. FEBS Lett. 564, 14–18 (2004).
Schneeberger, M., Gomis, R. & Claret, M. Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J. Endocrinol. 220, T25–T46 (2014).
Ong, Z. Y., Liu, J. J., Pang, Z. P. & Grill, H. J. Paraventricular thalamic control of food intake and reward: Role of glucagon-like peptide-1 receptor signaling. Neuropsychopharmacology 42, 2387–2397 (2017).
Sweeney, P. & Yang, Y. An excitatory ventral hippocampus to lateral septum circuit that suppresses feeding. Nat. Commun. 6, 10188 (2015).
Huang, E. J. & Reichardt, L. F. Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642 (2003).
Notaras, M. & van den Buuse, M. Brain-Derived Neurotrophic Factor (BDNF): novel Insights into regulation and genetic variation. Neuroscientist 25, 434–454 (2019).
Reichardt, L. F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. B 361, 1545–1564 (2006).
Hryciw, T. et al. The fibroblast growth factor receptor substrate 3 adapter is a developmentally regulated microtubule-associated protein expressed in migrating and differentiated neurons. J. Neurochem. 112, 924–939 (2010).
Minegishi, Y. et al. Prominent expression of FRS2β protein in neural cells and its association with intracellular vesicles. FEBS Lett. 583, 807–814 (2009).
The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, 523–531 (2023).
Xu, B. & Xie, X. Neurotrophic factor control of satiety and body weight. Nat. Rev. Neurosci. 17, 282–292 (2016).
Kernie, S. G., Liebl, D. J. & Parada, L. F. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 19, 1290–1300 (2000).
Wang, P. et al. A leptin–BDNF pathway regulating sympathetic innervation of adipose tissue. Nature 583, 839–844 (2020).
You, H., Chu, P., Guo, W. & Lu, B. A subpopulation of Bdnf-e1–expressing glutamatergic neurons in the lateral hypothalamus critical for thermogenesis control. Mol. Metab. 31, 109–123 (2020).
Xu, B. et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 6, 736–742 (2003).
Siu, J. J. et al. Molecular therapy of Melanocortin-4-Receptor obesity by an autoregulatory BDNF vector. Mol. Ther. Methods Clin. Dev. 7, 83–95 (2017).
Rios, M. et al. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol. Endocrinol. 15, 1748–1757 (2001).
Han, J. C. et al. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N. Engl. J. Med. 359, 918–927 (2008).
Gray, J. et al. Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int. J. Obes. 31, 359–364 (2007).
Yeo, G. S. H. et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat. Neurosci. 7, 1187–1189 (2004).
Gray, J. et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 55, 3366–3371 (2006).
Miranda-Lourenço, C. et al. Challenges of BDNF-based therapies: from common to rare diseases. Pharmacol. Res. 162, 105281 (2020).
Nagahara, A. H. & Tuszynski, M. H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov. 10, 209–219 (2011).
Bartus, R. T. & Johnson, E. M. Clinical tests of neurotrophic factors for human neurodegenerative diseases, part 1: where have we been and what have we learned? Neurobiol. Dis. 97, 156–168 (2017).
Liu, D. et al. Characterization of on-target adverse events caused by TRK inhibitor therapy. Ann. Oncol. 31, 1207–1215 (2020).
Nordvall, G., Forsell, P. & Sandin, J. Neurotrophin-targeted therapeutics: a gateway to cognition and more? Drug Discov. Today 27, 103318 (2022).
Parrado Fernandez, C. et al. Neuroprotective and disease-modifying effects of the Triazinetrione ACD856, a positive allosteric modulator of Trk-receptors for the treatment of cognitive dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 24, 11159 (2023).
Önnestam, K. et al. Safety, tolerability, pharmacokinetics and quantitative electroencephalography assessment of ACD856, a novel positive allosteric modulator of Trk-receptors following multiple doses in healthy subjects. J. Prev. Alzheimer’s Dis. 4, 778–789 (2023).
Montague, C. L. et al. Congenital leptin deficiency is associated with severe earlyonset obesity in humans. Nature 387, 903–908 (1997).
Clément, K. et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392, 398–401 (1998).
Krude, H. et al. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 19, 155–157 (1998).
Robert, S. et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet. 16, 303–306 (1997).
Ollmann, M. M. et al. Antagonism of Central Melanocortin receptors in vitro and in vivo by agouti-related protein. Science 278, 135–138 (1997).
Dahlström, M. et al. Identification of novel positive allosteric modulators of neurotrophin receptors for the treatment of cognitive dysfunction. Cells 10, 1871 (2021).
Gudbjartsson, D. F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015).
Jónsson, H. et al. Data descriptor: whole genome characterization of sequence diversity of 15,220 Icelanders. Sci. Data 4, 1–9 (2017).
Eggertsson, H. P. & Halldorsson, B. V. read_haps: using read haplotypes to detect same species contamination in DNA sequences. Bioinformatics 37, 2215–2217 (2021).
Kong, A. et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat. Genet. 40, 1068–1075 (2008).
Eggertsson, H. P. et al. Graphtyper enables population-scale genotyping using pangenome graphs. Nat. Genet. 49, 1654–1660 (2017).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, 1–10 (2015).
Elliott, P. & Peakman, T. C. The UK Biobank sample handling and storage protocol for the collection, processing and archiving of human blood and urine. Int. J. Epidemiol. 37, 234–244 (2008).
Halldorsson, B. V. et al. The sequences of 150,119 genomes in the UK Biobank. Nature 607, 732–740 (2022).
Li, S., Carss, K. J., Halldorsson, B. V. & Cortes, A. UK Biobank Whole-Genome Sequencing Consortium. Whole-genome sequencing of half-a-million UK Biobank participants. Preprint at medRxiv (2023) https://doi.org/10.1101/2023.12.06.23299426.
Eggertsson, H. P. et al. GraphTyper2 enables population-scale genotyping of structural variation using pangenome graphs. Nat. Commun. 10, 1–8 (2019).
Belyeu, J. R. et al. Samplot: a platform for structural variant visual validation and automated filtering. Genome Biol. 22, 161 (2021).
Sørensen, E. et al. Data resource profile: the Copenhagen Hospital Biobank (CHB). Int. J. Epidemiol. 50, 719–720E (2021).
Hansen, T. F. et al. DBDS Genomic Cohort, a prospective and comprehensive resource for integrative and temporal analysis of genetic, environmental and lifestyle factors affecting health of blood donors. BMJ Open 9, e028401 (2019).
Thorolfsdottir, R. B. et al. Variants at the Interleukin 1 Gene Locus and Pericarditis. JAMA Cardiol. 9, 165–172 (2023).
O’Connell, J. et al. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 10, e1004234 (2014).
Roden, D. et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther. 84, 362–369 (2008).
Pulley, J., Clayton, E., Bernard, G. R., Roden, D. M. & Masys, D. R. Principles of human subjects protections applied in an Opt-Out, De-Identified Biobank. Clin. Transl. Sci. 3, 42–48 (2010).
NashBio. AGD: The Alliance for Genomic Discovery. https://nashbio.com/agd/ (2025).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Lazaridis, I. et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513, 409–413 (2014).
Privé, F., Luu, K., Blum, M. G. B., McGrath, J. J. & Vilhjálmsson, B. J. Efficient toolkit implementing best practices for principal component analysis of population genetic data. Bioinformatics 36, 4449–4457 (2020).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 1–16 (2015).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Finer, S. et al. Cohort Profile: East London Genes & Health (ELGH), a community-based population genomics and health study in British Bangladeshi and British Pakistani people. Int. J. Epidemiol. 49, 20–21I (2020).
Nagai, A. et al. Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol. 27, S2–S8 (2017).
Kim, Y., Han, B. G., Moon, H. K., Song, J. H. & Yun, Y. min. Cohort profile: The Korean genome and epidemiology study (KoGES) consortium. Int. J. Epidemiol. 46, e20 (2017).
Feng, Y. C. A. et al. Taiwan Biobank: a rich biomedical research database of the Taiwanese population. Cell Genom. 2, 100197 (2022).
Gaziano, J. M. et al. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 70, 214–223 (2016).
Psaty, B. M. et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium design of prospective meta-analyses of genome-wide association studies from 5 Cohorts. Circ. Cardiovasc. Genet. 2, 73–80 (2009).
Loh, P. R. et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015).
Mantel, N. & Haenszel, W. Statistical aspects of the analysis of data from retrospective studies of disease. J. Natl. Cancer Inst. 22, 719–748 (1959).
Bulik-Sullivan, B. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Gold, L., Walker, J. J., Wilcox, S. K. & Williams, S. Advances in human proteomics at high scale with the SOMAscan proteomics platform. N. Biotechnol. 29, 543–549 (2012).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 53, 1712–1721 (2021).
Eldjarn, G. H. et al. Large-scale plasma proteomics comparisons through genetics and disease associations. Nature 622, 348–358 (2023).
Bergström, A. et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 367, eaay5012 (2020).
The World Factbook 2021. Washington, DC: Central Intelligence Agency. https://www.cia.gov/ (2021).
Office for National Statistics. Census 2021 data. https://www.ons.gov.uk/census (2021).
Becker, R. A., Wilks, A. R., Brownrigg, R., Minka, T. P. & Deckmyn, A. Draw Geographical Maps. https://cran.r-project.org/web/packages/maps/index.html (2023).
Pebesma, E. Simple features for R: standardized Support for Spatial Vector Data. R J 10, 439–446 (2018).
McLaren, W. et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010).
Acknowledgements
We thank all individuals who participated in the study and the staff at the Icelandic Patient Recruitment Center and the deCODE genetics core facilities. Collection and analysis of Icelandic pain data was in part supported by the European Commission to the painFACT project (H2020-2020-848099 to T.E.T.). This research has been conducted using FinnGen Data Freeze 10. We want to acknowledge the participants and investigators of the FinnGen study. S.B., K.B., P.C.H., H.B., and C.E. acknowledge the Novo Nordisk Foundation (grants NNF17OC0027594 and NNF14CC0001 to S.B.; NNF24SA0091838 to H.B.; AGR-2023-731-2304 to C.E.). For the NashBio study population, individual DNA samples selected from VUMC’s BioVU® biobank and structured clinical data from VUMC’s Synthetic Derivative (SD) were provided by NashBio and WGS data were generated using Illumina technology. The samples were provided to deCODE for WGS under the Alliance for Genomic Discovery (https://nashbio.com/agd), which is a collaboration between NashBio, Illumina, and eight pharmaceutical companies including deCODE’s parent company Amgen. The authors thank Million Veteran Program (MVP) staff, researchers, and volunteers, who have contributed to MVP, and especially participants who previously served their country in the military and now generously agreed to enroll in the study. (See https://www.research.va.gov/mvp/ for more details). The authors acknowledge the essential role of the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium in the development and support of this research. (See http://web.chargeconsortium.com for more details). The authors thank the investigators, the staff, and the participants of each contributing cohort in the CHARGE consortium publication from which these results were obtained. Genes & Health is/has recently been core-funded by Wellcome (WT102627, WT210561), the Medical Research Council (UK) (M009017, MR/X009777/1, MR/X009920/1), Higher Education Funding Council for England Catalyst, Barts Charity (845/1796), Health Data Research UK (for London substantive site), and research delivery support from the NHS National Institute for Health Research Clinical Research Network (North Thames). Genes & Health is/has recently been funded by Alnylam Pharmaceuticals, Genomics PLC; and a Life Sciences Industry Consortium of Astra Zeneca PLC, Bristol-Myers Squibb Company, GlaxoSmithKline Research and Development Limited, Maze Therapeutics Inc, Merck Sharp & Dohme LLC, Novo Nordisk A/S, Pfizer Inc, Takeda Development Centre Americas Inc. We thank Social Action for Health, Centre of The Cell, members of our Community Advisory Group, and staff who have recruited and collected data from volunteers. We thank the NIHR National Biosample Centre (UK Biocentre), the Social Genetic & Developmental Psychiatry Centre (King’s College London), Wellcome Sanger Institute, and Broad Institute for sample processing, genotyping, sequencing and variant annotation. This work uses data provided by patients and collected by the NHS as part of their care and support. We thank: Barts Health NHS Trust, NHS Clinical Commissioning Groups (City and Hackney, Waltham Forest, Tower Hamlets, Newham, Redbridge, Havering, Barking and Dagenham), East London NHS Foundation Trust, Bradford Teaching Hospitals NHS Foundation Trust, Public Health England (especially David Wyllie), Discovery Data Service/Endeavour Health Charitable Trust (especially David Stables), Voror Health Technologies Ltd (especially Sophie Don), NHS England (for what was NHS Digital) - for GDPR-compliant data sharing backed by individual written informed consent. Most of all we thank all of the volunteers participating in Genes & Health.
Author information
Authors and Affiliations
Consortia
Contributions
A.B.J., G.S., R.B.T., H.M.AE., G.M., U.T., D.F.G., G.T., H.H., P.S., and K.S. designed the study. A.B.J., G.S., R.B.T., H.M.AE., G.E., K.H.S.M., G.L.N., D.F.G., G.T., H.H., P.S., and K.S. interpreted the results. A.B.J., G.S., R.B.T., M.T., V.T., T.O., S.Rognvaldsson, E.E., G.E., R.F., E.V.I., G.M., K.H.S.M., A.O., P.I.O., F.P., A.Skaftason, L.S., A.Sturluson, G.A.T., M.O.U., A.Helgason, G.L.N., S.Ripatti, D.F.G., and G.T. contributed to the statistical and bioinformatics analyses. A.Sigurdsson, H.P.E., D.B., G.H.H., S.Kristmundsdottir, O.T.M., P.M., A.Sturluson, B.V.H., and D.F.G. contributed to the sequencing, genotyping, expression, and proteomics analyses. T.O., D.O.A., R.G.B., I.J., E.L.S., T.S., U.T., A.V., H.H., and K.S. coordinated and managed collection of samples and phenotype data in Iceland. A.B.J., G.S., R.B.T., V.T., T.O., S.Rognvaldsson, G.B., E.E., S.G., A.Helgadottir, A.E.L., A.O., F.P., L.S., H.S., U.S., U.D.T., T.E.T., M.O.U., G.B.W., G.T., V.S., H.H., P.S., and K.S. contributed to the definitions of phenotypes explored in the study. K.B., S.B., M.T.B., J.D., C.E., J.G., T.H., P.C.H., K.K.I., B.A.J., C.M., O.B.P., M.S., E.S., H.U., D.B.D.S., H.B., and S.R.O. handled collection, analysis, and supervision of data from Denmark. S.Knight, K.U.K., and L.D.N. handled collection and supervision of data from Intermountain, USA. M.T. and S.Ripatti analyzed and supervised data from the FinnGen study. A.B.J., G.S., R.B.T., T.O., S.Rognvaldsson, H.P.E., O.T.M., K.H.S.M., G.L.N., D.F.G., H.H., P.S., and K.S. wrote the manuscript. All authors contributed to the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors affiliated with deCODE genetics/Amgen, Inc. are employed by the company. H.B. receives lecture fees from MSD, BMS, Amgen, and Pfizer and has received a grant from the Novo Nordisk Foundation. C.M. is a Novo Nordisk stock owner. S.B. has ownership in Intomics A/S, Hoba Therapeutics Aps, Novo Nordisk A/S, Lundbeck A/S, and ALK albello A/S and managing board memberships in Proscion A/S and Intomics A/S. C.E. has received an unrestricted research grant from Novo Nordisk administered by the Aarhus University; no personal fees. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Nathalie Chami and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jonsdottir, A.B., Sveinbjornsson, G., Thorolfsdottir, R.B. et al. Missense variants in FRS3 affect body mass index in populations of diverse ancestries. Nat Commun 16, 2694 (2025). https://doi.org/10.1038/s41467-025-57753-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57753-2
