
Abstract
The prospective relationship between proteomics and inflammatory bowel disease (IBD) remains largely underexplored, presenting potential of therapeutic interventions. Using data from 48,800 IBD-free participants in the UK Biobank Pharma Proteomics Project (UKB-PPP), we assessed associations between 2923 plasma proteins and incident IBD risk using Cox analysis. Mendelian randomization (MR) meta-analysis, integrating cis-protein quantitative trait loci data from the UKB-PPP with IBD genome-wide association study data from the International Inflammatory Bowel Disease Genetics Consortium and FinnGen studies, identified causal proteins. Colocalization analysis strengthened evidence of shared common causal variants. Cohort study revealed associations of 673, 295, and 125 proteins with the risk of IBD, Crohn’s disease (CD), and ulcerative colitis (UC), respectively. MR and colocalization analyses prioritized IL12B, CD6, MXRA8, CXCL9, IFNG, CCN3, RSPO3, and IL18 as having causal and high colocalization evidence with IBD or its subtypes. Our findings advance understanding of IBD’s molecular etiology and highlight potential therapeutic targets.
Similar content being viewed by others

Introduction
Inflammatory bowel disease (IBD), a group of chronic inflammatory disorders of the intestine that include Crohn’s disease (CD) and ulcerative colitis (UC), has a rising global prevalence1. IBD significantly impacts the patient quality of life and may lead to various complications2. Early detection and treatment are crucial for improving patient outcomes and delaying disease progression. IBD is related to diverse environmental factors, such as smoking, unhealthy diet, and genetic factors, yet its precise etiology remains unclear3. Despite the availability of various treatment options, current therapies exhibit suboptimal efficacy, with clinical remission rates for most biologics and small molecules ranging between 30% and 50% after 1 year of treatment4. Measuring plasma levels of thousands of proteins, and when combined with genetic data, has proven valuable for identifying novel risk factors and potential therapeutic targets for several diseases, such as ischemic heart disease5, dementia6, heart failure7, and type 2 diabetes8.
Several case-control studies have reported differences in protein expression between patients with IBD and healthy controls (HC)9,10,11,12,13,14. For example, case-control studies by Eldjarn et al. based on the UK Biobank and Icelandic population have linked proteins such as PTGDS, CXCL11, and REG3A with IBD14. While these studies provide valuable insights, their cross-sectional design does not inform whether these differences occur before or after disease onset. To date, only two studies have investigated the association between baseline protein levels and the subsequent risk of developing IBD. The first study, conducted by Torres et al., revealed that certain proteins related to the complement cascade, lysosomal function, innate immunity, and glycosaminoglycan metabolism may exhibit changes 1–5 years before CD diagnosis15. However, due to the short follow-up period of the study, potential delays in seeking treatment, and influence of subclinical disease stages, it remains unclear whether these protein changes genuinely precede disease onset. Validation through large-scale, long-term prospective cohort studies is thus required. The second study by Chen et al. employed a prospective design with 72 UC patients and 150 HC and bidirectional Mendelian randomization (MR), and suggested that CXCL9, CCL11 and CASP8 might be causally linked to UC16. While providing valuable insights, the study was limited to only 92 inflammatory proteins and a relatively small sample size, constraining a comprehensive understanding of the proteome. Therefore, there is a critical need to integrate large-scale proteomics data from long-term prospective cohort studies with genetic research to obtain comprehensive evidence on proteins associated with the risk of incident IBD.
Advances in proteomics technologies, such as proximity extension assays using the Olink platform, have enabled the simultaneous measurement of thousands of proteins in large cohorts, facilitating longitudinal assessments of protein-disease associations. For instance, the UK Biobank Pharma Proteomics Project (UKB-PPP) utilized Olink Proximity Extension Assay to measure nearly 3000 proteins in over 50,000 participants, providing a broad and large-scale proteomics foundation for prospective studies that explore the association between a large number of proteins and the risk of developing IBD. However, while observational studies provide valuable insights into associations between proteins and disease risk, they are inherently limited in their ability to establish causal relationships due to unknown or unmeasured confounding factors. MR analysis leverages genetic variants as instrumental variables to make causal relationships between exposure and outcome. Since genetic variants are randomly assigned at conception and before disease onset, this approach effectively minimizes unknown confounding and reverse causation17. Recent advances in proteomics have identified a wealth of protein quantitative trait loci (pQTLs), laying the foundation for MR analyses of protein levels. Notably, cis-pQTLs, which are located near the encoding genes, are more likely to influence protein levels by directly affecting transcription or translation. In contrast, trans-pQTLs may influence protein levels through indirect mechanisms. The use of cis-pQTLs in MR analysis significantly reduces pleiotropy caused by indirect pathways18. Therefore, MR analyses leveraging cis-pQTL-related single nucleotide polymorphisms (SNPs) as instrumental variables for circulating proteins have emerged as a powerful approach to strengthen causal inference. However, there are currently no studies that have utilized UKB-PPP and cis-pQTLs MR analysis to identify causal proteins associated with IBD.
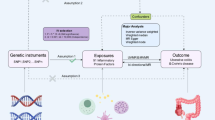
To investigate the association of the broad proteome with the risk of developing IBD, we integrated observational and genetic analyses. First, we evaluated the association between 2923 baseline plasma proteins and incident IBD risk in the UKB-PPP cohort, comprising 48,800 participants without IBD history at baseline. Second, we performed a two-sample MR meta-analysis using cis-pQTL data from the UKB-PPP alongside IBD genome-wide association studies (GWAS) data combined from the International Inflammatory Bowel Disease Genetics Consortium (IIBDGC) and FinnGen studies to assess causal relationships between candidate proteins and IBD, supplemented by reverse MR, Steiger filtering, and colocalization analysis. Moreover, we performed cross-platform replication MR analyses using cis-pQTLs from the deCODE and Fenland studies. Finally, downstream analyses, including protein-coding gene expression across 49 tissues for associations with IBD, single-cell expression analysis, single-gene knockout (KO) models, enrichment and pathway analyses, protein-protein interaction (PPI) studies, and druggability assessments, were conducted to elucidate potential mechanisms of candidate proteins and their viability as therapeutic targets for IBD (Fig. 1).

cis-pQTLs cis-protein quantitative trait loci, LD linkage disequilibrium, GWAS Genome-Wide Association Studies, IIBDGC International Inflammatory Bowel Disease Genetics Consortium, IBD inflammatory bowel disease, CD Crohn’s disease, UC ulcerative colitis, SNPs single nucleotide polymorphisms, MR Mendelian randomization, FDR false discovery rate, PPH4 posterior probability of Hypothesis 4.
Results
Participant characteristics
This study included 48,800 participants of European ancestry without a history of IBD at baseline. Baseline characteristics are detailed in Supplementary Table 1. During a median follow-up of 13.6 years (interquartile range, IQR: 12.9–14.3 years), totaling 640,799 person-years, 336 new cases of IBD during follow-up were reported, including 125 new cases of CD and 232 new cases of UC. Compared with participants who did not develop IBD during follow-up, those who developed IBD were older (mean baseline age: 58.37 ± 7.71 years vs. 57.10 ± 8.12 years), more inclined to smoke, more likely to be male, have lower education levels and household income, a higher likelihood of unemployment, higher Townsend deprivation index (TDI) scores, lower physical activity (PA) levels, poor diet, abnormal sleep duration ( < 7 h or ≥ 9 h), and higher body mass index (BMI).
Observational associations of proteins with IBD
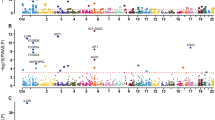
In univariate analyses, 1163 proteins were significantly associated with incident IBD [false discovery rate (FDR) < 0.05, Fig. 2a, Supplementary Data 1]. After adjusting for age, sex, assessment center, years of education, employment status, household income, and TDI in the initial adjusted Cox model, 800 proteins were found to be significantly associated with incident IBD risk (FDR < 0.05, Fig. 2b, Supplementary Data 1). Following further adjustments for smoking, alcohol consumption, PA, healthy diet, sleep duration, and BMI in the fully adjusted Cox model, 673 proteins remained significantly associated with incident IBD risk [FDR < 0.05, hazard ratios (HR) ranging from 0.76 to 1.44, Fig. 2c, Supplementary Data 1]. The association between proteins and IBD subtypes in unadjusted and initial adjusted models is shown in Figs. 2d, e and 2g, h. In the fully adjusted model, when investigating the association with IBD subtypes, 295 proteins were found to be significantly associated with incident CD (FDR < 0.05, HR from 0.60 to 1.68, Fig. 2f, Supplementary Data 1) and 125 with incident UC (FDR < 0.05, HR from 0.76 to 1.49, Fig. 2i, Supplementary Data 1). Comparison of HR for proteins significantly associated with CD and UC is shown in Supplementary Fig. 1.

Unadjusted any variable in estimating the hazard ratios of each protein for (a) inflammatory bowel disease (IBD), (d) Crohn’s disease (CD), and (g) ulcerative colitis (UC). Adjusted for age, sex, assessment center, education, employment status, household income, and TDI in estimating the hazard ratios of each protein for (b) IBD, (e) CD, and (h) UC. Additional adjusted for smoking status, alcohol consumption, PA, healthy diet, sleep during, and BMI in estimating the hazard ratios of each protein for (c) IBD, (f) CD, and (i) UC. Red, blue, and gray dots denote positive significant, inverse significant, and nonsignificant associations, respectively. FDR false discovery rate, HR hazard ratio. Source data are provided as Supplementary Data 1 and Source Data file.
We then performed sensitivity analyses. First, after excluding participants with protein levels below the limit of detection, 610 of 673 proteins significantly associated with IBD remained significant (Supplementary Data 2). Second, after additional adjusting for the first 10 genetic principal components, 672 of 673 proteins remained significant (Supplementary Data 3). Third, after excluding 6727 participants with a baseline history of other immune-mediated diseases (including 6663 participants who did not develop IBD and 64 participants who later developed IBD), 361 of 673 proteins remained significant (Supplementary Data 4). Fourth, after additional adjustments for other baseline chronic disease histories, 591 of 673 proteins remained significant (Supplementary Data 5). Finally, in the analyses stratified by the time between blood draw and diagnosis, 411 of 673 proteins remained significant in cases diagnosed within 2 years of the blood draw, primarily reflecting their potential as biomarkers for early detection. Moreover, 250 of these proteins remained significant in cases diagnosed more than 6 years after the blood draw, primarily reflecting their relevance to disease etiology (Supplementary Data 6).
For predicting the risk of IBD, in the testing dataset, the combined model of the traditional risk model and the simplified protein prediction model had an AUC of 0.68 [95% confidence interval (CI), 0.61–0.75], which was significantly better than the AUC of the traditional prediction model (0.62 [95% CI, 0.54–0.69]) (Supplementary Data 7, Supplementary Table 2, Supplementary Fig. 2). Similarly, for the CD and UC subtypes, in the testing dataset, the combined model of the traditional risk model and the simplified protein prediction model had AUCs of 0.64 (95% CI, 0.55–0.74) and 0.62 (95% CI, 0.53–0.72), also outperforming the AUCs of the traditional prediction models (0.59 [95% CI, 0.48–0.70] and 0.55 [95% CI, 0.47–0.64]) (Supplementary Data 7, Supplementary Table 2, Supplementary Fig. 2).
Mendelian randomization and colocalization analyses
Among the 673 proteins significantly associated with IBD identified via Cox regression, 555 proteins had available cis-pQTLs in the UKB-PPP GWAS14,19,20 (Supplementary Data 8). In the meta-analyses of MR results between 555 proteins from the UKB-PPP and IBD data from two genetic datasets (IIBDGC21 and FinnGen22), genetically determined levels of 18 proteins remained were significantly associated with IBD incidence after multiple corrections (FDR < 0.05, Fig. 3a, Supplementary Data 9). Furthermore, among the 295 proteins significantly associated with CD by Cox regression, 261 had available cis-pQTLs in the UKB-PPP GWAS. In the meta-analysis of MR results for these 261 proteins, 6 proteins were shown to be significantly associated with CD risk (FDR < 0.05), including three (IL12B, INFG, and SEPTIN8) overlapping with IBD and three (CXCL9, CCN3, and RSPO3) specific to CD (Fig. 3b, Supplementary Data 9). Additionally, among the 125 proteins significantly associated with UC by Cox regression, 114 had available cis-pQTLs in the UKB-PPP GWAS. In the meta-analysis of MR results for these 114 proteins, 4 proteins were shown to be significantly associated with UC risk (FDR < 0.05), including three proteins (CCL20, CCL7, and IL18) overlapping with IBD and one (NOS3) specific to UC (Fig. 3c, Supplementary Data 9).

The forest plots showed the results of observational analyses and MR analyses between candidate proteins and (a) inflammatory bowel disease, (b) Crohn’s disease, and (c) ulcerative colitis. Cox regression was used to evaluate the association between baseline plasma protein and the incidence of IBD and its subtypes during study follow-up in UKB-PPP study (baseline participants: n = 48,800; incident IBD events: n = 336; incident CD events: n = 125; incident UC events: n = 232) adjusted for age, sex, assessment center, education, employment status, household income, TDI, smoking status, alcohol consumption, PA, healthy diet, sleep during, and BMI. The measure of centre and error bars in observational analyses presented the HRs and 95% CI of IBD and its subtypes risk increase associated with per 1-SD higher protein level. MR analysis, including Wald ratio or inverse variance weighted method, was used to evaluate the association between cis-pQTLs from UKB-PPP and IBD and its subtypes from IIBDGC (with results shown in green) and FinnGen (with results shown in purple). Meta-analysis was used to combine MR analysis results of IIBDGC and FinnGen (with results shown in red). The measure of centre and error barsin in MR analyses presented the ORs and 95% CI of IBD and its subtypes per 1-SD higher protein level for these proteins from in MR analyses. All tests were two sided and adjusted for multiple comparisons, with an FDR < 0.05 considered as significant. UKB-PPP UK Biobank Pharma Proteomics Project, IIBDGC International Inflammatory Bowel Disease Genetics Consortium, HR hazard ratio, OR odds ratio, CI confidence interval. Source data are provided as Supplementary Data 1, 9, and Source Data file.
Comprehensive phenotypic assessments revealed no associations between these proteins and known confounding factors (Supplementary Data 10). For all proteins with more than two SNPs available, the MR-Egger intercept test indicated no strong evidence of significant horizontal pleiotropy (Supplementary Data 11). Reverse MR analysis showed no significant causal relationship between IBD, CD, or UC and the levels of 28 identified proteins (18 associated with IBD, 6 with CD, and 4 with UC) (Supplementary Data 12, Supplementary Table 3, Table 1). The Steiger filtering analyses confirmed the correct causal direction of changes in all 28 identified proteins to the development of IBD, CD, and UC (Table 1). Colocalization analyses showed that 3 proteins for IBD (IL12B, CD6, and MXRA8), 5 proteins for CD (CXCL9, IFNG, IL12B, CCN3, and RSPO3), and 1 protein for UC (IL18) had high colocalization evidence, which suggested that the effects on protein levels and disease are driven by the same causative variants (Supplementary Table 4, Table 1). Based on the above results, these proteins were classified into two tiers: eight unique proteins (IL12B, CD6, MXRA8, CXCL9, IFNG, CCN3, RSPO3, and IL18) passed all tests and were classified into tier 1 proteins, while other 14 proteins (CCL20, TNFRSF9, CCL7, IL1RL1, IL19, CCL13, CD72, EPHB4, SEPTIN8, LY9, IL10RA, IGLC2, ITGAV, and NOS3) were assigned to tier 2 proteins.
We conducted cross-platform replication MR analyses using cis-pQTLs from the deCODE23 and Fenland24 studies, which involved proteins detected by SomaScan. For proteins significantly associated in observational studies, MR analysis results between proteins from the deCODE study (using the SomaScan platform) and IBD demonstrated moderate correlations (Pearson correlations coefficients of 0.57, 0.60, and 0.67 for IBD, CD, and UC, respectively) when compared with MR results between proteins from the UKB-PPP (using the Olink platform) and IBD. Similarly, MR analysis results for protein GWAS data from the Fenland study (using the SomaScan platform) with IBD were moderately correlated (Pearson correlations coefficients of 0.64, 0.58, and 0.67 for IBD, CD, and UC, respectively) with those from the UKB-PPP (using the Olink platform) (Supplementary Data 13, 14, Supplementary Figs. 3, 4). Specifically, among the candidate proteins identified by the UKB-PPP, MR analyses confirmed consistent direction for all seven available proteins associated with IBD (IL12B, CCL7, IL1RL1, CD72, EPHB4, LY9, and MXRA8) in the deCODE or Fenland studies. For CD, three of four available proteins (CXCL9, IL12B, and RSPO3) showed consistent direction, and for UC, one available protein (CCL7) demonstrated a consistent direction (Supplementary Tables 5, 6 and Table 1).
Transcriptome analysis of protein-coding genes across different tissues
We performed expression quantitative trait loci (eQTL) MR analyses for the genes corresponding to the 22 candidate proteins across 49 tissues (Supplementary Data 15, Supplementary Fig. 5). Eight protein-coding genes (CD6, MXRA8, CCL20, IL18, IL1RL1, IGLC2, ITGAV, and NOS3) showed consistent MR results with the corresponding plasma proteins in relation to the risks of IBD or its subtypes. These genes demonstrated consistent associations with IBD risk across multiple blood or tissue-specific samples. For example, in sigmoid colon samples, the expression of the MXRA8 gene was negatively correlated with IBD risk, consistent with the effect observed in plasma proteins. In contrast, the genes TNFRSF9, IL19, CCL13, EPHB4, and SEPTIN8 exhibited varying impacts on IBD risk depending on the blood or tissue-specific sample. For instance, the expression of the SEPTIN8 gene in blood and most tissues (e.g., the colon) increased IBD risk, aligning with the role of SEPTIN8 protein in plasma. However, the expression of the SEPTIN8 gene in skin tissue decreased IBD risk. These findings highlight the potential for off-target effects when developing drugs targeting these genes in different tissues may present potential off-target effects.
Single-cell type expression analysis of candidate protein-coding genes
We conducted single-cell type expression analysis to evaluate the cell type-specific enrichment of the 22 candidate protein-coding genes in the colonic mucosa, as well as their differential expression levels between patients with IBD and HC. This analysis utilized a previously published single-cell RNA-seq dataset of human colonic mucosal tissues25. The cells were clustered into nine cell types: B cells, dendritic cells, endothelial cells, epithelial cells, macrophages, natural killer cells, smooth muscle cells, T cells, and tissue stem cells (Fig. 4a). Among the 22 protein-coding genes, expression data were available for all genes in IBD colon tissue, while in the HC group, expression data were available for all genes except IL12B and IL19 (Fig. 4b). The single-cell expression pattern for these 22 coding genes in each cell type for patients with IBD as well as HC is illustrated in Fig. 4c. Of the 22 protein-coding genes, 19 exhibited significant cell type-specific enrichment in colon tissue, with average |log2 fold change (FC)| > 0.25 and FDR < 0.05 (Fig. 4d). Further differential expression analysis revealed that the expression of CCL20 (enriched in macrophages and T cells), CD6 (T cells), CXCL9 (macrophages), IL18 (epithelial cells), and SEPTIN8 (endothelial cells) was each significantly more highly expressed in their respective enriched cell types in IBD compared to HC ( | log2FC | > 0.25 and FDR < 0.05, Fig. 4e, Supplementary Data 16), consistent with the results of observational and MR analyses.

a A total of nine cell types were identified in colonic mucosa tissue using graph-based clustering by the tSNE. b The t-SNE plot shows the expression of each candidate protein-coding gene across different cell types in the colon mucosa of IBD patients and healthy controls. c The bubble plot shows the expression proportion and levels of each candidate protein-coding gene across different cell types in the colon mucosa of IBD patients and healthy controls. d Cell-specific enrichment of each candidate protein-coding gene. e Comparison of each candidate protein-coding gene expression levels between IBD and health controls in the corresponding specifically enriched cell types. The Wilcoxon rank-sum test was used for analysis. All tests were two sided and adjusted for multiple comparisons. Log2FC > 0.25 and an FDR < 0.05 was considered as significant. The symbols * represent FDR < 0.05, ** represent FDR < 0.01, and *** represent FDR < 0.001. Source data are provided as Supplementary Data 16 and Source Data file.
PPI network, enrichment analysis, and druggability assessment
Our PPI network analysis revealed interactions among 18 of the 22 candidate proteins (Supplementary Fig. 6a). Among them, IFNG, IL18, CCL20, CXCL9, CCL7, and IL10RA are key proteins within the PPI network. These candidate proteins were primarily involved in biological processes such as signal transduction, inflammatory response, and immune response. They appear to contribute to IBD development and progression through pathways such as cytokine–cytokine receptor interactions, viral protein interactions with cytokines and cytokine receptors, JAK-STAT signaling pathway, and chemokine signaling pathway (Supplementary Fig. 6b, c, Supplementary Data 17). And we found that the interaction rate among our 22 selected candidate proteins (81.8%) was significantly higher than the interaction rate observed among random selections (11.7%, 95% CI: 0%–36.4%, Supplementary Data 18, Supplementary Fig. 7a). In addition, KO mouse models for these proteins identified several IBD-associated phenotypes, including immune system phenotypes (IL18, IL12B, TNFRSF9, CCL7, IL1RL1, IL19, CD72, IFNG, CD6, LY9, IL10RA, CXCL9, CCN3, and NOS3) and digestive/alimentary phenotypes (IL18, IL19, IFNG, IL10RA, and ITGAV) (Supplementary Data 19). And we found that the proportion of proteins with evidence from KO mouse models among our 22 candidate proteins (68.1%) was significantly higher than the proportion observed in random samples (36.4%, 95% CI: 18.2%–59.1%, Supplementary Data 20, Supplementary Fig. 7b).
To evaluate therapeutic potential, we searched the DrugBank database, the Open Targets database, and the Therapeutic Target Database for existing medications targeting these proteins (Table 1). Among these, olsalazine, which acts on IFNG, is an approved treatment for IBD. In addition, ustekinumab (an inhibitor of IL12B) and GSK-1070806 (an inhibitor of IL18) are currently undergoing clinical trials for IBD treatment. Furthermore, drugs targeting tier 1 proteins (CD6 and RSPO3) and tier 2 proteins (CCL20, TNFRSF9, IL1RL1, EPHB4, IL10RA, ITGAV, and NOS3) are being developed for other autoimmune diseases, cancers, and cardiovascular diseases in clinical trials, highlighting the potential for drug repurposing to treat IBD.
Moreover, we found that 18 of the 22 candidate proteins interact with known IBD drug targets. Specifically, 15 candidate proteins (IL12B, CD6, CCL20, IL10RA, ITGAV, EPHB4, IL18, IL19, LY9, IFNG, MXRA8, IL1RL1, TNFRSF9, CCL13, and CCL7) for IBD, three (IL12B, CCN3, and CXCL9) for CD, and four (IL12B, CCL20, NOS3, and CCL7) for UC were observed to interact with current drug targets. Detailed interactions and enrichment analyses between candidate proteins and known drug targets are detailed in Supplementary Fig. 8 and Supplementary Data 21, 22. And we found that the interaction rate between our identified candidate proteins and known IBD drug targets (81.8%) was significantly higher than the interaction rates of randomly selected proteins with known IBD drug targets (55.5%, 95% CI: 36.4–77.3%, Supplementary Data 23, Supplementary Fig. 7c).
Discussion
We performed plasma proteomics analyses, combined with prospective cohort and genetic studies, to investigate the causal roles of 2923 plasma proteins detected by Olink in IBD and its subtype. Using data from a prospective cohort study in the UK Biobank, we identified 673 proteins associated with IBD, 295 with CD, and 125 with UC. MR analyses further confirmed causal associations for 18 candidate proteins with IBD, six with CD, and four with UC. IL12B, CD6, MXRA8, CXCL9, IFNG, CCN3, RSPO3, and IL18 showed strong colocalization evidence and were classified as tier 1 proteins, while the remaining proteins were classified as tier 2 proteins. Single-cell expression analysis revealed significantly elevated expression levels of CCL20 in macrophages and T cells, CCL7 in smooth muscle cells, CD6 in T cells, CXCL9 in macrophages, IL18 in epithelial cells, and SEPTIN8 in endothelial cells from patients with IBD compared to HC. These proteins were predominantly enriched in biological processes such as signal transduction, inflammatory response, and immune response and may contribute to IBD development through pathways such as cytokine–cytokine receptor interaction, JAK-STAT signaling, and chemokine signaling. Druggability assessment found that clinical trials targeting IL12B, IFNG, and IL18 for IBD have been conducted. The tier 1 proteins CD6 and RSPO3, as well as several tier 2 proteins, have been targeted in drug development for other diseases and may serve as potential candidate targets for drug repurpose. These findings provide new insights into targeted drug development for IBD.
Among the tier 1 proteins identified in this study (IL12B, CD6, MXRA8, CXCL9, IFNG, CCN3, RSPO3, and IL18), IL12B, IFNG, and IL18 are already approved or under development as drug targets for IBD. Herein we identified five new potential drug targets (CD6, MXRA8, CXCL9, CCN3, and RSPO3) based on population-based prospective cohort studies, MR analyses, and colocalization evidence. Existing drug studies highlight that ustekinumab, an antibody therapy inhibiting IL-12 and IL-23 signaling, has been approved for treating moderate to severe CD since 2016 and active UC since 201926,27. Olsalazine, an anti-inflammatory agent targeting IFNG, blocks the production of cyclooxygenase-derived products of arachidonic acid metabolism and is used to treat IBD and UC28,29. GSK1070806, a humanized monoclonal antibody targeting IL-18, has undergone successful phase I clinical trials for IBD30,31. A previous cell-based experimental study suggested that elevated expression of CD6 and ALCAM in patients with IBD promotes CD4 + T-cell proliferation and differentiation into Th1 and Th17 cells, exacerbating intestinal mucosal inflammation in IBD32. MXRA8, a member of the transmembrane immunoglobulin superfamily, is specifically localized to the glial limitans and negatively associated with abdominal obesity in the elderly33. Some studies have reported elevated expression of CXCL9 in the serum of patients with CD34,35. A nested case-cohort study, including 72 UC cases and 140 controls, combined with MR analysis found that CXCL9 was causally associated with UC16. In a mouse model, high-dose dexamethasone treatment, which inhibits tumor progression, was found to significantly downregulate CXCL9 expression, a protein associated with immune responses36. Evidence from observational research, MR analysis, and Bayesian colocalization highlights CXCL9 as a potential drug target for CD. An animal study indicated that CCN3-induced chemokine secretion promotes immune cell recruitment and inhibits the M2 macrophage phenotype37. In addition, elevated serum CCN3 levels have been observed in patients with type 2 diabetes38. RSPO3 has been shown in mouse experiments to rapidly cause intestinal cancer by concomitantly stimulating stem cells and supportive niche cells while enhancing Wnt signaling pathway39. The combination of RSPO3 inhibition and paclitaxel treatment has been effective in targeting colorectal and other intestinal cancers40.
Among the 14 tier 2 proteins identified in this study (CCL20, TNFRSF9, IL1RL1, IL19, CCL7, CCL13, LY9, CD72, EPHB4, SEPTIN8, IL10RA, IGLC2, ITGAV, and NOS3), none have been developed as drug targets for IBD, marking them as potential new targets for IBD treatment. However, several proteins (CCL2041,42, IL1RL143,44,45,46,47,48, IL1949, CCL750, CCL1351, LY952, EPHB453,54, IL10RA55, ITGAV11, and NOS356) have been associated with IBD through genetic polymorphisms, mutations, mRNA expression, and protein expression in previous studies. Although CD7257, IGLC258, SEPTIN859, and TNFRSF960 lack direct evidence linking them to IBD, they are implicated in other immune system diseases. For instance, CCL20, a high-affinity ligand for CCR6, drives CCR6+ cell migration to inflamed tissues. Blocking CCR6-CCL20 interactions using antibodies or antagonists has been reported to prevent CCR6+ cell migration, reducing IBD severity41,42. The expression of IL1RL1, a receptor for interleukin-33, is upregulated in active UC compared to controls, and anti-TNF therapy modulates IL1RL1 levels, suggesting its potential as a therapeutic target for IBD45. Similarly, a cross-sectional study reported elevated abundance of IL19-producing cells in patients with active CD compared to active UC and non-inflammatory tissues49. Gene therapy targeting IL19 could improve mucosal delivery and treatment outcomes in intestinal diseases61. Chemokines, including CCL750 and CCL1351, have been associated with immune system diseases and appear to contribute to IBD by promoting adhesion molecule expression in epithelial cells and recruiting innate immune cells to inflamed sites62,63. LY9 is elevated in patients with untreated IBD and suppressed by corticosteroids52. Tesevatinib, an inhibitor of EPHB4, has shown promise in improving therapeutic outcomes in IBD mouse models by modulating immune responses and intestinal epithelial integrity53,54. Rare heterozygous missense variants in IL10RA, identified via exome sequencing, have been linked to early-onset IBD55. A case-control study reported lower serum ITGAV levels in patients with IBD and UC compared to HC11. NOS3 is implicated in IBD pathophysiology through its regulation of vascular homeostasis and immune responses56.
In terms of clinical relevance, for the tier 1 protein CD6, our cohort studies and MR analysis suggested that elevated plasma CD6 levels increase the risk of developing IBD. Moreover, CD6 exhibited consistent positive correlations between its effects in MR analyses at the transcriptomic level across most tissues and its corresponding effects in the plasma proteome. Single-cell expression analysis revealed that CD6 is predominantly enriched in colonic mucosal T cells, with higher expression levels in patients with IBD compared to HC. Interestingly, CD6 is also targeted by itolizumab, a drug used for treating other immune diseases. Itolizumab selectively targets the CD6–ALCAM pathway, reducing CD6 expression, inhibiting T-cell proliferation, and suppressing proinflammatory cytokine production64. Randomized clinical trials involving patients with psoriasis have established the efficacy and safety of this drug65. Thus, repurposing itolizumab for IBD treatment warrants further investigation. Furthermore, CD6 interacts with targets of current IBD drugs, including IFNG, the target of the aminosalicylate drug olsalazine, which may be involved in adaptive immune responses; ICAM1 and FCGR3B, targets of the integrin inhibitor natalizumab, likely contributing to cell adhesion; and TYK2, the target of the JAK inhibitor tofacitinib, potentially playing a role in T-cell proliferation regulation, thereby contributing to in the pathogenesis of IBD pathogenesis. MXRA8 exhibited consistent negative correlations between its effects at the transcriptomic level in MR analyses across tissues, such as the colon, and its effects in the plasma proteome. CCN3 is associated with NFKB1, a target of the aminosalicylate drug sulfasalazine, and may play a role be involved in inflammatory responses and signaling pathways. CXCL9 is enriched in macrophages within the colonic mucosa. CXCL9 expression levels in macrophages from patients with IBD are higher than in those in macrophages from HC. This observation may be explained by the excessive secretion of the pro-inflammatory factor CXCL9 by M1 macrophages, which contributes to inflammation and functional impairment of the intestinal mucosa66. CXCL9 is related to several drug targets, including NFKB1 and PTGS2 (targets of the aminosalicylate drug sulfasalazine); ICAM1, FCGR1A, and FCGR3B (targets of the integrin inhibitor natalizumab); JAK1 and JAK2 (targets of JAK inhibitors); and TNF (target of TNF inhibitors), and may be collectively involved in cellular responses to lipopolysaccharides, immune and inflammatory responses, and pathways such as Toll-like receptor and chemokine signaling. Our cohort studies and MR analyses indicated that elevated plasma levels of RSPO3 are associated with an increased risk of IBD. RSPO3 overexpression may result in hyperactivation of the Wnt/β-catenin signaling pathway, heightening the risk of intestinal inflammation and cancer development67. RSPO3 is a target of rosmantuzumab, which is for colorectal cancer treatment. Administering rosmantuzumab may alleviate inflammatory responses in patients with IBD by reducing RSPO3 levels and inhibiting overactivation of the Wnt signaling pathway.
The strengths of this study include assessing a substantial number of proteomic biomarkers and integrating a large prospective cohort study with MR analyses to validate causal associations between proteins and IBD, as well as its subtypes. However, several limitations should be noted. First, while we utilized one of the most extensive proteomic GWAS resources available, some proteins lack suitable instrumental variables, limiting the exploration of their causal associations and potential therapeutic value. Second, multiple testing corrections were performed using FDR, a less conservative approach than Bonferroni correction, which might increase the risk of false positives. Third, the study cohort, derived from the UK Biobank, predominantly comprises individuals of European ancestry who are middle-aged or older, potentially limiting the generalizability of our findings to other populations. Finally, although extensive mechanistic investigations were conducted on candidate proteins, further clinical trials are required to evaluate the efficacy and safety of these potential drug targets.
Through comprehensive analyses integrating observational and genetic approaches in large-scale proteomics studies, we identified 22 potential protein targets for IBD drug therapy. Among these, eight were prioritized as primary candidate proteins and the remaining 14 were classified as secondary candidate proteins for IBD treatment. Among primary candidate proteins, IL12B, IFNG, and IL18 have either been approved for IBD treatment or are undergoing clinical trials. Notably, CD6, MXRA8, CXCL9, CCN3, and RSPO3 represent new potential drug targets. Collectively, our findings offer novel insights into targeted therapeutics of IBD, underscoring the potential of proteomics-driven precision medicine in advancing IBD management.
Methods
A prospective cohort study in the UK biobank
Study design and population
This prospective cohort study was based on the UK Biobank68. In brief, 500,000 participants aged 40–69 years were enrolled from 22 assessment centers across England, Scotland, and Wales between 2006 and 2010, with multiple follow-ups. At baseline, participants provided sociodemographic, lifestyle, and health information, while blood samples were collected for genotyping and biochemical analyses. The UK Biobank study received ethical approval from the North West Multicenter Research Ethical Committee. All participants provided written informed consent. This research was conducted under UK Biobank application number 104277.
Proteomics assays
During the period from 2006 to 2010, participants completed the collection of baseline blood samples at the time of recruitment. For each participant, blood samples were collected in EDTA tubes, subsequently separated into plasma and stored at −80 °C. Between 2021 and 2022, the UKB-PPP consortium utilized the Olink™ Explore 3072 proximity extension assay for proteomics profiling analysis on baseline-collected plasma samples (between 2006 and 2010) from approximately 54,000 UK Biobank participants14,19,20. This analysis measured a total of 2941 proteins across eight panels, identifying 2923 unique proteins. The protein data processing involved several steps to ensure high data quality. The protein data analysis involves sample selection, data import excluding Olink controls, exclusion of individuals who withdrew consent or had unprocessed data, detection and removal of outlier samples, elimination of data with quality control or assay warnings, and identification followed by removal of probable sample swaps, as detailed in previous studies14,19,20. Additionally, extensive quality control measures and protein concentration standardization were performed, resulting in the generation of normalized protein expression (NPX) values for each protein per participant, NPX being Olink’s relative protein quantification unit on a log2 scale14,19,20.
Outcome ascertainment
Diagnostic cases were identified through linkage to various health-related records, including hospital inpatient data, death registry records, primary care data, and self-reported information in the UK Biobank. Patients with IBD were identified using the 10th edition of the International Classification of Diseases (ICD-10) codes K50 and K51, ICD-9 codes 555 and 556, and other codes that map specifically to self-report and primary care data (code details in Supplementary Table 7). Participants were followed until the date of incident IBD, death, loss to follow-up, or the censoring dates for Hospital Episode Statistics (i.e., Hospital Episode Statistics for England: October 31, 2022; Scottish Morbidity Records for Scotland: August 31, 2022; and Patient Episode Database for Wales: May 31, 2022), whichever occurred first. The date of the first record indicating IBD during the follow-up period was used to define the date of incident IBD in this study.
Covariate definitions
A series of demographic, socioeconomic, and behavioral factors were included as covariates in the analysis: age, sex, assessment center, years of education, employment, household income, TDI, smoking status, alcohol consumption, PA, healthy diet, sleep duration, and BMI. Years of education were categorized as ≤ 10, 11–15, or > 15 years. Employment status was categorized as employed or unemployed. Household income was categorized into four groups: <£18,000, £18,000–£30,999, £31,000–£51,999, and >£52,000. TDI scores were divided into quartiles as categorical variables. Smoking status was categorized as current, former, or never. Alcohol consumption was categorized as never, occasional, 1–2 times per week, 3–4 times per week, or daily. PA was assessed using the International Physical Activity Questionnaire and categorized as high, moderate, or low. A healthy diet, as defined by the American Heart Association, was categorized as high, moderate, or low. Sleep duration was categorized as < 7, 7.0–8.0, or ≥ 9 h/day. BMI was categorized according to WHO criteria: < 18.5 kg/m², 18.5–24.9 kg/m², 25.0–29.9 kg/m², and ≥ 30 kg/m². Missing covariate values were addressed with multiple imputation by chained equations.
Statistical analysis
Baseline proteomics data were available for 53,022 participants, focusing on 49,443 participants of European ancestry. After excluding individuals with a history of IBD at baseline (N = 643), the final analysis comprised 48,800 individuals. We normalized individual protein levels for subsequent analyses. Baseline characteristics of the included participants were described using means (standard deviations) for continuous variables and counts (proportions) for categorical variables.
Cox proportional hazards regression models were used to estimate the associations between protein levels and incident IBD and its subtypes to calculate HR and 95% CI. Model 1 was unadjusted for covariates. Model 2 was initially adjusted for age, sex, assessment center, years of education, employment status, household income, and TDI. Model 3 was fully adjusted for smoking status, alcohol consumption, PA, healthy diet, sleep duration, and BMI. Furthermore, we performed sensitivity analyses. First, Cox analysis was performed after excluding samples with values below the limit of detection. Second, additional adjustments were made for the first 10 genetic principal components in the Cox analysis. Third, we excluded participants with a baseline history of other immune-mediated diseases (code details in Supplementary Table 7). Fourth, additional adjustments were made for baseline histories of major chronic conditions, including hypertension, diabetes, hyperlipidemia, cardiovascular diseases, cancer, and other immune-mediated diseases (code details in Supplementary Table 7), in the Cox analysis. Finally, the association between protein levels and the risk of developing IBD was analyzed based on the time of diagnosis (diagnosed within < 2, 2–6, and > 6 years of follow-up).
To develop protein-based prediction models for IBD and its subtypes, the dataset was first randomly split into an 80% training subset and a 20% testing subset. The significant proteins identified by Cox proportional hazards regression in 80% training subset were input into the training subset’s eXtreme Gradient Boosting (XGBoost) classifier69. The importance of these proteins was visualized using Shapley Additive Explanations (SHAP) values, and a simplified protein prediction model was constructed using the top 10 most important proteins to facilitate clinical application70. The testing dataset was then used to assess and compare the AUC values of the traditional risk factor model, simplified protein prediction model, and combined model.
Multiple testing corrections were applied using the FDR method with the “fdrtool” package, considering the number of proteins analyzed71. A threshold of FDR < 0.05 was used to determine statistical significance. This approach controls for the risk of false positives and ensures a more reliable, stringent interpretation of results.
Genetic analysis and mechanism exploration
GWAS sources for protein instrumental variables
Protein instrumental variables for the primary analysis were sourced from the GWAS of the UKB-PPP14,19,20. Cross-platform replication MR analyses used instrumental variables from the GWAS of the deCODE23 and Fenland24 studies. Independent SNPs significantly associated with plasma protein expression levels at a genome-wide significance threshold (cis-pQTLs P < 5E − 8) were selected as instrumental variables. cis-pQTLs were defined as SNPs within 1 Mb of the gene encoding the protein. Independence was determined using the 1000 Genomes European panel by excluding LD (r2 < 0.001, clumping window > 10,000 kb). GWAS data for 2,940 proteins from 34,557 participants of European ancestry, measured on the Olink platform in the UKB-PPP, were obtained14,19,20. Subsequently, via independent and significant cis-SNPs for each protein, we identified 1976 proteins with cis-pQTLs suitable for subsequent two-sample MR analyses. Cross-platform replication MR analyses used proteins GWAS data from the deCODE study, involving 4,907 aptamers and 35,559 Icelandic individuals measured using the SomaScan platform23, and the Fenland study, involving 4,775 aptamers and 10,708 European participants, also measured using the SomaScan platform24.
GWAS sources for IBD and its subtypes
Summary-level GWAS data for IBD and its subtypes (CD and UC) were obtained from the IIBDGC and the FinnGen study. The IIBDGC dataset included 12,882 individuals with IBD (5,956 CD and 6,968 UC cases), all of European ancestry, confirmed through radiologic, endoscopic, and histopathologic evaluations21. The FinnGen dataset included 7,625 IBD cases (1,665 CD and 5,034 UC cases) diagnosed using ICD codes. There was no overlap between these data sources22.
Mendelian randomization analysis
We performed a two-sample MR analysis using cis-pQTLs derived from the UKB-PPP to evaluate associations between proteins and the risk of IBD and its subtypes from IIBDGC and the FinnGen study. For cross-platform replication MR analyses, we utilized cis-pQTLs from the deCODE and Fenland studies. When only one pQTL was available for a protein, the Wald ratio method was applied. For proteins with two or more genetic instruments, a fixed or random-effects inverse-variance weighting model was applied based on the presence of significant heterogeneity72. Horizontal pleiotropy was assessed using the MR-Egger intercept test73. The MR results for each protein across two outcome sources were combined via meta-analyses. MR analyses were performed using TwoSampleMR and MendelianRandomization packages in R.
To investigate reverse causality, we conducted sensitivity analyses using reverse MR. In this analysis, IBD and its subtypes were treated as exposures, and instrumental variables were selected using the same criteria as in the forward MR analysis. Given the higher explanatory power of instrumental variables from the IIBDGC21 compared to FinnGen22, summary-level data from the IIBDGC were used as the source for exposure to instrumental variables. Plasma protein GWAS data from the UKB-PPP14,19,20, deCODE23, and Fenland24 studies were used as outcomes. The inverse-variance weighting method assessed the reverse causal effects of IBD and its subtypes on each protein across the three sources, and meta-analyses combined the results, yielding overall estimates of reverse causal associations between IBD, its subtypes, and each protein. Additionally, Steiger filtering was employed to verify the directionality of associations between proteins and IBD, with P < 0.05 indicating that the effect direction is from exposure to an outcome. Specifically, Steiger filtering was used to remove proteins where SNPs explained a greater proportion of variation in the outcome than variation in the exposure74.
Colocalization analysis
To avoid the signals discovered by MR might arising from LD, we performed Bayesian colocalization analysis on the proteins that showed significant associations with IBD in both the cohort and MR analyses. Bayesian colocalization evaluates the probability that two traits share the same causal variant rather than the variant being shared coincidentally due to correlation through LD75. This analysis is crucial for strengthening the evidence of causal relationships between candidate proteins and IBD identified through MR analyses. The default parameters of the colocalization method were used to calculate posterior probabilities for five hypotheses: H0, a genetic variant in the region is not associated with either traits; H1, a genetic variant in the region is associated with Trait 1 (pQTL) but not Trait 2 (IBD GWAS); H2, a genetic variant in the region is associated with Trait 2 (IBD GWAS) but not Trait 1 (pQTL); H3, a genetic variant in the region is associated with both Trait 1 (pQTL) and Trait 2 (IBD GWAS) but with different causal variants; H4, a genetic variant in the region is associated with both Trait 1 (pQTL) and Trait 2 (IBD GWAS) and with the same causal variant. We defined proteins with a posterior probability of Hypothesis 4 (PPH4) ≥ 0.6 as high-support evidence of colocalization. This indicated that the two association signals were consistent with a shared causal variant, rather than being confounded by LD. In contrast, proteins with a posterior probability for PPH4 < 0.6 were considered as having low-support evidence for colocalization8.
Transcriptome analysis of protein-coding genes across different tissues
We proposed eQTL instruments using data from GTEx v.8 by performing conditional analysis on normalized gene expression in European ancestry individuals across 49 tissues, each with at least 70 samples76. To increase the coverage of protein-coding gene instruments across various tissues and to explore the tissue-specific associations and potential off-target effects of drug-target genes, we relaxed the significance threshold (P < 5E-5) within 1 Mb on either side of the encoded gene. Additionally, we performed LD clumping (r2 < 0.001, kb > 10,000) to identify cis-eQTLs for each encoded gene across different tissues.
Single cell-type expression analysis
To evaluate cell type-specific expression of target genes that have causal effects on IBD at the plasma protein level, we analyzed a previously published human colonic mucosa single-cell RNA-seq dataset (GSE214695) comprising 12 IBD cases and six HC25. Using the “Seurat” package77, we first performed data preprocessing and transformation based on the raw single-cell RNA-seq data. Genes with fewer than three counts in one cell and cells with fewer than 500 unique feature counts were removed. The NormalizeData and ScaleData functions were then used to normalize and scale the RNA transcripts per million. Subsequently, principal component analysis, t-distributed stochastic neighbor embedding, and graph-based clustering were applied to the merged Seurat object. Cell types were annotated using the “SingleR” package78. In total, expression profiles of 22,938 genes across 30,915 cells were included in the analysis. Differential expression analysis using the Wilcoxon rank-sum test was performed to compare gene expression levels between a specific cell type and other cell types, aiming to determine whether the identified IBD causal protein-coding genes were highly expressed in a particular cell type within the colonic mucosa. Furthermore, we conducted differential expression analysis using the Wilcoxon rank-sum test to compare the expression levels of candidate protein-coding genes in the specific enriched cell types between IBD cases and HC. Specific enriched cell types of the gene were identified based on an average |log2FC| greater than 0.25 and an FDR-adjusted P value of less than 0.05. This criterion was also applied to identify differentially expressed genes in the IBD and HC groups within the specifically enriched cell types.
PPI network, enrichment analysis, and druggability assessment
First, we performed PPI network analyses using the Search Tool for the Retrieval of Interacting Genes database to investigate interactions among the candidate proteins as well as between these proteins and the targets of approved IBD drugs. In addition, we randomly sampled 22 proteins (the number of candidate proteins identified in this study) from the 2923 proteins 1000 times. For each random selection, we calculated the interaction rates between the 22 randomly selected proteins and the interaction rates between the 22 randomly selected proteins and known IBD drug targets, and compared them with the interaction rates between our identified candidate proteins and the interaction rates between the candidate proteins and known IBD drug targets. Second, we used the Database for Annotation, Visualization and Integrated Discovery79 to perform gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses, characterizing biological processes and pathways enriched for the associated proteins. Third, we queried KO mouse models using the Mouse Genomics Informatics resource to evaluate associated phenotypes for the candidate proteins. In addition, we randomly sampled 22 proteins from the 2923 proteins 1000 times. For each random selection, we calculated the proportion of the 22 randomly selected proteins with evidence from KO mouse models of immune system/digestive/alimentary phenotype and compared it with the evidence from KO mouse models for the 22 identified candidate proteins. Fourth, to assess the druggability of the candidate proteins, we searched them in the DrugBank database80, the Open Targets database81 and the Therapeutic Target Database82.
Detailed descriptions of GWAS sources of protein, GWAS sources of IBD and its subtypes, prospective cohort study, and genetic analysis methods are provided in Supplementary Text 1–4.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Researchers can apply to use the UK Biobank resource for health-related research that is in the public interest (https://www.ukbiobank.ac.uk/enable-your-research/register). This research has been conducted using the UK Biobank application number 104277. The pQTL data were sourced from the UK Biobank Pharma Proteomics Project (UKB-PPP) (https://www.synapse.org/Synapse:syn51364943), the deCODE Health study (https://www.decode.com/summarydata/), and the Fenland study (https://www.omicscience.org/apps/pgwas). GWAS summary statistics for IBD and its subtypes can be accessed via the IIBDGC consortium (https://www.ibdgenetics.org/) and the FinnGen R9 study (https://www.finngen.fi/fi). Tissue-specific eQTL data were retrieved from the GTEx database (https://www.gtexportal.org/). Single-cell RNA-seq data were retrieved from colonic mucosa in IBD were obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE214695). Interacting Genes (STRING) database (https://cn.string-db.org/). Database for Annotation, Visualization and Integrated Discovery (DAVID, https://davidbioinformatics.nih.gov/). DrugBank database (https://go.drugbank.com). OpenTargets database (https://platform.opentargets.org/). Therapeutic Target Database (https://db.idrblab.net/ttd/). When using the dataset, authors should cite the original publication. Source data are provided with this paper.
Code availability
The code used in this study is openly available at: https://github.com/data-analysis-code/ProteomicCode. Data processing was completed using R software (version 4.1.0) and Python (version 3.10.0), with packages TwoSampleMR (version 0.5.8), MendelianRandomization (version 0.10.0), MRPRESSO (version 1.0), coloc (version 5.2.3), metafor (version 4.6-0), glmnet (version 4.1-8), fdrtool (version 1.2.17), dplyr (version 1.1.4), readr (version 2.1.5), stringr (version 1.5.1), tidyverse (version 2.0.0), forestplot (version 3.1.3), plyr (version 1.8.9), devtools (version 2.4.5),and remotes (version 2.5.0) in R, as well as numpy (version 1.26.4), pandas (version 2.2.2), lifelines (version 0.28.0), statsmodels (version 0.14.2), mne (version 1.7.0), shap (version 0.45.1), and xgboost (version 2.1.0) in Python.
References
Ng, S. C. et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet (Lond., Engl.) 390, 2769–2778 (2017).
Kaplan, G. G. The global burden of IBD: from 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 12, 720–727 (2015).
Danese, S., Sans, M. & Fiocchi, C. Inflammatory bowel disease: the role of environmental factors. Autoimmun. Rev. 3, 394–400 (2004).
Danese, S., Solitano, V., Jairath, V. & Peyrin-Biroulet, L. The future of drug development for inflammatory bowel disease: the need to ACT (advanced combination treatment). Gut 71, 2380–2387 (2022).
Mazidi, M. et al. Plasma Proteomics to Identify Drug Targets for Ischemic Heart Disease. J. Am. Coll. Cardiol. 82, 1906–1920 (2023).
Walker, K. A. et al. Large-scale plasma proteomic analysis identifies proteins and pathways associated with dementia risk. Nat. aging 1, 473–489 (2021).
Henry, A. et al. Therapeutic Targets for Heart Failure Identified Using Proteomics and Mendelian Randomization. Circulation 145, 1205–1217 (2022).
Yao, P. et al. Proteomic Analyses in Diverse Populations Improved Risk Prediction and Identified New Drug Targets for Type 2 Diabetes. Diab. Care 47, 1012–1019 (2024).
Iskandar, H. N. & Ciorba, M. A. Biomarkers in inflammatory bowel disease: current practices and recent advances. Transl. Res.: J. Lab. Clin. Med. 159, 313–325 (2012).
Starr, A. E. et al. Proteomic analysis of ascending colon biopsies from a paediatric inflammatory bowel disease inception cohort identifies protein biomarkers that differentiate Crohn’s disease from UC. Gut 66, 1573–1583 (2017).
Kalla, R. et al. Serum proteomic profiling at diagnosis predicts clinical course, and need for intensification of treatment in inflammatory bowel disease. J. Crohns Colitis 15, 699–708 (2021).
Deutschmann, C. et al. Identification of Chitinase-3-Like Protein 1 as a Novel Neutrophil Antigenic Target in Crohn’s Disease. J. Crohns Colitis 13, 894–904 (2019).
Lewis, J. D. The utility of biomarkers in the diagnosis and therapy of inflammatory bowel disease. Gastroenterology 140, 1817–1826.e1812 (2011).
Eldjarn, G. H. et al. Large-scale plasma proteomics comparisons through genetics and disease associations. Nature 622, 348–358 (2023).
Torres, J. et al. Serum Biomarkers Identify Patients Who Will Develop Inflammatory Bowel Diseases Up to 5 Years Before Diagnosis. Gastroenterology 159, 96–104 (2020).
Chen, J. et al. Bidirectional Mendelian Randomisation Analysis Provides Evidence for the Causal Involvement of Dysregulation of CXCL9, CCL11 and CASP8 in the Pathogenesis of Ulcerative Colitis. J. Crohns Colitis 17, 777–785 (2023).
Smith, G. D. & Ebrahim, S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J. Epidemiol. 32, 1–22 (2003).
Zhou, S. et al. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat. Med 27, 659–667 (2021).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 622, 329–338 (2023).
Dhindsa, R. S. et al. Rare variant associations with plasma protein levels in the UK Biobank. Nature 622, 339–347 (2023).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet 47, 979–986 (2015).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet 53, 1712–1721 (2021).
Pietzner, M. et al. Mapping the proteo-genomic convergence of human diseases. Science. (N.Y., NY) 374, eabj1541 (2021).
Garrido-Trigo, A. et al. Macrophage and neutrophil heterogeneity at single-cell spatial resolution in human inflammatory bowel disease. Nat. Commun. 14, 4506 (2023).
Benson, J. M. et al. Discovery and mechanism of ustekinumab: a human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. mAbs 3, 535–545 (2011).
Sands, B. E. et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med 381, 1201–1214 (2019).
Egan, L. J. & Sandborn, W. J. Inhibition of nuclear factor kappaB by sulfasalazine: a new target for inflammatory bowel disease therapy? Gastroenterology 115, 1295–1296 (1998).
Ito, R. et al. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin. Exp. Immunol. 146, 330–338 (2006).
GlaxoSmithKline. A. Single-blind, Randomised, Placebo-controlled Study to Investigate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single Doses of Intravenously Infused GSK1070806 in Healthy and Obese Subjects.: clinicaltrials.gov, 2017.
Birmingham, U. O. A Phase Ib/IIb, Randomised, Double Blind, Placebo-Controlled Trial to Investigate the Safety, Tolerability and Clinical Activity of Humanised Antibody GSK1070806 in the Treatment of Patients With Moderate-to-Severe Crohn’s Disease: clinicaltrials.gov, 2021.
Ma, C. et al. Critical Role of CD6highCD4+ T Cells in Driving Th1/Th17 Cell Immune Responses and Mucosal Inflammation in IBD. J. Crohns Colitis 13, 510–524 (2019).
Sathyan, S. et al. Plasma proteomic profile of abdominal obesity in older adults. Obes. (Silver Spring) 32, 938–948 (2024).
Zhong, W. M., Qian, X. H. & Jin, Z. W. Identification of potential predictive biomarkers and biological pathways and the correction with immune infiltration in the activation of Crohn’s disease. Immunogenetics 74, 527–537 (2022).
Boucher, G. et al. Serum Analyte Profiles Associated With Crohn’s Disease and Disease Location. Inflamm. Bowel Dis. 28, 9–20 (2022).
Xu, L. et al. High-Dose Dexamethasone Manipulates the Tumor Microenvironment and Internal Metabolic Pathways in Anti-Tumor Progression. Int J. Mol. Sci. 21, 1846 (2020).
Martinerie, C. et al. NOV/CCN3: A New Adipocytokine Involved in Obesity-Associated Insulin Resistance. Diabetes 65, 2502–2515 (2016).
Li, J. Y. et al. Serum CCN3 levels are increased in type 2 diabetes mellitus and associated with obesity, insulin resistance and inflammation. Clin. Chim. acta; Int. J. Clin. Chem. 494, 52–57 (2019).
Hilkens, J. et al. RSPO3 expands intestinal stem cell and niche compartments and drives tumorigenesis. Gut 66, 1095–1105 (2017).
Fischer, M. M. et al. RSPO3 antagonism inhibits growth and tumorigenicity in colorectal tumors harboring common Wnt pathway mutations. Sci. Rep. 7, 15270 (2017).
Allodi, M. et al. Probing the effects of MR120 in preclinical chronic colitis: A first-in-class anti-IBD agent targeting the CCL20/CCR6 axis. Eur. J. Pharmacol. 945, 175613 (2023).
Meitei, H. T., Jadhav, N. & Lal, G. CCR6-CCL20 axis as a therapeutic target for autoimmune diseases. Autoimmun. Rev. 20, 102846 (2021).
Seidelin, J. B. et al. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol. Lett. 128, 80–85 (2010).
Beltrán, C. J. et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 16, 1097–1107 (2010).
Pastorelli, L. et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc. Natl Acad. Sci. USA 107, 8017–8022 (2010).
Sponheim, J. et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am. J. Pathol. 177, 2804–2815 (2010).
Kobori, A. et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J. Gastroenterol. 45, 999–1007 (2010).
Latiano, A. et al. Associations between genetic polymorphisms in IL-33, IL1R1 and risk for inflammatory bowel disease. PLoS One 8, e62144 (2013).
Fonseca-Camarillo, G., Furuzawa-Carballeda, J., Granados, J. & Yamamoto-Furusho, J. K. Expression of interleukin (IL)-19 and IL-24 in inflammatory bowel disease patients: a cross-sectional study. Clin. Exp. Immunol. 177, 64–75 (2014).
Gaujoux, R. et al. Cell-centred meta-analysis reveals baseline predictors of anti-TNFα non-response in biopsy and blood of patients with IBD. Gut 68, 604–614 (2019).
Zwicker, S., Lira-Junior, R., Höög, C., Almer, S. & Boström, E. A. Systemic Chemokine Levels with “Gut-Specific” Vedolizumab in Patients with Inflammatory Bowel Disease-A Pilot Study. Int J. Mol. Sci. 18, 1827 (2017).
Hathout, Y. et al. Serum pharmacodynamic biomarkers for chronic corticosteroid treatment of children. Sci. Rep. 6, 31727 (2016).
Pfaff, D. et al. Involvement of endothelial ephrin-B2 in adhesion and transmigration of EphB-receptor-expressing monocytes. J. cell Sci. 121, 3842–3850 (2008).
Grandi, A. et al. Targeting the Eph/Ephrin System as Anti-Inflammatory Strategy in IBD. Front. Pharmacol. 10, 691 (2019).
Kelsen, J. R. et al. Exome sequencing analysis reveals variants in primary immunodeficiency genes in patients with very early onset inflammatory bowel disease. Gastroenterology 149, 1415–1424 (2015).
Kanwar, J. R., Kanwar, R. K., Burrow, H. & Baratchi, S. Recent advances on the roles of NO in cancer and chronic inflammatory disorders. Curr. Medicinal Chem. 16, 2373–2394 (2009).
Eiza, N., Sabag, A., Kessler, O., Toubi, E. & Vadasz, Z. Soluble CD72, is a T-cell activator probably via binding to CD6 in homeostasis and autoimmunity. Front Immunol. 15, 1367120 (2024).
Wang, W. et al. Multiregion single-cell sequencing reveals the transcriptional landscape of the immune microenvironment of colorectal cancer. Clin. Transl. Med. 11, e253 (2021).
Ridruechai, C. et al. Association analysis of susceptibility candidate region on chromosome 5q31 for tuberculosis. Genes Immun. 11, 416–422 (2010).
Shen, X. et al. 4-1BB Targeting Immunotherapy: Mechanism, Antibodies, and Chimeric Antigen Receptor T. Cancer Biother. radiopharmaceuticals 38, 431–444 (2023).
Li, M. C. et al. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World J. Gastroenterol. 10, 620–625 (2004).
Li, L. et al. CCL13 and human diseases. Front Immunol. 14, 1176639 (2023).
Wang, H. G. et al. Cepharanthine ameliorates dextran sulphate sodium-induced colitis through modulating gut microbiota. Microb. Biotechnol. 15, 2208–2222 (2022).
Rambaldi, B. et al. Phenotypic and functional characterization of the CD6-ALCAM T-cell co-stimulatory pathway after allogeneic cell transplantation. Haematologica 107, 2617–2629 (2022).
Lincheta, L. F. et al. Long-term therapy with itolizumab is safe and effective for patients with moderate to severe psoriasis: Results from an expanded-access program. Int Immunopharmacol. 134, 112225 (2024).
Uzzan, M. et al. Ulcerative colitis is characterized by a plasmablast-skewed humoral response associated with disease activity. Nat. Med 28, 766–779 (2022).
Hao, J. et al. Dysregulation of Wnt/β-catenin signaling contributes to intestinal inflammation through regulation of group 3 innate lymphoid cells. Nat. Commun. 15, 2820 (2024).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779 (2015).
Chen, T. & Guestrin, C. XGBoost: A Scalable Tree Boosting System. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. San Francisco, California, USA: Association for Computing Machinery, 2016:785–794.
Lundberg, S. M. et al. From Local Explanations to Global Understanding with Explainable AI for Trees. Nat. Mach. Intell. 2, 56–67 (2020).
Strimmer, K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24, 1461–1462 (2008).
Burgess, S., Bowden, J., Fall, T., Ingelsson, E. & Thompson, S. G. Sensitivity Analyses for Robust Causal Inference from Mendelian Randomization Analyses with Multiple Genetic Variants. Epidemiol. (Camb., Mass) 28, 30–42 (2017).
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 32, 377–389 (2017).
Hemani, G., Tilling, K. & Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet 13, e1007081 (2017).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383 (2014).
The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
Huang da, W., Sherman, B. T., & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–d1082 (2018).
Ochoa, D. et al. Open Targets Platform: supporting systematic drug-target identification and prioritisation. Nucleic Acids Res. 49, D1302–d1310 (2021).
Zhou, Y. et al. TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 52, D1465–d1477 (2024).
Acknowledgements
We thank all participants and investigators of the UK Biobank, the Inflammatory Bowel Disease Genetics Consortium, the FinnGen study, and Sun et al. GWAS analysis on proteome from UKB-PPP, Ferkingstad et al. GWAS analysis on proteome from deCODE Health study, and Pietzner. et al. GWAS analysis on proteome from Fenland study. This study was supported by the National Natural Science Foundation of China (grant numbers: 82173591 (M.L.), 82103912 (X.R.Y.) and 82204129 (T.C.Z.)), the Taishan Scholars Program of Shandong Province (tstp20230654 (M.L.) and tsqn202312328 (X.R.Y.)), the Excellent Youth Innovation Team of Shandong Provincial Higher Education Institutions (2022KJ012 (X.R.Y.)).
Author information
Authors and Affiliations
Contributions
M.L., X.Y., T.Z., and X.Z. designed and supervised the study. X.Z. participated in the data curation. X.Z. and H.Z. performed the data analyzes and prepared the tables and figures. H.Z., M.W. and J.M. contributed to suggestions on analysis strategy and findings interpretation. X.Z. wrote the original draft. M.L., X.Y., T.Z., and X.Z. contributed to project administration. All authors critically revised the content and contributed to editing the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Michel Georges, Urko Martinez Marigorta, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., Zhao, H., Wan, M. et al. Associations of 2923 plasma proteins with incident inflammatory bowel disease in a prospective cohort study and genetic analysis. Nat Commun 16, 2813 (2025). https://doi.org/10.1038/s41467-025-57879-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57879-3
This article is cited by
-
Potential drug targets for chronic obstructive pulmonary disease in the plasma proteome identified through Mendelian randomization
BMC Pulmonary Medicine (2025)
-
Associations between carotid artery intima-media thickness, traditional risk factors and proteins
npj Cardiovascular Health (2025)
