
Abstract
Atopic dermatitis is a highly heritable and common inflammatory skin condition affecting children and adults worldwide. Multi-ancestry approaches to atopic dermatitis genetic association studies are poised to boost power to detect genetic signal and identify loci contributing to atopic dermatitis risk. Here, we present a multi-ancestry GWAS meta-analysis of twelve atopic dermatitis cohorts from five ancestral populations totaling 56,146 cases and 602,280 controls. We report 101 genomic loci associated with atopic dermatitis, including 16 loci that have not been previously associated with atopic dermatitis or eczema. Fine-mapping, QTL colocalization, and cell-type enrichment analyses identified genes and cell types implicated in atopic dermatitis pathophysiology. Functional analyses in keratinocytes provide evidence for genes that could play a role in atopic dermatitis through epidermal barrier function. Our study provides insights into the etiology of atopic dermatitis by harnessing multiple genetic and functional approaches to unveil the mechanisms by which atopic dermatitis-associated variants impact genes and cell types.
Similar content being viewed by others

Introduction
Atopic dermatitis (AD) is one of the most common chronic conditions, affecting 15–20% of children and 5–10% of adults worldwide1,2. AD is characterized as a pruritic rash primarily on the flexural areas of the arms and legs, and can vary widely in severity and presentation3,4. Both genetic and environmental factors can predispose individuals to impaired epidermal barrier function and inflammation that, along with itch, drive the vicious cycle of AD5,6. Genetic studies published to date support a role for genetic defects in innate and adaptive immunity resulting in Th2 skewing as well as defects in skin barrier function, most notably loss-of-function variants in filaggrin (FLG)7,8. AD heritability has been estimated from twin studies to be 75–80%9,10,11, and there are currently more than 90 published GWAS loci that explain about 5–15% of heritability12,13,14, suggesting additional AD genetic loci are to be discovered.
As GWAS data in diverse populations becomes available, the human genetics field has moved toward multi-ancestry approaches. Multi-ancestry GWAS meta-analysis across diverse populations can increase the power to detect complex trait loci when the underlying causal variants are shared between ancestry groups. Our study is motivated by previous reports and our own experience meta-analyzing European cohorts and the desire to incorporate diverse populations of Asian and African ancestry. Moreover, we know that AD affects populations worldwide, and diverse populations can expand our understanding of the genetic architecture of AD.
Here, we report a large-scale multi-ancestry GWAS of 56,146 AD cases and 602,280 controls as well as ancestry-specific analyses. The cohorts include individuals of European (EUR), Asian (ASN), African (AFR), and American (AMR) ancestries. In total, we report 101 genome-wide significant loci associated with AD, including 16 loci that have not been previously reported as associated with AD or eczema. We characterize genetic factors for AD across multiple ancestral populations and perform fine-mapping and colocalization to identify putatively causal genes at genome-wide significant loci. Cell-type enrichment analysis identifies the disease-relevant cell types implicated by the GWAS signal, including keratinocytes, in which we perform functional experiments. Our findings highlight AD associated loci and link genes in these loci to cell types in key AD mechanistic nodes.
Results
Multi-ancestry and ancestry-specific GWAS meta-analyses identify novel AD loci
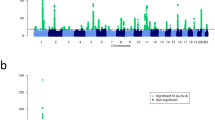
We performed GWAS meta-analysis on 12 cohorts comprising a total of 56,146 AD cases and 602,280 controls of European (EUR), Asian (ASN), African (AFR), American (AMR) and admixed ancestries (Fig. 1, Supplementary Data 1) to obtain ancestry-specific (EUR, EAS and AFR) and multi-ancestry (MULTI) GWAS summary statistics (Methods, Supplementary Data 2). Across all GWAS meta-analyses, we detected 101 genome-wide significant (P < 5e-08) non-overlapping loci (Supplementary Data 3), including 16 loci not reported as associated to AD or eczema (P < 5e−8) to date (Table 1, Fig. 2a); we will refer to these as novel. Of those, 7 loci have been identified as suggestive (P < =5e−6) in previous studies14,15,16 (Fig. 2b). Novel AD loci were identified in the two largest GWAS meta-analyses (MULTI and EUR) (Fig. 2a, Table 1). The 12 genome-wide significant loci identified in the ASN meta-analysis (Supplementary Fig. 1a) were located within the significant loci from the MULTI meta-analysis (Fig. 2b), however, comparison of the MULTI meta-analysis with and without the ASN cohorts confirms increased p-value significance for lead SNPs when the ASN samples are included (Supplementary Fig. 1b). No genome-wide significant loci at MAF > 0.01 were identified in the AFR GWAS meta-analysis despite the number of cases included (N = 7063); corresponding GWAS signal was seemingly well-controlled (λgc = 1.02) but underpowered (Supplementary Fig. 1, Supplementary Data 2), which may be due, among possible factors, to genetic admixture and differential environmental exposure17. To further assess the replicability of the 16 novel loci, we considered a lenient threshold (GWAS P < =5e−6) and identified 7/16 loci with suggestive signal across three studies14,15,16 (Table 1, Fig. 2b), six of which are reported in Budu-Aggrey et al.14. Compared to the GWAS meta-analyses generated herein, Budu-Aggrey included 16% (N = 65,107) more AD cases, but less diverse (30% less non-Europeans), also within European cohorts (85% less non-Finns). Among loci without suggestive signal, we identified rs541390276, a low-frequent variant (gnomAD 4.0 POPMAX MAF = 0.00049) with higher frequency in Finns (FinnGen R10 MAF = 0.0361). This variant has associations with asthma (FinnGen R10, P < 1.30e−7) and with dermatitis and eczema (FinnGen R10, P < 7.20e−10). Altogether, these observations indicate that, at least in part, the identified novel loci arise from more diverse – including within European ancestry level—cohorts utilized herein compared to other works.

Ancestry-stratified GWAS meta-analyses were utilized to identify and fine-map genome-wide significant GWAS loci for atopic dermatitis risk. GWAS signal was integrated with functional maps and evaluated for cell-type context-specific enrichment and QTL colocalization; genes were prioritized per GWAS locus. The expression of a subset of prioritized genes was functionally characterized in keratinocytes for differentiation, cell-subtype specificity, knock-out (KO) and Quantitative Trait Loci (QTL) signal. This figure contains illustrations from BioRender. Oliva, M. (2025) [https://BioRender.com/r58o311].

a Nominal p-values (y-axis) derived from the multiple ancestry-combined (MULTI) (top panel) and the European-ancestry stratified (EUR) (bottom panel) GWAS meta-analysis are plotted by corresponding genomic coordinate (x-axis). P-values are derived from a two-sided test for effect size in a fixed-effect inverse variance weighted approach (Methods). Novel associations are highlighted in green and annotated with the nearest gene. Associations that reached significance (P < 5e−08) in both MULTI and EUR GWAS meta-analyses are annotated in the top panel; associations that reached significance (P < 5e−08) in only the EUR GWAS meta-analysis are annotated in the bottom panel. b Top panel illustrates the presence of significant (P < 5e−08) AD GWAS loci (x-axis) across ancestry-stratified and -combined GWAS meta-analyses (y-axis); significant loci per ancestry endpoint are indicated with a darker color shade. P-values are derived from a two-sided test for effect size in a fixed-effect inverse variance weighted approach (Methods). Bottom panel illustrates overlap of significant AD GWAS loci with previously reported genome-wide significant (P < 5e−08) and suggestive (P < 5e−06) GWAS loci for AD/eczema, allergy, and asthma phenotypes (Supplementary Data 10, 11). Bold frame indicates the 16 novel AD GWAS loci reported herein, annotated with corresponding cytoband, lead variant and nearest gene. P-values are shown as reported in the EBI GWAS Catalog (Methods).
We quantified heritability—the proportion of phenotypic variance explained by genetics—in the European cohort to be 9.67% on a liability scale (Methods). This estimate agrees with previous reports11,13, but is substantially smaller than the 75–80% estimated heritability derived from twin studies9,10,11. This may be due to several factors, including contribution from genetic loci not captured by the European ancestry meta-analysis, rare variants with large effect sizes not captured by genotyping arrays, complex gene-gene or gene-environment interactions important to the genetic architecture of AD, or overestimation of twin heritability18.
To better understand the relationship between the AD-associated loci identified herein and previously published results on AD-related phenotypes, we assessed overlap with reported genomic associations for atopic march phenotypes other than AD: eczema, allergy, and asthma (Methods). Out of the 101 loci identified in our analysis, we observe that the majority (95/101) overlap significant or suggestive atopic-march associated loci (Fig. 2b), including 10 out of the 16 novel loci not previously reported to be associated with AD. The overlap supports the known shared genetic architecture of AD with other atopic-march phenotypes19,20 and pinpoints additional contributing loci. Of the 6 novel loci not previously reported in AD/eczema, allergy, or asthma GWAS, only the IL6ST locus - rs7731626 (5q11.2) - has reported disease associations in GWAS catalog, mainly with autoimmune phenotypes such as rheumatoid arthritis21, inflammatory disease21, multiple sclerosis22, and type 1 diabetes23. Combined, these results suggest that we have replicated genetic loci that play a role in atopic disease and identified novel loci that expand the current knowledge of the genetic architecture of AD.
Cell-type enrichment confirms key AD mechanistic nodes
To identify the disease-relevant cell types impacted by the identified AD relevant loci, we integrated GWAS signal with epigenetic and single-cell transcriptomic annotations. LD score regression (LDSC-SEG) was used to identify genomic annotations enriched for genetic trait heritability in the EUR GWAS, and bulk ATAC-seq data for cells isolated from peripheral blood from healthy donors (GSE118189, Methods)24. AD GWAS variants are enriched primarily in open chromatin of T-cell populations such Th1, Th2, Th17, and Treg cells. These findings support the known pathobiology of T-cell driven inflammation in AD25,26 (Fig. 3a).

a Significance of AD GWAS heritability enrichment (x-axis) in accessible chromatin per immune cell type (y-axis) of stimulated (red) and non-stimulated (gray) isolated immune cell ATAC-seq data annotations. P-values were derived from Linkage disequilibrium (LD) score regression in specifically expressed genes (LD-SEG) analysis (Methods). Red dashed line corresponds to FDR < 0.05 threshold. b Significance of AD GWAS signal enrichment (x-axis) in differentially expressed genes per skin cell type (y-axis). P-values correspond to MAGMA gene-program enrichment for AD associated variants with genes expressed in healthy and AD-affected skin profiled with sc-RNAseq. (Methods). Differential expression corresponds to Healthy (gray), Lesional (pink) and AD progression (red) programs. Healthy and Lesional programs are defined by genes highly expressed in a particular skin cell type compared to others. Disease progression program is defined as differential expressed genes between cells of the same type in AD-lesional relative to healthy tissue. Red and gray dashed line corresponds to FDR < 0.05 and P < 0.05 thresholds, respectively.
To explore disease-relevant cell types in skin, we utilized MAGMA to evaluate the enrichment of GWAS signal in skin cell types from the Human Cell Atlas skin dataset27, a single cell RNAseq dataset (sc-RNAseq) derived from healthy skin tissue (H) and AD lesional skin tissue (LS) (Methods). Cell-type-specific gene programs (differential gene expression in one cell type compared to other cell types in H or LS tissue) and disease progression (DP) programs (differential expression between cells of the same type in LS vs. H tissue) were constructed for 14 cell type categories to test for enrichment of GWAS signal (Methods). For cell type and DP programs, strong enrichment was identified in lymphocytes, including T-cells, NK cells, and innate lymphoid cells (ILCs), as well as macrophages and dendritic cells. These findings underscore the contributions of both innate and adaptive immune cell types in the inflammatory node of AD biology. Enrichment in keratinocytes pinpoints another key mechanistic node in AD biology, the skin barrier. This signal is significantly enriched in LS skin from AD patients and in the DP program, but not in healthy skin (Fig. 3b). Refinement of the keratinocyte cluster into defined sub-populations shows strongest enrichment in differentiated keratinocytes in LS and DP programs followed by undifferentiated and proliferating keratinocytes in DP program and inflammatory differentiated KCs in LS program (Supplementary Fig. 2). The specific keratinocyte enrichment patterns underscore the role for AD GWAS implicated genes in epidermal differentiation and barrier function in the upper layers of the epidermis.
To investigate the genetic contribution of the AD GWAS signal to the cell types identified in Fig. 3b, we clustered the gene program scores for the 146 genes with MAGMA Bonferroni-corrected p-value < 2.7e−06 for the 14 cell-type clusters and identified distinct clustering by immune and non-immune cell types in the skin (Supplementary Fig. 3). Additionally, the DP cell types frequently clustered separately from the H and LS cell types, indicating different roles for AD GWAS genes in H and LS states compared to DP. An exception is for the keratinocytes and melanocytes, where the LS and DP programs cluster together. Enrichment of the GWAS signal in keratinocytes and clustering of LS and DP keratinocyte gene programs supports further exploration of AD GWAS genes influencing keratinocytes and the role in barrier function.
Integration of functional annotations identifies putative causal genes at AD GWAS loci
To identify putative causal genes and variants at each locus, we performed the following analyses. First, we performed fine-mapping on 78/101 loci that reached genome-wide significance (P < 5e−08) in EUR (75/78) or ASN (11/78) meta-GWAS analyses (excluding MHC region). We identified 13 credible set variants annotated as moderate or high impact in 9 genes (Supplementary Data 4) including 3 coding variants in the FLG, TESPA1, and NLRP10 loci with posterior inclusion probability (PIP > 0.9 (Supplementary Data 4). Notably, for the fine-mapped variants in TESPA1 (rs183884396, PIP = 0.99) and NLRP10 (rs59039403, PIP = 1), the minor alleles are enriched in specific ancestral populations. TESPA1_rs183884396-G allele is >14x more frequent in the Finnish population compared to non-Finnish Europeans and NLRP10_rs59039403-G has an allele frequency in East Asian populations of 12.4% compared to <0.1% in EUR populations, demonstrating the value of cohorts from diverse ancestries to identify genetic signal for AD. Second, we identified coding variants in LD with the lead variants from the 3 GWAS meta-analyses performed with genome-wide significant results (P < 5e−8), excluding the MHC region (r2 > 0.6, MULTI, EUR, EAS samples in 1KG Phase 3; Methods) and annotated the coding variants predicted to impact gene function. Twenty-seven coding variants in 19 genes were identified across the MULTI, EUR, and ASN meta-analyses (Supplementary Data 5). The identified genes include reported causal or putatively causal AD genes such as FLG28 and NLRP1028 as well as genes with reported eczema, dermatitis or immunodeficiency phenotypes in OMIM: SIK3, IL7R and RTEL1, respectively. Combined, the genes with coding variation in the fine-mapped credible sets and in LD with the GWAS lead variants identify potentially causal AD genes.
Next, we performed colocalization with Quantitative Trait Loci (QTLs) to infer molecular consequences of the AD GWAS variants. We compiled and harmonized a QTL catalog of 297 full summary statistics of expression (eQTL), splicing (sQTL), protein (pQTL), and DNA methylation (mQTL) maps maximizing the inclusion of immune cell types relevant to AD (Supplementary Data 6). In total, we identified 3195 colocalizations (PP4 > 0.75) across the majority (86/100) of AD-associated non-MHC loci including 13/16 novel loci (Supplementary Data 7, Fig. 4a). While for most (85%, 73/86) QTL-associated loci, colocalization(s) at gene expression level (eQTL) were identified, 13/86 loci lacked eQTL associations but were supported by other molecular phenotypes (Supplementary Fig. 4). Among those, we identified genes related to pathways relevant to AD, e.g. IL-22 genes, and less characterized genes, e.g. CLEC16A, uncharacterized in AD but reported to be a master regulator of autoimmunity29. It has been shown that multiple regulatory effects for the same gene often mediate the same complex trait associations, and that QTLs derived from different molecular phenotypes have an independent contribution to complex traits30,31. We quantified QTL support per gene (Fig. 4b) and identified CRAT, IL6R, IL7R, and INPP5D as supported by all QTL molecular phenotypes, and by multiple cells/tissue QTL endpoints. While the CRAT locus GWAS lead variant is located in an intronic region of the uncharacterized protein-coding gene ENSG00000235007, the AD risk allele rs1107329-C is associated (P = 6.9e−44, b = 0.25) with increased CRAT protein abundance in plasma and increased levels of 2-methylmalonylcarnitine32. (Fig. 4c). CRAT encodes carnitine O-acetyltransferase; CRAT transcript levels and acetylcarnitines are reported to be altered in skin33 and serum of AD patients34, respectively. These results indicate that rs1107329-C increased AD risk may derive from genetic impact on carnitine metabolism.

a Number of AD GWAS loci with identified QTL colocalizations (PP4 > 0.75), stratified by QTL molecular phenotype and GWAS ancestry and novelty endpoints, colored in red—if previously reported—or blue—if novel. Bold or pale color hue represents loci identified in the multi-ancestry or European ancestry AD GWAS, respectively. PP4 corresponds to the posterior probability of whether a shared causal variant exists in the region (Methods). b Number of genes with identified QTL colocalizations (PP4 > 0.75), stratified by QTL molecular phenotype type. The inset heatmap represents the number of QTL endpoints per QTL type (x-axis) for each of the four genes supported by all QTL types (y-axis); barplot shows aggregated cross-QTL support per gene. c Genotype-phenotype association p-values of the CRAT locus. Panels illustrate CRAT pQTL signal in ARIC EA plasma (top) and GWAS signal for AD (middle) and GWAS signal for increased levels of 2-methylmalonylcarnitine (bottom). d Genotype-phenotype association p-values of the CD207 locus. Panels illustrate CD207 eQTL signal in GTEx skin not exposed to sun (top), ImmuNexUT myeloid dendritic cells (mDC) (middle) and GWAS signal for AD (bottom). Lead GWAS variant mapped to AD risk allele is typed and illustrated by a triangle-shaped point pointing upwards, indicating allele association with positive phenotype effect. Linkage disequilibrium between loci is quantified by squared Pearson coefficient of correlation (r2). P-values correspond to nominal GWAS and QTL associations, derived from multiple regression two-sided t-tests.
The usage of diverse cell and tissue QTL endpoints can aid the prioritization of cell of origin for a candidate gene. For example, we identified CD207 colocalization instances exclusively in skin and myeloid dendritic cell (DC) eQTL endpoints, with the AD risk allele rs4852714-A associated with decreased and increased expression of CD207 in skin and DCs, respectively (Fig. 4d). In skin, CD207 is exclusively expressed in Langerhans cells (LCs), which are epidermal resident DCs of the myeloid lineage. These results pinpoint skin-resident dendritic cells as the causal cell of origin type of rs4852714-A increased AD risk and demonstrate the need of skin QTL maps at cell-type resolution to confidently assess the effect directionality of AD risk alleles on impacted genes in causal cell-type contexts.
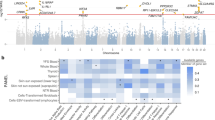
Finally, to prioritize genes at each locus, we scored each candidate gene within the locus by aggregated support from multiple lines of evidence, including variant-to-gene predictions, QTL evidence, coding variant genes, AD phenotype annotations (Methods, Supplementary Data 8, Supplementary Fig. 5). Top-scoring genes in novel loci include ITK and BATF (Fig. 5a). ITK encodes IL-2 inducible T-cell kinase, colocalizes exclusively in T-cells, is upregulated in AD lesional skin33,35 and has been suggested as a potential target for the therapy of T-cell-mediated inflammatory skin diseases35. Autosomal recessive mutations in ITK cause Lymphoproliferative Syndrome 1, a primary immunodeficiency characterized by early childhood Epstein-Barr virus associated immune dysregulation manifesting in lymphoma and autoimmune disorders. BATF encodes basic leucine zipper ATF-like transcription factor, it colocalizes in CD8+ memory T-cells and is upregulated in AD lesional skin33. AD GWAS associations with the BATF gene family member BATF3 have been reported36. In mice, the Batf/Batf3 interaction controls Th2-type immune response through regulation of IL-4 production37. Given their role in immune response, T-cell function, and reported links to immune diseases, ITK and BATF may play a role in AD biology and warrant further investigation.

a Supporting evidence by candidate per GWAS locus. b Scheme of ligand-receptor and proximal components of IL-6 and IL-22 signaling pathway. Corresponding expression and protein QTL associations mapped to the lead AD GWAS variant risk allele are shown. This figure contains illustrations from BioRender. Oliva, M. (2025) [https://BioRender.com/d11f365]. c Genotype-phenotype association p-values of the IL6ST, IL22RA2, ITK and BATF locus. First panel illustrates ITK ENST00000522616 sQTL signal in T helper 2 cells (Th2), second panel illustrates BATF eQTL signal in CD8+ memory T-cells, third panel illustrates IL6ST eQTL signal in T follicular helper cells (Tfh), forth panel illustrates IL22RA2 pQTL signal in plasma. For all panels, bottom part illustrates GWAS signal for AD. Lead GWAS variant mapped to AD risk allele is typed and indicated by a triangle-shaped point, pointing upwards or downwards for positive or negative effect size, respectively. Linkage disequilibrium between loci is quantified by squared Pearson coefficient of correlation (r2). P-values correspond to nominal GWAS and QTL associations, derived from multiple regression two-sided t tests.
Among high-scoring, prioritized genes we found multiple instances of receptor–ligand interactions for circulating cytokines and TNF-superfamily members, including genes involved in IL-6 and IL-22 signaling pathways (Fig. 5b, c). The variant rs7731626 is located within an intron of ANKRD55 and colocalizes with T-cell eQTLs for both ANKRD55 and IL6ST, which encodes IL-6 receptor complex protein gp130. We, and others38, prioritize IL6ST as the putative causal gene, linking the locus to the IL-6 signaling pathway that plays a key role in autoimmune and chronic inflammatory diseases. We identified additional IL-6 pathway genes – IL6, IL6R (IL-6 receptor), and SOCS3 (JAK/STAT inhibitor) – as prioritized candidates for corresponding AD GWAS loci. In the IL-22 pathway, we identified both IL22 and IL22RA2 as prioritized AD candidates. IL22RA2 is the gene prioritized at the most significant novel AD GWAS locus (P = 2.57e−10) and has support from both cis and trans pQTL signals in plasma. The trans IL22RA2 pQTL variant rs4265380 is located at the prioritized gene RUNX3 locus. RUNX3 has been previously associated with psoriasis39 and may modulate frequency of Th17 and Th22 cells40; RUNX3 and IL22RA2 interactions have been reported to be involved in macrophages IL-22 mediated intestinal inflammatory response in mouse leading to colitis41. The prioritization approach captures known and uncharacterized AD molecular associations, aids the prioritization of putative AD-causal genes and cellular contexts, and provides insight into gene sets and pathways contributing to AD pathobiology.
Integration of colocalization and functional assays identifies keratinocyte-specific AD-linked genes
Skin barrier defects, a key feature of AD, primarily affect keratinocytes42, and we observed AD genetic signal enrichment for genes differentially expressed in keratinocytes from AD patients (Fig. 3). Hence, we hypothesized that a fraction of the colocalized genes impact AD by altering keratinocyte-specific gene expression programs and mechanisms. To explore our hypothesis, we selected 22 colocalized genes associated with keratinocyte-specific signatures (Methods, Supplementary Data 9). We defined this set as “AD keratinocyte-linked gene candidates” and profiled them by four complementary functional assays. Firstly, to evaluate their enhanced expression in keratinocyte populations, we generated sc-RNAseq profiles of epidermal cells from seven human body sites (Methods, Supplementary Fig. 6). Secondly, to investigate their involvement in keratinocyte differentiation, we generated a three-dimensional epidermal model and generated bulk-RNA-Seq profiles from seven differentiation timepoints (Methods, Supplementary Fig. 7). Thirdly, we explored their response to the AD-relevant cytokine pathways IL-13 and IL-2243, considered the two major effector cytokines in AD pathogenesis44,45, by silencing each gene in keratinocyte models and characterizing the expression of cytokine IL-22 and IL-13 pathway proxy genes under different treatment conditions: no treatment, stimulation with IL-22, IL-13, or both (Methods). Finally, to assess whether AD risk alleles affect transcript abundance in keratinocytes, we mapped cis eQTLs from keratinocyte cell lines derived from 50 human subjects (Methods).
We observed that, compared to non-prioritized candidates (Supplementary Fig. 8), per-locus prioritized AD keratinocyte-linked genes (Fig. 6) tend to yield significant effects across assays. Considering differential expression by keratinocyte cell population, we identified CEBPA as enhanced in differentiated keratinocytes, AQP3 and RGS14 in non-keratinized keratinocytes, and RORA and ANK3 in keratinized populations. In epidermal models, CEBPA, AQP3, RORA and ANK3 were identified as strongly positively associated with keratinocyte differentiation (Fig. 6). While cytokine pathway signal differs by treatment, all tested prioritized candidates—except RGS14—show nominally significant (t test, P < 0.01) signal in at least one treatment condition (Supplementary Fig. 9b). Despite the limited power of the keratinocyte eQTL map, all tested prioritized candidates show nominally significant (t test, P < 0.05) eQTL signal, confirming that AD risk alleles impact candidate genes expression in keratinocytes (Fig. 6, Supplementary Data 9).

1st panel, Keratinocyte subtype differential expression (x-axis) by gene (y-axis). Differential expression values correspond to expression log fold change (FC) between a total of 96,194 keratinocyte and 178,640 non-keratinocyte cells, mean-averaged across body sites (n = 7 sites, Methods), and whiskers represent the standard deviation of the mean. 2nd panel, Differential expression (x-axis) of gene (y-axis) as a function of epidermal raft differentiation. Differential expression values correspond to log expression fold change as a function of differentiation timepoints (n = 7 timepoints, Methods), and whiskers represent the 95% confidence interval of the value. 3rd panel, Differential expression of pathway (x-axis) proxy genes by candidate gene (y-axis). Differential expression values correspond to the standardized mean difference (SMD) of the expression of genes that are proxies of IL-22 and IL-13 pathways between the presence or absence of siRNA targeting corresponding gene candidate. SMD values were derived from across-proxy-genes per-pathway meta-analysis (n = 3 proxies, Methods). Whiskers represent the 95% confidence interval of the value. 4th panel, Keratinocyte eQTL effect size (x-axis) by candidate gene (y-axis) (n = 50 subjects). Effects are mapped to AD risk alleles of the corresponding GWAS locus lead variants. Whiskers represent the 95% confidence interval of the value. CEBPA effect size is not shown due to insufficient AD risk allele frequency (Methods).
Combined, resources generated herein provide valuable mechanistic insights for uncharacterized AD genomic associations. One example is the novel ANK3 locus. ANK3 is known to play a role in neurodevelopmental disorders46, but its role in atopic dermatitis has not yet been described. We hypothesize that ANK3 could be associated with AD immunopathogenesis by playing a role in keratinocyte-mediated inflammatory programs, as we observe that ANK3 expression is enhanced in keratinized populations (granular layer), and the corresponding transcript silencing increases IL-13 pathway proximal gene expression in IL-13 + IL-22 co-stimulated keratinocytes (Fig. 6, Supplementary Fig. 9). The AD risk allele rs11817236-A may impact ANK3 in a cell-type specific manner; we observed decreased expression in keratinocytes (Fig. 6), but increased expression is reported in specific T-cell populations47. Together, these results highlight the utility of performing a comprehensive QTL-GWAS colocalization approach and integrating this data with cell-type relevant functional assays, to elucidate the potential mechanism by which genes not previously linked to AD could play a role in the disease by altering keratinocyte function.
Discussion
We present a large-scale, AD GWAS meta-analysis leveraging multi-ancestry cohorts, including N = 13,183 non-European subjects affected by AD. We identified 101 loci associated with AD, including 16 novel loci not previously reported. While the inclusion of subjects from diverse ancestry backgrounds contributed to the overall multi-ancestry GWAS meta-analysis signal, we did not identify novel ancestry-specific GWAS loci in non-European cohorts. Fine-mapping of the ancestry-specific EUR and ASN GWAS meta-analyses identified variants with high posterior probability of being causal, including missense variants in TESPA1 and NLRP10 enriched in Finnish and East Asian populations, respectively. These findings support that larger AD cohorts in underrepresented non-European populations are required to increase the power to detect genome-wide significant variants, and that fine-mapping of ancestry-specific AD GWAS signal can identify putatively causal genes and variants. Furthermore, previous studies of AD and asthma in African Americans suggest that applying a stricter phenotype definition may aid discovery of novel signal48,49, and existing differential AD-triggering environmental exposures17 may contribute to explain the higher prevalence of AD for this community that is seemingly not explained by genetics50. To improve the characterization of AD genetic architecture, future efforts should not only continue the diversification and expansion of AD GWAS cohorts, but also focus on the refinement of phenotype definition and control for environmental exposure.
Exploring the genetic relationship between AD and atopic march phenotypes may help us to better understand the genetic architecture of disease. Atopic dermatitis often precedes the development of atopic march, defined as disease progression of AD to asthma and allergic rhinitis, which is associated with more severe and persistent disease19. We found that most of the identified AD loci overlap with previously reported atopic march GWAS loci, supporting the largely shared genetic architecture across atopic diseases. Both the replicated and novel AD loci highlight genomic regions that expand the understanding of AD biology.
Integration of multi-omic QTL sources from diverse biotypes to prioritize disease causal genes comprises the most extensive QTL-GWAS colocalization effort for AD genetic risk signals to date. Inclusion of complementary molecular phenotypes with cell-type specific QTL sources enabled the identification of AD-gene links otherwise missed. To prioritize genes at the novel and known loci, we leveraged multiple, complementary variant-to-gene approaches and functional annotations to derive a prioritization score. We focused on the biological interpretation of candidate genes at novel loci (for example ITK and BATF) and featured multiple prioritized genes in the IL-6 and IL-22 pathways, including IL6ST, IL22RA2 and SOCS3, located in novel AD loci.
Cell-type enrichment analysis identified immune cells, particularly T-cells, as the top enriched cell type in the AD genetic signal, supporting the known pathobiology of T-cell driven inflammation in AD. Additionally, enrichment analysis in skin sc-RNAseq identified keratinocytes as a significant cell-type contributor, revealing AD genetic signal enrichment in sc-RNAseq-derived keratinocyte expression signatures and supporting the known role for skin barrier alterations in AD. To functionally characterize the keratinocyte signal on a selected gene candidate set, we generated a comprehensive array of skin and keratinocyte functional assays, including a cross-body epidermal sc-RNAseq atlas, keratinocyte multi-timepoint differentiation expression data, keratinocyte-derived eQTLs and KOs across AD-relevant stimulation contexts in interleukin pathways. The integrated cross-assay results pinpoint RORA, CEBPA, AQP3, and ANK3 as strong AD-linked candidates and highlight keratinocyte-subtype and context specificities. Yet, we acknowledge that future work to characterize the prioritized keratinocyte genes’ role in the complex crosstalk of epidermal differentiation and cytokine response is warranted. Importantly, the array of unique keratinocyte-derived resources generated herein can be further utilized by the scientific community to better understand AD pathobiology linked to the disruption of the epidermal barrier.
In conclusion, we leveraged multiple genetic and functional approaches to understand the mechanisms by which AD-associated variants impact genes and cell types. We identified novel AD susceptibility loci, prioritized potentially causal genes, and pinpointed cellular contexts that contribute to the genetic architecture of AD. The provided resources can be utilized to further characterize the contribution of genetic signal to AD pathobiology and enable future efforts to identify AD-associated genes that may be transferrable into clinically actionable targets for atopic dermatitis.
Methods
Ethics
The study adhered to all applicable regulations governing the use of human participants and was conducted in accordance with the principles of the Declaration of Helsinki. Participants in the FinnGen study provided informed consent for biobank research, with the study protocol (No. HUS/990/2017) approved by the Coordinating Ethics Committee of the Hospital District of Helsinki and Uusimaa (HUS). The UK Biobank received ethical approval from the North West Multi-center Research Ethics Committee (approval number: 11/NW/0382), with all participants giving informed consent. The CHOP cohort study was approved by the Institutional Review Board of Children’s Hospital of Philadelphia (IRB# 4886). Informed consent was obtained from all CHOP subjects or, if subjects were less than 18 years of age, from a parent and/or legal guardian, with assent from the child if 7 years or older. BioBank Japan participants provided written, informed consent approved by ethics committees of the Institute of Medical Sciences, the University of Tokyo and RIKEN Center for Integrative Medical Sciences. Skin biopsies were obtained from volunteer patients following protocols approved by the University of Michigan Institutional Review Board, written informed consent was obtained from all subjects.
Study populations
In this study, we included genome-wide association study summary statistics derived from genetic and phenotype data from FinnGen, UK BioBank, and Children’s Hospital of Philadelphia and previously published GWAS from EAGLE consortium and BioBank Japan. The detailed information for each study is described below.
FinnGen
The FinnGen research project [www.finngen.fi] was launched in 2017 with the aim to improve human health through genetic research. The project combines genome information with digital health care data from national registries: the genotype data are linked to national hospital discharge, death, cancer, and medication reimbursement registries using the national personal identification numbers. In the current analysis, we included 166,390 Finnish participants from FinnGen Data Freeze 10. Cases were obtained from ICD-8/9/10 diagnosis codes for atopic dermatitis, excluding other forms of dermatitis, from inpatient, outpatient or primary care registries and we required that a case also had a prescription code for AD medications from purchase or reimbursement registries. Controls excluded individuals with any dermatitis ICD codes or AD comorbidities (asthma and allergic rhinitis). The association analysis included 20,115 cases and 146,275 controls. GWAS was performed in the FinnGen Sandbox using the Scalable and Accurate Implementation of GEneralized mixed model (SAIGE v0.36.3.2) including sex, age, genotyping batch and the first 10 genetic principal components. SNPs with minor allele count (MAC) > 5 and imputation quality score > 0.6 were kept in the association analysis.
UK Biobank
The UKBB is a large and population-based prospective cohort of approximately 500,000 participants aged 40–69 years recruited between 2006 and 2010 in the United Kingdom. For the European ancestry analysis, we only included participants with European ancestry defined as Caucasian by the UKBB Field 22006. Atopic dermatitis cases were defined using ICD-9/10 codes for atopic dermatitis, excluding other forms of dermatitis, from hospital in-patient data and primary care and also had a prescription code for AD. Controls excluded individuals with any dermatitis ICD-9/10 codes or AD comorbidities (asthma and allergic rhinitis). The UKBB European ancestry association analysis included 10,470 cases and 210,720 controls.
For individuals of African and Central South Asian, we utilized the Pan-UK Biobank project [pan.ukbb.broadinstitute.org] assignment of UKBB participants to 6 continental ancestries. This resulted in identification of 6636 individuals of African ancestry and 8876 individuals of Central-South Asian ancestry in the UKBB project. Similar to the above-described case-control definitions, cases were defined using ICD-9/10 codes for atopic dermatitis, excluding other forms of dermatitis, from hospital in-patient data and primary care. Controls excluded individuals with any dermatitis ICD-9/10 codes or AD comorbidities (asthma and allergic rhinitis). The UKBB African ancestry association analysis included 146 cases and 4799 controls and the Central South Asian ancestry association analysis included 376 cases and 5594 controls (Supplementary Data 1).
GWAS for the UKBB European, African, and Central South Asian cohorts was performed using mixed logistic regression model including sex, age, the first 10 genetic principal components and a genetic relatedness matrix. To fit the model we used SAIGEgds (v1.6.0), an R package that implements the Scalable and Accurate Implementation of GEneralized mixed model (SAIGE) method on a Genomic Data Structure (GDS) file format to optimize computational efficiency. SNPs with minor allele count (MAC) > 10 and imputation quality score > 0.7 were kept in the association analysis.
Children’s Hospital of Philadelphia (CHOP)
The cohort from the Center for Applied Genomics is composed of approximately 85,000 juvenile subjects below 18 years of age and of a diverse set of ancestries. Subjects were recruited at Children’s Hospital of Philadelphia starting in 2006 and continuing to the present; genotyping of subjects has occurred across the same timeline. Cases were obtained from ICD-9 diagnosis codes for atopic dermatitis (6918, 6918B), excluding individuals with other forms of dermatitis. Controls excluded individuals with any dermatitis ICD-9 codes (690–698).
Subjects in the study were genotyped on multiple versions of Illumina genotyping arrays. These arrays fell into four families (HumanHap 550/610, Infinium Omni 2.5, Infinium OmniExpress, and Infinium Global Screening Array). Chips within families were merged, filtered for genotype missingness (0.05), individual missingness (0.02) and minor allele frequency (0.05). Genotypes were imputed against the TOPMed reference panel using the TOPMed imputation server51,52,53. Imputed genotypes were filtered for imputation quality using the R2 metric (>= 0.6). Post filtering imputed datasets were merged on common SNPs. The final dataset contained 41,180,882 variants.
Ancestry was assigned using PCA. Merged genotypes were filtered for minor allele frequency (0.01) and then pruned for linkage disequilibrium using PLINK (v1.9) (indep-pairwise 500 50 0.05)54. The PCA dataset contained 157,203 SNPs. PCA was performed using flash-pca (v2.0)55. The first three principal components were visualized using the plot3D function from the rgl library in R v4.2.3. Subjects were grouped based on observed centers of density into European, African, East Asian, South Asian, and Hispanic/American ancestries. Subjects that fell outside of those five groupings were aggregated into a sixth group, designated “Unassigned”. Genotype files for each ancestry were separated, and another ancestry-specific PCA was performed as above to identify any further outliers.
The association analysis included individuals of African ancestry (6917 cases and 11,080 controls), European ancestry (1590 cases and 21,499 controls), East Asian ancestry (219 cases, 692 controls), South Asian ancestry (123 cases, 1045 controls), American ancestry (237 cases, 1884 controls) and unassigned mixed ancestry (869 cases, 4927 controls). GWAS was performed Scalable and Accurate Implementation of GEneralized mixed model (SAIGE v1.1.4) including sex, age, genotyping batch and the first 10 genetic principal components. SNPs with minor allele count (MAC) >= 5 and imputation quality score >= 0.6 were kept in the association analysis.
EAGLE consortium
The EArly Genetics and Lifecourse Epidemiology (EAGLE) Consortium AD meta-analysis published by Paternoster et al in ref. 12, performed fixed-effect GWAS meta-analysis on ~21,000 cases and 95,000 controls. Cases were defined as described in the paper12, briefly each cohort defined cases by a mixture of self-report, doctor diagnosis, and/or ICD codes. Summary statistics were generated for the discovery cohort using GWAMA56 and were downloaded from the EBI GWAS Catalog (study accession GCST003184, excludes 23&Me and non-European cohorts), totaling 10,788 cases and 30,047 controls of European ancestry.
BioBank Japan
BioBank Japan collaboratively collects DNA and serum samples from 12 medical institutions in Japan and recruited approximately 200,000 patients with a diagnosis of at least one of 47 diseases. Mean age at recruitment is 63 years. Sakaue et al. 57 performed GWAS analysis on 220 phenotypes, including atopic dermatitis. Summary statistics for atopic dermatitis GWAS were downloaded from the BioBank Japan portal (https://pheweb.jp/downloads). The GWAS summary statistics include 4,296 AD cases and 163,807 controls run using SAIGE (v.0.37) and included age, age2, sex, age × sex, age2 × sex and the top 20 principal components as covariates. Cases were defined by ICD-10 code L20 and controls were individuals without a L20 ICD-10 code in their medical history.
GWAS meta-analysis
Multi-ancestry GWAS statistics were obtained by fixed-effect inverse variance weighted meta-analysis of the summary statistics (beta values) from FinnGen, UKBB_EUR, UKBB_AFR, UKBB_CSA, EAGLE, BBJ and the 6 CHOP cohorts, using GWAMA (v2.2.2)56 (Supplementary Data 1). Ancestry-stratified GWAS statistics were obtained by performing an analogous meta-analysis strategy considering cohorts stratified by continental populations: EUR, ASN, AFR (Supplementary Data 2). In total, GWAS signal from 4 ancestry endpoints—multi-ancestry, EUR, ASN, and AFR populations—were generated. Subsequently, we identified autosomal loci with GWAS signal, i.e. genomic windows containing independent GWAS signals, across ancestry endpoints. For that, we first constructed a reference dataset of best-guess genotypes from UK Biobank (UKBB)58 by considering imputed dosages of variants with info score > 0.3 and MAF > 0.1%, selecting genotypic data corresponding to 15,000 randomly selected or to 2000 ancestry-matched unrelated UKB samples, to generate multi-ancestry or ancestry-stratified genotype panels, respectively. We then filtered variants with missingness > 5% and Hardy–Weinberg equilibrium test P < 1 × 10−7. For each of the 4 GWASs, we used the PLINK (v1.9) ref ‘clumping’ algorithm to select top-associated variants (P < 5 × 10−8) and corresponding LD-linked variants at r2 > 0.05 with the top associated variant within ±1 Mb, utilizing the GWAS-matching ancestry-stratified or multi-ancestry UKB genotype data. We determined the genomic span of each LD-based clump and added 1 kb up- and downstream as buffer to the region. If any of these windows overlapped, we merged them together into a single (larger) locus.
To determine a set of non-redundant GWAS loci across ancestry endpoints, we selected all multi-ancestry derived clumps, and complemented this set with non-overlapping clumps identified in a single ancestry. The resulting GWAS hit loci set is composed of genomic regions with suggestive GWAS signal in at least one ancestry endpoint. For each GWAS locus, considering the ancestry endpoint were the GWAS hit was identified, the smallest p-value per locus was defined as the proxy GWAS lead variant for that locus. The final set is composed of 101 GWAS loci.
Comparison of the 101 loci with published literature was performed for atopic march phenotypes: atopic eczema/eczema (EFO_0000274, HP_0000964, allergic disease (MONDO_0005271) and asthma (MONDO_0004979) studies reported in the EBI GWAS catalog (https://www.ebi.ac.uk/gwas; 2024-09-26). To include not only genome-wide significant (P < 5e−08) but also suggestive (P < =5e -06) GWAS signal, we considered both reported entries for all atopic march GWAS studies (Supplementary Data 10) and full summary statistics from the largest GWAS per disease group: Budu-Aggrey14 (atopic eczema/eczema), Han59 (asthma) and Ferreira20 (allergic disease) (Supplementary Data 11). Overlap analysis was performed in R with data.table foverlaps() function. For the 101 genomic loci, start and stop positions windows were set to ±0.5 Mb from the lead variant and intersected with reported variant positions from the EBI GWAS catalog, and with variant positions from the aforementioned GWASs. Novel AD loci are defined as loci not overlapping previously published atopic eczema/eczema GWAS P < 5e−08.
LDSC heritability
LDSC regression (v1.0.1) was applied to estimate the variant-based heritability (h2SNP) of AD from the European ancestry GWAS meta-analysis. h2SNP was estimated on liability scale using population prevalence (--pop-prev) 0.15 and sample prevalence (--samp-prev) 0.095 in the meta-analyzed sample (4-cohort EUR meta-analysis 42,963 cases/451,435 controls).
Fine-mapping
Statistical fine-mapping was performed using the SuSiE (susieR v0.12.35) with GWAS summary statistics from the European, Asian and African meta-analyses and LD reference panels calculated from UKBB for EUR, AFR and EAS individuals as classified by the Pan-UKBB project [pan.ukbb.broadinstitute.org]. We defined fine-mapping regions based on a 1 Mb window around each lead variant and excluded the major histocompatibility complex (MHC) region from analysis due LD structure in the region. The model allowed up to 10 causal variants per region and 95% credible sets (CS) were calculated with posterior inclusion probabilities (PIP) of each variant reported. In loci with multiple causal variants identified, there will be multiple 95% CS. All variants in the 95% credible sets were annotated with VEP (v110, [http://grch37.ensembl.org/Homo_sapiens/Tools/VEP]) using default criteria to select one block of annotation per variant.
LDSC – cell-type specificity
LDSC-SEG (v1.0.1) was used to identify genomic annotations enriched for AD trait heritability. Bulk ATAC-seq data (GSE118189 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE118189]) for cells isolated from peripheral blood from healthy donors was used to test for enrichment in open chromatin regions. These isolated cells were cultured in vitro with and without stimulation as described by Calderon et al. 24. Briefly, T cells were stimulated with Dynabeads conjugated with CD3/CD28 antibodies and IL-2; B cells were stimulated with anti-IgG/IgM antibodies and IL-4; Monocytes were stimulated with LPS; and NK cells were stimulated with CD2 and CD355 coated beads as well as IL-2. Together there are 222 samples for 32 types of immune cells, 20 of which have data for both unstimulated and stimulated status. Each of cell-type/ATAC-seq bed files was added to the baseline model independently when building the regression model and testing for enrichment.
Generation of cell-type-specific and disease programs
We scored genes for skin cell-type-specific programs by testing for differential expression by cell-type in healthy and lesional AD cells independently, utilizing a sc-RNAseq dataset derived from skin in the Human Cell Atlas27. Briefly, sc-RNAseq data of skin biopsies from 5 healthy controls (HC) and 4 atopic dermatitis (AD) patients were analyzed. The skin biopsies were separated into epidermis and dermis before dissociated and enriched for various cell fractions (keratinocytes, fibroblasts, and endothelial cells) and immune cells (myeloid and lymphoid cells) to up sample rare cell-types. In total, the HC group skin samples sc-RNAseq dataset includes 195,739 cells and AD lesion group sc-RNAseq dataset includes 63,512 cells). Cells were clustered using UMAP dimensionality reduction and Leiden graph-based method then annotated by comparing differentially expressed genes between clusters to published bulk transcription profiles or protein expression of defined cell types. Four major groups of cell types (lymphoid cells, myeloid cells, keratinocytes, and other non-immune cells) were identified and further clustered in subsequent rounds of feature selection, embedding, visualization and clustering. To generate cell-type-specific programs, we first identified genes specifically expressed in one cell type compared to other cell types in HC or AD sc-RNAseq datasets. To generate disease progression programs, we identified differential expression between cells of the same type in AD vs. HC datasets.
Gene-program enrichment analysis
We adapted MAGMA v1.09b to evaluate the association of disease-association statistics with the cell-type-specific gene programs and disease progression gene programs. In step 1, we performed a gene-level analysis based on a multiple linear principal components regression model to aggregate the association statistics of multiple markers to each gene associated with the AD phenotype. For each gene g the gene p-value pg from step1 was converted to a Z-value zg with the probit function. In step 2, we tested whether the genes in a gene-set are more strongly associated with the phenotype of interest than other genes. The competitive gene-program analysis tested whether the genes in the cell-type-specific and disease programs are associated with a particular cell type. The p-values from gene-program analysis were transformed to values between 0 and 1 using the min-max normalization resulting in a relative weighting of genes in each program. We corrected for multiple testing by cell type and disease program to identify significant program-cell-type enrichment (Benjamini–Hochberg FDR < 0.05). Clustering of gene program scores was performed for the 146 AD GWAS genes from the gene-level analysis that passed a more stringent multiple testing correction for all genes in the genome (Bonferroni P < 2.7E−06 = 0.05/18,471 genes) using the R program pheatmap with Pearson correlation (complete) as the clustering distance method for both rows and columns.
Determination of QTL-AD-GWAS colocalized loci
To investigate possible associations between cis-genetically regulated molecular phenotypes (QTLs) and Atopic Dermatitis (AD), we compiled an exhaustive QTL map collection and employed a colocalization approach.
Compilation of QTL full-summary statistics maps
To maximize the expectation of identifying Atopic Dermatitis putatively causal molecular links, we compiled an exhaustive collection of cis quantitative trait loci (QTL) mappings (maps) derived from several molecular phenotypes (MPs): gene (eQTLs), splicing phenotypes (sQTLs), DNA methylation (mQTLs) and protein abundance (pQTLs).
The QTLs originate from widely different contexts, i.e., tissue and cell types, stimuli, and developmental states; we considered a total of 297 cis QTL maps with full statistics available and genome-wide molecular phenotype tests. The majority (94%) of QTL maps are derived from gene expression or splice phenotypes (e/sQTLs), 4% are derived from DNA methylation (mQTLs) and 2% from protein (pQTLs) abundances. Details of QTL maps are provided in Supplementary Data 6. A total of 157 eQTL maps were obtained from bulk-tissue or isolated cells, 127 of which from 31 different studies included in the eQTL Catalogue60 ([https://www.ebi.ac.uk/eqtl/], version 5, April 2022). Thirty additional bulk-tissue and isolated-cell eQTL maps were obtained from additional sources: two meta-analyzed eQTL maps derived from blood (eQTLGen61, [https://eqtlgen.org/cis-eqtls.html]) and induced pluripotent stem cells (iPSC) (i2QTL62, [https://doi.org/10.5281/zenodo.4005576]), and 28 maps derived from isolated immune cells (ImmuNexUT63, [https://humandbs.biosciencedbc.jp/en/hum0214-v8#E-GEAD-420]). In addition, 14 immune cell eQTL maps derived from single cell RNA-Seq (sc-eQTLs) were obtained (OneK1K47, [https://onek1k.org/]). Considering splicing phenotypes, we included 109 sQTL maps derived from transcript abundances included in the v5 eQTL Catalogue60. Considering DNA methylation, a total of 11 mQTL maps were obtained. We included 9 maps from eGTEx sources: breast mammary tissue, colon transverse, kidney cortex, lung, muscle skeletal, ovary, prostate, testis, and whole blood (eGTEx30,64, [https://gtexportal.org/home/downloads/egtex]), one additional muscle skeletal (FUSION65, [https://www.ebi.ac.uk/birney-srv/FUSION/]) and one brain (ROSMAP66, [http://mostafavilab.stat.ubc.ca/xQTLServe/]) cis mQTL maps. Considering protein abundance, we included six pQTL maps from plasma (SomaScan deCODE 202167, [https://download.decode.is/form/folder/proteomics], SomaScan Sun et al. 68, [https://www.ebi.ac.uk/gwas/downloads/summary-statistics]; SomaScan and Olink FinnGen [https://www.finngen.fi/en/access_results]; ARIC EUR and AFR SomaScan 202269 [http://nilanjanchatterjeelab.org/pwas]).
Colocalization of AD GWAS loci with QTLs
For each of the 100 AD-associated loci not overlapping with the MHC region, we identified overlapping (>1 bp) molecular phenotype (MP) cis-region loci from each QTL map, considering GWAS and molecular phenotype cis-QTL analyzed variants. For each overlapping MP-GWAS region pair, we applied coloc v5.52 to QTL along with GWAS summary statistics, only if the locus contained >=1 variant with nominal QTL P < 1e−05 and GWAS P < 5e−08. Prior probabilities of a variant yielding a) a QTL association (p1), b) a GWAS association (p2) and c) a QTL and a GWAS association (p12) were set to p1 = 1e−04, p2 = 1e−04, p12 = 1e−06. Only the regions with at least 50 variants in common between the GWAS and MP loci were tested for colocalization. Both for QTLs and GWAS statistics, colocalization was performed on effect size (effect size) and associated standard error (effect size s.e.) values, except for ImmuNexUT eQTLs; colocalization was performed on p-values and minor allele frequency (MAF) values. We defined suggestive support for QTL-AD-GWAS colocalization at posterior probability PP4 > 0.75. For mQTLs, CpG probe identifiers were mapped to genes according to regulatory region annotations from EPIC.hg38.manifest.tsv.gz and HM450.hg38.manifest.tsv.gz [https://zwdzwd.github.io/InfiniumAnnotation].
Variant-to-gene mapping and prioritization of high-confidence genes
Given GWAS hit loci, candidate genes per locus can be prioritized by combining evidence across molecular resources70. Here we performed variant-to-gene mapping and aggregated functional annotation from various sources to generate scores that aim to represent the likelihood of a gene to be causally involved in AD. Genes with the highest aggregated score are named ‘prioritized genes’. Variants were mapped to genes using the combination of multiple methods: nearest gene, Open Targets Variant-to-Gene (V2G), DEPICT, fine-mapping and colocalization. Nearest gene for each genome-wide significant loci was annotated by GREAT (v4.0.4)71,72 using the lead variant, association rule: Single nearest gene: 1,000,000 bp max extension. Open Targets Variant-to-Gene (V2G)73 was used to assign lead variants to genes by selecting the gene with highest overall V2G score. DEPICT (Data-driven Expression-Prioritized Integration for Complex Traits)74 was run on all variants with P < 5e-08 using 1KG EUR LD reference panel to prioritize most likely causal genes at associated loci based on functional annotation. We identified 102 genes with significant (FDR < 0.05) DEPICT score, across 59 loci. VEP (v110) annotation of fine-mapped credible sets identified 9 genes with moderate or high impact coding variants (Supplementary Data 4). And we utilized VEP to annotate 60 genes containing coding variants in LD (r2 > 0.6) with corresponding GWAS locus lead variant from the MULTI and EUR GWAS meta-analyses (1KG EUR LD reference) and the ASN GWAS meta-analysis (1KG EAS LD reference) (Supplementary Data 5). Considering colocalization results per GWAS locus, we annotated all genes with at least one significant colocalization (PP4 > 0.75, as described in above colocalization methods) derived from sQTL, eQTL and pQTL maps (Supplementary Data 7). Considering deCODE and FinnGen trans pQTL summary statistics (SomaScan deCODE 202167, [https://download.decode.is/form/folder/proteomics]; SomaScan and Olink FinnGen [https://www.finngen.fi/en/access_results]) corresponding to GWAS loci lead variants, we annotated genes with trans pQTL signal (Supplementary Data 8). That is, if lead variant of GWAS locus 1 had a nominally significant (P < 5e-08) trans pQTL signal associated to a protein encoded in GWAS locus 2, the GWAS locus 2 gene encoding such protein would be annotated with trans pQTL signal.
All genes implicated by any of the above strategies were then annotated with OMIM entries associated with reported phenotypes involving the skin or immune system involvement (Supplementary Data 8). With this approach, we annotated 57 genes with a potential role in skin or immune processes. Additionally, genes reported in literature with coding variants in AD patients were compiled and annotated against the gene list (see reference publications in Supplementary Data 12) And finally, genes with significant differential expression (False Discovery Rate, FDR ≤ 5% and |log2(Fold Change)|≥1) from Tsoi et al. 33 comparisons of AD lesional skin to control (healthy) skin bulk RNAseq were annotated (n = 3264 DEGs, see Supplementary Data 13). The amount of evidence across all sources was added in an unweighted fashion to generate aggregated scores for 498 unique genes across 101 GWAS loci (Supplementary Data 8).
Selection and prioritization of AD keratinocyte-linked gene candidates
We selected genes with =>1 s/eQTL colocalization signal (PP4 > 0.75), where the AD risk allele increases the expression of the gene or transcript in at least one s/eQTL endpoint. We integrated skin cell enrichment metrics (Supplementary Data 2 of Dusart et al.75) and narrowed down this set by selecting genes with evidence of correlation with keratinocyte-representative transcripts, e.g. with mean correlation with keratinocyte reference transcripts > 0.30, and that being higher than the mean correlation with any non-keratinocyte reference transcript set. The selected set is composed of 22 genes: AQP3, NAB1, CEBPA, IL2RB, RORA, GRID2IP, RGS14, RTF1, LIME1, ZFYVE21, SLC2A4RG, SCAMP3, IL22RA2, WNK1, CLIP1, KIAA2013, LMAN2, MAP3K14, NDUFA4L2, ANK3, CHRAC1, PCDH1. We refer to this set as “AD keratinocyte-linked gene candidates”; genes that are candidates to play a causal role in Atopic Dermatitis through a pathogenic effect in keratinocytes.
Characterization of keratinocyte subtype specificity signal using a cross-body sc-RNA-Seq skin dataset
To assess the robustness of the keratinocyte-specific gene expression of the keratinocyte-linked colocalized genes, we generated a sc-RNAseq dataset composed of disease unaffected epidermal samples from 96 skin biopsies: 18 from published datasets, with processed data available at GEO under accession IDs GSE173706 and GSE249279 and 78 from newly generated datasets. Raw data for all 96 samples is available at SRA under accession id PRJNA1054546. The Seurat object derived from the harmonized RNAseq dataset, and curated cell-type signature genes utilized to define cell types, are available at Figshare [https://doi.org/10.25452/figshare.plus.c.7282969]. For all included samples, detailed biopsy sample and protocol details are described at Figshare [https://doi.org/10.25452/figshare.plus.c.7282969. Overall, biopsies originated from seven body sites (face, scalp, axilla, palmoplantar, arm, leg, and back). Skin biopsies were taken from the unaffected tissue of skin disease patients and healthy donors. Generation of single-cell suspensions for scRNA-seq was performed as follows: skin biopsies were incubated overnight in 0.4% dispase (Life Technologies) in Hank’s Balanced Saline Solution (Gibco) at 4 °C. Epidermis and dermis were separated. Epidermis was digested in 0.25% Trypsin-EDTA (Gibco) with 10 U/mL DNase I (Thermo Scientific) for 1 h at 37 °C, quenched with FBS (Atlanta Biologicals), and strained through a 70 μM mesh. Dermis was minced, digested in 0.2% Collagenase II (Life Technologies) and 0.2% Collagenase V (Sigma) in plain medium for 1.5 h at 37 °C and strained through a 70 μM mesh. The epidermal and dermal cells were combined in 1:1 ratio. Libraries were then sequenced on the Illumina NovaSeq 6000 sequencer to generate 150 bp paired-end reads. Data processing including quality control, read alignment (hg38), and gene quantification was conducted using the 10X Cell Ranger software. The samples were then merged into a single expression matrix using the cellranger aggr pipeline. In total, across body sites, 274,834 cells were profiled, including 96,194 keratinocytes. Seurat v3.0. was utilized to normalize, scale, and reduce the dimensionality of the data. We filtered out low-quality cells containing less than 200 genes per cell as well as greater than 5000 genes per cell. Cells containing more mitochondrial genes than the permitted quantile of 0.05 were removed. We removed ambient RNA using R package SoupX v1.6.2. Doublets were removed using scDblFinder v1.12.0. Principal components (PC) were obtained from the topmost 2000 variable genes, and the Uniform Manifold Approximation and Projection (UMAP) dimensional reduction technique was applied to the 30 topmost variable PC-reduced dataset. Batch effect correction was performed utilizing harmony v1.0, using donor as batch. After batch correction, cells were clustered using shared nearest neighbor modularity optimization-based clustering. Cluster marker genes were identified with FindAllMarkers; cluster corresponding cell type was identified by comparing marker genes to curated cell-type signature genes (Supplementary Fig. 6). Differential expression by keratinocyte subtype was performed with Seurat (v4.3.0) FindMarkers function by comparing keratinocyte subtype to non-keratinocyte clusters. The log fold-change of the average expression between a keratinocyte subtype cluster compared to the rest of clusters is utilized as keratinocyte-subtype gene expression statistic.
Characterization of keratinocyte-differentiation gene expression signal using 3-D human epidermal tissue cultures
We investigated the gene expression patterns of the keratinocyte-linked colocalized genes in keratinocyte differentiation occurring in the formation of 3-D human epidermal raft cultures. Normal human epidermal keratinocytes were isolated from epidermis (n = 3) and grown using J2-3T3 mouse fibroblasts as a feeder layer originally described by Rheinwald and Green76. 3-D human epidermal raft cultures seeded in collagen hydrogels were prepared using three distinct donor pools as described previously77 and grown at an air-liquid interface for 12 days in E-Medium (DMEM/DMEM-F12 (1:1), 5% Fetal Bovine Serum, adenine (180 µM), Bovine pancreatic insulin (5 µg/ml), Human apo- transferrin (5 µg/ml), triiodothyronine (5 µg/ml), L-Glutamine (4 mM), Cholera toxin (10 ng/ml), Gentamicin (10 µg/ml), Amphotericin B (0.25 µg/ml)). At day 9 at an air-liquid-interface to allow for epidermal maturation, the epidermal rafts (RHE) were treated with 0.1% BSA/phosphate-buffered saline (Sigma Aldrich, St Louis, MO) for 72 Hrs. Epidermal tissues were separated at the stages from Sub-confluent stage to 3-D raft on day 12 (Sub-confluent, Day 0-Confluent, Day 3-Confluent, Day 3-Raft, Day 6-Raft, Day 9-Raft, Day 12-Raft) from the collagen scaffold and lysed in QIAzol for RNA isolation. RNA samples were sent to the University of Michigan Advanced Genomics Core for RNA sequencing. Libraries for RNA-Seq were generated from polyadenylated RNA and sequenced at six libraries per lane on the Illumina Genome Analyzer IIx. We used Tophat278 to align RNA-seq reads to the human genome, using annotations of GENCODE as gene model79. HTSeq was used to quantify gene expression levels80. Normalization was performed by DESeq281. The processed RNA-Seq data can be found in Figshare [https://doi.org/10.25452/figshare.plus.c.7282969]. Differential expression by timepoint was performed with limma82 on FPKM values on non-lowly expressed genes (zFPKM > -3, in at least 2 timepoints). Timepoint was modeled as a quantitative variable (Sub-confluent=1, Day 0-Confluent=2, Day 3-Confluent=3, Day 3-Raft=4, Day 6-Raft=5, Day 9-Raft=6, Day 12-Raft=7), and the function duplicateCorrelation was utilized to model technical replicate effects. The estimated log-fold change attributable to timepoint is utilized as keratinocyte-differentiation gene expression statistic.
Characterization of candidate gene silencing in IL-13 and IL-22 pathways
To characterize the effects of silencing AD keratinocyte-linked genes on IL-13 and IL-22 pathways, which are implicated in AD pathogenesis43,83, we knocked-out (KO) candidate genes with silencing RNAs (siRNA) in N/TERTs84 immortalized keratinocytes cells, and evaluated the expression of interleukin pathway proxy genes S100A9, S100A8, S100A7 (IL-22) and CCL26, CISH, HSD3B1 (IL-13). Keratinocytes were plated in 96 well plate (20,000 cells/well) and incubated at 37 °C with 5% CO2 overnight. 100 μM Accell siRNAs for AD keratinocyte-linked gene candidates (Supplementary Data 9) were prepared in 1x siRNA buffer (Dharmacon# B-002000-UB-100). 1 μl of 100 μM siRNA was diluted with 100 μl accel delivery medium (Dharmacon # B-005000) for each well of 96 well plate. The growth medium was removed from the cells, 100 μl of the delivery mix with siRNAs was added to each well, and the plate was incubated at 37 °C with 5% CO2. Accell Non-targeting Control siRNA (Dharmacon # D-001910-01-05) was used as a negative control. After 24 h, cells were either stimulated with 10 ng/ml of IL-13 (R&D Systems # 213-ILB), 20 ng/ml of IL-22 (R&D Systems # 782-IL) or co-stimulated with IL-13 (10 ng/ml) and IL-22 (20 ng/ml). After 24 h of stimulation, cells were harvested for RNA preparation. RNAs were isolated from cell cultures using Qiagen RNeasy plus kit (Cat # 74136). Reverse transcription was performed using a High-Capacity cDNA Transcription kit (ThermoFisher # 4368813). qPCR was performed on a QuantStudio 5 Real-time PCR system (Applied Biosystems) with TaqMan Universal PCR Master Mix (ThermoFisher # 4304437) using TaqMan primers. RPLP0 (ThermoFisher # Hs99999902_m1) was used as a loading control. Three technical replicates were considered. Knockdown efficiency was validated by the TaqMan primer of each of the siRNA target genes (Supplementary Data 14). KO efficiency values are illustrated in Supplementary Fig. 9. The protocol failed for LIME1 and MAP3K14, which were not considered in further analyses. The differential expression of proxy genes between the presence or absence of siRNA targeting corresponding gene candidate was evaluated in each condition by t-test, considering the three technical replicates, and derived standardized mean difference (SMD) effect size and corresponding sampling variance were generated with f(x) ‘escalc’ from ‘metafor’ v.4.0. SMD values were meta-analyzed across markers per pathway, considering results from IL-13 + IL-22 treatment, with metafor::rma function.
Mapping of keratinocyte eQTLs
We mapped eQTLs in keratinocyte cell lines stimulated with IFNa (5 ng/ml), derived from N = 50 subjects, for which RNA-Seq profiles and genotype data were generated85. Gene expression values were first normalized by DESeq2, and PEER was used to account for latent confounding factors. The genotype data was generated by the Illumina Infinium CoreExome array, and imputation was performed using 1000 Genomes Project (GRCh37/hg19) as reference panel. Cis (±1 Mb from gene transcription start site) eQTL were mapped using FastQTL v2.0 by fitting a linear regression model (p ∼ g + C) where p is the gene expression vector, g is the genotype vector, and C is a matrix of 10 PEER factors derived from gene expression; eQTLs signal was assessed by the effect size corresponding to the term g. Full summary statistics are provided [https://doi.org/10.25452/figshare.plus.c.7282969]. The eQTL mappings restricted to 22 variant-gene pairs corresponding to AD keratinocyte-linked gene candidates - 20 AD GWAS loci and 22 genes - are provided in Supplementary Data 9. These eQTL effects were estimated considering the index variant of the AD GWAS hit corresponding to the keratinocyte-linked colocalized gene.
Correlation of keratinocyte assays’ readouts with candidate priority status
The set of 22 keratinocyte-linked genes was profiled by four different functional assays in keratinocytes (see corresponding Method sections above). We hypothesized that keratinocyte-linked gene candidates prioritized as more likely to be causal would yield more signal across assays, indicative of their active role in key keratinocyte pathways. To test this hypothesis, we first classified 5/22 candidates (AQP3, CEBPA, RORA, RGS14, ANK3) as ‘prioritized’, by having top aggregated scores per locus and accounting for more than half of eQTL colocalization instances per locus (Supplementary Data 9, Fig. 5). We then compared aggregated corresponding assay readouts with the remaining 17/22 non-prioritized candidates (Supplementary Fig. 7) and assessed significant differences in statistics distribution by means of Mann–Whitney test. Indeed, the statistics for prioritized genes are significantly larger than for non-prioritized genes in all assays: in IL-13 and IL-22 pathways gene silencing assays (Mann–Whitney P = 0.01 on absolute SMD values), in keratinocyte eQTLs (Mann-Whitney P = 3.58e−15 on absolute eQTL effect size values), in keratinocyte-differentiation gene expression signal (Mann–Whitney P = 1.18e−03 on absolute log-fold change values) and in keratinocyte subtype specificity signal (Mann–Whitney P = 2.82e−06 on absolute log-fold change values).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
We have placed data to perform main analyses, and to generate figures, at https://doi.org/10.25452/figshare.plus.28385684. The raw skin sc-RNAseq data is available at SRA under accession id PRJNA1054546, including 18 biopsies from published datasets, with processed data available at GEO under accession IDs GSE173706 and GSE249279 and 78 from newly generated datasets. Bulk ATAC-seq data for cell-type enrichment analysis is available at GEO under accession ID GSE118189. The Seurat object from the processed sc-RNAseq dataset, cis eQTL summary statistics from keratinocyte cell lines, and gene expression changes from 3-D epidermal raft cultures are available at https://doi.org/10.25452/figshare.plus.c.7282969. Full GWAS summary statistics are available at the GWAS Catalog under accession IDs: GCST90503108, GCST90503109, GCST90503110, GCST90503111.
Code availability
The code to perform analyses, and to generate figures, is available at Figshare repository https://doi.org/10.25452/figshare.plus.28385684 and corresponds to the v.1.0 release of the github repository https://github.com/AbbVie-ComputationalGenomics/AtDermQTLGWAS_manuscript/releases/tag/AtDermQTLGWAS_manuscript.
References
Weidinger, S. & Novak, N. Atopic dermatitis. Lancet 387, 1109–1122 (2016).
Bieber, T. Atopic dermatitis. N. Engl. J. Med. 358, 1483–1494 (2008).
Chovatiya, R. & Silverberg, J. I. The heterogeneity of atopic dermatitis. J. Drugs Dermatol. 21, 172–176 (2022).
Schuler, C. F. T., Billi, A. C., Maverakis, E., Tsoi, L. C. & Gudjonsson, J. E. Novel insights into atopic dermatitis. J. Allergy Clin. Immunol. 151, 1145–1154 (2023).
Sanders, K. M. & Akiyama, T. The vicious cycle of itch and anxiety. Neurosci. Biobehav. Rev. 87, 17–26 (2018).
Caubet, J. C. & Eigenmann, P. A. Allergic triggers in atopic dermatitis. Immunol. Allergy Clin. North Am. 30, 289–307 (2010).
Irvine, A. D., McLean, W. H. & Leung, D. Y. Filaggrin mutations associated with skin and allergic diseases. N. Engl. J. Med. 365, 1315–1327 (2011).
Palmer, C. N. et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 38, 441–446 (2006).
Bataille, V., Lens, M. & Spector, T. D. The use of the twin model to investigate the genetics and epigenetics of skin diseases with genomic, transcriptomic and methylation data. J. Eur. Acad. Dermatol. Venereol. 26, 1067–1073 (2012).
Elmose, C. & Thomsen, S. F. Twin studies of atopic dermatitis: interpretations and applications in the Filaggrin Era. J. Allergy (Cairo) 2015, 902359 (2015).
Zheng, J. et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017).
Paternoster, L. et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet. 47, 1449–1456 (2015).
Sliz, E. et al. Uniting biobank resources reveals novel genetic pathways modulating susceptibility for atopic dermatitis. J. Allergy Clin. Immunol. 149, 1105–1112.e9 (2022).
Budu-Aggrey, A. et al. European and multi-ancestry genome-wide association meta-analysis of atopic dermatitis highlights importance of systemic immune regulation. Nat. Commun. 14, 6172 (2023).
Hirota, T. et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat. Genet. 44, 1222–1226 (2012).
Baurecht, H. et al. Genome-wide comparative analysis of atopic dermatitis and psoriasis gives insight into opposing genetic mechanisms. Am. J. Hum. Genet. 96, 104–120 (2015).
Martinez, A., de la Rosa, R., Mujahid, M. & Thakur, N. Structural racism and its pathways to asthma and atopic dermatitis. J. Allergy Clin. Immunol. 148, 1112–1120 (2021).
Matthews, L. J. & Turkheimer, E. Three legs of the missing heritability problem. Stud. Hist. Philos. Sci. 93, 183–191 (2022).
Marenholz, I. et al. Meta-analysis identifies seven susceptibility loci involved in the atopic march. Nat. Commun. 6, 8804 (2015).
Ferreira, M. A. et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat. Genet. 49, 1752–1757 (2017).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
International Multiple Sclerosis Genetics, C. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365 (2019).
Robertson, C. C. et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat. Genet. 53, 962–971 (2021).
Calderon, D. et al. Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat. Genet. 51, 1494–1505 (2019).
Weidinger, S., Beck, L. A., Bieber, T., Kabashima, K. & Irvine, A. D. Atopic dermatitis. Nat. Rev. Dis. Prim. 4, 1 (2018).
Schuler, C. F. T. et al. Genetic and immunological pathogenesis of atopic dermatitis. J. Invest. Dermatol. 144, 954-968 (2024).
Reynolds, G. et al. Developmental cell programs are co-opted in inflammatory skin disease. Science 371 (2021).
Tanaka, N. et al. Eight novel susceptibility loci and putative causal variants in atopic dermatitis. J. Allergy Clin. Immunol. 148, 1293–1306 (2021).
Pandey, R., Bakay, M. & Hakonarson, H. CLEC16A—an emerging master regulator of autoimmunity and neurodegeneration. Int. J. Mol. Sci. 24 (2023).
Consortium, G. T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Oliva, M. et al. DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits. Nat. Genet. 55, 112–122 (2023).
Schlosser, P. et al. Genetic studies of paired metabolomes reveal enzymatic and transport processes at the interface of plasma and urine. Nat. Genet. 55, 995–1008 (2023).
Tsoi, L. C. et al. Atopic dermatitis is an IL-13-dominant disease with greater molecular heterogeneity compared to psoriasis. J. Invest. Dermatol. 139, 1480–1489 (2019).
Ottas, A. et al. Blood serum metabolome of atopic dermatitis: Altered energy cycle and the markers of systemic inflammation. PLoS ONE 12, e0188580 (2017).
von Bonin, A. et al. Inhibition of the IL-2-inducible tyrosine kinase (Itk) activity: a new concept for the therapy of inflammatory skin diseases. Exp. Dermatol. 20, 41–47 (2011).
Pasanen, A. et al. Identifying atopic dermatitis risk loci in 1,094,060 individuals with subanalysis of disease severity and onset. J. Invest. Dermatol. 144, 2417–2425 (2024).
Kuwahara, M. et al. Bach2-Batf interactions control Th2-type immune response by regulating the IL-4 amplification loop. Nat. Commun. 7, 12596 (2016).
Lessard, S. et al. Leveraging large-scale multi-omics evidences to identify therapeutic targets from genome-wide association studies. BMC Genomics 25, 1111 (2024).
Tsoi, L. C. et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet 44, 1341–1348 (2012).
Fu, D. et al. Downregulation of RUNX3 moderates the frequency of Th17 and Th22 cells in patients with psoriasis. Mol. Med. Rep. 13, 4606–4612 (2016).
Hantisteanu, S. et al. Runx3 prevents spontaneous colitis by directing the differentiation of anti-inflammatory mononuclear phagocytes. PLoS ONE 15, e0233044 (2020).
Das, P. et al. Keratinocytes: an enigmatic factor in atopic dermatitis. Cells 11 (2022).
Napolitano, M., di Vico, F., Ruggiero, A., Fabbrocini, G. & Patruno, C. The hidden sentinel of the skin: an overview on the role of interleukin-13 in atopic dermatitis. Front. Med. (Lausanne) 10, 1165098 (2023).
Gupta, R. K., Fung, K., Figueroa, D. S., Ay, F. & Croft, M. Integrative keratinocyte responses to TWEAK with IL-13 and IL-22 reveal pathogenic transcriptomes associated with atopic dermatitis. J. Invest. Dermatol. 144, 1071–1074.e6 (2023).
Humeau, M., Boniface, K. & Bodet, C. Cytokine-mediated crosstalk between keratinocytes and T cells in atopic dermatitis. Front Immunol. 13, 801579 (2022).
Iqbal, Z. et al. Homozygous and heterozygous disruptions of ANK3: at the crossroads of neurodevelopmental and psychiatric disorders. Hum. Mol. Genet. 22, 1960–1970 (2013).
Yazar, S. et al. Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease. Science 376, eabf3041 (2022).
Sleiman, P. M. et al. Variants of DENND1B associated with asthma in children. N. Engl. J. Med. 362, 36–44 (2010).
Almoguera, B. et al. Novel locus for atopic dermatitis in African Americans and replication in European Americans. J. Allergy Clin. Immunol. 143, 1229–1231 (2019).
Abuabara, K. et al. Genetic ancestry does not explain increased atopic dermatitis susceptibility or worse disease control among African American subjects in 2 large US cohorts. J. Allergy Clin. Immunol. 145, 192–198.e11 (2020).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Fuchsberger, C., Abecasis, G. R. & Hinds, D. A. minimac2: faster genotype imputation. Bioinformatics 31, 782–784 (2015).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Abraham, G., Qiu, Y. & Inouye, M. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics 33, 2776–2778 (2017).
Magi, R. & Morris, A. P. GWAMA: software for genome-wide association meta-analysis. BMC Bioinform. 11, 288 (2010).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Han, Y. et al. Genome-wide analysis highlights contribution of immune system pathways to the genetic architecture of asthma. Nat. Commun. 11, 1776 (2020).
Kerimov, N. et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet 53, 1290–1299 (2021).
Vosa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Bonder, M. J. et al. Identification of rare and common regulatory variants in pluripotent cells using population-scale transcriptomics. Nat. Genet. 53, 313–321 (2021).
Ota, M. et al. Dynamic landscape of immune cell-specific gene regulation in immune-mediated diseases. Cell 184, 3006–3021 e17 (2021).
Consortium, G. T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Taylor, D. L. et al. Integrative analysis of gene expression, DNA methylation, physiological traits, and genetic variation in human skeletal muscle. Proc. Natl Acad. Sci. USA 116, 10883–10888 (2019).
Ng, B. et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci. 20, 1418–1426 (2017).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 53, 1712–1721 (2021).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Zhang, J. et al. Plasma proteome analyses in individuals of European and African ancestry identify cis-pQTLs and models for proteome-wide association studies. Nat. Genet. 54, 593–602 (2022).
Sobczyk, M. K. et al. Triangulating molecular evidence to prioritize candidate causal genes at established atopic dermatitis loci. J. Invest Dermatol 141, 2620–2629 (2021).
Tanigawa, Y., Dyer, E. S. & Bejerano, G. WhichTF is functionally important in your open chromatin data? PLoS Comput. Biol. 18, e1010378 (2022).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Ghoussaini, M. et al. Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 49, D1311–D1320 (2021).
Pers, T. H. et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 6, 5890 (2015).
Dusart, P. et al. A tissue centric atlas of cell type transcriptome enrichment signatures. bioRxiv https://doi.org/10.1101/2023.01.10.520698 (2023).
Rheinwald, J. G. & Green, H. Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature 265, 421–424 (1977).
Arnette, C., Koetsier, J. L., Hoover, P., Getsios, S. & Green, K. J. In vitro model of the epidermis: connecting protein function to 3d structure. Methods Enzymol. 569, 287–308 (2016).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Anders, S., Pyl, P. T. & Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Lopez, D. V. & Kongsbak-Wismann, M. Role of IL-22 in homeostasis and diseases of the skin. APMIS 130, 314–322 (2022).
Dickson, M. A. et al. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 20, 1436–1447 (2000).
Zhang, H. et al. 818 Multi-condition TWAS for inflammatory skin disorders highlights roles of genetic signals in cytokine-stimulated keratinocytes. J. Investig. Dermatol. 143, S140 (2023).
Acknowledgements
The authors thank Xiuwen Zhang and John Lee for their support of UKBB data analysis. We acknowledge Jacob Degner, Emily King, Jeff Waring, and Zoltan Dezso for their critical review of the manuscript. This research was carried out using the UK Biobank resource under application number 26041. We acknowledge the BioBank Japan Project for access to summary statistics. We want to acknowledge the participants and investigators of the FinnGen study. The FinnGen project is funded by two grants from Business Finland (HUS 4685/31/2016 and UH 4386/31/2016) and the following industry partners: AbbVie Inc., AstraZeneca UK Ltd, Biogen MA Inc., Bristol Myers Squibb (and Celgene Corporation & Celgene International II Sàrl), Genentech Inc., Merck Sharp & Dohme Corp, Pfizer Inc., GlaxoSmithKline Intellectual Property Development Ltd., Sanofi US Services Inc., Maze Therapeutics Inc., Janssen Biotech Inc, Novartis AG, and Boehringer Ingelheim. Following biobanks are acknowledged for delivering biobank samples to FinnGen: Auria Biobank (www.auria.fi/biopankki), THL Biobank (www.thl.fi/biobank), Helsinki Biobank (www.helsinginbiopankki.fi), Biobank Borealis of Northern Finland (https://www.ppshp.fi/Tutkimus-ja-opetus/Biopankki/Pages/Biobank-Borealis-briefly-in-English.aspx), Finnish Clinical Biobank Tampere (www.tays.fi/en-US/Research_and_development/Finnish_Clinical_Biobank_Tampere), Biobank of Eastern Finland (www.ita-suomenbiopankki.fi/en), Central Finland Biobank (www.ksshp.fi/fi-FI/Potilaalle/Biopankki), Finnish Red Cross Blood Service Biobank (www.veripalvelu.fi/verenluovutus/biopankkitoiminta) and Terveystalo Biobank (www.terveystalo.com/fi/Yritystietoa/Terveystalo-Biopankki/Biopankki/). All Finnish Biobanks are members of BBMRI.fi infrastructure (www.bbmri.fi). Finnish Biobank Cooperative FINBB is the coordinator of BBMRI-ERIC operations in Finland. The Finnish biobank data can be accessed through the Fingenious® services (https://site.fingenious.fi/en/) managed by FINBB. J.E.G., M.K.S., and L.C.T. are supported by NIH-P30-AR075043. National Eczema Association (AD NEA23-CERG202) supported HV’s studies.
Author information
Authors and Affiliations
Contributions
B.R.G. and M.O. conceived the study with contributions of M.K.S., M.E.M., H.H., J.E.G., and K.M.S. B.R.G. conceived and performed the meta-GWAS and derived downstream analyses, M.O. conceived and performed QTL colocalization and derived downstream analysis. F.T. conceived and performed the cell-type enrichment analysis, including the ideation of the MAGMA approach. B.R.G. and M.O. conceived and performed candidate gene assessment analysis. M.E.M., supervised by H.H., generated the CHOP GWAS and A.H.S., D.W., J.T.G., A.H.S., H.V., L.Y., and F.D.M. contributed with QC and interpretative analyses. M.K.S., supervised by J.E.G., conducted the siRNA and 3-D epidermal model experiments. C.H. contributed to the profiling of keratinocyte-linked genes to the interpretation of the siRNA experiments. R.B., J.M.K., supervised by L.C.T., generated the keratinocyte eQTLs, and the skin sc-RNAseq experiment and corresponding data. R.U., M.T.P., and Q.L. contributed to quality control, and interpretation of the keratinocyte and skin datasets. B.R.G. and M.O. led the writing and editing of the manuscript and supplement; all authors contributed to the editing of the manuscript and supplement. B.R.G. and M.O. coordinated analyses of all contributing authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
B.R.G., M.O., K.M.S. are employees of AbbVie. F.T. and C.H. were employees of AbbVie at the time of the study. J.E.G. (University of Michigan) has received research support from AbbVie, Janssen, Almirall, Prometheus Biosciences/Merck, BMS/Celgene, Boehringer Ingelheim, Galderma, Eli Lilly, and advisor to Sanofi, Eli Lilly, Galderma, BMS, Boehringer Ingelheim. M.K.S., R.U., M.T.P., Q.L., R.B., J.M.K., L.C.T. are employees of University of Michigan and have no funding to disclose. M.E.M., A.H.S., F.D.M., D.W., J.T.G, L.Y., H.V., H.H. are employees of the Children’s Hospital of Philadelphia and have no funding to disclose. The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.
Peer review
Peer review information
Nature Communications thanks Chikashi Terao, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Oliva, M., Sarkar, M.K., March, M.E. et al. Integration of GWAS, QTLs and keratinocyte functional assays reveals molecular mechanisms of atopic dermatitis. Nat Commun 16, 3101 (2025). https://doi.org/10.1038/s41467-025-58310-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58310-7
