
Abstract
Conjugated bile acids (BAs) are multi-functional detergents in the gastrointestinal (GI) tract produced by the liver enzyme bile acid-CoA:amino acid N-acyltransferase (BAAT) and by the microbiome from the acyltransferase activity of bile salt hydrolase (BSH). Humans with inflammatory bowel disease (IBD) have an enrichment in both host and microbially conjugated BAs (MCBAs), but their impacts on GI inflammation are not well understood. We investigated the role of host-conjugated BAs in a mouse model of colitis using a BAAT knockout background. Baat−/− KO mice have severe phenotypes in the colitis model that were rescued by supplementation with taurocholate (TCA). Gene expression and histology showed that this rescue was due to an improved epithelial barrier integrity and goblet cell function. However, metabolomics also showed that TCA supplementation resulted in extensive metabolism to secondary BAs. We therefore investigated the BSH activity of diverse gut bacteria on a panel of conjugated BAs and found broad hydrolytic capacity depending on the bacterium and the amino acid conjugate. The complexity of this microbial BA hydrolysis led to the exploration of bsh genes in metagenomic data from human IBD patients. Certain bsh sequences were enriched in people with Crohn’s disease particularly that from Ruminococcus gnavus. This study shows that both host and microbially conjugated BAs may provide benefits to those with IBD, but this is dictated by a delicate balance between BA conjugation/deconjugation based on the bsh genes present.
Similar content being viewed by others

Introduction
Inflammatory bowel disease (IBD) is a chronic inflammatory disease of the gastrointestinal (GI) tract, including Crohn’s disease (CD) and ulcerative colitis (UC)1. IBD has become a global disease with a rising prevalence, especially in the Western world, affecting more than 2.39 million individuals in North America2. Although myriad therapies are available, including aminosalicylates, corticosteroids, immunomodulators and biologics, these existing treatments only provide temporary remission for most patients and mostly target symptoms of the disease not its underlying causes3. The etiology of IBD is still unclear, but evidence suggests that a multitude of factors are responsible for its pathogenesis, including genetic predispositions, environmental triggers, mucosal immune dysregulation, and intestinal microbiota dysbiosis4,5. Mounting evidence shows that the trillions of microorganisms that inhabit the human gut play an important role in the occurrence and development of IBD, but the mechanisms of their contribution to disease development remains mostly unknown6.
Previous studies have highlighted microbially-derived metabolites as bioactive in IBD, particularly bile acids (BAs) produced by the host liver and then modified by the microbiome7,8,9. BAs are steroidal natural products that are synthesized in the liver from cholesterol10. The two primary BAs, cholic acid (CA) and chenodeoxycholic acid (CDCA), are conjugated with either taurine or glycine in a reaction catalyzed by bile acid CoA:amino acid N-acyltransferase (BAAT)11. These human conjugated primary BAs are then released into the gallbladder before postprandial secretion into the small intestine where they facilitate fat digestion and absorption of cholesterol and vitamins12. About 95% of secreted BAs are reabsorbed in the terminal ileum and returned to the liver in a mechanism known as enterohepatic circulation13,14. The remaining 5% of BAs are transformed by the gut microbiome to form a diverse pool of secondary BAs in the gastrointestinal tract13. The critical first step in the metabolism of BAs by the gut microbiome is catalyzed by bile salt hydrolase (BSH, EC 3.5.1.24, also known as choloylglycine hydrolase), a homotetrameric enzyme that deconjugates acyl- bound glycine and/or taurine from the sterol core of the primary BAs15. This enzymatic step is considered a “gateway reaction” to subsequent microbial BAs modifications, which are diverse and control the host’s overall BA pool11,16. BSHs are identified across all major bacterial and archaeal phyla in the gut, but their catalytic efficiencies and substrate preferences vary15,17. Two recent studies revealed that BSH also has aminoacyltransferase activity that conjugates diverse amino acids to cholic acid, to generate the microbial conjugated bile acids (MCBAs), which further increase the complexity and diversity of the human BAs pool18,19,20. Some of these novel MCBAs were found to activate the transcription factors Pregnane X Receptor and Aryl Hydrocarbon Receptor, indicating that they may have a role in regulating metabolic homeostasis7. The recent expansion of conjugated BA diversity from the microbiome brings into question the role these compounds may have in gut inflammation and IBD.
Some studies have demonstrated that dysregulation of BA metabolism and BSH activity was directly related to IBD7,21,22. BA profiling of GI samples from patients has consistently shown increased levels of primary BAs and decreased secondary BAs8,23,24. More recent studies have shown that MCBAs are also elevated in the inflamed gut7,20, which is somewhat of an enzymatic contradiction, because BSH is responsible for hydrolyzing the abundant primary BAs while simultaneously producing MCBAs from its acyl transfer activity18. Complicating the matter further, patients with active IBD had been shown to have decreased levels of BSH activity in their fecal samples22. Clearly, the balance between conjugation and deconjugation of host and microbial enzymes is complex and there is a knowledge gap surrounding the role of BSHs and conjugated BAs in IBD. Filling that gap may lead to the development of novel therapeutics using conjugated BAs or specific BSHs for treatment of this chronic inflammatory disease.
This study investigated the implications of BA conjugation in a mouse model of GI inflammation. Our initial experiments utilized a novel BAAT knockout mouse model with highly reduced conjugated BAs to determine if the absence of these molecules would alter disease pathology. It was found that conjugated BAs are important for limiting pathology in this mouse model, with indications of a mechanism via improved epithelial barrier function. In light of these findings and new knowledge of the diversity of conjugated BAs7,20, we investigated the hydrolytic capacity of 17 bacteria on diverse BAs and explored the BSH sequence space in publicly available metagenomic datasets from subjects with IBD. Our results show that conjugated BAs are important for reducing pathology in an inflamed mammalian gut, but that the balance between hydrolysis and conjugation is complex and dependent on the BSH sequences present in the microbiome.
Results
Baat −/− KO mice have heightened pathology compared to wild-type mice under DSS treatment
As an approach to investigate the roles of BA conjugation in gut inflammation, we employed a recently developed Baat−/− KO mouse model that has highly reduced conjugated BAs, particularly reduced TCA (full characterization of Baat−/− KO model available in refs. 25,26). The animal experiment was carried out according to the procedure shown in Fig. 1a. Mice in the DSS groups were treated with 2.5% DSS for 7 days and were then humanely euthanized on day 7. Daily measurement of body weight showed that Baat−/− KO mice lost more weight compared with WT animals during the course of the experiment (Fig. 1b). The DAI score (Fig. 1c), which evaluates colitis severity based on body weight loss, rectal bleeding, and stool status, was significantly higher in the KO-DSS group compared to the WT-DSS group after 7 days (p < 0.05), indicating a greater impact of DSS on Baat−/− KO mice. Additionally, the measurement of colon lengths demonstrated that Baat−/− KO group mice displayed significantly (p < 0.05) shorter colons compared to the WT-DSS group (Fig. 1d, e). There was no significant difference in histologic score, epithelial damage, nor mucosal depth in Baat−/− KO mice compared with WT mice under DSS treatment (Fig. 1f, g). Taken together, the results indicated that Baat−/− KO mice showed enhanced signs of DSS-induced colitis compared to the wild-type mice, without a notable difference in histopathology.

a Experimental animal model in the Baat−/− KO mice versus wild-type mice under DSS treatment for 7 days (n = 4–5). b Weight changes during the experiment. c Daily DAI scores. d Example images of the colons from each group of mice. e The colon lengths of each animal. f Representative photomicrographs of H&E-stained (original magnification, ×100, scale bar = 100 μm). g Histology scores with representative H&E staining. Data in (b, c) expressed as the mean ± SEM; n = 4–5 animals per group; all boxplots are the interquartile range, center line is the median, with the whiskers denoting minima and maxima; statistics analyzed by one-way ANOVA with Tukey’s post hoc, and *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
TCA supplementation rescues severe phenotypes of Baat −/− KO mice under DSS-induced colitis
Because our previous study found that the Baat−/− KO mice had 27-fold lower taurine-conjugated BAs than WT in their liver25, we hypothesized that TCA plays a vital role in the heightened host response to DSS in these animals. To test this, we supplemented Baat−/− KO mice with chow containing 0.3% TCA with and without DSS treatment (Fig. 2a). After 6 days of treatment with 2.5% DSS, mice in the DSS-TCA treatment group lost less weight compared to those only exposed to DSS (Fig. 2b). Moreover, TCA supplementation resulted in a more rapid recovery of body weight during the 7-day recovery period post-DSS treatment than for mice only exposed to DSS (p < 0.05, Fig. 2b). Additionally, TCA supplementation had a positive effect on the length of the mouse colon (Fig. 2c, d, p < 0.05) and reduced the impact of DSS treatment on the spleen index (spleen weight: body weight ratio) (Fig. 2e, p < 0.05). The DAI score on day 13 in the DSS-TCA group was also significantly lower compared to the DSS group (Fig. 2f, p < 0.05). While the overall histologic scores were not significantly different (Figure S1a, b), the number of goblet cells counted by AB staining was higher in DSS-TCA group compared to the DSS group (p = 0.062), with visual detection of goblet cells and mucus staining returning in mice receiving TCA supplementation (Fig. 2g, h).

a Experimental animal design using Baat−/− KO mice fed with 0.3% TCA under DSS-induced colitis (n = 6-8). (b) Weight changes during the experiment (Data expressed as the mean ± SEM). c Example images of the colons from each group of mice. d The colon lengths of each animal. e Spleen index (spleen weight: body weight ratio). f DAI scores on day 13. g Representative photomicrographs of alcian blue-periodic acid-Schiff (AB-PAS)-stained colon sections (original magnification, ×100, scale bar = 100 μm). h Goblet cell counts from each group of mice. i Representative photomicrographs of MUC-2 staining colon sections (scale bar = 50 μm). j Quantification of MUC2 staining. (k–n) mRNA expression of MUC-2, ZO-1, FXR and TGR5 in colon. All boxplots are the interquartile range, center line is the median, with the whiskers denoting minima and maxima; n = 6–8 animals per group. Weight changes, colon lengths, spleen index, DAI scores, AB-PAS staining, goblet cell counts, and mRNA expression data were tested by one-way ANOVA with Tukey’s post hoc. MUC-2 staining data analyzed by Kruskal-Wallis test followed by Dunn’s test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
TCA treatment increased the expression of mucosal integrity and BA receptors markers in Baat −/− KO mice on DSS
In light of the histological analysis indicating improved goblet cell function and mucus production, we explored the gene and protein expression changes related to cell barrier integrity and mucus physiology in the colons in DSS mice supplemented with TCA by RT-qPCR and immunohistochemistry (IHC). The protein and gene expression of MUC-2 was increased in the DSS-TCA group compared to the DSS group (Fig. 2i-k, p < 0.05), which is consistent with the alcian blue-periodic acid-Schiff (AB-PAS) goblet cell counts. Additionally, we found that the gene expression of intercellular tight junction genes, including Claudin-1, Occludin-1, and ZO-1 was higher in the TCA-supplemented groups compared to the non-supplemented groups, though only ZO-1 reached significance at an alpha-level below 0.05 (Fig. 2i and Figure S2a, b, Claudin-1 p = 0.09). Further, IHC showed that the mice treated with DSS and TCA had a higher ZO-1 protein levels than the DSS control group mice (Figure S1c, d, p < 0.05).
To gain further insight into the mechanism by which TCA affects intestinal inflammation, we also measured mRNA expression levels of pro-inflammatory factors and bile acid-related receptors (Figure S2c, d). Tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β) gene expression was higher in DSS treated animals as expected, however, the DSS-TCA and DSS groups were not significantly different from each other (p > 0.05). Expression of BA receptors Farnesoid X Receptor (FXR) and Takeda G-protein Receptor 5 (TGR5, also known as GPBAR1) were significantly increased in the TCA treatment groups compared to untreated groups (p < 0.05) and were significantly higher in the TCA group than in the TCA-DSS group (Fig. 2m, n, p < 0.05).
Microbiome changes from TCA treatment of Baat −/− KO mice on DSS
To further investigate the effects of TCA supplementation on Baat−/− KO mice under DSS-induced colitis, the cecum and feces were collected for 16S rRNA gene amplicon sequencing. Microbiome community changes among the treatment groups were similar in the cecum and fecal data (Figure S3 and S4), we therefore focused more detailed analysis on the cecum profiles. TCA treatment slightly decreased the microbiome alpha-diversity and species richness compared to untreated animals (p > 0.05) and DSS treatment decreased these measures further (p < 0.05, Fig. 3a). TCA supplementation during DSS treatment prevented the DSS-induced loss in microbiome diversity, making its measure more similar to the TCA only treated group (Fig. 3a, p < 0.05). The PCoA analysis results showed that the experimental groups had significantly different microbiome profiles from each other with the DSS and non-DSS groups being distinctly different. Interestingly, TCA supplementation during DSS treatment shifted the microbiome profiles towards the that of the non-DSS animals, indicating some recovery of the microbiome structure (Fig. 3b, as indicated by PERMANOVA testing p < 0.05). Analysis of taxa changes at the phylum level between DSS and DSS-TCA treatments showed a reduction in the relative abundance of Firmicutes_A and Bacteroidota, and an increase in the relative abundance of Proteobacteria in the DSS treatment (Figure S5a). At the genus level, the relative abundance of Akkermansia was increased in the DSS-TCA treatment compared to DSS treatment in both the cecum and feces, but this did not reach statistical significance (p = 0.28, Figure S5b). TCA supplementation significantly diminished the DSS-induced loss in the relative abundance of Lachnospiraceae family (p < 0.05, Fig. 3c), a group known to metabolize BAs and produce secondary BAs such as deoxycholate (DCA).

a Microbiome alpha diversity determined by the Shannon index; n = 4-5 animals per group (b) The principal coordinate analysis (PCoA) based on Bray-Curtis dissimilarity shows the microbiome β-diversity; n = 4–5 animals per group. c Relative abundance of Lachnospiraceae. d–g The principal coordinate analysis (PCoA) based on Bray-Curtis dissimilarity of liver, serum, cecum, and feces. h Average relative abundance of TCA, GCA, CA, DCA, oxoCA, CA-MCY, CA-MCYO, CA-MCYO2, and MCBAs. Heatmap values (Z-score by rows) represent mean abundance of each BA in each group. Grey color indicates values that were not detected. The number of mice in the microbiome and metabolome results was n = 4–5 mice per group. All boxplots are the interquartile range, center line is the median, with the whiskers denoting minima and maxima. Microbiome data analyzed by one-way ANOVA with Tukey’s post hoc, and *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Metabolites data analyzed by Kruskal-Wallis followed by Dunn’s post hoc, # indicated the significant differences between HC and TCA groups (p < 0.05), × indicated the significant differences between HC and DSS groups (p < 0.05), and * indicated the significant differences between DSS and DSS-TCA groups (p < 0.05).
DSS and TCA treatment alter conjugated and secondary BA profiles in Baat −/− KO mice
We used untargeted metabolomics to profile changes in the metabolome under TCA supplementation in the DSS Baat−/− KO mouse experiment. Although TCA supplementation had no significant effect on overall metabolite structure of the liver or serum (Fig. 3d, e, as indicated by PERMANOVA analysis p > 0.05), it had a pronounced impact on cecum and fecal metabolomes (Fig. 3f, g, as indicated by PERMANOVA analysis p < 0.05). Interestingly, the beta diversity results of the metabolomes showed that the TCA supplemented groups were more similar to each other regardless of DSS treatment, though the effect of DSS was also strong (Fig. 3f, g).
As expected, supplementation of the control and DSS treated animal’s diet with TCA significantly increased its abundance in liver, serum, and cecum, but not in the fecal samples (p > 0.05, Fig. 3h). Similar to reports in human IBD literature8, the induction of gut inflammation by DSS alone increased TCA abundance in feces (Fig. 3h, p > 0.05). The other primary conjugated BA glycocholic acid (GCA), only detected in cecum and feces, was also higher in samples from the DSS treatment group compared to the combined DSS-TCA treatment or the non-DSS treatments (Fig. 3h, p < 0.05). Accordingly, the abundance of CA was markedly increased by TCA treatment in the liver, serum, cecum, and feces of both control and DSS treated animals (Fig. 3h, p < 0.05). TCA treatment also increased the abundance of secondary BAs produced by the microbiome including DCA in serum, cecum, and feces (Fig. 3h) and the oxidized secondary BAs (oxoCAs) in cecum and feces (Fig. 3h, p < 0.05). This indicated that TCA was being hydrolyzed by the intestinal microbiome in the colon to generate CA, DCA and oxoCAs, though other aspects of BA metabolism and signaling may contribute to increased CA. This finding contrasted that from the human IBD literature, where there is abundant TCA in those with IBD but secondary BAs, such as DCA, are not increased8.
Due to the growing diversity of BA conjugation biochemistry from the host and its microbiome including newly discovered cysteamine conjugates that are antagonists of FXR25,27, we investigated differences in the abundances of some of the novel conjugated BAs in response to DSS and TCA treatments. The abundance of the host produced CA-methylcysteamine (CA-MCY), CA-methylcysteamine sulfoxide (CA-MCYO) and CA-methylcysteamine sulfodioxide (CA-MCYO2) was generally increased in the inflamed gut of mice on DSS, with statistical significance found in the fecal samples (HC group compared to DSS group p < 0.05, Fig. 3h). TCA supplementation subsequently reduced the levels of these unique cysteamine-conjugated BAs particularly CA-MCYO and CA-MCYO2 in the feces (p < 0.05, Fig. 3h). MCBAs, including CysCA, GluCA and LysCA, were also significantly higher in the feces of mice on DSS treatment (p < 0.05, Fig. 3h), another trend matching that observed in people with IBD7,20. TCA decreased MCBAs under DSS-induced colitis, though this did not reach statistical significance between the DSS and DSS-TCA groups.
Highly variable hydrolytic activity of gut bacteria on conjugated BAs in vitro and in vivo
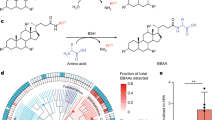
Collectively, our multi-omics profiling of Baat−/− KO mice pointed to the importance of diverse conjugated BAs in promoting health in the inflamed mammalian gut; however, there was also evidence for extensive hydrolysis of the supplemented TCA and production of various secondary BAs by the microbiome, which could compromise the beneficial effects. Therefore, to better understand the relationships between altered conjugated and secondary BA profiles and microbiome structure in a model of GI inflammation, we explored the BSH hydrolysis activity among diverse gut bacteria on diverse conjugated BAs. A total of 17 gut bacterial strains belonging to 9 different genera (strain list in supplementary data S1) and a fresh community of gut microbes from mouse fecal samples were evaluated for their ability to hydrolyze TCA, GCA and varied MCBAs in vitro. Genome sequence searching determined that 14 of these strains contained an annotated bsh gene and 3 (Clostridium hylemonae, Clostridium scindens and Peptostreptococcus anaerobius) did not. The strains and fecal community were separately cultured for 48 h with each of the 10 conjugated BAs at 1 mM concentration then analyzed for hydrolysis by UPLC-MS/MS. We observed an overall low level of conjugate hydrolysis, which expectedly, included those strains without a known bsh gene (Fig. 4a). However, C. scindens was able to hydrolyze diverse conjugated BAs (particularly those with small amino acid conjugates GCA, SerCA and AlaCA), even though it does not have a known BSH enzyme in its genome. This is similar to its MCBA production activity as reported in ref. 18. Among bacteria containing the bsh gene, we observed variable levels of hydrolysis that depended on the amino acid conjugate. Enterococcus faecalis, Lactiplantibacillus plantarum, C. perfringens and Enterocloster bolteae showed robust hydrolysis of the conjugates, but with some limits in their ability to hydrolyze GluCA and ThrCA. Bacteroides fragilis exhibited a strong propensity for the host-produced TCA, GCA, with some ability to hydrolyze the MCBAs SerCA and AlaCA. In addition, in vitro culture of a mouse fecal community also showed strong hydrolysis of TCA, SerCA, TyrCA, and AlaCA, but GluCA, ThrCA, and LeuCA were more recalcitrant (Figure S6). Ruminococcus gnavus (newly Mediterraneibacter gnavus) and E. clostridioformis, which were enriched in CD patients (see below section on BSH characterization in IBD), exhibited a weak ability to hydrolyze conjugated BAs. Overall, these in vitro experiments showed that TCA, GCA, SerCA and AlaCA were more prone to hydrolysis whereas GluCA, and ThrCA were more resistant. These varied hydrolytic capacities of different gut bacteria may explain both observations in our murine experiments, that linked an altered gut microbiome and secondary BA production in the inflamed murine gut, and evidence in the literature, showing altered conjugated BA profiles from the host and its microbiome in patients with IBD7.

a The abundances of conjugated cholic acids are represented as colors corresponding to log10transformed peak areas (area under the curve, AUC). Values represent the mean from n = 3 replicates of log10 areas changes compare to medium after 48 h in vitro culture. Phylogenetic tree of 17 Bacteroidetes strains using GTDB-tk based on whole bacteria genome. b Experimental animal design in wild-type C57BL/6 J mice fed with a final dose of 50 mg/kg TCA or GluCA peanut butter pellets under DSS-induced colitis (n = 10 each group). c–e Relative abundance of CA, DCA and oxocholic acid abundance in feces. Fecal metabolites data was analyzed by Kruskal-Wallis followed by Dunn’s post hoc, and *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All boxplots are the interquartile range, center line is the median, with the whiskers denoting minima and maxima.
Given that GluCA was more resistant to hydrolysis than TCA by bacteria in vitro, as well as its reported enrichment in CD patients7, we further compared the differences in the effects of TCA and GluCA supplementation on BA hydrolysis in DSS induced colitis model. Wild type C57BL/6 J mice in DSS-TCA and DSS-GluCA groups mice were fed peanut butter pellets infused with 50 mg/kg of TCA or GluCA every day for 17 days. On day 10, mice in the DSS groups were treated with 2.5% DSS for 7 days and then all animals were humanely euthanized on day 17 (Fig. 4b). In this C57BL/6 J wild type background, there was no significant difference among the DSS, DSS-TCA and DSS-GluCA groups in terms of disease signs including weight loss, colon length or spleen index (Figure S7a-c, p > 0.05). Metabolomics analysis showed that TCA significantly increased the abundance of CA and other secondary BAs (oxo-BAs), but GluCA treatment did not (Fig. 4c, d, p < 0.05). DCA showed a similar trend, but this did not reach statistical significance (Fig. 4e). This suggests that TCA was more readily hydrolyzed by the gut microbiome to produce secondary BAs than GluCA treatment, mirroring the effects shown from bacterial cultures in vitro.
BSHs characterization in IBD
Our in vivo and in vitro data show that conjugate BAs, particularly TCA, are important for reducing pathology in the inflamed murine gut, but the abundance of both conjugate BAs and secondary BAs are dictated by the activity of BSH enzymes from the gut microbiome, which have highly varied activity depending on the gene sequence and BA conjugate encountered. Therefore, we mined the bsh gene sequences and abundance from 774 samples of 6 publicly available metagenomics datasets (Table S2) for comparison among disease types. Subjects with either Crohn’s or ulcerative colitis had lower Shannon diversity indices and observed sequences of BSHs than healthy individuals (Fig. 5a, b, p < 0.05). The alpha-diversity of BSHs was significantly lower in CD and UC patients than in healthy controls (Fig. 5a, b), but there was some variation depending on the cohort analyzed (Figure S8a, b). Beta-diversity of BSH profiles was also different between IBD patients and healthy controls (as indicated by PERMANOVA analysis of the collective cohorts, p < 0.05), with some variation within cohorts (Fig. 5c and Figure S9). Furthermore, the healthy controls exhibited the highest cumulative relative abundance of BSHs, while the CD patients exhibited the lowest (Fig. 5d; Figure S10). Our results suggest reduced BSHs diversity and abundance in IBD patients, especially those with CD, and the collective sequence space of BSH genes in the microbiome are unique among IBD subtypes.

a, b The alpha diversity of BSHs in IBD patients and healthy controls, which was measured using the Shannon index and Observed BSHs. c The β-diversity of BSHs was measured by principal coordinate analysis (PCoA) based on Bray-Curtis dissimilarity. d The cumulative relative abundance of BSHs in IBD patients and healthy controls. e–g Taxonomic characterization of BSHs at phylum, family, and genus levels. h–k Examples of significant differing BSHs in IBD compared to healthy controls. Data was analyzed by two-sided Wilcox rank-sum test followed by BH correction, and *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, sample sizes available in methods. All boxplots are the interquartile range, center line is the median, with the whiskers denoting minima and maxima.
A BSH phylogenetic tree was built using the amino sequences of the 244 identified BSHs (Figure S11 and supplementary data S2) analyzed for enrichment in particular bacterial BSH sequences. The BSHs were assigned to 76 genera from 4 phyla, including Actinomycetota, Bacillota, Euryarchaeota, and Pseudomonadota (Fig. 5e). The majority of BSHs belonged to the Lachnospiraceae, Oscillospiraceae, and Clostridiaceae families (Fig. 5f), and the Blautia, Clostridium, Ruminococcus, Gemmiger, Roseburia, and Eubacterium genera (Fig. 5g). The abundance changes of BSHs in patients with CD (n = 207) and UC (n = 186) compared to healthy controls (n = 381) were tested via Wilcox rank-sum test followed by BH correction. The results showed that there were 218 and 159 BSHs significantly (p < 0.05) changed in CD and UC patients, respectively, compared to healthy controls. Among these, the proportion of BSHs with increased relative abundance in patients with CD and UC was 14.68% (32/218) and 29.56% (47/159), respectively. This reflected that the relative abundance of BSHs was mostly decreased in the patients with IBD. The BSHs from Lachnospiraceae were significantly decreased in patients with CD compared to healthy controls (Fig. 5h, p < 0.05). Additionally, the BSHs from unclassified Clostridium, unclassified Bacillota and Boudabousia were significantly decreased in the CD patients compared to the healthy controls across the 6 IBD cohorts (p < 0.05). Notably, the relative abundance of the BSH gene of an unknown Ruminococcus spp. was significantly reduced in CD patients (Fig. 5i), however, Ruminococcus gnavus BSH (found in all cohorts except for HMP and PRJNA42990) was significantly increased (Fig. 5j, p < 0.05). The gene from the Lachnospiraceae member E. clostridioformis was also significantly increased in LLdeep, PRISM and PRJEB1220 cohorts compared to the compared to healthy controls (Fig. 5k, p < 0.05). Collectively, this analysis of data from subjects with IBD shows the varied BSH sequence space between diseased and healthy subjects maybe responsible for the altered conjugated BA profiles which have been reported in the IBD literature8,23,24.
Discussion
BAs are hepatically synthesized cholesterol derivatives with profound effects on systemic metabolism, such as lipid digestion, vitamin absorption and immune regulation28. Recent research has shown that BA chemical diversity was greatly underappreciated, particularly that of conjugated BAs7,20,29. Thus, our understanding of the role of BAs in GI disease is incomplete, making it challenging to establish clear causation between altered BA profiles and a diseased state, which is a characteristic of virtually all GI disorders under study7,20,29. For example, clinical studies of IBD have shown a dysregulation of BAs metabolism, with a consistently observed increase in primary conjugated BAs, including TCA and GCA, and a decrease in secondary BAs, such as DCA8,23. However, the causal relationships between BA dysregulation and IBD, the role of the ever-expanding diversity of these molecules, and the mechanisms by which they impact disease are poorly understood.
To begin to fill this knowledge gap, we employed the novel Baat−/− KO mouse model that presents with highly reduced primary BAs to assess their effects on DSS-induced colitis. Our first experiments showed that Baat−/− KO mice exhibited more severe colitis signs compared to WT mice under 2.5% DSS induced colitis, with shorter colon length and more weight loss, indicating the host-conjugated TCA plays an important role in this inflammatory condition. To verify this, we fed Baat−/− KO mice TCA supplemented food which largely decreased signs of colitis from developing. Though further work on cytokine profiling and immune cell responses are needed in this KO mouse model, there was preliminary evidence that this improvement was due to the preservation of intestinal epithelial integrity, not necessarily a reduction in inflammatory signaling. Colitis-induced damage to the epithelial barrier was limited by TCA supplementation, as seen by the thicker mucus layer of the colon compared to mice only subjected to DSS, higher staining of mucus filled goblet cells, maintenance of mucin expression levels, and rescued expression of tight junction markers, particularly ZO-1. The mucus layer of the intestines plays a crucial role in maintaining gut health and protecting the epithelium from harmful factors30,31. The MUC-2 glycoprotein, which is synthesized in goblet cells and a major component of the gut mucus layer32, is altered in IBD, with UC patients excreting less during the active phase33. Similarly, tight junctions genes, which play a central role in epithelial barrier regulation34, are also down-regulated in IBD patients35. In addition, a significant increase in the mRNA expression levels of the BA receptors FXR and TGR5 with TCA supplementation was observed, which was irrespective of DSS treatment. FXR is a nuclear receptor mainly found in intestinal epithelial cells, hepatocytes, and immune cells, and TGR5 belongs to the superfamily of G protein-coupled receptors and is expressed in epithelial cells21. Studies found that activation of FXR alleviated IBD by improving the function of the intestinal mucosal barrier, and TGR5 activation in intestinal stem cells may promote regeneration of enterocytes21,36. The metabolomics analysis suggested that TCA supplementation led to a significant increase of DCA. DCA has been shown to be a potent agonist of FXR and TGR537. Sinha et al. showed that supplementation with DCA ameliorated intestinal inflammation in a mouse model of colitis, which was in part dependent on TGR523. The pathological and multi-omics analysis of Baat−/− KO mice in the DSS model demonstrates that conjugated BAs are important for reducing pathology from inflammation and the likely mechanism is through their maintenance of the mucus barrier.
Several previous studies have demonstrated that intestinal microbiota plays a critical role in IBD where there is a dysbiosis and overall loss of microbial diversity compared to healthy individuals, particularly during flares of disease symptoms8. Our study in mice replicated this microbial diversity loss, which was then rescued by TCA supplementation. Furthermore, we found a higher relative abundance of Lachnospiraceae in response to TCA supplementation, irrespective of DSS treatment. The bacteria belonging to the Lachnospiraceae family, whose abundance is also reduced in patient with IBD38, are known to manipulate BAs including the 7α-dehydroxylating bacteria, which are responsible for converting primary BAs into secondary BAs39. Additionally, TCA supplementation increased the relative abundance of the mucus-layer-degrading bacteria Akkermansia in cecum and feces, but only under DSS-induced colitis. Akkermansia, which normally colonizes the mucus layer of the intestine, is widely recognized for its protective role in IBD and its ability to degrade mucus40. Studies have demonstrated that Akkermansia can alleviate DSS-induced colitis by repairing epithelial damage and strengthening the mucosal barrier41. This is an intriguing finding, as it indicates that this conjugated BA may facilitate microbial niche recovery in an inflamed gut, including microbes residing in a niche comprised of a recovering mucosal layer. The mechanism of TCA induced microbial diversity in these experiments remains unknown but could be due to antimicrobial properties of BAs reshaping microbial interactions or simply a manifestation of the improved GI epithelial barrier health. Due to the varied antimicrobial activity of these molecules18, the effect of conjugated BAs on gut microbial diversity in an inflamed environment is an important area for further research.
One of the more interesting findings in this study was the increase in conjugated BAs of diverse sources in tandem with the increase in secondary BAs. It has long been established that patients with IBD have elevated liver-conjugated BAs (TCA and GCA) in their gut and recent evidence suggests microbial conjugates are also increased7,8,20. Accordingly, diverse conjugated BAs, including MCBAs and those BA-methylcysteamine (BA-MCY), made from the complicated action of the human vanin 1 (VNN1) enzyme39, were also elevated in the inflamed animals in this study. This demonstrates that GI inflammation in humans and mice selects for the production of diverse forms of conjugated BAs beyond just TCA and GCA. However, the IBD literature shows that this is at the expense of lowered secondary BAs, likely due to reduced BSH activity from the microbiome22. In part to better understand this discrepancy, we explored the hydrolytic capacity of BSH enzymes from diverse gut bacteria exposed to chemically synthesized MCBAs compared to TCA and GCA. We found generally lower hydrolytic capability than might be expected, even when including bacteria with a bsh gene annotated in their genome. This suggests our current understanding of BSH function is still limited, particularly its substrate specificity. There are many possible explanations for the low hydrolysis of conjugated BAs observed here, which include the inability of certain gut microbial species to import these compounds and hydrolyze them and steric hindrance within the BSH active site for particular conjugated BAs. More research is needed to translate the identification of a bsh gene in a microbial genome to activity of the enzyme on conjugated BAs in vivo. Furthermore, acyl-conjugate chemical diversity altered the hydrolytic efficiency of BSH, demonstrating its substrate specificity at the acyl-site also varies. Enterococcus faecalis, L. plantarum, C. perfringens and E. bolteae showed robust hydrolysis of the conjugated BAs, and our previous study found that these bacteria were also robust MCBAs producers18. Surprisingly, C. scindens, without a clear bsh gene, could hydrolyze GCA, SerCA and AlaCA, which indicates there may be other enzymes capable of performing hydrolase functions on conjugated BAs yet to be identified. We also found that TCA, GCA, SerCA and AlaCA were more readily hydrolyzed by bacteria than other conjugated BAs. Foley et al. found that the deconjugation of AlaCA and SerCA from Lactobacillaceae BSH was robust, matching that of GCA and TCA42. They suggested that because alanine and serine are small amino acids, akin to glycine, that steric constraints may limit deconjugation of larger ones33. GluCA and ThrCA were less susceptible to hydrolysis by the bacteria tested here, implicating side chain hydrophilicity may also limit hydrolysis. These in vitro results translated in vivo, where mice self-administered peanut butter pellets infused with TCA and GluCA under DSS-induced colitis had varied production of secondary BAs, with GluCA limiting their production. Further work on the hydrolytic susceptibility of the diverse BAs now found in humans7 and their implications for GI health are needed, especially considering this study shows that conjugated BAs can alter outcomes of GI inflammation.
The findings here that TCA improves pathology in DSS induced colitis, but its presence in the gut is dictated by a balance between host conjugation and the varied hydrolytic capacities of the gut microbiota, led us to investigate the BSH sequence space in metagenomic data from patients with IBD. It was found that BSH diversity and abundance is significantly decreased in patients with IBD compared to the healthy controls, particularly those from the Lachnospiraceae; however, BSHs from Ruminococcus gnavus and E. clostridioformis were significantly increased. Comparing this finding to our hydrolytic measurements and MCBA conjugation from previous studies18 revealed these two bacteria were robust producers of MCBAs but had limited hydrolysis of conjugated BAs. This in vivo to in vitro comparison demonstrates the importance of understanding the hydrolytic capacity of diverse conjugated BAs and the role of a dysbiotic microbiome in shaping the BA profile in an inflamed human gut. It may also help explain why patients with IBD have high amounts of host-conjugated BAs and MCBAs, which is somewhat of an enzymatic paradox, because the former are eliminated and the later produced by activity of the same BSH enzyme18,19. Collectively, our study points to an intricate balance between BA conjugation by the host, deconjugation and reconjugation by the microbiota and a healthy mucus barrier in IBD. Our pathological findings mirror that of Li et al.33, who showed that conjugated BAs improve barrier integrity in gut epithelial cells and that inhibiting BSH has a similar effect in vivo. Both this study and ours points to the potential to manipulate the conjugated BA profile and gut bacteria that shape it to benefit patients with chronic inflammatory GI disease.
As the understanding of conjugated BA diversity grows, including the fact that some are produced by the same enzyme that hydrolyzes them, it is becoming clear that we have an overall poor understanding of the mechanisms behind their production in the human gut, which can involve host and microbial enzymes performing molecular crosstalk with BA receptors in the epithelium16. The need to parse out these mechanisms is heightened by the principal finding of this study, that at least one prevalent conjugated BA is important for improving pathology in an inflamed GI tract. It is possible that other forms of conjugated BAs, including the MCYs or MCBAs, may have even stronger effects, but the novelty and lack of commercial availability of these molecules limits current ability to experiment. TCA, GCA and the diverse other forms of conjugated BAs could be promising therapeutics for subjects with gut inflammation, particularly those with lower levels of these molecules natively, which can be readily assayed by mass spectrometry. To translate new knowledge of these molecules to medical use for any disease state, the balance between their conjugation and deconjugation in the gut must be better understood.
Methods
Animals
All animal procedures were approved by the Michigan State University Institutional Animal Care and Use Committee and conformed to NIH guidelines. Mice were housed in a temperature and humidity-controlled room under a 12-h cycle of light and darkness. Cage changes were performed weekly in a laminar flow hood. In our initial experiment, 9 wild-type (WT) and 9 Baat−/− KO C57BL/6 mice (6–8 weeks of age) were bred from heterozygote parents in-house, genotyped by PCR25, and WT and Baat−/− KO mice were randomly assigned to four groups (n = 4 WT-healthy control (HC), 5 WT-DSS, 4 KO-HC and 5 KO-DSS groups). The mice in the DSS group were administered 2.5% dextran sodium sulfate (DSS, molecular weight range: 35,000–50,000, Thermo Scientific, USA) daily in drinking water for 7 days to induce colitis. Mice were weighed daily and on day 7 all mice were euthanized through anesthesia using isoflurane followed by cervical dislocation. At necropsy, colon lengths were measured, and spleens were weighed for comparisons across treatment groups.
In a follow-up taurocholic acid (TCA) supplementation experiment, Baat−/− KO mice were subjected to a treatment with TCA or mock control. For this study, 27 Baat−/− KO mice (6-8 weeks of age) were randomly assigned to four treatment groups (n = 6 HC, 6 TCA, 7 DSS, and 8 DSS-TCA). For DSS treatment groups, 2.5% DSS was administered in drinking water for 6 days, followed by 7 days of normal, non-DSS-treated water. HC and TCA group mice were given regular water throughout the experiment. Mice in TCA treatment groups were fed with food containing 0.3% TCA (ENVIGO, USA), while mice in the HC and DSS groups were given the same food but without TCA. Body weight was recorded daily, and fecal samples were collected by temporarily placing the mice in clean plastic cups on day 13 prior to sacrifice. On day 13 all mice were euthanized through anesthesia using isoflurane followed by cervical dislocation. Colon, liver, spleen, and cecal content were then harvested after mice were euthanized. Colon lengths were measured, and spleens were weighed.
In a third experiment, to explore the degree of hydrolysis of an MCBA (glutaminocholic acid, Glu-CA) compared to TCA in the inflamed gut, C57BL/6 J wildtype mice 6-8 weeks of age were purchased from Jackson Laboratories and acclimated in the new facility for 1 week (note: Baat−/− animals were not used in this study to better reproduce the native condition). Then, mice were singly housed in cages and acclimated to the singly housed environment for 3 days. After this acclimation period, mice were fasted for 12 h and then given a plain peanut butter pellet for training (fasted training). The peanut-butter training is part of the peanut butter feeding method, a procedure developed to efficiently and accurately administer BAs to mice18,43. All mice that passed the training were selected to the next stage of training (non-fasted training), where they were given a plain pure peanut butter pellet under normal chow feed supply for three consecutive days. The mice that successfully completed training were included in subsequent study. Forty male mice were randomly assigned to four groups (HC, DSS, DSS-TCA, and DSS-GluCA groups) with 10 mice per group. Mice in HC and DSS groups were treated with two 10 mg peanut butter pellets every day for 17 days, and mice in DSS-TCA and DSS-GluCA groups were weighed and treated with two 10 mg peanut butter pellets with 25 mg/kg TCA or GluCA every day for 17 days. For the DSS-treated groups, 2.5% DSS was added to the drinking water for 7 days after the 10 days normal water. Mice were weighed every day and fecal samples were collected before sacrifice. On day 17 all mice were euthanized through anesthesia using isoflurane followed by cervical dislocation. Spleen and colon were then harvested after mice were euthanized. Colon lengths were measured, and spleens were weighed.
Tissues collected included serum, liver, cecum, and feces and were frozen immediately by liquid nitrogen and stored at −80 °C until they were thawed for use. Assessment of spleen inflammation index was performed by the spleen weight (g) normalized by its body weight (g). Disease activity index (DAI) was assessed by measuring the following: body weight, rectal bleeding, and stool status were scored daily. Rectal bleeding: 0 = no bleeding, 2 = slight bleeding, and 4 = gross bleeding; diarrhea: 0 = well-formed stools, 2 = soft and pasty stools, and 4 = watery stools.
Histological assessment of colon inflammation
Colon specimens were fixed overnight in 10% formalin at room temperature, sectioned at 5 μm, paraffin-embedded, and stained with hematoxylin and eosin (H&E). A subset was stained with alcian blue-periodic acid-Schiff (AB-PAS) reaction to highlight mucin-containing goblet cells. An independent board-certified veterinary anatomic pathologist who was blinded to the treatment evaluated the slides. A modified scoring system was used to determine a histologic score, including degree of inflammatory cell infiltration (normal=0 dense inflammatory infiltrate= 3) and changes to crypt architecture (normal=0, severe crypt distortion with loss of entire crypts=3). In addition, epithelial injury was determined by % of surface erosions, mucosal depth was measured, goblet cell depletion was determined (depleted=0, Normal=1), and goblet cell counts were obtained for the subset stained with AB-PAS.
Immunohistochemical analysis
Colon sections were deparaffinized followed by heat induced epitope retrieval utilizing TRIS/EDTA buffer (pH 9.0; Scytek labs, USA), and treated with hydrogen peroxide in methanol for 30 min to block endogenous peroxidase activity. Immunohistochemistry was performed on the Biocare intelliPath FlexTM automated platform (Biocare Medical, USA) using ProMARKTM detection reagents, with AutoWash buffer rinses between steps. Non-Specific Proteins were blocked with Rodent Block M (Biocare Medical, USA) for 20 min followed, by incubation in primary antibodies (MUC-2 antibody (1:500, ab272692, Abcam, USA) and ZO-1 antibody (1:500, ab221546, Abcam, USA)) for 1 h, and Rabbit-on-Rodent HRP-Polymer (Biocare Medical, USA) for 30 min. The reaction was developed with AEC (Biocare Medical, USA) for 5 min followed by counter stain with CATHE hematoxylin (Biocare Medical, USA) for 5 min. Post staining slides were rinsed in distilled water, dehydrated in ethanol, cleared in Xylene and cover slipped with Optic Mount 1 media (Mercedes Scientific, USA). The intensity of MUC-2 and ZO-1 staining was analyzed using ImageJ software. The quantitative intensity of MUC-2 and ZO-1 staining was calculated using the Deconvolution2 plugin in ImageJ software. All images were processed identically.
RNA extraction and quantitative real-time PCR
RNA from colon samples of mice was extracted by the Quick-RNA Midiprep Kit (Zymo, USA) according to the manufacturer’s instructions. Total RNA was reverse transcribed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, USA). RT-qPCR was performed using SYBR Green Master Mix (Applied Biosystems, USA) in CFX96 System (BIO-RAD, USA) using cDNA under following conditions: 50 °C for 2 min, 95 °C for 10 min, then 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The mRNA expression of mouse genes was analyzed with the specific oligo nucleotide primers information is listed in the Table S1. The relative mRNA expression was calculated using the comparative cycle method (2−ΔΔCt). β-actin served as an internal reference gene.
Microbial 16S rRNA gene sequencing
DNA from cecum and feces was extracted using the Quick-DNA Fecal/Soil Microbe Miniprep kit (Zymo, USA) according to the manufacturer’s instructions. To test extraction efficacy, the full length 16S rRNA genes were amplified using primers 27 f (5’-AGAGTTTGATCMTGGCTCAG-3’) and 1492r (5’-TACGGYTACCTTGTTACGACTT-3’) and analyzed via agarose gel electrophoresis. Subsequent microbiome sequencing was performed on amplifiable samples using Illumina compatible primers 515 f and 806r to amplify the V4 hypervariable region of the 16S rRNA gene. Sequencing was performed on the MiSeq platform at the Michigan State University RTSF Genomics Core following the protocol previously described by ref. 25.
Raw sequences data were analyzed using QIIME2 (version 2022.08)44. Sequences were filtered based on quality to generate amplicon sequence variants (ASVs) via the DADA2 method45. Taxonomy was assigned using the q2-feature-classifier against the Greengene2 database (version 2022.10)46. Sequences were rarefied to 9000 reads per sample and core diversity metrics, including Shannon index, observed ASVs, and Bray-Curtis dissimilarity, were calculated using QIIME2.
Liver, serum, cecum and fecal metabolite extraction
Phosphate buffered saline (PBS) was added to liver, cecum and feces samples (3:1, v:w) and homogenized via bead bashing at 20 Hz for 30 s with 1 min of rest three times using a Bead Ruptor (Omni International, GA). Then 50 μL of liver, cecal or fecal homogenate and 50 μL of serum was added to 150 μL ice cold methanol at a final concentration of 60% methanol followed by overnight incubation at 4 °C. Extracted metabolites were stored at −80°C prior to mass spectrometry analysis.
LC-MS/MS metabolomics
Extract samples were spun down at 12,000 g for 5 min, and the supernatant was transferred to 96-well plates followed by dilution 1:1 (v:v) in 50% methanol containing 2.5 μg/mL phenolsulfonphthalein (as an internal standard) for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Liquid chromatography was performed using a Vanquish Autosampler (Thermo Scientific, USA) and an Acquity UPLC BEH C-18 column, 2.1 mm × 100 mm (Waters, USA). Mass spectrometry was performed using a Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific, USA). All analyses used a 10 µL injection volume, 0.4 mL/min flow rate, and 60 °C column temperature. Samples were eluted using a linear solvent gradient of water (A) and acetonitrile (B), each containing 0.1% formic acid, across a 12-minute chromatographic run as follows: 0-1 min, 2% B; 1–8 min, 2–100% B; 8–12 min, 100% B; 10–12 min, 2% B. Data was acquired using electrospray ionization in positive mode. MS1 data were collected using a 35,000 resolution, AGC target of 1e6, maximum iteration time of 100 ms, and a scan range set from 100 to 1500 m/z during (min 1–10). Data-dependent MS2 spectra were collected for the top 5 most abundant peaks identified in MS1 survey scans. Files were converted to mzXML format and submitted to the Global Natural Products Social Molecular Networking Database (GNPS, gnps.ucsd.edu) for molecular networking and spectral library matching47.
Bacterial and mouse fecal community culturing exposed to diverse BAs
All strains used in this study are listed in supplementary data S1 in the supplementary material. All bacterial strains were cultured in Reinforced Clostridial Medium (RCM), which contained 10 g of proteose peptone, 10 g of beef extract, 5 g of yeast extract, 10 g of lactose, 5 g of sodium chloride, 3 g of sodium acetate, 2 g of K2HPO4, 0.25 g of calcium chloride, 0.2% Tween 80, and 0.5 g of L-cysteine in 1 L distilled water. The medium was adjusted to a final pH of 7.0 and autoclaved at 121 °C for 15 min.
All strains and the mouse fecal community were grown at 37 °C under anaerobic conditions. Starting from -80 °C freezer stocks, bacterial strains were first grown to an optical density at 600 nm (OD600) of at least 1.0 in 15 mL polypropylene tubes containing RCM medium. These 50 μL cultures were then used to inoculate 450 μL of RCM medium with supplemented BAs in 1-mL 96-deep-well plates (Thermo Scientific, USA). The medium contained one of each of the 10 BAs, TCA, glycocholic acid (GCA), alanocholic acid (AlaCA), serocholic acid (SerCA), phenylalanocholic acid (PheCA), threonocholic acid (ThrCA), tryptophanocholic acid (TyrCA), glutaminocholic acid (GluCA), leucocholic acid (LeuCA), and aspartocholic acid (AspCA), at 1 mM each. Fresh fecal samples from 5 WT C57BL/6 mice in different cages were collected. Mice fecal samples were homogenized in the 15 mL tube with 4 mL anaerobic Brain Heart Infusion (BHI, Sigma-Aldrich USA) medium, and cultured for 24 h. Subsequently passaging was then performed in 15 mL tubes with 4 mL BHI medium at a dilution of 1:4 every 24 h for a total of 2 passages. 50 μL passaged mice fecal bacteria community (OD600 = 0.6) was inoculated to 450 μL BHI medium with the 10 BA conjugations mentioned above, at 1 mM each in 1-mL 96-deep-well plates (Thermo Scientific, USA), and cultured for 48 h. Plates were sealed with a rubber mat and incubated at 37 °C for 48 h under anaerobic conditions. Metabolites were extracted as previously described for UPLC-MS/MS analysis25. Briefly, 200 μL of culture was added to 300 μL ice cold methanol at a final concentration of 60% methanol followed by overnight incubation at 4 °C. Extracted metabolites were stored at −80 °C prior to mass spectrometry analysis.
Reference BSH sequences collection
The query BSH protein sequences were collected from the National Center for Biotechnology Information (NCBI) using the keywords “bile salt hydrolase” and “choloylglycine hydrolase”. The source was limited to bacteria and archaea, and the amino acid sequence length was 300 to 400. A total 3058 BSH sequences were obtained and then filtered for non-redundancy using CD-HIT (version 4.8.1, default parameter) at a 95% sequence identity threshold, and the final output 778 BSHs sequences were used as a representative set of BSHs for further analysis. The taxonomy information of BSHs was obtained from the NCBI taxonomy browser.
IBD metagenomic sequence data collection
Metagenomic sequence data was downloaded from NCBI Sequence Read Archive (SRA) database under the accession number PRJNA00072 (PRISM and LLDeep)9, PRJEB1537148, PRJNA42999049, PRJEB122050, and PRJNA389280 (HMP2)51. The metadata information was obtained from the materials provided in corresponding papers. For the longitudinal data, only the first time point per individual was selected.
A total of 774 samples were included in the analysis. Data from PRJNA0072 were acquired from two different cohorts, which were PRISM and LLDeep. From PRISM cohort, 34, 68, and 51 fecal samples were analyzed from Healthy subjects (HC), those with Crohn’s disease (CD), or ulcerative colitis (UC), respectively. From LLDeep cohort, 22, 20, and 23 samples of HC, CD, and UC, respectively. From PRJEB15371, 53, and 26 samples of HC, and CD, respectively. From PRJNA429990, 15, 41, and 25 samples of HC, CD, and UC, respectively. From PRJEB1220, 236, 12, and 67 of HC, CD, and UC, respectively. From HMP2 cohort, 21, 40, and 20 of HC, CD, and UC, respectively.
Metagenomic data processing and BSHs abundance analysis
The raw sequencing data from the publicly available IBD cohorts was quality controlled using Metawrap (version 1.3.2) with default parameters52. The clean reads from each sample were individually assembled to contiguous sequences (contigs) using Megahit (version 1.1.3). After assembly, open reading frames (ORFs) in the contigs were predicted with Prodigal (version 2.6.3). The non-redundant gene set was clustered by CD-HIT (version 4.8.1) at a 95% sequence identity threshold. The clean reads from each sample were compared to the non-redundant gene set by BWA (version 0.7.17). The relative abundance of each gene in the sample was calculated based on the number of reads mapped to each gene by BBtools53. The BSHs were identified by taking the referenced 778 BSHs as query sequences and using DIAMOND (version 0.9.36) BLASTP with an e-value = 1e−5 and 40% sequence identity as the cutoff values. To ensure the accuracy and reliability of the statistical analysis, the putative BSHs present in less than 10% of all samples were excluded, and a pseudo-count of 1×10-10 was added to avoid nonfinite values. The cumulative BSHs relative abundance was calculated by summing the relative abundance of all BSHs in each sample.
BSHs diversity characterization
Alpha-diversity metrics, including the Shannon index and observed BSHs calculated by vegan package (version 2.6-4) in R (version 4.3.0), were used to evaluate the diversity of BSHs in IBD patients and healthy controls. Beta-diversity was evaluated by calculating the Bray-Curtis distance using vegan package in R, to quantify the differences of BSHs among samples. Moreover, the permutational multivariate analysis of variance (PERMANOVA) was used to investigate the differences in BSHs composition between IBD patients and healthy controls by vegan package in R. All identified BSHs were aligned by their amino acid sequences with the multiple sequence alignment MUSCLE and a phylogenetic tree was built using the maximum likelihood method in the software MEGA (version 11.0). The phylogenetic tree was visualized and decorated by iTOL54.
Statistical analysis
Data was expressed as means ± standard errors of the means (SEM) and boxplots show the median, interquartile ranges, minimum and maximum with individual data points visualized. For the murine experiments, statistical significance between groups for weight changes, colon lengths, spleen index, DAI, mRNA expression and microbiome diversity metrics among groups were analyzed by one-way ANOVA followed by Tukey’s post-hoc test. The statistical significance differences of the IHC staining and metabolome data were calculated using Kruskal-Wallis followed by Dunn’s post-hoc test. Statistically significant differences of BSHs between IBD patients and healthy control were determined by Wilcox rank-sum test followed by Benjamini-Hochberg (BH) correction. Beta-diversity of the microbiome, metabolome and BSHs data was quantified using principal coordinate analysis (PCoA) based on Bray-Curtis dissimilarity. PERMANOVA testing was used to determine if there were significant differences among the groups. All analyses were performed in R (version 4.3.0), and p values less than 0.05 (p < 0.05) was considered statically significant, as specified in the text and figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Mass spectrometry data are publicly available within the MassIVE database (gnps.ucsd.edu) under MassIVE ID MSV000095705. 16S rRNA gene amplicon data were deposited in the NCBI under project number PRJNA1152983. Source data are provided with this paper.
References
Flynn, S. & Eisenstein, S. Inflammatory bowel disease presentation and diagnosis. Surgical Clin. North Am. 99, 1051–1062 (2019).
Lewis, J. D. et al. Incidence, prevalence, and racial and ethnic distribution of inflammatory bowel disease in the United States. Gastroenterology 165, 1197–1205.e2 (2023).
Burisch, J. et al. The cost of inflammatory bowel disease in high-income settings: a lancet gastroenterology & hepatology commission. Lancet Gastroenterol. Hepatol. 8, S2468125323000031 (2023).
Caruso R., Lo B. C. & Núñez G. Host–microbiota interactions in inflammatory bowel disease. Nat. Rev. Immunol. 20, 411–426 (2020).
Neurath, M. F. et al. Host–microbiota interactions in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 20, 411–426 (2019).
Li, M. et al. Gut microbial metabolome in inflammatory bowel disease: from association to therapeutic perspectives. Computational Struct. Biotechnol. J. 20, 2402–2414 (2022).
Gentry, E. C. et al. Reverse metabolomics for the discovery of chemical structures from humans. Nature 626, 419–426 (2024).
IBDMDB Investigators, Lloyd-Price, J. et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019).
Franzosa, E. A. et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol 4, 293–305 (2019).
Hsu C. L. & Schnabl B. The gut–liver axis and gut microbiota in health and liver disease. Nat. Rev. Microbiol. 21, 719–733 (2023).
Guzior, D. V. & Quinn, R. A. Review: microbial transformations of human bile acids. Microbiome 9, 140 (2021).
Collins S. L., Stine J. G., Bisanz J. E., Okafor C. D. & Patterson A. D. Bile acids and the gut microbiota: metabolic interactions and impacts on disease. Nat. Rev. Microbiol. 21, 236–247 (2022).
Ridlon J. M. & Gaskins H. R. Another renaissance for bile acid gastrointestinal microbiology. Nat. Rev. Gastroenterol. Hepatol. 21, 348–364 (2024).
Fleishman, J. S. & Kumar, S. Bile acid metabolism and signaling in health and disease: molecular mechanisms and therapeutic targets. Sig Transduct. Target Ther. 9, 97 (2024).
Song, Z. et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 7, 9 (2019).
Mohanty I. et al. The changing metabolic landscape of bile acids – keys to metabolism and immune regulation. Nat. Rev. Gastroenterol. Hepatol. https://doi.org/10.1038/s41575-024-00914-3 (2024).
Yao, L. et al. A selective gut bacterial bile salt hydrolase alters host metabolism. eLife 7, e37182 (2018).
Guzior D. V., et al. Bile salt hydrolase acyltransferase activity expands bile acid diversity. Nature. 626, 852–858 (2024).
Rimal B. et al. Bile salt hydrolase catalyses formation of amine-conjugated bile acids. Nature. 626, 859–863 (2024).
Quinn, R. A. et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 579, 123–129 (2020).
Pan, Y. et al. Novel approaches in IBD therapy: targeting the gut microbiota-bile acid axis. Gut Microbes 16, 2356284 (2024).
Khodakivskyi P. V. et al. Noninvasive imaging and quantification of bile salt hydrolase activity: from bacteria to humans. Sci. Adv. 7, eaaz9857 (2021).
Sinha, S. R. et al. Dysbiosis-induced secondary bile acid deficiency promotes intestinal inflammation. Cell Host Microbe 27, 659–670.e5 (2020).
Yang, Z.-H. et al. Altered profiles of fecal bile acids correlate with gut microbiota and inflammatory responses in patients with ulcerative colitis. WJG 27, 3609–3629 (2021).
Neugebauer, K. A. et al. Baat Gene knockout alters post-natal development, the gut microbiome, and reveals unusual bile acids in mice. J. Lipid Res. 63, 100297 (2022).
Alrehaili, B. D. et al. Bile acid conjugation deficiency causes hypercholanemia, hyperphagia, islet dysfunction, and gut dysbiosis in mice. Hepatol. Commun. 6, hep4.2041 (2022).
Won, T. H. et al. Host metabolism balances microbial regulation of bile acid signalling. Nature 638, 216–224 (2025).
Fogelson, K. A., Dorrestein, P. C., Zarrinpar, A. & Knight, R. The gut microbial bile acid modulation and its relevance to digestive health and diseases. Gastroenterology 164, S0016508523001610 (2023).
Mohanty, I. et al. The underappreciated diversity of bile acid modifications. Cell 187, 1801–1818.e20 (2024).
Kang, Y., Park, H., Choe, B.-H. & Kang, B. The role and function of mucins and its relationship to inflammatory bowel disease. Front Med 9, 848344 (2022).
Stange, E. F. & Schroeder, B. O. Microbiota and mucosal defense in IBD: an update. Expert Rev. Gastroenterol. Hepatol. 13, 963–976 (2019).
Luis, A. S. & Hansson, G. C. Intestinal mucus and their glycans: a habitat for thriving microbiota. Cell Host Microbe 31, 1087–1100 (2023).
Li, D. K. et al. Inhibition of microbial deconjugation of micellar bile acids protects against intestinal permeability and liver injury. Sci. Adv. 8, eabo2794 (2022).
Kuo, W.-T. et al. The tight junction protein ZO-1 Is dispensable for barrier function but critical for effective mucosal repair. Gastroenterology 161, 1924–1939 (2021).
Keita, Å. V. et al. Gut barrier dysfunction—a primary defect in twins with crohn’s disease predominantly caused by genetic predisposition. J. Crohn’s. Colitis 12, 1200–1209 (2018).
Sorrentino, G. et al. Bile acids signal via TGR5 to activate intestinal stem cells and epithelial regeneration. Gastroenterology 159, 956–968.e8 (2020).
Long, X.-Q. et al. Bile acids and their receptors: potential therapeutic targets in inflammatory bowel disease. World J. Gastroenterol. 29, 4252–4270 (2023).
Berry, D. & Reinisch, W. Intestinal microbiota: a source of novel biomarkers in inflammatory bowel diseases? Best. Pract. Res. Clin. Gastroenterol. 27, 47–58 (2013).
Hu, J. et al. Gut microbiota-mediated secondary bile acids regulate dendritic cells to attenuate autoimmune uveitis through TGR5 signaling. Cell Rep. 36, 109726 (2021).
Pittayanon, R. et al. Differences in gut microbiota in patients with vs without inflammatory bowel diseases: a systematic review. Gastroenterology 158, 930–946 (2020).
Qu, S. et al. Akkermansia muciniphila alleviates dextran sulfate sodium (DSS)-induced acute colitis by NLRP3 activation. Microbiol Spectr. 9, e00730–21 (2021).
Foley M. H. et al. Bile salt hydrolases shape the bile acid landscape and restrict Clostridioides difficile growth in the murine gut. Nat. Microbiol. 8, 611–628 (2023).
Zapata R. C., Zhang D., Chaudry B. & Osborn O. Self-administration of drugs in mouse models of feeding and obesity. J. Vis. Exp. 172, 10 (2021).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
McDonald D. et al. Greengenes2 enables a shared data universe for microbiome studies. 42, 715–718 (2024).
Wang, M. et al. Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat. Biotechnol. 34, 828–837 (2016).
He Q. et al. Two distinct metacommunities characterize the gut microbiota in Crohn’s disease patients. GigaScience; 6, 1–11 (2017).
Weng, Y. J. et al. Correlation of diet, microbiota and metabolite networks in inflammatory bowel disease. J. Dig. Dis. 20, 447–459 (2019).
MetaHIT Consortium, Nielsen, H. B. et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32, 822–828 (2014).
Schirmer, M. et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol 3, 337–346 (2018).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158 (2018).
Bushnell, B., Rood, J. & Singer, E. BBMerge – accurate paired shotgun read merging via overlap. PLoS ONE 12, e0185056 (2017).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Acknowledgements
The authors would like to thank MSU mass spectrometry and metabolomics core for their help. This work was funded by the Biochemistry and Molecular Biology Department Team-Up Grant and the Tetrad Grant Program of Michigan State University and the National Institute of Diabetes and Digestive and Kidney Diseases grant #1R01DK140854 awarded to PI Quinn.
Author information
Authors and Affiliations
Contributions
Y.F. performed the experiments, collected the data, data analysis, and wrote the paper; S.L.R., K.H., C.T.G., M.O., D.V.G., C.M., C.B. and V.E.W. reviewed & edited the paper; V.E.W. performed the H&E analysis; R.A.Q. conceived the designed, analysis, and revised the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ipsita Mohanty, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, Y., Guzior, D.V., Okros, M. et al. Balance between bile acid conjugation and hydrolysis activity can alter outcomes of gut inflammation. Nat Commun 16, 3434 (2025). https://doi.org/10.1038/s41467-025-58649-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58649-x
