
Abstract
Double-stranded RNA (dsRNA) binding proteins (dsRBPs) play crucial roles in various cellular processes, especially in the innate immune response. Comprehensive characterization of dsRBPs is essential to understand the intricate mechanisms for dsRNA sensing and response. Traditional methods have predominantly relied on affinity purification, favoring the isolation of strong dsRNA binders. Here, we adopt the proteome integral solubility alteration (PISA) workflow for characterizing dsRBPs, resulting in the observation of 18 known dsRBPs and the identification of 200 potential dsRBPs. Next, we focus on zinc finger protein 385 A (ZNF385A) and discover that its knockout activates the transcription of interferon-β in the absence of immunogenic stimuli. The knockout of ZNF385A elevates the level of endogenous dsRNAs, especially transcripts associated with retroelements, such as short interspersed nuclear element (SINE), long interspersed nuclear element (LINE), and long terminal repeat (LTR). Moreover, loss of ZNF385A enhances the bioactivity of 5-Aza-2’-deoxycytidine (5-AZA-CdR) and tumor-killing effect of NK cells. Our findings greatly expand the dsRBP reservoir and contribute to the understanding of cellular dsRNA homeostasis.
Similar content being viewed by others


Introduction
Double-stranded RNAs (dsRNAs) adopt an A-form duplex structure, characterized by narrow major grooves and wide minor grooves1,2. In most cases, dsRNAs are non-self molecules generated during virus infections and are best known for their roles in activating innate immune response in host cells3,4. The sensing of dsRNAs is commonly mediated by three types of machineries: (i) pattern-recognition receptors, such as RIG-I-like receptors (RLRs), toll-like receptor 3 (TLR3), NOD-, LRR-, and pyrin domain-containing proteins, trigger the type-I interferon (IFN-I) signaling and inflammatory pathway; (ii) protein kinase R (PKR, also known as EIF2AK2) phosphorylates eukaryotic initiation factor-2α, thereby blocking the global translation; (iii) oligoadenylate synthases (OASes) produce 2′-5′-linked oligoadenylates that activate RNaseL for global RNA degradation5
Apart from exogenous dsRNA originating from viral infections, endogenous dsRNAs can also be formed through the base pairing of complementary sequences within nuclear or mitochondrial transcripts6,7. These self-derived duplexes can be immunogenic and must be strictly regulated. For example, cell-encoded adenosine deaminase acting on RNA (ADAR1) catalyzes the A-to-I editing on the paring regions to melt the integrity of the duplexes thereby preventing their recognition by the IFN-I activating machinery8. Mitochondrial membranes serve as a barrier, preventing the leakage of mitochondrial RNAs into the cytoplasm9. In physiological perturbations, e.g., gene mutations, dysregulated epigenetic control, changes to RNA modification, and splicing inhibition, self-duplexes are accumulated and lead to the onset of diseases such as inflammatory disorders or neurodegeneration10,11,12. On the other hand, the activation of endogenous expression of RNA duplexes can be beneficial in cancer therapy, as they can elicit anti-cancer effects by inducing IFN-I signaling. For instance, the DNA methyltransferase inhibitor 5-Aza-2’-deoxycytidine (5-AZA-CdR) has been reported to promote transcription of various types of transposable elements and induce a viral mimicry effect to activate dsRNA sensors13,14. In addition, inhibition of lysine-specific histone demethylase 1 (LSD1) has been demonstrated to promote the accumulation of endogenous dsRNA, exhibiting a synergistic effect when combined with programmed death-1 (PD-1) blockade15.
Given the pivotal roles played by dsRNA binding proteins (dsRBPs) in various stages of dsRNA recognition and response, a comprehensive characterization of these proteins is imperative for elucidating the biological functions of dsRNAs and the underlying mechanisms. Many approaches have been developed for the isolation and profiling of single-stranded RNA (ssRNA) binding proteins. These approaches typically involve stabilizing the protein-RNA complexes by ultraviolet (UV) crosslinking, followed by isolation by different methods, such as oligo (dT) beads16, metabolic RNA labeling17, and orthogonal organic phase separation18,19. Subsequently, mass spectrometry is employed to decipher the bound proteins. However, as dsRBPs primarily interact with the sugar-phosphate backbone and cannot be efficiently crosslinked by UV light, conventional methods are not suitable for the characterization of dsRBPs2. Of note, this feature also accounts for the lack of sequence preference of RNA binding for known dsRBPs. Currently, the method for dsRBP identification is limited to affinity purification followed by mass spectrometry (AP-MS) using ploy(I:C) conjugated beads, which favors the detection of high affinity binders and may miss weaker interactions that are biologically important20,21. Therefore, a high-throughput approach based on alternative working principles is required to provide complementary information for the identifications of dsRBPs.
Cellular thermal shift assay (CETSA) is widely used to study thermal stability alteration of proteins upon ligand binding and enables detection of drug-target engagement22. The integration of CETSA with mass spectrometry, known as thermal proteome profiling (TPP), allows high-throughput thermal profiling of cellular proteins and comprehensive assessment of drug targets23,24. In addition to drug molecules, TPP has been recently applied to study the binding of other types of ligands such as metabolites25,26 and proteins27, or impacts of post‐translational modifications28. In TPP, cells or the lysates are heated to different temperatures and labeled with isobaric tandem mass tags (TMT) for the subsequent quantification analysis. To improve the throughput, proteome integral solubility alteration (PISA) was developed by combining samples across a temperature gradient after thermal treatment. Instead of establishing the melting curves, ΔSm (a proxy of the difference between areas under the melting curves) and fold changes (FCs) of protein abundance are used to evaluate the alteration of solubility that may be caused by stimuli such as heating29. Later, Li et al. reported that the use of three heating temperatures, 52, 53, and 54 oC, could increase the detection sensitivity and analysis throughput30. TMT labeling followed by data-dependent acquisition (DDA) has been widely used for protein quantification in the PISA assay29,30,31,32,33,34. Recently, data independent acquisition (DIA)-based quantification was also utilized for PISA35. Moreover, Emery-Corbin et al. applied multiple overlapping thermal gradients in conjunction with DIA-MS, resulting in satisfactory proteome coverage and sensitivity36. Other efforts have also been made to further increase the throughput, such as the matrix thermal shift assay37 and the matrix-augmented pooling strategy38.
In this study, we propose that dsRNA binding may cause solubility change of the associated proteins and employ a PISA-DIA workflow to characterize dsRBPs. Using this approach, we observed solubility alteration for 218 proteins following incubation with poly(I:C), including 18 known dsRBPs. The data provided a pool of dsRBP candidates for understanding the intricate mechanisms of dsRNA sensing and response. Next, we performed CETSA and pull-down assays to validate the dsRNA-binding capability of multiple detected proteins, including zinc finger protein 385 A (ZNF385A), which exhibited the largest fold change among the identified potential dsRBPs. Interestingly, we found that the knockout (KO) of ZNF385A induced IFN-I response and led to accumulation of endogenous dsRNA. Through TurboID proximity labeling, ZNF385A was found to bind with RNA splicing-associated proteins, such as polypyrimidine tract-binding protein 1 (PTBP1) and TAR-DNA-binding-protein 43 (TARDBP, also known as TDP43), suggesting that ZNF385A might regulate the levels of endogenous dsRNA through RNA splicing. Through dsRNA immunoprecipitation and sequencing (dsRIP-seq), we discovered that majority of duplex-containing transcripts enriched in ZNF385A KO cells were associated with retroelements, specifically short interspersed nuclear element (SINE), long interspersed nuclear element (LINE), and long terminal repeat (LTR), which predominantly located within introns. Further functional study revealed that ZNF385A depletion enhanced the effects of 5-AZA-CdR treatment and natural killer (NK) cell-mediated cytotoxicity, implying the potential value of targeting ZNF385A in cancer treatment.
Results
The PISA workflow for dsRBP identification
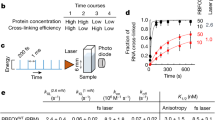
The recognition of dsRNA and the modulation of downstream signaling is carried out by a network of dsRBPs that often undergo conformational changes or oligomerization upon dsRNA binding, which may impact the biophysical properties of the associated proteins2,12. Therefore, we propose that profiling the dsRNA-induced protein solubility change may be exploited to characterize dsRBPs. Low molecular weight (LMW) poly(I:C), a widely used dsRNA surrogate39,40,41, was employed in this study. Of note, the known dsRBPs primarily interact with the dsRNA backbone, and the interactions are thus largely sequence independent12,42. It should be noted that the length of dsRNA can affect binding. As compared to high molecular weight (HMW) poly(I:C), LMW poly(I:C) better represents the relatively small size of endogenous dsRNA molecules12,43. Additionally, the majority of the known dsRBPs sense dsRNA duplexes with length less than 1 kb12. To validate our hypothesis, we performed a CETSA experiment ranging from 45 to 56 oC on two known dsRBPs, i.e., Staufen-1 (STAU1) and protein activator of PKR (PRKRA), in clarified HeLa cell lysates incubated with or without the dsRNA analog, poly(I:C). As a result, we observed that both dsRBPs exhibited markedly increased solubility at multiple temperatures after binding with poly(I:C) (Supplementary Fig. 1a).
Here we adopted a PISA-DIA workflow to characterize dsRBPs. The experiment was conducted using HeLa cells, a widely used cell model for RBP16,44,45 and dsRNA6,46 studies, and it could serve as an appropriate cellular model for investigating the regulation of dsRNA pathways in cancer. We were able to detect a number of classic dsRBPs from these cells (Supplementary Fig. 1b). In the PISA experiment, four temperature zones were employed, including PISA1 (45, 46, 47 oC), PISA2 (48, 49, 50 oC), PISA3 (51, 52, 53 oC), and PISA4 (54, 55, 56 oC). As shown in Fig. 1, HeLa cell lysates were incubated with or without poly(I:C) and the aliquots were heated at 12 different temperatures ranging from 45 to 56 oC. The samples were then centrifuged and equal volume of soluble fractions were taken from each tube and pooled in four groups before subjected to MS analysis.

The HeLa cells were disrupted using PBS with four freeze-thaw cycles. The extracts were incubated with poly(I:C) or the same volume of PBS for 30 min at RT and divided into 12 aliquots, each heated at the specified temperatures. Following centrifugation, the supernatants were combined into four tubes according to the experimental design. Four biological replicates were conducted. After digestion with trypsin, the peptide samples were analyzed by LC-MS/MS. ΔSm was measured as the difference in integral abundances of the protein between the poly(I:C) treated and untreated samples.
Characterization of dsRBPs by PISA
By using DIA-based proteomics, 5461 (PISA1), 5334 (PISA2), 4831 (PISA3), and 4554 (PISA4) proteins were quantified across four biological replicates (Fig. 2a and Supplementary Data 1). The number of detected proteins inversely correlated with temperature, as higher temperature resulted in more protein denaturation and precipitation. Principal component analysis (PCA) and correlation analysis revealed that poly(I:C) did not dramatically change the proteome landscape (Supplementary Fig. 2a, b), indicating that poly(I:C) only affected a small fraction of cellular proteins. Using P value < 0.01 and |log2FC | > 0.58 as cutoff, 20 from PISA1 (17 increased and 3 decreased), 61 from PISA2 (27 increased and 34 decreased), 116 from PISA3 (61 increased and 55 decreased), and 71 from PISA4 (26 increased and 45 decreased) were determined to be significantly altered in solubility after incubated with poly(I:C) (Fig. 2a and Supplementary Data 1).

a Volcano plots represent the quantitative analysis of proteins across four biological replicates in four PISA assays. Proteins with significantly increased soluble amount are shown as red dots, while proteins with decreased soluble amount are displayed as blue dots. Log2FC was plotted on the x-axis and −log10 P value was plotted on the y-axis. P values were calculated by an unpaired two-sided t-test with no adjustment. PISA proteome integral solubility alteration. FC fold change. Molecular function (b) and biological process (c) analysis of proteins with increased solubility identified by PISA, with P < 0.05 as cutoff and the top 15 terms are displayed. The P values were calculated using the hypergeometric distribution. d InterPro-annotated domain enrichment analysis with P < 0.05 as the cutoff. The P values were calculated using the hypergeometric distribution. e A scatter plot displaying protein abundance changes incubated with or without poly(I:C) at 37 oC (n = 2 independent experiments). Proteins with |log2FC| > 0.58 are shown in blue (decreased) and red (increased). Ctrl, control. f Molecular function analysis of proteins with increased solubility with the top 10 terms displayed. The P values were calculated using the hypergeometric distribution.
Among the identified proteins, we observed 18 known dsRBPs, such as ADAR1, interferon-inducible double-stranded RNA-dependent protein kinase activator A (PRKRA), and STAU1 (Fig. 2a), suggesting that the established strategy was effective in identifying dsRBPs. The majority of the known dsRBPs showed increased abundance in one or more PISA experiments, suggesting that dsRNA binding enhanced their solubility, while only one known dsRBP, metadherin (MTDH), exhibited decreased solubility as detected by PISA2 (Fig. 2a and Supplementary Fig. 3a). Despite the similarity in protein detection coverage among the four PISA experiments (Supplementary Fig. 3b), the proteins observed with altered solubility displayed significant complementarity (Supplementary Fig. 3c, d), suggesting that the PISA strategy incorporating different temperature zones could increase the identification coverage of potential dsRBPs when applied with a limited proteome depth (~4000 proteins), DIA acquisition, and no fractionation prior to nLC-MS/MS. For example, a known dsRBP, zinc finger NFX1-type containing 1 (ZNFX1), was only identified with increased intensity in PISA3, and another known dsRBP, MTDH, was only identified in PISA2 (Fig. 2a and Supplementary Fig. 3a). Some of the identified potential dsRBPs, such as DEAD-box helicase (DDX24), poly(ADP-ribose) polymerase 1 (PARP1), tripartite motif containing 5 (TRIM5), and ubiquitin carboxyl-terminal hydrolase 38 (USP38) were also uniquely detected by one PISA experiment (Supplementary Fig. 3e). Among the four experiments, PISA3 (51, 52, 53 oC) detected the largest number of potential dsRBPs, and the identified proteins displayed greater FCs in PISA3. This phenomenon could be attributed to the fact that heating the samples at temperatures between the original Tm and the shifted Tm induced by dsRNA binding could cause the most dramatic difference in their abundances. Taken the detected known dsRBPs for example, their original Tm values were between 47.1 and 61.5 oC, with a median value of 49.6 oC (Supplementary Fig. 3f), and heating the samples at temperatures slightly higher than original Tm values could result in larger FCs between the control and poly(I:C)-treated group. Of note, the known dsRBPs exhibited different levels of alteration. For example, adenosine deaminase RNA specific B1 (ADARB1), PRKRA, DHX9, STAU1, and STAU2 displayed greater FCs than the others (Supplementary Fig. 3a), suggesting that dsRNA binding may alter the structure of different proteins to distinct degrees. Besides the known dsRBPs, the PISA strategy detected dozens of other proteins showing increased poly(I:C)-induced solubility, such as poly(rC)-binding protein 2 (PCBP2), heterogeneous nuclear ribonucleoprotein K (HNRNPK), NF-κB-repressing factor (NKRF), and ZNF385A (Supplementary Fig. 3e). In total, we identified 88 proteins with increased solubility and 130 proteins with decreased solubility induced by dsRNA (Supplementary Data 1).
Next, we carried out a functional enrichment analysis of the identified proteins using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. The molecular function enrichment analysis showed that “double stranded RNA binding” was significantly enriched for the proteins with enhanced solubility (Fig. 2b and Supplementary Data 2). In addition, several proteins were related to siRNA binding, e.g., PRKRA, DExH-box helicase 9 (DHX9), and endoribonuclease Dicer (DICER1), and helicase activity, e.g., DDX24 and DDX18. Moreover, multiple DNA binding proteins, e.g., PARP1, poly(rC)-binding protein 1 (PCBP1), and PCBP2, exhibited increased solubility when incubated with poly(I:C), implying their possible roles of dsRNA binding (Fig. 2b and Supplementary Data 2). Biological process analysis revealed significant enrichment in processes such as ribosome biogenesis, ncRNA processing and viral genome replication (Fig. 2c and Supplementary Data 2). Cellular component analysis indicated that the proteins with increased solubility were prominently associated with ribosomes (Supplementary Fig. 4a). Pathway enrichment analysis revealed that the proteins with increased solubility involved in pathways related to ribosome and RIG-I-like receptor signaling (Supplementary Fig. 4b). Domain analysis indicated enrichment of dsRNA binding domain (dsRBD), domain associated with zinc fingers (DZF), and helicase (Fig. 2d). Meanwhile, proteins with decreased solubility exhibited association with focal adhesion (Supplementary Fig. 4c) and endocytosis (Supplementary Fig. 4d). Since the majority of the known dsRBPs showed increased solubility upon poly(I:C) incubation, our subsequent study primarily focused on proteins with increased solubility.
To further elucidate the characteristics of potential dsRBPs identified through our approach, we conducted a biophysical property analysis encompassing hydrophobicity, isoelectric point, sequence length, and molecular weight. First, the proteins with increased solubility were more basic compared to the human proteome (Supplementary Fig. 4e), supporting the critical role of electrostatic interaction in mediating dsRNA binding, which was typically sequence independent and facilitated by positively charged surfaces47,48. Interestingly, the proteins with increased solubility were slightly less hydrophobic compared to the proteome (Supplementary Fig. 4e). Furthermore, the median size of the proteins with increased solubility was larger than that of the human proteome (Supplementary Fig. 4e). Collectively, the PISA approach confirmed 18 known dsRBPs and identified a set of dsRNA binding candidates, providing a rich resource to further explore the biological functions of dsRNA and the molecular mechanisms.
In the PISA experiment, we noticed that some proteins exhibited abundance alteration in the soluble fraction at relatively low heating temperature (PISA1), e.g., STAU1, PRKRA, and EIF2AK2 (Fig. 2a). We speculated that the binding of dsRNA could enhance the solubility of these proteins under physiological conditions, given that dsRNAs are hydrophilic molecules. To test such hypothesis, we examined the solubility of cellular proteins affected by poly(I:C) at 37 oC. Using |log2FC | > 0.58 as cutoff in both of the two biological replicates, the abundance of 35 proteins were observed to be increased in the soluble fraction and 111 proteins were decreased after the incubation with poly(I:C) (Fig. 2e and Supplementary Data 3). Interestingly,poly(I:C) significantly increased the abundance of 14 classic dsRBPs at 37 oC, e.g., ADAR, ADARB1, STAU1, STAU2, and DHX9 (Fig. 2e and Supplementary Fig. 5a), suggesting that dsRNA binding could markedly increase the solubility of their associated proteins under the physiological temperature. In addition, the solubility of additional 21 proteins, such as putative heat shock protein HSP 90-beta 4 (HSP90AB4P), hydroxylysine kinase (HYKK), and phosphatidylinositol 3-kinase regulatory subunit gamma (PIK3R3), was observed to be increased (Supplementary Data 3). Moreover, 15 proteins displaying increased solubility at 37 oC were detected by PISA, e.g., PRKRA, STAU1 and STAU2, and 12 proteins showing decreased solubility at 37 oC were identified by PISA, e.g., Yin Yang 1 (YY1) and superkiller complex protein 2 (SKI2) (Supplementary Fig. 5b).
Biophysical property analysis revealed that proteins with increased or decreased solubility both exhibited higher isoelectric point values compared to the proteome, and the proteins with decreased solubility were more hydrophilic (Supplementary Fig. 5c). Molecular function enrichment analysis revealed that “double stranded RNA binding” was significantly enriched for the proteins with enhanced solubility induced by poly(I:C) (Fig. 2f). Proteins with decreased abundance in the soluble fractions after incubation with poly(I:C) at 37 oC were significantly associated with the ribosome constituent and ubiquitin protein ligase binding (Supplementary Fig. 5d). Interestingly, several proteins involved in innate immunity, e.g., NF-κB inhibitor interacting Ras-like 2 (NKIRAS2), interferon-stimulated gene 15 (ISG15), and nuclear factor-kappa-B-inhibitor alpha (NFKBIA), were detected with decreased solubility after incubated with poly(I:C) (Supplementary Data 3).
Validation of the identified potential dsRBPs
To further validate the identification, we employed CETSA to examine several proteins that exhibited altered abundance in the PISA assay. β-actin was examined as the negative control and it displayed no alteration after poly(I:C) incubation (Fig. 3a). The selected known dsRBPs, i.e., PRKRA, STAU1, and EIF2AK2, all exhibited higher solubility at 37 oC and larger Tm values after incubated with poly(I:C) (Fig. 3b–d). Additionally, two identified potential dsRBPs, NKRF and ZNF385A, also exhibited higher solubility at 37 oC and were stabilized by poly(I:C) in CETSA (Fig. 3e, f). NKRF has been reported to negatively regulate the activity of NF-κB signaling and function as a transcriptional repressor of target genes, such as IFNβ and IL849,50. ZNF385A is an RNA-binding Cys2 His2 (C2H2) zinc finger protein and is involved in the regulation of cell cycle and apoptosis51. However, none of these two proteins have been implicated in dsRNA binding and their roles in dsRNA biology require further investigation. Next, we further validated the dsRBP candidates by dsRNA pull-down assays using poly(C) and poly(dA:dT) as ssRNA and dsDNA controls, respectively (Fig. 3g, h). The known dsRBPs, i.e., STAU1, PRKRA, and EIF2AK2, all displayed strong binding with poly(I:C), and STAU1 also bound to poly(C) (Fig. 3g, h). ZNF385A specifically bound to poly(I:C). NKRF bound to poly(I:C), poly(C), and poly(dA:dT).

a–f CETSA analysis results of β-actin (a), PRKRA (b), STAU1 (c), EIF2AK2 (d), NKRF (e), and ZNF385A (f). The left subpanels show the western blots revealing the thermostability of the proteins heated from 37 to 67 oC. The middle subpanels show the western blots and bar graphs revealing the change of protein abundance in the soluble fraction at 37 oC. A representative of three independent experiments is displayed. Data represent the means ± s.d. of triplicate technical measurements (****P < 0.0001, by unpaired two-tailed Student’s t-test, “ns” indicates not significant). The right subpanels display melting curves for the proteins of interest. Tm values were fitted by GraphPad Prism 9.5.0. For quantification, measurements on two gels were calibrated using the two samples at 37 oC with or without poly(I:C) to avoid the variation between gels. g, h Poly(I:C) pull-down assay. g Biotinylated ploy(dA:dT) or poly(I:C) was incubated with HeLa cell lysates and then isolated with streptavidin-conjugated beads. h Similarly, poly(I:C) or poly(C) agarose beads were incubated with HeLa cell lysates and then isolated by centrifugation. The pull-down samples were then analyzed by western blotting for PRKRA, STAU1, EIF2AK2, ZNF385A, and NKRF. Results are representative of two independent experiments. Source data are provided as a Source Data file.
Then, we sought to validate some of the potential targets decreased in the PISA assay, including MTDH, MAP kinase-activated protein kinase 3 (MAPKAPK3), TRAF-type zinc finger domain containing 1 (TRAFD1), NKIRAS2, and zinc finger protein 787 (ZNF787) (Supplementary Fig. 6a, b). MTDH, annotated as a dsRBP in the GO database, displayed strong binding with poly(I:C) as compared with poly(C) and poly(dA:dT). MAPKAPK3 bound with poly(C) and poly(I:C). TRAFD1, NKIRAS2, and zinc figure protein ZNF787, bound with both poly(I:C) and poly(dA:dT). The results suggest that dsRNA binding may reduce the solubility of certain targets.
Since ZNF385A exhibited the largest FC among the discovered potential targets with increased solubility in the PISA experiment and specific binding towards dsRNA in the pull-down assay, we focused on ZNF385A in the following experiments. We used AlphaFold352 to predict the structure of the complex formed by ZNF385A and dsRNA. ZNF385A contains three C2H2-type zinc fingers (aa 74-98, aa 201-225, and aa 261-285), and the predicted results revealed that dsRNA was surrounded by these zinc fingers, possibly through hydrogen bond formed between the dsRNA backbone and the S85, S212, T223, H279, R284, A209, and R286 residues (Fig. 4a). Then, we constructed seven FLAG-tagged plasmids for ZNF385A by mutating or deleting its zinc finger domains (Fig. 4b). The poly(I:C) pull-down assay further confirmed that ZNF385A bound to dsRNA through the zinc finger domains (Fig. 4c). Interestingly, mutating all the zinc fingers in ZNF385A (Full-mutant) abolished its dsRNA-binding activity, while it could still bind to dsRNA when one of the three zinc fingers was mutated or deleted (the other six mutants) (Fig. 4c), suggesting that these three zinc fingers were capable of binding to dsRNA independently.

a The structure of ZNF385A-dsRNA complex predicted by AlphaFold3 (https://alphafoldserver.com/). The augmented plots show side chains forming key contacts, with hydrogen bonds shown as red dashed lines. Segments that contributed to the aa 74-98, aa 201-225, and aa 261-285 structures are in orange, green, and pink, respectively. b The constructed plasmids for dsRNA pull-down assay. c The poly(I:C) pull-down assay of the FLAG-tagged mutants and truncations of ZNF385A using HeLa cells. Poly(I:C) agarose beads were incubated with the cell lysates and then isolated by centrifugation. The FLAG-tagged proteins in the input and pull-down samples were detected by western blotting, with GAPDH used as an internal standard. Results are representative of two independent experiments. Source data are provided as a Source Data file.
ZNF385A depletion induced IFN-I response
To explore the biological implications of the interaction between ZNF385A and dsRNA, we investigated whether ZNF385A could impact IFN-I signaling by conducting a CRISPR/Cas9 KO assay using two single guide RNAs (sgRNAs) in three human cell lines (HEK293T, HeLa, and ASPC1 cells). The results from western blotting and DNA sequencing confirmed that ZNF385A was successfully knocked out (Fig. 5a–c, Supplementary Fig. 7a–c). Intriguingly, RNA isolation and quantitative real-time polymerase chain reaction (RT-qPCR) results revealed that IFNβ was significantly up-regulated upon ZNF385A KO in all three cell lines (Fig. 5d–f). Consistently, interferon stimulated genes (ISGs), including ISG15, retinoic acid-inducible gene I (RIG-I), and melanoma differentiation-associated gene 5 (MDA5), were also up-regulated (Fig. 5d–f). The up-regulated protein expression of RIG-I and MDA5 was also observed in HeLa and ASPC1 cell lines after ZNF385A KO (Fig. 5g). Gain-of-function mutations of MDA5 (GOF-MDA5) exhibit aberrantly enhanced signaling activity in response to endogenous dsRNAs and cause a variety of immune disorders53,54. Here we observed that ZNF385A KO further activated IFN-I response in cells expressing GOF-MDA5 (G495R) in cells (Fig. 5d–f). Furthermore, depletion of ZNF385A in a non-tumorigenic human lung epithelial cell line, BEAS.2B, activated IFN-I response as evidenced by the increased mRNA expression of IFNβ, ISG15, RIG-I, and MDA5 (Supplementary Fig. 8a–c). The results indicate the broad role of ZNF385A in the regulation of type-I interferon signaling across cells of different origins. Next, we performed a rescue assay by overexpressing ZNF385A in the KO cells, and the results showed that ZNF385A overexpression restored the mRNA level of IFNβ to the normal levels (Supplementary Fig. 9). In summary, ZNF385A can modulate the expression of IFNβ and may exert a negative regulatory role in IFN-I signaling.

Validation of ZNF385A KO by western blotting in HEK293T (a), HeLa (b), and ASPC1 (c) cells. Results are representative of two independent experiments. d–f The relative mRNA levels of IFNβ, ISG15, RIG-I and MDA5 in HEK293T (d), HeLa (e), and ASPC1 (f ) cells transfected with the gain-of-function mutant of MDA5 (G495R, GOF-MDA5) or the empty vector (EV). Data represent the means ± s.d. of triplicate independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, by unpaired two-tailed Student’s t-test, “ns” indicates not significant). g Western blots of RIG-I and MDA5 in HeLa and ASPC1 cell lines. Source data are provided as a Source Data file.
ADAR1 is an RNA-editing enzyme that catalyzes adenosine-to-inosine conversions in dsRNAs, and the deficiency of ADAR1 results in inflammatory phenotypes characterized by spontaneous cytokine production, including type I IFNs55. To explore the crosstalk between ZNF385A and other dsRNA signaling pathways, we investigated the role of ADAR1 on type-I interferon signaling in ZNF385A WT and KO cells. Upon ADAR1 knockdown, the mRNA levels of IFNβ and ISG15 were further upregulated in ZNF385A KO cells (Supplementary Fig. 10a). Consistently, ADAR1 overexpression decreased the mRNA levels of IFNβ and ISG15 in ZNF385A KO cells (Supplementary Fig. 10b).
ZNF385A KO promoted endogenous dsRNA accumulation
To further explore the molecular mechanism by which ZNF385A modulates IFN-I signaling, we examined whether it could affect the level of endogenous dsRNA. We assessed the level of endogenous dsRNAs in WT and ZNF385A KO cells by a dot blotting assay using the anti-dsRNA J2 antibody, an antibody widely used to recognize viral or cellular dsRNA longer than 40 bp without sequence specificity56. We found that ZNF385A KO induced dsRNA accumulation in the cell (Fig. 6a–c). Furthermore, the J2 dot blotting assay showed that overexpressing ZNF385A reduced the abundance of dsRNA in ZNF385A KO cells to a level comparable to that of WT cells (Supplementary Fig. 9c). Next, we performed J2 immunofluorescence imaging using three cell lines, including HeLa, ASPC1, and BEAS.2B. The results confirmed that the cellular dsRNA level was increased, and they were localized in both cell nucleus and cytoplasm (Fig. 6d–f). In addition, RNase-III digestion markedly diminished the fluorescence intensity (Fig. 6d–f), suggesting that the observed signal was indeed from dsRNA.

The analysis of dsRNA in HEK293T (a), HeLa (b), and ASPC1 (c ) cells by dot blotting. WT, wild type. KO, knockout. A representative of three independent experiments is displayed. Data represent the means ± s.d. of triplicate technical measurements (****P < 0.0001, by unpaired two-tailed Student’s t-test). Representative images showing the immunostaining of intracellular dsRNA in WT and ZNF385A KO HeLa (d), ASPC1 (e), and BEAS.2B (f) cells, with or without RNase III treatment (mean ± s.d., n = 50 individual cells, ****P < 0.0001, by unpaired two-tailed Student’s t-test). Scale bar, 10 μm. A representative of three independent experiments is displayed. g The workflow of dsRNA immunoprecipitation and RNA-sequencing (dsRIP-seq). h A heatmap showing the J2-isolated transcripts enriched in KO_dsRIP samples. i A pie plot showing the retrotransposons associated with transcripts enriched in KO_dsRIP samples. SINE, short interspersed nuclear element. LINE, long interspersed nuclear element. LTR, long terminal repeat. j–m The distribution of sequencing reads of dsRIP-seq data. Four representative genes, FTX (j), ADARB2 (k), TRPC6P2 (l), and LINC01446 (m), are displayed. n–p Relative RNA levels of Alu, ERVK, and ERVL in HEK293T (n), HeLa (o), and ASPC1 (p ) cells. Data represent the means ± s.d. of triplicate independent experiments by unpaired two-tailed Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001). Source data are provided as a Source Data file.
To explore the composition of accumulated dsRNAs after ZNF385A KO, we performed dsRNA immunoprecipitation using the J2 antibody followed by RNA-sequencing (Fig. 6g). PCA showed that the expression profiles of the four samples, including WT_input, KO_input, WT_dsRIP and KO_dsRIP, formed distinct clusters (Supplementary Fig. 11a), indicating the marked difference between WT and ZNF385A KO cells. The duplex-containing RNAs enriched in ZNF385A KO cells were identified according to the criteria reported in a previous study57, with dsRNAs overexpressed in ZNF385A KO cells also considered for the analysis (Supplementary Fig. 11b), resulting in a total of 934 dsRNA conformation-containing genes enriched in ZNF385A KO cells (Fig. 6h, Supplementary Data 4). We examined the other genes that did not fit the criteria, such as nuclear receptor binding factor 2 (NRBF2) and N-acetylglucosamine-1-phosphodiester alpha-N-acetylglucosaminidase (NAGPA), and they exhibited no change between WT and KO samples in both input and dsRIP data (Supplementary Fig. 11c, d), indicating that they were not affected by ZNF385A KO. Interestingly, RepeatMasker analysis revealed that the transcripts enriched in dsRNA immunoprecipitates from ZNF385A KO cells were predominantly derived from genes harboring retrotransposons, including the SINE, LINE, and LTR (Fig. 6i). These retrotransposons account for more than 75% of all transposons in the associated genes, and this percentage was much higher than that of the human genome (~42.2%), implying the substantial contribution of retrotransposons for the formation of dsRNAs regulated by ZNF385A. For example, long non-coding RNA five prime to Xist (FTX) with L1 (Fig. 6j), adenosine deaminase RNA specific B2 (ADARB2) with Alu (Fig. 6k), short transient receptor potential channel 6 pseudogene 2 (TRPC6P2) with ERVK (Fig. 6l), and LINC01446 with ERVL (Fig. 6m) were highly enriched in the KO_dsRIP samples. Alu belongs to the short interspersed repetitive element class of non-long terminal repeat (non-LTR) retrotransposons, and L1 is the most abundant class of LINE, while ERVK and ERVL are representatives of LTR retrotransposons in the human genome58. RNA structures from these regions were predicted using the RNAfold webserver (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi), showing the formation of dsRNA structure in the regions containing retroelements (Supplementary Fig. 11e–h).
Consistently, ZNF385A KO induced higher expression of three selected endogenous duplex elements as examined by RT-qPCR, including Alu retrotransposons and two human endogenous retrovirus genes, ERVK and ERVL (Fig. 6n–p). The elevated expression of retroelements has been shown to induce innate immune response mediated by dsRNA sensors13,59. Our results implicate that the activation of IFN-I signaling observed in ZNF385A KO cells is likely attributed to the increased expression level of endogenous dsRNAs.
To further investigate the underlying molecular mechanism, we employed Turbo-ID proximity labeling to identify the interacting proteins for ZNF385A. HeLa cells were transfected with V5-TurboID-nuclear export signal (NES) (cytoplasmic control), V5-TurboID-nuclear localization signal (NLS) (nuclear control), and V5-TurboID-ZNF385A plasmids, respectively. After biotin labeling and isolation, liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) analysis was performed to identify the bound proteins (Fig. 7a). Western blotting confirmed the expression of the fusion proteins and showed decent biotin labeling efficiency (Fig. 7b). The PCA and correlation analysis displayed distinct clusters formed by proteome profiles of each group (Supplementary Fig. 12a, b). To eliminate contaminations, proteins identified in the WT cells and proteins specifically observed in NES and NLS control cells were pooled to generate a list of background proteins (Supplementary Fig. 12c, Supplementary Data 5). Using P < 0.05 and FC > 4 as cutoff, 33 proteins were detected to be specifically enriched in ZNF385A group (Supplementary Fig. 12d). GO enrichment analysis revealed that these proteins were significantly associated with RNA splicing (Supplementary Fig. 12e). Among these proteins, four were simultaneously enriched in the comparisons of ZNF385A-vs-WT, ZNF385A-vs-NES, ZNF385A-vs-NLS, including TARDBP, PTBP1, YTH domain-containing family protein 2 (YTHDF2), and ZNF385A itself, and were considered as highly confident identifications (Fig. 7c). Amongst, TARDBP and PTBP1 displayed higher FCs compared to YTHDF2 (Fig. 7c). Therefore, we focused on PTBP1 and TARDBP for further investigation into their biological functions of binding with ZNF385A.

a The workflow of TurboID proximity labeling for identifying ZNF385A-interacting proteins. b Western blotting validation of TurboID-based proximity labeling. Results are representative of two independent experiments. c Comparison of the significantly enriched proteins from ZNF385A vs WT, ZNF385A vs NLS, and ZNF385A vs NES groups shown by a Venn plot. WT, wild type. NLS, nuclear localization signal. NES, nuclear export signal. ID, identity. FC, fold change. d Anti-FLAG co-IP assay was performed to validate the binding of PTBP1 and TARDBP to ZNF385A. EV, empty vector. Results are representative of two independent experiments. e Anti-PTBP1 co-IP assay was performed to explore the interaction between PTBP1 and TARDBP in WT and ZNF385A KO cells. Results are representative of two independent experiments. f The pie chart displaying the types and percentages of DNA regions for the retrotransposons enriched in the KO_dsRIP group. np, non- protein. pr, protein. CDS, coding sequence. UTR, untranslated Region. g The bar plot for the origins (intronic and non-intronic) of retrotransposons, including SINE, LINE, and LTR, enriched in ZNF385A KO_dsRIP samples. SINE, short interspersed nuclear element. LINE, long interspersed nuclear element. LTR, long terminal repeat. h Reads detected from CELF2 as a representative transcript containing intron-embedded retrotransposons. i–k J2-enriched retrotransposon-containing introns in CELF2 are predicted to form double-stranded secondary structures, including Alu (i), ERVL (j), and L1 (k). Source data are provided as a Source Data file.
First, we performed the anti-FLAG co-immunoprecipitation (co-IP) assay using HeLa cells expressing FLAG-tagged ZNF385A to verify the binding of PTBP1 and TARDBP to ZNF385A, as well as the interaction between PTBP1 to TARDBP. As expected, both PTBP1 and TARDBP could bind to ZNF385A (Supplementary Fig. 13a, b, Fig. 7d). Interestingly, we found that the interaction between PTBP1 and TARDBP was ZNF385A-dependent (Fig. 7e). Moreover, poly(I:C) pull-down assay revealed that both TARDBP and PTBP1 were associated with dsRNA (Supplementary Fig. 13c). These results suggested that ZNF385A, PTBP1, TARDBP, and dsRNA may form a complex. Previous studies have indicated that splicing disorder leads to the export of unspliced transcripts containing intronic dsRNA related to retrotransposons, and dsRNA-sensing pathways can respond to such global aberrations of RNA splicing60,61. In the absence of ZNF385A, however, the RNA splicing process might be disrupted, resulting in the abnormal accumulation of endogenous dsRNAs.
In accordance with such a hypothesis, analysis of the dsRIP-seq data revealed that 71.24% of the retrotransposons associated with the dsRNAs enriched in ZNF385A KO cells were localized within the introns (Fig. 7f). In detail, all three major types of the endogenous retrotransposons displayed high preference to the intronic derived regions, with 55.4%, 85.2%, and 80.9% for LINE, SINE and LTR, respectively (Fig. 7g). For example, the detected reads for CUGBP Elav-like family member 2 (CELF2) were mapped to annotated retroelements localized in the intron region (Fig. 7h). The RNA structures predicted by RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) showed the formation of dsRNA conformation in these enriched regions (Fig. 7i–k).
In addition, the TurboID results showed that besides TARDBP and PTBP1, ZNF385A potentially interacted with dozens of proteins enriched in the RNA splicing process, such as DEAD/H box 5 (DDX5), heterogeneous nuclear ribonucleoprotein F (HNRNPF), and Gem-associated protein 5 (GEMIN5) (Supplementary Fig. 12d, e), implying that ZNF385A may be involved in the regulation of RNA splicing. To further explore the potential impact of ZNF385A KO on RNA splicing, we performed the quantitative analysis of genes by mapping to the introns using the input data. Intriguingly, 953 genes mapped with introns were upregulated in ZNF385A KO cells and only 181 were down-regulated, indicating the widespread accumulation of intron-retained transcripts following ZNF385A KO (Supplementary Fig. 14a). To gain insights into the nature of these increased intron-retained transcripts, we performed RepeatMasker analysis and found that retrotransposons accounted for more than 65% of all transposons (Supplementary Fig. 14b), which is markedly higher than their occurrence in the human genome (~42.2%).
Collectively, these findings imply that ZNF385A KO may lead to abnormal retention of introns, thereby contributing to the retrotransposon-derived dsRNA accumulation in the ZNF385A KO cells.
ZNF385A depletion synergized with the bioactivity of 5-AZA-CdR and enhanced natural killer cell-mediated cytotoxicity
The biological functions and clinical impacts of ZNF385A have been largely unexplored. As the activation of IFN pathways has been implicated in cancer immunity, we question whether ZNF385A is associated with cancer progression. We performed a pan-cancer expression analysis of ZNF385A using the cancer genome atlas program (TCGA) datasets and found that it was up-regulated in 21 types of cancers, such as pancreatic adenocarcinoma (PAAD), kidney renal clear cell carcinoma (KIRC), and cervical squamous cell carcinoma (CESC) (Supplementary Fig. 15a). Next, we investigated the role of ZNF385A in immune infiltration. Correlation analysis demonstrated that the expression of ZNF385A was inversely correlated with the presence of activated NK cells in various cancers, such as PAAD and CESC (Supplementary Fig. 15b). Moreover, ZNF385A displayed a positive correlation with tumor-associated macrophages in 21 cancer types and immunosuppressive Treg cells in 10 cancer types (Supplementary Fig. 15b). Our findings suggest that ZNF385A may play a pivotal role in tumor progression through promoting immune suppression.
Next, we explored whether the effects of ZNF385A on IFN-I response could be exploited in cancer treatment. Previous studies have demonstrated that 5-AZA-CdR, a common chemotherapeutic agent inhibiting DNA methyltransferases, triggers the accumulation of endogenous dsRNA by suppressing DNA methylation and exerts its therapeutic efficacy in part through the activation of a broad range of dsRNA sensors (e.g., MDA5, TLR3 and OASes)13,14. Interestingly, we observed an increased level of IFNβ mRNA in all three ZNF385A KO cell lines compared to WT cells following treatment with 5-AZA-CdR (Fig. 8a–c), suggesting a potential opportunity to enhance the efficacy of 5-AZA-CdR for cancer treatment through co-targeting ZNF385A.

The relative mRNA levels of IFNβ, ISG15, RIG-I, and MDA5 in HEK293T (a), HeLa (b) and ASPC1 (c) cells treated with 0.3 μM of 5-AZA-CdR for 24 h. WT, wild type. KO, knockout. DMSO, dimethyl sulfoxide. 5-AZA-CdR, 5-Aza-2’-deoxycytidine. Data represent the means ± s.d. of triplicate independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, by unpaired two-tailed Student’s t-test). d The CCK8 assay was performed to assess the impact of ZNF385A KO on NK92-mediated cell cytotoxicity in HEK293T, HeLa, and ASPC1 cells. IFNβ protein was added at 10 ng/mL. NK92, natural killer-92. IFNβ, interferon β. Data represent the means ± s.d. of four biological replicates (**P < 0.01, ***P < 0.001, ****P < 0.0001, by unpaired two-tailed Student’s t-test, “ns” indicates not significant). e NK92 cell-mediated cytotoxicity was analyzed in WT and ZNF385A-KO ASPC1 cells using the calcein acetoxymethyl ester (Calcein-AM) method. Representative images are shown in the left panel. Scale bar = 400 μm. Percentages of Calcein AM-positive cells in different groups are presented in the right panel. Data represent the means ± s.d. of triplicate independent experiments (*P < 0.05, **P < 0.01, **** P < 0.0001, by unpaired two-tailed Student’s t-test, “ns” indicates not significant). f The depletion of ZNF385A enhanced endogenous dsRNA accumulation, thereby promoting the bioactivity of 5-AZA-CdR and NK cell-mediated cytotoxicity. DNMT DNA methyltransferase, DNMTi DNA methyltransferase inhibition, HERV human endogenous retrovirus, Me methylation. Source data are provided as a Source Data file.
Since IFNβ promotes NK cell cytotoxicity against tumor cells62,63 and the overexpression of ZNF385A negatively correlates with NK cell infiltration in the tumor microenvironment (TME) (Supplementary Fig. 15b), we asked whether ZNF385A in target cells modulated NK cell-mediated cytotoxicity. We first performed the NK92 killing assay using cell counting kit-8 (CCK8) in three cell lines, all of which exhibited moderately increased sensitivity to NK cell-mediated killing when ZNF385A was knocked out (Fig. 8d, WT + NK92 vs. KO + NK92). Furthermore, IFNβ treatment enhanced NK cell killing in ZNF385A-expressing cells (Fig. 8d, WT + NK92 vs. WT + IFNβ + NK92), but had minimal additional effect on NK92 activity in ZNF385A KO cells (Fig. 8d, KO + NK92 vs. KO + IFNβ + NK92), suggesting that ZNF385A KO functioned likely through activating IFNβ production. Taken together, our findings imply that ZNF385A may suppress tumor cell IFNβ expression to reduce NK killing. In addition, we also performed calcein acetoxymethyl ester (Calcein-AM) cell viability assay, and similar results were obtained and showed that ZNF385A KO enhanced NK cell-mediated cytotoxicity (Fig. 8e).
Discussion
dsRBPs participate in gene regulation, RNA processing, RNA interference, and innate immune response2,64,65. The origin of dsRNAs is not solely confined to viral infection, as they can also be endogenously generated from retroelements and mitochondrial DNA12,66. The systematic identification of dsRBPs can provide insights into the life cycle of dsRNAs, encompassing their biosynthesis, processing, interactions with proteins, and ultimate fate. However, comprehensive profiling of the global landscape of dsRBPs remains challenging, as the current approach for characterizing dsRBPs is limited to AP-MS and the traditional methods for profiling ssRBPs are inadequate for identifying dsRBPs because of inefficient UV cross-linking.
Here, we adopted PISA for high throughput profiling of dsRBPs based on their solubility alteration upon substrate binding. Compared to the affinity purification-based approaches, our method does not require dsRNA labeling. In addition, the procedure is straightforward and provides information regarding the physicochemical properties of the observed proteins. This method may be applied to discover interacting proteins for various other types of RNA molecules, especially non-coding RNAs, such as siRNA, lncRNA, and circRNA, as well as DNA. However, there are several limitations for this strategy, and some known dsRBPs were not detected in this study. First, dsRNA binding does not necessarily alter the solubility or thermal stability of all the target proteins, and certain target proteins may bind to dsRNAs with a low stoichiometry, resulting in minimal solubility alteration. Second, this method detects the targets without any enrichment and may lose some of the potential dsRBPs with low abundance. Stimulating the cells with type I INFs to boost the expression of some proteins involved in dsRNA signaling prior to PISA analysis may further enhance the sensitivity of detection. Additionally, we implemented PISA not in its full workflow that employs TMT labeling, peptide fractionation, and deep DDA analysis but adapted it to our proteomics workflows using DIA without fractionation, resulting in a much lower proteome depth than reported in other PISA studies30,67. This could have limited the detection and quantification of additional dsRBPs. Moreover, for dsRBPs that bind with dsRNAs at relatively low stoichiometry, their solubility changes may not be captured as significant intensity alterations. In light of the findings in this study, it is worthwhile to explore the other chemical proteomic approaches, such as limited proteolysis (LiP) and the stability of proteins from rates of oxidation (SPROX), for their applications in characterizing dsRBPs. Techniques with different working principles may be capable of probing distinct subsets of the proteome and providing complementary information to further expand the landscape of dsRBPs68,69,70.
Based on the PISA profiling, we discovered ZNF385A as a potential dsRBP. ZNF385A is previously known as an RNA-binding protein containing three C2H2 zinc finger domains. It is also a multifunctional transcription factor involved in diverse biological processes, including transcriptional regulation, cell differentiation, neural development, tumor suppression, and immune regulation51,71. However, the roles of ZNF385A in dsRNA binding and the underlying mechanisms have yet to be elucidated. Here, we found that ZNF385A KO led to endogenous dsRNA accumulation. In line with this, loss of ZNF385A enhanced the abundance of endogenous retrotransposons, a major source of immunogenicity. Our findings suggest that the functional mechanism of ZNF385A differs substantially from that of ADAR1. Loss-of-function mutations in ADAR1 leads to elevated immunogenicity of endogenous duplex elements, rather than altering their abundance8,55. Intriguingly, we observed that up-regulated dsRNA induced by ZNF385A KO was likely under the control of ADAR1 editing, implying that ADAR1 may work in concert with ZNF385A to maintain cellular dsRNA homeostasis.
Mechanistic study uncovered that ZNF385A may participate in the regulation of cellular dsRNA processing and innate immune surveillance through RNA splicing. Two splicing-associated proteins, PTBP1 and TARDBP, were identified as ZNF385A-binding partners in this study. Aberrant RNA splicing can result in unspliced transcripts with retained introns, which may form double-stranded structures and induced an innate immune response when transported to the cytoplasm60,61. A previous study reported that the loss of TARDBP was associated with endogenous dsRNA accumulation72. In addition, PTBP1 and TARDBP function as splicing factors that ensure proper RNA processing, and their repression has been reported to increase the retention of introns in transcripts73,74. Furthermore, we observed that PTBP1 and TARDBP interacted with each other in a ZNF385A-dependent manner. This evidence prompted us to assume that the depletion of ZNF385A may modulate dsRNA homeostasis via the interaction with PTBP1 and TARDBP. However, additional experiments are required to uncover the precise molecular mechanism.
Pan cancer analysis of ZNF385A expression in TCGA datasets revealed that ZNF385A could be a potential biomarker overexpressed and associated with poor prognosis in multiple cancers. The activation of IFNβ induced by the depletion of ZNF385A could be further exploited for cancer treatment in combination with 5-AZA-CdR, which is a clinically approved DNA methyltransferase inhibitor. Additionally, the increased NK cell killing ability regulated by the loss of ZNF385A might be utilized to develop strategies to improve NK cell-mediated immunotherapy. In vivo experiments are warranted to further evaluate the therapeutic potential of targeting ZNF385A.
In conclusion, we applied the PISA assay on lysates to discover dsRBPs, and the results greatly broaden the landscape of the dsRBPs and promote the understanding of molecular mechanism involving dsRNAs. Through functional investigation, we uncover the role of a the discovered dsRBP, ZNF385A, in regulating the activation of IFNβ via the accumulation of endogenous dsRNAs (Fig. 8f). The depletion of ZNF385A enhanced the IFN-activating effect of 5-AZA-CdR and NK cell-induced cytotoxicity, suggesting that inducing intrinsic dsRNA stress by targeting ZNF385A in combination therapies holds promise for accelerating cancer treatment.
Methods
Reagents and materials
The LMW poly(I:C), poly(I:C)-biotin and poly(dA: dT) were purchased from InvivoGen (San Diego, CA, USA). Poly(C), poly(I), poly(C)-agarose, CNBr-activated agarose 4B, dithiothreitol (DTT), iodoacetamide (IAA), and urea were purchased from Sigma (St. Louis, MO, USA). Protease inhibitor cocktail tablets were purchased from Roche Diagnostics (Indianapolis, IN, USA). The sequencing grade modified trypsin was purchased from Promega (Madison, WI, USA). LC-MS grade acetonitrile was from Merck (White-house Station, NJ, USA). Mouse monoclonal antibody against GAPDH (1:10000, Cat#60004-1-Ig, clone name: 1E6D9) and β-actin (1:10000, Cat#66009-1-Ig, clone name: 2D4H5), DYKDDDDK tag recombinant antibody (1:20000, 66008-4-Ig, clone name: 8H6A10), rabbit IgG control polyclonal antibody (Cat#30000-0-AP), rabbit polyclonal antibody against PTBP1 (1:2000, Cat#12582-1-AP), TARDBP (1:2000, Cat#10782-2-AP), TRAFD1 (1:1000, Cat#27741-1-AP), NKIRAS2 (1:1000, Cat#10174-1-AP), MAPKAPK3 (1:1000,Cat#15424-1-AP), ADAR1 (1:1000, Cat#14330-1-AP), MTDH (1:1000, Cat#13860-1-AP), ZNF787 (1:1000, Cat#31934-1-AP), EIF2AK2 (1:5000, Cat#18244-1-AP), and ZNF385A (1:1000, Cat#26288-1-AP) were purchased from Proteintech (Wuhan, China). Rabbit polyclonal antibody against NKRF (1:1000, Cat#A4853), mouse monoclonal antibody against V5-tag (1:1000, Cat#AE017, clone name: AMC0506), HRP-conjugated mouse anti-rabbit IgG light chain (1:5000, Cat#AS061, clone name: AMC0531), rabbit monoclonal antibody against PRKRA (1:1000, Cat#A8779, clone name: ARC1299), RIG-I (1:1000, Cat#A3689, clone name: ARC0824), MDA5 (1:1000, Cat#A2419, clone name: ARC0760), and STAU1 (1:1000, Cat#A4619, clone name: ARC1054) were purchased from ABclonal (Wuhan, China). HRP-conjugated goat-anti-mouse IgG (H + L) (1:3000, Cat#abs20001) and HRP-conjugated goat-anti-rabbit IgG (H + L) (1:3000, Cat#abs20002) were purchased from Absin (Shanghai, China). Puromycin was purchased from Invitrogen (Carlsbad, CA, USA). Biotin DecaLabel™ DNA Labeling Kit was purchased from Thermo Fisher (Waltham, MA, USA). Phosphate buffered saline (PBS) was prepared by using 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4 to buffer at pH 7.4. Deionized water from Milli-Q purification system (Millipore, Billerica, MA, USA) was used throughout the study.
Cell culture
HeLa, HEK293T, ASPC1, and BEAS.2B cells were purchased from American Type Culture Collection (ATCC) and were maintained with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (100 µg/mL) in a 5% CO2 incubator at 37 oC. NK92 cells were also purchased from ATCC and cultured with the specialized NK-92 medium supplemented with 20 U/mL of IL-2 (Procell, Wuhan, China).
Preparation of cell lysates
HeLa cells were washed with ice-cold PBS for three times and then transferred into a centrifugation tube. The cells were collected by centrifugation at 1000 rpm for 3 min, flash-frozen in liquid nitrogen, and lysed by PBS supplemented with protease inhibitors cocktail by four cycles of freezing cells in liquid nitrogen and thawing at 25 oC. Then, the lysate was centrifuged at 20,000 × g, 4 oC for 20 min, and the soluble fraction was collected. The total protein concentration was determined using a bicinchoninic acid assay (BCA) kit (Thermo Fisher Scientific, Waltham, MA, USA) and then adjusted to 1 mg/mL for subsequent steps.
PISA assay
The cell lysates were incubated with 10 μg/mL poly(I:C) or same volume of PBS and incubated at room temperature (RT) for 30 min with gentle shaking. Four biological replicates were performed. Next, 100 μL of the sample was distributed into twelve PCR tubes. The samples were heated in a thermocycler for 3 min across a temperature gradient from 45 to 56 oC. Following heating, samples were equilibrated at RT for 5 min. These samples were centrifuged at 21,000 × g for 2 h at 4 oC to remove aggregated proteins. An equal volume from each PCR tube was combined into four samples, including 45, 46, and 47 oC for PISA1; 48, 49 and 50 oC for PISA2; 51, 52 and 53 oC for PISA3; 54, 55 and 56 oC for PISA4. Next, the protein concentration of the soluble fractions was determined by BCA, and the samples were digested with trypsin following the filter-aided sample preparation (FASP) method using Amicon Ultra-0.5 centrifugal filters (Merck Millipore, Darmstadt, Germany) with 10 kDa molecular weight cut-off. Briefly, proteins were reduced with 10 mM DTT for 45 min at RT, alkylated with 40 mM IAA for 45 min in the dark at RT, and quenched with 40 mM DTT for 45 min at RT, followed by three washes with 50 mM ammonium bicarbonate and digestion with trypsin at an enzyme to protein ratio of 1:50 at 37 oC for 16 h. The digested peptides were collected by centrifugation, and the samples were dried with a SpeedVac concentrator (Savant Instruments, Farmingdale, NY, USA). Prior to MS analysis, the tryptic peptide samples were dissolved in 0.1% formic acid (FA), and the peptide concentration was measured by Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA).
High-pH reversed-phase high performance liquid chromatography fractionation of peptides
To build the data dependent acquisition (DDA) based spectral library, 100 μg of HeLa tryptic peptides were reconstituted in solvent A (0.01% v/v ammonium hydroxide) and loaded onto a 4.6 mm × 250 mm XBridge Peptide BEH C18 Column with 5 µm beads (Waters, Milford, MA, USA). Peptides were separated at a flow-rate of 700 μL/min using a Rigol L-3000 Series high performance liquid chromatography (HPLC) (Rigol, Beijing, China) with a non-linear gradient of solvent B (98% ACN, 2% solvent A) as follows: 5–35% (30 min), 35–95% (2 min), 95% (5 min), 95–5% (2 min), and then holding at 5% solvent B for 6 min. Fractions were collected in 1.5 mL centrifuge tubes every minute from 7 min to 36 min. Collected fractions were dried in a SpeedVac vacuum concentrator, resuspended in 0.1% FA, and combined into 10 samples by pooling every three fractions distanced by 10, such as #1, #11, and #21.
CETSA
Cell lysates were obtained and processed as described in section 4.3 and 4.4. After incubation with poly(I:C), an equal volume from the sample was dispensed into 22 PCR tubes. Samples were heated in a thermocycler for 3 min to different temperatures (37, 40, 43, 46, 49, 52, 55, 58, 61, 64, 67 oC). Then samples were equilibrated at RT for 5 min and centrifuged at 100,000 × g for 20 min to remove aggregated proteins. Next, 40 μL of the resulting soluble fraction was collected and mixed with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer, boiled and analyzed by western blotting.
Poly(I:C) pull-down assay
For the pull-down assay with poly(I:C) agarose, 20 μL bed volume of poly(C) agarose was mixed with 40 μg of poly(I) in buffer containing 50 mM Tris (pH 7.0) and 150 mM NaCl. The mixture was mixed gently for 12 h at 4 oC. Next, the beads were washed three times with lysis buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), and 0.2% Nonidet P-40, 1.5 mM MgCl2, RNase inhibitor, and protease inhibitor cocktail. Cell lysates were prepared by disruption in the lysis buffer for 30 min on ice. Lysates were clarified by centrifugation at 16,000 × g for 20 min and the protein concentrations were determined with a BCA assay. Next, 2 mg of extracted cellular protein was incubated with poly(I:C) agarose for 4 h with rotation at 4 oC. The control poly(C) agarose and blank control CNBr-activated agarose 4B were processed in parallel. Finally, the beads were washed with lysis buffer for five times and then mixed with the SDS-PAGE sample loading buffer, and the eluted proteins were analyzed by western blotting.
To generate poly(dA:dT)-biotin, poly(dA:dT) was labeled using the Biotin DecaLabel DNA Labeling Kit (Thermo Scientific) in accordance with the manufacturer’s instructions. In brief, 1 μg of poly(dA:dT) was mixed with 10 μL of 5 × reaction buffer containing decanucleotide and adjusted to 44 μL with nuclease free-water. The contents of the tube were boiled for 10 min and then cooled on ice. Next, 5 μL of biotin labeling mix and 5 units of Klenow fragment exonuclease was added. The tube was then incubated at 37 oC for 20 h, and the reaction was terminated by the addition of 1 μL of 0.5 M EDTA (pH 8.0). The cellular proteins were extracted from HeLa cells by the lysis buffer (40 mM Tris, 120 mM NaCl, 1% Triton X-100) supplemented with 1× protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) and 1 U mL−1 RNase inhibitor. The total protein concentration of the extract was measured with BCA. Then, 40 μL of Dynabeads™ MyOne™ Streptavidin C1 (Invitrogen) were washed and incubated with poly(dA:dT)-biotin, poly(I:C)-biotin or left untreated at 4 oC for 2 h with rotation. Next, the beads were added to cell lysates and incubated at 4 oC for 4 h, separately. Then, the beads were washed five times by the lysis buffer and further analyzed by western blotting.
Construction of ZNF385A mutants
We constructed N-terminal FLAG-tagged plasmids for ZNF385A in a pCDNA3.1+ vector by either deleting or mutating its zinc finger domains. For the deletion, the three zinc finger domains (74-98, 201-225, and 261-285 aa) were truncated individually. For the mutation, we introduced the substitutions (R82E, K94D, R97E, K207D, K221D, K226D, K269D, K277D, R283E, R284E, and R286E) either within one of the three zinc fingers or across all sites together. The constructed plasmids were then transfected into HeLa cells for 48 h. Next, the cells were washed three times with ice cold PBS, harvested using the lysis buffer, and subjected to the poly(I:C) pull-down assay as described in Section 4.7. The FLAG tag was detected by western blotting, with GAPDH used as the internal standard.
CRISPR/Cas9-based gene KO and ADAR1 knockdown
The two sgRNA sequences targeting ZNF385A (Supplementary Table 1) were selected using the sgRNA designer WU CRISPR (https://crisprdb.org/wu-crispr/). The two sgRNA target sequences were individually cloned into the pSpCas9(BB)2A-Puro (PX459) V2.0 vector (Addgene, Watertown, MA, USA) by inserting synthesized oligo-DNA primers corresponding to the target sequence into the BbsI restriction sites. The constructed plasmids were transfected into sub-confluent HEK293T cells, HeLa cells, ASPC1 cells, and BEAS.2B cells grown in DMEM supplemented with 10% FBS. Following a 48 h recovery period, cells were cultured in the medium supplemented with 2 µg/mL puromycin for another 48 h for puromycin resistance screening, and then the selection reagent was removed from the cells. Once recovered, cells were collected and the genomic DNA was extracted with a TIANamp Genomic DNA kit (TIANGEN, Beijing, China). The fragment containing the target sequences of the two sgRNAs was amplified by PCR with the designed primers (Supplementary Table 2), and the products were examined with 2% agarose gel electrophoresis. The target bands were cut out and purified with an agarose Gel DNA Extraction Kit Ver.4.0 (Takara, Otsu, Japan) for sequencing.
For siRNA mediated gene knockdown, cells were transfected with siRNA targeting ADAR1 (siADAR1) or a negative control siRNA (siNC) using the Lipofectamine 2000 (Life Technologies, Carlsbad, California, USA), according to the manufacturer’s instructions. siRNAs were synthesized by Sangon Biotech (Shanghai, China), and the sequences were listed in Supplementary Table 3.
TurboID proximity labeling
HeLa cells at approximately 80% confluency were transiently transfected with the V5-TurboID-ZNF385A plasmid using Lipofectamine 2000 (Life Technologies, Carlsbad, California, USA) in serum-free media. V5-TurboID-NES and V5-TurboID-NLS plasmids were used as the cellular localization controls, and the ligase-omitted sample was used as the blank control. After 6 h, the Lipofectamine 2000-containing media was replaced with fresh serum-containing media. Three biological replicates were performed. Cells were harvested and labeled with 500 μM biotin for 20 min at 37 oC after 24 h following transfection. The labeling was stopped by transferring the cells to ice and washing five times with ice-cold PBS. Cells were lysed in 1 mL RIPA lysis buffer on ice for 20 min and scraped off from the culture dishes using a cell scraper and then transferred into 1.5 mL microcentrifuge tubes. Lysates were clarified by centrifugation at 10,000 rpm for 10 min at 4 oC.
To isolate biotinylated proteins, 40 μL of streptavidin beads slurry (10 mg/mL) of Dynabeads™ MyOne™ Streptavidin C1 (Invitrogen) were washed twice with RIPA buffer before incubated with the clarified lysates containing 3 mg of protein per sample with rotation at 4 oC overnight. The beads were subsequently washed twice with 1 mL of RIPA lysis buffer, once with 1 mL of 1 M KCl, 1 mL of 0.1 M Na2CO3, 1 mL of 2 M urea in 10 mM Tris-HCl (pH 8.0), and twice with 1 mL RIPA lysis buffer. To completely remove the potential detergent, proteins bound to streptavidin beads were washed twice with 200 μL of 50 mM Tris-HCl buffer (pH 7.5) followed by two washes with 2 M urea in 50 mM Tris-HCl (pH 7.5) buffer. The beads were then processed for LC-MS/MS analysis as described by Kelvin et al.75. Briefly, the beads were incubated with 80 μL of 2 M urea/50 mM Tris-HCl containing 1 mM DTT and 0.4 μg trypsin for 1 h at RT with shaking. After 1 h, the supernatant was transferred to a fresh tube. The streptavidin beads were washed twice with 60 μL of 2 M urea in 50 mM Tris-HCl buffer (pH 7.5), and the washes were combined with the on-bead digest supernatant. The eluate was reduced with 10 mM DTT for 30 min at RT with shaking and alkylated with 40 mM IAA for 45 min in the dark at RT with shaking. An additional 0.5 μg of trypsin was added to the sample and the digestion was allowed to continue overnight at 37 oC with shaking. Finally, samples were acidified by adding FA to 1%, desalted with C18 StageTips, and dried in SpeedVac.
LC-MS/MS analysis
The digested peptides were analyzed by a Fusion Lumos mass spectrometer coupled with an Easy-nLC 1000 system (Thermo Fisher Scientific). Briefly, peptides were loaded onto a 150 μm × 2 cm self-packed C18 trap column (particle size 3 μm, Dr. Maisch GmbH, Germany) and separated on a 150 μm × 30 cm self-packed C18 analytical column (particle size 1.9 μm, Dr. Maisch GmbH). The column was operated at 50 oC and at a flow rate of 300 nL/min. HPLC solvent A consisted of 100% water and 0.1% FA, and solvent B was composed of 80% ACN, 20% water, and 0.1% FA. The gradient of solvent B was set as follows: 8% to 12% (10 min), 12% to 27% (69 min), 27% to 45% (28 min), 45% to 95% (3 min), and holding at 95% for 10 min.
For the PISA assay, the instrument was operated in the DIA mode. The cycle time was 3 s, and each cycle consisted of a MS1 scan of 350–1000 m/z with 60,000 resolution and automatic gain control (AGC) target of 1 × 106, followed by 38 MS2 scans of 200–2000 m/z with 30,000 resolution and AGC target of 5 × 105. Variable isolation windows were applied as previously described76. Briefly, 38 variable isolation windows were applied as follows: 350–390 m/z, 390–420 m/z, 420–450 m/z, and 30 isolation windows stepped by 15 m/z in the range of 450–900 m/z, followed by another 5 windows as 900–950 m/z, 950–1000 m/z, 1000–1050 m/z, 1050-1125 m/z, 1125–1200 m/z.
For the proximity labeling samples and spectral library construction samples, the instrument was operated in DDA mode. The full scan was acquired from m/z 350 to 1 500 with a resolution of 120,000, AGC as 5 × 105, and maximum injection time as 50 ms. MS/MS scans were performed with a resolution of 30,000, AGC target as 1 × 105, and maximum injection time as 50 ms. The selected ions were isolated using the quadrupole analyzer in a window of 1.6 m/z and fragmented by higher energy collisional dissociation fragmentation with normalized collision energy of 35%. Dynamic exclusion time was set to 30 s.
Bioinformatics and statistical analysis
For DDA-based spectrum library generation, the FragPipe computational platform (v13.0) with MSFragger (v3.0) and Philosopher (v3.2.9) components was used. Raw MS files from bRPLC fractions were converted to the open mzML format using the MSConvert utility of the Proteowizard (v3.0.20196) software suite. For the MSFragger analysis, MS/MS spectra were searched against Homo sapiens FASTA sequence database downloaded from Swiss-Prot (20,375 sequence entries, released in September 2020). Mass search tolerance was set as 10 ppm for MS and MS/MS. Peptide length was set as 7–30, and peptide mass was set as 300–1800 Da. Cysteine carbamidomethylation (+57.0215 Da) was specified as a fixed modification, and methionine oxidation (+15.9949 Da) was set as a variable modification. Up to two missed cleavages for trypsin digestion were allowed. The search results were further processed using the Philosopher pipeline. First, MSFragger output files were processed using PeptideProphet to compute the posterior probability of correct identification for each peptide to spectrum match. The protein groups assembled by ProteinProphet were filtered to 1% precursor and protein-level false discovery rate (FDR) using the target-decoy strategy. In brief, reversed protein sequences were appended to the original databases as decoys. The final output named as “con_lib.csv” was imported to DIA-NN for DDA-based spectral library. The acquired data were analyzed using DIA-NN (v1.8) using a protein database composed of the Homo sapiens FASTA database downloaded from Swiss-Prot (20,375 sequence entries, released in September 2020). The processing parameters were set as follows: Trypsin/P with maximum one missed cleavage; protein N-terminal M excision; Carbamidomethyl on C as a fixed modification; no variable modifications; peptide length from 7 to 30; precursor charge 1-4; precursor m/z from 300 to 1800; fragment m/z from 200 to 1800. For DDA data, MaxQuant (version 2.1.0.0) was employed to perform label-free quantification. Peak lists were generated and searched against the Swiss human protein database as described above. The processing parameters were set as follows: search type, Standard for DDA data; Carbamidomethyl on C as a fixed modification; Oxidation on M and Acetyl at protein N-terminus as variable modifications; Trypsin/P with maximum two missed cleavages; peptide mass tolerance in first search, 20 ppm; PSM and protein FDRs were both set to 0.01.
For the data from PISA, protein quantification was performed in R (version 4.3.2) using the output from DIA-NN. Two or more prototypic peptides were required for protein identification. Proteins were quantified using the “Precursor.quantity” intensities. The quantification matrix was generated by the “diann” R package. Proteins with less than 50% missing values were retained for quantification. The data matrix was normalized by the quantile method, log2 transformed, and imputed by the “imputeLCMD” package. Unpaired two-tailed t-test was performed for statistical analysis. Proteins with P < 0.01 and |log2FC | > 0.58 were considered to be significantly changed. PCA was performed using the “FactoMineR” package, and Pearson correlation analysis was conducted with the “corrplot” package. Volcano plot was generated by the “ggplot2” package. For GO and KEGG pathway enrichment analysis, “ggplot2” and “clusterProfiler” packages were used. The domain enrichment analysis was performed by DAVID 6.8 (https://david.ncifcrf.gov/). Melting curves in CETSA were fitted using the R package “NPARC”. The pan-cancer expression analysis of ZNF385A in the TCGA dataset was performed by the “UCSCXenaShiny” R package. The correlation between ZNF385A expression and immune cell infiltration was analyzed using the “TCGAplot” R package.
For the prediction of the structure of ZNF385A-dsRNA complex, AlphaFold3 (https://alphafoldserver.com/) was used by taking the full length of the sequence of ZNF385A and dsRNA with random sequence (5′-GGUAGACGCUUCGGCGUUUGCC-3′) as input. The UCSF Chimera X (version 1.7.1) software was used for visualization. The Hydrogen bonds and contact residues were labeled with default parameters set in UCSF Chimera X.
dsRNA dot blotting
Total cellular RNA was extracted using the TRIzol reagent (Thermo Scientific) according to the manufacturer’s protocol. Isolated RNA was dotted on a 0.22 μm polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). Then, RNA was UV cross-linked using 125 mJ/cm2 at 254 nm and unbound RNA was washed off using the Tris buffered saline with 0.1% Tween 20 (TBST). The membrane was blocked for 1 h using 5% skim milk in TBST at RT and incubated with anti-dsRNA J2 antibody (Scions, Hungary) overnight at 4 oC. Next, the membrane was incubated with the secondary antibody for 1 h at RT. Following washing with TBST for three times, the membrane was visualized using the enhanced chemiluminescence solution.
J2 immunofluorescence imaging
HeLa, ASPC1, and BEAS.2B cells and the corresponding ZNF385A KO cells were used for J2 immunofluorescence imaging. First, cells were washed with PBS, fixed in 4% paraformaldehyde for 15 min at RT, and then permeabilized with PBS containing 0.25% Triton X-100 for 10 min at RT. The negative control cells were treated with 4U RNase III (NEB, Cat#E6146) in reaction buffer at 37 oC for 4 h after permeabilization following the manufacturer’s instruction. Next, cells were blocked with 1% BSA in PBS for 1 h and incubated with monoclonal anti-dsRNA antibody J2 at 4 oC overnight. After incubation, cells were washed with PBS with 0.05% tween 20 for three times and then incubated with the secondary antibody (Alexa Fluor 568 goat anti-mouse IgG, Invitrogen) for 1 h at RT in the dark. Finally, the cells were incubated with 250 ng/mL DAPI for 10 min and mounted with coverslip. Cells were imaged and analyzed using a Nikon Eclipse Ti-E inverted microscope with the NIS-Element AR 4.50 software. J2 intensity was normalized to mean intensity in the WT cells for each experiment.
dsRNA immunoprecipitation (J2 dsRIP)
The J2 dsRIP experiment was performed according to the reported procedure57. Briefly, the protein G Dynabeads (Thermo Fisher Scientific) were washed and resuspended in the dsRIP lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 3 mM MgCl2, 0.5% IGEPAL CA-630). For each sample, 25 μL of beads were pre-bound to 5 μg of anti-dsRNA mAb (J2) by incubation for 2 h at 4 oC. HeLa WT and KO cells were collected and washed with ice cold PBS for three times. Then, 3 × 107 cells for each group were lysed in 0.6 mL dsRIP lysis buffer for exactly 5 min on ice. Total RNA was harvested from the rest of cells using Trizol for both groups to serve as input samples and stored at −80 oC. After that, the cell lysates were spin down by centrifugation at 13,000 × g for 10 min at 4 oC. For immunoprecipitation, 25 μL of J2-bound Protein G Dynabeads were added to the lysate and incubated for 1 h at 4 oC with rotation. Then, the beads were washed four times with dsRIP lysis buffer, and J2-bound dsRNA was harvested directly from beads with TRIzol with a ratio of 1:5 of chloroform added.
dsRIP-seq library preparation and next-generation sequencing
The library generation and dsRIP-seq were performed according to the previous protocol57. Briefly, both the input and dsRIP samples were used for library generation. Sequencing libraries were prepared using the Ribo-clean rRNA Depletion Kit (Vazyme, China, Cat# RN416) and KCTM Digital Stranded mRNA Library Prep Kit (Wuhan Seqhealth Co., Ltd. China) following the manufacturer’s instruction. Subsequently, the libraries were quantified by Qubit 3.0 and sequenced on a DNBSEQ-T7 platform (MGI) using the PE150 sequencing mode to generate paired-end reads.
dsRIP-seq data analysis
Raw sequencing data was first filtered by fastp (version 0.23.1), where low-quality reads were discarded and the reads with adapter sequences were trimmed. Clean reads were further treated to eliminate duplication bias introduced in library preparation and sequencing. In brief, clean reads were first clustered according to the UMI sequences, grouping reads with the same UMI sequence into the same cluster. Reads within each cluster were compared using pairwise alignment, and then reads with over 95% sequence identity were extracted to a new sub-cluster. After all sub-clusters were generated, multiple sequence alignment was performed to get one consensus sequence for each sub-cluster. These steps eliminated errors and biases introduced by PCR amplification or sequencing. The sequences were then used for standard mRNA-seq analysis. They were mapped to the human genome hg38 using STAR software (version 2.7.6a). Reads mapped to the exon regions of each gene were counted by featureCounts (Subread-1.5.1; Bioconductor) and RPKM values were calculated. Differentially expressed genes between groups were identified using the edgeR package (version 4.0.16), and a P-value cutoff of 0.05 and a FC cutoff of 2 were used. The dsRNAs enriched in ZNF385A KO cells were determined according to a reported protocol57 as illustrated in Supplementary Fig. 11b. Briefly, dsRIP-enriched transcripts were identified by comparing dsRIP and input signals (Filter1). Next, Filter 2 and Filter 3 were applied to identify the increase of dsRNA conformation in KO cells. In parallel with Filter 2 and Filter 3, Filter 4 was used to find dsRNA-containing transcripts with elevated abundance in KO cells. Annotation for repeat elements was performed using RepeatMasker (open-4.0.9). “Bedtools intersect” was applied to perform an intersection analysis between genes and transposable elements77. The regions of genes were further divided into CDS, np-Exon, np-intron, pr-intron, 5′ UTR, and 3′ UTR. IGV78 (version 2.19.1) was used to visualize read counts against the genome. RNA structure prediction was conducted using the RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) with default parameters.
Co-IP
The cells were harvested and lysed with Triton X-100 lysis buffer (40 mM Tris, 120 mM NaCl, and 1 % Triton X-100) supplemented with the protease inhibitor cocktail. Antibody against the protein of interest or normal IgG was incubated with protein A/G beads (MCE, Shanghai, China) at 4 oC for 12 h. The total protein concentrations of the cell lysates were measured with BCA. Then, 2 mg of the cell lysates were incubated with antibody-conjugated beads at 4 oC for 12 h. Proteins in the immunoprecipitates were analyzed by western blotting.
Western blotting
The proteins were resolved by SDS-PAGE and transferred to the 0.22 µm PVDF membrane. The blots were incubated with primary antibodies and then developed by enhanced chemiluminescence after incubated with a horseradish peroxidase-conjugated secondary antibody. GAPDH and β-actin were used as internal standards. The band intensities were measured using ImageJ (version 1. 54 g) for quantitative comparisons.
RT-qPCR
Total RNA was isolated from the cells using TRIzol reagent (Invitrogen), and complementary DNA was synthesized using the FastQuant RT kit (Sangon Biotech, Shanghai, China) according to the manufacturer’s instructions. The DNA contamination was avoided by a gDNA remover step, in which samples were incubated with DNase I at 42 oC for 2 min. Quantitative RNA expression analysis was performed on a QuantStudio 1 Plus real-time PCR system (Applied Biosystems, USA) using the 2 × HQ SYBR qPCR Mix (Low ROX) (Zoman, Beijing, China) following the manufacturer’s instructions. Each sample was analyzed in triplicates, and the relative RNA expression FCs were calculated with 2−ΔΔCT and normalized to a housekeeping gene, GAPDH. The stability of GAPDH as a housekeeping gene was evaluated using the geNorm79 program, based on the principle that the expression ratio of two ideal internal control genes is identical in all samples regardless of the experimental condition or cell type. Three other commonly used housekeeping genes, including RPL13, 18S, and β-actin, were used for the measure. The primer sequences used in this study were listed in Supplementary Table 4.
NK92 cell-mediated tumor cell killing assay
The tumoricidal activity of NK92 cells was evaluated using a CCK-8 Kit (GplBio, Montclair, CA, USA). Briefly, the WT and ZNF385A KO cells were seeded into 96-well plates with a density of approximately 5 × 103 cells/well for 12 h. Next, NK92 cells were added at the 1:2 Effect: Target (E:T) ratios and co-cultured in 0.1 mL complete medium (RPMI 1640 medium with 10% fetal bovine serum) for 24 h. Afterward, NK 92 cells in the suspension were gently washed out by PBS. Subsequently, CCK-8 reagent was added to each well, and the plates were incubated for 4 h at 37 oC. Finally, the optical density was measured at 450 nm by a plate reader.
The Calcein-AM assay was also performed to evaluate the NK92 cell-mediated tumor cell killing. First, the WT and ZNF385A KO cells were incubated with 10 ng/mL of IFNβ for 24 h at 37 oC in a cell incubator. Then, the cells were detached using 0.25% trypsin-EDTA and collected by centrifugation at 1000 rpm for 3 min. Subsequently, the cells were washed with PBS for three times, labeled using 10 mM Calcein AM (Beyotime, Shanghai, China) for 30 min at RT in the dark, and then washed 5 times with PBS. Afterward, 5 × 103 labeled cells/well were seeded into 96-well plates. Meanwhile, NK92 cells were added to the wells at a 1:5 E:T ratios. The cells were co-incubated at 37 oC in an atmosphere of 95% air and 5% CO2 for 12 h. Cells with complete lysis were used as the negative control, achieved by incubation with 0.5% Triton X-100 for 1 h. Finally, fluorescence images of the Calcein AM-labeled cells were obtained using a fluorescence microscope at 10 × magnification.
Statistics and reproducibility
At least three biological replicates were conducted in the biological assays. Prism version 9.5.0 (GraphPad) was used for statistical analyses and graph generation. For the comparisons, unpaired two-tailed Student’s t-test was performed between two groups unless otherwise stated. The P value less than 0.05 was considered statistically significant. All experiments were repeated at least twice with similar results.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX80 partner repository with the dataset identifier PXD051654. The raw and processed dsRIP-seq datasets generated in this study have been deposited into Gene Expression Omnibus (GEO) under the series entry number of GSE289807. Source data are provided with this paper.
References
Reich, D. P. & Bass, B. L. Mapping the dsRNA World. Cold Spring Harb. Perspect. Biol. 11, a035352 (2019).
Hur, S. Double-stranded RNA sensors and modulators in innate immunity. Annu Rev. Immunol. 37, 349–375 (2019).
Schultze, J. L. & Aschenbrenner, A. C. COVID-19 and the human innate immune system. Cell 184, 1671–1692 (2021).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Schlee, M. & Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 16, 566–580 (2016).
Dhir, A. et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242 (2018).
Kassiotis, G. & Stoye, J. P. Immune responses to endogenous retroelements: taking the bad with the good. Nat. Rev. Immunol. 16, 207–219 (2016).
Liddicoat, B. J. et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120 (2015).
Harapas, C. R. et al. Organellar homeostasis and innate immune sensing. Nat. Rev. Immunol. 22, 535–549 (2022).
Heneka, M. T., Kummer, M. P. & Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477 (2014).
de Reuver, R. & Maelfait, J. Novel insights into double-stranded RNA-mediated immunopathology. Nat. Rev. Immunol. 24, 235–249 (2024).
Chen, Y. G. & Hur, S. Cellular origins of dsRNA, their recognition and consequences. Nat. Rev. Mol. Cell Biol. 23, 286–301 (2022).
Chiappinelli, K. B. et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986 (2015).
Mehdipour, P. et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 588, 169–173 (2020).
Sheng, W. et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174, 549–563.e19 (2018).
Castello, A. et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406 (2012).
Bao, X. et al. Capturing the interactome of newly transcribed RNA. Nat. Methods 15, 213–220 (2018).
Queiroz, R. M. L. et al. Comprehensive identification of RNA–protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat. Biotechnol. 37, 169–178 (2019).
Yan, S. et al. Characterization of RNA-binding proteins in the cell nucleus and cytoplasm. Anal. Chim. Acta 1168, 338609 (2021).
Pennemann, F. L. et al. Cross-species analysis of viral nucleic acid interacting proteins identifies TAOKs as innate immune regulators. Nat. Commun. 12, 7009 (2021).
Girardi, E. et al. Proteomics-based determination of double-stranded RNA interactome reveals known and new factors involved in Sindbis virus infection. RNA 29, 361–375 (2023).
Molina, D. M. et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 (2013).
Savitski, M. M. et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784 (2014).
Franken, H. et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 10, 1567–1593 (2015).
Huber, K. V. M. et al. Proteome-wide drug and metabolite interaction mapping by thermal-stability profiling. Nat. Methods 12, 1055–1057 (2015).
Sridharan, S. et al. Proteome-wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat. Commun. 10, 1155 (2019).
Becher, I. et al. Pervasive protein thermal stability variation during the cell cycle. Cell 173, 1495–1507.e18 (2018).
Potel, C. M. et al. Impact of phosphorylation on thermal stability of proteins. Nat. Methods 18, 757–759 (2021).
Gaetani, M. et al. Proteome integral solubility alteration: a high-throughput proteomics assay for target deconvolution. J. Proteome Res. 18, 4027–4037 (2019).
Li, J., Van Vranken, J. G., Paulo, J. A., Huttlin, E. L. & Gygi, S. P. Selection of heating temperatures improves the sensitivity of the proteome integral solubility alteration assay. J. Proteome Res. 19, 2159–2166 (2020).
Zhang, X., Lytovchenko, O., Lundström, S. L., Zubarev, R. A. & Gaetani, M. Proteome integral solubility alteration (PISA) assay in mammalian cells for deep, high-confidence, and high-throughput target deconvolution. Bio Protoc. 12, e4556 (2022).
Gaetani, M. & Zubarev, R. A. Proteome integral solubility alteration (PISA) for high-throughput ligand target deconvolution with increased statistical significance and reduced sample amount. Methods Mol. Biol. 2554, 91–106 (2023).
Saei, A. A. et al. Multifaceted proteome analysis at solubility, redox, and expression dimensions for target identification. Adv. Sci. 11, e2401502 (2024).
Sabatier, P., Beusch, C. M., Meng, Z. & Zubarev, R. A. System-wide profiling by proteome integral solubility alteration assay of drug residence times for target characterization. Anal. Chem. 94, 15772–15780 (2022).
Batth, T. S. et al. Streamlined analysis of drug targets by proteome integral solubility alteration indicates organ-specific engagement. Nat. Commun. 15, 8923 (2024).
Emery-Corbin, S. J. et al. Improved drug target deconvolution with PISA-DIA using an extended, overlapping temperature gradient. Proteomics 24, e2300644 (2024).
Ruan, C. et al. Matrix thermal shift assay for fast construction of multidimensional ligand–target space. Anal. Chem. 94, 6482–6490 (2022).
Ji, H. et al. Target deconvolution with matrix-augmented pooling strategy reveals cell-specific drug-protein interactions. Cell Chem. Biol. 30, 1478–1487.e7 (2023).
Harris, P. et al. Double-stranded RNA induces molecular and inflammatory signatures that are directly relevant to COPD. Mucosal Immunol. 6, 474–484 (2013).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 (2010).
Wang, Y. et al. Mitochondria-localised ZNFX1 functions as a dsRNA sensor to initiate antiviral responses through MAVS. Nat. Cell Biol. 21, 1346–1356 (2019).
Masliah, G., Barraud, P. & Allain, F. H.-T. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol. Life Sci. 70, 1875–1895 (2013).
Lee, K., Ku, J., Ku, D. & Kim, Y. Inverted Alu repeats: friends or foes in the human transcriptome. Exp. Mol. Med. 56, 1250–1262 (2024).
Castello, A. et al. Comprehensive identification of RNA-binding domains in human cells. Mol. Cell 63, 696–710 (2016).
Xiao, Y. et al. Profiling of RNA-binding protein binding sites by in situ reverse transcription-based sequencing. Nat. Methods 21, 247–258 (2024).
Paget, M. et al. Stress granules are shock absorbers that prevent excessive innate immune responses to dsRNA. Mol. Cell 83, 1180–1196.e8 (2023).
Li, X. et al. Structural basis of double-stranded RNA recognition by the RIG-I like receptor MDA5. Arch. Biochem. Biophys. 488, 23–33 (2009).
Lu, C. et al. The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure 18, 1032–1043 (2010).
Huang, L. et al. Down-regulation of miR-301a suppresses pro-inflammatory cytokines in Toll-like receptor-triggered macrophages. Immunology 140, 314–322 (2013).
Zhou, Q. et al. MicroRNA 29 targets nuclear factor-κB–repressing factor and claudin 1 to increase intestinal permeability. Gastroenterology 148, 158–169.e8 (2015).
Das, S. et al. Hzf determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell 130, 624–637 (2007).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Rice, G. I. et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet 46, 503–509 (2014).
Ahmad, S. et al. Breaching self-tolerance to Alu duplex RNA underlies MDA5-mediated inflammation. Cell 172, 797–810.e13 (2018).
Chung, H. et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172, 811–824.e14 (2018).
Blango, M. G. & Bass, B. L. Identification of the long, edited dsRNAome of LPS-stimulated immune cells. Genome Res. 26, 852–862 (2016).
Gao, Y., Chen, S., Halene, S. & Tebaldi, T. Transcriptome-wide quantification of double-stranded RNAs in live mouse tissues by dsRIP-Seq. STAR Protoc. 2, 100366 (2021).
Kazazian, H. H. Mobile elements: drivers of genome evolution. Science 303, 1626–1632 (2004).
Roulois, D. et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973 (2015).
Wu, Y. et al. Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. EMBO J. 37, e99017 (2018).
Bowling, E. A. et al. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell 184, 384–403.e21 (2021).
Barnes, S. A. et al. Type I interferon subtypes differentially activate the anti-leukaemic function of natural killer cells. Front Immunol. 13, 1050718 (2022).
Makowska, A. et al. Interferon beta increases NK cell cytotoxicity against tumor cells in patients with nasopharyngeal carcinoma via tumor necrosis factor apoptosis-inducing ligand. Cancer Immunol. Immunother. 68, 1317–1329 (2019).
Jin, L. et al. The molecular mechanism of dsRNA processing by a bacterial Dicer. Nucleic Acids Res. 47, 4707–4720 (2019).
Nicholson, A. W. Ribonuclease III mechanisms of double-stranded RNA cleavage. Wiley Interdiscip. Rev. RNA 5, 31–48 (2014).
Cottrell, K. A., Andrews, R. J. & Bass, B. L. The competitive landscape of the dsRNA world. Mol. Cell 84, 107–119 (2024).
Knyazeva, A. et al. A chemical inhibitor of IST1-CHMP1B interaction impairs endosomal recycling and induces noncanonical LC3 lipidation. Proc. Natl. Acad. Sci. 121, e2317680121 (2024).
Ruan, C. et al. Proteome-wide deconvolution of drug targets and binding sites by lysine reactivity profiling. Anal. Chem. 94, 3352–3359 (2022).
Piazza, I. et al. A machine learning-based chemoproteomic approach to identify drug targets and binding sites in complex proteomes. Nat. Commun. 11, 4200 (2020).
Bailey, M. A., Martyr, J. G., Hargrove, A. E. & Fitzgerald, M. C. Stability-based proteomics for investigation of structured RNA–protein interactions. Anal. Chem. 96, 3044–3053 (2024).
Nakamura, H. et al. Cooperative role of the RNA-binding proteins Hzf and HuR in p53 activation. Mol. Cell Biol. 31, 1997–2009 (2011).
Dunker, W. et al. TDP-43 prevents endogenous RNAs from triggering a lethal RIG-I-dependent interferon response. Cell Rep. 35, 108976 (2021).
Iannone, C. et al. PTBP1-activated co-transcriptional splicing controls epigenetic status of pluripotent stem cells. Mol. Cell 83, 203–218.e9 (2023).
Tziortzouda, P., Van Den Bosch, L. & Hirth, F. Triad of TDP43 control in neurodegeneration: autoregulation, localization and aggregation. Nat. Rev. Neurosci. 22, 197–208 (2021).
Cho, K. F. et al. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat. Protoc. 15, 3971–3999 (2020).
Jiang, N. et al. A data-independent acquisition (DIA)-based quantification workflow for proteome analysis of 5000 cells. J. Pharm. Biomed. Anal. 216, 114795 (2022).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Thorvaldsdóttir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, research0034.1 (2002).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Acknowledgements
The authors would like to thank Ms. Yan Gao and Prof. Xiangyang Zhang at the Instrumental Analysis Center of School of Pharmaceutical Science and Technology at Tianjin University for their assistance with the mass spectrometric analysis. This work was supported by grants from the National Natural Science Foundation of China (22474087 to R. C.; 92354303 to K.M.; 22293030 and 22293032, to G.L.), Tianjin Natural Science Foundation (22JCYBJC00550 to R.C.), Open Project Program of the State Key Laboratory of Medical Proteomics (SKLP-O202406 to R.C.), the research program from Jinnan Hospital, Tianjin University (Tianjin Jinnan Hospital) (JNYYKY-202302 to X.M.), and Tianjin Research Innovation Project for Postgraduate Students (2021YJSB192 to N.J.).
Author information
Authors and Affiliations
Contributions
R.C. conceived and designed the project. N.J. carried out the mass spectrometry and biology experiments with assistance by H.K., Y.L., W.Q., K.C., H.X., and G.L.; N.J. conducted the structure analysis instructed by K.M.; R.C. and X.M. supervised the study. The manuscript was written by N.J., R.C., and X.M.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Stephanie DeWitte-Orr, who coiewed with Sarah Poynter and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, N., Yang, H., Lei, Y. et al. Characterization of dsRNA binding proteins through solubility analysis identifies ZNF385A as a dsRNA homeostasis regulator. Nat Commun 16, 3433 (2025). https://doi.org/10.1038/s41467-025-58704-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58704-7
