
Abstract
Patients with peritoneal metastasized colorectal cancer (PM-CRC) have a dismal prognosis. We hypothesized that an immunosuppressive environment in the peritoneal cavity underlies poor prognosis. We define the composition of the human peritoneal immune system (PerIS) using single-cell technologies in 18 patients with- and without PM-CRC, as well as in matched peritoneal metastases (n = 8). Here we show that the PerIS contains abundant immunosuppressive C1Q+VSIG4+ and SPP1+VSIG4+ peritoneal-resident macrophages (PRMs), as well as monocyte-like cavity macrophages (mono-CMs), which share features with tumor-associated macrophages, even in homeostasis. In PM-CRC, expression of immunosuppressive cytokines IL10 and VEGF increases, while simultaneously expression of antigen-presenting molecules decreases in PRMs. These intratumoral suppressive PRMs originate from the PerIS, and intraperitoneal depletion of PRMs in vivo using anti-CSF1R combined with anti-PD1 significantly reduces tumor burden and improves survival. Thus, PRMs define a metastatic site-specific immunosuppressive niche, and targeting PRMs is a promising treatment strategy for PM-CRC.
Similar content being viewed by others


Introduction
Metastases to the peritoneal cavity are commonly observed in gastrointestinal malignancies, such as in patients with colorectal cancer (CRC)1. Peritoneal metastases (PMs) are associated with a dismal prognosis, as evidenced by a median overall survival of less than 12 months2. Only in a minority of patients, cytoreductive surgery (CRS) combined with heated intraperitoneal chemotherapy (HIPEC) is a treatment option, but recurrence rates are high, and so is associated morbidity3. Improving survival and reducing disease burden in patients with peritoneal metastasized CRC (PM-CRC) is a large unmet clinical need.
Immune checkpoint blockade (ICB) has gained much excitement over the past decade because of its unparalleled potential to treat certain types of metastasized cancer, like melanoma and non-small-cell lung cancer4,5, however, only a minority of patients with CRC respond adequately to ICB. Much attention in the field goes to dissecting the biology underlying therapy resistance, including tumor-intrinsic mechanisms such as lack of neoantigen presentation as observed in microsatellite stable (MSS) cancers. Interestingly, however, peritoneal metastasized cancer is often associated with therapy resistance to ICB, even in CRC with microsatellite instability (MSI)6,7, and irrespective of the origin of the primary tumor site (e.g., colon, stomach, pancreas)1,6,8. This suggests that in peritoneal metastases, tumor cell-extrinsic mechanisms, such as the presence of an immunosuppressive niche, play a role in therapy resistance. Interestingly, it was recently demonstrated that PM-CRC is highly enriched for the MSS CMS4 subtype9,10; a molecular subtype of CRC that is typically characterized by infiltration of tumor-associated macrophages (TAMs) and Tregs and exclusion of cytotoxic T cells11,12.
To define whether peritoneal cavity immune cells contribute to a site-specific immunosuppressive niche that underlies therapy resistance in PM-CRC, we studied the composition of the human peritoneal immune system (PerIS) and its role in defining the immune environment in PM-CRC, using Cellular Indexing of Transcriptomes and Epitopes sequencing (CITE-seq) and single-cell RNA-sequencing (scRNA-seq). We investigated peritoneal fluid (PF) immune cells in a series of both healthy subjects and in patients with PM-CRC, as well as the tumor immune microenvironment (TIME) in PM. We found that even in homeostatic conditions, the PerIS is defined by high abundance of immunosuppressive C1QA+VSIG4+ and SPP1+VSIG4+ peritoneal resident macrophages (PRMs), which share features with TAMs. In PM-CRC, these PRMs increased expression of immunosuppressive cytokines, including IL10 and VEGF, while expression of antigen-presenting molecules was downregulated. Intriguingly, SPP1+VSIG4+ PRMs, with features of lipid-associated and regulatory TAMs, were the most abundant TAMs in peritoneal metastases. In a separate patient cohort, macrophage immunosuppressive gene set scores showed increased infiltration of macrophages in PM as compared to matched liver metastases and primary CRC. Finally, we demonstrate in pre-clinical in vivo studies that simultaneous depletion of macrophages using anti-CSF1R with anti-PD1 therapy significantly reduced disease burden and improved survival. Therefore, targeting peritoneal macrophages is a promising strategy to enhance ICB therapy in PM-CRC.
Results
The peritoneal immune system is defined by immunosuppressive peritoneal cavity resident macrophages
To provide a comprehensive and unbiased characterization of the human PerIS, we employed CITE-seq to simultaneously quantify the transcriptome as well as the protein expression of 134 cell-surface exposed proteins on live cells from PF obtained from patients that underwent surgery for achalasia, a disorder of the esophagus in which no immunological alterations of the peritoneal cavity are expected. These PF samples are therefore considered healthy controls (HC, n = 5; 14,441 cells). To confirm tissue-specificity, we compared PF CITE-seq transcriptome data with scRNA-seq data on peripheral blood mononuclear cells (PBMCs; 13,242 cells) from matched patients (Supplementary Fig. 1a and Supplementary Table 1). Although some overlap of PF-derived immune cells was observed with PBMC-derived immune cell subsets, we also observed PF-unique clusters demonstrating that these peritoneal immune cells have a distinct transcriptional profile (Fig. 1A and Supplementary Fig. 1B). By harnessing both the gene and protein expression profiles, we successfully resolved the 4 main immune lineages using markers CD3/CD3D, CD11b/ITGAM, CD56/NCAM1 and CD20/MS4A1, thereby identifying T cells, myeloid cells, natural killer (NK)/innate lymphoid cells, and B cells respectively (Supplementary Fig. 1C). Overall, there was a good correlation detected between protein and gene expression for the canonical markers (Supplementary Fig. 1D). Clusters represented tissue-specific immune cells with no patient specific subsets (Supplementary Fig. 1E). This multi-omic single-cell approach was particularly useful for immunophenotyping rare immune cell subsets that displayed more optimal resolution at either protein level, such as CD141 (THBD) for conventional dendritic cells 1 (cDC1), or gene level, such as IL7R (CD127) and CTLA4 (CTLA4) for innate lymphoid cells and CD4 regulatory T cells, respectively (Supplementary Fig. 1F, G).

A Gene expression Uniform Manifold Approximation and Projection (UMAP) overlay of color coded PBMC (red) derived immune cells (n = 13,242 cells) and PF (blue) derived immune cells (n = 14,441 cells) using Harmony for batch correction95. B Gene and protein weighted nearest neighbor (GP-WNN) UMAP color coded PF derived immune cells (n = 14,441 cells) identifying 27 subsets. C GP-WNN UMAP feature plots of reclustered MNPs in PF of HCs showing log transformed expression of scRNA markers used for myeloid subset identification DCs: CLEC4C, XCR1, and CD1C and CITE-seq CD1C; mononocytes + macrophages: ITGAM, MARCO, CD14, CCR2, and CD163 and CITE-Seq (CD14, CD192 and CD163). D GP-WNN UMAP color coded PF derived monocytes + macrophages (n = 3,937 cells) identifying 4 clusters. E GP-WNN UMAP feature plot of reclustered monocytes + macrophages PF of HCs showing log transformed expression of VSIG4. F GP-WNN UMAP feature plots of reclustered monocytes + macrophages in PF of HCs showing log transformed expression of scRNA markers CD163, VCAN, CCR2, C1QA, SPP1, and CD163 (CITE-Seq). G Boxplot showing the annotation and subsequent proportion of monocytes + macrophages subsets relative to total MNPs in PF of HCs (n = 5 donors) (does not add to 100% as DC subsets are not included in the graph). Box: 25th to 75th percentiles; line: median; whiskers: minimum and maximum values. UMAP Uniform Manifold Approximation and Projection, scRNA-seq single cell RNA sequencing, CITE-seq cellular indexing of transcriptomes and epitopes sequencing, HC healthy controls, PBMC peripheral blood mononuclear cell, PF peritoneal fluid, Mono-macs monocytes + macrophages, MNPs mononuclear phagocytes, T T cells, NK/ILC natural killer cells/innate lymphoid cells, B B cells, TCM T central memory, Treg regulatory T, CTL cytotoxic T lymphocyte, TEM T effector memory, MAIT mucosal-associated invariant T, GDT gamma delta T cells, NKT natural killer T, CDC1s conventional dendritic cells type 1, CDC2s conventional dendritic cells type 2, PDCs plasmacytoid dendritic cells, DCs dendritic cells, Class classical, mono-CM monocyte-like cavity macrophage, CM cavity macrophage. Source data are provided as a Source Data file.
Compared to PBMCs, the PerIS harbored fewer T and B cells, similar numbers of NK/innate lymphoid cells, and a larger proportion of myeloid cells (Supplementary Fig. 2A, B). In total, we identified 27 different immune cell types in the human PerIS (Fig. 1B and Supplementary Fig. 2C). Monocytes-macrophages were the most abundant immune cells of the PerIS accounting for ~30% of all immune cells (Supplementary Fig. 2D). Given the high abundance of macrophages within the PerIS and their putative role in shaping the immune environment in cancer13, we further phenotyped this immune subset by reclustering only mononuclear phagocytes (MNPs) (Fig. 1C). As all MNPs express ITGAM, dendritic cells (DCs) were separated from monocytes and macrophages based on CLEC4C, XCR1 and CD1C/CD1C, which were subsequently used to identify the plasmacytoid dendritic cells (PDCs) and conventional dendritic cells type 1 (CDC1s) and 2 (CDC2s), respectively (Fig. 1C). Virtually all monocyte and macrophage subpopulations were defined based on the expression of CD14, CD163, MARCO and CCR2 (Fig. 1C). Unsupervised clustering of the PF-derived monocyte and macrophage populations resulted in four clusters (Fig. 1D). Interestingly, all clusters showed expression of V-set and immunoglobulin domain-containing 4 (VSIG4)14 (Fig. 1E), a complement receptor of the immunoglobulin superfamily (CRIg) that is specifically expressed in resting tissue-resident macrophages15,16. Cluster 1 expressed both monocyte-like markers CD14 and CCR2 on both transcriptional and protein level (Supplementary Fig. 2E), macrophage marker VCAN, and tissue resident marker VSIG4 on transcriptional level (Fig. 1E, F), suggesting that cluster 1 represented peritoneal monocyte-like cavity macrophages (mono-CMs) as described recently17. Indeed, CITE-seq demonstrated that this VCAN+CCR2+ cluster was CD163dim at protein level, confirming that MARCO+CD163cite-dimVCAN+VSIG4+CCR2+ were mono-CMs17 (mean: 28.1%; cluster 1). In addition, higher entropy was observed for cluster 1 relative to the other clusters, suggesting higher differentiation potential18 compared to clusters 2 and 4, which represented more terminally-differentiated macrophages (Supplementary Fig. 2F). Unlike cluster 1, cluster 3 displayed lower entropy and was annotated as a second subset of mono-CMs expressing C1Q (MARCO+CD163cite-dimVCAN+C1QA+VSIG4+CCR2− C1Q+ mono-CMs; mean: 4.7%; cluster 3). Cluster 2 presented a more pronounced expression of CD163 (CITE-seq) and C1QA, but lacked the overt expression of VCAN and CCR2, thereby distinguishing it from the mono-CMs, which we therefore named C1Q+ cavity macrophages (CMs) MARCO+CD163cite-brightVCAN−C1QA+VSIG4+CCR2−. These C1Q+ CMs were the most abundant macrophages (mean: 30.9%). Lastly, we identified a subset of macrophages co-expressing SPP1 (MARCO+CD163cite-brightSPP1+VSIG4+CCR2- SPP1+ CMs; mean: 10.5%; cluster 4) (Fig. 1F, G). While the macrophage expression of SPP1 and C1QA was described to be mutually exclusive19, co-expression was reported in TAMs and correlates with worse overall survival in breast cancer patients20 and resistance to ICB in melanoma patients21. Furthermore, C1QA, combined with co-expression of TREM2, a gene that was expressed in the majority of PRMs (Supplementary Fig. 2G), has also been associated with immunosuppression22,23. Having established the presence of immunosuppressive PRMs in the homeostatic PerIS and given the fact that macrophages lack unifying nomenclature, we employed marker gene analyzes to identify differential expressed genes (DEG) analyzes to further characterize these macrophage subsets. As expected, we found high expression of marker genes used for subset classification (SPP1, C1QA, VCAN, and CCR2). SPP1+ CMs expressed NRP1 and LGMN, while C1Q+ mono-CMs expressed FLT1 and EZR. C1Q+ CMs are characterized by FN1 and LYVE1 expression, and mono-CMs expressed FGL2 and MNDA, all of which genes are involved in promoting immunosuppression, tumor growth, and migration (Supplementary Fig. 2H and Supplementary Data 1)24,25,26,27,28,29,30,31. Moreover, gene ontology (GO) analyzes32,33,34 found no major differences between the subsets apart from enhanced cell-cell signaling and regulation of lipid transport in SPP1+ CMs (Supplementary Data 2). However, using a recently published proposed model on TAM signatures, we classified all of our identified macrophage subsets in the PF of HCs accordingly35. Most subsets were found to match the lipid-associated (LA), resident-tissue macrophage like (RTM), regulatory (Reg) and proangiogenic (Angio) TAMs signatures, while inflammatory-cytokine enriched (INFLAM) or IFN-primed (IFN), associated with pro-inflammatory characteristics, were underrepresented in PRMs in healthy controls (Fig. 2A). Indeed, genes associated with pro-inflammatory functions including IL1B and IDO1 were downregulated in bona fide CMs36,37,38. By contrast, anti-inflammatory genes FOLR2, MRC1, TREM2, and MERTK were highly expressed in both C1Q+ CMs and SPP1+ CMs23,38,39,40 (Fig. 2B). The four macrophage subsets could be further characterized using our 134-cell-surface CITE-seq panel, showing unique protein expression profiles. Interestingly, especially the CD163cite-bright C1Q+ CMs and SPP1+ CMs show marked expression of CD169, CD304, HLA-DR, and CD112, respectively. This is associated with IL10 production, stimulating angiogenesis while functioning as the VEGFR2 co-receptor, increased antigen presentation, and regulatory T cell differentiation via binding of TIGIT, and may thereby contribute to the immunosuppressive peritoneal niche41,42,43 (Fig. 2C).

A Radarplot showing TAM median Ucell score signature per macrophage subset in PF. B Dot plot of selected genes encoding pro- (red) or anti-inflammatory (green) genes in all four monocytes + macrophages subsets. C Heatmap showing the row-scaled mean log2transformed expression of top 5 unique marker proteins per monocytes + macrophages subset. D GP-WNN UMAP feature plots of reclustered mononocytes + macrophages showing log transformed expression of C1QA and SPP1 in healthy PF, colon, and liver tissue. E Boxplot analysis of monocytes + macrophages, C1Q+ CMs, and SPP1+ CMs comparing PF (n = 5 donors), colon (n = 5 donors), and liver (n = 5 donors). Box: 25th to 75th percentiles; line: median; whiskers: minimum and maximum values. Statistics: Mann–Whitney test, two-tailed. *p = 0.032, **p = 0.0079. UMAP Uniform Manifold Approximation and Projection, scRNA-seq single cell RNA sequencing, CITE-seq cellular indexing of transcriptomes and epitopes sequencing, HC healthy controls, PF peritoneal fluid, Mono-macs monocytes + macrophages, mono-CM monocyte-like cavity macrophage, CM cavity macrophage, RTM resident-tissue macrophages, Angio pro-angiogenic, IFN interferon-primed, INFLAM inflammatory-cytokine enriched, LA lipid-associated, Prolif proliferating, Reg immune regulatory, pro-inflam pro-inflammatory, anti-inflam anti-inflammatory. Only statistically significant differences are shown (*p ≤ 0.05; **p ≤ 0.01). Source data are provided as a Source Data file.
To understand whether the abundance of macrophages could provide a tumor-promoting niche that contributes to poor prognosis and therapy resistance in peritoneal metastasized cancer, we compared abundance of macrophages in PF with macrophage abundance in both colon- (Gut Cell Atlas)44 and liver tissue (Liver Cell Atlas; GSE192742)45, being the primary cancer site in CRC and the most common site of disseminated CRC, respectively (Supplementary Fig. 2I and Supplementary Fig. 3A–D). Interrogating healthy, non-cancer bearing donors, the relative abundance of macrophages in the PF was approximately 9 and 18 times higher than in colon and liver, respectively (Fig. 2D, E). Interestingly, while C1Q+ macrophages were present in healthy colon- and liver tissue albeit in lower abundance (8x and 5x respectively, compared to peritoneal fluid), SPP1 expression was not found in homeostatic colon- and liver-derived macrophages from the selected donors (Fig. 2E).
Taken together, the homeostatic PerIS is characterized by a large number of PRMs that harbor anti-inflammatory characteristics. In addition, the homeostatic peritoneal cavity also harbors a subset of SPP1+ macrophages, which shares features that are typically attributed to TAMs and which we did not detect in normal colon and liver tissue (Gut Cell Atlas44 and Liver Cell Atlas45).
Peritoneal resident macrophages shape an immunosuppressive niche for PM-CRC
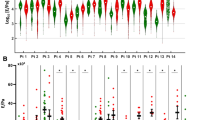
Given the TAM-like phenotype of PRMs in homeostasis, we next defined whether PRMs retained their immunosuppressive phenotype in the presence of peritoneal metastases in thirteen PM-CRC patients, using scRNA-seq of PF-immune cells (Supplementary Table 1). Patients were prospectively included upon macroscopically visible PM during diagnostic laparoscopy and had a median Peritoneal Carcinomatosis Index (PCI) score of 13. The majority of PM was mismatch repair proficient (92.3%), as expected12. Only two (15.4%) patients received prior systemic chemotherapy 3 months before the sample was obtained. All four macrophage subsets were identified in the PerIS of PM-CRC patients but clustered differently from their HC counterparts, indicating PM-specific transcriptional differences (Fig. 3A). No patient specific clusters were found (Supplementary Fig. 4A). Although no significant differences in abundance were observed in macrophage subsets between HC and PM-CRC patients (Fig. 3B), differential gene expression analysis demonstrated that PRMs maintained their polarized immunosuppressive nature. In fact, anti-inflammatory macrophage markers such as VEGFA and IL10, both of which have been associated with a pro-tumorigenic environment46,47,48,49, were significantly higher expressed in PM-CRC as compared to HC, in mono-CMs, C1Q+ CMs, and SPP1+ CMs. By contrast, PF from PM-CRC patients presented lower expression of genes encoding proteins involved in antigen presentation, such as HLA-A, HLA-C and HLA-DRB1, in all but the C1Q+ CMs (Fig. 3C, D, Supplementary Fig. 4B). KEGG pathway analysis confirmed reduced antigen processing and presentation, as well as cytokine-cytokine receptor interaction pathways in PM-CRC macrophages (Supplementary Fig. 4C and Supplementary Data 3). In addition to transcriptional changes in PRMs, we also observed significantly increased protein levels of the immunosuppressive cytokines IL10 and VEGFA (Fig. 3E), as well as another important anti-inflammatory cytokine TGF-β50 (Supplementary Fig. 4D) in the PF of PM-CRC patients compared to HC. The significance of this observation was demonstrated by the fact that protein levels of VEGFA and IL10 levels, as well as TGF-β, positively correlated with PCI score (Fig. 3F and Supplementary Fig. 4E). In addition, both IL10 and VEGFA were predominantly expressed in macrophages in the PF of HC and PM-CRC patients (Fig. 3G, H), although VEGFA was also slightly expressed by DCs, granulocytes and non-immune populations (Fig. 3G, H). These findings demonstrate a highly immunosuppressive tumor niche in the peritoneal cavity, with abundance of PRMs that define an immune-privileged phenotype in PM-CRC.

A UMAP color-coded PF-derived macrophage subsets (n = 8661 cells) comparing HC (left) and PM-CRC (right) identifying 4 macrophage subsets. B Stacked bar chart analysis of macrophage subsets as a proportion of macrophages comparing PF of HC to PM-CRC. C Heatmap showing effect size comparing PM-CRC with HC (Wald statistic) as obtained using DESeq2 of significant altered genes in either HC (turquoise) or PM-CRC (brown) of macrophage subsets from selected KEGG pathways. DESeq2 Wald test derived p values. D Bar chart showing mean with SEM of absolute gene expression levels of HLA-DRB1, HLA-C, IL10, and VEGFA in all macrophage subsets in PF comparing HC (n = 5 donors) with PM-CRC (n = 13 patients). Statistics: Wald test using DESeq2. Multiple comparison adjustments and adjusted p values are used for testing. HLA-DRB1: **p = 0.007; *p = 0.014; HLA-C: *p = 0.022; ***p = 0.0003; IL10: *p = 0.035; 0.037; 0.039; VEGFA: **p = 0.007; 0.008. E Normalized Protein Expression (NPX) levels of IL-10 and VEGFA, using Olink technology in PF comparing HC (n = 5 donors) to PM-CRC patients (n = 12 patients). Line at median. Statistics: Mann–Whitney test, two-tailed. ***p = 0.0006; 0.0003. F Correlation plot of NPX levels of IL-10 and VEGFA in the PF of PM-CRC patients (n = 21 patients) and PCI score. Statistics: Spearman correlation. *p = 0.043; 0.030 (two-tailed). G UMAP color-coded PF and PM-CRC (non-)immune subsets (n = 35,329 cells). H Feature plots of (non-)immune subsets identified in g, showing log-transformed expression of scRNA markers IL10 and VEGFA. UMAP Uniform Manifold Approximation and Projection, HC healthy controls, PM-CRC peritoneal metastasized colorectal cancer, CMs cavity macrophages, mono-CMs monocyte-like cavity macrophages, rec receptor, KEGG Kyoto Encyclopedia of Genes and Genomes, PCI Peritoneal Cancer Index, NPX Normalized Protein Expression, NK/ILC natural killer cells/innate lymphoid cells, Mono-macs monocytes + macrophages. Only statistically significant differences are shown (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001). All UMAPs in this figure were generated using only gene expression data. Source data are provided as a Source Data file.
Peritoneal-resident immunosuppressive macrophages infiltrate PMs
To determine whether PRMs constitute the tumor immune microenvironment in PM-CRC, we analyzed PF and matched PMs of eight patients with isolated PM-CRC (Supplementary Fig. 1a). In line with our recent studies, the majority (7/8; 87.5%) had microsatellite stable (MSS) CRC12 (Supplementary Table 1). Phenotyping identified 34 immune subsets (6459 cells) within PM-CRC (Supplementary Fig. 5A–C), of which the top three most abundant subsets were the CD8 T effector memory cells (mean: 13.1%), SPP1+ CMs (mean: 8.5%) and CD4 regulatory T cells (mean: 8.0%) (Supplementary Fig. 5D). Phenotyping of the monocyte and macrophage subsets in PMs at single cell level demonstrated that PMs harbored similar mono-CM and CM subsets as detected in the PF, as well as both classical (cluster 1) and non-classical (cluster 2) monocytes (Fig. 4A and Supplementary Fig. 5A, C). Interestingly, the SPP1+ CMs, which we identified as a peritoneal macrophage subset, were the most abundant macrophage within the TIME of peritoneal metastases (mean 47.7%, Fig. 4B), although also other mono-CMs and C1Q+ CMs were found within PM-CRC (Fig. 4B). To visualize macrophages within PM, we performed IHC for CD163, which was uniformly expressed on peritoneal macrophages and mono-CMs (Supplementary Fig. 5E). We demonstrated that CD163+ macrophages infiltrated both invasive margin and tumor centers (Supplementary Fig. 5F), while CD8+ T cells were excluded from the PM tumor centers and instead accumulated at the invasive margins (Supplementary Fig. 5G).

A UMAP color coded for PMs from PM-CRC patients derived mono-mac subsets (n = 8 patients; 1,747 cells). B Boxplot of proportion of macrophage subsets out of total macrophages in PMs from PM-CRC patients (n = 8 patients). Box: 25th to 75th percentiles; line: median; whiskers: minimum and maximum values. Statistics: Mann–Whitney test, two-tailed. *p = 0.038, **p = 0.0019; 0.003, ***p = 0.0002. C Feature plot showing VSIG4 expression in PM derived monocytes + macrophages subsets. D UMAP color coded for monocytes or macrophages for HC (n = 5 patients) as well as the PM-CRC patients (n = 8 patients) of PBMC-, PF-, and, from PM-CRC only, PM-derived monocytes and macrophages (n = 17,930 cells) using Harmony for batch correction95. E UMAP color coded for tissue and disease state of monocytes and macrophages for HC (n = 5 patients) as well as the PM-CRC patients (n = 8 patients) of PBMC-, PF- and, from PM-CRC only, PM-derived monocytes and macrophages (n = 17,930 cells) using Harmony for batch correction95. F Dendrogram based on the Euclidian pairwise distances calculated between monocytes and macrophage samples obtained from HC (n = 5 patients) and PM-CRC patients (n = 8 patients). G Schematic overview of experimental set-up of PKH26 in vivo macrophage labeling (Created in BioRender. Vermeulen, J. (2025) https://BioRender.com/oi0rimk). H Gating strategy used to identify peritoneal PKH26+ macrophages: Plots are representative for their respective group. I Boxplot showing the percentage of PKH26+ F4/80+ macrophages in Spleen, PF, and PM, 2 weeks after tumor cell injection without (−, n = 1 mouse) or with (+, n = 6 mice) PKH26 linker injection. Box: 25th to 75th percentiles; line: median; whiskers: minimum and maximum values. UMAP Uniform Manifold Approximation and Projection, CMs cavity macrophages, mono-CMs monocyte-like cavity macrophages, mono-macs monocyte-macrophage, Spl spleen, PF peritoneal fluid, PM peritoneal metastasis, HC healthy controls, PM-CRC peritoneal metastasized colorectal cancer, i.p. intraperitoneal. (ns = non-significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001). All UMAPs in this figure were generated using only gene expression data. Source data are provided as a Source Data file.
To address whether PM macrophage infiltration was an environment- rather than a tumor-specific feature, we assessed PF macrophage abundance as well as PM macrophage infiltration in 6 gastric cancer patients with PMs (PM-GC). All macrophage subsets found in the PerIS of PM-CRC patients were also identified in the PerIS of PM-GC patients (Supplementary Fig. 6A–F). In addition, we confirmed that also in PM-GC, SPP1+ CMs as well as C1Q+ CMs were highly abundant in tumors (Supplementary Fig. 6G, H).
To define whether immune cells in PM were PerIS-derived, we first assessed expression of tissue resident marker VSIG4 within the myeloid subsets, which showed that the majority of myeloid cells express VSIG4, except for classical and non-classical monocytes, which are likely blood-derived (Fig. 4C). Reclustering monocytes and macrophages from PBMCs, PF and PM from both HCs and PM-CRC patients together indicated separate clusters for monocytes and macrophages with notable separation between PF- and PM-derived macrophages, which is to be expected as macrophages undergo differentiation within the TIME51 (Fig. 4D, E). Indeed, through unsupervised clustering analysis, we demonstrated that the PM-derived macrophages are more similar to the PF-derived macrophages compared to PBMC-, PF-, or PM-derived monocytes (Fig. 4F). We therefore hypothesized that PM-derived macrophages originate from the PF. To prove this, we performed an in vivo mouse macrophage labeling experiment using the PKH26 linker, which enabled us to trace PRMs. We found that 2 weeks after CT26 colorectal cancer cell injection, up to 61% of F4/80+ macrophages in PM-CRC were PKH26+, and thus directly peritoneal-derived (Fig. 4G–I). Collectively, this demonstrates that PRMs actively infiltrate peritoneal metastases52,53.
Peritoneal-resident immunosuppressive macrophages contribute to the peritoneal metastatic TIME
We next investigated potential mechanisms by which PRMs were attracted into PM by investigating expression levels of macrophage receptors that have been shown to be critical for macrophage recruitment into cancers54. In human PM, we observed expression of CCR1, CCR5, CSF1R, and CXCR3, on SPP1+ CMs and C1Q+ CMs in PMs, as well as on mono-CMs (Fig. 5A). In addition to cell surface receptors on macrophages, we analyzed the presence of ligands in the PF, and identified increased levels of the key macrophage chemoattractants CCL3 (MCP-1α), CSF-1 and CXCL9 in PM-CRC patients compared to HC, suggestive for the involvement of the CCL3-CCR1/CCR5, the CSF1-CSF1R and the CXCL9-CXCR3 axis in PF PRMs (Fig. 5B).

A Dot plots of selected genes encoding receptors of key macrophage chemoattractants. B Boxplot showing the normalized protein expression (NPX) levels using Olink technology in PF comparing HC (n = 5 donors) to PM-CRC patients (n = 12 patients). Line at median. Statistics: Mann–Whitney test, two-tailed. ***p = 0.0003; 0.0006; 0.0003. C Radarplot showing TAM median Ucell score signature per macrophage subset in PM. D Dot plots representing the 5 most differentially expressed TAM-associated genes per macrophage subset. Size represents percentage expressing cells and color represents the median gene expression. E Heatmap of bulk RNA-sequencing data (GSE190609) of paired patients’ primary tumor (PT), liver metastasis (LM), and peritoneal metastasis (PM) of colorectal cancer showing Z-score of immunosuppressive macrophage markers. F Paired patient analysis of gene set score in (E) (n = 12 patients with PT and PM, 6 patients also had LM). Statistics: paired t-test, two-tailed. *p = 0.026; 0.013. CMs cavity macrophages, mono-CMs monocyte-like cavity macrophages, mono-mac monocyte-macrophage, PM peritoneal metastasis, PT primary tumor, LM liver metastasis, HC healthy controls, PM-CRC peritoneal metastasized colorectal cancer, RTM resident-tissue macrophages, Angio pro-angiogenic, IFN interferon-primed, INFLAM inflammatory-cytokine enriched, LA lipid-associated, Prolif proliferating, Reg immune regulatory. (ns = non-significant; *p ≤ 0.05; ***p ≤ 0.001). Source data are provided as a Source Data file.
Macrophages in PM showed similar transcriptional TAM signatures as in PF35 and could mainly be characterized as lipid-associated and regulatory-like TAMs (Fig. 5C). Interestingly, both C1Q+ mono-CMs and SPP1+ CMs, accounting for the vast majority (mean: 78.7%) of TAMs in PM, had the most immunosuppressive phenotype and particularly showed increased expression of FN1 and CD74, respectively, both genes associated with cancer progression and dampened anti-tumor immune responses55,56 (Fig. 5D).
Lastly, we set out to define whether infiltration of PRMs shapes a peritoneal-unique TIME, which is distinct from the TIME in other metastases or primary tumors. In an independent metastatic CRC cohort, we compared the expression of a macrophage immunosuppressive gene signature (MARCO, C1QA, VSIG4, CD163, and SPP1) in PMs with matched primary CRC and liver metastases10. In line with our results, immunosuppressive gene set scores were significantly higher in PM as compared to matched primary CRC and matched liver metastases10 (Fig. 5E, F), demonstrating that PM are characterized by increased influx of immunosuppressive macrophages. Thus, blocking influx of suppressive macrophages by targeting these macrophage receptors could be promising treatment strategy for PM-CRC.
Improved overall survival in mice after macrophage-depleting therapy combined with immune checkpoint blockade
Given the potential role of PRMs in poor outcome of PM-CRC, we next studied whether peritoneal macrophage depletion would be a therapeutic strategy to overcome resistance to ICB in a pre-clinical PM-CRC model, in which CT26 colorectal cancer cells were intraperitoneally injected in BALB/c mice57. As (1) we demonstrated that CSF1R is broadly expressed in human PRMs, and csf1r expression has been demonstrated on mouse peritoneal macrophages17,58, and (2) we observed increased levels of CSF1 in PF of PM-CRC patients as compared to controls (Fig. 5B), we used anti-CSF1R as a macrophage depleting strategy, and anti-PD1 to activate suppressed anti-tumor T cells. Effective depletion of peritoneal macrophages was confirmed in anti-CSF1R-treated groups by flow cytometry in both PMs (Fig. 6A, B) and PF (Supplementary Fig. 7A) and in PMs by immunofluorescence for CD206 (Fig. 6C, D). Anti-CSF1R/anti-PD1 treatment was accompanied by increased intratumor levels of activated FSChigh CD8+ T cells (Supplementary Fig. 7B) and increased levels of intratumor CD8+ T cells as demonstrated by flow cytometry (Supplementary Fig. 7C, D) and CD8 IF (Fig. 6E, F). Subgroup analysis of mice that either reached their humane endpoint or were sacrificed after 21 days showed that anti-CSF1R/anti-PD1 combination therapy, but not anti-PD1 alone, resulted in a reduction of tumor burden, including significantly decreased levels of ascites and peritoneal tumor burden as measured by the modified PCI score for murine studies (mPCI57) (Fig. 6G–I and Supplementary Fig. 7E). Most importantly, these improved clinical parameters were accompanied by improved survival rates in both a prevention (Fig. 6J, K) and a treatment (Fig. 5J, L) model, in anti-CSF1R/anti-PD1-treated mice as compared to mice that received anti-CSF1R, anti-PD1 monotherapy or vehicle controls (Fig. 6J–L). Interestingly, depletion of macrophages combined with anti-PD1 resulted in complete remission with absence of ascites in almost half of the mice (3/7) compared to anti-PD1 monotherapy (1/7) and anti-CSF1R monotherapy or vehicle control (both 0/7) (Fig. 6M, N and Supplementary Fig. 7F). Together, these data demonstrate that the combination of macrophage depleting therapy with anti-PD1 immune checkpoint blockade is an effective treatment strategy for PM-CRC in these models.

A Gating strategy used to identify peritoneal macrophages: CD11b+F4/80+. Plots are representative for their respective group. B Quantification of flow cytometry data of % peritoneal macrophages out of myeloid cells in PMs after 21 days. Mice received control (n = 7 mice), anti-CSF1R (n = 5 mice), anti-PD1 (n = 4 mice) or anti-CSF1R/anti-PD1 combination therapy (n = 3 mice). Bar chart showing mean with SEM. Statistics: Kruskal–Wallis. *p = 0.027, 0.046 C IF of CD206+ macrophages in PM. D Quantification of CD206+ IF staining in PMs. Data shown as mean number of CD206+ cells per HPF with SEM. (control n = 5 mice; anti-CSF1R n = 5 mice; anti-PD1 n = 4 mice; anti-CSF1R/anti-PD1 n = 4 mice; minimally 3 high-power field (HPF) per mouse). Statistics: Kruskal–Wallis. *p = 0.038, **p = 0.005 E IF of CD8+ T cells in PM split per different treated groups. F Quantification of CD8+ IF staining in PM. Data shown as mean number of CD8+ cells per HPF with SEM. (control n = 5 mice; anti-CSF1R n = 5 mice; anti-PD1 n = 4 mice; anti-CSF1R/anti-PD1 n = 3 mice; minimally 3 HPF per mouse). Statistics: Kruskal–Wallis. G, H Subgroup analysis of mice that either reached their humane endpoint (control: n = 10 mice; anti-CSF1R: n = 6 mice; anti-PD1: n = 4 mice and anti-CSF1R/anti-PD1: n = 1 mouse) or were sacrificed after 21 days (anti-CSF1R/anti-PD1: n = 2 mice). Data shown as mean with SEM. Bar chart of ascites (G) Statistics (G): Fisher’s exact test. *p = 0.0016; 0.0035. Bar chart of mPCI (H). Statistics (H): Kruskal–Wallis. *p = 0.013. I Representative pictures of vehicle control (upper panel) and combination therapy (lower panel). J Schematic overview of in vivo experiments. Experiment 1: anti-CSF1R at t = 0 days. Experiment 2: anti-CSF1R at t = 7 days (Created in BioRender. Vermeulen, J. (2025) https://BioRender.com/hb9202t). K Survival analysis after 4 weeks (see J; exp 1). Statistics: Log-rank test. l Survival analysis after 6 weeks (see J; exp 2). Statistics: Log-rank test. M Stacked bar chart analysis showing proportion of mice achieving complete remission after 42 days. N Representative picture of combination therapy showing complete remission. Mφ macrophage, α anti, CFS1R colony stimulating factor 1 receptor, PD1 programmed cell death protein 1, HPF high-power field, PM peritoneal metastasis, mPCI modified peritoneal cancer index57, CR clinical remission, i.p. intraperitoneal. Pre-clinical model: intraperitoneal injection of CT26 colorectal cancer cell line in BALB/c mice, followed by administration of anti-CSF1R and anti-PD1. (*p ≤ 0.05; **p ≤ 0.01). Source data are provided as a Source Data file.
Discussion
In the current study, we determined the phenotypical characteristics of the human peritoneal immune system to find potential immunomodulatory treatment strategies for PM-CRC.
First, our characterization of the PerIS in healthy controls demonstrated that, under homeostatic conditions, the peritoneal cavity contains abundant levels of resident macrophages, T cells, and NK cells. Neutrophils appeared to be underrepresented in peritoneal fluid, but this may be a technical issue of the single-cell platform that was used, rather than a biological phenomenon, as previously also reported59. While it should be noted that we focus on immune cells in this study, we acknowledge the importance of non-immune cells like tumor-associated fibroblasts or other stromal cells as well60,61.
Given the high abundance of macrophages in the PF, we investigated their phenotype using scRNA-seq and CITE-seq and demonstrated that the majority of PF macrophages have an immunosuppressive phenotype based on their expression level of MARCO+CD163cite-brightC1QA+VSIG4+CCR2-, with high expression of immunosuppressive genes APOE, FOLR2, MRC1, TREM2, and MERTK23,38,39,40. Interestingly, the peritoneal cavity in healthy controls also contained a subset of macrophages expressing SPP1, a subset that is typically restricted to tumor-associated macrophages of various cancers19,22. SPP1 encodes phosphoprotein 1, also referred to as osteopontin, a secreted phosphorylated glycoprotein that characterizes a subset of TAMs whose intratumoral presence is associated with poor clinical outcome in CRC and lung carcinomas. SPP1+ macrophages in tumors negatively correlate with tumor lymphocyte infiltration and are associated with CD8+ T cell suppression as well as evasion of anti-tumor immune responses62,63,64. Indeed, SPP1+ macrophages interact with cancer-associated FAP+ fibroblasts, possibly through TGF-β and interleukin-1, increasing fibrosis, which further limits T cell infiltration62. In addition, SPP1-expressing TAMs facilitate chemo- and radiation resistance by activating the CD44 receptor, PI3K/AKT signaling, and induction of epithelial-mesenchymal transition64,65,66,67. Lastly, SPP1+ macrophages in peritoneal fluid showed high expression of CD112, a protein that links to TIGIT and thereby induces immunosuppressive CD4+ regulatory T cell differentiation43. Taken together, these functions of SPP1+ macrophages facilitate tumor growth and prevent effective anti-tumor immune responses68.
Next to SPP1+ macrophages, we observed high abundance of C1Q+ CMs and C1Q+ mono-CMs in the homeostatic PerIS. C1Q+ macrophages have also been linked to poor prognosis. For example, recent studies in human and mouse malignant pleural effusion indicate that C1Q-expressing TAMs undermine effective anti-tumor effects of CD8+ T cells by enhancing fatty acid metabolism, thereby activating peroxisome proliferator-activated receptor gamma, which ultimately leads to transcription of immune inhibitory molecules as TREM2, Tim-3, SIRPα, PD-1, and PD-L169.
Second, we demonstrated that PRMs in the setting of PM-CRC show increased immunosuppressive nature, as demonstrated by downregulation of antigen presentation machinery particularly in C1Q+ and SPP1+ macrophages. This is important as human leukocyte antigen-DR (HLA-DR) molecules are essential to generate targeted immune responses70. Indeed, presence of MHC IIlow TAMs is correlated with disease progression in hepatocellular carcinoma murine model71. Apart from decreased antigen presentation, macrophages can promote cancer cell growth by secreting factors that promote angiogenesis, such as VEGFA, or factors that inhibit T cell activity, including IL10 and TGFB50,72,73. While TGFB was not differentially expressed in macrophages, we observed higher expression of VEGFA and VEGFR1 on C1Q+ macrophages, and increased IL10 expression in C1Q+ and SPP1+ macrophages, which was accompanied by increased levels of VEGF and IL10 in peritoneal fluid of PM-CRC patients, cytokines that have been associated with poor prognosis in patients with peritoneal metastasized gastric cancer74.
Third, we have shown a direct link between the local immune compartment in the peritoneal cavity and the TIME in peritoneal metastases. While an outstanding question in the field is to what extend tissue-resident immune cells dictate the tumor immune infiltrate, we here demonstrate that intratumoral macrophages share features with PF macrophages and also express tissue-resident marker VSIG4, suggesting peritoneal origin. The hypothesis that local peritoneal macrophages define the tumor environment was further strengthened by the observation that PM-derived macrophages are transcriptionally more similar to PF-derived macrophages than blood-derived monocytes. Indeed, SPP1+ CMs, C1Q+ CMs, and C1Q+ mono-CMs subsets that were observed in PF were also found in PM of PM-CRC patients, as well as in both PF and PM in an independent cohort of PM-GC patients. Lastly, presence of peritoneal-labeled macrophages within peritoneal tumors in mice suggests that these PRMs actively shape the TIME of PM. In line, this has previously also been demonstrated in the setting of liver metastases, in which circulating PKH-labeled macrophages infiltrated liver metastases and promoted tumor growth52. Interestingly, comparing immunosuppressive macrophage gene sets scores from a separate series of patients with matched PM, liver metastases, and primary colon cancers, we show that peritoneal cancers are enriched for immunosuppressive macrophages. Taken together, this demonstrates that peritoneal macrophages likely shape a distinct metastatic niche, which may be involved in poor survival of peritoneal metastatic disease.
Finally, we tested this hypothesis in vivo in pre-clinical mouse models for PM-CRC. A unique opportunity in peritoneal metastasized disease is the ability to directly interfere with the local immune microenvironment by administration of intraperitoneal immunomodulatory treatments, which is fundamentally different from metastases to other organs. Using anti-CSF1R, we effectively depleted peritoneal fluid macrophages, which in the human PF broadly expressed CSF1 receptor. It is likely that treatment with anti-CSF1R antibody also depleted macrophages that line the peritoneal surface75,76,77, and these have been shown to be tumor-promoting75,76. Future studies will be needed to disentangle the distinct role of the different macrophages75,76,77. Macrophage depleting strategies have previously been shown to be effective in peritoneal metastasized ovarian and gastric cancer78,79,80. Interestingly, several clinical trials are currently ongoing, studying the use of TAM targeting agents like anti-CSF1R, in combination with either ICB or backbone chemotherapy, and show tolerable safety profiles in humans13. Treatment with FDA-approved CSF1R activation inhibitor emactuzumab depletes TAMs, increases lymphocyte infiltration, and improves patient outcome in patients suffering diffuse-type giant cell tumors81,82. Interestingly, CSF1R+ TAMs were reduced upon emactuzumab treatment in multiple solid tumors, including colorectal cancer82. We utilized in vivo immunocompetent mouse models to validate and test our findings. Successful macrophage depletion resulted in increased levels of activated FSChigh CD8T cells and an increased number of intratumor CD8+ T cells, corroborating findings of macrophage depleting strategies in mouse cancer models53,83,84. More importantly, targeting macrophages either before or after tumor cell implantation, combined with immune checkpoint blockade, significantly reduced disease severity and improved survival in a pre-clinical mouse model for PM-CRC. Although survival data based on mouse discomfort remains difficult to directly link to tumor burden, we feel that mostly, albeit not exclusively, mouse discomfort is correlated to tumor burden as mice treated with anti-CSF1R/anti-PD1 have significant lower ascites formation (Fig. 6g) and tumor burden (Fig. 6h). As anti-CSF1R potentially depletes all macrophages, including pro-inflammatory anti-tumor macrophages, it would be interesting to investigate in future studies whether anti-inflammatory PRMs can be selectively targeted, for example through metabolic reprogramming85.
In conclusion, the PerIS is a unique immune compartment containing high abundance of immunosuppressive resident macrophages, and while this is likely an evolutionary important feature in the setting of for example, peritonitis86, this can be deleterious in the setting of peritoneal metastases, as cancer cells may hijack this feature to favor their growth. Targeting PRMs is a potential weak spot of peritoneal metastases, and is thus an exciting strategy to combat peritoneal metastasized cancers and improve outcomes for patients with this devastating condition.
Methods
Patients
Patients were enrolled in the study according to Dutch research guidelines of the Federation of Dutch Medical Scientific Societies (FMDSS), as described in “Human Tissue and Medical Research: Code of Conduct for Responsible Use”. The Medical Ethical Committee (METC) of the Amsterdam UMC approved the study protocol: 2019_224. Patients were informed and provided informed consent prior to sampling. Patients did not receive any form of compensation.
Peritoneal flushes were obtained perioperatively. Participating patients were subject to either laparoscopic or laparotomic abdominal surgery, depending on the surgical indication of the procedure. Between 2019 and 2024, a total of 23 patients (5 achalasia (HC), 13 PM-CRC, and 6 PM-GC) were enrolled in this study at the Amsterdam UMC. Patients who underwent surgery for achalasia (n = 5), a rare disorder of the esophagus, in which no immunological alterations of the peritoneal cavity are to be expected, were included to study the peritoneal immune system in homeostasis. For PM-CRC and PM-GC, adult patients (>18 years old) with histologically-proven PM (n = 13 and n = 6, respectively) were included. Exclusion criteria were presence of other intra-abdominal diseases (i.e., endometriosis, pancreatitis, appendicitis, cholecystitis, cholangitis, perforation of the GI tract, bleeding of the GI tract or corpus alienum), or abdominal surgery shorter than 6 months ago. The sex of participants was considered in the study design and was determined based on self-report.
Sample collection
Peritoneal flushes were obtained perioperatively during abdominal surgery by flushing milky spot-enriched areas like the diaphragm and omentum, similar as what has been done in mice87,88. Peritoneal immune cells within the mesothelial lining may have been missed, and additionally, it should be noted that differences in previous reported human studies on the peritoneal immune system may differ because in these studies, single cell suspensions were usually obtained from peritoneal fluid, such as peritoneal dialysis effluent or ascites89,90,91. Potential blood contamination was minimized while gaining access to the abdominal cavity, after which the peritoneal cavity was flushed with 1 L of 0.9% NaCl saline solution at body temperature. The omentum and paracolic gutters (1/3) as well as both sides of the diaphragm (1/3 right and 1/3 left) were flushed. The peritoneal flush was removed by suction 2 min after infusion and stored in clean plastic canisters. Afterwards, the peritoneal fluid was quickly transferred to a glass canister and stored on ice. Periprocedural, peripheral blood (6 mL) was drawn from the patient and collected in EDTA tubes and kept on room temperature. Peritoneal tumor samples were taken perioperatively at discretion of the operating surgeon and transferred to transferring medium (RPMI 1640 + 10% FCS) for processing into single cell suspensions or 4% PFA/PBS for tissue embedding.
PBMC preparation for scRNA-seq
Whole blood was transferred to an empty 50 mL tube, and the EDTA tube was flushed with PBS (RT) and added to a total of 17 mL. Then, a total of 13 mL Ficoll-Paque® PLUS (GE/VWR) was added gently, and the sample was centrifuged at 876 × g for 20 min (RT). The PBMC layer was collected, transferred to a new tube, and PBS (RT) was added to a total of 30 mL, after which the sample was centrifuged at 493 × g for 7 min (RT). The supernatant was discarded, and the sample was washed with ice-cold PBS 0.1% BSA and centrifuged at 493 × g for 7 min (4 °C). Ten mL of erythrocyte lysis buffer (self-made) was added and kept on ice for 10 min after the sample was washed with 10 mL ice-cold PBS 0.1% BSA, forced through a 70 µm cell strainer (Greiner), and centrifuged at 493 × g for 7 min (4 °C). The sample was washed again with 10 mL ice-cold PBS 0.1% BSA and centrifuged at 493 × g for 7 min (4 °C). Finally, the sample was resuspended in 1 mL of ice-cold PBS 0.1% BSA and counted using the hemocytometer trypan blue exclusion method.
Peritoneal fluid preparation for scRNA-seq
Tubes (50 mL) were coated with FCS, after which the total volume of peritoneal fluid was transferred into these tubes, forced through a 100 µm cell strainer (Greiner), and centrifuged at 493 × g for 7 min (4 °C). Supernatant was discarded, and pellets of all 50 mL tubes were pooled with ice-cold PBS 0.1% BSA, and the sample was centrifuged at 493 × g for 7 min (4 °C).
Ten mL of erythrocyte lysis buffer (self-made) was added and kept on ice for 10 min after the sample was washed with 10 mL ice-cold PBS 0.1% BSA and the suspension was forced through a 70 µm cell strainer (Greiner) and centrifuged at 493 × g for 7 min (4 °C). The sample was washed again with 10 mL ice-cold PBS 0.1% BSA and centrifuged at 493 × g for 7 min (4 °C). Finally, the sample was resuspended in 1 mL of ice-cold PBS 0.1% BSA and counted using the hemocytometer trypan blue exclusion method.
Peritoneal tumor dissociation and preparation for scRNA-seq
To isolate single cells for scRNA-seq, tumors were transferred to a 6-well plate and washed with ice-cold PBS. Tumors were cut into small pieces using a scalpel blade and transferred to a 50 mL tube and 10 mL warm digestion medium (RPMI 1640, 1.5 mg/ml Collagenase (Sigma-Aldrich), 20 µg/ml DNAse I (Roche)) was added and the sample was placed at 37 °C for 30 min (mice) or 45 min (human) in a prewarmed VWR® Incubating Mini Shaker (400 rpm). To stop the digestion process, 10 mL of cold wash medium (RPMI 1640 + 10% FCS) was added, the tube was shaken vigorously, and the suspension was forced through a 70 µm cell strainer (Greiner) and centrifuged at 493 × g for 7 min. The pellet was washed in PBS 0.1% BSA and centrifuged again at 493 × g for 7 min (4 °C). The pellet was then resuspended in 10 mL red blood cell lysis buffer (self-made) and incubated for 10 min on ice. Ten mL cold PBS 0.1% BSA wash medium was added, and the suspension was centrifuged at 493 × g for 7 min (4 °C) and washed once more, and finally resuspended in 1 mL PBS 0.1% BSA. Cell numbers and viability were assessed using hemocytometer trypan blue exclusion, and brought to a concentration of 25 × 106 cells/mL for subsequent sorting and kept on ice.
scRNA-seq library preparation
Sorted DAPI- live cells were centrifuged at 493 × g for 5 min (4 °C), and supernatant was removed. Cells were diluted in 0.1% BSA/PBS to 1000 cells/μL and kept on ice until further processing. Directly after this, libraries (loading of 7200 cells) were prepared according to Chromium Next GEM Single Cell 3ʹ Reagent Kits v3.1 (PN-1000121). Briefly, Gel Beads-in-emulsion (GEMs) were generated by combining barcoded Single Cell 3′ v3.1 Gel Beads, a Master Mix containing cells, and Partitioning Oil onto Chromium Next GEM Chip. Immediately following GEM generation, the Gel Bead was dissolved, primers were released, and any co-partitioned cell was lysed. Primers were mixed with the cell lysate and a mastermix containing reverse transcription (RT) reagents. Next, Silane magnetic beads were used to purify the first-strand cDNA from the post GEM-RT reaction mixture. The full-length cDNA was amplified via PCR to generate sufficient mass for library construction. After end repair, A-tailing, adaptor ligation, and PCR amplification, the final mRNA libraries were sequenced on the Illumina HiSeq4000 and NovaSeq6000 at the Core Facility Genomics, Amsterdam UMC. The mRNA libraries were sequenced to 150 M per GEM-well.
CITE-seq library preparation
Sorted DAPI- live cells were centrifuged at 493 × g for 5 min (4 °C), and supernatant was removed. Cells were transferred to an Eppendorf in 50 μL of 0.1% BSA/PBS and incubated with Human TruStain FcXTM Fc blocking reagent (Biolegend) on ice for 10 min. Subsequently, cells were transferred to the TotalSeqTM—B Human Universal Cocktail, V1.0 (BioLegend) and incubated on ice for 30 min. The cells were washed twice with 0.1% BSA/PBS and centrifuged at 493 × g for 5 min (4 °C). Cell suspensions and library preparation were performed as described previously under “scRNA-seq library preparation” (loading of 7200 cells).
Statistics and reproducibility
The scRNA-seq experiments were performed on 5 patients with achalasia (HC), 13 patients with PM-CRC, and 6 patients with PM-GC. No statistical method was used to predetermine sample size, and no data were excluded from the analyzes. The Investigators were blinded to mouse allocation during experiments and outcome assessment.
scRNA-seq data analysis
Analyzes were orchestrated using Snakemake92 version 7.21. Raw reads were aligned to the human reference genome GRCh38 and processed using the CellRanger 7.0.0 software from 10X Genomics to generate unique molecular identifier (UMI) counts. The filtered gene expression matrices were imported into R version 4.1.1 and processed using the Seurat R package93 version 4.1.1. Gene counts were normalized by SCTransform94, while CITE-seq counts were normalized using CLR. Comparisons where PBMC, PF, and/or PM were combined were processed using Harmony to remove batch effects95 for subsequent unsupervised clustering analyzes. Unsupervised clustering analyzes were performed using the Louvain method on either the gene expression alone (PBMCs) or on the integrated gene and protein expression (PF) using the weighted nearest neighbor approach. Subsequent visualizations were conducted using the uniform manifold approximation projection (UMAP) algorithm calculated on the principal components96 derived from gene expression data, or the weighted nearest neighbor graph calculated from both gene and protein expression when interrogating PF from HC. Cells were initially annotated by lifting over annotations from a public PBMC dataset97. A subsequent curation was conducted by manually checking canonical markers. Debris and dead cells were identified based on a high percentage of mitochondrial reads in combination with a low number of unique genes identified. Since we were interested in the immune composition of the PerIS and TIME, all CD45- non-immune cells, apoptotic, proliferating, dead/dying cells, multiplets, and neutrophils were removed from the dataset. Neutrophils were excluded from subsequent analysis because (i) comparison with PBMCs was difficult since PBMCs, but not PF, had undergone Ficoll treatment, and (ii) the 10X genomics platform generally yields low transcripts due to low RNA content and high amounts of RNases.
Differential abundance analyzes were conducted by performing pairwise t-tests as implemented in the propeller function from speckle98 version 0.99.7 when comparing between samples, whereas Mann-Whitney tests were implemented to compare proportions between cell types. Multi-sample differential expression analyzes were conducted using the pseudobulk method99, where we summed reads per cell type per sample and conducted differential expression analyzes using DESeq2100 package version 1.34. Gene set enrichment analyzes were conducted using the fgsea package version 1.26. Cellular classification of TAMs was conducted using UCell101 version 2.4, where comparative analyzes were conducted through a mixed model approach with a random effect per donor using the lme4 package for general omnibus testing and nlme package for pairwise analyzes.
Public scRNA-seq datasets
The Colon Immune atlas of the publicly available dataset “Gut Cell Survey” (gutcellatlas.org)44 was used to compare the colonic immune compartment to our data. In total, 5 healthy patients (2 males and 3 females; patients IDs: 290b, 298c, 302c, 390c, and 417c; regions: cecum, sigmoid colon, and transverse colon) were included. In total, 27,619 cells were used in our comparative analysis. The mRNA libraries in this dataset were sequenced using only Chromium Next GEM Single Cell 3ʹ Reagent Kits v2. The cecum, sigmoid and transverse colon were included in the analysis, and the mesenteric lymph nodes were excluded. Further analysis of the data was performed as described previously under “scRNA-seq data analysis”. Cell subset annotations were manually curated and matched to our own dataset for comparability purposes.
The Human Liver atlas of the publicly available dataset “Liver Cell Atlas” (GSE192742; https://www.livercellatlas.org)45 was used to compare the liver immune compartment to our data. In total, 5 selected histologically healthy (absence of fibrosis), lean patients (5 females; patient IDs: H14, H16, H21, H22, and H25) were included. In total, 29,037 cells were used in our comparative analysis. The mRNA libraries in this dataset were sequenced using only Chromium Next GEM Single Cell 3ʹ Reagent Kits v3.1. Further analysis of the data was performed as described previously under “scRNA-seq data analysis”. Cell subset annotations were manually curated and matched to our own dataset for comparability purposes.
Immune subsets and subsequent macrophages were annotated using the same genes as per Fig. 1C, F. Dotplots and feature plots presenting the expression of several marker genes for the public datasets are included in the manuscript (Supplementary Fig. 3A–D).
Cell culture
Cell line CT26 was cultured in Dulbecco’s modified Eagle’s medium with L-glutamine, 15 mM HEPES (Thermo Fisher Scientific), supplemented with 8% fetal bovine serum (Life Technologies), penicillin, and streptomycin. The cell line was authenticated and regularly tested for mycoplasma infection.
Animal experiments
All in vivo experiments were approved by the animal experimentation committee at the Amsterdam UMC (in Amsterdam under the nationally registered license number AVD11800202013801) and performed according to national guidelines. Female BALB/cOlaHsd mice (6–12 weeks old) were purchased from Envigo. The mice were housed on a 12 h light-dark cycle at 20–26 °C with 30–70% humidity. Animals were randomly assigned to experimental groups.
Intraperitoneal tumor growth and treatment
To generate intraperitoneal tumors, 1 × 105 CT26 cells in medium were mixed at a 1:1 ratio with Matrigel (Corning) and injected intraperitoneally in BALB/c (CT26) mice. Anti-mouse CSF1R (BioXCell, clone AFS98) or vehicle control (phosphate-buffered saline, Fresenius Kabi) treatment was started at the same day as tumor cell injection and administered 3x week intraperitoneally for 21 days, 400 μg/injection. Anti-mouse PD1 (BioXCell, clone J43) or vehicle control (phosphate-buffered saline, Fresenius Kabi) treatment was started 7 days after tumor cell injection and administered 3x week intraperitoneally for 16 days, 100 μg/injection. In the second model, treatment initiation of all groups was only started 7 days after tumor cell injection. Mice were euthanized after 3 weeks for immune cell analysis, or when a humane endpoint was met for survival analysis, with a maximum of 16 weeks. Humane end points were determined as more than 15% weight gain within 2 days or signs of discomfort (e.g., decreased activity, reduced self-grooming, hunched posture, isolation from the group). The predefined humane endpoints were not exceeded. Tumors were harvested and scored according to the modified Peritoneal Carcinomatosis Index (mPCI), which evaluates tumor burden across seven anatomical regions of the murine peritoneal cavity: right and left subphrenic, (sub)hepatic, subgastric, small bowel/mesentery, pelvic, and back area. Each region was assigned a score from 0 to 3 based on the number and size of tumor nodules (0 = no macroscopic tumors, 1 = tumor nodules ≤2 mm, 2 = tumor nodules between 2 and 5 mm or >5 tumor nodules, 3 = tumor nodules ≥5 mm or >10 tumor nodules present), with a total possible score of 2157.
Peritoneal macrophage labeling
PKH26 Red Fluorescent Cell Linker Kit for Phagocytic Cell Labeling (Sigma–Aldrich; 500 µl of 5 µM solution) was used to selectively label cells with phagocytic capabilities, and in the peritoneal cavity, selectively stains peritoneal macrophages102. BALB/c mice were injected intraperitoneally with PKH26 or PBS control, after which, 2 days later CT26 cells were injected intraperitoneally, as described under “Intraperitoneal tumor growth and treatment”. Mice were sacrificed 16 days after initial PKH26 dye injection and PF, spleen, and peritoneal tumors were processed and analyzed using flow cytometry, as described in their respective parts, “Mouse peritoneal fluid, spleen, and tumor preparation for flow cytometry and Flow cytometry”, respectively.
Mouse peritoneal fluid, spleen, and tumor preparation for flow cytometry
Directly after euthanasia using CO2 (no cervical dislocation to prevent intra-abdominal blood contamination), peritoneal lavage with 5 mL ice-cold PBS 0.1% BSA was performed. The suspension was kept on ice, forced through a 100 µm cell strainer (Greiner), and centrifuged at 493 × g for 7 min (4 °C). Peritoneal tumors were dissociated as described in “Peritoneal tumor dissociation and preparation for scRNA-seq”. Spleens were dissociated when actively forced through a 100 µm cell strainer (Greiner) and centrifuged at 493 × g for 7 min (4 °C). Subsequent processing was the same for peritoneal fluid, spleen, and tumor samples.
Supernatant was discarded, and 10 mL RBC lysis buffer (Thermo Fisher Scientific) was added for 5 min, after which the sample was washed with 10 mL ice-cold PBS 0.1% BSA, forced through a 70 µm cell strainer (Greiner), and centrifuged at 493 × g for 7 min (4 °C). The sample was washed again with 10 mL ice-cold PBS 0.1% BSA and centrifuged at 493 × g for 7 min (4 °C). Finally, the sample was resuspended in 1 mL ice-cold PBS 0.1% BSA, counted using the hemocytometer trypan blue exclusion method, and brought to a concentration of a maximum of 25 × 106 cells/mL for subsequent flow cytometry analysis.
Flow cytometry
Murine PF, spleen, and PM were included for flow cytometry analysis. After successful isolation of single cell suspension, cells were washed in PBS and stained with LIVE/DEADTM Fixable Blue Dead Cell Stain Kit (Invitrogen, 1:1000). Cells were then centrifuged at 493 × g for 7 min (4 °C) and washed in PBS 0.1% BSA and incubated with TruStain FcX™ (anti-mouse CD16/32; clone 93, Biolegend, 1:100) for Fc receptor blocking. Afterwards, cells were centrifuged at 493 × g for 7 min (4 °C) and washed in PBS 0.1% BSA and subsequently stained for surface markers belonging to either a T-cell panel or myeloid panel using the following antibodies: CD8a; PerCP/Cy5 (53–6.7, Biolegend, 1:100), CD3; PE/Dazzle594 (17A2, Biolegend, 1:200), CD4; PE (GK1.5, Biolegend, 1:100); CD11b; Pacific blue (M1/70, Biolegend, 1:100), F4/80; PE/Cy7 (BM8, eBioscience, 1:100), CD19; 6D5 (APC/Cy7, Biolegend, 1:100), CD45; AF647 (30-F11, Biolegend, 1:200), CD45; AF700 (30-F11, Biolegend, 1:800), CSF1R; APC (AFS98, Biolegend, 1:200), F4/80; APC (BM8, Biolegend, 1:100). Acquisition was performed on the BD LSR Fortessa™, where large peritoneal macrophages (LPMs) were identified as live/dead-CD45+CD3−CD19−CD11b+F4/80+. Analysis was performed using FlowJo (Treestar) and R. The gating strategy of Figs. 4H, 6A and Supplementary Fig. 7C can be found in Supplementary Fig. 8A, B, C. For the PKH26 dye, laser was excited at 561 nm and detected using 585/15 band pass, as recommended by the manufacturer.
Immunohistochemistry (human peritoneal tumors)
Immunohistochemical stainings were performed on 5 μm-thick sections of paraffin-embedded tissues with the Ventana Benchmark Ultra immunostainer (Roche/Ventana, Tucson, USA). For detection and visualization of the proteins, the Optiview detection kit, including DAB and CuSO4 (Roche, Basel, Switzerland), was used. After Immunohistochemical staining, the sections were dehydrated with baths of ethanol and cleared with xylene. All sections were mounted with Tissue Tek ® coverslipping film (Sakura Finetek Europe B.V., Alphen aan den Rijn, The Netherlands). The following primary antibodies were used: CD8 (Agilent, 1:50) and CD163 (Cell Marque, 1:25). Pictures were made using Leica DM6 (Leica Microsystems), using the N PLAN 40x/0.65 objective, PCO edge 4.2 camera, and LEICA LAS-X acquisition software was used. Adobe Illustrator (Adobe) was used as processing software to crop images.
Immunofluorescence
Directly after isolation, tumors from mice sacrificed 19–23 days after tumor cell injection were fixed overnight in 4% formaldehyde, embedded in paraffin, and sectioned. Immunofluorescence stainings were performed on 5 μm-thick sections of paraffin-embedded tissues. Sections were deparaffinized with xylene and gradually rehydrated in ethanol. Slides were boiled in 0.01 mol/L sodium citrate buffer (pH 6) for 10 min at 120 °C in an autoclave for antigen retrieval. Slides were blocked for 30 min at room temperature in PBT. Then, slides were incubated overnight with primary antibody diluted in the blocking buffer. Slides were washed with PBS, and secondary antibody diluted in blocking buffer was added for 1 h at room temperature. Slides were mounted using Vectashield antifade mounting medium with DAPI (Vector Laboratories). The following primary antibodies were used: CD8 (ThermoFischer, 1:100) and CD206 (Abcam, 1:400). For both, donkey anti-rabbit AF568 (Invitrogen, 1:200) was used as secondary antibody. Pictures were made using Leica DM6 (Leica Microsystems), using the N PLAN 40x/0.65 objective, PCO edge 4.2 camera, the DAP and Y3 filter blocks, and Leica LAS-X acquisition software was used. Adobe Illustrator (Adobe) was used as processing software to crop images.
Olink
In peritoneal fluid of both healthy controls and patients suffering PM-CRC, the abundance of 92 immuno-oncology proteins was analyzed using the OLINK Target 96 Immuno-Oncology assay. An additional cohort (n = 9; total n = 21) was used for validation and correlation analysis. In short, a unique pair of oligonucleotide-labeled antibody probes binds to the targeted protein, which results in proximity-dependent DNA polymerization. The subsequent sequence is detected and quantified by real-time PCR. Protein concentrations are normalized and transformed using internal extension controls to adjust for intra-run variation. Data are provided as Normalized Protein Expression (NPX), with higher values indicating higher protein expression. Samples for HC and PM-CRC were randomized on the plate to avoid batch effects.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw sequencing data generated during this study have been made deposited in the European Genome-phenome Archive (EGA) under controlled access to comply with the local privacy laws. PBMC and PF scRNA-seq and CITE-seq (PF) data from healthy controls can be found under accession codes: EGAD50000000250, EGAD50000000251, and EGAD50000000252, respectively [https://www.ega-archive.org/studies/EGAS50000000174]. PF and PM scRNA-seq data from PM-CRC patients can be found under accession codes: EGAD50000000248 and EGAD50000000249, respectively [https://ega-archive.org/studies/EGAS50000000173]. Researchers are allowed to apply for access to the data access committee governing this data under the condition that the data will be used for research purposes only and that no attempts will be made to identify the participants. Source data are provided as a Source Data file. Data from the Colon Immune atlas were obtained from the “Gut Cell Survey’ (gutcellatlas.org). Data from the Liver Cell Atlas was obtained from GSE192742. [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE192742]. Source data are provided with this paper.
Code availability
All computational scripts used to prepare and analyze the data can be found on https://github.com/ND91/TIPRJ0000015_MAPS_CRC (https://doi.org/10.5281/zenodo.15064487).
Change history
05 May 2025
In the version of the article initially published, the reference 103 was incorrectly cited. It has been removed from the HTML and PDF versions of the article.
References
Franko, J. et al. Prognosis of patients with peritoneal metastatic colorectal cancer given systemic therapy: an analysis of individual patient data from prospective randomised trials from the Analysis and Research in Cancers of the Digestive System (ARCAD) database. Lancet Oncol. 17, 1709–1719 (2016).
Ploug, M., Graversen, M., Pfeiffer, P. & Mortensen, M. B. Bidirectional treatment of peritoneal metastasis with Pressurized IntraPeritoneal Aerosol Chemotherapy (PIPAC) and systemic chemotherapy: a systematic review. BMC Cancer 20, 105 (2020).
Bijelic, L., Yan, T. D. & Sugarbaker, P. H. Failure analysis of recurrent disease following complete cytoreduction and perioperative intraperitoneal chemotherapy in patients with peritoneal carcinomatosis from colorectal cancer. Ann. Surg. Oncol. 14, 2281–2288 (2007).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Cho, B. C. et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 23, 781–792 (2022).
Fucà, G. et al. Ascites and resistance to immune checkpoint inhibition in dMMR/MSI-H metastatic colorectal and gastric cancers. J. Immunother. cancer 10, e004001 (2022).
Alouani, E. et al. Efficacy of immunotherapy in mismatch repair-deficient advanced colorectal cancer in routine clinical practice. An AGEO study. ESMO Open 8, 101574 (2023).
Thomassen, I. et al. Incidence, prognosis, and possible treatment strategies of peritoneal carcinomatosis of pancreatic origin: a population-based study. Pancreas 42, 72–75 (2013).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356 (2015).
Laoukili, J. et al. Peritoneal metastases from colorectal cancer belong to Consensus Molecular Subtype 4 and are sensitised to oxaliplatin by inhibiting reducing capacity. Br. J. Cancer 126, 1824–1833 (2022).
Kleber, J. et al. Immune profile of patients with peritoneal carcinomatosis selected for CRS-HIPEC therapy. Cancer Immunol. Immunotherapy CII 72, 3867–3873 (2023).
Lenos, K. J. et al. Molecular characterization of colorectal cancer related peritoneal metastatic disease. Nat. Commun. 13, 4443 (2022).
Mantovani, A., Allavena, P., Marchesi, F. & Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 21, 799–820 (2022).
Bain, C. C. et al. Rate of replenishment and microenvironment contribute to the sexually dimorphic phenotype and function of peritoneal macrophages. Sci. Immunol. 5, eabc4466 (2020).
Helmy, K. Y. et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 124, 915–927 (2006).
Vogt, L. et al. VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J. Clin. Investig. 116, 2817–2826 (2006).
Han, J. et al. Human serous cavity macrophages and dendritic cells possess counterparts in the mouse, with a distinct distribution between species. Nat. Immunol. 25, 155–165 (2023).
Teschendorff, A. E. & Enver, T. Single-cell entropy for accurate estimation of differentiation potency from a cell’s transcriptome. Nat. Commun. 8, 15599 (2017).
Cheng, S. et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809.e723 (2021).
Nalio Ramos, R. et al. Tissue-resident FOLR2(+) macrophages associate with CD8(+) T cell infiltration in human breast cancer. Cell 185, 1189–1207.e1125 (2022).
Xiong, D., Wang, Y. & You, M. A gene expression signature of TREM2(hi) macrophages and γδ T cells predicts immunotherapy response. Nat. Commun. 11, 5084 (2020).
Revel, M., Sautès-Fridman, C., Fridman, W. H. & Roumenina, L. T. C1q+ macrophages: passengers or drivers of cancer progression. Trends Cancer 8, 517–526 (2022).
Katzenelenbogen, Y. et al. Coupled scRNA-Seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell 182, 872–885 (2020).
Casazza, A. et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer cell 24, 695–709 (2013).
Shen, L. et al. M2 tumour-associated macrophages contribute to tumour progression via legumain remodelling the extracellular matrix in diffuse large B cell lymphoma. Sci. Rep. 6, 30347 (2016).
Qian, B. Z. et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J. Exp. Med. 212, 1433–1448 (2015).
Khan, K., Long, B., Deshpande, G. M. & Fox, P. L. Bidirectional tumor-promoting activities of macrophage ezrin. Int. J. Mol. Sci. 21, 7716 (2020).
Zhang, L. et al. Fibronectin 1 derived from tumor-associated macrophages and fibroblasts promotes metastasis through the JUN pathway in hepatocellular carcinoma. Int. Immunopharmacol. 113, 109420 (2022).
Elfstrum, A. K. et al. LYVE-1-expressing macrophages modulate the hyaluronan-containing extracellular matrix in the mammary stroma and contribute to mammary tumor growth. Cancer Res. Commun. 4, 1380–1397 (2024).
Galpin, K. J. C. et al. FGL2 promotes tumour growth and attenuates infiltration of activated immune cells in melanoma and ovarian cancer models. Sci. Rep. 14, 787 (2024).
Meng, Y. et al. Myeloid cell-expressed MNDA enhances M2 polarization to facilitate the metastasis of hepatocellular carcinoma. Int. J. Biol. Sci. 20, 2814–2832 (2024).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Ashburner, M. et al. Gene ontology: a tool for the unification of biology. the gene ontology consortium. Nat. Genet. 25, 25–29 (2000).
Aleksander, S. A. et al. The gene ontology knowledgebase in 2023. Genetics 224, iyad031 (2023).
Ma, R. Y., Black, A. & Qian, B. Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 43, 546–563 (2022).
Lopez-Castejon, G. & Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 22, 189–195 (2011).
Munn, D. H. & Mellor, A. L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 34, 137–143 (2013).
Unuvar Purcu, D. et al. Effect of stimulation time on the expression of human macrophage polarization markers. PLoS ONE 17, e0265196 (2022).
Puig-Kröger, A. et al. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 69, 9395–9403 (2009).
Cai, B. et al. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci. Signal. 11, eaar3721 (2018).
Yeung, S. T., Ovando, L. J., Russo, A. J., Rathinam, V. A. & Khanna, K. M. CD169+ macrophage intrinsic IL-10 production regulates immune homeostasis during sepsis. Cell Rep. 42, 112171 (2023).
Veglia, F., Sanseviero, E. & Gabrilovich, D. I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 21, 485–498 (2021).
Riquelme, P. et al. TIGIT(+) iTregs elicited by human regulatory macrophages control T cell immunity. Nat. Commun. 9, 2858 (2018).
James, K. R. et al. Distinct microbial and immune niches of the human colon. Nat. Immunol. https://www.nature.com/articles/s41590-020-0602-z (2020).
Guilliams, M. et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 185, 379–396.e338 (2022).
Azizi, E. et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308.e1236 (2018).
Ruffell, B. & Coussens, L. M. Macrophages and therapeutic resistance in cancer. Cancer Cell 27, 462–472 (2015).
Marchesi, M. et al. HLA-dependent tumour development: a role for tumour associate macrophages? J. Transl. Med. 11, 247 (2013).
Roelands, J. et al. Transcriptomic and immunophenotypic profiling reveals molecular and immunological hallmarks of colorectal cancer tumourigenesis. Gut 72, 1326–1339 (2023).
Mariathasan, S. et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018).
Qian, B. Z. & Pollard, J. W. Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 (2010).
Hossain, M., Shim, R., Lee, W. Y., Sharpe, A. H. & Kubes, P. Gata6(+) resident peritoneal macrophages promote the growth of liver metastasis. Nat. Commun. 13, 4406 (2022).
Chow, A. et al. Tim-4(+) cavity-resident macrophages impair anti-tumor CD8(+) T cell immunity. Cancer Cell 39, 973–988.e979 (2021).
Argyle, D. & Kitamura, T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front. Immunol. 9, 2629 (2018).
Zhou, W. H. et al. The overexpression of fibronectin 1 promotes cancer progression and associated with M2 macrophages polarization in head and neck squamous cell carcinoma patients. Int. J. Gen. Med. 15, 5027–5042 (2022).
Figueiredo, C. R. et al. Blockade of MIF-CD74 signalling on macrophages and dendritic cells restores the antitumour immune response against metastatic melanoma. Front. Immunol. 9, 1132 (2018).
Bastiaenen, V. P. et al. A mouse model for peritoneal metastases of colorectal origin recapitulates patient heterogeneity. Lab Investig. 100, 1465–1474 (2020).
Hume, D. A. & MacDonald, K. P. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 119, 1810–1820 (2012).
Salcher, S. et al. High-resolution single-cell atlas reveals diversity and plasticity of tissue-resident neutrophils in non-small cell lung cancer. Cancer Cell 40, 1503–1520.e1508 (2022).
Valkenburg, K. C., de Groot, A. E. & Pienta, K. J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 15, 366–381 (2018).
Zhao, Y. et al. Stromal cells in the tumor microenvironment: accomplices of tumor progression? Cell Death Dis. 14, 587 (2023).
Qi, J. et al. Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat. Commun. 13, 1742 (2022).
Matsubara, E. et al. SPP1 derived from macrophages is associated with a worse clinical course and chemo-resistance in lung adenocarcinoma. Cancers 14, 4374 (2022).
Klement, J. D. et al. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Investig. 128, 5549–5560 (2018).
Qian, J. et al. Cancer-associated mesothelial cells promote ovarian cancer chemoresistance through paracrine osteopontin signaling. J. Clin. Investig. 131, e146186 (2021).
Nakamura, T. et al. Osteopontin-integrin α(v)β(3) axis is crucial for 5-fluorouracil resistance in oral squamous cell carcinoma. FEBS Lett. 589, 231–239 (2015).
Ostheimer, C., Bache, M., Güttler, A., Reese, T. & Vordermark, D. Prognostic information of serial plasma osteopontin measurement in radiotherapy of non-small-cell lung cancer. BMC Cancer 14, 858 (2014).
Nasir, I. et al. Tumor macrophage functional heterogeneity can inform the development of novel cancer therapies. Trends Immunol. 44, 971–985 (2023).
Zhang, S. et al. C1q(+) tumor-associated macrophages contribute to immunosuppression through fatty acid metabolic reprogramming in malignant pleural effusion. J. Immunother. Cancer 11, e007441 (2023).
Mosaad, Y. M. Clinical role of human leukocyte antigen in health and disease. Scand. J. Immunol. 82, 283–306 (2015).
Wang, B. et al. Transition of tumor-associated macrophages from MHC class II(hi) to MHC class II(low) mediates tumor progression in mice. BMC Immunol. 12, 43 (2011).
Valković, T. et al. Correlation between vascular endothelial growth factor, angiogenesis, and tumor-associated macrophages in invasive ductal breast carcinoma. Virchows Arch. 440, 583–588 (2002).
Ruffell, B. et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26, 623–637 (2014).
Song, H. et al. Macrophages on the peritoneum are involved in gastric cancer peritoneal metastasis. J. Cancer 10, 5377–5387 (2019).
Zhang, N. et al. LYVE1+ macrophages of murine peritoneal mesothelium promote omentum-independent ovarian tumor growth. J. Exp. Med. 218, e20210924 (2021).
Etzerodt, A. et al. Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J. Exp. Med. 217, e20191869 (2020).
Uderhardt, S., Martins, A. J., Tsang, J. S., Lämmermann, T. & Germain, R. N. Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell 177, 541–555.e517 (2019).
Weiss, J. M. et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J. Clin. Investig. 128, 3794–3805 (2018).
Moughon, D. L. et al. Macrophage blockade using CSF1R inhibitors reverses the vascular leakage underlying malignant ascites in late-stage epithelial ovarian cancer. Cancer Res. 75, 4742–4752 (2015).
Che, K. et al. Macrophages reprogramming improves immunotherapy of IL-33 in peritoneal metastasis of gastric cancer. EMBO Mol. Med. 16, 251–266 (2024).
Cassier, P. A. et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol. 16, 949–956 (2015).
Ries, C. H. et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25, 846–859 (2014).
Zhu, Y. et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74, 5057–5069 (2014).
Peranzoni, E. et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 115, E4041–e4050 (2018).
Liu, Y. et al. Metabolic reprogramming in macrophage responses. Biomark. Res. 9, 1 (2021).
Leendertse, M. et al. Peritoneal macrophages are important for the early containment of Enterococcus faecium peritonitis in mice. Innate Immun. 15, 3–12 (2009).
Ray, A. & Dittel, B. N. Isolation of mouse peritoneal cavity cells. J. Vis. Exp. 28, 1488 (2010).
Grootjans, J. et al. Epithelial endoplasmic reticulum stress orchestrates a protective IgA response. Science 363, 993–998 (2019).
Liu, M., Silva-Sanchez, A., Randall, T. D. & Meza-Perez, S. Specialized immune responses in the peritoneal cavity and omentum. J. Leukoc. Biol. 109, 717–729 (2021).
Huang, X. Z. et al. Single-cell sequencing of ascites fluid illustrates heterogeneity and therapy-induced evolution during gastric cancer peritoneal metastasis. Nat. Commun. 14, 822 (2023).
Liao, C.-T. et al. Peritoneal macrophage heterogeneity is associated with different peritoneal dialysis outcomes. Kidney Int. 91, 1088–1103 (2017).
Mölder, F. et al. Sustainable data analysis with Snakemake. F1000Res 10, 33 (2021).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Healy, J. & McInnes, L. Uniform manifold approximation and projection. Nat. Rev. Methods Primers 4, 82 (2024).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Phipson, B. et al. propeller: testing for differences in cell type proportions in single cell data. Bioinformatics 38, 4720–4726 (2022).
Murphy, A. E. & Skene, N. G. A balanced measure shows superior performance of pseudobulk methods in single-cell RNA-sequencing analysis. Nat. Commun. 13, 7851 (2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Andreatta, M. & Carmona, S. J. UCell: robust and scalable single-cell gene signature scoring. Comput Struct. Biotechnol. J. 19, 3796–3798 (2021).
Gautier, E. L., Ivanov, S., Lesnik, P. & Randolph, G. J. Local apoptosis mediates clearance of macrophages from resolving inflammation in mice. Blood 122, 2714–2722 (2013).
Acknowledgements
We wish to thank all the patients and staff from all the units that participated in the study. In particular, Susan Kenter, Core facility Genomics. Noortje van Gils, CITE-seq protocol. Pim Koelink, Jacqueline Vermeulen, Stephanie de Wit, Sofía Frigerio, and Lisanne Nijman support with laboratory experiments. Toni van Capel, Berend Hooibrink, Kim Brandwijk-Paarlberg and Daisy Picavet-Havik, Core facility Advanced Microscopy and Flow Cytometry and Wim Vos, Pathology. Figures 4g, 6j and Supplementary Fig. 1a were created with BioRender.com. This work is supported by The New York Stem Cell Foundation (LV), grants from the European Research Council (ERC-CoG 101045612 - NIMICRY) (LV), ZonMw (Vici 09-15018-21-10029) (LV) and Dutch Cancer Society grant (KWF 13435 / 2021-1) (LV), KWF YIG 13915 (JG), NWO ZonMw Veni Grant 09150161810115 (JG), NWO ZonMw Vidi Grant 09150172210058 (JG), Top Institute for Knowledge and Innovation grant ImPACT (JG), donation by mr H.J.M. Roels through Oncode Institute (JG). LV is a New York Stem Cell Foundation—Robertson Investigator.
Author information
Authors and Affiliations
Contributions
J.S., S.B., J.V., R.F., H.K., D.P., N.M., L.X., M.S., C.J., K.L., and J.T. performed experiments. J.S. and R.F. collected patient material and clinical data. J.S., A.L.Y., S.B., J.V., R.F., H.K., and K.L. analyzed data. A.L.Y., J.V., K.L., and J.V.G. provided bioinformatics input. J.S., A.L.Y., S.B., K.L., L.V., and J.G. conceived, designed, and directed the research. J.S., A.L.Y., S.B., J.V., R.F., H.K., L.X., M.S., N.v.G., O.K., M.W., C.J., K.L., J.G.V., S.G., S.D., J.T., G.D., L.V., and J.G. provided scientific input. J.S., A.L.Y., S.B., L.V., and J.G. wrote the manuscript. All authors approved the content of the manuscript.
Corresponding author
Ethics declarations
Competing interests
A.L.Y. received consultancy fees from Janssen, Johnson & Johnson, DeciBio, and was previously employed by GSK. None of the aforementioned entities had relation to the content of this publication. L.V. received consultancy fees from Bayer, MSD, Genentech, Servier, and Pierre Fabre, but these had no relation to the content of this publication. L.V. is an employee of Genentech Inc. J.G. has a collaboration with Roche. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jo Van Ginderachter, Rudolf Oehler, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Saris, J., Li Yim, A.Y.F., Bootsma, S. et al. Peritoneal resident macrophages constitute an immunosuppressive environment in peritoneal metastasized colorectal cancer. Nat Commun 16, 3669 (2025). https://doi.org/10.1038/s41467-025-58999-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58999-6
