
Abstract
Idiopathic pulmonary fibrosis (IPF) is a severe lung disease occurring throughout the world; however, few clinical therapies are available for treating this disorder. Overactivated fibroblasts drive abnormal fibrosis accumulation to maintain dynamic balance between inflammation and extracellular matrix (ECM) stiffness. Given pulmonary cell can regenerate, the lung may possess self-repairing abilities if fibrosis is removed via clearance of overactivated fibroblasts. The aim of this study was to evaluate the therapeutic activity of transient antifibrotic chimeric antigen receptor (CAR) T cells (generated via a novelly-designed lipid nanoparticle-messenger RNA (LNP-mRNA) system) and explore the regeneration mechanisms of lung in a male mouse model of bleomycin-induced pulmonary fibrosis. Here we found that fibrosis-induced ECM stiffening impaired alveolar epithelial cell compensation. The proposed LNP-mRNA therapy eliminated overactivated fibroblasts to rescue pulmonary fibrosis. The restored ECM environment regulated the cellular profile. The elevated plasticity of AT2 and Pclaf+ cells increased AT1 cell population via polarization. Apoe+ macrophages and increased numbers of effector T cells were shown to reestablish pulmonary immunity. Hence, LNP-mRNA treatment for fibrosis can restore pulmonary structure and function to similar degrees to those of a healthy lung. This therapy is a potential treatment for IPF patients.
Similar content being viewed by others


Introduction
Idiopathic pulmonary fibrosis (IPF) is a fibrosing interstitial lung disease of unknown etiology1, demonstrating the high incidence and mortality2. Given the limited therapeutic methods on pulmonary fibrosis, numerous studies have explored antifibrotic approaches and its pathogenesis. The release and excessive accumulation of collagen fibers caused by overactivated fibroblasts, which are marked with fibroblast activation protein (FAP), lead to pathological changes in the extracellular matrix (ECM) and persistent inflammation, resulting in a vicious cycle of cellular activity and extracellular fibrosis3,4,5,6,7. Compared with traditional drugs that block signaling pathways8, chimeric antigen receptor (CAR)-T therapy has shown promising prospects for the treatment of fibrosis9. In recent years, lipid nanoparticle (LNP)-mRNA system has been used to generate CAR-T cells in vivo, which effectively inhibited cancer and cardiac fibrosis10,11,12. However, studies on the application of the LNP-mRNA system for the treatment of pulmonary fibrosis are lacking.
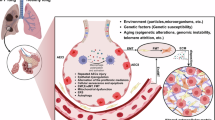
The objective of this study was to evaluate the therapeutic activity of transient FAP-targeted CAR-T (FAPCAR-T) cells generated in vivo via a novelly designed LNP-mRNA system and the lung regeneration mechanism in a mouse model of bleomycin (BLM)-induced pulmonary fibrosis. We hypothesized that the persistent accumulation of fibrosis elevated ECM stiffness and impaired lung regeneration by supporting the inflammatory environment and changing the cell profile. To test the hypothesis, we carried out a comprehensive analysis of proteomics and single-cell sequencing (scRNA-seq) in the young/aged mouse model of BLM-induced pulmonary fibrosis before and after the LNP-mRNA treatment. It was found that the stiffening of the ECM impaired the differentiation of alveolar type II (AT2) epithelial cells into type I (AT1) cells13,14, which damaged the self-repairing abilities of alveolar structure. The LNP-mRNA therapy eliminated pulmonary fibrosis and restored the ECM environment, which increased the plasticity of AT2 and Pclaf+ cells to rescue AT1 cell population (Fig. 1). Moreover, pulmonary immunity was reestablished by Apoe+ macrophages and increased numbers of effector T cells. Overall, the optimized LNP-mRNA system generated FAPCAR-T cells in vivo, cleared pulmonary fibrosis, and promoted lung recovery.

Overactivated fibroblasts induce pulmonary fibrosis and increase ECM stiffness, which hinders AT2 to AT1 differentiation resulting in PATS accumulation and fibrosis progression. Transient FAPCAR-T cells generated in vivo via LNP-mRNA can eliminate overactivated fibroblasts and promote lung regeneration via alveolar epithelial polarization. (BLM: bleomycin; LNP: lipid nanoparticle; ECM: extracellular matrix; AT1/2: alveolar epithelial type I/II; PATS: pre-alveolar type I transitional cell state). Created in BioRender65.
Results
LNP-mRNA system
We optimized the LNP component to improve transfection efficiency by replacing cholesterol with an equal amount of β-sitosterol (Fig. 2A). A Previous study proposed that β-sitosterol promotes the endosomal escape of lipid particles15. Different ratios of β-sitosterol to LNPs (0-40%) had negligible effects on the physical properties of LNPs (Fig. S1A). We tested the expression of luciferase mRNA in 293 T, fibroblast, and normal liver cells after incubating LNPs at different β-sitosterol concentrations for 24 h (Fig. S1B). LNPs with 30% β-sitosterol and 10% cholesterol demonstrated the highest mRNA expression and were defined as βLNP.

A Schematic diagram of traditional and β-sitosterol modified targetable LNP (βLNP) structures. B STEM images of membrane structure of βLNP and βLNP with CD5 surface modification (Scale bar: 25 and 50 nm). C Diagram of FAPCAR 2.1 and 2.2 IVT mRNA. Optimized TMD replaced CD28 TMD in FAPCAR 2.2 sequence. D IHC staining for FAP of mouse lung at 0, 14 and 28 days after BLM modeling. (Scale bar: 50 μm) arrow: FAP-positive overactivated fibroblasts. E FAPCAR 2.1 and FAPCAR 2.2 receptor expression on mouse T cell surface after 24 h incubation with IgG/βLNP or CD5/βLNP (n = 5). F Targeted lysis assay after 36 h co-culturing transient CAR-T with 3T3 cells (n = 3). G Cytokine production after 48 h co-culturing FAPCAR-T with FAP-3T3 cells (effector: target ratio=10: 1; n = 3). H PKH26 labeled CD5/βLNP accumulated in both spleen and liver and IgG/βLNP in the liver only. I Colocalization of CD5/βLNPs (PKH26 marked in red) and CD3e positive T cells (green) in the spleen for 24 h after i.v. injection (Scale bar: 100 and 20 μm). J Dose- and time-dependent distribution and expression changes of luciferase mRNA delivered by βLNPs in mice (n = 4). K Flow cytometry assay and quantification of FAPCAR 2.1 and 2.2 mRNA expressions for 24 h after CD5/βLNPs injection (10 μg) compared with saline injection (n = 5). ‘n’ means independent biological replications. The values are means ± SDs. *P < 0.05. P-value determined by one-way ANOVA followed by Tukey’s multiple comparisons test (E, G, K). (CAR: chimeric antigen receptor; IVT: in vitro transcription; TMD: transmembrane domain; IHC: immunohistochemistry; ORF: open reading frame; FAP: fibroblast activation protein; STEM: scanning transmission electron microscopy). Source data are provided as a Source Data file.
To endow particles with a targeted recognition capability, CD5 antibodies were added to βLNPs via SATA-maleimide chemistry to minimize T-cell depletion by CD5 compared with other antibodies (Fig. S1C). Transmission electron microscopy (TEM) scanning revealed no significant difference between βLNPs and CD5-modified βLNP (CD5/βLNP), both of which had bilayer membranes of ~4 nm and core regions with dense nucleotides (Fig. 2B). The CD5 antibodies had a thickness of ~14 nm on CD5/βLNP membrane (Fig. 2B), which was consistent with the results of nanoparticle tracking analysis (Fig. S1D). After coincubating freshly isolated and activated mouse T cells with particles encapsulated with EGFP or luciferase mRNA for 24 h, the CD5/βLNP group exhibited significantly higher EGFP or luciferase expression rates than did the unmodified βLNP group (Fig. S1E). In addition, the presence of few CD5 antibodies on βLNPs did not result in false-positive T cells population during flow cytometry analysis (Fig. S1F). To visualize LNP adhesion to mouse T cells, CD5/βLNPs labeled with PKH26 were coincubated for 4-8 h, which revealed strong adhesion at 4 h and visible endocytosis at 8 h (Fig. S1G).
FAPCAR-T cells generated via the CD5/βLNP-mRNA system
IVT (in vitro transcription) mRNA comprised a 5’ cap structure, optimized UTR domains, a Kozak sequence, a modified poly(A) tail and an ORF domain encoding the CAR sequence (Fig. 2C). In addition to the traditional CAR design with a CD28 transmembrane domain (TMD), we introduced a de novo TMD design following a programmable membrane protein (proMP) strategy16. Previous studies have shown that transmembrane fragments derived from CD28 have the potential to recruit additional costimulatory signals, which causes the overactivation of CAR-T cells17,18. This effect may be beneficial in cancer treatment but could cause nonessential cell activation and cytokine release in nononcology applications. Fibroblast activation protein (FAP) is a marker of activated fibroblast prominence. The high expression of FAP was detected in lung tissue at 14 and 28 days after the administration of bleomycin (Fig. 2D). Based on these findings, we designed two FAP-targeted CAR sequences with a CD28 TMD (FAPCAR 2.1) and with an optimized TMD (FAPCAR 2.2).
There was ~80% expression of FAPCAR 2.1/2.2 on T cell membrane after the coincubation of CD5/βLNP-mRNA with primary mouse T cells for 24 h (Fig. 2E, S2A). CD5/βLNP delivered ~6 × 109 copy number of FAPCAR-mRNAs (normalized to GAPDH) to T cells within 24 h (Fig. S2B), most of which disappeared within 96 h (Fig. S2C). The cytotoxicity assay with NIH 3T3 cells revealed E:T ratio-dependent lysis of transient FAPCAR-T cells to target cells that only expressed FAP (Fig. 2F). Moreover, CD5/βLNP-FAPCAR 2.2 led to an ~30% reduction in cytokine release compared with that of CD5/βLNP-FAPCAR 2.1 without altering lysis capacity (Fig. 2G), indicating the superiority of the optimized TMD and the increased suitability for pulmonary fibrosis treatment. Moreover, CD5/βLNP accumulated in both spleen and liver, whereas IgG/βLNP only accumulated in the liver for 24 h after PKH26-labeled βLNPs were injected into mice (Fig. 2H). Analysis of splenic sections showed CD5/βLNP mainly colocalized with T cells (CD3e positive) and rarely colocalized with B cells (CD19) or DCs (CD11c). In addition, partial colocalization with F4/80 indicated the phagocytosis of exogenous LNPs by macrophages (Fig. 2I, S2D). We subsequently evaluated luciferase mRNA expression at different doses. In the spleen and liver, a greater injection quantity was accompanied by increased expression; however, expression in both of these organs were systematically cleared after 96 h (Fig. 2J, S2E). Then, the in vivo transfection of FAPCAR mRNA was analyzed by flow cytometry. The results revealed that ~3% of primary T cells were programmed into FAPCAR-T cells 24 h after injection with 10 μg of CD5/βLNPs-FAPCAR, which was reduced by 50% after 72 h. The CD5-targeted-LNPs had no selective bias toward CD4- or CD8-positive T cells (Fig. 2K, S2F, S2G).
CD5/βLNP-FAPCAR 2.2 therapy reversed pulmonary fibrosis
Treatment with CD5/βLNPs-FAPCAR 2.2 successfully generated FAPCAR-T cells in vivo. We subsequently evaluated antifibrotic responses in young (8-week-old) and aged (16-month-old) mouse models, which were established with one-time tracheal instillation of bleomycin. CD5/βLNP-FAPCAR 2.2, IgG/βLNP-FAPCAR 2.2, or saline was injected into BLM mice at postoperative days 7 and 14. A commercial benchmark drug known as Pirfenidone (PFD) was also introduced for treatment via oral gavage (100 mg/kg/day)19 (Fig. 3A). HE staining revealed severe lung fibrosis and alveolar abnormalities beginning on day 7 postoperatively. There was a gradual extension of fibrotic area and parenchymatous lung tissue during the fibrosis progression (Fig. S3A). After CD5/βLNP-FAPCAR 2.2 treatment, the body weight and lung coefficient recovered rapidly, and the survival rate was highest in all of the groups that received BLM surgery (Fig. 3B–D). In the CD5/βLNP group (but not in the control or PFD groups), we observed an accumulation of CD3+ T cells at postoperative day 15 (24 h after the second injection) (Fig. S3B). We further generated a tdTomato reporter gene within FAPCAR mRNA to observe transfection. In the spleen and fibrotic lung, CD3 and tdTomato coexpressing T cells were identified (Fig. 3E), providing evidence for CD5/βLNP transfection and FAPCAR T cell infiltration into fibrotic lung tissue. Moreover, flow cytometry revealed that the number of FAPCAR-positive T cells was increased in both spleens and lungs after CD5/βLNP-FAPCAR 2.2 treatment (Fig. 3F, S3C, D) but not after IgG/βLNP treatment or in healthy animals. Importantly, the FAP+ fibroblast population decreased significantly in the CD5/βLNP group, whereas PFD treatment had no effects on this indicator (Fig. 3G, S3E). Following the standard guidelines for lung fibrosis assessment20, micro-CT scan was initially performed before animals were sacrificed at postoperative day 28. The imaging results demonstrated regional consolidation and diffuse shadows in the BLM+saline group, as well as increased Hounsfield unit (HU) density in the pulmonary area, whereas these symptoms were significantly relieved by the CD5/βLNP-FAPCAR 2.2 treatment (Fig. 3H). Reconstructed 3D lung models indicated a significant reduction in fibrotic tissue (red area) after treatment, and the increase of air volume in the lung demonstrated functional recovery (Fig. 3I, S3F). Then the lung was fully perfused before harvesting. Exterior images of the entire lung revealed that fibrotic lungs showed red lesion due to interstitial red blood cell leakage caused by tissue damage, whereas the lungs in the healthy group and in the CD5/βLNP treatment group were white (Fig. 3J). Consistently, immunohistochemistry (IHC) staining of FAP-positive cells showed a remarkable reduction of overactivated fibroblasts after CD5/βLNP-FAPCAR 2.2 therapy (Fig. S3G). Histological analysis (as assessed via Masson and picrosirius red staining) highlighted a significant improvement in extracellular matrix fibrosis of the CD5/βLNP treatment group than the saline, IgG/βLNP or PFD groups (Fig. 4A–C). Furthermore, hydroxyproline (HYP) assessment revealed that the collagen content approached healthy levels after CD5/βLNP-FAPCAR 2.2 treatment (Fig. 4D). The Mechanical Testing assessment revealed that the mean elasticity modulus of the whole-lung tissue was remarkable decreased after CD5/βLNP-FAPCAR 2.2 treatment (Fig. 4E). Overall, injecting CD5/βLNP-FAPCAR 2.2 significantly reduced the number of FAP-positive cells and rescued pulmonary fibrosis while treatment with PFD provided limited relief from fibrosis, which was possibly due to the short duration of the medication.

A Schematic diagram of fibrosis model establishment and treatment strategy in young or aged mice. B, C Young mouse BW changes (B) and survival curve (C) of control and BLM groups treated with IgG/βLNP-FAPCAR 2.2, CD5/βLNP-FAPCAR 2.2 (10 μg) and saline (n = 10). The survival rate was analyzed using the log-rank (Mantel-Cox) test. D Lung coefficients (lung weight (mg)/BW (g)) (n = 5,5,10,10,10,7). E tdTomato reporter gene expressed in T cells of spleen and lung at 24 h after the second CD5/βLNP-FAPCAR 2.2 injection (Scar bar: 10 μm). F Flow cytometry plots for FAPCAR expression in CD3+ pulmonary T cells of BLM+Saline and CD5/βLNP-FAPCAR 2.2 treatment groups and the ratio of FAPCAR expression to CD3+ pulmonary T cells (n = 4). G Flow cytometry analysis of FAP+Pdgfra+ fibroblasts in BLM+Saline and CD5/βLNP-FAPCAR 2.2 treatment groups, ratio of FAP+Pdgfra+ fibroblasts to all lung cells, and ratio of Pdgfra+ fibroblasts (both FAP+ and FAP—) to all lung cells (n = 4). H, I Representative micro-CT images and mean HU density (H), and lung air volume (mm3) (I) computed according to CT results at postoperative 28 days (n = 4,4,7,7,7,7). J Digital photos of lung tissue. ‘n’ means independent biological replications. The values are the means ± SDs. *P < 0.05. P-value determined by one-way ANOVA followed by Tukey’s multiple comparisons test (D, F, H, I) and two tailed unpaired Student’s t-test (G). (BW: body weight; HU: Hounsfield unit). Source data are provided as a Source Data file. Created in BioRender (A, B)65.

A Masson and picrosirius red staining of fibrotic areas (Scale bar: 100 and 200 μm). B, C Quantified changes of fibrotic areas (B) and Ashcroft scale (C) (n = 5,5,12,10,10,9). D Hydroxyproline (HYP) assay for quantification of collagen content. (n = 4,4,10,10,10,7). E Elasticity modulus assay of the entire lung tissue in healthy, fibrotic, and treatment groups by the Mechanical Testing System (n = 4,4,5). F–H CD5/βLNP-FAPCAR 2.2 anti-fibrotic assay in the fibrotic model of aged (16 month) mice: BW (F), survival rate (G), and lung coefficients (H) (n = 4,4,8,8,8). The survival rate was analyzed using the log-rank (Mantel-Cox) test. I–M Digital photos (I), micro-CT images (J, K) (n = 4), quantification of fibrosis area using the Masson staining (L) (n = 4,4,8,8,7) and HYP assay (M) (n = 4,4,7,7,7) showing the significant reduction of fibrosis after CD5/βLNP-FAPCAR 2.2 therapy. ‘n’ means independent biological replications. The values are the means ± SDs. *P < 0.05. P-value determined by one-way ANOVA followed by Tukey’s multiple comparisons test (B–E, H, I, K–M). Source data are provided as a Source Data file. Created in BioRender (F)65.
Antifibrotic response in aged mice
The incidence of IPF is positively correlated with aging and is more prevalent among the elderly population2. To further verify the role of CD5/βLNP-FAPCAR 2.2 in inhibiting pulmonary fibrosis, we generated a fibrosis model in aged mice (16-month-old). The survival rate was significantly higher and lung coefficients were lower in the CD5/βLNP group than those in the BLM+Saline and IgG/βLNP groups, albeit the CD5/βLNP group of aged mice did not regain body weight as fast as that of young mice (Fig. 4F–H). The micro-CT images showed that BLM-induced pulmonary fibrosis was more severe in aged mice compared to the young when they received the same dose, i.e., larger area of regional consolidation, higher HU density, and less air volume in the lung (Fig. 4I–K, S4A). The flow cytometry results showed that the population of FAP-positive fibroblasts was reduced, demonstrating the successful generation of transient FAPCAR-T cells (Fig. S4B). Moreover, Masson staining and HYP quantification revealed a significant decrease in fibrosis and collagen content in aged mice after CD5/βLNP-FAPCAR 2.2 treatment (Fig. 4L, M, S4C). These results suggest that CD5/βLNP-FAPCAR 2.2 therapy can inhibit pulmonary fibrosis in aged mice.
The toxicity or damage to organs was evaluated in both young and aged mice after CD5/βLNP-FAPCAR 2.2 treatment. Standard blood test showed negligible effects of CD5/βLNP-FAPCAR 2.2 treatment on red blood cells and platelets (Table S1, S2). The slight increase of lymphocytes (LYMPH#) may be caused by the active mobilization of immune T cells from the spleen (Table S1). Biochemical parameters for liver, kidney and heart as well as histological analysis showed no injury caused by the CD5/βLNP-FAPCAR 2.2 treatment (Table S3, S4 and Fig. S3I, S4D). There was no significant difference in the spleen weight rate (spleen weight normalized to body weight) between the control and CD5/βLNP groups (Fig. S3H, S4E). These findings demonstrate the safety and efficacy of CD5/βLNP-FAPCAR 2.2 therapy.
Characteristics of proteins in fibrotic lung
We performed the principal component analysis (PCA) of three time instances (postoperative day 0, 14 and 28 after BLM administration) (4 mice per group), which revealed significant changes in proteomes with time in BLM mice (Fig. 5A). The K-means clustering algorithm divided 4126 DEPs (differentially expressed proteins) of the whole groups into 6 clusters, which was consistent with heatmap results. Based on the GOBP analysis, clusters were annotated as DNA repair, immune system process, ECM organization, intracellular signal transduction, protein transport, and lipid metabolic process (Fig. 5B). In particular, cluster 3 was characterized by a continuous increase in ECM protein expression, which involved in the upregulation of collagen and fibronectin expression (e.g., Fn1, Fbln1, Col4a2, Col18a1) after the BLM administration (Fig. 5C, D). Moreover, upregulated expression of Lox family promoted the cross-linking of ECM components, suggesting the increased ECM stiffness (Fig. S5A). Previous studies have demonstrated that elevated tension inhibited AT2 to AT1 differentiation and induces abnormal accumulation of pre-alveolar type I transitional cell state (PATS) (marked by the increased expression of KRT8 and CLDN4, Fig. S5B)21,22,23. This resulted in alveolar tissue damage, as well as accelerated fibrotic development.

A Proteomic strategies of fibrotic tissues at postoperative day 0, 14 and 28 after BLM administration and PCA analysis of three time instances in BLM mice (n = 4, 4, 4). B–D Protein clusters obtained from K-means algorithm and showed in heatmap, in which 4126 DEPs were enriched into 6 clusters. Cluster annotation obtained from GOBP results (B); Dot plot showing top-10 GOBP terms enriched in proteins of cluster 3 (C); and ECM relevant proteins (D) at BLM postoperative day 0, 7 and 14. E Proteomic strategies of saline or CD5/βLNP-FAPCAR 2.2 treatment for BLM mice. PCA analysis showed the significant difference between BLM+Saline and BLM + CD5/βLNP groups (n = 4, 4). F Dot plot showing the top-5 GOBP, GOCC and GOMF enriched terms of downregulated DEPs corresponding to Fig. (E). G Schematic diagram of fibroblast clearance and ECM stiffness regulation. H Representative immunofluorescence images of PATS (Red: KRT8; Green: Sftpc; Blue: DAPI) (Scar bar: 50 μm). I Dot plot of GO enrichment from upregulated DEPs corresponding to Fig. (E). J Network projection of epithelial polarization pathways. ‘n’ means independent biological replications. P-value determined by Fisher’s exact test (C, F, I) and one-way ANOVA (D). (DEP: differentially expressed protein; BP: biological process; CC: cellular component; MF: molecular function). Created in BioRender (A, E, G) 65.
Proteomics analysis was demonstrated between fibrotic and recovered lungs due to CD5/βLNP-FAPCAR 2.2 treatment (4 mice per group). PCA revealed significant differences in DEPs between BLM+saline and CD5/βLNP groups (Fig. 5E). The top DEPs that were downregulated by the CD5/βLNP-FAPCAR 2.2 treatment were enriched in the regulation of ECM, inflammation, and immunity (Fig. 5F, S5C–F), such as downregulated expression of ECM-related proteins (Lox, Ctsd, and Ctsz)24 and upregulated expression of anti-inflammatory proteins (Prx, Hpdg, and Ifitm3). These findings indicate that the inhibition of the inflammatory environment and ECM remodeling process was promoted by using CD5/βLNP-FAPCAR 2.2.
Alveolar epithelial polarization after antifibrotic therapy
Proteomic analysis showed the clearance of fibrosis in the ECM after CD5/βLNP-FAPCAR 2.2 treatment. The Mechanical Testing assessment showed a significant reduction in the elastic modulus of lung tissue after treatment (Fig. 4E). These findings demonstrated the reduction of extracellular tissue tension (Fig. 5G), which could contribute to the differentiation of AT2 to AT1. Furthermore, the immunofluorescence results showed a significant decrease in the number of PATS after treatment (Fig. 5H, S5G), suggesting that the differentiation of AT2 to AT1 had returned to normal. The differentiation of AT2 to AT1 cells is associated with dramatic changes in cell shape and structure, which are generally considered to be epithelial polarization processes. Such transformation is typically accompanied by many changes in the expression of signaling and structural proteins. We observed that the upregulated DEPs were mainly enriched in cytoskeleton and intercellular connectivity signaling pathways after CD5/βLNP-FAPCAR 2.2 treatment, all of which were related to cell polarization (Fig. 5I, S6A–C). Hence, we depicted the polarization molecular map based on the KEGG analysis (Fig. 5J). Here, epithelial cells are assumed to be the key contributors to the pathway enrichment in lung recovery. To verify this hypothesis, it is required to exclude macrophage polarization or epithelial-mesenchymal transformation (EMT) process because both processes might occur during the disease progression. The expression of EMT markers25 was analyzed by the western blotting (Fig. 6A). Mesenchymal cells, transformed from epithelial cells, expressed low level of E-cadherin (an adhesion factor) and high level of Vimentin. Expressions of these markers were close to the normal level after CD5/βLNP-FAPCAR 2.2 treatment. Hence, EMT was inhibited by the treatment and did not contribute to polarization signaling enrichment. On the other hand, the enrichment genes relevant to macrophages decreased after treatment in proteomics data (Fig. 6B). Consistent with the decrease in gene expression, the flow cytometry results revealed the increased number of macrophages marked with F4/80 in inflammatory fibrotic lungs, which decreased after CD5/βLNP-FAPCAR 2.2 treatment (Fig. 6C). The analysis of the subphenotype revealed that the treatment decreased the number of M2 macrophages that were identified as the profibrotic phenotype (Fig. 6D, E and S6D, E). A subgroup characterized by double-positive expression (CD86+/CD163+) increased in both CD5/βLNP and PFD treatment groups (Fig. S6F). Given recent findings that M1/M2 phenotypes are not mutually exclusive in macrophages26, the cells in the treatment groups may undergo a state shift. Similar results were also observed in the BLM model of aged mice (Fig. S6G, H). Overall, the decrease of total macrophage population and M2 subphenotype excluded the macrophages polarization from the pathway enrichment after CD5/βLNP-FAPCAR 2.2 treatment.

A Western blot images and quantification of EMT marker genes and Cdc42 protein expressions (n = 4). B Proteomic heatmap for macrophage markers. C Flow cytometry analysis for percentages of macrophage (marked by F4/80) in lung cells (n = 4). D Flow cytometry plot for a comparison of M1/M2 phenotypes between BLM+Saline and CD5/βLNP groups. CD86: marker for M1 phenotype; CD163: marker for M2 phenotype. E Representative immunofluorescence images of macrophages in lung tissue. (Cyan: F4/80; Red: iNOS; Green: CD163; White: DAPI) (Scar bar:20 μm). F, G Representative immunofluorescence images showing the position of polarization factors, Cdc42 and Pkcz(aPKC) in cells (Red: Cdc42; Green: Pkcz; White: DAPI) (Scar bar: 10 μm and 5 μm). H Representative immunofluorescence images of key molecules in regulating epithelial polarization. (Blue: DAPI; Red: Calponin; Green: Pkcz zeta; White: Cldn 5) (Scale bar: 20 μm and 10 μm). I Relative abundance of proteins relevant to angiogenesis (Vegfa, Eng) and carbon dioxide regulation (Ca4, Ca13) (n = 4). ‘n’ means independent biological replications. The values are the means ± SDs. *P < 0.05. P-value determined by one-way ANOVA followed by Tukey’s multiple comparisons test (A, C) and two-tailed unpaired Student’s t-test (I). (EMT: epithelial-mesenchymal transformation). Source data are provided as a Source Data file.
For epithelial cell polarization, the signaling axis formed by Cdc42 and aPKC (Pkc-zeta) has been considered the key to polarization regulation27. ECM signals are sensed and transmitted by Crb3, which recruits Cdc42-aPKC-PAR6 to form the complex at the apical domain. This complex represents a key step in excluding proteins from the apical domain, which promotes the polarity distribution of proteins between apical, lateral and basement membranes via regulating cytoskeleton27,28,29. To verify the mechanism, we initially detected Cdc42 expression level using western blotting. The expression of Cdc42 in lung tissue increased after CD5/βLNP-FAPCAR 2.2 treatment (Fig. 6A). Moreover, immunofluorescence results showed that Cdc42 and PKCz were colocalized on the cell membrane and the colocalization tended to be on one side rather than the uniform distribution throughout the cell membrane after treatment (Fig. 6F, G). The cytoskeleton-associated tight junction was enhanced (Fig. 6H). Furthermore, upregulation of angiogenic markers (Eng, Vegfa) and carbon dioxide transport proteins (Ca4, Ca13) (Fig. 6I) denoted that the increased polarization of proteins drove the self-repair of impaired structure and function of BLM lungs after clearance of overactivated fibroblasts by FAPCAR 2.2 T cells.
Cellular profile for lung regeneration
The proteomics results suggested that the protein pathways were enriched in ECM remodeling and epithelial polarization, implying that multiple cell types might be involved in the signal regulation. To explore the lung regeneration process following successful fibrosis clearance, we mapped cellular profile by the scRNA-seq analysis of lungs between BLM+Saline and CD5/βLNP groups via the 10×Genomics platform (Fig. 7A). Unbiased cell capture and characterization with established cell markers divided 47,129 cells into 21 clusters. We observed that most of cells belonged to the immune system, including T cells (CD3e+), NK cells (Nkg7+), B cells (CD79a+) and immune monocytes (Cst3+). In addition, several small populations were identified as pulmonary parenchymal cells, including alveolar cells (Sftp+), endothelial cells (Pecam+), and fibroblasts (Dcn+) (Fig. 7B, S7A, B). Compared with those in the BLM group, the numbers of alveolar cells, fibroblasts, T cells and macrophages significantly changed in the CD5/βLNP group, suggesting that the cellular profile was regulated by the CD5/βLNP-FAPCAR 2.2 treatment (Fig. 7C, D).

A Study design for mouse lung single-cell sequencing (n = 2, independent biological replicates). B Uniform manifold approximation and projection (UMAP) plot for clusters and annotation of impartial captured lung cells. C, D A comparison of cluster components (BLM+Saline vs. BLM + CD5/βLNP-FAPCAR 2.2) (C) and sample distribution of significantly changed clusters (D). E UMAP embedding of fibroblast clusters. F A comparison of fibroblast cell clusters (BLM+Saline vs. BLM + CD5/βLNP-FAPCAR 2.2). G Expression projection of fibrotic and overactivated fibroblast marker genes on UMAP. H, I UMAP plot and annotation for re-clustered alveolar cells (H) and a comparison of cluster states (BLM+Saline vs. BLM + CD5/βLNP-FAPCAR 2.2) (I). J Dot plot showing representative markers for each alveolar cell cluster. K Expression projection of specific alveolar epithelial cell markers. L, M PAPCs marker genes in the entire pulmonary cell clusters (L) and highlighted PAPCs marker genes in UMAP plots that re-clustered with alveolar cells (M). N RNA velocity analysis for clusters including alveolar cells and PAPCs. (PAPCs: proliferating alveolar progenitor cells). Created in BioRender (A)65.
Fibroblasts were comprised of three distinct clusters, including inflammatory, fibrotic and mesothelial cells, which is consistent with recent studies30,31. Cluster 0 was identified as inflammatory fibroblasts with high expressions of Cxcl12 and Cxcl13. Fibrotic fibroblasts were characterized by Fbln2 and other pathological ECM genes. Notably, the two clusters overexpressing Col1a1 and Col3a1 were significantly reduced after treatment. Moreover, mesothelial fibroblasts were characterized by Msln and remained unchanged in all groups (Fig. 7E, F, S7C–E). In addition, FAP was expressed in both fibrotic and inflammatory clusters. Most of cells in the two clusters were shown in the BLM group, suggesting that FAPCAR-T cells effectively eliminated the target cells without affecting other cell populations (Fig. 7G).
Alveolar cells are the key factors for lung regeneration, as suggested above (Fig. 7H). AT1 cells were classified into two clusters, i.e., ‘AT1’, which expressed a higher level of Ager, and ‘Hopxhigh AT1’, which specifically expressed high Hopx. Hopx was involved in early developmental and differentiation pathways32. And in terms of Krt8 expression in this cluster, Hopxhigh AT1 has been considered an intermediate statement. Moreover, cluster 2 was identified as PATS that highly expressed Krt8 and Sftpc. The population of Cxcl2+ was characterized by the high expression of genes relevant to inflammation, fibrosis and lipid metabolism. This cluster almost disappeared after treatment, which indicated that these cells could be highly inflammatory damaged cells that temporarily appeared during the development of pulmonary fibrosis (Fig. 7I–K, S7F, G). All of the above-mentioned clusters, identified as intermediated statements or associated with inflammation, were reduced after CD5/βLNP-FAPCAR 2.2 treatment, consistent with the proteomics analysis (Fig. 7I, S7G). Importantly, we identified cluster 12 in UMAP of the entire pulmonary cells as proliferating alveolar progenitor cells (PAPCs) (Fig. 7B), a significantly elevated cluster relevant to alveolar cells. This population was characterized by the specific expression of Pclaf, Mki67 and Top2a (Fig. 7L, S7H, S8A). Previous research highlighted that PAPCs played a pivotal role in alveolar cell lineage plasticity as a progenitor cell reservoir and were indispensable for lung regeneration33. To verify the functionality of PAPCs, we re-clustered PAPCs with alveolar cells (Fig. 7M) and performed the cell lineage trajectory inference using the RNA velocity and Slingshot. The developmental trajectory of PAPCs was found toward to alveolar cells (Fig. 7N, S8B). Moreover, AT1 cells were the differentiation endpoint of AT2, Hopxhigh AT1 and PATS clusters. These results suggested that alveolar cell lineage plasticity was upregulated and AT2 or PAPCs played a positive role in differentiation into AT1 cells during the lung recovery.
Apoe+ macrophages and T cell types are critical for the reconstitution of pulmonary immunity
In addition to structural restoration, the reconstitution of immunity is also an important step in organ regeneration such that we analyzed macrophages and T cells. First, the macrophages were divided into 10 clusters, of which 4 clusters (clusters 2, 4, 5, 7) steadily decreased, and 3 clusters (clusters 0, 3, 8) increased after CD5/βLNP-FAPCAR 2.2 treatment. Clusters 0, 1, 2, 4, 5, and 6 were enriched for alveolar macrophage (AM) markers (Ear1, Ear2, Chil3, Plet1). The remaining clusters were monocyte-derived macrophages (mono-M) that expressed Ccr5, CD33, Sell, and Itgam34,35 (Fig. 8A–C, S8C). Clusters 2, 4, and 5 were identified as profibrotic lipid-associated macrophages (LAMs) because of high expression of profibrotic genes (Fn1, Spp1, CD44) and lipid metabolism genes (Lpl, Trem2, Fabp4, Fabp5). Clusters 7 and 9 expressed inflammatory genes (Il1b, S1009a, S1008a) and typical bone marrow marker gene (CD33), indicating their monocyte lineage (Fig. 8C, S8D). Cluster 7 also participated in profibrotic processes. The decreased macrophages (clusters 2, 4, 5, 7) suggested that FAPCAR-T cells disrupted the viscous inflammatory-fibrosis cycle via the clearance of overactivated fibroblasts, which was consistent with the flow cytometry and proteomics results.

A UMAP plot for re-clustered macrophage clusters (n = 2, independent biological replicates). B Cell percentages in different clusters. C Highlighted marker genes of alveolar, fibrotic and lipid associated macrophages. D CellphoneDB analysis between macrophage cluster pairs. E The significantly changed receptor-ligand pairs in the cluster 1/3 communication. P-value determined by Bimod test. F, G Pseudotime (F) and RNA velocity (G) analysis for developmental rate of Apoe+ mono-macrophages. H–J UMAP plots for re-clustered T cell clusters (n = 2): clusters annotation (H), a comparison of two groups (BLM+Saline vs. BLM + CD5/βLNP-FAPCAR 2.2) (I) and characteristic gene expression (J). K KEGG enrichment results for a combination of clusters 0,1,6 and a combination of clusters 7,8,9. L, M Pseudotime (L) and Slingshot (M) analysis of differentiation pathways starting from Tn cells. N Expression level of inducible T-Cell costimulatory (Icos) and interleukin-17a (Il17a), which increased with developmental trajectories. (AM: alveolar macrophages; Mono-M: monocyte-derived macrophages; Tn: naïve T cell; DNT: double negative T cell; Tm: memory T cell; Tc: cytotoxic T cell; Th: T helper cell; Treg: regulatory T cells).
The population of the increased expression after treatment (clusters 0, 3, 8) may be involved in tissue reconstruction process. Clusters 3 and 8 had common features of high Apoe expression and CD11b positive, which were significantly elevated after CD5/βLNP-FAPCAR 2.2 treatment (Fig. S8C, D). Cluster 8 was also enriched with the highest MHC genes expression, indicating mature and activated features in the macrophage lineage. The two clusters expressed lipid related genes, but exhibited the low Lpl expression, which distinguished them from other LAMs that were mainly associated with fibrosis or inflammation. Therefore, we explored potential effects of the Apoe+ mono-M population on tissue regeneration. Since Apoe is a type of secreted protein, we first evaluated cellular communications. CellphoneDB analysis revealed strong interaction between alveolar and Apoe+ macrophages (Fig. 8D). Trem2 has been reported to be a receptor that triggers differentiation and development of myeloid cells36. Protein interaction analysis showed that Apoe and Trem2 ligand pairs appeared only in the cluster 1/3 group (Fig. 8E). The findings suggested that Apoe+ macrophages could help stimulate differentiation and development of myeloid cells. To further confirm the findings, pseudotime analysis revealed that clusters expressing high monocyte markers served as root cells, and macrophages differentiated through the intermediate cluster 3 to produce the activated mature innate immunologic cells or the mature alveolar macrophages (Fig. 8F, S8E). Accordingly, RNA velocity analysis showed the development path from the Apoe+ mono-M to AM clusters (Fig. 8G, S8F). Overall, fibrosis-related alveolar macrophages were significantly reduced by the CD5/βLNP-FAPCAR 2.2 treatment, and Apoe+ mono-M population participated in quantitative supplementation of alveolar macrophages to maintain their phenotype for the reconstruction of immunity.
For T cells, clusters 7-9 were identified as the mature effector cell populations (Tc1 and Th1) and CD4+ memory T cells (Tm) by the annotation with marker genes, respectively. Re-clustering showed the significantly increased cell number in the three clusters after CD5/βLNP-FAPCAR 2.2 treatment. Naive T cells (Tn, clusters 0, 1, 6) were characterized by Lef1, Ccr7, and Sell and were more abundant in the BLM group (Fig. 8H–J, S8G, H)37. KEGG enrichment revealed that Tn cells were enriched in the Jak-Stat pathway and stem cell differentiation, whereas effector cell populations were enriched in the Rap1 signaling pathway, cytokine release pathway and TCR activation pathway (Fig. 8K). Based on Pseudotime, Slingshot and RNA velocity, Tn cells could be classified as stem cells. These analyses further revealed different developmental trajectories of Tn cells: differentiation into a group that highly expresses Ifng and Gzma or into Th17 and γδ T cells (Fig. 8L–N, S8I, J). Moreover, Ly6a, which is a marker of TCR-dependent activation, was highly expressed in Th1 cells. These results indicated that a large number of effector cells accumulated in lung tissue after CD5/βLNP-FAPCAR 2.2 treatment because of T cell immune activation, which partially resulted from transient FAPCAR-T recruited in the fibrotic lung. Notably, this therapy resulted in new clusters of memory T cells, which were completely absent in the BLM group (Fig. 8I).
Discussion
In this study, we optimized the LNP-mRNA system to transfect T cells in vivo to generate antifibrotic transient FAPCAR-T cells. In the pulmonary fibrotic model of young and aged mice, CD5/βLNPs-FAPCAR 2.2 treatment cleared FAP+ overactivated fibroblasts and minimized fibrotic area. Based on the proteomics and scRNA-seq analysis, we evaluated protein expression and cell characteristics after treatment. The enriched polarization was the key pathway for alveolar epithelial regeneration. Furthermore, the changes in cell profile after treatment demonstrated that the enhanced plasticity of alveolar cells and the supplementation of AT1 cells were the main contributors to lung regeneration. Apoe+ monocyte-derived macrophages supplied alveolar macrophages and helped to maintain their phenotype stability via the Apoe-Trem2 axis; moreover, together with the increased number of effector T cells, these immune cells reestablished pulmonary immunity after treatment.
Fibrosis is a highly dynamic process associated with overactivated fibroblasts in organs, such as the heart, lung and kidney8,38,39. Transient FAPCAR-T cells generated in vivo via the LNP-mRNA system cleared overactivated fibroblasts and have previously been shown to inhibit the development of cardiac fibrosis12. Here, the optimized CD5/βLNPs-FAPCAR 2.2 is proposed for the treatment of pulmonary fibrosis. Unlike traditional molecular drugs, such as oral Pirfenidone treatment that inhibits TGF-β40,41, transient FAPCAR-T cells directly clear overactivated fibroblasts. In our study, PFD did not inhibit fibrosis significantly, which was possibly due to the short duration of the medication (with normal dosing involving 6 weeks of administration in a mouse model and 3-6 months in clinical trials19,42,43). In contrast, the rapid suppression of fibrosis during a short-term time period was an advantage of CD5/βLNPs-FAPCAR 2.2 therapy. A successful LNP-mRNA system requires fewer toxic components and higher transfection efficiency. Previous studies have suggested that mRNA expression transfected via LNP can be improved by replacing cholesterol44 with equal amount of β-sitosterol, which is a plant cholesterol analog that lowers blood pressure and lipids as a medicine15,45. This study showed that a ratio of 3:1 (β-sitosterol to cholesterol in βLNP) was associated with the highest expression of mRNAs (>6 times greater than that of traditional cholesterol LNPs). To prevent liver’s clearance and improve transfection efficiency, antibody-based (i.e., T cell membrane markers, CD3, CD4, CD8, CD5, etc.) membrane modifications can guide LNP retention in specific extrahepatic tissues46,47,48,49. We selected CD5/βLNPs because CD5 is expressed by T cells but is not required for T cell effector function46,50. The novelly-designed CD5/βLNPs-FAPCAR 2.2 generated transient CAR-T cells in spleen and resulted in a low release of cytokines.
Tracheal administration of bleomycin resulted in pulmonary fibrosis, which increased lung ECM stiffness in both young and aged mice. The elevated mechanical tension that resulted from a stiffened ECM diminished AT2 to AT1 differentiation and increased the abnormal accumulation of PATS. This mechanism impaired the replenishment of alveolar epithelial cells and resulted in the deterioration of fibrosis23,51. CD5/βLNPs-FAPCAR 2.2 treatment rescued pulmonary fibrosis both in young and aged mice. This effect decreased ECM stiffness and changed the pathological environment around the epithelial cells. Cell membrane receptors transmit the change of pathological signals into epithelial cells to cause cascade reactions while integrin-based adhesions translate mechanical signals to affect actin aggregation and movement52,53,54. This mechanism regulates epithelial cell polarization, which plays an important role to maintain physiological functions, such as tissue barrier and gas exchange in healthy epithelial cells. The CD5/βLNPs-FAPCAR 2.2 therapy restored the AT2 to AT1 differentiation, which facilitated the reestablishment of cell polarity and restored the self-repair ability of epithelial cells. Moreover, the increased expression of Cdc42, in conjunction with aPKC, affected the distribution of protein polarization in epithelial cells. Interestingly, Cdc42 is indispensable in AT2, whereby it allows for differentiation into AT1 rather than remaining in the PATS state23.
The specific structure and cell types allow the lung to recover more easily after the elimination of fibrosis, although pulmonary fibrosis is prone to form large scars compared with hearts with severe diffuse fibrosis. The scRNA-seq analysis provides the first insight into the changes of pulmonary cell profile after CD5/βLNPs-FAPCAR 2.2 treatment. The clusters of fibroblasts, alveolar cells, macrophages and T cells were regulated, and the composition changes of these cells tended to promote lung regeneration. The CD5/βLNP-FAPCAR 2.2 treatment eliminated the fibroblast populations with collagen production while preserving mesothelial fibroblasts. ATI cells were supplemented via AT2 or PAPCs differentiation, which promoted alveolar regeneration after treatment. In addition, we investigated the unique role of Apoe+ macrophages in promoting monocyte-derived macrophage differentiation and maintaining the phenotype stability of alveolar macrophages. Previous studies have shown that these effects were regulated by cholesterol metabolism and macrophage colony-stimulating factors via Apoe55. This study further denoted the importance of Apoe-Trem2 signaling pathway in connecting Apoe+ mono-M and AMs. Apoe+ macrophages can also reduce pulmonary fibrosis directly via phagocytosis of Collagen I, which depended on the increased glycolysis and LPR1 binding56. Overall, Apoe+ macrophages play a positive role in the reduction of pulmonary fibrosis and the reestablishment of immunity environment. On the other hand, the presence of Tm cells, the increase in the number of Th and Tc cells possibly resulted from the increased differentiation of Tn cells after treatment. Transient FAPCAR-T cells might partially introduce foreign effector T cells, which were difficult to distinguish from Tn-differentiated effector cells. Hence, this phenomenon requires further investigation. Overall, these changes in T cells benefits for persistent inflammatory resistance.
In this study, the relatively early intervention with CD5/βLNP-FAPCAR 2.2 had significant antifibrotic effects on both young and aged mice. The treatment of BLM mice with severe pulmonary fibrosis (e.g., mice who were investigated from day 28 after the administration of bleomycin) still requires further investigation. These findings may help in selecting an appropriate therapeutic plans of CD5/βLNPs-FAPCAR 2.2 for IPF patients with various degrees of fibrosis in the subsequent clinical trials.
In conclusion, CD5/βLNP-FAPCAR 2.2 therapy can rescue BLM-induced pulmonary fibrosis in mice and further promote lung regeneration via alveolar epithelial cell polarization. These findings may provide more information on potential improvements in lung function in IPF patients.
Methods
Animal preparation
The animal care and experimental protocols conformed to the recommendations in the 8th Edition of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH, revised 2011) and were in accordance with the Animal Management Rules of China (Documentation 55, 2001, Ministry of Health, China). The study was approved by the Animal Research Committee of Shanghai Jiao Tong University.
C57BL/6 J young mice (males, 8 weeks) and aged mice (males, 16 month) were used to generate the pulmonary fibrosis model. Bleomycin (2 mg/kg, 40 μL solutions, per animal) was dripped into the lungs through the trachea under anesthesia. The animals were kept in the Laboratory Animal Center of Shanghai Jiao Tong University and housed at 21‒25°C, with 40‒50% humidity, a 12 h light:dark cycle, and free access to drinking water and standard feed. Lipid nanoparticles (LNP) with mRNAs were injected into the tail vein of model animals at postoperative 7 and 14 days after tracheal administration of bleomycin. Animals were observed every day. Body weight was recorded every 3 days.
mRNA preparation
mRNAs were prepared with the in vitro transcription (IVT). FAPCAR 2.1 design was based on the second-generation CAR sequence principle. CAR domains were comprised of CD8α leader, anti-muFAP scFV, CD8α hinge, CD28 transmembrane (TMD) and 4-1BB & CD3ζ intracellular signal. Amino acid sequences of protein fragments were extracted and backtracked to DNA sequences according to the functional annotation on Uniprot.org. The light chain (VL) and heavy chain (VH) amino acid sequences of the anti-muFAP scFV segment (clone73.3) were derived from a previous study57. The VH is linked distal to the VL (kappa) by a 3-time repeated G4S linker and then genetic codes are optimized for the mouse species. Four cysteine (Cys) are introduced to form disulfide bonds for scFv stability. Two Cys are placed at both ends of the linker (C terminus of VL and N terminus of VH) and two in VL-VH domain (L100-H44). The domains of FAPCAR 2.2 were the same as FAPCAR 2.1 except that CD28 TMD was replaced by a TMD designed with programmable membrane proteins (proMPs) strategy16. The complete DNA sequence was synthesized by the company (Sangon Biotech, Shanghai, China). RISR-RIAD peptide is known to maintain T cell activity, which was added to FAPCAR 2.1 and 2.2 sequence tails using the P2A ligand peptides58. tdTomato report gene was added using same strategy to track FAPCAR expression in T cells. The ex vivo synthesized sequences of FAPCAR were cloned into IVT template plasmid as the ORF region which also carried T7 promoter, modified 5’ UTR, kozak sequence, 3’ UTR, and a 110 nt-long poly (A) tail59,60,61. Cloning and recombination of plasmids and fragments were performed using the One Step Cloning Kit provided by Vazyme (#C112, Nanjing, Jiangsu, China). The plasmid was linearized by the restriction enzyme (Thermo Fisher, Waltham, MA, USA) and used as the template to produce mRNA with T7 High Yield RNA Transcription kit (#DD4202, Vazyme). N1-me-pseudo UTP was used during transcription to replace traditional UTP. Capping strategy was performed using the CAG trimer (Vazyme) in transcription. mRNA was purified in nuclease-free environment with magnetic RNA clean beats (Vazyme) and analyzed by agarose gel and finally stored in −80°C.
Formulation and characterization of LNP
LNP was generally comprised of 4 lipid components, i.e., ionizable lipid D-Lin-MC3-DMA (MCE, NJ, USA), distearoylphosphatidylcholine (DSPC), cholesterol and DMG-PEG 2k (Millipore Sigma, St. Louis, MO, USA) at molar ratio of 50:10:38:2. We designed an optimized LNP by replacing cholesterol with equal amount of β-sitosterol, which was defined as the X% βLNP (X% is the ratio of β-sitosterol to LNP). mRNAs were encapsulated in LNPs. Briefly, in the LNP-mRNA system, the ratio of lipid nitrogen to nucleic acid phosphate was 3:1 and the mass ratio of total LNP to mRNA was about 10:1. mRNAs were dissolved in 20 mM citric acid buffer (pH=4) and LNPs were in ethanol at a concentration of 10 mg/mL with 3:1 volume ratio of the two solutions. The syringes containing mRNA solution and lipid solution were connected to a tee union connector (0.05 in thru hole, #P-728; IDEX, Oak Harbor, WA) with HPLC peek tube (0.02 in ID for mRNA and 0.01 in ID for lipid solution, IDEX). Another peek tube (0.04 in ID) was used to collect products into a 50 mL tube. The laboratory encapsulation total flow rate was 16 mL/min (12 mL/min for mRNA solution and 4 mL/min for lipid solution)62. Prepared suspension was immediately diluted 100 times with PBS containing 25 mM EDTA. (pH=7.4) and then concentrated to mRNA of ~1 μg/μL. Diameter, polydispersity, zeta potential and encapsulation efficiency of LNP-mRNA particles were determined using the NanoBrook Omni (Brookhaven), Zetaview (Particle Metrix, Germany) and Ribogreen assay (Thermo Fisher).
Preparation of antibody-targeted particles
The chemically modified PEG lipid, DSPE-PEG 2k-maleimide (#880126 P, Merck), was added to the lipid component at 1:3 molar ratio of DSPE-PEG 2k-maleimide to DMG-PEG 2k. Procedure for sulfhydryl modification required reaction of rat anti-mouse CD5 or isotype IgG antibodies (Biolegend, San Diego, CA, USA) with 20-fold excessive SATA (N-succinimidyl S-acetylthioacetate) (Millipore Sigma) at room temperature for 30 min. Subsequently, N-Hydroxylamine (0.5 M) was added to the antibodies at volume ratio of 1:10 for 2 h to generate sulfhydryl groups. Unreacted components were removed promptly by Zeba desalting columns (Thermo Fisher). Antibodies and LNPs (molar ratio of 1:2 of antibody:maleimide) were mixed and incubated under argon at 4 °C overnight. 500 KD ultrafiltration tube was used to remove unbounded antibodies. All LNP-mRNAs were filtered using 0.22 μm filter before injection.
Cell culture and bioluminescence analysis
T cells, isolated from mice, were activated by CD3/CD28 Streptamer kit (#6-8920, iba, Goettingen, Germany) for 30 min and cultured in RPMI 1640 medium (#8123340, Gibco) containing 10% FBS (#A5669701,Gibco), 1% penicillin/streptomycin, 50 μM β-mercaptoethanol (Beyotime, Shanghai, China), 1 mM sodium pyruvate (Beyotime) and 100 U/mL interleukin-2 (#CK24, Novoprotein, Suzhou, Jiangsu, China). Mouse embryonic fibroblast cell line NIH/3T3 (#CRL-1658, ATCC), 293 T cells (#CRL-3216, ATCC) and normal liver cell NCTC1469 (#CCL-9.1, ATCC) were cultured in DMEM medium (#6123047, Gibco) with 10% FBS. Hepatic parenchymal cell AML12 (#CRL-2254, ATCC) was cultured in special DMEM media with 10% FBS, 0.5% ITS-G and 40 ng/ml dexamethasone. All cells were cultured in 37 °C with 5% CO2.
Transduced cells were generated by completely mixing lipid nanoparticles encapsulated with EGFP, luciferase or FAPCAR mRNAs with cultured cells (5 μg LNPs in 3×105 cultured cells) and then incubating for 24 h. The mRNA expression was analyzed by using the flow cytometry or bioluminescence intensity.
Flow cytometry
T Cells were centrifuged to remove culture medium and re-suspended with staining buffer. FAPCAR expression level in T cells was analyzed with anti-mouse IgG (H + L) and F(ab’)2 Fragment (PE Conjugate) (#8887S, Cell Signaling Technology, Boston, MA, USA) antibodies. Mouse spleen was carefully milled and filtered through 70 μm mesh to form single-cell suspension in cold PBS. Mouse lung tissues were minced in the digest solution and processed into single cell suspensions using a 70 μm filter membrane. FVS 700 (live/death dye, #564997, BD Bioscience, CA, USA), CD3e (#562600, BD Bioscience), CD8a (#557654, BD Bioscience), F4/80 (#565411, BD Bioscience), CD86 (#558703, BD Bioscience), CD163 (#12-1631-82, Thermo Fisher), FAP (#FAB9727G, R&D system) and Pdgfra (#12-1401-81, Thermo Fisher) were used in experiments. The cell analysis was performed by using the Cytoflex (Beckman Coulter, Brea, CA, USA) and BD LSRFortessa (BD Bioscience). Gating strategy was used for screening lymphocytes (FSC-A/SSC-A); FSC-A/FSC-H for selecting single cells; and FVS 700 for excluding dead cells from lymphocytes (Fig. S9). Plot images were analyzed by the FlowJo software (BD Bioscience).
Histology and Immunohistochemistry
Mouse tissues were fixed in 4% paraformaldehyde for 24 h. All samples were embedded in paraffin and sectioned into 8 μm slices. Before staining, sections were treated with xylene and dehydrated with ethanol. The H&E, Masson’s trichrome, and Picrosirius red staining was performed according to the manufacturer’s instruction. All stained sections were washed with water, dehydrated with ethanol, and sealed with neutral resin after xylene treatment.
Sections of citrate antigen retrieval were incubated overnight with anti-mouse FAP antibody (#ab218164, Abcam, Cambridge, UK). The hematoxylin-eosin staining was demonstrated. Slides were digitally scanned using the Slide Scanner at 40× (VS200, Olympus, Shinjuku, Tokyo, Japan) and analyzed by Olyvia (ver.4.1, Olympus). Quantification of fibrosis area was performed using the ImageJ software. The advanced Ashcroft score was used to grade fibrotic degree63,64. Two analysts were blinded about treatment strategies and the scores were averaged for statistics.
Ex vivo cytotoxicity assay
NIH/3T3 cells, transfected with FAP protein, were used as target cells. 7×103 cells were pre-adhered to 96-well plate. 24 h before coincubation, mouse T cells (effector cell) were transfected with IgG/βLNP-FAPCAR 2.1 or CD5/βLNP-FAPCAR 2.1&2.2 while 3T3 cells were transfected by βLNP-luciferase. Effector cells and target cells were mixed and cultured according to E:T = 1:1, 2:1, 5:1 or 10:1. NIH/3T3 cells were the blank control. After 36 h coincubation, cells were washed with cold PBS and evaluated for luciferase luminescence according to the manufacturer’s instruction (#RG051S, Beyotime). The reduction of bioluminescence intensity (BLI) indicated lysis of target cells. Lysis efficiency was calculated as: %lysis = (1−(BLI of test well /BLI of only target cells well)) ×100%.
Elasticity modulus assay
Lung tissue elasticity modulus were analyzed using the Mechanical Testing System (MACH-1 V500C, Biomomentum Inc., Laval, Canada). 0.25 N sensor was used in a 10% compression experiment. The formula is written as: elasticity modulus (gf/mm2)= Slope × Sample height /(cross-sectional area × 100).
Immunofluorescence staining and small animal live imaging system
PKH26 (Millipore Sigma) was used for pre-labeling particles following the manufacturer’s instruction. To observe the adhesion efficiency of CD5 modified βLNP, labeled particles were added to T cell culture medium for 4 and 8 h. T cells were washed with PBS to remove suspended and unadhered CD5/βLNP, fixed with 4% paraformaldehyde, and stained by DAPI. Moreover, labeled particles were injected to tail vein. The mouse was sacrificed after 24 h. Heart, liver, spleen, lung, and kidney were collected and analyzed by using fluorescence mode (551/567 nm of excitation/emission) of small animal live imaging system (#IVIS spectrum, PerkinElmer, MA, USA).
Immunofluorescence staining was performed for frozen sections or paraffin section. CD3e (#78588 T, Cell Signaling Technology), CD11c (#64804, Cell Signaling Technology), CD19 (#3574, Cell Signaling Technology), F4/80 (#30325, Cell Signaling Technology), CD163 (#ab316218, Abcam), iNos (#CL594-18985, Proteintech), Krt8 (#ab53280, Abcam), Sftpc (#ab90716, Abcam), Cdc42 (#10155-1-AP, Proteintech), Claudin 5 (#ab131259, Abcam), Calponin (#CL594-13938, Proteintech, Chicago, IL, USA) and PKC zeta (aPKC; #CL488-26899, Proteintech) were used for experiments. Images were acquired with Nikon Ti-E A1R HD25 microscopy.
Biodistribution/expression of different doses of IgG or CD5/βLNP encapsulating luciferase mRNA were evaluated by animal live imaging. 150 mg/kg D-luciferin (#ST196, Beyotime) was intraperitoneally injected to mice. Images were taken from supine and lateral positions separately using bioluminescence mode at postoperative 15 min. Images were shown as radiance (p/sec/cm2/sr) and regions of interest (ROI) were quantified by total flux (photons/s).
Micro-CT scan
Micro-CT analysis was performed for 27 days after bleomycin administration. Mice were anesthetized with isoflurane and fixed in prone position. Micro-CT data were acquired using Bruker Skyscan 1276 (Bruker, Billerica, MA, USA) with the parameters as follow: 100 KV, 80 μA, and 18.4 μm/slice. CT data were exported by the DataViewer software (Bruker). Mean Hounsfield unit (HU) density and lung air volume were analyzed to characterize lung health under various treatments. The total volume of lung air was computed using the CTAn software (Bruker).
Hydroxyproline (HYP) assay
Fresh lungs of young or aged fibrotic mice were fully perfused with PBS. The tissue was minced in tube and treated with the hydrochloric acid at 95 °C for 6 h. The hydroxyproline assay kit (sigma) was used according to the manufacturer’s instructions.
Blood routine examination and biochemical parameters
Fresh blood was collected by cardiac puncture when termination at postoperative 28 days. The samples were divided into two parts and stored in either anticoagulant (EDTA-K2) or non-anticoagulant tubes. The blood in the anticoagulant tube was tested for physiological indexes of red blood cells, hemoglobin, white blood cells and platelets by blood cell analyzer (XN-1000V, Sysmex, Kobe, Japan). Samples in the non-anticoagulant tube were solidified at room temperature for 1 h, and serum was collected. The automated biochemical analyzer (cobas C311, Roche, Basel, Switzerland) was used to detect LNP-associated organ damages, including liver (ALT, AST, ALB2, ALP), heart (CK, CKMB), and kidney function (UA2, CREA2, UREA/BUN).
PCR strategy
The construction of IVT template plasmids was performed using Gibson assembly method (#C112, CloneExpress II One Step Cloning kit, Vazyme). The template fragments and synthetic sequences were amplified by high-fidelity DNA polymerase (R045A, Takara, Kyoto, Japan). The primers are shown in Supplementary Table 2. The PCR program was set as: 98 °C for 3 min, 98 °C for 10 s, 55–60 °C (temperature and time were set according to primers), 72 °C for 30 s, and a total of 35 cycles. The amplified products were recombined and transformed on ice using DH5α competent cells, based on which the sequence for the plasmid was extracted and confirmed.
To analyze copy number of T cells, total RNA was extracted and purified using TRIzol-chloroform-isopropanol protocol. cDNA was synthesized by reverse transcription using 1 μg total RNA by HiScript III 1st Strand cDNA Synthesis Kit (#R312-01, Vazyme). mRNAs from in vitro transcription were used as standard RNA, then transcribed into standard cDNA under the same conditions after gradient dilution. qPCR was performed with SYBR Green Supermix (TaKaRa) with the protocol as follow: 95 °C for 5 min, repeating 40 cycles of 95 °C for 5 s and 60 °C for 45 s using StepOnePlus real-time PCR system (Applied Biosystem, Foster City, CA, USA). Primers are provided in Supplementary table 2. The standard RNA copy number was computed following the formula: copy number (copies/μL) = 6.02 × 1023×((concentration ng/μL)×10-9/ base number × 340). The standard copy number curve was fitted with the CT value of the qPCR and the log value of the standard RNA copy number. Then sample copy number was computed with standard curve and normalized with GAPDH.
Data-independent acquisition (DIA) Proteomics assay
Proteins were extracted using the SDT lysis buffer (4% SDS, 100 mM DTT, 100 mM Tris-HCl, pH 8.0) and then quantified. Proteins were digested with trypsin (Promega) at a 50:1 ratio overnight at 37 °C using the FASP method. Peptides were collected by centrifugation, desalted with C18 StageTips, and quantified with OD280.
LC-MS/MS were performed on an Orbitrap Astral mass spectrometer coupled with Vanquish Neo UHPLC system (Thermo Fisher Scientific). Peptides from each sample were loaded into a column (50 cm Low-Load µPAC™ Neo HPLC Column, Thermo Scientific) at a flow rate of 2.2 μL/min. Then eluted over 8 minutes with buffer B increasing linearly from 4% to 99% at 1.25 μL/min. The DIA method included a survey scan (380–980 m/z, resolution 240,000, AGC 500%, 5 ms injection) and MS/MS scans (150–2000 m/z, 2 m/z isolation window, AGC 500%, 3 ms injection, normalized collision energy 25, cycle time 0.6 s). Full MS and DIA spectra were recorded in profile and centroid modes, respectively.
The DIA MS data were analyzed using DIA-NN 1.8.1. The presence of one or more unique peptide segments indicated that the protein was detected. The data searched against the UniProtKB reviewed (Swiss-prot) database (uniprot-Mus musculus (Mouse) [10090]-88473-20230717.fasta). The maximal 1 missed cleavage sites and the mass tolerance of 10 ppm for precursor ions and 10 ppm for fragment ions were defined for database search. Carbamidomethylation of cysteines was defined as fixed modification, while acetylation of protein N-terminal, oxidation of Methionine was set as variable modifications for database searching. At most one variable modification is set up for each peptide segment. Peptide length range was set from 7-30. Charge of peptide was from 1 to 4. The fragment ion m/z range was from 150-2000. The database search results were filtered and exported with <1% false discovery rate (FDR) at peptide-spectrum-matched level, and protein level, respectively. The protein sample treatments and LC-MS/MS experiments were supported by Bioprofile Inc. (Shanghai, China).
Bioinformatic analysis were carried out with DIA-NN 1.8.1, Microsoft Excel and R statistical computing software. Anova was used to calculate p values across multiple groups, and p < 0.05 was defined as significant, clustered in different expression protein (DEP). A total of 11,993 proteins were identified in the study of pulmonary fibrosis progression, of which 4126 were DEPs. The sample mass spectrometry intensities in dataset were normalized for each row with the formula: (x-mean)/sd. Normalized values in each row had the same mean and standard deviation. The normalized raw data were then assessed using the Z-score (the distance from the sample point to the population mean) to distinguish the distribution across the groups. Positive values indicated upregulation relative to the overall mean, while negative values indicated downregulation. The p value and fold change (FC) were used in comparation between two groups. Student t test was used for calculating p value. Proteins with expression fold change>1.5 (up and down) and p < 0.05 were identified as the different expression protein (DEP). A total of 11,840 proteins were identified in BLM+Saline vs BLM + CD5/βLNPs-FAPCAR 2.2 group, of which 1309 were DEPs. To annotate the sequences, information was extracted from UniProtKB/Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology (GO). GO and KEGG enrichment analyses were carried out with the Fisher’s exact test (P < 0.01), and FDR correction for multiple testing was also performed.
scRNA-seq library preparation
Fibrotic lungs were obtained from BLM+Saline and BLM + CD5/βLNP-FAPCAR 2.2 young mice (two samples for one group). were dissociated using a dissociation solution. And single-cell suspension samples were subsequently prepared by filtration, red cell lysis and dead cells removement. Cell viability was assessed using trypan blue staining, with a required viability threshold of >85%. For scRNA-seq library preparation, cells were loaded onto the 10× Genomics Chromium instrument and 10× Genomics Chromium Single-Cell 3’kit V3 reagents were used according to the manufacturer’s protocols. The samples were pooled into libraries and sequenced on the Illumina NovaSeq 6000 at a minimum depth of 20,000 reads per cell. The experiments were supported by Bioprofile Inc. (Shanghai, China).
scRNA-seq data preparation and analysis
The Cell Ranger pipeline was used for processing all samples after sequencing (10× Genomics, V3.1.0). Raw sequencing data were demultiplexed and fastq files were generated using Illumina bcl2fastq software V 2.20. The scRNA-seq data were aligned to the Ensembl_v105_mus_musculus and row count matrices were imported to the R package Seurat V3.1.1. Cells were excluded due to the gene number (<500), UMI counts (<8000) and mitochondrial gene percentage (<25%). We used DoubletFinder package for individual samples to remove doublets and 47,129 cells were obtained after filtering. Clustering was then run using the functions FindVariableFeatures, FindNeighbors and Findclusters with resolution = 0.8 and dims = 1:20. The output UMAP graph were generated by DimPlot. Marker genes were identified using FindAllMarkers R package focusing on genes expressed by more than 10% of cells within a cluster and with a log fold change greater than 0.26. GO analysis was performed using the function FindMerkers. Merge function was used to create one merged Seurat object of BLM and CD5/βLNP treatment groups. This merged object was processed with differential gene expression and GO term analysis per cluster. Cells were manual annotated as a specific cluster for a marker after qualitative evaluation of feature plot and violin plots. Clusters of macrophage, T cell, alveolar cell and fibroblast were re-clustered with the same strategy. Clusters were manual reviewed and annotated and CD3+ clusters were excluded from macrophages and alveolar cells. We performed pseudotime analysis on the UMAP embeddings using Monocle 2. Slingshot analysis were using the function Slingshot R package, specifying starting cluster as “AT2” or “Tn cells”. We used the CellphoneDB and NicheNet default pipeline to predict enriched ligand-receptor pairs signaling to macrophages. The regulatory potential of top 20 ligand-receptor pairs (showed top 13) was represented in a dot plot.
Statistics and reproducibility
Statistical analysis was performed by the GraphPad Prism v.10 and data were presented as mean ± SD (standard deviation). P-value of <0.05 was considered to be statistically significant. Differences between two and multiple populations were assessed using the two-tailed Students t-test and one-way ANOVA with Tukey’s post-hoc test respectively. Survival rate of animals was analyzed with the log-rank (Mantel-Cox) test. All experiments were performed three or more times independently under identical or similar conditions. Immunofluorescence staining and electron microscopy assays were performed for three independent biological replicates and presented as representative images in this work.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings from this study are available within the manuscript and its supplementary information. The scRNA-seq data have been deposited in the Gene Expression Omnibus database under accession code GSE283267. The proteomics data have been submitted to ProteomeXchange via the PRIDE database with Project accession code PXD053333. Source data are provided with this paper.
References
Al-Tamari, H. M. et al. FoxO3 an important player in fibrogenesis and therapeutic target for idiopathic pulmonary fibrosis. EMBO Mol. Med. 10, 276–293 (2018).
Sgalla, G., Biffi, A. & Richeldi, L. Idiopathic pulmonary fibrosis: Diagnosis, epidemiology and natural history. Respirology 21, 427–437 (2016).
Wolters, P. J., Collard, H. R. & Jones, K. D. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev. Pathol. 9, 157–179 (2014).
Richeldi, L., Collard, H. R. & Jones, M. G. Idiopathic pulmonary fibrosis. Lancet 389, 1941–1952 (2017).
Parimon, T. et al. Potential mechanisms for lung fibrosis associated with COVID-19 infection. QJM 116, 487–492 (2013).
Chanda, D. et al. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 65, 56–69 (2019).
Savin, I. A., Zenkova, M. A. & Sen’kova, A. V. Pulmonary fibrosis as a result of acute lung inflammation: molecular mechanisms, relevant in vivo models, prognostic and therapeutic approaches. Int J. Mol. Sci. 23, 14959 (2022).
Henderson, N. C., Rieder, F. & Wynn, T. A. Fibrosis: from mechanisms to medicines. Nature 587, 555–566 (2020).
Aghajanian, H. et al. Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433 (2019).
Reinhard, K. et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 367, 446–453 (2020).
Kranz, L. M. et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401 (2016).
Rurik et al. CAR T cells produced in vivo to treat cardiac injury. Science 375, 91–96 (2022).
Kadur Lakshminarasimha Murthy, P. et al. Human distal lung maps and lineage hierarchies reveal a bipotent progenitor. Nature 604, 111–119 (2022).
Zacharias, W. J. et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 555, 251–255 (2018).
Patel, S. et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat. Commun. 11, 983 (2020).
Elazar, A. et al. De novo-designed transmembrane domains tune engineered receptor functions. Elife 11, e75660 (2022).
Brudno, J. N. et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat. Med 26, 270–280 (2020).
Majzner, R. G. et al. Tuning the antigen density requirement for CAR T-cell Activity. Cancer Discov. 10, 702–723 (2020).
Oku, H. et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur. J. Pharm. 590, 400–408 (2008).
Jenkins, R. G. et al. An official american thoracic society workshop report: Use of animal models for the preclinical assessment of potential therapies for pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 56, 667–679 (2017).
Mao, Y. et al. Deep spatial proteomics reveals region-specific features of severe COVID-19-related pulmonary injury. Cell Rep. 43, 113689 (2024).
Kobayashi, Y. et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 22, 934–946 (2020).
Wu, H. et al. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells. Cell 180, 107–121(2020).
Yoo, Y. et al. Therapeutic potential of targeting cathepsin S in pulmonary fibrosis. Biomed. Pharmacother. 145, 112245 (2022).
Bartis, D., Mise, N., Mahida, R. Y., Eickelberg, O. & Thickett, D. R. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax 69, 760–765 (2014).
Yuan, F. et al. Roles of the m6A Modification of RNA in the glioblastoma microenvironment as revealed by single-cell analyses. Front Immunol. 13, 798583 (2022).
Buckley, C. E. & St Johnston, D. Apical-basal polarity and the control of epithelial form and function. Nat. Rev. Mol. Cell Biol. 23, 559–577 (2022).
Apodaca, G., Gallo, L. I. & Bryant, D. M. Role of membrane traffic in the generation of epithelial cell asymmetry. Nat. Cell Biol. 14, 1235–1243 (2012).
Hayase, J. et al. The WD40 protein Morg1 facilitates Par6-aPKC binding to Crb3 for apical identity in epithelial cells. J. Cell Biol. 200, 635–650 (2013).
Tsukui, T., Wolters, P. J. & Sheppard, D. Alveolar fibroblast lineage orchestrates lung inflammation and fibrosis. Nature 631, 627–634 (2024).
Tsukui, T. et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat. Commun. 11, 1920 (2020).
Bourque, J., Kousnetsov, R. & Hawiger, D. Roles of hopx in the differentiation and functions of immune cells. Eur. J. Cell Biol. 101, 151242 (2022).
Kim, B. et al. PCLAF-DREAM drives alveolar cell plasticity for lung regeneration. Nat. Commun. 15, 9169 (2024).
Lv, J. et al. Dynamic atlas of immune cells reveals multiple functional features of macrophages associated with progression of pulmonary fibrosis. Front Immunol. 14, 1230266 (2023).
Sanin, D. E. et al. A common framework of monocyte-derived macrophage activation. Sci. Immunol. 7, eabl7482 (2022).
Chavakis, T., Alexaki, V. I. & Ferrante, A. W. Jr Macrophage function in adipose tissue homeostasis and metabolic inflammation. Nat. Immunol. 24, 757–766 (2023).
Zheng, L. et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474 (2021).
Younesi, F. S., Miller, A. E., Barker, T. H., Rossi, F. M. V. & Hinz, B. Fibroblast and myofibroblast activation in normal tissue repair and fibrosis. Nat. Rev. Mol. Cell Biol. 8, 617–638 (2024).
Odell, I. D. et al. Epiregulin is a dendritic cell-derived EGFR ligand that maintains skin and lung fibrosis. Sci. Immunol. 7, eabq6691 (2022).
King, T. E. Jr. et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med 370, 2083–2092 (2014).
Richeldi, L. et al. Trial of a preferential phosphodiesterase 4B inhibitor for idiopathic pulmonary fibrosis. N. Engl. J. Med 386, 2178–2187 (2022).
Yoon, H. Y., Kim, H., Bae, Y. & Song, J. W. Pirfenidone and risk of lung cancer development in IPF: a nationwide population-based study. Eur. Respir. J. 7, 2401484 (2024).
Inomata, M. et al. Pirfenidone inhibits fibrocyte accumulation in the lungs in bleomycin-induced murine pulmonary fibrosis. Respir. Res 15, 16 (2014).
Eygeris, Y., Gupta, M., Kim, J. & Sahay, G. Chemistry of lipid nanoparticles for RNA Delivery. Acc. Chem. Res 55, 2–12 (2022).
Eygeris. Y., Patel. S., Jozic. A., Sahay. G. Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery. Nano Lett 20, 4543–4549 (2020).
Kheirolomoom, A. et al. In situ T-cell transfection by anti-CD3-conjugated lipid nanoparticles leads to T-cell activation, migration, and phenotypic shift. Biomaterials 281, 121339 (2022).
Xue, L. et al. High-throughput barcoding of nanoparticles identifies cationic, degradable lipid-like materials for mRNA delivery to the lungs in female preclinical models. Nat. Commun. 15, 1884 (2024).
Parhiz, H. et al. PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J. Control Release 291, 106–115 (2018).
Ishida, T., Iden, D. L. & Allen, T. M. A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett. 460, 129–133 (1999).
Kim, D., Wu, Y., Li, Q. & Oh, Y. K. Nanoparticle-mediated lipid metabolic reprogramming of t cells in tumor microenvironments for immunometabolic therapy. Nanomicro Lett. 13, 31 (2021).
Strunz, M. et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 11, 3559 (2020).
Humphrey, J. D., Dufresne, E. R. & Schwartz, M. A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 15, 802–812 (2014).
Kechagia, J. Z., Ivaska, J. & Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 20, 457–473 (2019).
Chaudhuri, O., Cooper-White, J., Janmey, P. A., Mooney, D. J. & Shenoy, V. B. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 584, 535–546 (2020).
Theobald, H. et al. Apolipoprotein E controls Dectin-1-dependent development of monocyte-derived alveolar macrophages upon pulmonary β-glucan-induced inflammatory adaptation. Nat. Immunol. 25, 994–1006 (2024).
Cui, H. et al. Monocyte-derived alveolar macrophage apolipoprotein E participates in pulmonary fibrosis resolution. JCI Insight 5, e134539 (2020).
Wang, L. C. et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res 2, 154–166 (2014).
Newick, K. et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase a localization. Cancer Immunol. Res 4, 541–551 (2016).
Corbett, K. S. et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 586, 567–571 (2020).
Von. Niessen. A. G. O. Optimization of RNA Cancer Vaccines Using 3’UTR Sequences Selected for Stabilization of RNA. Universitätsbibliothek Mainz https://doi.org/10.25358/openscience-5189 (2016).
Trepotec, Z., Geiger, J., Plank, C., Aneja, M. K. & Rudolph, C. Segmented poly(A) tails significantly reduce recombination of plasmid DNA without affecting mRNA translation efficiency or half-life. RNA 25, 507–518 (2019).
Maier, M. A. et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 21, 1570–1578 (2013).
Hübner et al. Standardized quantification of pulmonary fibrosis in histological samples. Biotechniques 44, 507–511 (2008).
Ashcroft, T., Simpson, J. M. & Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 41, 467–470 (1988).
Yan, J. https://BioRender.com/c53i130 (2025).
Acknowledgements
This work was supported by the National Key Research and Development Program of China 202lYFA1000200 and 2021YFA1000203 (YL. Huo) and Shenzhen Science and Technology R&D Grant KQTD20180411143400981 (YL. Huo). Image items used in this work were created in BioRender: https://BioRender.com/c53i130.
Author information
Authors and Affiliations
Contributions
Conceptualization: Yan J., Shou J., Huo Y. Methodology: Yan J., Wang S.-Y., Su Q., Zou M.-W., Zhou Z.-Y., Shou J. Investigation: Yan J., Wang S.-Y., Su Q., Zhou Z.-Y. Visualization: Yan J. Funding acquisition: Huo Y. Supervision: Huo Y. Writing—original draft: Yan J., Huo Y. Writing—review & editing: Yan J., Huo Y.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yan, J., Wang, SY., Su, Q. et al. Targeted immunotherapy rescues pulmonary fibrosis by reducing activated fibroblasts and regulating alveolar cell profile. Nat Commun 16, 3748 (2025). https://doi.org/10.1038/s41467-025-59093-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59093-7
This article is cited by
-
CAR-engineered cell therapies: current understandings and future perspectives
Molecular Biomedicine (2026)
-
In vivo CAR cell therapy: from bench to bedside
Journal of Hematology & Oncology (2025)
-
Treatment Strategies for Acute Exacerbation of Interstitial Lung Disease Associated with Systemic Autoimmune Rheumatic Diseases in Chinese Patients: A Scoping Review
Rheumatology and Therapy (2025)
