
Abstract
MicroRNA-mediated post-transcriptional regulation of lung alveolar type 2 (AT2) and AT1 cell differentiation remains understudied. Here, we demonstrate that the let-7 miRNA family plays a homeostatic role in AT2 quiescence by preventing the uncontrolled accumulation of AT2 transitional cells and promoting AT1 differentiation. Using mouse and organoid models, we show that genetic ablation of let-7a1/let-7f1/let-7d cluster (let-7afd) in AT2 cells prevents AT1 differentiation and leads to KRT8 transitional cell accumulation in progressive pulmonary fibrosis. Integration of AGO2-eCLIP with RNA-sequencing identified direct let-7 targets within an oncogene feed-forward regulatory network, including BACH1/EZH2/MYC, which drives an aberrant fibrotic cascade. Additional CUT&RUN-sequencing analyses revealed that let-7afd loss disrupts histone acetylation and methylation, driving epigenetic reprogramming and altered gene transcription in profibrotic AT2 cells. This study identifies let-7 as a central hub linking unchecked oncogenic signaling to impaired AT2 cell plasticity and fibrogenesis.
Similar content being viewed by others


Introduction
Lung respiratory diseases rank as major global health challenges and are among the leading causes of death worldwide. Lung regeneration after injury is facilitated by epithelial progenitor stem cells (PSC)s found in airway and alveolar regions1,2,3. These cells are increasingly recognized as crucial factors in the onset of human lung diseases. Interstitial lung diseases (ILD)s are respiratory disorders characterized by excessive accumulation of extracellular matrix (ECM) and fibrotic tissue in alveoli leading to respiratory failure4. This condition is linked to abnormalities in facultative epithelial PSCs, especially alveolar type 2 (AT2) cells5,6. AT2 cells are crucial for lung alveolar tissue repair, serving dual roles: as PSCs that can differentiate into alveolar type 1 (AT1) cells and as homeostatic cells (hAT2) essential for surfactant production7,8,9. Recent studies in some lung injury models found that hAT2 cells become primed (pAT2), self-renew and give rise to heterogeneous population of KRT8+ transitional cells (hereafter referred to as ADI, alveolar differentiation intermediates) which can become AT1 cells10,11,12,13. Studies have shown that ADIs are characterized by the activation of P53, TGFβ, epithelial mesenchymal transition (EMT) signaling pathways and their pathological accumulation promotes lung injury and fibrosis10,11,12,13. A study identified a murine ADI cluster 7 subset (ADI-7) associated with stalled AT1 differentiation13. In the lungs of patients with Idiopathic Pulmonary Fibrosis (IPF), the most common endotype of ILDs, fibrogenic cells resembling murine ADIs -termed “aberrant basaloid” (AB) cells- persist and appear impaired in AT1-transdifferentiation11,12,13,14.
Studies in human IPF tissues and animal models have shown reduced expression of let-7 microRNA (miRNA) family members15,16, as well as TGFβ/SMAD3 mediated repression of let-7d in epithelial cells15. Systemic inhibition of let-7 function with an antagomiR was found to trigger thickening of alveolar septa and remodeling15. Let-7 is recognized as a tumor suppressor which prevents the proliferation and metastasis of cancer stem cells via unrestrained induction of multiway hub transcription factor (TF) oncogenes such as EZH2, MYC, and KRAS17,18,19,20. Also, let-7 has been shown to act as an orchestrator of key pathways and processes involved in pulmonary fibrosis, including PI3K/AKT/MTOR and EMT20,21.
This study delves into the molecular and cellular roles of the let-7 family in AT2 cells, underscoring its vital role in lung tissue homeostasis, protection against injury, and fibrosis. We revealed the crucial role of let-7 as a molecular brake to uncontrolled expansion, activation of AT2 PSCs and formation of ADIs. Loss of let-7 activity in AT2 cells also hampers cell fate where ADIs are prevented from transitioning into AT1s. Mechanistic studies highlight let-7 as a gatekeeper of physiologic expression levels of a profibrotic oncogene gene regulatory network (OGRN) comprised of BACH122, EZH223, and MYC24 in preservation of AT2 cell plasticity and lung homeostasis.
Results
Time-dependent downregulation of let-7 family expression during peak formation of ADIs after bleomycin-induced lung injury
We examined let-7 expression dynamics after bleomycin-induced lung injury in mice (GSE195773)25. Our analysis of this published small RNA-seq dataset revealed transient downregulation of let-7 family members at 7- and 14-days relative to controls followed by return to baseline by 21-days post-bleomycin (Supplementary Fig. 1a). Total let-7 activity decreased approximately 25% after 7-days post-bleomycin in lungs of mice vs controls (Supplementary Fig. 1b). Approximately 75% of total activity was comprised from members: let-7a, let-7b, let-7c, let-7d, and let-7f. These let-7 members are transcribed, in part, from conserved let-7b/let-7c2 (let-7bc2) and let-7a1/let-7f1/let-7d (let-7afd) gene clusters26 and contribute upwards of 20% and 55% respectively of total let-7 activity in naive lung (Supplementary Fig. 1b,c). The temporal downregulation of let-7 expression coincides with the transient appearance and peak formation of injury associated KRT8+ ADIs following bleomycin injury 11,12. Based on these observations, we hypothesized that let-7 family governs ADI cell formation in alveolar regeneration.
Depletion of let-7afd in AT2 cells causes spontaneous lung injury acutely
To understand the mechanism of let-7 function in AT2 cells, we utilized our let-7bc2 and let-7afd conditional knockout (KO) mice26. We employed let-7bc2f/f;Sftpc-CreERT2/+, let-7afdf/f;Sftpc-CreERT2/+ mice with and without the R26R-LSL-tdTomato reporter (Sftpc-tdT) for phenotypic analysis – hereafter referred to as let-7bc2AT2 and let-7afdAT2 mice (Supplementary Fig. 1c). Quantitative RT-PCR (qPCR) from flow sorted Sftpc-tdT+ AT2 cells confirmed >90% excision of let-7bc2 or let-7afd clusters after intraperitoneal tamoxifen administration (iTAM) (Supplementary Fig. 1d, e).
To assess the consequence of deletion of let-7bc2 or let-7afd clusters, lung histology and physiologic phenotypic analysis were carried out following 6-days of iTAM. Genetic loss of let-7afd in AT2 cells led to decreased arterial oxygen saturation (SpO2) following iTAM (Fig. 1a). Approximately 28% of let-7afdAT2 mice developed a pulmonary hemorrhage phenotype discerned by gross lung dissections and hematoxylin and eosin (H&E) stained sections (Fig. 1b, c). The histologic analysis of let-7afdAT2 mice revealed diffuse bleeding into the alveolar spaces and immune cell infiltration relative to controls (Fig. 1c). The let-7bc2AT2 mice exhibited a milder temporal drop in SpO2 relative to let-7afdAT2 mice but no hemorrhage phenotype (Fig. 1a, Supplementary Fig. 1f).

a Quantitative measurement of Sp02 from control, let-7bc2AT2, let-7afdAT2 mice (n = 5 per group) following iTAM. Data are mean ± s.d. *let-7bc2AT2 vs control, #let-7afdAT2 vs control, §let-7bc2AT2 vs let-7afdAT2, P values were obtained by one-way ANOVA with Tukey’s multiple comparison test. Not significant (NS). Representative lung dissections (b) and H&E-stained lung sections (c) from indicated mice 6-days after iTAM. Scale bars: 5 mm (b); 2 mm upper panel; 50 μm lower panel (c). Arrowheads indicate leukocytes. d GSEA plot shows significant induction of cycling epithelial genes in let-7afd−/− Sftpc-tdT+ AT2 cells compared to controls (n = 3 samples per group). e RNA-seq derived heatmap shows differentially expressed AT2 transition markers in whole lungs or Sftpc-tdT+ AT2 cells of let-7afdAT2 mice compared to controls after 6 days of iTAM. Adjusted p values were derived by two-sided Wald test and BH correction for multiple testing with BaseSpace workflow. f Transcript expression in Sftpc-tdT+ AT2 cells after 6 days of iTAM was examined by RNA-seq (n = 3 samples per group). Data are mean ± s.e.m. Adjusted p values vs control were calculated as noted in panel (e). Representative immunostaining (g) and quantification (h) of KI67+SFTPC+Sftpc-tdT+ cells in lungs of let-7afdAT2 vs control mice 14-days after iTAM. KI67 (gray), SFTPC (green), Sftpc-tdT (red), DAPI (blue). Arrows indicate KI67+SFTPC+Sftpc-tdT+ cells (scale bar 25 μm). (n = 3 mice per group). i Schematic representation to establish AT2 organoid cultures in mice. Created in BioRender. King, K. (2025) https://BioRender.com/x04u478. j Brightfield images show let-7afd−/− vs control AT2 organoids cultured in AMM. Scale bars 1 mm. k Quantification of CFE and spheroid diameters of organoids in panel (j). (n = 5 per group). Each dot represents one mouse. l GO BP enrichment analysis were calculated from significantly upregulated genes on RNA-seq from let-7afd−/− compared to control Sftpc-tdT+ AT2 cells (n = 3 samples per group) or organoid cultures (n = 2 mice per group). Adjusted p values were determined with a one-sided hypergeometric test with FDR correction with ShinyGo58. h, k Data are mean ± s.e.m. P values by unpaired two-tailed Student’s t test. a–c, g, h, j, k Representative of three independent experiments. Source data are provided as a source data file.
Deletion of let-7afd in AT2 cells stimulates progenitor stem cell proliferation and formation of ADI cells acutely in the lung
We performed bulk RNA sequencing (RNA-seq) of whole-lung or flow cytometric sorting of Sftpc-tdT+ AT2s respectively between let-7afdAT2 and controls following 6-days of iTAM. Gene set enrichment analysis (GSEA) showed significant induction of cell cycle related genes (e.g., Mki67), pAT2 (e.g., Hif1a) and ADI/ADI-7 (e.g., Cldn4, Ctgf) identity genes upon deletion of let-7afd in lungs and purified AT2 cells (Fig. 1d–f, Supplementary Fig. 2a–c). Lungs and purified AT2 cells from let-7afdAT2 mice also exhibit downregulation of hAT2 markers (Supplementary Fig 2a–c). Deletion of let-7afd led to downregulation of AT1 identity genes in sorted AT2 cells (Supplementary Fig. 2b). We additionally confirmed increased cycling AT2 lineage cells in lungs of let-7afdAT2 mice following 14-days of iTAM by immunofluorescence (IF) detection of nuclear KI67 in SFTPC+ Sftpc-tdT+ cells (Fig. 1g, h).
To determine if transcriptomic changes arise, in part, due to intrinsic phenotypic changes in AT2 cells, we carried out colony forming efficiency (CFE) measurements in combination with RNA-seq on let-7afdAT2 and control AT2 cell-derived organoids grown in AT2 maintenance medium (AMM)27 (Fig. 1i). CFE and spheroid diameters were significantly increased in let-7afdAT2 cells (Fig. 1j, k, Supplementary Fig. 2d). GSEA and gene ontology (GO) biological process enrichment analysis respectively revealed a striking parallel between let-7afdAT2 organoids and Sftpc-tdT RNA-seq datasets in the induction cell cycle genes (Fig. 1l, Supplementary Fig. 2e). The let-7afdAT2 organoids exhibited upregulation of pAT2 and ADI markers (Supplementary Fig. 2e). However, in contrast with sorted let-7afd−/− AT2 cells where hAT2 and AT1 markers appeared repressed, cultured let-7afd−/− AT2 organoids maintain similar marker expression as compared to controls (Supplementary Fig. 2e, f). The let-7afd−/− organoids did not exhibit significant induction of basal cell identity markers suggesting they are not redirected towards airway cell fate (Supplementary Fig. 2f). Collectively, this data indicates that acute loss of let-7afd expression triggers the uncontrolled proliferative expansion of AT2 PSCs and the formation of ADI cells in injured lungs acutely.
Integrated biochemical and transcriptomics analysis in the generation of a let-7 mRNA targetome in AT2 cells
To validate and identify functional targets of let-7, we performed enhanced Argonaute 2 UV-cross-linking immunoprecipitation-sequencing with enrichment of let-7 targets (AGO2-eCLIP+let-7)28 in WT mice following 6-days of bleomycin injury (Fig. 2a). The libraries yielded 13,027 chimeric let-7:mRNA called peaks in the 3’untranslated region (3’UTR) of genes (Fig. 2b, Supplementary Data 1). Motif analysis further showed highly significant and specific enrichment of let-7 “seed” region binding motifs within chimeric let-7 peaks (Fig. 2c). Collapsing of chimeric let-7 peaks into individual transcription units yielded 2009 target genes in acute bleomycin-injured lungs (Fig. 2d).

a Schematic of experimental design for AGO2-eCLIP+let-7 analysis after bleomycin treatment. b Fractional representation of let-7:mRNA chimeric reads of let-7 members. c The let-7 binding motif is highly enriched by HOMER. d Total number of genes with let-7:mRNA chimeric peaks. e Sylamer shows overrepresentation of let-7 binding motifs in let-7afd−/− AT2 cells. f Venn Diagram shows gene totals of let-7 AT2 cell targetome. g Heatmaps show relative expression of significantly upregulated and validated let-7 gene targets in let-7afd−/− vs control Sftpc-tdT+ AT2 cells (n = 3 samples per group) or AT2 organoids (n = 2 mice per group) by bulk RNA-seq. Adjusted p value < 0.05 vs controls using a two-sided Wald test and BH correction. h KEGG pathway enrichment analysis for the let-7 targetome was computed with a one-sided hypergeometric test and FDR correction via Enrichr59. i ChIP-Seq regulatory network analysis indicates significant TF footprints of hub targets of let-7 in let-7afd−/− Sftpc-tdT+ cells and organoids. A two-tailed hypergeometric test was used as previously described61. j Functional pathway and protein interactome network shows significant connectivity of let-7 hub targets. TFs (blue), enzymes (orange). Network q value was calculated with EnrichmentMap in Cytoscape. k Schematic alignments of let-7 “seed” region with target mRNA sequences in mice and humans. l UCSC genome browser tracks from AGO2-eCLIP+let-7 indicates binding of let-7 to targets. Purple triangles indicate let-7 motifs. White asterisks indicate aligned site in (k). m QPCR analysis for indicated let-7 targets in let-7afd−/− vs control Sftpc-tdT+ AT2 cells 14-days post-iTAM (samples per group: n = 3, Ezh2; n = 4, Bach1; n = 4 Myc). Data are mean ± s.e.m. P values by two-tailed unpaired Student’s t test. n Line plots show smoothed relative expression of hub genes across the ADI pseudotime trajectories after bleomycin injury in published dataset12. Expression values were estimated by fitting a generalized additive model across pseudotime trajectory. o The expression pattern for 217 of 394 let-7 targets is based on inferred likelihood of detection. p Jaccard overlap scores were calculated between let-7 targetome and epithelial cells defined in GSE14125912. Source data are provided as a Source Data file.
To enrich for functional targets of let-7 in AT2 cells, we integrated the AGO2-eCLIP+let-7 targetome with the sorted Sftpc-tdT+ and/or organoid transcriptome datasets. Initially, we applied Sylamer29 on let-7afd−/− transcriptome data to ascertain let-7 post-transcriptionally represses mRNA targets. Sylamer showed prominent miR-mediated mRNA destabilization since the motifs corresponding to let-7 were enriched within the 3’UTRs of induced genes in let-7afd−/− Sftpc-tdT+ cells compared to controls (Fig. 2e). Next, we made use of Venn Diagram intersection analysis to select biochemically validated, significantly upregulated, and experimentally validated let-7 target genes in sorted Sftpc-tdT+ and organoid transcriptome datasets. The integrated approach revealed a let-7 AT2 targetome comprised of 394 genes (Fig. 2f, g, Supplementary Data 2).
Pathway discovery of let-7 mRNA targetome identifies leading edge genes associated with stem cell renewal, cell growth, and cell differentiation
Functional enrichment analysis of the let-7 targetome highlighted diverse networks of let-7 targets associated with an array of cellular programs including stem cell renewal, cell growth, and cell differentiation (Supplementary Data 2, 3). Interestingly, KEGG enrichment analysis showed the top pathways as miRNAs in cancer, focal adhesion, ECM-receptor interaction, and P53 signaling (Fig. 2h, Supplementary Data 3). MiRNAs typically govern cell programs upstream of transcription factors (TF) and multiway hub genes in gene regulatory networks (GRN)18,19,20,29. Annotation of the let-7 targetome revealed TFs which includes gene activators (e.g. E2f1, Hif1a, Myc), gene repressors and context dependent inducers/repressors of gene expression (e.g., Bach1, Ezh2, Foxp2) (Supplementary Data 1–3). Major hub genes, Kras and Nras were also identified as targets of let-7 (Supplementary Data 1–3). ChIP-Seq regulatory analysis on transcriptome datasets showed significant enrichment for MYC, E2F1, E2F6, and BACH1 targets in genes upregulated after depletion of let-7afd (Fig. 2i, Supplementary Data 4). Targets of EZH2 were overrepresented in downregulated genes upon loss of let-7afd (Fig. 2i). Intriguingly, the same is also true for the EZH2 partner proteins SUZ12/EED and core components of the Polycomb Repressive Complex 2 (PRC2) which mediate tri-methylation of histone H3 on lysine 27 (H3K27me3)30,31, implying that deletion of let-7afd promotes EZH2 mediated epigenetic silencing (Fig. 2i, Supplementary Data 4). Additionally, loss of let-7afd promotes the induction of KRAS pathway in AT2 cells (Supplementary Fig. 3a). Network analysis revealed strong connectivity of EZH2 with hub targets implicating the existence of a let-7 dependent oncogenic GRN (OGRN) in AT2 cells (Fig. 2j). We note the unique properties of OGRN genes as sufficient drivers of uncontrolled cell growth and failed terminal differentiation in disease30,32,33. We annotated the location of the let-7 chimeric peaks and binding motifs for let-7 within 3’UTRs (Fig. 2k, l, Supplementary Fig. 3b, c, Supplementary Data 1, 2). Bach1, E2f1, Ezh2, Myc, Kras, and Nras contain evolutionarily conserved sites and were previously identified as direct targets of let-7 in non-AT2 cells17,18,19,20,34. Hif1a emerged as a direct target of let-7; however, the binding site is poorly conserved in humans (Supplementary Fig. 3b, c). We confirmed significant increased mRNA expression of OGRN genes by qPCR detection in sorted let-7afd−/− Sftpc-tdT+ cells following 14 days of iTAM (Fig. 2m, Supplementary Fig. 3d).
Transcriptional convergence of alveolar and PSCs show enrichment of let-7 targetome genes in ADI cells
We evaluated the expression dynamics of the let-7 targetome during AT2 cell “bridging” into ADIs following bleomycin injury from a published scRNA-seq dataset12. At the same time, we also incorporated expression data for MHC-II+ club cells because they were associated with production of AT2 intermediate cells in bleomycin injury. Interestingly, Bach1, Ezh2, Kras, Myc, and Nras expression levels were increased along the differentiation trajectory from AT2 towards ADIs (Fig. 2n, Supplementary Fig. 3e). Conversion of MHC-II+ club cells into KRT8+ ADI showed a more nuanced pattern for Ezh2 and Bach1 indicated by an increase followed by a reduction of expression (Fig. 2n). Most let-7 OGRN genes are upregulated in WT AT2 cells after 5-days of bleomycin treatment, suggesting they are co-regulated during lung regeneration (Supplementary Fig. 3f). Many let-7 targets in our dataset exhibit a gradual increase and peak in expression in ADIs relative to both AT2 and MHC-II+ club cells (Fig. 2o). The let-7 targetome exhibits greater enrichment in AT1 cells and transitional AT2 cells than hAT2 cells (Fig. 2p). Together, these results support the notion that let-7 serves as a braking mechanism in generation of pAT2/ADIs following lung injury and alveolar regeneration upstream of a stress-inducible OGRN. These data prompted us to explore the phenotypic consequences of chronic absence of let-7afd in pulmonary fibrosis.
Chronic deletion of let-7afd in AT2 cells induces progressive and spontaneous lung fibrosis
At 1-month of iTAM (Supplementary Fig. 4a), both male and female let-7bc2AT2 and let-7afdAT2 mice showed spatially heterogenous disruption of the distal lung architecture marked by enlargement of alveolar spaces and increased presence of alveolar and peribronchial leukocytes and hemosiderin-laden macrophage infiltrates (Fig. 3a–c, Supplementary Fig. 4b, c). Approximately 30% of let-7afdAT2 mice furthermore exhibit heterogenous disorganization of alveoli including septal wall thickening and the appearance of interstitial cells (Fig. 3a, b). The let-7afdAT2 mice additionally show isolated areas of alveolar septal destruction, fibroblastic foci, and collagen deposition primarily at the periphery of the lung (Fig. 3a, b, d, e). A parallel histologic evaluation of let-7bc2AT2 mice at 1-month of iTAM indicates a milder phenotype without lung collagen deposition (Fig. 3a, b, d, e). When the let-7afdAT2 mice were followed for 2-months of iTAM, they exhibit resolution of pulmonary fibrosis but retain enlargement of alveolar spaces vs controls (Supplementary Fig. 4d–g). Pulmonary biomechanics and spirometry measurements further showed that the let-7afdAT2 mice exhibit a transient decrease in compliance and inspiratory capacity after 1-month, but fully recover by 2-months of iTAM (Supplementary Fig. 4h, i).

a, b, f, g Representative Masson’s trichrome-stained sections of lung lobes at 1-month iTAM (a, b) or 6-months (f, g) booster-iTAM. Scale bars: 2 mm upper panels; 50 μm lower panels (n = 16 mice per group). c Differential cell counts from BALF of mice is shown (n = 8, control; n = 8, let-7afdAT2; n = 7, let-7bc2AT2). Macrophages (Mac), Neutrophils (Neu). Ashcroft score was used to evaluate lung injury after 1 month iTAM (d) or 6 months booster iTAM (h). Samples per group: (1-month: n = 5, control; n = 5, let-7afdAT2; n = 4, let-7bc2AT2); (6-months: n = 10, control; n = 10, let-7afdAT2; n = 5, let-7bc2AT2). Data are mean ± s.e.m. P values were determined by Kruskal–Wallis with Dunn’s correction. Hydroxyproline levels from lungs of indicated groups of mice at 1-month iTAM (e) or 6-months booster iTAM (i). 1-month iTAM (n = 4, control; n = 4, let-7bc2AT2; n = 5, let-7afdAT2); 6-months of booster iTAM (4 mice per group). c, e, i Data are mean ± s.e.m. P values were obtained by one-way ANOVA with Tukey’s multiple comparison test. a–i Representative of three independent experiments. Not significant (NS). Source data are provided as a Source Data file.
To determine if continual Cre/loxP deletion of let-7 clusters maintains pulmonary fibrosis over 1-month, let-7bc2AT2 and let-7afdAT2 mice received monthly iTAM boosters for up to 6-months (Supplementary Fig. 5a). With this regimen, the pulmonary inflammation, heterogeneous disruption of alveolar architecture and fibrotic phenotype persisted up to 6-months in >90% of let-7afdAT2 mice (Fig. 3f–i). In contrast, the let-7bc2AT2 mice displayed a milder ILD phenotype relative to let-7afdAT2 mice but without collagen deposition (Fig. 3f–i). Pulmonary biomechanics demonstrated the let-7afdAT2 mice exhibit more robust restrictive impairment in lung function vs controls and let-7bc2AT2 mice (Supplementary Fig. 5b). To discern the transcriptome of the let-7afdAT2 mice, we carried out RNA-seq on whole lungs of mice at 6-months of booster iTAM. Differential gene expression analysis and GSEA indicated significant induction of inflammation and fibrosis pathways in lungs of let-7afdAT2 mice (Supplementary Fig. 5c). Overall, these data establish that let-7afd deletion in AT2 cells models progressive spontaneous pulmonary fibrosis.
Ablation of the let-7afd in AT2 stimulates the persistence of ADI cells
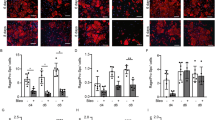
Based on this data, we hypothesized that AT2 cell loss of let-7afd drives the proliferative expansion of fibrogenic AT2 intermediates. We validated the presence of supernumerary AT2 lineage cells in lungs of let-7afdAT2 mice compared to controls after 1-month of iTAM and 5-months of booster iTAM respectively by IF quantification of Sftpc-tdT+ cells (Fig. 4a). Analysis of contiguous lung H&E sections for fibroblastic aggregates and IF detection of KRT8 revealed the spatial co-occurrence of ADIs in damaged areas with mesenchymal cell expansion and alveolar destruction in let-7afdAT2 mice (Fig. 4b). Further quantitative IF analysis confirmed the persistence of KRT8+ ADIs in lung parenchyma of let-7afdAT2 mice at 1- and 5-months of deletion (Fig. 4c, d). Cells expressing KRT8 exhibited distinct cell shape including elongated squamous morphology as recently described for ADIs (Fig. 4b, c)11,12. IF also confirmed increased Sftpc-tdT+ labeled cells co-expressing KRT8 or CLDN4 in let-7afdAT2 mice compared to controls (Fig. 4e, f). Reinforcing our hypothesis, at 6-months of booster iTAM lungs of let-7afdAT2 mice furthermore exhibit prolonged expression of ADI and ADI-7 cell markers (Fig. 4g, h).

a Quantification of Sftpc-tdT+ cells from total DAPI cells in alveolar regions of let-7afdAT2 mice compared to control mice at 1- or 5-months of booster iTAM (n = 3 mice per group). b Representative H&E-stained (upper panels) and immunostaining (lower panels) serial sections of lung after 1 month of iTAM in let-7afdAT2 compared to control mice. IF shows KRT8 (green), SFTPC (red), DAPI (blue). Scale bars: 50 μm. c Representative immunostaining of KRT8 (green), SFTPC (red), DAPI (blue) at 1 month (upper panels) or 5 months of booster iTAM (lower panels) in let-7afdAT2 vs control mice. Arrowheads point to KRT8+SFTPC+ cells. Scale bars: 25 μm. d Quantification of KRT8 MFI from alveolar regions excluding lung airways in panel (c) (n = 3 mice per group). e Representative immunostaining of Sftpc-tdT+ traced KRT8+ (upper panels) or CLDN4+ (lower panels) cells in lungs of let-7afdAT2 mice compared to controls at 1-month post-iTAM. KRT8 (green), CLDN4 (gray), Sftpc-tdT (red), DAPI (blue). Arrowheads point to KRT8+ or CLDN4+ Sftpc-tdT+ cells. Scale bars: 25 μm. f Quantification of Sftpc-tdT+ CLDN4+ from total Sftpc-tdT+ cells at 1-month post-iTAM (n = 3 mice per group). g, h GSEA and heat map plots derived from RNA-seq show significant induction of ADI genes in lungs of let-7afdAT2 vs control mice with 6-months of booster iTAM (n = 4 per group). Adjusted p value < 0.05 vs controls were obtained using a two-sided Wald test and BH correction for multiple testing. a, d, f Data are mean ± s.e.m. P values were determined by unpaired two-tailed Student’s t test. a–f Representative of three independent experiments. Source data are provided as a Source Data file.
Deficiency of let-7afd AT2 cell hypertrophy and stimulates the expression of profibrotic genes
To determine whether deletion of let-7afd promotes AT2 cell hypertrophy and/or other maladaptive ultrastructural cell changes associated with clinical ILD5, we made use of transmission electron microscopy (TEM) in lungs of mice after 3-months of booster iTAM. TEM stereological analysis revealed the let-7afdAT2 mice exhibit enlarged or hypertrophic AT2 cells with increased area relative to controls (Fig. 5a, b). Notably, let-7afd−/− AT2 cells also displayed increased number and total cell area of lamellar bodies (LBs) with disorganized lamellae (Fig. 5a, b). The morphometric measurements of circularity furthermore show cell shape elongation of let-7afd−/− AT2 cells compared to controls (Fig. 5a, b). Remarkably, let-7afdAT2 mice also exhibit binucleated AT2 cells (11.1%, 5 of 45 cells) a unique feature not evidenced in control mice (0%, 0 of 44 cells) (Fig. 5a).

a Representative TEM image shows an enlarged let-7afd−/− AT2 cell with increased abnormal lamellar bodies (LB)s and more than one nucleus (n = 3 mice per group). The white asterisk point to individual nucleus. An elongated let-7afd−/− AT2 cell is shown on right. Scale bars: 2 μm. b TEM quantification of cell area and LB cell area (μm2), LB number per μm2 of cell area, and cell circularity index in each AT2 cell. Each dot represents one AT2 cell (control AT2 cells n = 16; let-7afd−/− AT2 cells = 16, from a total of 3 mice per group. c Functional network of let-7 targets associated with PI3K/AKT/MTOR and/or EMT. Network adjusted q value was computed by EnrichmentMap in Cytoscape. d Expression levels of let-7 hub targets were evaluated by qPCR after 2 months booster-iTAM from let-7afd−/− and control sorted Sftpc-tdT+ cells (n = 4 samples per group from pools of 2 mice). e Let-7 target gene expression was determined by RNA-seq in lungs of let-7afdAT2 and control mice after 6-months of booster iTAM (n = 4 mice per group). Data are mean ± s.e.m. is plotted relative to control samples set to a mean of 1.0. Adjusted p values were derived by two-sided Wald test and BH correction for multiple testing with BaseSpace workflow. ****q < 0.0001, **q < 0.01 correspond to: Bach1 (0.000245), E2f1 (0.00692), Hif1a (1.2E-08), and Myc (0.000000278). Not significant (NS). f Representative IF images show increased numbers of BACH1+ (top panels), EZH2 (middle panels), and MYC+ (bottom panels) Sftpc-tdT+ cells respectively (indicated by yellow arrows) in lungs of let-7afdAT2 vs control mice lungs after 1-month of iTAM. BACH1 (gray); EZH2 (gray); MYC (gray), Sftpc-tdT (red); AGER (green); DAPI (blue). Scale bars: 25 μm. Quantification of BACH1+ (g), EZH2+ (h), and MYC+ (i) Sftpc-tdT+ cells in total Sftpc-tdT+ cells (n = 3 mice per group). b, d, g–i Data are mean ± s.e.m. P values were obtained by unpaired two-tailed Student’s t test. d, f–i Representative of three independent experiments. Source data are provided as a Source Data file.
We hypothesized that let-7 with its vast OGRN targets and impact on cell size and shape might orchestrate fibrogenesis upstream of PI3K/AKT/MTOR and ECM/EMT pathways. Correspondingly, analysis of our RNA-seq datasets revealed that absence of let-7afd activates these profibrotic pathways in AT2 cells (Supplementary Fig. 6a, b, Supplementary Data 3). Integrative pathway annotation of the let-7 AT2 cell targetome highlights a functionally interconnected protein interactome network, consisting of over 50 gene targets of let-7 directly or indirectly involved in PI3K/AKT/MTOR and EMT pathways (Fig. 5c). Notably, this interactome includes genes associated with fibrosis, such as TFs (Myc)24, receptors (Itgb3, Plaur), ligands (Ctgf, Col5a2), enzymes (Nras, Kras) and cytoskeletal proteins (Tpm1) (Fig. 5c, Supplementary Fig. 6c–e, Supplementary Data 1–3). Most OGRN genes (e.g., Bach1, Ezh2) were included in the network given their inductive roles in tissue fibrosis22,23 and engagement of PI3K/AKT/MTOR33,35 and EMT21,36 pathways respectively (Fig. 5c). To further explore the potential contribution of OGRN in pulmonary fibrosis, we sought to determine whether they remained significantly induced in let-7afdAT2 mice with fibrosis. Consistent with our acute phenotypic analysis, transcript levels of most OGRN genes remained upregulated in sorted AT2 cells and lungs of let-7afdAT2 mice with fibrosis over controls (Fig. 5d, e). Furthermore, let-7afdAT2 mice with fibrosis exhibit increased numbers of BACH1+, EZH2+ or MYC+ Sftpc-tdT traced cells than controls (Fig. 5f–i, Supplementary Fig. 6f–h). These findings suggests that absence of let-7afd drives unchecked expression of OGRN at the top of the hierarchy and ECM structural proteins downstream.
Genetic loss of let-7afd makes AT2 cells vulnerable to DNA damage, senescence, and apoptosis and impedes AT2 to AT1 terminal differentiation
Studies have shown that in different models of lung injury and fibrosis, AT2 cells can undergo cell senescence and/or apoptosis related to disruptions in quality control programs5. Furthermore, ADI cells are inherently prone to DNA damage and cell senescence during normal injury repair and in IPF11. Additionally, unchecked expression of BACH1, EZH2, or MYC in non-cancer cells can trigger DNA damage, senescence, and/or apoptosis. We examined lungs of mice with γH2AX as a marker of DNA damage in conjunction with the β-galactosidase activity assay for cell senescence. We also assessed for apoptotic AT2 cells in lungs of mice by IF detection of active-CASP3. At 1-month of deletion, lung immunostaining showed significant accumulation and frequency of γH2AX+SFTPC+ and active-CASP3+Sftpc-tdT+ labeled AT2s in let-7afdAT2 mice compared to controls (Fig. 6a–c). Correspondingly, let-7afdAT2 mice showed robust accumulation of β-galactosidase substrate X-gal in lung parenchyma, including AT2s indicating induction of a widespread cellular senescence program (Fig. 6d). Congruent with these findings, sorted Sftpc-tdT cells and whole lungs of let-7afdAT2 mice exhibit induction of DNA repair, senescence, senescence-associated secretory phenotype (SASP), and apoptotic genes than controls (Fig. 6e, f, Supplementary Fig. 7a–c). Annotation of the let-7 targetome identified Casp3 and Serpine1 as potentially important mediators of these cell processes (Supplementary Fig. 7d, e, Supplementary Data 1–3).

a Immunostaining for γH2AX (green), SFTPC (red), DAPI (blue) upper panels; active CASP3 (green), SFTPC (red), AGER (gray), DAPI (blue) second row panels; HOPX (green), Sftpc-tdT (red), DAPI (blue) third row panels; AGER (green), Sftpc-tdT (red), DAPI (blue) lower panels in lungs of let-7afdAT2 and control mice after 1 month of iTAM. Arrows indicate γH2AX+SFTPC+ cells, active-CASP3+SFTPC+ cells, HOPX+Sftpc-tdT+, and AGER+Sftpc-tdT+cells respectively. Scale bars: 25 μm. Quantification of γH2AX+SFTPC+ cells (b) or active-CASP3+ SFTPC+ cells (c) from total SFTPC+ cells (n = 3 mice per group). d Representative β-galactosidase staining in lungs of let-7afdAT2 and control mice after 1-month of iTAM. Arrowheads point to AT2 cells in thickened alveolar septa (n = 6 mice per group). Scale bars: 50 μm. Indicated genes were detected by qPCR from let-7afd−/− and control Sftpc-tdT+ cells after 14-days of iTAM (e) or 2-months (f) of booster iTAM (n = 4 samples per group from pools of 2 mice). g Quantification of HOPX+Sftpc-tdT+ cells from total Sftpc-tdT+ cells in lungs of let-7afdAT2 and control mice at 1-month after iTAM (n = 3 mice per group). b, c, e–g Data are mean ± s.e.m. P values were determined by unpaired two-tailed Student’s t test. a–g Representative of three independent experiments. Source data are provided as a Source Data file.
We posited that the persistence of ADIs in let-7afdAT2 mice might relate to impaired AT1 trans-differentiation. Thus, we tested whether let-7afd is necessary for differentiation of AT2s into AT1s with lung IF with AT1 cell markers AGER and HOPX. The frequency of Sftpc-tdT+ cells co-expressing AT1 markers increased in let-7afdAT2 mice vs controls after 1-month of deletion as an indication of AT1-transdifferentiation (Fig. 6a, g). However, consistent with the alveolar destruction and net loss of AT1 cells, AT1 identity markers were lower in lungs of let-7afdAT2 mice than controls (Supplementary Fig. 7f, g). Club epithelial cell markers were also downregulated suggesting that let-7afd deletion also promotes injury in the airway (Supplementary Fig 7f, g).
To ascertain whether loss of let-7afd blunts AT2 to AT1 cell differentiation in an intrinsic manner, we compared the formation of ADIs and AT1 differentiation by IF in control and let-7afd−/− AT2 cell organoids grown in AT1 cell differentiation medium (ADM) (Fig. 7a). On day 7 of ADM culture, CLDN4+ and KRT8+ cells and expression levels were enhanced significantly in let-7afd−/− organoids over controls which corroborates the increased ADIs in lungs of let-7afdAT2 mice (Fig. 7b, c, Supplementary Fig. 8a, b). In further support of abnormal AT1 trans-differentiation, let-7afd−/− organoids exhibited reduced AGER+, HOPX+, AQP5+ cells and expression in contrast with controls (Fig. 7d–f, Supplementary Fig. 8c–e). Of note, let-7afd−/− organoids also exhibit reduced expression and fewer LGALS3+ (a late ADI marker) cells vs controls which highlight a late block in AT1 differentiation (Fig. 7d, g, Supplementary Fig. 8f). By comparison, SFTPC+ cells and levels were enhanced in let-7afd−/− organoids suggesting that absence of let-7 promotes not only the accumulation of ADIs but also reinforces retention of AT2 identity (Fig. 7d, h, Supplementary Fig. 8g). To ascertain whether let-7afd depletion promotes DNA damage or death in differentiation impaired let-7afd−/− organoids, we examined levels of γH2AX and active CASP3 by IF. The number of γH2AX+ and active CASP3+ cells were increased in let-7afd−/− AT2 cultures compared with controls (Supplementary Fig. 8e, h, i). Thus, let-7afd serves as a pivotal brake to the formation of lung injury inducing ADI cells, which are stalled in AT1 cell differentiation. Furthermore, loss of let-7afd renders AT2 cells prone to senescence and apoptosis in a cell intrinsic manner which could aggravate pulmonary fibrosis37.

a Schematic representation of experimental design to examine the role of let-7afd in AT2 to AT1 differentiation in cultured AT2 organoids. Created in BioRender. King, K. (2025) https://BioRender.com/x04u478. b, d Representative immunostaining for ADI, AT1 markers and BACH1, EZH2 expression in let-7afd−/− vs control AT2 organoids grown as in panel (a). (n = 3 mice per group). Panel (b) shows KRT8 (green), DAPI (blue). Panel (d) shows AGER (green), SFTPC (red), DAPI (blue) top row panels; HOPX (green); LGALS3 (red); DAPI (blue) second row panels; BACH1 (red), KRT8 (green), DAPI (blue) third row panels; EZH2 (red), KRT8 (green), DAPI (blue) bottom row panels. Scale bars: 25 μm. Quantification of KRT8+ (c), AGER+ (e), HOPX+ (f), LGALS+ (g), SFTPC+ (h), BACH1+ (i), or EZH2+ (j) cells respectively in total DAPI cells (n = 3 mice per group). Data are mean ± s.e.m. P values were determined by unpaired two-tailed Student’s t test. Each circle represents a mouse. Representative of three independent experiments. k Line plots show smoothed relative expression of selected let-7 hub targets in the ADI to AT1 trajectory defined in published dataset12. Expression values were estimated by fitting a generalized additive model across pseudotime trajectory12. KRT8 peak is represented by a dashed line. Source data are provided as a Source Data file.
To explore potential mechanisms of impaired AT2 to AT1 trans-differentiation of let-7afd−/− AT2 cultures, we examined BACH1 and EZH2 expression by IF in ADM cultures. Interestingly, let-7afd−/− cultured AT2 organoids exhibited higher levels and frequency of BACH1+ and EZH2+ cells as compared to controls (Fig. 7d, i, j, Supplementary Fig. 8j, k). Both BACH1 and EZH2 have been associated with impaired epithelial cell differentiation upon overexpression23,38. Analysis of bleomycin-induced injury scRNA-seq dataset12 revealed a gradual decrease in dynamic expression BACH1/EZH2 and targetome as ADIs move in the direction of AT1 cell differentiation (Fig. 7k, Supplementary Fig. 8l). Taken together these analyses lend support that loss of let-7afd hampers AT2 cell “bridging” into AT1 cells, in part, via ectopic expression of BACH1/EZH2.
Let-7afd orchestrates AT2 profibrotic dysfunction epigenetically via acetylation and methylation of histone H3 on lysine 27 (H3K27)
Pathologic overexpression of EZH2, BACH1, or MYC has been associated with epigenetic induction and repression of genes involved in cell state transitions and cell identity at the level of acetylation39,40,41 and methylation31,42 of H3K27 and other histones. We therefore hypothesized that deletion of let-7 reprograms fibrogenic AT2s by promoting the deposition of H3K27ac on previously silent enhancers resulting in activation of cell growth and ADI genes, while simultaneously repressing key genes via H3K27me3 deposition.
To test this, we conducted cleavage under targets and release using nuclease (CUT&RUN)-DNA sequencing on chromatin to identify differential H3K27ac and H3K27me3 modifications in flow sorted let-7afd−/− compared to control Sftpc-tdT+ cells on day 6 after iTAM treatment (Fig. 8a, Supplementary Fig. 9a, Supplementary Data 5, 6). In let-7afd−/− AT2 cells, we observed increased H3K27ac peaks relative to control cells, which showed significant enrichment of cell cycle genes (Fig. 8b, Supplementary Data 7). Conversely, increased H3K27me3 marks in let-7afd−/− AT2 cells localized to development-associated genes (Supplementary Fig. 9b, Supplementary Data 7). To identify potential TF regulatory networks, we performed motif analysis via HOMER. Several TF motifs, including BACH1, were significantly associated with H3K27ac modifications (Fig. 8c, Supplementary Data 8).

a Experimental design scheme to carry out CUT&RUN-seq from sorted Sftpc-tdT+ cells obtained from let-7afdAT2 or control mice (n = 2 mice per group). GO BP (b) and HOMER transcription factor motif (c) enrichment of enriched H3K27ac peaks in let-7afd−/− compared to control Sftpc-tdT+ cells. Adjusted p values were derived by hypergeometric testing and BH correction for multiple testing by HOMER (b) or GREAT web-based tool (c)65. d Integration analysis of CUT&RUN-seq with RNA-seq in let-7afd−/− vs control Sftpc-tdT+ cells show correlation with transcription. Gene totals of congruent-expression change directionality (blue+red) and incongruent directionality (gray) are shown for H3K27ac and H3K27me3. Pie chart and ORA gene enrichment analysis indicate a significant fraction of upregulated let-7 targetome genes exhibit H3K27ac marks suggestive of increased enhancer activity in let-7afd−/− vs control Sftpc-tdT+ cells. P value was calculated with a one-tailed hypergeometric test. e–g UCSC tracks show H3K27ac and H3K27me3 peaks in selected genes in let-7afd−/− Sftpc-tdT+ cells compared to controls. Enriched H3K27ac peaks in let-7afd−/− Sftpc-tdT+ cells vs controls are shaded in red. The enriched H3K27me peaks in let-7afd−/− Sftpc-tdT+ cells are highlighted in blue. ENCODE candidate cis-regulatory elements (cCREs). h Representative immunostaining of H3K27me3+ cells in let-7afd−/− vs control AT2 organoids in ADM (n = 3 per group). H3K27me3 (red), KRT8 (green), DAPI (blue). Scale bars: 25 μm. i Quantification of H3K27me3+ nuclear MFI expression. Data are mean ± s.e.m. Each dot represents one cell (n = 149 cells per genotype, 3 mice per group). Data are mean ± s.e.m. P value was determined by an unpaired two-tailed Student’s t test. Representative of three independent experiments. Source data are provided as a Source Data file.
To focus on differentially induced and repressed genes where H3K27 marks have a functional impact, we integrated the CUT&RUN-seq analyses with our bulk transcriptomic dataset from sorted let-7afd−/− vs control AT2 cells (Fig. 8d, Supplementary 9c, Supplementary Data 9, 10). This analysis revealed a positive correlation between enhanced H3K27ac modifications and let-7afd-dependent gene activation in let-7afd−/− AT2 cells vs controls (Fig. 8d). Notably, H3K27ac modifications were positively correlated with the let-7 targetome including Bach1, Ezh2, Hif1a, and Myc (Fig. 8d, e, Supplementary Fig. 9d, Supplementary Data 5, 9). Additionally, canonical ADI cell markers such as Cldn4 displayed increased H3K27ac modifications in let-7afd−/− AT2 cells vs controls (Fig. 8f, Supplementary Data 5, 9). Interestingly, deletion of let-7afd also resulted in combined H3K27ac and H3K27me3 co-modifications in Sftpc and Bmp1 which may indicate expression fine-tuning via bivalent chromatin (Fig. 8g, Supplementary Data 5, 6, 10). Of note, the antifibrotic and cell growth suppressor target of EZH2, Bambi43, and other important genes exhibit increased H3K27me3 deposition and reduced expression in let-7afd−/− AT2 cells compared to controls (Fig. 8g, Supplementary Fig. 9c, e, f, Supplementary Data 6, 10). To investigate whether the let-7afd deletion promotes H3K27me3, we examined subcellular localization by IF in cultured AT2 organoids. H3K27me3 nuclear expression levels were significantly enhanced in let-7afd−/− organoids over controls (Fig. 8h, i). In conclusion, these epigenomic data highlight the integral role of let-7afd chromatin remodeling, where aberrant enhancer H3K27ac activity and repressive H3k27me3 modifications lead to uncontrolled transcription of cell growth and ADI marker genes.
Human Aberrant Basaloid (AB) cells from IPF patients exhibit enrichment of the let-7 interactome
We speculated that reduced let-7 activity in lung epithelial cells of IPF patients might contribute to accumulation of AB cells. To investigate this hypothesis, we analyzed a published single-cell RNA sequencing dataset from human IPF lungs14. Few targets of let-7 were upregulated in IPF AT2 relative to control AT2 cells (Fig. 9a, b, Supplementary Data 11). On the other hand, AB cells exhibited significant enrichment of the murine let-7 interactome relative to AT1 and AT2 cells from IPF patients (Fig. 9a, b, Supplementary Data 11). Furthermore, upregulated targets of let-7 in AB cells compared to control AT2s figure prominently in fibrosis pathways including ECM and EMT (Fig. 9c, Supplementary Data 12). Relative to AT1 and AT2 cells, AB cells furthermore exhibit enrichment of EMT genes (Fig. 9d). The let-7 OGRN genes, ARID3A, BACH1, EZH2, NRAS were significantly upregulated in AB cells compared to control AT2 cells while FOXP2 and MYC were repressed (Fig. 9e and Supplementary Data 11). We also evaluated for consensus target signatures of EZH2 and BACH1 in IPF. This analysis found that AB cells show increased EZH2 and BACH1 target signatures compared to IPF AT2 cells (Fig. 9f, g). Our analyses suggest the let-7 OGRN axis might contribute to the formation of AB cells in IPF; however further studies will be required for more direct evidence.

a UCell Enrichment scores for the murine let-7 targetome in AT2, AB, and AT1 cells from control (Ctrl) or IPF lungs. b Broken circle plots indicate the total number and proportions of let-7 targets which are significantly upregulated (UP), downregulated (DOWN), or non-significant (NS) between Ctrl and IPF cells. Not present (n.p.) indicates gene targets missing in published dataset GSE13683112. c Gene process enrichment analysis for upregulated let-7 targets in AB cells compared to control AT2 cells was computed with a one-tailed hypergeometric test and FDR correction using Metascape60. d The Hallmark EMT pathway enrichment scores in control vs IPF AT2, AB and AT1 cell samples. e Dot plots show the relative expression of selected let-7 targets and identity markers in basal, AT2, AB, and AT1 cells from control and IPF patients. The size of each dot represents the percentage of cells in the cluster expressing the gene, while the color represents the mean expression of gene in cell cluster. f, g Consensus targets of BACH1 and EZH2 were evaluated for enrichment in AT2, AB and AT1 cells in control and IPF samples. High confidence targets (HCT)s. a, d, f, g Data are mean ± s.e.m half violin plots for cell values. P values were obtained with a one-way ANOVA with Sidak post-hoc test (AT2 Ctrl, n = 2655; AT2 IPF, n = 496; AB, n = 448; AT1 Ctrl, n = 502; AT1 IPF, n = 176). Expression data was derived from published scRNA-seq dataset GSE13683114. Source data are provided as a Source Data file.
Discussion
Our study lends support to the importance of let-7 miRNA in AT2s as a molecular gatekeeper in lung homeostasis, injury, and healing. We identified transient and coordinated downregulation of let-7 family after bleomycin-induced lung injury, indicating a tightly regulated post-transcriptional response which paves the way for the emergence of repair-associated AT2s. This interpretation aligns with our studies indicating that AT2-cell specific genetic ablation of let-7afd removes a “brake” on AT2 activation resulting in uncontrolled cell growth and accumulation of ADIs in pulmonary hemorrhage which precedes a temporary chronic fibrotic remodeling phase. Additionally, we discovered that let-7 not only restricts AT2 cells from transitioning into ADI intermediates but it is also crucial for exiting this state. Interestingly, with a booster Cre/loxP deletion protocol, we observed that ADIs and pulmonary fibrosis were maintained in let-7afdAT2 mice. We speculate that extensive cell death and senescence of let-7afd−/− AT2s may secondarily recruit airway epithelial PSCs12,44,45,46 which can repair the parenchyma.
The let-7 miRNA family exerts broad cellular effects by silencing hundreds of target mRNAs in GRNs20. Here we demonstrate that let-7 simultaneously regulates an extensive GRN in AT2 cells, including oncogenes such as BACH1, EZH2, and MYC previously linked to uncontrolled cell growth and fibrosis22,23,24,33,47. Our data favors that this OGRN is persistently upregulated in AT2 cells due to impaired let-7 post-transcriptional regulation which would support progressive pulmonary fibrosis disease. We note that the let-7 OGRN likely functions as a powerful coherent or synergistic feedforward-loop because these genes converge on similar phenotypic disturbances in diseased epithelial cells. Impressively, OGRN let-7 targets are enriched in cycling and pAT2 and ADIs but their expression decreases as cells progress toward AT1 identity. With permanent loss of let-7afd, our data indicates that AT2 cell identity is held within ADI state, where the let-7 OGRN genes are most highly expressed.
The leading-edge targets of let-7, such as EZH2, BACH1, MYC, are associated with epithelial stem cell growth, transformation, and enhanced metastatic potential involving processes which include EMT30,32,36. Moreover, published studies on let-7 have shown that its loss stimulates cell growth and EMT programs, while its overexpression limits these processes20,21. Intriguingly, BACH1, EZH2, and MYC have also been associated with partial EMT in cancer, where epithelial cells do not fully transition into mesenchymal cells but exhibit a continuum of epithelial and mesenchymal traits which potentiate metastasis48. Lineage tracing and scRNA-seq studies indicate that AT2 transitional cells do not transform into fibroblasts via EMT10,11,12,13; instead, they adopt a mixed epithelial and mesenchymal gene expression program. Recent studies showed that the accumulation of AT2 transitional cells can drive lung injury and ILD11,12,13. Given these observations, we propose a model where loss of let-7afd contributes to partial EMT in transitional AT2s via the OGRN.
Prior studies have highlighted the importance of BACH1, EZH2, and MYC on heritable chromatin remodeling leading to phenotypic changes in cells39,40,41. Overexpression of BACH1, EZH2, or MYC promote histone modifications including H3K27ac at active enhancers to facilitate oncogenic cell signaling39,40,41. Furthermore, recent studies have shown that BACH1 and EZH2 can promote H3K27me3 epigenetic silencing cooperatively in cells42,49. According to our data, the loss of let-7afd enhances H3K27ac modifications which may lead to opening of chromatin and increased accessibility of pioneer TFs to the enhancers of let-7 targetome, thereby boosting not only mRNA and protein production post-transcriptionally but also transcription output. This likely amplifies the positive feedback loop with Bach1, Ezh2, Myc themselves acting as coherent chromatin remodeling TF drivers in the establishment of a profibrotic AT2 epigenetic landscape. However, the specific roles of BACH1, EZH2 and other OGRN targets as necessary and/or sufficient mediators in the persistence of ADIs and pulmonary fibrosis remain largely undetermined. Nonetheless, recent research on Ezh2 has shown that its deletion hampers the growth of lung epithelial cells and reduces the emergence of cycling epithelial cells and KRT8+ transitional epithelial cells in murine organoids50.
Our genetic studies to reduce let-7 activity in AT2 cells relied primarily on phenotypic studies of let-7afd but did not provide a mechanistic perspective on the contribution of let-7bc2 and we did not examine the role of other clusters in AT2 cells. Another limitation of our study is that although we demonstrated dynamic changes in AT2 cells upon loss of let-7afd by integrated approaches, we did not achieve transcriptomic and epigenomic depth at a single cell resolution which would have allowed more insight on the role of let-7 on AT2 cell plasticity and alveolar cell niche.
In conclusion, our study provides insights into let-7 as a braking mechanism to oncogene dysregulation in AT2 cell-driven pulmonary fibrosis. Our observation that AB cells from IPF patients express higher levels of the let-7 targetome offers the enticing possibility that targeted delivery of let-7 could be used as a treatment. We point that several reports indicate the effectiveness of targeted let-7 delivery in mitigating fibrosis51,52, and pharmacologic targeting of EZH2 is employed against both cancer and fibrosis53,54.
Methods
Mice and tamoxifen administration
Animals were housed in specific pathogen-free environment with access to standard chow (PicoLab Select Rodent 50 IF/6F 5V5R) and water ad libitum, with a constant temperature (21–24 °C) and humidity of (40-60%) on a 14-h/10h-light/dark cycle. Mice of both sexes were used for experiments. We recently described the creation of the C57BL/6 isogenic let-7bc2fl/fl or let-7afdfl/fl mice26. Mice were crossed to Sftpctm1(cre/ERT)Blh (Sftpc-CreERT2), Rosa26R-CAG-LSL-tdTomato (R26tdT) purchased from JAX and genotyped by PCR. For all phenotypic studies, mice aged 8–12 weeks old were given an initial iTAM regimen defined as 4 doses of 160 mg/kg tamoxifen (Sigma, T5649) dissolved in corn oil (Sigma, C8267) by intraperitoneal injections (i.p.) every other day. In some chronic phenotypic experiments, mice received iTAM boosters following four weeks after the initial iTAM. The boosters consisted of 4 doses of 80 mg/kg iTAM every other day and these were administered monthly. Let-7bc2f/f, let-7afdf/f or wild-type (WT) Sftpc-tdT mice were used as controls and treated with iTAM in parallel with experimental mice. Mice were genotyped by PCR from ear clippings with published primers26. The mice were euthanized via i.p. administration of a commercial euthanasia solution followed by bilateral opening of thorax. All studies with mice were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee and followed the National Research Council Guide for the Care and Use of Laboratory Animals.
Physiologic measurements
Arterial oxygen saturation (SpO2) measurements were collected using the STARR Life Sciences MouseOx Plus (Oakmont, Pennsylvania) on awake mice with a neck cuff sensor to obtain five readings over a 30 min interval. The mice were shaved on the neck and allowed 45 min to acclimate without restrains with a neck cuff sensor prior to good quality recordings. The lung respiratory biomechanics values were obtained with the flexiVent (Scireq) system55. Mice undergoing flexiVent procedure were subjected to anesthesia, followed by tracheostomy and cannulation with an 18 G metal cannula. The mice were placed under mechanical ventilation at a respiratory rate of 150 breaths/min, a tidal volume of 10 mL/kg and a PEEP set at 3cmH2O in enclosed chamber for pulmonary function measurements following manufacturer guidelines. The average of three measurements with a coefficient of determination ≥0.95 were calculated for each mouse.
Lung histology and histomorphometry analysis
Mouse lungs were perfused with sterile PBS via right ventricle cardiac perfusion. For formalin-fixed paraffin embedding tissues, lungs were inflated with instillation of 4% paraformaldehyde (PFA) via a tracheal cannula at 25-cm H2O pressure for 15 min and then tied by nylon suture. The lungs were then transferred into 50 mL polypropylene tube containing PFA for overnight fixation at 4 °C. Then lung lobes were separated, transferred into tissue cassettes, and then immersed in 70% ethanol at 4 °C. Samples were provided to the Human Tissue Acquisition and Pathology (HTAP) Core at BCM for paraffin embedding, cutting of 5μM thick sections, and staining by H&E or Masson’s trichrome. The PFA-fixed lung cryosections were obtained in similar manner, but after the overnight fixation step in PFA at 4 °C, the lungs were equilibrated with 30% sucrose for 1-2 days, prior to embedding in OCT. Samples were then cut into 7–10 uM thick sections for the analysis. β-galactosidase (X-gal) lung staining was performed on frozen lung sections using the Senescence Cells Histochemical Staining Kit (Sigma, CS0030) following manufacturer instructions. Hydroxyproline Colorimetric Assay Kit (Abcam, ab222941) was used to measure collagen in lung lobes following kit instructions and optical density was measured using a Varioskan LUX plate reader (Thermo Fisher Scientific, VL0000D0).
Bronchoalveolar fluid (BALF) lavage and Prussian blue staining
Bronchoalveolar lavage for measurements of total and differential cell counts was done as we described26. Prussian Blue staining of BALF cytospins was performed using the Iron Stain Kit (Sigma, HT20-1KT) following kit instructions.
Immunostaining
For immunostaining of paraffin embedded lung tissues or organoid sections, antigen retrieval was performed with 1X BioCare Diva Decloaker RTU (BioCare, DV2004G1) buffer in a Decloaking Chamber NxGen (BioCare, DC2012) at 95 °C or 110 °C for 15 min. Slides were then allowed to cool in antigen retrieval buffer for 10 min before washing in distilled water. Both paraffin embedded and thawed cryo-sectioned slides were permeabilized with 0.3% Triton X-100 in 1% Normal Goat Serum (Abcam, ab7481) for 10 min. Antigen blocking was performed in 5% Normal Goat Serum for 1 h. Samples were then incubated overnight at 4 °C with the following primary antibodies diluted in 1% Normal Goat Serum: Rat anti-Ki67 conjugated FITC (1:50, Invitrogen, 11-5698-82), Mouse anti-active/pro-caspase3 (1:20, Invitrogen, MA1-91637), Mouse anti-EZH2 (1:100, Invitrogen, 14-9867-82), Rabbit anti-EZH2 (1:200, Cell Signaling, 5246), Rabbit anti-BACH1 (1:200, Novus Biological, NBP2-55113), Mouse anti-γH2AX (1:200, Novus Biological, NB100-74435), Rabbit anti-CLDN4 (1:200, Invitrogen, 36-4800), Rabbit anti-pro-SFTPC (1:200, Millipore, AB3786), Rabbit anti-RFP/tdTomato (1:200, Rockland, 600-401-379), Rat anti-Krt8/TROMA-I (1:20, DSHB, TROMA-I-s), Rat anti-Galectin-3/LGALS3 (1:500, Cedarlane, CL8942AP), Rat anti-RAGE/AGER (1:100, R&D Systems, MAB1179-100), Rabbit anti-H3K27me (1:200, Invitrogen, MA5-11198), Rabbit anti-Aqp5 (1:100, Invitrogen, PA5-36529), and Mouse anti-HopX (1:200, Santa Cruz, sc-398703). The next day, samples were incubated for 1 h at room temperature in the following secondary antibodies diluted in 1% Normal Goat Serum with DAPI (1:10000, Sigma, D9542): Alexa Fluor 488 goat anti-mouse (1:2000, Thermo Fisher Scientific, A11029), Alexa Fluor 488 goat anti-Rabbit (1:2000, Thermo Fisher Scientific, A-32731), Alexa Fluor 488 goat anti-Rat (1:2000, Thermo Fisher Scientific, A11006), Alexa Fluor 555 goat anti-Mouse (1:2000, Thermo Fisher Scientific, A32727), Alexa Fluor 555 goat anti-Rabbit (1:2000, Thermo Fisher Scientific, A21428), Alexa Fluor 555 goat anti-Rat (1:2000, Thermo Fisher Scientific, A21434), Alexa Fluor 647 goat anti-Rabbit (1:2000, Thermo Fisher Scientific, A21244), Alexa Fluor 647 goat anti-Rat (1:2000, Thermo Fisher Scientific, A21247), Alexa Fluor 790 Goat anti-Rabbit (1:2000, Thermo Fisher Scientific, A11369). In some cases, paraffin embedded lung sections were treated after secondary staining with TrueBlack Lipofuscin Autofluorescence Quencher (Biotium, 23007) for 1 min following product instructions to block autofluorescence. Slides were mounted with ProLong Glass Antifade Mountant (Thermo Fisher Scientific, P36980).
Lung cell suspensions and purification of AT2 cells
Isolation of Sftpc-tdT+ lineage derived AT2s was performed as previously described with minor modifications27,56. Briefly, mouse lungs were perfused with cold PBS via right ventricle of the heart and then inflated with 1 mL digestion solution containing 5 units/mL dispase (Corning, 354235), 450 units/mL collagenase type I (Gibco, 17100-017), 0.33 units/mL DNase I (Sigma, 10104159001), and 1X antibiotic-antimycotic (Thermo Fisher Scientific, A5955-100ML) in Advanced DMEM/F-12 (Thermo Fisher Scientific, 12634028). Lung lobes were then minced into 1-2 mm3 pieces, transferred into 4 mL digestion solution, and incubated at 37 °C with rotation for 40 min. Samples were dissociated by vigorously mixing by P1000 pipette tip until no large pieces remained. Samples were filtered through a 70 µm strainer, pelleted, and then resuspended in 1 mL RBC Lysis Buffer (Invitrogen, 00-4333-57) for 1-min before washing with 5 mL Advanced DMEM/F-12 with 1X antibiotic-antimycotic. Lung cell suspensions were stained with Live/Dead Fixable Blue stain (Invitrogen, L34962), Alexa Fluor 700 anti-mouse CD45.2 (Biolegend, 109822), Brilliant Violet 711 anti-mouse EpCam (Biolegend, 118233), Alexa Fluor 700 anti-mouse CD31 (Biolegend, 102444), Brilliant Violet 421 anti-mouse Pdgfrα (Biolegend, 135923) antibodies and incubated for 30 min in dark on ice. Cells were suspended in ice cold 1X PBS with 2% FBS and filtered through a 40 µm strainer followed by sorting of AT2 cells with Sftpc-tdT tracer on a FACSAria (BD Biosciences). The purity of sorted cells was >97% and it was confirmed via immunostaining with SFTPC (Supplementary Fig. 10). In initial AT2 organoid culture experiments, lung cell suspensions were incubated on antibody-coated dishes at 37 °C with 5% CO2 for 45 min to remove adherent cells and immune cells as an intermediate step prior to flow sorting of AT2 cells as detailed above. This intermediate step was done by coating 100 mm tissue culture dishes with 42 µg anti-Mouse CD45 (BioLegend, 103101) and 16 µg anti-Mouse CD32 (BioLegend, 156402) in 7 mL PBS for 24–48 h at 4 °C as previously described56.
RNA extraction and qPCR
Cell pellets or lung lobes from individual mice were snap frozen in 350 µl of 1:100 β-mercaptoethanol in the RLT Lysis Buffer from the RNAeasy mini plus Kit (Qiagen, 74134). For extraction of RNA in bulk RNA-seq lineage labeled Sftpc-tdT+ AT2 cells (~700,000) were pooled from three mice per genotype by flow sorting as indicated above. Cell samples were thawed and vortexed while lung samples were homogenized for total RNA extraction intended for qPCR or bulk RNA-seq. The RNAeasy mini plus Kit (Qiagen, 74134) was used for removal of genomic DNA and RNA isolation. Quality control was performed with NanoDrop 2000C Spectrophotometer (Thermo Fisher Scientific). Equal amount of input RNA was used for preparation of cDNA with the High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Thermo Fisher Scientific, 4374966). Pre-designed Taqman probes were used with TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific, 4444557). Gapdh served to normalize gene expression by ΔΔCT method. The qPCR was conducted and analyzed on ViiA 7 System (Applied Biosystems). The following Taqman probes were used: Bach1 (Mm01344527_m1), Casp3 (Mm01195085_m1), Cdkn2a (Mm00494449_m1), E2f1 (Mm00432939_m1), Ezh2 (Mm00468464_m1), Kras (Mm00517492_m1), Myc (Mm00487804_m1), Nras (Mm03053787_s1), Trp53 (Mm01731287_m1), Gapdh (Mm99999915_g1). The validated and published pri-let-7b/c2 and pri-let-7a1/d/f1 transcript Taqman probes26 were custom designed by Applied Biosystems (44411114, areptx2, arfvmhy).
AT2 3D organoid cultures and differentiation
Culture of 3D organoids was carried out as previously described with minor modifications27,57. Lineage labeled Sftpc-tdT+ AT2 cells were purified from 8 to 10-week-old mice on day 6-day after iTAM treatment. AT2 cells were resuspended in thawed Cultrex UltiMatrix Matrigel (Biotechne, BME001-05) at 1,000 cells/µL. For each biological replicate, organoids were initiated in each well of 6-well plate into five 20 µl domes containing ~20,000 cells and incubated at 37 °C with 5% CO2 for 20 min without media. When domes hardened, 2 mL of alveolar maintenance media (AMM) without Interleukin-1β was added to each well. The organoids were fed AMM without Interleukin-1β every other day and grown for 10–14-days for subsequent analysis. For RNA-seq, the domes were washed with 1 mL sterile PBS and then incubated in 1 mL Cultrex Organoid Harvesting Solution (Biotechne, 3700-100-01) on ice for 1 h. Domes were gently detached from 6-well plates by spatula, resuspended in 10 mL PBS, pooled for each biological replicate, and then pelleted at 300 × g for 10 min at 4 °C. Cells were washed and pelleted several times with 10 mL sterile PBS to remove Matrigel. In AT2 to AT1 differentiation experiments, organoids were switched from AMM into AT2-Differentiation medium (ADM) on day 1027,57. ADM media composition: 5 ng/mL Human EGF (Gibco, PHG0313), 1 ng/mL Mouse FGF10 (R&D Systems, 6224-FG), 5 µg/mL Heparin (Sigma-Aldrich, H3149), 1x B-27-supplement (Thermo Fisher Scientific, 17504044), 1x Antibiotic-Antimycotic (Thermo Fisher Scientific, A5955), 1x Glutamax (Thermo Fisher Scientific, 35050061), 1.25 mM N-Acetyl-L-Cysteine, 10% Fetal Bovine Serum, in Advanced DMEM/F-12. The cells were then incubated for 7-days in ADM to promote AT1 differentiation27,57. For immunostaining analysis, the AT2 organoids were fixed, sectioned, and stained as described above. Organoid CFE and alveolosphere size diameters were quantified using FIJI (ImageJ, v2.14.0).
Bulk RNA-seq processing, pathway enrichment, and regulatory network analysis
Purified RNA (>800 ng) from each of the samples was used for the bulk RNA-seq analysis. RNA samples with RIN scores greater than 7 were selected for RNA-seq with the Agilent Bioanalyzer 2100. Samples were submitted to the Genomic and RNA Profiling Core at BCM for RNA-seq and library preparation. Nonstrand-specific, polyA+ -selected RNA-seq libraries were generated using the Illumina TruSeq protocol. Libraries were sequenced to a median depth of 35–40 million 100-bp single-end reads on a NovaSeq sequencer (Illumina). The FASTQ files underwent quality control metrics (trimming), read mapping, and gene annotation (GRCm38/mm10 genome) with default parameters with the RNA-seq Alignment v2.0.2 pipeline from Illumina’s BaseSpace (https://basespace.illumina.com/). The RNA-Seq Differential Expression v1.0.1 workflow (Illumina BaseSpace) was used with default parameters to extract gene counts and to obtain differentially expressed genes (DEGs) with an adjusted p value false discovery rate (FDR) < 0.05 between control and experimental samples using a two-sided Wald test and Benjamini–Hochberg (BH) correction for multiple testing. Gene Ontology (GO) Biological Process (BP) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were carried out with web-based tools ShinyGo58, Enrichr (https://maayanlab.cloud/Enrichr/)59 and Metascape (https://metascape.org/)60 to obtain adjusted p values with one-sided hypergeometric tests and FDR correction for multiple testing. The gene set enrichment analysis (GSEA) was performed with GSEA v4.2.3 Mac application using standard settings. The epithelial cell marker gene sets used for GSEA were obtained from Mouse Lung Injury & Regeneration web tool (https://schillerlabshiny.shinyapps.io/Bleo_webtool/)12 (Supplementary Data 13). The Jaccard overlap scores for the let-7 targetome were calculated by taking the size of the intersect and dividing it by the size of the union from epithelial cell types defined in GSE14125912. AT2 and ADI pseudotime trajectory values were obtained from Strunz et al.12, Heatmaps and plots were generated with R (v2022.12.0). Published raw data files corresponding to total RNA-seq of purified AT2 cells from control vs bleomycin treated mice (n = 3 mice per group) (GSE115730)6 were extracted from NCBI SRA toolkit and then processed for differential gene expression. To predict transcription factors whose function is impacted by let-7, ChIP-Seq regulatory network analysis was performed on the let-7afd−/− RNA-seq datasets with the Signaling Pathways Project knowledgebase as described below61. Briefly, we used the ChIP-Seq consensome gene lists from ChIP-Atlas, in which mouse (or human) transcription factor or regulatory genes are ranked based upon their average MACS2 occupancy across all publicly archived datasets for selection of high confidence targets (HCT). We then organized the ranked RNA-seq datasets gene lists into percentiles to generate the node ChIP-Seq consensomes. The 95th percentiles of all consensomes HCTs was used as the input for the intersection analysis. Statistical analysis for ChIP-Seq enrichment was carried out with a one-sided hypergeometric test with BH correction to generate q-values61.
Small RNA-seq data analysis
The published raw data files corresponding to small RNA-seq from lungs of mice after vehicle or bleomycin treatment (GSE195773)25 were extracted with the NCBI SRA toolkit. The SRA data files were analyzed with miRNA Analysis v1.0.0 application workflow (Illumina BaseSpace). To obtain mature let-7 miRNA normalized counts, standard settings were used corresponding to murine GRCm38/mm10 genome and miRbase DB v21 database. Plots were generated with R (v2022.12.0) by normalizing bleomycin treated samples to untreated samples for each timepoint.
CUT&RUN sequencing, processing, and data analysis
We utilized published CUT&RUN sequencing protocols as described below62,63. Lineage labeled Sftpc-tdT+ AT2 cells (~100,000) were flow sorted from mice as indicated above. Cells were pooled from two mice per genotype for CUT&RUN sequencing (n = 2). Cells were washed with wash buffer (50 ml total H2O with 20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, and one Roche Complete protein inhibitor tablet (Millipore, 11873580001). Cells were then incubated with activated concanavalin-A beads (Bang Laboratories, L200731C) for fifteen minutes at room temperature. Slurry was then separated with a Miltenyi MACSiMAG separator (Miltenyi 130-092-168), washed once, and resuspended in wash buffer containing 0.05% digitonin (Digitonin Wash buffer, Millipore, 300410). Digitonin wash buffer was used from here on out and cells kept on ice. The samples were placed on chilled tube rack kept on ice when not rotating for remainder of protocol. The samples were incubated with primary antibodies overnight at 4 °C rotating and then washed three times and incubated on rotator with Digitonin wash buffer containing pAG-MNase (Epicypher 15-1016) for one hour at 4 °C. Antibodies used were Rabbit anti-H3K27ac (Cell Signaling Technologies, 8173; 1:50 concentration), Rabbit anti-H3K427me3 (Cell Signaling Technologies, 9733; 1:50 concentration), and Rabbit anti-mouse IgG (Jackson ImmunoResearch, 315-005-003; 1:100). After, samples were washed three times they were resuspended in 100uL Digitonin wash buffer and chilled on ice rack for two minutes. 2μL of 100 mM CaCl2 was added to each sample to catalyze MNase cleavage. Cells were then kept at 4 °C for 40 minutes. Cleavage was stopped by adding equal volume stopping buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 0.05% Digitonin, 0.05 mg/mL glycogen, 5 µg/mL RNase A, 2 pg/mL heterologous spike-in DNA (Epicypher 18-1401), and samples were heated to 37 °C for 20 min for MNase/Histone/DNA complex release into supernatant. Slurry was separated with magnetic stand, and supernatant with DNA was collected into new tubes. DNA was purified with phenol chloroform extraction (Thermo Fisher Scientific, 15593031) and resuspended in ultrapure water (Invitrogen, 10-977-023). The samples were then submitted for library generation and sequencing to Admerahealth (South Plainfield, NJ).
The amount of DNA was determined by Qubit 2.0 DNA HS Assay (ThermoFisher, Massachusetts, USA) and quality was assessed by Tapestation High sensitivity D1000 DNA Assay (Agilent Technologies, California, USA). The KAPA HyperPrep kit (Roche, Basel, Switzerland) was used for library preparation. Assessment of library quality and quantity was done with Qubit 2.0 DNA HS Assay (ThermoFisher, Massachusetts, USA), Tapestation High Sensitivity D1000 Assay (Agilent Technologies, California, USA) for sequencing on an Illumina HiSeq 4000 (Illumina, California, USA) with a read length configuration of 150 paired-end (PE).
CUT&RUN sequencing data was trimmed for low quality reads and Illumina adapters using the trim galore (0.6.10) package (https://github.com/FelixKrueger/TrimGalore). Data was mapped using bowtie2 against the mouse genome University of California Santa Cruz (UCSC) genome browser build mm10. Peaks were called using MACS2 with IgG serving as control in each sample. Differential peaks between control and let-7afd−/− samples were determined using DiffReps v1.55.6 (https://github.com/shenlab-sinai/diffreps), with a significance cutoff and FDR adjusted q value < 0.05 and fold change exceeding 1.5x for H3K27ac peaks and a q value of <0.10 with fold change exceeding 1.5x for H3K27me3 peaks. Signal tracks were generated using DeepTools v2.1.0 (https://deeptools.readthedocs.io/en/2.1.0/index.html) and BEDTOOLS v2.31.0 suite (https://bedtools.readthedocs.io/en/latest/content/bedtools-suite.html) was used for annotation of differential peaks overlapping genes +/−10kb from its genebody64. GO terms for differential peaks were determined using GREAT v4.0.4 to compute binomial p values with two-sided hypergeometric test and BH correction65. Enriched motifs were derived using the findMotifsGenome.pl HOMER tool66. Data peaks were visualized with the UCSC genome browser.
AGO2 eCLIP and let-7 interactome network analysis
To obtain lung samples for AGO2-eCLIP+let-7, eight-week-old WT C57BL/6 mice were administered with constant flow of 3.5% isoflurane during intratracheal administration of 4U/kg bleomycin. The mice were then euthanized 6-days after treatment. Mouse lungs were cleared by cardiac perfusion and then inflated with 1 mL RNAse free PBS prior to harvesting and snap freezing with liquid nitrogen. Samples were provided to EclipseBio (San Diego, California) for generation of AGO2-eCLIP+let-7 libraries. The AGO-eCLIP+let-7 was performed in duplicate from 2 lungs of mice with chimeric ligation of miRNA and mRNA as previously described but with following modifications for lung sample processing and the enrichment of let-7 targets28,67. Briefly, approximately 20 × 106 cells from the lung were plated on a dish then UV crosslinked, then pelleted and suspended in 1 ml volume of eCLIP lysis mix. The lysates were then sonicated (QSonica Q800R2) for 5 min, 30 s on/30 s off with an energy setting of 75% amplitude, followed by digestion with RNase-I (Ambion). A primary mouse monoclonal AGO2/EIF2C2 antibody (sc-53521, Santa Cruz Biotechnology) was pre-coupled for 1 h with Sheep Anti-Mouse IgG Dynabeads (Thermo Fisher Scientific, 11202D) and then added to the homogenized lysate for a 2 h immunoprecipitation (IP) at 4 °C. After the IP, a 2% portion of the sample was taken as paired Input and the remainder of lysate washed with eCLIP high stringency wash buffers. The miRNA:mRNA chimeric ligation step was done at room temperature for 1 h with T4 RNA ligase (NEB). The IP samples were then treated with alkaline phosphatase (FastAP, Thermo Fisher) and then T4 PNK (NEB) for ligation of barcoded RNA adapters and isolation of AGO2-RNA complexes28,67. To generate AGO2-eCLIP libraries enriched for let-7 chimeras, 5’ biotinylated ssDNA probes antisense to murine let-7a-5p, let-7b-5p, let-7d-5p, let-7f-5p miRNAs were ordered from Integrated DNA Technologies and resuspended in nuclease-free water to a final concentration of 100 µM. Each probe was bound individually to Capture Beads (Eclipse Bioinnovations) and afterward mixed. The resulting probe-bound bead mixture was added to the reserved AGO2-eCLIP enrichment fractions and allowed to hybridize over a 1.5-h period, with incrementally decreasing temperatures. Beads were washed to remove nonspecific RNAs and afterward, bound RNAs were eluted from beads using a DNase treatment. Eluted RNA was purified using an RNA Clean and Concentrator-5 Kit (Zymo Research, cat. #R1015/R1016) and taken through reverse transcription, DNA adapter ligation, and PCR amplification to generate sequencing libraries. Library sequencing was performed as SE122 on the NextSeq 2000 platform. BedGraph files of let-7 chimeric peaks were generated for visualization with UCSC genome browser.
The AGO2-eCLIP+let-7 processing, and bioinformatics analysis was done by Eclipsebio (San Diego, California) with a proprietary analysis pipeline v1 developed from several published eCLIP publications28,67. The original bioinformatic pipelines are available (https://github.com/yeolab/eclip; https://github.com/YeoLab/chim-eCLIP). A brief description follows: The umi_tools (v1.1.1) tool was used to prune Unique molecular identifier (UMIs) from reads. Next, cutadapt (v3.2) was used for trimming 3’ adapters. Repetitive elements and rRNA sequences were removed and non-repeat mapped reads mapped to murine GRCm38/mm10 using STAR (v2.7.7a). PCR duplicates were removed with umi_tools (v1.1.1). miRbase (v22.1) miRNAs were reverse mapped to reads with bowtie (v1.2.3) and miRNA portion of each read trimmed. STAR (v2.7.7a) was used to map the miRNA portion of each read to the genome. PCR duplicates were trimmed, and miRNA target cluster peaks were identified with CLIPper (v2.1.0). Individual clusters were annotated with corresponding miRNAs names and peaks annotated to transcripts GENCODE release M25 (GRCm38.p6). Peaks present in both biological samples were selected for downstream analysis. De novo HOMER (v4.11)66 motif analysis was also applied to confirm the let-7 seed region as the most enriched motif in dataset. Let-7 chimeric peaks with overlap to the 3’UTR regions of protein coding genes were designated as the AGO2-eCLIP let-7 interactome. The Sylamer test was performed with standard parameters on ranked list derived from a sorted list of upregulated to downregulated genes in bulk RNA-seq dataset68. Markov-correction was then used in Sylamer test to remove tetramer composition biases in the growing window. This was followed by a one-sided hypergeometric test with Bonferroni multiple-testing correction to compute the enrichment of miRNA binding motifs across the ranked gene list68. Upregulated (p < 0.05) genes in let-7afd−/− Sftpc-tdT and/or cultured organoids were overlayed with AGO2-eCLIP let-7 interactome and experimentally validated and published let-7 targets in miRTarbase database69 with Excel software (Microsoft). Pathway enrichment analysis for the let-7 interactome was carried out with Enrichr59 and Metascape60. Protein-protein interaction networks and q values were derived from Metascape (using parameter of Combined Core) and then visualized and organized with EnrichmentMap/Cytoscape (v3.10.1).
Human IPF Atlas scRNA-seq integration
A published scRNA-seq dataset (GSE136831) deposited in IPF Cell Atlas from control and IPF patients was used14. The cell annotations for cell clusters were provided by the authors14. Cell-level expression for individual genes was obtained with R package Seurat70. Gene signature scores were generated using the UCell method. Violin plots for individual genes or gene signatures in specific cell types were generated using the R statistical package v4.4.0. Dotplot expression of individual gene in selected cell types were generated using Seurat70. The Hallmark EMT and ENCODE Consensus ChIP-X gene targets for EZH2 used for UCell enrichment were obtained from Enrichr59. BACH1 high confidence transcriptional targets were derived with the ChIP-Seq regulatory network toolkit61.
Transmission electron microscopy (TEM) and analysis
We utilized a published protocol for fixation6, processing and embedding of lung tissues for preservation of AT2 cell lamellar body (LB) ultrastructure. Mice were euthanized, followed by an incision of the IVC artery and descending aorta to allow for cardiac draining of blood. Lungs were inflated with 1.2 mL of freshly prepared room temperature TEM fixative (4% paraformaldehyde, 2.5% glutaraldehyde, 0.02% picric acid in 0.1 M sodium cacodylate buffer, pH 7.3), tie sutured and then fixed overnight at 4 °C. Strips of lung tissue were dissected, washed in 0.1 M sodium cacodylate buffer, fixed in a solution of 1% OsO4 and 1.5% potassium ferricyanide for 1 hr with gentle agitation, washed several times with 0.1 M sodium cacodylate buffer and then incubated overnight at 4 °C. Samples were then washed with ddH20, stained with 1.5% uranyl acetate in the dark for 1 h, dehydrated with a graded series of ethanol, infiltrated with and embedded in EMbed812 epoxy resin (#14120, Electron Microscopy Sciences) and then sectioned at 100 nm thickness with a Leica EM UC7 ultramicrotome. Images were collected with a JEOL JEM-1400Flash TEM operating at 120 kV of accelerating voltage and equipped with a high-contrast pole piece and an AMT NanoSprint15 Mk-II sCMOS camera. Images were examined with FIJI (ImageJ, v2.9.0) to quantify AT2 cell area (μm2), LB area (μm2), LB number/cell area (#/μm2) and AT2 cell circularity.
Microscopy, image acquisition, and quantification
Slides were imaged on a Leica DM6000B (Leica, Wetzlar, Germany) using cameras CoolSNAP HQ2 (Teledyne Photometrics, USA) or Infinity3-6UR (Teledyne Lumenera, USA). A Keyence BZ-X800 microscope (Keyence, USA), an Olympus IX83 microscope (Olympus, USA), and a Nikon A 1Rs confocal microscope were used for imaging. Image processing was done with BZ-X Series Advanced Analysis software (Keyence) or cellSens image deconvolution software (Olympus). Brightfield color images captured by Leica microscope where white balance with FIJI (ImageJ, v2.14.0), and then stitched using AIVIA software (Leica, v11.0). Organoid domes were imaged on 6-well plates with a BioTek LionHeart LX microscope (Agilent, USA). Images were captured with 10X objective and stitched with built-in software. Cell counts were done manually based on markers and DAPI with FIJI software. Mean Fluorescence Intensity (MFI) was measured by FIJI software. Images of interest where then prepared using Adobe Photoshop and Illustrator. Mean linear intercept (MLI) measurements were carried out with the Measure MLI plugin for ImageJ v1.52 (https://med.nyu.edu/research/nolan-lab/software)71. Briefly, ten fields of view (1555 µm × 1555 µm) in alveoli devoid of bronchi or vasculature were randomly selected per mouse to calculate the MLI. Ashcroft Scoring was performed using ten randomly chosen fields of view per mouse lung which were then assessed by a system of grades on a 0–8 scale as described earlier72. Briefly, each field was compared to an image atlas and a histology grade characterization table and then scored by closest match. The mean of all fields were then recorded per mouse72.
Statistical analysis
Values are represented as indicated in figure legends. P values were calculated using two-tailed unpaired two-tailed Student’s t test or one-way ANOVA, as indicated in figure legends. A p value < 0.05 was considered statistically significant. Determination of sample size for animal experiments were based on pilot tests. Investigators were not blinded in the allocation of experiments. No data was excluded from analysis. Mice of both genders were randomly assigned into experimental or control groups for experiments. Statistical analyses were performed using GraphPad Prism (v9.5.1) except for the RNA-seq and CUT&RUN-seq which were done as described above.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the Article, Supplementary information, or the Source Data file. AGO2-eCLIP+let-7, bulk RNA-seq, and CUT&RUN-seq raw and processed data has been deposited in the NCBI Gene Expression Omnibus database under accession code GSE287951 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE287951]. The WT scRNA-seq dataset used for pseudotime expression trajectories of alveolar PSCs following bleomycin injury are available at GEO (GSE141259) [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141259]. The small RNA-seq used for let-7 expression after bleomycin injury is available at (GSE195773) [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE195773]. The AT2 cell WT RNA-seq dataset following bleomycin injury is available at (GSE115730) [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115730]. The scRNA-seq IPF cell atlas dataset used are available at GEO (GSE136831) [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136831]. Source data are provided with this paper.
References
Basil, M. C. et al. The cellular and physiological basis for lung repair and regeneration: past, present, and future. Cell Stem Cell 26, 482–502 (2020).
Hogan, B. & Tata, P. R. Cellular organization and biology of the respiratory system. Nat. Cell Biol. https://doi.org/10.1038/s41556-019-0357-7 (2019).
Basil, M. C., Alysandratos, K. D., Kotton, D. N. & Morrisey, E. E. Lung repair and regeneration: Advanced models and insights into human disease. Cell Stem Cell 31, 439–454 (2024).
Wuyts, W. A. et al. Idiopathic pulmonary fibrosis: best practice in monitoring and managing a relentless fibrotic disease. Respiration 99, 73–82 (2020).
Katzen, J. & Beers, M. F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Invest. 130, 5088–5099 (2020).
Chung, K. P. et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 10, 3390 (2019).
Barkauskas, C. E. et al. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Invest. 123, 3025–3036 (2013).
Nabhan, A. N., Brownfield, D. G., Harbury, P. B., Krasnow, M. A. & Desai, T. J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359, 1118–1123 (2018).
Zacharias, W. J. et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 555, 251–255 (2018).
Choi, J. et al. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell 27, 366–382.e367 (2020).
Kobayashi, Y. et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 22, 934–946 (2020).
Strunz, M. et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 11, 3559 (2020).
Wang, F. et al. Regulation of epithelial transitional states in murine and human pulmonary fibrosis. J. Clin. Invest. 133, https://doi.org/10.1172/JCI165612 (2023).
Adams, T. S. et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 6, eaba1983 (2020).
Pandit, K. V. et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 182, 220–229 (2010).
Kim, S. et al. Integrative phenotyping framework (iPF): integrative clustering of multiple omics data identifies novel lung disease subphenotypes. BMC Genomics 16, 924 (2015).
Kim, H. H. et al. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 23, 1743–1748 (2009).
Kong, D. et al. Loss of let-7 up-regulates EZH2 in prostate cancer consistent with the acquisition of cancer stem cell signatures that are attenuated by BR-DIM. PLoS One 7, e33729 (2012).
Johnson, S. M. et al. RAS is regulated by the let-7 microRNA family. Cell 120, 635–647 (2005).
Ma, Y., Shen, N., Wicha, M. S. & Luo, M. The roles of the Let-7 family of microRNAs in the regulation of cancer stemness. Cells 10, https://doi.org/10.3390/cells10092415 (2021).
Jin, B. et al. Let-7 inhibits self-renewal of hepatocellular cancer stem-like cells through regulating the epithelial-mesenchymal transition and the Wnt signaling pathway. BMC Cancer 16, 863 (2016).
Liu, Y. & Zheng, Y. Bach1 siRNA attenuates bleomycin-induced pulmonary fibrosis by modulating oxidative stress in mice. Int. J. Mol. Med. 39, 91–100 (2017).
Le, H. Q. et al. An EZH2-dependent transcriptional complex promotes aberrant epithelial remodelling after injury. EMBO Rep. 22, e52785 (2021).
Qin, H. et al. C-MYC induces idiopathic pulmonary fibrosis via modulation of miR-9-5p-mediated TBPL1. Cell Signal 93, 110274 (2022).
Strobel, B. et al. Time and phenotype-dependent transcriptome analysis in AAV-TGFbeta1 and Bleomycin-induced lung fibrosis models. Sci. Rep. 12, 12190 (2022).
Erice, P. A. et al. Downregulation of Mirlet7 miRNA family promotes Tc17 differentiation and emphysema via de-repression of RORgammat. Elife 13, https://doi.org/10.7554/eLife.92879 (2024).
Konishi, S., Tata, A. & Tata, P. R. Defined conditions for long-term expansion of murine and human alveolar epithelial stem cells in three-dimensional cultures. STAR Protoc. 3, 101447 (2022).
Van Nostrand, E. L. et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514 (2016).
Rodriguez, A. et al. Requirement of bic/microRNA-155 for normal immune function. Science 316, 608–611 (2007).
Batool, A., Jin, C. & Liu, Y. X. Role of EZH2 in cell lineage determination and relative signaling pathways. Front. Biosci. 24, 947–960 (2019).
Comet, I., Riising, E. M., Leblanc, B. & Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 16, 803–810 (2016).
Dhanasekaran, R. et al. The MYC oncogene - the grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 19, 23–36 (2022).
Zhang, X. et al. Bach1: function, regulation, and involvement in disease. Oxid. Med. Cell Longev. 2018, 1347969 (2018).
Hou, W., Tian, Q., Steuerwald, N. M., Schrum, L. W. & Bonkovsky, H. L. The let-7 microRNA enhances heme oxygenase-1 by suppressing Bach1 and attenuates oxidant injury in human hepatocytes. Biochim. Biophys. Acta 1819, 1113–1122 (2012).
Yang, R. et al. E2F7-EZH2 axis regulates PTEN/AKT/mTOR signalling and glioblastoma progression. Br. J. Cancer 123, 1445–1455 (2020).
Igarashi, K., Nishizawa, H., Saiki, Y. & Matsumoto, M. The transcription factor BACH1 at the crossroads of cancer biology: from epithelial-mesenchymal transition to ferroptosis. J. Biol. Chem. 297, 101032 (2021).
Yao, C. et al. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 203, 707–717 (2021).
Sato, M. et al. BACH1 promotes pancreatic cancer metastasis by repressing epithelial genes and enhancing epithelial-mesenchymal transition. Cancer Res. 80, 1279–1292 (2020).
Wei, Z. et al. MYC reshapes CTCF-mediated chromatin architecture in prostate cancer. Nat. Commun. 14, 1787 (2023).
Kim, J. et al. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 25, 2808–2820.e2804 (2018).
Niu, C. et al. BACH1 recruits NANOG and histone H3 lysine 4 methyltransferase MLL/SET1 complexes to regulate enhancer-promoter activity and maintains pluripotency. Nucleic Acids Res. 49, 1972–1986 (2021).
Wei, X. et al. Bach1 regulates self-renewal and impedes mesendodermal differentiation of human embryonic stem cells. Sci. Adv. 5, eaau7887 (2019).
Weber, F., Treeck, O., Mester, P. & Buechler, C. Expression and Function of BMP and Activin Membrane-Bound Inhibitor (BAMBI) in Chronic Liver Diseases and Hepatocellular Carcinoma. Int. J. Mol. Sci. 24, https://doi.org/10.3390/ijms24043473 (2023).
Hu, Y. et al. Airway-derived emphysema-specific alveolar type II cells exhibit impaired regenerative potential in COPD. Eur. Respir. J. 64, https://doi.org/10.1183/13993003.02071-2023 (2024).
Lv, Z. et al. Alveolar regeneration by airway secretory-cell-derived p63(+) progenitors. Cell Stem Cell 31, 1685–1700.e1686 (2024).
Liu, Q. et al. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat. Genet. 51, 728–738 (2019).
Shen, Y. et al. c-Myc promotes renal fibrosis by inducing integrin alphav-mediated transforming growth factor-beta signaling. Kidney Int. 92, 888–899 (2017).
Luond, F. et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell 56, 3203–3221.e3211 (2021).
Yuan, Z. et al. Knockdown of Bach1 protects periodontal bone regeneration from inflammatory damage. J. Cell Mol. Med. 27, 3465–3477 (2023).
Byrd, A. L. et al. Dysregulated polycomb repressive complex 2 contributes to chronic obstructive pulmonary disease by rewiring stem cell fate. Stem Cell Rep. 18, 289–304 (2023).
Sun, L. et al. Exosomal miRNA Let-7 from menstrual blood-derived endometrial stem cells alleviates pulmonary fibrosis through regulating mitochondrial DNA damage. Oxid. Med. Cell Longev. 2019, 4506303 (2019).
Huleihel, L. et al. Let-7d microRNA affects mesenchymal phenotypic properties of lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 306, L534–L542 (2014).
Duan, R., Du, W. & Guo, W. EZH2: a novel target for cancer treatment. J. Hematol. Oncol. 13, 104 (2020).
Bao, X. et al. Inhibition of EZH2 prevents acute respiratory distress syndrome (ARDS)-associated pulmonary fibrosis by regulating the macrophage polarization phenotype. Respir. Res. 22, 194 (2021).
McGovern, T. K., Robichaud, A., Fereydoonzad, L., Schuessler, T. F. & Martin, J. G. Evaluation of respiratory system mechanics in mice using the forced oscillation technique. J. Vis. Exp., e50172, https://doi.org/10.3791/50172 (2013).
Chen, Q. & Liu, Y. Isolation and culture of mouse alveolar type II cells to study type II to type I cell differentiation. STAR Protoc. 2, 100241 (2021).
Katsura, H. et al. Human lung stem cell-based alveolospheres provide insights into SARS-CoV-2-mediated interferon responses and pneumocyte dysfunction. Cell Stem Cell 27, 890–904 e898 (2020).
Ge, S. X., Jung, D. & Yao, R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629 (2020).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Ochsner, S. A. et al. The Signaling Pathways Project, an integrated ‘omics knowledgebase for mammalian cellular signaling pathways. Sci. Data 6, 252 (2019).
Skene, P. J., Henikoff, J. G. & Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 13, 1006–1019 (2018).
Le, D. T. et al. BATF2 promotes HSC myeloid differentiation by amplifying IFN response mediators during chronic infection. iScience 26, 106059 (2023).
Trevino, L. S. et al. Epigenome environment interactions accelerate epigenomic aging and unlock metabolically restricted epigenetic reprogramming in adulthood. Nat. Commun. 11, 2316 (2020).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Krivdova, G. et al. Identification of the global miR-130a targetome reveals a role for TBL1XR1 in hematopoietic stem cell self-renewal and t(8;21) AML. Cell Rep. 38, 110481 (2022).
van Dongen, S., Abreu-Goodger, C. & Enright, A. J. Detecting microRNA binding and siRNA off-target effects from expression data. Nat. Methods 5, 1023–1025 (2008).
Huang, H. Y. et al. miRTarBase update 2022: an informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 50, D222–D230 (2022).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Crowley, G. et al. Quantitative lung morphology: semi-automated measurement of mean linear intercept. BMC Pulm. Med. 19, 206 (2019).
Hubner, R. H. et al. Standardized quantification of pulmonary fibrosis in histological samples. Biotechniques 44, 514–507 (2008).
Acknowledgements
We thank Drs. David Corry and Farrah Kheradmand for valuable scientific suggestions and for generous sharing of lab equipment. M. Sayeeduddin for histology at the Tissue Acquisition and Pathology Core (funded in part by P30 CA125123); and Joel M. Sederstrom for flow cytometry at the BCM and Cell Sorting Core (funded in part from NIH (CA125123 and RR024574); we thank the Texas Children’s Hospital William T Shearer Center for Human Immunobiology and Dr. Ronald Parchem for access to microscope equipment. Bioinformatics analysis was partially supported by The Cancer Prevention Institute of Texas (CPRIT) grants RP210227 and RP200504, NIEHS grants P30 ES030285 and P42 ES027725. This work was supported by grants from the NHLBI (R01HL140398, HL167814 to A.R.; HL155672 to K.Y.K.; F31 HL164287 to B.T.T.), and NIGMS (T32 GM136554 to M.S.).
Author information
Authors and Affiliations
Contributions
M.J.S., M.S. and A.R. conceptualized experiments, interpreted data, and wrote the manuscript. M.J.S., M.S., P.A.E., B.T.T, S.L.L. carried out experiments. M.D.M. carried out the TEM imaging. A.F.C. provided microscope image acquisition expertise. M.E.R-E., R.S.K. L.M.S., S.C.W., S.A.O., C.C., A.R. performed bioinformatic data analysis. K.Y.K. provided expertise and supervision of epigenomics experiments. A.R., A.E., I.O.R., N.J.M., C.C., provided expertise and co-supervised bioinformatic data analysis.
Corresponding author
Ethics declarations
Competing interests
F.R. is currently an employee at Vertex Pharmaceuticals. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Seasock, M.J., Shafiquzzaman, M., Ruiz-Echartea, M.E. et al. Let-7 restrains an epigenetic circuit in AT2 cells to prevent fibrogenic intermediates in pulmonary fibrosis. Nat Commun 16, 4353 (2025). https://doi.org/10.1038/s41467-025-59641-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59641-1
