
Abstract
Catalytic enantioselective three-component aminative difunctionalization of readily available 1,3-dienes offers a straightforward methodology to fast access significant and complex chiral allylic amines. Nevertheless, compared to the widely studied two-component reactions, the three-component reactions, especially using anilines—very common bulk feedstock chemicals as aminating reagents are underdeveloped. More importantly, the limited examples of enantioselective three-component aminative difunctionalization of 1,3-dienes with anilines only showed 1,2-selectivity; and the corresponding 1,4-regioselectivity remains unknown. Here, we report a copper-catalyzed enantioselective radical three-component 1,4-perfluoroalkylamination of 1,3-dienes with anilines and perfluoroalkyl reagents, efficiently providing an array of valuable perfluoroalkylated chiral allylic amines in good to excellent yields with excellent enantioselectivity. Mechanistic investigations, including controlled experiments and DFT studies, elucidate the origination of the regioselectivity and enantioselectivity, and suggest a radical reaction pathway involving an asymmetric cross-coupling between allylic radical and copper-stabilized nitrogen radical species to construct C–N bond enantioselectively.
Similar content being viewed by others
Introduction
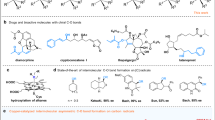
Chiral allylic amines are useful synthetic building blocks and essential structural element in natural products, and pharmaceuticals1,2,3. Perfluoroalkyl groups, such as the trifluoromethyl group, have been confirmed to be a crucial unit that can lead to improved bioavailability, metabolic stability, and efficacy4,5. Some trifluoromethylated chiral allyl amines have been demonstrated to be promising pharmaceuticals. For instance, DPC-083 is an orally available non-nucleoside reverse transcriptase inhibitor with broad inhibitory effects on HIV6; compound I is a potential inhibitor and inactivator of γ-aminobutyric acid aminotransferase (Fig. 1a)7. Therefore, the development of effective and streamlined synthetic methods for perfluoroalkyl-containing chiral allylic amines is of great importance, especially those starting from bulk feedstock chemicals.

a Examples of biological and pharmaceutical active molecules with perfluoroalkylated chiral allylic amine substructure. b Transition metal-catalyzed asymmetric aminative difunctionalization of 1,3-dienes. c Transition metal-catalyzed asymmetric 1,2-three-component aminativedifunctionalization of 1,3-dienes with aniline derivatives. d This work: enantioselective three-component radical 1,4-perfluoroalkylamination of 1,3-dienes with aniline derivatives.
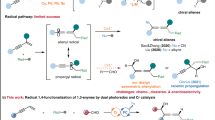
1,3-Dienes are common bulk feedstock chemicals. A transition metal-catalytic asymmetric three-component aminative difunctionalization of 1,3-dienes, due to simultaneously introducing an amino and another functional groups in one-step with high regio- and stereoselectivity, is a promising and straightforward strategy to access structurally diverse chiral allylic amines8,9,10. To date, by leveraging the pre-prepared aminating reagents, such as dienyl amine, N-aryl amide, aryl urea, aminal, diaziridinone, or dioxazolone, enantioselective arylamination11,12,13,14,15,16, alkenylamination17, aminomethylamination18, diamination19,20,21 and oxyamination22 have been successfully achieved with the vast majority being two-component annulative reactions. The corresponding three-component transformations are rare. Moreover, aniline and its derivatives, as readily available chemical materials and ubiquitous units in functional materials, are highly charming aminating reagents in the enantioselective aminative difunctionalization of 1,3-dienes (Fig. 1b). Very recently, by using a structurally fixed 1,3-diene as the substrate, Zhang and coworkers16 disclosed a Pd-catalyzed three-component enantioselective 1,2-arylamination of 1,3-cyclohexadiene with aryl iodide and anilines via a Heck-type insertion to form aryl π-allyl palladium intermediate followed by nucleophilic attack of aniline (Fig. 1c).
Radical reactions, featuring high activity and good functional group compatibility, have attracted much attention23,24. With the rapid development of catalytic asymmetric synthesis, remarkable and impressive advances have been made in radical enantioselective transformations25,26,27. Radical-initiated asymmetric difunctionalization of 1,3-dienes, through the first radical addition onto 1,3-diene to form allylic radical, then combining with metal to generate π-allyl metal species and final reductive elimination or nucleophilic/electrophilic attack allowing various functional groups to be introduced, has emerged as a promising and robust platform for a fast buildup of complex enantioenriched allylic compounds28. In this regards, asymmetric 1,2-diamination29, alkylamination30, alkylesterification31,32, alkylcyanation33, amidocyanation34, dialkylation35,36, arylalkylation37, alkylsulfonylation38, and azidooxygenation39 of 1,3-dienes, have been successfully achieved. Impressively, Gong and coworkers40 recently realized photoinduced Pd-catalyzed enantioselective 1,2-carboamination of 1,3-dienes with aliphatic amines and aliphatic C − H bonds, where three examples of electron-poor anilines were reported (Fig. 1c). Despite these great advances, reports of enantioselective three-component aminative difunctionalization of 1,3-diene with anilines is still quite limited. More importantly, the corresponding enantioselective 1,4-selectivity remains unexplored. As our continuous research interests on asymmetric radical reactions41,42,43,44,45,46,47, herein, we disclosed a Cu-catalyzed enantioselective perfluoroalkylamination of 1,3-dienes with aniline derivatives and perfluoroalkyl reagents, efficiently synthesizing various perfluoroalkyl-containing chiral allylic amine derivatives (Fig. 1d). This reaction shows impressively unique 1,4-regioselectivity and the excellent enantioselectivity. DFT investigations elucidated the precise control of the selectivity and revealed that the asymmetric formation of C–N bond involved an enantioselective radical cross-coupling of allylic radical and copper-stabilized nitrogen-centered radical species.
Results
Reaction optimization
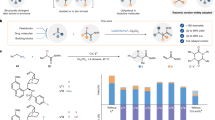
We choose a reaction of (E)-buta-1,3-dien-1-ylbenzene (1a) with N-methylaniline (2a) and Togni reagent (3a) as the model reaction. Initially, the racemic trifluoromethylamination was carried out in the presence of copper catalyst without ligand. Intriguingly, only 1,4-addition product 4 was formed in 38% yield and no 1,2-adduct was observed (Table 1, entry 1). Then, the asymmetric version was carefully investigated (Table 1 and Supplementary Table 1-4). When chiral bisoxazoline L1 as the ligand was added to the model reaction, we were pleased to find that 4 was obtained in 78% isolated yield and 84:16 er (entry 2). Other chiral bisoxazoline ligands, such as L2 and L3, could not improve the yield and the enantioselectivity of 4 (entries 3 and 4, and Supplementary Table 1). In the absence of the copper catalyst, no reaction occurred (entry 5). Reducing the reaction temperature was beneficial to raise the enantioinduction, but it required an extension of the reaction time (entry 6 and Supplementary Table 2). The reaction in a mixed solvent of CH3CN and EtOAc (EA) (1:4) gave the best result (entries 7–9 and Supplementary Table 3), that the desired 1,4-adduct 4 was obtained in 84% isolated yield and 93:7 er (entry 9). Screening other copper catalysts was also conducted, and vaguely inferior yield under otherwise identified conditions were obtained (entries 10–12 and Supplementary Table 4). Distinctly, during the whole optimization, the competitive 1,2-adduct, which was often predominated in literatures of 1,3-diene enantioselective difunctionalization, was not detected.
Substrate scope
After establishing the optimal reaction conditions, the generality of this methodology was examined. Firstly, the scope of 1,3-diene components was investigated. As highlighted in Fig. 2a, a series of enantioenriched trifluoromethyl-containing allylic amines 4–30 were smoothly obtained under the optimized reaction conditions. Remarkably, directly employing a Z/E mixture of 1,3-diene 1b-1v as the substrates could also generate the corresponding (E)-selective products 5–25. Specifically, ortho-methyl, fluoro-, and chloro-substituted phenyl-1,3-dienes were transformed to products 5, 6 and 7 in moderate yields and high enantioselectivity. When meta-substituted aromatic dienes were used as the substrates, the desired products 8–13 were formed in up to almost quantitative yield with high enantioselectivity. For para-substituted aromatic dienes, such as alkyl and halogen atom (F, Cl, and Br), chiral trifluoromethyl-contained allyl amines 14–17 were obtained in good to excellent yields with moderate enantioselectivity. In addition to monosubstituted aromatic 1,3-dienes, polysubstituted aromatic 1,3-dienes were also examined under the optimal conditions. Disubstituted aromatic 1,3-dienes were suitable substrates, forming corresponding chiral allyl amines 18–22 in moderate to almost quantitative yields with moderate to high enantioselectivities. The trisubstituted aromatic 1,3-diene was effective substrate, furnishing corresponding product 23 in good result. Furthermore, the fused aromatic ring substituted 1,3-dienes were also tested. The desired reactions proceeded smoothly, affording the corresponding products 24 and 25 in good results. Disubstituted 1,3-diene also worked very well, forming allylic amine 26 in approximate quantitative yield and high enantioselectivity. Additionally, 1,3-dienes 1x, 1 y, 1z and 1aa derived from drugs borneol, naproxen, diacetonefructose and ibuprofen were smoothly converted into the expected trifluoromethyl-contained chiral allylic amines 27, 28, 29 and 30, respectively, with good yields and satisfied stereoselectivity. These results showcased the robustness and usefulness of the reaction. An alkyl-substituted 1,3-diene, such as 1-benzyl-1,3-diene, was also subjected to the trifluoromethylamination reaction, but only a trace amount of the desired 1,4-addition product was observed, along with an inseparable mixture.

a Scope of 1,3-dienes. b Scope of anilines. Reaction conditions: 1 (0.25 mmol), 2 (0.1 mmol), 3a (0.2 mmol), Cu(CH3CN)4PF6 (10 mol%), (R, R)-L1 (12 mol%) in 2 mL mixed solvent of CH3CN and EA (1:4) at −10 °C for 72 h under nitrogen. Yields were isolated ones. Er values were determined by HPLC analysis. Dr values were determined by 1H NMR analysis. aReaction for 5 days.
The scope of aniline derivatives was probed (Fig. 2b). A various of chain secondary aromatic amines, such as para- or meta-electron-donating or -withdrawing group substituted N-methyl aromatic amines were tolerated with the reaction to furnish desired chiral trifluoromethyl-containing allyl amines 31–44 in good to excellent yields and excellent er. N-Methyl 3,4-disubstituted aromatic amines were capable substrates, affording the expected products 45–48 in good yields with good er value. Also, N-methyl fused aromatic amines can yield the expected products 49 and 50 with excellent yield and good stereoselectivity. Besides methyl, ethyl, n-butyl, 3-phenylpropyl, and 4-phenylbutyl as the aliphatic chain of the secondary amines, were compatible for the reaction to give 51–60 in good yields and excellent er. Cyclic secondary aromatic amine was also competent substrate, giving rise to the expected product 61 in good yield with excellent enantioselectivity. Besides secondary amines, primary amines, such as aniline and the derivatives worked smoothly to produce 62–66 in moderate to good yields and excellent enantioselectivity. Diphenylamine was also attempted, but the substrates were completely remained. When aliphatic amines, such as dibenzylamine, benzylamine, morpholine and N-methyl benzylamine, were employed to react with 1-(2,5-dimethyphenyl)-1,3-diene under standard conditions, the 1,3-diene was recovered, accompanied by unisolated mixture.
Various perfluoroalkyl iodides were also investigated (Fig. 3). When employing lauroyl peroxide (LPO) as the oxidant under slightly modified conditions, various iodinated perfluoroalkanes can successfully participate in the asymmetric perfluoroalkylamination. The reaction of iodinated perfluorobutane with N-methyl aniline and various substituted aromatic 1,3-dienes furnished corresponding perfluorobutyl-containing allylic amines 67–76 in good to excellent yields and high enantioselectivity. The iodinated perfluorobutane and N-methyl aniline can reaction with 1,3-dienes originated from drug molecules diacetonefructose and borneol smoothly to deliver to promising products 77 and 78 in excellent yields and more than 25:1 diastereoselectivity. The reaction of iodinated perfluorobutane with 1,3-diene and other N-alkyl aromatic amines or primary aromatic amine worked well, and the desired products 79–84 were smoothly obtained in good yields and higher values. Additionally, 2-iodo-1,1,1-trifluoroethane, perfluorohexyl-, heptyl-, decanyl-, and hepta-fluoroisopropyl-iodide were well-tolerated, affording corresponding products 85–89 in good to excellent yields with high enantioselectivity. In all cases, only one isomer was observed.

Reaction conditions: 1 (0.25 mmol), 2 (0.1 mmol), 3 (0.2 mmol), LPO (0.2 mmol), Cu(CH3CN)4PF6 (10 mol%), (R, R)-L1 (12 mol%) in 1 mL DCM at −20 °C for 72 h under nitrogen atmosphere. Er values were determined by HPLC analysis. Dr values were determined by 1H NMR analysis.
Synthetic applications
To verify the practicality of this methodology, a gram-scale synthesis was first conducted, and the 1,4-trifluoromethylamination product 19 was readily gained with high efficiency and high enantioselectivity (Fig. 4a). The C–C double bond of allylic amine 19 can be smoothly oxidized by K2OsO4∙2H2O to afford a chiral syn-diol compound 90 in good yield under mild reaction conditions (Fig. 4b). The absolute configuration of the major diastereoisomer 90 was confirmed as (2 R, 3 R, 4 R) by X-ray crystallographic analysis (Supplementary Table 7). Since after the oxidation the substitution pattern affected the priority of the groups at the chiral carbon atom of the C–N bond in oxidative product 90, stemming from chiral allylic amine 19, enabled us to assign the absolute configuration of 19 produced in the asymmetric 1,4-trifluorometylamination of 1,3-diene as (S) (Supplementary Section VII), and those of the others were assigned by analogy. The N-acylation of product 63 could deliver the optically pure product 91 (Fig. 4c).

a Gram-scale synthesis. b Oxidation of C–C double bond. c N–H Acylation.
Mechanistic investigations
To probe the mechanism of the asymmetric transformation, some controlled experiments were conducted (Fig. 5). First, a radical trap experiment was performed by using 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) as the radical inhibitor. The model reaction was found to be completely suppressed, and the trifluoromethyl-TEMPO adduct 92 was obtained in 67% yield (Fig. 5a and Supplementary Section VIII. A). Next, a radical clock reaction with (2-(buta-1,3-dien-1-yl)cyclopropyl)benzene (1ab) was carried out, and the reaction furnished the ring-opened homoallylic amine 93 in 44% yield (Fig. 5b and Supplementary Section VIII. B). These results suggested that an allylic radical intermediate is possibly involved in this transformation. Furthermore, a linear relationship between the product and catalyst er value was founded, implying a likely 1:1 copper-to-ligand ratio in the enantioselectivity-determining transition states (Fig. 5c and Supplementary Section VIII. C).

a Radical trap experiment. b Radical clock experiment. c Nonlinear er relationship between catalyst and product.
DFT Computational investigations
To obtain a deeper understanding of the copper-catalyzed asymmetric 1,4-perfluoroalkylamination reaction of 1,3-diene, several elementary steps involving regioselectivity and stereoselectivity were scrupulous evaluated through density functional theory (DFT) calculations, using 1p, 2d, and 3a as the model substrates. Viewing the DFT calculated results (Supplementary Fig. 4) and literatures48,49,50, the initial trifluoromethylation involved a SET process between Cu(I) catalyst and Togni reagent formed a Cu(II)–O species and a trifluoromethyl radical, and subsequent trifluoromethyl radical adding to 1,3-diene to generate allylic radical 2AC. The resultant L*Cu(II)–OCOAr species Com1 underwent ligand exchange/deprotonation with aniline derivative 2 d to produce formal three-coordinate L*Cu(II)–N species (Supplementary Fig. 5). Furthermore, the spin density analysis of the L*Cu(II)-NRAr’ showcased that considerable spin density located on N atom (0.55 e−) and less spin density on copper (0.04 e−) (Fig. 6a). These results implied that the formal L*Cu(II)−NRAr’ complex could be viewed as a L*Cu(I)–NRAr’ radical species INT-651, consistent with Fu and Peters’ report where a three-coordinate Cu(I)–N radical was recognized52. Given that an enantioselective cross-coupling between carbon-centered radical and copper-stabilized nitrogen-centered radical has been recently demonstrated to construct asymmetric C–N bond by Fu and Peters group52, as well as our group43. Thus, the coupling of INT-6 with the allylic radical 2AC via the transition state TS5-S to furnish S-type 33-S product was evaluated, and the Gibbs activation energy (ΔG‡) is calculated to be 10.8 kcal/mol (Fig. 6b, right, black line). Additionally, the energy of the transition state TS5-R was calculated (Fig. 6b, right, red line), which is 2.2 kcal/mol higher than TS5-S. Furthermore, we found that in the favorable transition state TS5-S, the CF3 moiety of radical 2AC is oriented closer to the phenyl group of the chiral ligand and could contribute significant C–H···F and C–F···π interactions that stabilized TS5-S, thereby enhancing the enantioselectivity (Fig. 6c and Supplementary Fig. 6). Additionally, the independent gradient model based on Hirshfeld partition analysis was performed to identify potential intermolecular interactions among the ligand, 2AC and INT-6 (IGMH, illustrated in Fig. 6d and Supplementary Fig. 7). In TS5-S, multiple noncovalent interactions, namely C–H···F, C–F···π, C–H···O, and C–H···π, were distinctly observed. In contrast, only the C–H···π interaction was observed in TS5-R. These results elucidate the preferential formation of the S-configured product, providing a rationale for the observed 95:5 enantiomeric ratio favoring (S)-33. An alternative pathway involving the copper-allyl intermediate through the reductive elimination process, specifically from intermediate INT-6 to transition state TS8-S, was also investigated. This pathway exhibited a ΔG‡ of 15.0 kcal/mol, which is 4.2 kcal/mol higher than that of the radical cross-coupling process, making it energetically less favorable (Supplementary Fig. 5). It should be noted that this asymmetric C–N bond formation represents a rare example of enantioselective cross-coupling of allylic radical and metal-stabilized nitrogen-centered radical.

a Spin density calculation of INT-6 species. b Gibbs energy profiles of copper catalytic cycles providing S-type (black line) and R-type (red line) 1,4-adduct (right) and S-type (black line) and R-type (red line) 1,2-adduct (left) involves cross-coupling process of two radical intermediates. c Optimized structures and relative energies of transition states TS5-S, TS5-R and TS5’-S. d Schematics for the transition-state structures for enantio- and regio-selectivity and the corresponding independent gradient model based on IGMH.
To elucidate the origin of the regioselectivity, the radical coupling C–N bond formation leading to the 1,2-adduct was also calculated. This process proceeded through transition states TS5’-S with a ΔG‡ of 13.9 kcal/mol (Fig. 6b, left black line), which was higher than TS5-S (ΔG‡ = 10.8 kcal/mol, affording the 1,4-addition product) by 3.1 kcal/mol. Further structural analysis (Fig. 6c and Supplementary Fig. 6) revealed that in TS5’-S, the CF3 group of 2AC is oriented toward the cavity of the chiral ligand, in contrast to its orientation in TS5-S. This results in the disruption of the stabilizing noncovalent interactions, including the C–H···F hydrogen bonding and C–H···π, which are mediated by the CF3 group and the aromatic phenyl ring of the ligand. In TS5-S, the CF3 group is proximal to the five-membered heterocyclic ring of ligand and the methyl moiety of 4-(tert-butyl)-N-methylaniline (2d). The favorable arrangement maximizes the noncovalent interactions between the catalyst and substrates, which were further confirmed by IGMH analysis (Fig. 6d and Supplementary Fig. 7). These favorable noncovalent interactions are served as the primary driving force for the unusual 1,4-regioselectivity observed in the radical cross-coupling process.
Based on the controlled experiments and DFT investigations, we proposed a plausible mechanism for this transformation as outlined in Fig. 7. Firstly, a single-electron transfer (SET) between Togni reagent 3a and Cu(I) in the presence of chiral ligand afforded CF3 radical species and chiral Cu(II)–O complex Com1. Subsequently, CF3 radical added to 1,3-diene 1p to afford allylic radical 2AC. Meanwhile, interaction of Com1 and aniline derivative 2 d formed a chiral Cu(I)–NRAr’ radical complex INT-6, which enantioselectively coupled with 2AC to finally provide trifluoromethylated chiral allylic amine 33, along with the regeneration of Cu(I) species into the next catalytic recycle. This transformation represents a scarce enantioselective radical 1,4-difunctionalization of 1,3-dienes.

A plausible reaction mechanism for copper-catalyzed enantioselective radical 1,4-trifluoromethylamination of 1,3-dienes with trifluoromethyl reagents and anilines.
Discussion
In summary, we have successfully developed a Cu-catalyzed enantioselective three-component radical 1,4-perfluoroalkylamination of 1,3-dienes with aniline derivatives and perfluoroalkyl reagents. This reaction renders the efficient assembly of a series of crucially important chiral perfluoroalkyl-containing allylic amines in distinctive regioselectivity and excellent enantioselectivity. The experimental and DFT studies provided reasonable explanations for the excellent regioselectivity and enantioselectivity of the elementary C–N bond formation in the proposed catalytic cycle and revealed that the asymmetric formation of C–N bond involved an unequaled enantioselective cross-coupling between allylic radical and copper-stabilized nitrogen-centered radical. This strategy blazes an alternative way towards chiral allylic compounds and provides alternative for the regioselective and enantioselective transformation of conjugated dienes.
Methods
General procedure for enantioselective 1,4-trifluoromethylamination of 1,3-dienes
Into a nitrogen-filled glove box, a vial (15.0 mL) equipped with a magnetic stir bar was charged with Cu(CH3CN)4PF6 (0.01 mmol), L1 (0.012 mmol). Anhydrous acetonitrile (0.4 mL) and ethyl acetate (1.6 mL) were added, and the reaction mixture was stirred for 15 min until it changes from colorless to green. Then 1 (2.5 equiv.), 2 (0.1 mmol), and 3a (2.0 equiv.) were added sequentially. Finally, the tube was sealed with a Thermo Scientific PTFE screw cap equipped with a septum, removed from the glovebox. The reaction mixture was stirred at −10 °C for 72–96 h, until the reaction was complete as indicated by TLC. The reaction mixture was then quenched with water, extracted with CH2Cl2 (3 × 10 mL) and the combined organic layers were concentrated in vacuo. The resulting crude product was purified by flash column chromatography on silica gel to give the corresponding product.
General procedure for enantioselective 1,4-polyfluoromethylamination of 1,3-dienes
Into a nitrogen-filled glove box, a vial (15.0 mL) equipped with a magnetic stir bar was charged with Cu(CH3CN)4PF6 (0.01 mmol), L1 (0.012 mmol). Anhydrous dichloromethane (1 mL) was added, and the reaction mixture was stirred for 15 min until it changes from colorless to green. Then 2 (0.1 mmol), 1 (2.5 equiv.), LPO (2.0 equiv.) and 3 (2.0 equiv.) were added sequentially. Finally, the tube was sealed with a Thermo Scientific PTFE screw cap equipped with a septum, removed from the glovebox. The reaction mixture was stirred at −20 °C for 72 h, until the reaction was complete as indicated by TLC. The reaction mixture was then quenched with water, extracted with CH2Cl2 (3 × 10 mL), and the combined organic layers were concentrated in vacuo. The resulting crude product was purified by flash column chromatography on silica gel to give the corresponding product.
Data availability
Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2351254 (90). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of the study are available within the article and its Supplementary Information, or from the corresponding author upon request. Source data are provided with this paper.
References
Johannsen, M. & Jorgensen, K. A. Allylic amination. Chem. Rev. 98, 1689–1708 (1998).
Skoda, E. M., Davis, G. C. & Wipf, P. Allylic amines as key building blocks in the synthesis of (E)-alkene peptide isosteres. Org. Process Res. Dev. 16, 26–34 (2012).
Patick, A. K. et al. In vitro antiviral activity of AG7088, a potent inhibitor of human rhinovirus 3C protease. Antimicrob. Agents CH 43, 2444–2450 (1999).
Muller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881–1886 (2007).
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320–330 (2008).
Corbett, J. W. et al. Expanded-spectrum nonnucleoside reverse transcriptase inhibitors inhibit clinically relevant mutant variants of human immunodeficiency virus type 1. Antimicrob. Agents CH 43, 2893–2897 (1999).
Lu, H. & Silverman, R. B. Fluorinated conformationally restricted γ-aminobutyric acid aminotransferase inhibitors. J. Med. Chem. 49, 7404–7412 (2006).
Li, G., Huo, X., Jiang, X. & Zhang, W. Asymmetric synthesis of allylic compounds via hydrofunctionalisation and difunctionalisation of dienes, allenes, and alkynes. Chem. Soc. Rev. 49, 2060–2118 (2020).
Wu, X. & Gong, L.-Z. Palladium(0)-catalyzed difunctionalization of 1,3-dienes: from racemic to enantioselective. Synthesis 51, 122–134 (2019).
Xiong, Y., Sun, Y. & Zhang, G. Recent advances on catalytic asymmetric difunctionalization of 1,3-dienes. Tetrahedron Lett. 59, 347–355 (2018).
Overman, L. E. & Rosen, M. D. Terminating catalytic asymmetric Heck cyclizations by stereoselective intramolecular capture of η3-allylpalladium intermediates: total synthesis of (−)-spirotryprostatin B and three stereoisomers. Tetrahedron 66, 6514–6525 (2010).
Chen, S.-S., Meng, J., Li, Y.-H. & Han, Z.-Y. Palladium-catalyzed enantioselective heteroannulation of 1,3-dienes by functionally substituted aryl iodides. J. Org. Chem. 81, 9402–9408 (2016).
Chen, S.-S., Wu, M.-S. & Han, Z.-Y. Palladium-catalyzed cascade sp2 C−H functionalization/intramolecular asymmetric allylation: from aryl ureas and 1,3-dienes to chiral indolines. Angew. Chem. Int. Ed. 56, 6641–6645 (2017).
Zhang, T. et al. Pd(II)-catalyzed asymmetric oxidative annulation of N-alkoxyheteroaryl amides and 1,3-dienes. Org. Lett. 21, 2048–2051 (2019).
Flubacher, D. & Helmchen, G. Enantioselective domino heck-allylic amination reactions. Tetrahedron Lett. 40, 3867–3868 (1999).
Wang, J. et al. Pd-catalyzed enantioselective three-component carboamination of 1,3-cyclohexadiene. J. Am. Chem. Soc. 146, 21231–21238 (2024).
Kagechika, K., Ohshima, T. & Shibasaki, M. Asymmetric Heck reaction-anion capture process. A catalytic asymmetric synthesis of the key intermediates for the capnellenols. Tetrahedron 49, 1773–1782 (1993).
Liu, Y., Xie, Y., Wang, H. & Huang, H. Enantioselective aminomethylamination of conjugated dienes with aminals enabled by chiral palladium complex-catalyzed C–N bond activation. J. Am. Chem. Soc. 138, 4314–4317 (2016).
Du, H., Yuan, W., Zhao, B. & Shi, Y. Catalytic asymmetric diamination of conjugated dienes and triene. J. Am. Chem. Soc. 129, 11688–11689 (2007).
Zhao, B., Du, H., Cui, S. & Shi, Y. Synthetic and mechanistic studies on Pd(0)-catalyzed diamination of conjugated dienes. J. Am. Chem. Soc. 132, 3523–3532 (2010).
Wu, M.-S., Fan, T., Chen, S.-S., Han, Z.-Y. & Gong, L.-Z. Pd(II)-catalyzed asymmetric oxidative 1,2-diamination of conjugated dienes with ureas. Org. Lett. 20, 2485–2489 (2018).
Li, X., Song, H., Yu, S., Mi, R. & Li, X.-X. Rhodium-catalyzed enantioselective 1,4-oxyamination of conjugated gem-difluorodienes via coupling with carboxylic acids and dioxazolones. Angew. Chem. Int. Ed. 62, e202305669 (2023).
Yan, M., Lo, J. C., Edwards, J. T. & Baran, P. S. Radicals: reactive intermediates with translational potential. J. Am. Chem. Soc. 138, 12692–12714 (2016).
Studer, A. & Curran, D. P. Catalysis of radical reactions: a radical chemistry. Perspect. Angew. Chem. Int. Ed. 55, 58–102 (2016).
Mondal, S. et al. Enantioselective radical reactions using chiral catalysts. Chem. Rev. 122, 5842–5976 (2022).
Gu, Q.-S., Li, Z.-L. & Liu, X.-Y. Copper(I)-catalyzed asymmetric reactions involving radicals. Acc. Chem. Res. 53, 170–181 (2020).
Wang, F., Chen, P. & Liu, G. Copper-catalyzed radical relay for asymmetric radical transformations. Acc. Chem. Res. 51, 2036–2046 (2018).
Wang, P.-Z., Xiao, W.-J. & Chen, J.-R. Recent advances in radical-mediated transformations of 1,3-dienes. Chin. J. Catal. 43, 548–557 (2022).
Du, H., Zhao, B., Yuan, W. & Shi, Y. Cu(I)-catalyzed asymmetric diamination of conjugated dienes. Org. Lett. 10, 4231–4234 (2008).
Liu, Z.-L., Yan, J.-L., Chen, K., Xiang, H.-Y. & Yang, H. Enantioselective 1,2-carboamination of 1,3-dienes with N-hydroxyphthalimide (NHP) esters enabled by a photoinduced Pd catalysis. Org. Lett. 26, 8762–8767 (2024).
Zhu, X. et al. Asymmetric radical carboesterification of dienes. Nat. Commun. 12, 6670 (2021).
Chen, J. et al. Photoinduced copper-catalyzed asymmetric C−O cross-coupling. J. Am. Chem. Soc. 143, 13382–13392 (2021).
Lu, F.-D., Lu, L.-Q., He, G.-F., Bai, J.-C. & Xiao, W.-J. Enantioselective radical carbocyanation of 1,3-dienes via photocatalytic generation of allylcopper complexes. J. Am. Chem. Soc. 143, 4168–4173 (2021).
Forster, D., Guo, W., Wang, Q. & Zhu, J. Dual photoredox and copper catalysis: enantioselective 1,2-amidocyanation of 1,3-dienes. ACS Catal. 13, 7523–7528 (2023).
Xiong, Y. & Zhang, G. Enantioselective 1,2-difunctionalization of 1,3-butadiene by sequential alkylation and carbonyl allylation. J. Am. Chem. Soc. 140, 2735–2738 (2018).
Schwarz, J. L., Huang, H.-M., Paulisch, T. O. & Glorius, F. Dialkylation of 1,3-dienes by dual photoredox and chromium catalysis. ACS Catal. 10, 1621–1627 (2020).
Zhan, X. et al. Catalytic asymmetric cascade dearomatization of indoles via a photoinduced Pd-catalyzed 1,2-bisfunctionalization of butadienes. Angew. Chem. Int. Ed. 63, e202404388 (2024).
Liu, Z.-L. et al. Photoinduced, palladium-catalyzed enantioselective 1,2-alkylsulfonylation of 1,3-dienes. ACS Catal. 14, 3725–3732 (2024).
Li, G.-Q. et al. Photoinduced copper-catalyzed asymmetric three-component radical 1,2-azidooxygenation of 1,3-dienes. Angew. Chem. Int. Ed. 63, e202405560 (2024).
Ruan, X.-Y. et al. Photoinduced Pd-catalyzed enantioselective carboamination of dienes via aliphatic C−H bond elaboration. J. Am. Chem. Soc. 146, 12053–12062 (2024).
Yuan, X. et al. Asymmetric radical oxyboration of β-substituted styrenes via late-stage stereomutation. Angew. Chem. Int. Ed. 62, e202313770 (2023).
Miao, H., Guan, M., Xiong, T., Zhang, G. & Zhang, Q. Cobalt-catalyzed enantioselective hydroamination of arylalkenes with secondary amines. Angew. Chem. Int. Ed. 62, e202213913 (2023).
Liang, Y.-J. et al. Copper-catalyzed hydroamination of polyfluoroalkyl substituted alkenes via asymmetric radical cross-coupling access to α-chiral tertiary alkylamines. Chem. Catal. 2, 2379–2390 (2022).
Qin, T. et al. Cobalt-catalyzed asymmetric alkylation of (hetero)arenes with styrenes. Angew. Chem. Int. Ed. 61, e202201967 (2022).
Qin, T. et al. Cobalt-catalyzed radical hydroamination of alkenes with N-fluorobenzenesulfonimides. Angew. Chem. Int. Ed. 60, 25949–25957 (2021).
Yang, S. et al. Copper-catalyzed asymmetric aminocyanation of arylcyclopropanes for synthesis of γ-amino nitriles. ACS Catal. 9, 716–721 (2019).
Zheng, G., Li, Y., Han, J., Xiong, T. & Zhang, Q. Radical cascade reaction of alkynes with N-fluoroarylsulfonimides and alcohols. Nat. Commun. 6, 7011 (2015).
Zhu, R. & Buchwald, S. L. Enantioselective functionalization of radical intermediates in redox catalysis: copper-catalyzed asymmetric oxytrifluoromethylation of alkenes. Angew. Chem. Int. Ed. 52, 12655–12658 (2013).
Liang, Z., Wang, F., Chen, P. & Liu, G. Copper-catalyzed in-termolecular trifluoromethylthiocyanation of alkenes: convenient access to CF3-containing alkyl thiocyanates. Org. Lett. 17, 2438–2441 (2015).
Wu, F.-P., Yuan, Y. & Wu, X.-F. Copper-catalyzed 1,2-trifluoromethylation carbonylation of unactivated alkenes: efficient access to β-trifluoromethylated aliphatic carboxylic acid derivatives. Angew. Chem. Int. Ed. 60, 25787–25792 (2021).
The oxidation state of Cu in TSI-S and TSI-R was analyzed by a bonding analysis based on localized orbital bonding analysis (LOBA), see: Thom, A. J. W., Sundstroma, E. J. & Head-Gordon, M. LOBA: a Localized Orbital Bonding Analysis to Calculate Oxidation States, with Application to a Model Water Oxidationcatalyst. Phys. Chem. Chem. Phys. 11, 11297–1130 (2009).
Cho, H., Suematsu, H., Oyala, P. H., Peters, J. C. & Fu, G. C. Photoinduced, copper-catalyzed enantioconvergent alkylations of anilines by racemic tertiary electrophiles: synthesis and mechanism. J. Am. Chem. Soc. 144, 4550–4558 (2022).
Acknowledgements
We thank the financial support from the National Natural Science Foundation of China (22371037 to Y. L. and 22193012 to Q. Z.).
Author information
Authors and Affiliations
Contributions
Y.L. and Q.Z. directed the projects; Y.L. and Q.Z. designed the experiments; X.S. and Y.Z. performed experiments; L.Z. performed DFT calculations; all authors contributed to data analysis; Y.L. and L.Z. wrote the manuscript with feedback from the other authors; Q.Z. reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shangguan, X., Zhu, L., Zhang, Y. et al. Copper-catalyzed enantioselective three-component radical 1,4-perfluoroalkylamination of 1,3-dienes. Nat Commun 16, 4939 (2025). https://doi.org/10.1038/s41467-025-60227-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60227-0
