
Abstract
Oncogenic alterations in fibroblast growth factor receptor (FGFR)-family proteins occur across cancers, including pediatric gliomas. Our genomic analysis of 11,635 gliomas across ages finds that 5.3% of all gliomas harbor FGFR alterations, with an incidence of almost 9% in pediatric gliomas. Alterations in FGFR proteins are differentially enriched by age, tumor grade, and histology, with FGFR1 alterations associated with glioneuronal histologies. Leveraging isogenic systems, we confirm FGFR1 alterations to induce downstream Mitogen Activated Protein Kinase (MAPK) and mTOR signaling pathways, drive gliomagenesis, activate neuronal transcriptional programs and exhibit sensitivity to MAPK pathway and pan-FGFR inhibitors. Finally, we perform a retrospective analysis of clinical responses in children diagnosed with FGFR-altered gliomas and find that treatment with currently available inhibitors is largely associated with stability of disease. This study provides key insights into the biology of FGFR1-altered gliomas, therapeutic strategies to target them and associated challenges that still need to be overcome.
Similar content being viewed by others
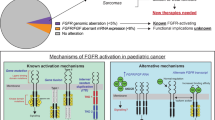

Introduction
Fibroblast growth factor receptor (FGFR) family proteins are commonly altered across many human cancers, including but not restricted to adult gliomas, cholangiocarcinomas, gastric, breast, lung, and bladder cancers1,2,3. The presence of FGFR alterations in these tumors has motivated the development of pan-FGFR inhibitors, of which there are four that are FDA approved for use in adult cancers. FGFR alterations have also been reported in pediatric gliomas4,5,6,7,8, however, their incidence across glioma subtypes and optimal strategies to target them have not been fully elucidated.
Somatic driver events involving FGFR1 and FGFR2 have been described in pediatric low-grade gliomas (pLGG) and pediatric high-grade gliomas (pHGGs), and rearrangements in FGFR3 were first reported in adult gliomas4,5,6,7,8. These alterations include recurrent rearrangements in FGFR1 or FGFR3, most frequently fused to TACC1 or TACC3, respectively, with FGFR1::TACC1 rearrangements occurring in pediatric gliomas and FGFR3::TACC3 in adult glioblastomas (GBMs)9,10,11. FGFR2 rearrangements have also been reported in pLGG, including with the fusion partners CTNNA3, INA, KIAA1598, and OPTN, which are found most commonly in pediatric low-grade neuroepithelial tumors12,13. FGFR inhibition has been evaluated in trials of adult patients with FGFR3::TACC3 rearranged GBM14. However, these studies were associated with limited responses, likely due to the aggressive nature of GBM, as well as dose-limiting toxicities and drug resistance2,15,16. Therefore, such agents have not been significantly studied in clinical trials for pediatric FGFR-altered low-grade gliomas but have potential to yield more promising results given the less aggressive nature of these tumors.
pLGGs are associated with high overall survival rates, but severe life-long co-morbidities, as the current standard of care treatment for children with LGGs includes surgical resection, and multi-agent chemotherapy17. pLGGs most commonly have driver alterations in the Mitogen-activated protein kinase (MAPK) pathway, with BRAF alterations being the most prevalent5,6,18. Targeted therapies, including MAPK-targeted therapies and BRAF inhibitor therapies, have therefore been evaluated in clinical trials for patients with recurrent disease. Such efforts have yielded promising results with recent FDA approvals of the RAF inhibitor tovarafenib for recurrent BRAF-altered pLGGs, and the combination of dabrafenib/trametinib19,20,21 for pLGGs with BRAF V600E mutations. Given that these drugs have primarily been directed towards patients with BRAF-altered gliomas, it is still unclear if agents such as MAPK inhibitors will be effective in the unique setting of FGFR-altered gliomas. The ideal therapeutic strategy to target these FGFR-altered pediatric gliomas remains unknown.
To address this, we applied an integrative functional genomic approach to systematically evaluate the role of FGFR drivers in pediatric gliomas. We found that 5.3% of all pediatric and adult gliomas harbor FGFR alterations, with an incidence of 8.9% in pediatric gliomas. FGFR1 alterations were most frequent in pediatric gliomas, particularly pLGGs, and were sufficient to induce gliomagenesis. Generating isogenic neural stem cell model systems, we found FGFR1-altered model systems to be sensitive to pan-FGFR inhibition in vitro, with modest results observed in vivo. Notably, these findings are consistent with early experience in pediatric patients treated with currently available MEK or FGFR inhibition, providing key insights into the design of therapeutic approaches for children with FGFR-altered pLGG.
Results
Gliomas harbor a diverse landscape of alterations in FGFR family members and are associated with specific patterns of age and co-mutations
FGFR alterations in pediatric gliomas have been described; however, their incidence and association with tumor subtypes and other clinical features have not been evaluated across large cohorts of patients4,5,6,7. To address this, we examined a cohort of 11,635 gliomas from pediatric (0–17 years; n = 1344) and adult (≥18 years; n = 10,276) patients with genomic sequencing results to identify any tumors with alterations in FGFR1-4 genes. We identified 619 FGFR-altered gliomas (5.3%, 619/11,635) across three independent datasets (DFCI, KiTZ, and Foundation Medicine), with an incidence of 8.9% in pediatric patients (120/1,344) (Supplementary Fig. S1a and Supplementary Data S2)3,22. Two of these datasets represented institute cohorts that were prospectively generated in the clinical setting, including gliomas at DFCI (between 2013 and 2019) or within the Molecular Neuropathology 2.0 Study (MNP) led by KiTZ22. The third dataset includes gliomas that were sequenced by Foundation Medicine from samples referred for clinical sequencing by multiple institutes.
Pediatric and adult gliomas harbored recurrent but highly diverse classes of alterations in different members of the FGFR family. Alterations in FGFR1 (50.8%, 315/619) and FGFR3 (42.6%, 264/619) were by far the most common, with FGFR2 (5%, 31/619) and FGFR4 (1.45%, 9/619) being altered in only a small subset of tumors. FGFR1 alterations were more prevalent in pediatric patients (88% of all FGFR alterations) relative to adult patients (42% of all FGFR alterations; Fisher’s exact p < 0.0001). FGFR2 alterations also trended to being enriched in pediatric gliomas (9% of all FGFR-altered gliomas) relative to adult gliomas (4.2% of all FGFR-altered gliomas, Fisher’s exact p: 0.058), while FGFR3 alterations occurred almost exclusively in adult gliomas (52% of adult FGFR alterations versus 4% of pediatric, Fisher’s exact p < 0.0001). We did not observe any significant associations between patient sex and the altered FGFR gene (Supplementary Fig. S1b) (Fisher’s exact p = 0.994).
FGFR-altered gliomas occur in all locations of the brain; however, different FGFR family members exhibit anatomical predilection. Overall, 69% of all FGFR-altered tumors (81/117) in the DFCI and KiTZ cohorts (which had tumor location data available) were hemispheric tumors, with thalamic involvement (13/117) representing the next most frequent location at 11% (Supplementary Fig. S1c, Supplementary Data S2). However, while FGFR2- and FGFR3-altered gliomas are predominantly hemispheric tumors, only 58% of all FGFR1-altered gliomas (46/79) were hemispheric. Instead, FGFR1 alterations were also commonly found in midline gliomas that occur in the thalamus and brainstem, and the cerebellum (Supplementary Fig. S1c).
FGFR1 alterations included both single-nucleotide variants (SNVs) (41.2% of all FGFR-altered gliomas) and structural variants (SVs) (9.7% of all FGFR-altered gliomas, Fig. S1d–f and Supplementary Data S2). SNVs in FGFR1 are predicted to activate the FGFR1 kinase domain (FGFR1 N546K and K656E mutations) (Supplementary Fig. S1d) and have been previously reported in cohorts of pediatric and adult gliomas, in addition to extracranial tumors23,24,25,26,27. We also identified four other putative driver mutations of unknown significance, of which three were in the kinase domain (T141R, N546D, T657S, T658P).
The most common FGFR1 SVs included both gene rearrangements (27/619) that induce expression of a fusion protein (4.36% of all FGFR-altered gliomas), or tandem duplication (ITD) of the FGFR1 kinase domain (13/619), which occurred in 2.1% of all FGFR-altered gliomas, a similar incidence to prior reports (Supplementary Fig. S1e, f and Supplementary Data S2)4,5,6. FGFR1 rearrangements most frequently involved the TACC1 fusion partner, as has been previously reported9,10,11. This fusion protein is predicted to retain exons 1-17 of FGFR1, including its membrane and kinase domains, fused with exons 7-13 of the C-terminal TACC1 protein (Supplementary Fig. S1e). Our cohort also included FGFR1 alterations with three additional fusion partners, one that our group has previously reported (CLIP2) in pediatric gliomas27, one previously reported in small cell lung cancer (PLEKHA2)28, and one that has not been reported (ARHGEF18). PLEKHA2, the gene that encodes the protein Plekstrin Homology Domain Containing A2, was found in two gliomas from two different cohorts (Supplementary Data S2).
The FGFR1-ITD, as previously reported4,29,30, centers on the FGFR1 kinase domain itself, resulting in its amplification (Supplementary Fig. S1f). It is important to note that the FGFR1-ITD cannot be identified using current pipelines within the Foundation Medicine cohort. Taking this into account, the incidence of FGFR1-ITDs increased to 11% of all of the FGFR-altered tumors within the DFCI and KiTZ datasets (Supplementary Data S2). FGFR1-ITDs occurred almost exclusively in pLGG, however, our cohort did include one pediatric patient with an FGFR1-ITD in the setting of a MYCN-activated high-grade glioma (Fig. 1a, b and Supplementary Data S2). In addition to these, 3.2 % of FGFR-altered gliomas harbored copy-number variants such as gains or duplications.

a Heatmap showing hierarchical clustering of the methylation data (top 10,000 differentially methylated probes; ward D2; Euclidean distance) for the 425 patients in the KiTZ cohort. This heatmap shows beta-values as indicated by the color scale. Cluster 1 and 2, 2A and 2B are labeled on the heatmap. Metadata highlighted include if the tumor was FGFR-altered, which FGFR gene was altered or co-altered, which alteration type, the methylation family prediction, tumor grade, and if it was a glioneuronal tumor. b Co-mutation plot summarizing alterations in the FGFR genes and other recurrently mutated genes across 120 FGFR-altered pediatric tumors in the three cohorts (DFCI, KiTZ, FM). The co-mutation plot is ordered by FGFR alteration type, patient age, and grade. N/A- data not available. FM- Foundation Medicine. c Horizontal bar plots depicting the top 5 significant Gene Ontology C5 (MsigDB) terms enriched (ranked by significance) in FGFR1- (n = 16) or BRAF-altered (n = 130) gliomas. Negative log(FDR) values from the GSEA analysis are shown on the x-axis. Source data are provided in Supplementary Data S2 and S3 and the Source Data File.
Alterations involving FGFR2 and FGFR3 were primarily SVs (Supplementary Fig. S1g, h), including rearrangements or copy-number alterations. As previously reported, CTNNA3 and TACC3 were the most frequent rearrangement partners for FGFR2 and FGFR3, respectively9,10. We also found 11 fusion partners, to our knowledge, that have not been previously reported across any cancers (Supplementary Data S2). In addition, we found another two fusion partners that have been previously reported in extra-cranial tumors, but not in the context of brain tumors31,32. These include SHTN1 and GKAP1, the genes that encode the proteins Shootin 1 and G Kinase Anchoring Protein 1, respectively (Supplementary Data S2).
We also detected SNVs in FGFR2 and FGFR3. FGFR2 SNVs were observed in 3% of all FGFR-altered gliomas. These include previously reported2,33 alterations that most commonly occur in the kinase region (A648T, K659E, R664W) of FGFR2, followed by the extracellular region (A97T, R203H, R251Q, P303L, D336N), and transmembrane domain (D336N) (Supplementary Fig. S1i). SNVs in FGFR3 represented 4.3% of all FGFR-altered gliomas, and the mutations spanned multiple regions of the gene. The K650E mutation in the kinase region was the most common mutation in our cohorts, and this has been reported in multiple cancers, including GBM (Supplementary Fig. S1j)33. We observed rare SNVs or copy-number alterations in FGFR4 in nine adult patients, accounting for 1.45% of the cohort of FGFR-altered gliomas. These mutations have not been reported in gliomas but have been in other cancers (Supplementary Fig. S1k)33,34,35.
Finally, we found distinct associations between the frequency of SVs for each FGFR family member and pediatric and adult gliomas. In children, SVs involving FGFR1 occurred in both low-grade and high-grade gliomas. Within the DFCI and KiTZ cohorts, in which all tumors underwent review by a pediatric neuropathologist, 34% of all FGFR1 rearrangements and/or ITDs occurred in the context of pLGGs, while 8% in the pHGGs. In contrast, gliomas with FGFR2 SVs were exclusively observed in pLGG, while FGFR3-rearranged gliomas were primarily observed in adult high-grade gliomas (glioblastoma IDH wild-type) (Supplementary Fig. S2a, b). FGFR1-ITD and FGFR2 SVs were only found in pediatric gliomas, whereas FGFR1/3 gain, FGFR3 fusion, and FGFR4 alterations were almost exclusively found in adult gliomas (Supplementary Fig. S2a).
Taken together, we conclude that FGFR alterations are found across a heterogeneous group of gliomas, spanning both pediatric and adult patients.
Pediatric glioneuronal gliomas are enriched for FGFR alterations
We next focused on delineating associations between FGFR alterations and subtypes of gliomas within all the gliomas in the KiTZ cohort for which DNA methylation profiles were available (n = 425, Fig. 1a and Supplementary Fig. S2c–e). Applying unsupervised hierarchical clustering to further dissect the heterogeneity within FGFR-altered pediatric gliomas, we observed two broad clusters of gliomas, which we defined as Cluster 1 and Cluster 2. Only 5% of all gliomas in Cluster 1 (4/78) harbored FGFR alterations, and these were almost all high-grade gliomas (96%, 75/78), of which 27% were histone-mutant gliomas (21/78). Cluster 2 is separated into two further subgroups. The first group (2A) was heavily enriched for pilocytic astrocytomas (98.5%, 207/210, Fisher’s exact p < 0.0001), and only 3.8% (8/210) of these included FGFR alterations. In contrast, the second group (2B) had the highest incidence of FGFR alterations, accounting for 13% (18/137) of gliomas within this group. This subcluster of gliomas included the vast majority of all glioneuronal tumors within the entire cohort (37%, 51/137; Fisher’s exact p < 0.0001).
FGFR-altered tumors within this cohort included those that had been histologically diagnosed as DNET (n = 10), Pilocytic astrocytoma (n = 5), Rosette-forming glioneuronal tumors (n = 2), and high-grade gliomas including Anaplastic astrocytoma (n = 3), GBM (n = 1), H3K27M mutant DMG (n = 2), or anaplastic pilocytic astrocytoma (n = 3). Four tumors included were challenging to diagnose by pathology. Two were given ‘descriptive’ diagnoses, and the other two were initially diagnosed as CNS neuroblastoma or medulloblastoma. These four tumors were reclassified according to their methylation profiling as DNET, ganglioglioma, disseminated leptomeningeal glioneuronal tumor, and high-grade glioma, respectively.”
In addition, glioneuronal tumors identified in the DFCI and KiTZ cohorts were specifically enriched in FGFR1-altered pediatric gliomas (Fisher’s exact p = 6e-11), with glioneuronal FGFR1-altered tumors making up 5.3% of all the pediatric tumors.
Together, these analyses suggest that FGFR1 alterations in children are most frequently found in glioneuronal subtypes of LGGs. This observation is consistent with previous reports of FGFR alterations in several glioneuronal tumors, including DNET, extra-ventricular neurocytoma, and PLNTY12,23,24,26,36,37.
FGFR1-altered pediatric gliomas frequently harbor additional genetic events
One of the most distinctive aspects of pLGGs with FGFR1 SNVs was that they frequently co-occurred with other genetic events. This is in contrast with BRAF-altered pLGGs or pLGGs driven by other alterations, which are thought to largely represent single-driver tumors4,38. Within pLGGs in the DFCI and KiTZ cohorts, 35% of all gliomas with FGFR1 SNVs (15/40) harbored at least one additional somatic event, either involving FGFR1 itself (Supplementary Fig. S1d) or additional driver alterations predicted to activate mTOR or MAPK signaling. In our cohorts, there were four pLGG tumors that harbored co-occurring mutations in the MAPK/mTOR pathway including NF1 (n = 1), PTPN11 (n = 1), PTPN11 + PIK3CA (n = 1), and PIK3R1 (n = 1) (Fig. 1b and Supplementary Data S2)4,5,6,18,24,39. Intriguingly, the N546K driver mutation is found in three of these four tumors. Overall, the SNVs in these additional genes consisted of pathogenic SNVs in PTPN11, including G503R and E76Q SNVs that are in located in the phosphatase domain and the SH2 domain of the protein, respectively. In contrast, only 5% of pLGGs with FGFR1 SVs (2/40) harbored co-occurring alterations. FGFR1 SNVs in pediatric HGGs also co-occurred with other driver events that have been described in pediatric HGGs, however, these differed from those observed in pLGGs and included histone mutations, copy-number alterations, including MDM4 and CDK4 amplifications, and deletions in CDKN2A/B (Fig. 1b and Supplementary Data S2)7,8,18.
FGFR1-altered pLGGs with hotspot kinase mutations also harbored additional mutations in FGFR1 itself (11/40, 28%) (Fig. 1b and Supplementary Data S2). Of these 11 gliomas, one glioma harbored co-occurring FGFR1 T658P + T657S mutations, one glioma harbored the N546K SNV, while the remaining nine harbored the K656E kinase mutation. The glioma with the N546K mutation had a co-occurring SNV involving the kinase domain of FGFR1, including K656N. Similarly, gliomas with the K656E hotspot kinase SNV harbored additional SNVs within the kinase domain itself, including G660C, D652G, K638R, I651M, K656N, D652M, and N546S (Supplementary Fig. S1d and Supplementary Data S2).
Together, these analyses lead us to conclude that FGFR1-altered pLGGs represent the most frequent subgroup of FGFR-altered gliomas in children. Furthermore, we also find that these gliomas harbor significant heterogeneity within their genomic and clinical profiles. We focused our subsequent studies on these FGFR1-altered pLGGs with the goal of understanding how each FGFR1 alteration enhances glioma formation, and the clinical implications associated with these oncogenic mechanisms.
Expression profiles of FGFR-altered gliomas identify neuronal signatures and lineage associations to normal stages of brain development
Our observation that FGFR1-altered gliomas are predominantly classified as glioneuronal tumors by DNA methylation profiling raises the possibility that they may also express neuronal differentiation and signaling pathway genes. To investigate this, we interrogated transcriptome-wide expression profiles of 146 pLGG, 16 FGFR1-altered (9 SVs and 7 SNVs) and 130 BRAF-altered tumors (88 SVs, 42 SNVs)4,40,41 to identify gene signatures that are differentially expressed in FGFR-altered gliomas (Supplementary Data S3). Indeed, within this heterogeneous dataset that encompasses multiple histological datasets, with almost tenfold more gliomas with BRAF compared to FGFR alterations, we observed significant enrichment of gene sets and pathways in FGFR-altered gliomas. Gene set enrichment analysis (Gene Ontology C5 gene sets in MsigDB)42,43 of the 206 genes significantly upregulated in FGFR1-altered tumors revealed enrichment of neuronal gene sets specifically involving the synapse and neurotransmission (Fig. 1c and Supplementary Data S3). We observed similar enrichment of neuronal gene sets within the ‘Cell Type’ signatures (C8 gene sets in MsigDB)42,43, including GABAergic neurons and dopaminergic neurons (Supplementary Fig. S2f). In addition to upregulation of neuronal gene sets, GSEA analysis including gene sets within the C2 curated database also revealed enrichment of signatures associated with the regulation of cell differentiation programs, including of polycomb repression and promoter bivalency in stem and progenitor cells (Supplementary Fig. S2g). In contrast, BRAF-altered gliomas showed enrichment of gene sets that regulate immune response and extracellular matrix/matrisome pathways (Fig. 1c and Supplementary Fig. S2g), including those involved with differentiation of stromal and fibroblast cell types44 (Supplementary Fi. S2f).
These data provide evidence that FGFR1-altered gliomas are associated with clear enrichment of neuronal expression signatures compared to their BRAF-altered counterparts. This differential lineage enrichment between FGFR1 and BRAF-altered pLGGs may be reflective of developmental origins of the tumors, tumor location, or the consequence of different driver mutations.
Given the striking association between different FGFR family drivers across different classes of pediatric and adult gliomas, we first hypothesized that these differences reflect differences in the developmental origins of each glioma. We tested this by investigating temporal and spatial expression of each FGFR family member in prenatal development through postnatal life, across brain regions and cell types. We first profiled expression of each FGFR gene across time (prenatal to adult brain) and brain regions, using two human lifespan datasets of bulk RNA-sequencing, the Evo-devo atlas45 and Brainspan46 (Fig. 2a and Supplementary Figs. S3a–d and S4a, b).

a Expression of FGFR1 in bulk RNA-seq across lifespan in human forebrain (n = 55) (left) and hindbrain (n = 59) (right). X-axis denotes sample age, measured in natural log of weeks post conception, and x-axis tick values correspond to weeks in prenatal points and years in postnatal points. Vertical dashed line depicts birth. Y-axis depicts RPKM expression values. b, c UMAP representation of human embryo cells 4–5.5 weeks post conception. N = 7 embryos, 185k cells. b Left: Cells colored by neural progenitor cells annotated in the original study colored green. c Right: Cells colored by normalized expression of FGFR1. Color scale midpoint is set to the midpoint of maximum and minimum expression. d Detection rate (% of cells with expression >0) of FGFR1 expression in broad cell types of first trimester human brain. e Top 15 cell clusters by detection rate (% of cells with expression of >0) of Fgfr1 expression in developing mouse pons and forebrain. Cell clusters grouped by broad cell type.
Expression of both FGFR1 and FGFR3 was found in early development and exhibited distinct temporal patterns as development proceeded. In the forebrain, FGFR1 was expressed early in the prenatal brain and decreased towards birth (Fig. 2a and Supplementary Fig. S3a). In the cerebellum, in contrast, while prenatal patterns of FGFR1 expression mirrored those of the forebrain, postnatal expression continued to increase through life, a result corroborated across datasets47 (Fig. 2a and Supplementary Fig. S3a). In turn, FGFR3 displayed similar patterns for the forebrain and hindbrain, with higher expression in the first weeks of prenatal development (Supplementary Figs. S3c and S4b). FGFR2 and FGFR4 showed very low expression with minimal changes over developmental time (Supplementary Fig. S3b, d and S4a). These data suggest that FGFR genes have unique spatiotemporal expression during brain development, and further investigation will be needed to understand if these patterns have correlations with FGFR-altered brain tumors and the specific alteration type they harbor.
We next hypothesized that the association of FGFR1 alterations with multi-lineage glioneuronal tumors suggests that they may arise from a specific type of undifferentiated neural progenitor cell. We therefore assessed FGFR1 expression in single-cell RNA expression atlases of the human embryo 4–5.5 pcw48, human brain in the first trimester of gestation49, as well as murine atlases of the developing forebrain and pons50,51. Indeed, we found that FGFR1 expression was not restricted to any one cluster or cell type in the brain and instead was observed across multiple lineages, including glial cells, neuronal cells, neural and glial progenitors, and non-neuroectoderm cells across both human and mouse datasets (Fig. 2b–e, and Supplementary Fig. S4c).
We also evaluated associations between expression of FGFR2 and FGFR3 across development. FGFR2 was expressed highly in the spinal cord (Supplementary Fig. S4d) and enriched within multiple cell types, including the choroid plexus cells, oligodendrocytes, and astrocytes, both pre- and postnatally in single-cell mouse forebrain and pons (Supplementary Fig. S4e). FGFR3 is more abundantly expressed than FGFR1 and FGFR2 in bulk data but does not appear to demonstrate lineage specificity. Very early in development, before initiation of gliogenesis in humans and correlating with the prenatal peak observed in bulk datasets, FGFR3 is expressed in neural progenitors (Fig. 2b and Supplementary Fig. S4c, f). Later on, FGFR3 is most strongly detected in astrocytes, across regions and across species, with expression sustained in this lineage in the adult brain (Supplementary Fig. S4g, h). This is intriguing as FGFR3 alterations are predominantly found within glioblastoma in adults.
Overall, the presence of FGFR1 expression in both glial and neuronal cell types in the developing brain may suggest its expression is associated with early progenitor cells with similarity to phenotypes seen in glioneuronal tumors.
Expression of FGFR1 alterations is sufficient to induce growth factor independence
We next sought to evaluate the role of each FGFR alteration in driving gliomagenesis. Unfortunately, patient-derived FGFR1-altered pLGGs cannot be propagated ex vivo. We therefore generated a panel of isogenic primary mouse neural stem cell (mNSC) and TERT-immortalized human neural stem cell (ihNSC) models that were transduced to express FGFR1 alterations and other driver alterations that are found in pLGGs (Fig. 3a). In total, we generated a comprehensive collection of nine lines, including FGFR1 N546K (F1-N546K), FGFR1-ITD (F1-ITD), FGFR1::TACC1 (F1::TACC1), PTPN11 SNV (PTPN11 alone), as well as a line with co-mutations in FGFR1 and PTPN11 (F1-N546K + PTPN11). These were compared to lines with wild-type FGFR1 (F1-WT), BRAF drivers common in pLGGs (KIAA1549::BRAF fusion or BRAF V600E SNV), and GFP or HcRed (Vector control) as the control (Fig. 3a and Supplementary Fig. S5a–c).

a Overview of the generation of the isogenic NSC lines. Mouse or hTERT-immortalized human NSCs were virally transduced with each of the FGFR or BRAF alterations, or control vectors, followed by a second transduction containing the co-occurring PTPN11 mutation or control vectors. Created in BioRender. Apfelbaum, A. (2024) https://BioRender.com/p48w741. b Bar plots depicting the percentage of cumulative doubling growth rate at day 14 for each mNSC cell line grown with no growth factor (no gf)/with growth factor (+gf). Values and error bars depict the mean ± SEM of three independent biological replicates. Significance calculated by a Welch’s two-sided t test compared to the vehicle control. *p < 0.05, **p < 0.01, ***p < 0.001. c Same as in b but for the ihNSC lines. Representative immunoblots (densitometry of three biological replicates in Supplementary Fig. S5j–o) of downstream MAPK and PI3K/mTOR signaling pathway effectors for d mNSC and e ihNSC models. Phosphorylated proteins represent activated signaling pathways. pERK is a readout for MAPK signaling, and pAKT and pS6 are readouts for PI3K/mTOR signaling. Vector Control and PTPN11 alone are grown in the presence of growth factor, while lines harboring FGFR1 and BRAF drivers are grown without growth factor supplementation. Abbreviations for models= F1-N546K: FGFR1 N546K SNV, F1-N546K + PTPN11: FGFR1 N546K SNV + PTPN11 E69K SNV, F1-ITD: FGFR1-ITD, F1::TACC1: FGFR1::TACC1, V600E: BRAF V600E SNV, K::B: KIAA1549::BRAF. Source data provided in the Source Data File.
We first evaluated whether expression of FGFR1 alterations was sufficient to render our mNSCs and ihNSCs growth factor independent, a hallmark of transformation. Wild-type mNSCs and ihNSCs require supplementation of growth factors, including epidermal growth factor (EGF) and fibroblast growth factor (FGF2) to maintain stemness and proliferative potential. We leveraged this model system to withdraw EGF and FGF2 from each of our engineered cell lines and measured cell proliferation in the presence and absence of growth factors. All murine and human models grew robustly in the presence of growth factors (Supplementary Fig. S5d–i). mNSC models driven by FGFR1 alterations were able to maintain proliferation in the absence of growth factors and showed similar growth to BRAF-positive controls (Fig. 3b and Supplementary Fig. S5f, h). Significantly less growth was seen in FGFR1 WT, PTPN11 SNV, or vector control (Fig. 3b and Supplementary Fig. S5d). We also observed similar trends in the ihNSC where all FGFR1 alterations are sufficient to induce growth-factor independence, which was similar to BRAF while cells transduced with PTPN11 SNV or vector control exhibited slow growth in the absence of growth factor (Fig. 3c and Supplementary Fig. S5e, g, i).
FGFR1-driven NSCs sustain downstream MAPK and mTOR signaling
Growth factor independence supports the hypothesis that each FGFR1 alteration is sufficient to maintain MAPK and mTOR activation to transform NSCs. First, we assessed the levels of total and phosphorylated effector kinases (ERK, ribosomal protein S6, and AKT) using immunoblots and observed activation of both pathways across our panels of FGFR1 and BRAF-altered NSCs (Fig. 3d, e and Supplementary Fig. S5j–o). Importantly, the level of activation observed in FGFR1-altered models in the absence of growth factor was similar to that observed with vector control lines in the presence of growth factors for both the mNSC and ihNSC models, with no statistically significant differences found between the FGFR1-expressing models and vector controls (Fig. 3d, e and Supplementary Fig. S5j–o). While we observed a trend of higher ERK activation in the BRAF-altered models, this did not reach statistical significance (Supplementary Fig. S5k, n).
We further validated these findings across our models at both the transcriptomic and phosphoproteomic levels. First, we generated transcriptome-wide RNA-sequencing of our mNSCs and ihNSCs (Supplementary Fig. S6a–e) and assessed the level of MAPK activation by measuring expression of a transcriptional gene set that has been associated with MAPK-pathway activation (MAPK Pathway Activating Score (MPAS)52). Across our panel of mouse isogenic models, we did not observe any statistically significant differences between the models, further supporting our conclusion that the FGFR1- or BRAF-transformed models sustain MAPK signaling to similar levels as the vector controls passaged with full GF supplementation (Supplementary Fig. S7a). However, we did observe differences across the human NSC models, with those transduced to express each FGFR or BRAF variant and cultured in the absence of GF exhibiting increased activation of MAPK signaling compared to non-transformed vector control cells (Supplementary Fig. S7b).
Next, we assessed induction of phosphorylation of proteins associated with MAPK and mTOR activation, applying proteome-wide phosphoproteomics to our ihNSC models. Consistent with our prior finding across our immunoblots, we observed similar upregulation of phosphorylation of AKT1, ERK1, and ERK2 and S6 in the FGFR1-transduced NSCs (grown in no growth factor) compared to NSCs transduced with vector control (grown in growth factor) (Supplementary Fig. S7c). Similarly, we observed minimal differences in levels of activation of each pathway in the FGFR-driven models compared to NSCs transduced with each BRAF oncogene (Supplementary Fig. S7c).
Integrating each of these experiments, we conclude that expression of each FGFR1 variant in NSCs is sufficient to maintain activation of MAPK and mTOR signaling in the absence of growth-factor supplementation.
Neuronal transcriptional programs are enriched in FGFR1-altered mNSC models
We next sought to determine whether our panel of FGFR1-transformed NSCs transcriptionally resembles other features associated with human FGFR1-altered pLGGs, compared to those with BRAF alterations. We performed these analyses within each panel of isogenic NSCs, as our transcriptome-wide RNA-sequencing analysis revealed initial differences between our mouse and human isogenic models (Supplementary Fig. S6a–e). Applying PCA analysis, we found that the FGFR1-driven mouse neural stem cell models separated clearly from the BRAF-driven models, regardless of FGFR1 variant that they were transduced to express (Supplementary Fig. S6a, b). However, in the human NSC models, lines transduced to express SNVs in either BRAF or FGFR1 clustered more closely together, compared to those transduced to express SVs (Supplementary Fig. S6b, c). Within this group, there was clear separation between NSCs expressing each FGFR1 SV from those transduced to express the KIAA1549::BRAF fusion.
Within the mNSC lines, we found a total of 659 genes differentially expressed between the FGFR1- and BRAF-altered lines (Base mean > 50, FDR < 0.05, absolute LFC > 1.5) (Supplementary Data S4). Of these, 319 genes were upregulated in the FGFR1-altered models. The most differentially expressed gene was Hs6sp2, which encodes Heparan Sulfate 6-O Sulfotransferase 2 and is important for efficient signaling of FGFR family receptors53. Multiple genes involved in neuronal development, including Epha3, Emb, Ednrb, and Ntrk2, were also within the top 25 most differentially expressed genes in FGFR1 models (Fig. 4a).

a Heatmap of the top 25 genes differentially expressed (DE) in the FGFR1 vs BRAF-altered mNSC lines. Gene names highlighted in orange represent genes involved in neuronal development. b Horizontal bar plots depicting the top 5 significant Gene Ontology C5 (MsigDB) Terms enriched (ranked by significance) in FGFR1 (n = 4) or BRAF- (n = 2) altered mNSC lines. c Venn diagram depicting the overlap of the top 20 significantly enriched C5 gene sets in the FGFR1-altered patient tumors and FGFR1-altered mNSC lines (n = 5 gene sets intersected). Significance was calculated by performing a two-sided Fisher’s exact test (p = 2.6e-11). Red text in the overlap list highlights gene sets that were also enriched in the FGFR1 SV-altered ihNSC lines. d UMAP embedding of all FGFR1-altered mNSC lines, colored by line. N = 4 samples, 19,009 cells. e UMAP embeddings of the FGFR1-altered mNSCs colored by the GO Neurogenesis gene set. Legend depicts gene set score. N = 4 samples, 19,009 cells. Abbreviations for models= F1-N546K: FGFR1 N546K SNV, F1-N546K + PTPN11: FGFR1 N546K SNV + PTPN11 E69K SNV, F1-ITD: FGFR1-ITD, F1::TACC1: FGFR1::TACC1, V600E: BRAF V600E SNV, K::B: KIAA1549::BRAF. Source data provided in Supplementary Data S4 and the Source Data File.
Indeed, applying gene set enrichment analysis, we found that expression programs within mNSCs transduced with the FGFR1 oncogenes were significantly enriched with gene sets associated with neuronal differentiation when compared to BRAF-expressing cells, similar to our prior analyses across the human pLGG samples (Figs. 1c and 4b and Supplementary Data S4). In addition to neuronal program gene sets, we also found enrichment of gene sets associated with neuronal cell signatures, and polycomb repressive and bivalent marked genes (Supplementary Fig. S8a, b). In contrast, within the BRAF-altered cell lines, we saw enrichment of extracellular matrix (ECM) programs, adhesion and motility terms, and stromal and endothelial cell signatures (Fig. 4B; Supplementary Fig. S8c, d and Supplementary Data S4), also similar to our prior findings in BRAF-altered patient gliomas (Fig. 1c, and Supplementary Fig. S2f, g).
Human immortalized NSCs transduced to express FGFR1 SVs likewise exhibited upregulation of neuronal signatures compared to those expressing the KIAA1549::BRAF fusion. Given the minimal differences between ihNSCs transduced to express FGFR1 or BRAF SNVs, we restricted this analysis to models transduced to express FGFR1::TACC1 or the FGFR1-ITD, comparing to the KIAA1549::BRAF expressing line. Within this analysis, we found a total of 626 genes differentially expressed between the FGFR1 SV and BRAF-altered SV lines (Base mean > 50, FDR < 0.05, absolute LFC > 1.5). Next, we applied the same gene set enrichment analysis to the FGFR1 SV-enriched genes and identified significant enrichment in similar neuronal gene sets and polycomb repressive and bivalent marked genes as previously identified in the mNSCs and patient data (Supplementary Fig. S8c). In the BRAF SV line, there was similar enrichment of extracellular matrix programs, collagen gene sets, and stromal cell signatures as we identified in the mNSCs and patient data (Supplementary Fig. S8d).
We next sought to determine how closely our isogenic models recapitulated the differences we had found between human FGFR1-altered and BRAF-altered pLGGs. First, we evaluated whether there was significant overlap between gene sets enriched in our FGFR1- or BRAF-expressing isogenic models compared to the human FGFR1- or BRAF-altered pLGGs. Within the gene sets included in the C2, C5, and C8 GSEA databases, we observed significant overlap between enriched gene sets in the FGFR1 models compared to the BRAF models in our mNSCs and the patient tumors (p = 1.5e-19, p = 2.6e-11, p = 7.5e-15) (Fig. 4c and Supplementary Figs. S8e and S9c). Notably, the overlapping gene sets included multiple neuronal gene sets, in addition to those that regulate the polycomb repressive complex (Fig. 4c and Supplementary Figs. S8e and S9c). In each case, many of these gene sets were also enriched in our FGFR1-SV expressing isogenic ihNSCs. In addition, we found significant overlap of enriched gene sets in our BRAF-altered lines with the BRAF-altered patient tumors, specifically cell adhesion, ECM, and stromal gene sets (p = 1.3e-3, p = 6.1e-14, p = 2.3e-9) (Supplementary Fig. S9a, b, d). Taken together, we conclude that our isogenic FGFR1 models recapitulate increased expression of neuronal gene sets that are also observed in FGFR1-altered pLGGs.
This expression of neuronal expression programs within the FGFR1 models is heterogenous. We evaluated the heterogeneity of transcriptional signatures identified in the FGFR1-altered cell lines and patient tumors using RNA-sequencing of the FGFR1-altered mNSC lines (n = 19,009 cells). Using Uniform Manifold Approximation and Projection (UMAP) embedding for dimension reduction analysis, we found that the different FGFR1-altered mNSC lines clustered separately from each other (Fig. 4d), independent from expression of cell-cycle signatures within individual cells (Supplementary Fig. S9e). FGFR1-altered cells exhibited heterogenous expression of the GO neurogenesis gene set, both within and across cell lines (Fig. 4e). The FGFR1 SV-altered lines had significantly higher expression of the GO neurogenesis gene set compared to the FGFR1 SNV-altered lines, suggesting different FGFR1 alterations may influence these neuronal transcriptional states differently (Supplementary Fig. S9f).
These data suggest that our isogenic FGFR1-altered mNSCs express similar cell programs as the patient FGFR1-altered gliomas, with enrichment of gene sets associated with neuronal differentiation, development, and signaling compared to BRAF-altered glioma models.
FGFR1 alterations are sufficient to drive tumor formation in vivo
Having confirmed that our models recapitulated expression programs observed in human pLGGs, we next sought to evaluate whether each FGFR1 alteration was sufficient to induce gliomagenesis. We orthotopically injected the isogenic mNSC models into the brains of SCID mice and monitored for signs of tumor formation (Supplementary Fig. S10a). Mice injected with mNSCs transduced with vector control or PTPN11 E69K alone cells did not form any tumors (Fig. 5a, b). Intracranial injection of mNSC transduced to express our positive controls (BRAF V600E and KIAA1549::BRAF) was sufficient to induce glioma formation, with BRAF V600E resulting in more consistent and rapid tumor formation than the KIAA1549::BRAF fusion and FGFR1 alterations (Fig. 5b).

a Representative axial MRI images of gliomas following intracranial injection of the isogenic mNSC lines expressing oncogenes shown. Red arrow points to gliomas in the right hemisphere of the brain. b Kaplan–Meier survival curves of mice harboring intracranial allografts of mNSCs transduced to express each alteration. Ten mice were injected with each cell line. Significance was calculated by performing a Log-rank (Mantel-Cox) test performed. ***p < 0.0001. c Bar graph showing the percentage of mice with tumor hemorrhage by MRI assessment. d Representative H&E images showing glioma formation at low magnification (1.5x, scale bar = 1 mm) and high magnification (40x, scale bar = 100 μm), with H&E slides and Ki-67 staining (one representative image of one tumor per condition per stain/magnification- quantified in e–h). e–h For the box and whisker plots, the whiskers with error bars represent the minima and maxima values. The middle line represents the center value (50th percentile), and the bounds of the box represent the 25th to 75th percentiles of the data. AI-based digital quantification of e tumor maximal diameter, f cellularity, and g Ki-67 positivity in hotspot regions and h whole tumors using U-net on the Visiopharm platform. See Methods for detailed quantification schema. Tumor n: 4 (V600E, K::B), 5 (F1-N546K, F1-N546K + PTPN11, F1-ITD), or 6 (F1::TACC1). i Distribution of tumor border’s growth pattern (infiltrative, circumscribed, or mixed) for each group. Tumor n: F1-N546K = 5, F1-N546K + PTPN11 = 5, F1::TACC1 = 6, F1-ITD = 5, V600E = 4, K::B = 4). j AI-based digital quantification of vasculature and hemorrhage percentage per tumor area in each group using DenseNet on the HALO AI platform. Data presented as mean ± SEM. Tumor n: 4 (V600E, K::B), 5 (F1-N546K, F1-N546K + PTPN11, F1-ITD), or 6 (F1::TACC1). Abbreviations for models= F1 N546K: FGFR1 N546K SNV, F1-N546K + PTPN1: FGFR1 N546K SNV + PTPN11, F1-ITD: FGFR1-ITD, F1::TACC1: FGFR1::TACC1, V600E: BRAF V600E, K::B: KIAA1549::BRAF. Source data provided in the Source Data File.
MRI imaging was used to track glioma formation during this study (Fig. 5a and Supplementary Fig. S10a). A total of 78 mice (58 with FGFR1 or BRAF alterations) were imaged during the study, and gliomas were confidently detected in 72% mice implanted with mNSCs driven by FGFR1 or BRAF alterations. The overall tumor penetrance for BRAF V600E transduced mNSCs was 100%, while the penetrance of KIAA1549::BRAF was 90%. Injection of mNSCs transduced to express either of the FGFR1 SVs (FGFR1::TACC1 or FGFR1-ITD) was also sufficient to induce glioma formation with an overall penetrance of 100%. The overall penetrance of tumor formation following injection of NSCs transduced to express the FGFR1 N546K mutation alone was 80%, while the penetrance for NSCs transduced to express both the FGFR1 N546K and PTPN11 mutation was 90%.
Intracranial hemorrhage was noted on MRI imaging in ~45% of all gliomas and occurred least frequently in gliomas driven by BRAF V600E or by FGFR1 N546K alone (Fig. 5c). Within the FGFR1-altered gliomas that underwent MRI (n = 39) intracranial hemorrhage was detected in 50% (5/10) of gliomas with co-occurring mutations in FGFR1 N546K + PTPN11E69K, 50% (5/10) in FGFR1::TACC1-altered gliomas, and 66.66% (6/9) in FGFR1-ITD-altered gliomas (Fig. 5c). In contrast, only 10% (1/10) of the FGFR1 N546K gliomas had intracranial hemorrhage. Within the BRAF-altered gliomas that underwent MRI (n = 19), intracranial hemorrhage was detected in 30% (3/10) of BRAF V600E tumors and 66.66% (6/9) in gliomas that harbor the KIAA1549::BRAF fusion (Fig. 5c).
We observed differences in overall survival of mice following intracranial injection of each of these isogenic model systems (Log-rank Mantel Cox, p < 0.0001). Mice injected with BRAF V600E-expressing NSCs had the shortest overall survival compared to all other conditions (median survival of 56 days, p value < 0.001), while mice harboring allografts of the FGFR1 N546K + PTPN11 mutant NSCs survived the longest (median survival of 141 days) (Fig. 5b). Interestingly, mice harboring allografts transduced to express the FGFR1 SVs (FGFR1-ITD and FGFR1::TACC1) exhibited a trend to shorter survival compared to those with the FGFR1 SNVs (82 and 94.5 days respectively, compared to 141 for the double mutant and 115.5 days for the FGFR1 N546K alone) (Fig. 5b).
Neuropathological and AI-aided digital pathology examination of the tumor histology and immunostaining showed that mNSC FGFR1 and BRAF-altered tumors all had pathological features consistent with a glioma (Fig. 5d–h and Supplementary Figs. S10b–g, and S11a, b). Cells had glial cytology in many regions, but all showed mitotic activity and moderate to severe atypia most consistent with higher grade, not lower grade, gliomas. No specific features of low-grade gliomas were noted (Rosenthal fibers, eosinophilic granular bodies, biphasic appearance, ganglionic neurons). The tumors all showed glial lineage marker expression (GFAP and OLIG2) and AI-aided quantification of glial markers and the proliferation marker (Ki67) showed wide-ranging differences in their expression patterns across genotypes, but none were statistically significant (Fig. 5d–h and Supplementary Fig. S10b–d). All genotypes expressed GFAP and OLIG2 in a subset of tumor cells, and the average Ki67 proliferation index across all tumors was 24% ranging from 1.13% to 59.5%. We did not observe significant differences in the morphological features of tumor diameter or cell density between different FGFR1 genotypes (Fig. 5e–h and Supplementary Fig. S10c, d).
We next sought to evaluate activation of mTOR and MAPK signaling within these intracranial allografts, probing with antibodies for pS6 and pERK, respectively (Supplementary Fig. S10e–g). All tumors expressed pS6; however, the FGFR1-ITD tumors had significantly less positive staining compared to the FGFR1::TACC1 fusion and BRAF-altered lines (FGFR1::TACC1 vs FGFR1-ITD, p = 0.001, BRAF V600E vs FGFR1-ITD, p = 0.0004, and KIAA1549::BRAF vs FGFR1-ITD, p = 0.0066), Supplementary Fig. S10e, f). All tumors expressed pERK to a relatively similar level across genotypes, with no significant differences (Supplementary Fig. S10e, g). We conclude that our isogenic intracranial allografts maintain activation of both pathways, consistent with their in vitro counterparts.
The tumors did show evidence of distinct growth patterns with pathologist-scored assessment of tumor border infiltration relative to adjacent normal brain tissue (Supplementary Fig. S11c). FGFR1-ITD gliomas were scored as diffuse with infiltrative borders and did not have areas of circumscribed tumor. In contrast, BRAF V600E gliomas were all scored as completely circumscribed with pushing borders. The other glioma genotypes all harbored mixed growth patterns (Fig. 5i).
Given our prior observations of intracranial hemorrhage on MRI imaging, we also quantified hemorrhage within these samples by assessing the percentage of hemorrhage relative to vasculature on H&E within each glioma using an AI-based tissue segmentation classifier that we trained on the HALO AI platform (Supplementary Fig. S10d). While we did not observe statistically significant differences between each driver, gliomas expressing KIAA1549:BRAF, FGFR1:TACC1 and FGFR1 N546K + PTPN11 trended to higher proportions of hemorrhage relative to the amount of vasculature tissue (fold change hem/vasc = 1.44, 3.6 and 7.4), while those with BRAF V600E gliomas had the lowest (fold change hem/vasc = 0.32) (Fig. 5j). Altogether, these data demonstrate the ability of our isogenic mNSC models to induce gliomagenesis in allograft models and highlight potential differences in biological growth across genotypes.
pan-FGFR inhibitors represent therapeutic opportunity for FGFR1-altered gliomas
Small molecule inhibitors targeting the MAPK and mTOR pathways have rapidly emerged as novel therapeutic approaches for children with pLGGs, including recent FDA approvals for the use of pan-RAF inhibitors, or combination MEK and Type 1 BRAF inhibition, for gliomas that harbor BRAF alterations19,20,54. However, the activity of these agents in FGFR1-altered tumors is unknown, and the most efficacious path to precision medicine approaches for children diagnosed with FGFR1 alterations pLGGs is unclear. We thus evaluated these agents across our isogenic models.
We first evaluated the in vitro efficacy of a panel of MEK (trametinib), RAF (belvarafenib or tovorafenib), and mTOR (everolimus) inhibitors across our panel of isogenic mouse and human NSCs (Fig. 6a–d Supplementary Fig. S12a–d and Supplementary Data S5). We found that our oncogenic BRAF mNSC models exhibited sensitivity to trametinib and the RAF inhibitors (Fig. 6a–c), with minimal activity observed with single-agent everolimus (Fig. 6d). Focusing more closely on the MEK and RAF inhibitors, we found that the FGFR1-altered NSCs were less sensitive to these agents compared to the BRAF-altered models (Fig. 6a–c and Supplementary Fig. S12a–c). However, we did also observed some variability in sensitivity within the FGFR1-altered lines, with models harboring FGFR1 SNVs exhibiting greater sensitivity than the FGFR1 SVs.

Dose response curves in the FGFR1 and BRAF-altered mNSC models for a trametinib (MEKi), b belvarafenib (RAFi), c tovorafenib (RAFi), d everolimus (mTORi), e infigratinib (FGFRi), f erdafitinib (FGFRi). Data presented as mean ± SEM of 3 biological replicates. g Model summarizing the in vivo drug study. 300,000 F1::TACC1-driven mNSC cells were injected into the right hemisphere of mice. After gliomas were detected, mice were treated with single or combination doses of infigratinib, everolimus, and trametinib. MRI was performed prior to drug treatment and weekly after start of drug treatment. Cohort 1 (n = 31) was treated after 24 or 25 days post cell injections, and Cohort 2 (n = 19) was treated after 30 or 31 days post cell injections. Created in BioRender. Apfelbaum, A. (2025) https://BioRender.com/p65k638. h Dot plot showing the volume of tumors in cohort 1 (n = 31) and cohort 2 (n = 19) prior to treatment. Volume assessed by MRI. Lines represent the median. Significance was calculated by performing a two-sided Mann Whitney test performed. ****p < 0.0001. i Kaplan–Meier survival curves for mice in Cohort 1 by each treatment group (treated after 24/25 days-dotted line). Significance was calculated by performing a Log-rank (Mantel-Cox) test (Vehicle vs. infigratinib: p = 0.025, Vehicle vs. TI: p = 0.038, Vehicle vs. EI: p = 0.025). j Kaplan–Meier survival curves for mice in Cohort 2 by each treatment group (treated after 30/31 days-dotted line). Abbreviations for models= F1-N546K: FGFR1 N546K SNV, F1-N546K + PTPN11: FGFR1 N546K SNV + PTPN11 E69K SNV, F1-ITD: FGFR1-ITD, F1::TACC1: FGFR1::TACC1, V600E: BRAF V600E SNV, K::B: KIAA1549::BRAF. Source data provided in the Supplementary Data S5 and the Source Data File.
In contrast, all FGFR1-altered models were more sensitive to pan-FGFR inhibitors than BRAF-altered models. We performed dose response curves in our FGFR1 and BRAF-altered lines using four FDA-approved pan-FGFR inhibitors (infigratinib, erdafitinib, pemigatinib, futibatinib) and then calculated the area under the curve for each treatment (Fig. 6e, f and Supplementary Fig. S12e–l). As expected, we observed minimal responses to FGFR inhibition in isogenic models that expressed the BRAF oncogenes. However, all four of the pan-FGFR inhibitors exhibited activity across our FGFR1-altered models, with IC50s in the nanomolar range. Overall, all FGFR-expressing models treated with pan-FGFR inhibitors exhibited lower AUCs compared to BRAF-expressing lines (0.36 ± 0.02 vs 0.91 ± 0.03, p < 0.0001 in mNSC, 0.64 ± 0.03 vs 0.92 ± 0.01, p < 0.0001 in ihNSC) (Fig. 6e, f and Supplementary Fig. S12k–n). Intriguingly, we also observed a spectrum of responses among FGFR1-altered lines, with the SVs being the most sensitive compared to the SNV alone or SNV double mutant (0.3 ± 0.01 vs 0.43 ± 0.03, p < 0.0001 in mNSC, 0.58 ± 0.02 vs 0.74 ± 0.01, p < 0.0001 in ihNSC) (Fig. 6e, f and Supplementary Fig. S12k–n).
Our prior characterization of our model systems had revealed simultaneous activation of MAPK and mTOR pathway signaling in the absence of growth factor (Fig. 3d, e). This is particularly relevant for FGFR1-mutated gliomas that harbor co-mutations such as PTPN11, which can activate these pathways downstream of the FGFR1 receptor. Moreover, studies in other tumor contexts have revealed mTOR activation as a potential resistance mechanism for pan-FGFR inhibition55,56. We therefore hypothesized that combination approaches with agents to simultaneously inhibit MAPK or mTOR activity may enhance the efficacy of FGFR inhibitors.
We first tested this hypothesis in vitro by assessing synergy across our FGFR1-altered mNSC lines with the FGFR inhibitor infigratinib in combination with either the MEK inhibitor trametinib or the mTOR inhibitor everolimus (Supplementary Fig. S13a, b). BRAF V600E-altered mNSC models were included as a negative control. We observed no reproducible combinations of doses between trametinib and infigratinib that exhibited synergy across the FGFR1-altered lines (Supplementary Fig. S13b). In contrast, combinations of infigratinib with the mTOR inhibitor everolimus exhibited synergy (Bliss synergy score >10) at low doses of each drug (Supplementary Fig. S13a), but not in our negative control BRAF V600E model. These data support FGFR and mTOR inhibition as a potential combination therapy strategy in pLGGs.
Diverse FGFR1-altered gliomas drive differential in vivo therapy effects of FGFR inhibitors
We next evaluated the efficacy of FGFR inhibition in vivo, leveraging the brain penetrant agent infigratinib, both as single agent therapy, and in combination with either trametinib or everolimus. Dose tolerability studies were first performed to identify drug doses that were not toxic (Supplementary Fig. S14a, b). Next, the striatum of SCID mice were injected with intracranial allografts of FGFR1::TACC1-expressing mNSCs and observed for glioma formation using MRI imaging, upon which mice were randomized to daily oral gavage treatment with either infigratinib (15 mg/kg), everolimus (5 mg/kg), trametinib (2 mg/kg), infigratinib + trametinib (15 + 2 mg/kg), or infigratinib + everolimus (15 + 5 mg/kg) (Fig. 6g).
The first cohort of mice with detectable gliomas initiated treatment on days 24 or 25 post injection, with a mean tumor volume as assessed by MRI of (5.65 ± 5.9 mm3) (Fig. 6h). Within this cohort, treatment of mice with FGFR1::TACC1 gliomas with infigratinib (either as single agent or in combination) significantly prolonged survival compared to mice treated with either vehicle, trametinib or everolimus alone (p: 0.025, p: 0.025, p: 0.039, respectively) (Fig. 6i). The median survival of mice treated with infigratinib was 16 days post initiation of treatment, compared to 9, 7 or 7 days with vehicle treated, everolimus or trametinib alone. However, no effects of combination therapy were seen compared to infigratinib alone (Fig. 6i).
The second cohort of mice with detectable gliomas initiated treatment 6 days later. However, these mice harbored significantly larger gliomas compared to those treated in the first cohort (5.65 mm3 vs 21.1 mm3, p value < 0.0001) (Fig. 6h), even though we had not detected gliomas by MRI imaging at the same time as Cohort 1. In this context, and with rapidly growing gliomas, we did not observe any survival advantage in any treatment arm, including with infigratinib. The median survival post initiation of treatment for mice treated with infigratinib within Cohort 2 was only 10 days, compared to 6.5 days for mice treated with vehicle control alone (Fig. 6j).
Finally, to assess the pharmacodynamic phenotype associated with treatment, we treated an additional cohort of mice with each treatment and harvested their tumors 49 h after initiation of treatment, which correspond to a 1-h time-point after the third scheduled dose (Supplementary Fig. S14c). First, we applied the AI-based cell segmentation approach on the H&E-stained tumor tissue to evaluate differences in cellular density with each treatment (Supplementary Fig. S14d). We found significant reductions in density of tumors harvested from mice treated with infigratinib compared to vehicle controls, either alone (7 ± 0.8 vs 5 ± 0.4 103 cells/mm2, p = 0.048) or in combination of with trametinib (7 ± 0.8 vs 4.2 ± 0.24 103 cells/mm2, p = 0.0039) or everolimus (7 ± 0.8 vs 4.9 ± 0.32 103 cells/mm2, p = 0.04), while there were no differences in the overall diameter of the tumors (Supplementary Fig. S14d–f). Next, we evaluated levels of Ki67 as a marker of proliferation, and pS6 and pERK as markers of mTOR and MAPK activity. At this early time point, only mice treated with the combination of infigratinib and trametinib had significantly reduced staining of Ki67 relative to vehicle controls (38 ± 3.6 % vs 19 ± 1%, p = 0.006). There was noticeable variability in the level of staining of pS6 and pERK in the vehicle-treated mice. Despite this, we observed trends for reduced levels of pS6 and pERK across all arms that included infigratinib compared to vehicle controls (Supplementary Fig. 14j and Supplementary Data S6 average level of staining). However, these differences only reached statistical significance in the infigratinib-treated cohort for pS6 (p = 0.017).
Together, these findings suggest a potential therapeutic benefit for single-agent FGFR-inhibition in FGFR1::TACC1 expressing gliomas, however, with a potential window of opportunity with respect to glioma size and aggressiveness of growth.
Human FGFR1-altered pLGGs exhibit moderate responses to the current generation of MEK and FGFR inhibitors
There are currently no FDA-approved treatments nor open clinical trials for children with FGFR-altered gliomas, and the optimal therapeutic approach remains unclear. This is particularly relevant in the setting of current MAPK pathway inhibitors that have entered the clinical arena for pLGGs. We thus sought to perform a multi-institute retrospective analysis of patients diagnosed with FGFR-altered pLGGs, focusing on responses to standard chemotherapy or following treatment with MEK or FGFR inhibitors. This analysis included both unpublished cases integrated with a meta-analysis of published reports.
First, we performed a retrospective review of the 15 children with FGFR1-altered pLGGs diagnosed at DFCI that were included in our initial genomics analysis. Of these 15 patients, at a median follow-up of 5.86 years, six had an initial near-total or gross-total resection without subsequent progression, and three had an initial subtotal resection followed by a more definitive resection at progression without a need for systemic treatment (Fig. 7a and Supplementary Data S7). The remaining six children, all of whom had residual disease, required further treatment with systemic chemotherapy (five with carboplatin/vincristine and one patient with single-agent carboplatin) (Fig. 7a). One patient had a partial response to therapy, while four patients had a best response of stable disease. The remaining patient was not evaluable (Fig. 7a and Supplementary Data S7).

a Pie chart showing the number of patients with FGFR-altered gliomas within the DFCI cohort that did (n = 6) or did not require further treatment (n = 9) following surgical management and had gross (n = 9) or sub-total (n = 6) resections (n = 15 patients total). b Bar graph depicts the % of FGFR1-altered pediatric glioma patients treated with chemotherapy (n = 6) or targeted inhibitors with either FGFR- or MEK-inhibition, broken down by low-grade (pLGG) or high-grade (pHGG) histology (pLGG: MEKi n = 8, FGFRi n = 24; pHGG: FGFRi n = 11). Chemotherapy-treated patients were from the DFCI cohort, targeted inhibitor-treated patients were from the DFCI cohort, published literature, and multi-institutional case studies. SD stable disease, PR partial response, PD progressive disease, CR complete response. Source data provided in Supplementary Data S7.
Across the larger meta-analysis, 43 episodes of targeted inhibitor treatment in 41 patients (32 patients with pLGGs) were identified, including patients treated with the pan-FGFR inhibitors erdafitinib32,57,58,59 or debio134760, or MEK inhibition61,62,63,64. Two of these patients were treated with both an FGFR and MEK inhibitor as single agents at different timepoints. Of the 24 patients with pLGGs treated with FGFR inhibition, four children had a partial response, five had progressive disease, while the remaining 15 patients had stable disease documented as their best response (Fig. 7b). Six of the eight patients with FGFR-altered pLGGs treated with MEK inhibition had stable disease as their best response, with progressive disease and a partial response documented in the seventh and eighth patients (Fig. 7b and Supplementary Data S7). To note, varying response criteria were used across the DFCI cohort and included publications, including RAPNO-LGG and RANO criteria, as well as individualized response criteria65,66.
Overall, combining with patients treated with high-grade gliomas, the observed responses to targeted (FGFR or MEK) inhibitor therapy in pediatric FGFR-altered gliomas have been modest. Of 43 treatment courses identified, there were seven cases (16.2%) with partial responses, 24 (55.8%) with stable disease, and 11 (25.6%) with progressive disease documented as the best response observed.
Discussion
FGFR alterations are common across gliomas, particularly in pediatric patients. We find FGFR1 to be the most frequently altered FGFR family member in pediatric gliomas and highlight the heterogeneity of somatic drivers affecting them. Importantly, while we show glioma-associated FGFR1 alterations to be sufficient to drive gliomagenesis, we also highlight the need to optimize the efficacy of agents that target FGFR kinases themselves, or downstream activation of the MAPK and/or mTOR signaling pathways. While these FGFR driver events present the promise of precision medicine approaches, the path forward to the most effective strategies to target FGFR1 alterations remains elusive, even with the clinical development of pan-FGFR inhibitors.
Our finding that FGFR-altered gliomas exhibit heterogeneity across locations in which they arise has important clinical implications, particularly related to accurate diagnosis. While FGFR2 and FGFR3-altered gliomas most commonly occur in the cerebral hemispheres, almost 20% of all FGFR1-altered tumors arise in the midline structures, including the thalamus and brainstem. Given that these tumors are frequently diagnosed through stereotactic biopsies that can yield smaller amounts of tissue, it is imperative that all sequencing panels used in their molecular work-up are sufficient to detect each of the FGFR1 driver events. Finally, it is equally important that tools and algorithms used for variant calling have sufficient sensitivity to detect the FGFR1 internal kinase duplications, which is currently a limitation. For example, the analysis of the tumors included from Foundation Medicine in this cohort likely underestimates the frequency of these alterations.
FGFR-altered gliomas also exhibit tight associations with patient age, with FGFR1 and FGFR2 alterations occurring predominantly in pediatric gliomas, while FGFR3 occurs most frequently in adult gliomas. The reason for this remains unclear. Our analysis of expression patterns of these proteins across development also suggests specific associations with brain development, which may account for these differences. However, this is speculative, and further work is required to understand the interplay between expression of FGFR proteins during normal development and the implications for gliomagenesis.
Generation of primary pLGG cell line models has been a major challenge for our field, with the inability to propagate glioma cells in vitro or as patient-derived allografts67. Those that have been published have BRAF alterations, which excludes many of the other alterations, particularly FGFR1. To overcome this, we have generated multiple isogenic models of both mouse and human neural stem cells that are driven by FGFR1 and BRAF alterations. While they have the limitation that they are not patient-derived models, our isogenic systems provide a powerful tool to study each driver alteration and therapeutic approaches, both in vitro and as brain allografts in immunodeficient mice. A limitation of our models is that the control cell lines (Vector Control or FGFR1 WT lines) are grown in the presence of growth factor, making it more difficult to compare to the growth factor-independent FGFR1- and BRAF-altered lines. Thus, as of now, most of our analyses compare FGFR and BRAF-altered groups. However, it would be good to extend in the future to alternative control models that are grown in the same conditions. While the intracranial gliomas grow as “high-grade” tumors in the immune-deficient context, they also appear to recapitulate some key features of the human pLGGs. A particularly intriguing finding is the increased rate of intracranial hemorrhage that we observed in some of our FGFR1-altered and BRAF fusion gliomas, which has also been reported in the setting of children with pLGGs68,69. Further work is required to validate this initial observation and to determine the mechanism through which FGFR1-altered gliomas increase the risk of hemorrhage.
We confirm that FGFR1 alterations in pLGGs exhibit genetic heterogeneity. Both SVs and SNVs are sufficient to generate activating driver events across pediatric gliomas. While SVs involving FGFR proteins frequently occur as single driver events, similar to the majority of pLGGs overall, two hotspot SNV mutations (N546K and K565E) commonly co-occur with other mutations in FGFR1 itself, or in addition to other genes that activate MAPK and/or mTOR signaling (for example mutations in PTPN11 and PI3K pathway members). Intriguingly, we observed differences in the patterns of co-occurring alterations. FGFR1 N546K mutant gliomas harbor additional mutations that occur primarily in the extracellular domain, while FGFR1 K656E mutant gliomas have FGFR1 co-mutations that exclusively co-occur in the kinase domain (Supplementary Fig. S1D). Patterns of co-occurring mutations within the extracellular domain or the FGFR1 kinase may reflect different processes through which the FGFR1 SNVs enhance gliomagenesis, which will be important to dissect to inform optimal therapeutic approaches. For example, mutations in the extracellular domain may influence ligand binding and specificity, compared to kinase mutations that may further contribute to its aberrant activation. Finally, gliomas with FGFR1 SNVs that did not harbor second SNVs in FGFR1 itself frequently co-occurred with other alterations along the MAPK and/or mTOR pathways, including PTPN11, NF1, and PIK3CA.
It is important to recognize that while FGFR1-altered gliomas can harbor PTPN11 and NF1 alterations, these can also represent germline variants. A limitation of our current study is that clinical cohorts included did not include germline sequencing, making it difficult to determine the incidence of concurrent germline alterations. However, in the clinical context, it will be important to consider the possibility of germline variants for these patients. In particular, the diagnosis of FGFR1-altered gliomas with concurrent alterations may represent the first clue of an underlying congenital disorder such as Neurofibromatosis 1 (loss of NF1) or Noonan’s syndrome (NS), which is associated with germline PTPN11 mutations in ~50% of patients70.
The role of concurrent PTPN11 mutations in shaping the phenotype of FGFR1-altered pLGGs remains unclear. Despite the association of PTPN11 mutations with NS, it is rare to observe gliomas that only harbor a single alteration in PTPN11. Indeed, in our models, the PTPN11 E69K variant was insufficient to transform NSCs or induce glioma formation as a single driver event. Intriguingly, prior reports have indicated enrichment of low-grade glioneuronal histologies in patients with NS who develop brain tumors, a feature that we have also associated with FGFR1-altered gliomas71,72,73. In most studies focused on gliomas that arise in patients with NS, gliomas are largely diagnosed by morphology and neuronal marker staining, with most sequencing studies limited to characterizing variants within PTPN11 itself70,72. However, where additional profiling of somatic alterations present in gliomas that occur in the context of NS has been performed, variants within FGFR1 co-occurring with alterations in PIK3CA or PIK3R1 have been reported71,73. In addition, another study has reported a co-occurring alteration in KRAS73. These observations lead us to hypothesize that in patients with NS, a second somatic hit along the MAPK or PI3K pathway (potentially including FGFR1) is likely needed to initiate gliomagenesis. However, additional work is required to test this.
Our study sheds several insights into the clinical implications of FGFR1 alterations in the setting of pLGGs. Currently, pLGGs of different histological and molecular subtypes are treated with the same chemotherapy protocols, most commonly including vincristine and carboplatin. Our findings clearly support FGFR-altered gliomas as being distinct from those driven by BRAF alterations. Consistent with prior reports, we found that FGFR1 alterations are enriched in glioneuronal pLGG types5,23,24,26,36,37, suggesting a potential link between FGFR1 expression and neuronal programs. This is further supported by our finding that overexpression of the FGFR1 alterations promotes more neuronal transcriptional states compared to the BRAF alterations.
The therapeutic implications of these differences in underlying cell state are not well understood. For example, it has been reported that MEK inhibition in GBMs is associated with increased neuronal differentiation74. An alternate hypothesis is that MAPK pathway inhibition selects for pre-existing glioneural cell states that harbor primary resistance. Indeed, our isogenic FGFR1 NSC models exhibit less sensitivity to MAPK pathway inhibitors compared to those that harbor BRAF alterations. Further work is necessary to delineate these mechanisms, which will be essential to inform strategies to optimize the use of these inhibitors in the clinical setting. Indeed, the role of other pathways activated by FGFR alterations beyond the MAPK and AKT pathways in modulating glioma growth remains to be determined.
Our study also confirms the inherent therapeutic challenges associated with FGFR1-altered pLGGs. Our clinical cohort suggests modest responses to both conventional chemotherapy and treatment with MEK inhibitors, a finding that is partially reinforced by our model systems that exhibited less sensitivity to MAPK pathway inhibition in the setting of FGFR1 alterations. There are currently no clinical trials evaluating the efficacy of FGFR inhibitors for children with pLGGs. However, our meta-analysis of the use of some of these FGFR inhibitors in the clinical setting has also revealed modest responses, with stable disease often reported as the best response. It should be emphasized that this is a small cohort collated from multiple studies with heterogeneously treated patients, with variable tumor histologies, co-alterations, tumor locations, degrees of resection, prior therapies, treatment dosages, and response criteria, making it impossible to draw definitive comparative conclusions between therapeutic approaches.
While penetrance across the blood-brain-barrier likely contributes to the lack of responses, our in vivo studies using a brain-penetrant inhibitor were also associated with transient responses, and only in mice in which treatment was initiated with smaller glioma sizes. It is possible that systemic toxicity has required the use of suboptimal doses, both in our mouse experiments and more broadly in the early-phase clinical trials. Indeed, it is important to note that the use of FGFR inhibitors in some of these trials was associated with a significant rate of bone complications, including fractures32. Inhibitors with even higher levels of brain penetrance are likely required.
Finally, it should be highlighted that for pLGGs in particular, where the goal of treatment is often stabilization rather than eradication of disease, these modest responses may still be clinically meaningful. These results therefore do not discount the potential clinical utility of chemotherapy or targeted inhibitors in FGFR-altered gliomas, however, they highlight that future work is essential to optimize the use and efficacy of FGFR inhibitors, to allow dosing using schedules that minimizes long-term toxicities, and ultimately to better characterize the role for targeted therapies in treatment of FGFR-altered gliomas.
Methods
Ethics statement
Ethics approval was granted by relevant human IRB and/or animal research committees (IACUC) of Dana-Farber Cancer Institute (DFCI), Brigham and Women’s Hospital, and Boston Children’s Hospital. All patients and/or the parents or legal guardians of pediatric patients provided informed written consent prior to collection of tissue samples (DFCI Protocol #10-417) or were studied under specific IRB waiver of informed consent (DFCI Protocol #10-043) as de-identified samples
DFCI Patient cohort information
This study was performed under DFCI IRB-approved protocols DFCI 10-043, DFCI 11-104, and 10-417 (Supplementary Data S2). The DFCI protocol 10-043, which includes a waiver of informed consent, allowed for a retrospective analysis of glioma samples, including pathology and genomic assays, in addition to collection of de-identified clinical data. In addition, of the 87 FGFR-altered patients spanning pediatric and adult patients included in our study, 77 consented under DFCI 10-417 or DFCI 11-104 for data release (see data availability and Supplementary Data S2).
To assess the landscape of FGFR-altered gliomas, clinical data and variant tumor calls were obtained through our institutional Precision Medicine Program sequencing database, which contained clinical and research data from 2514 primary CNS neoplasms included in this cohort. FGFR-altered gliomas underwent pathology and cytogenetic review for confirmation of fusion events, identification of focal copy number events (<10 MB), and absence of fusion and identification of recurrent mutations within the cohort (>2). The review process identified 87 tumors with likely pathogenic FGFR alterations, referred to as the FGFR-altered cohort.
Oncopanel Sequencing:
The DFCI cohort underwent next-generation targeted exome sequencing (OncoPanel) of cancer-related genes performed using Illumina-based methods as previously described in ref. 75. As this study was a retrospective analysis, some variability between cases exists due to changes in the design of this assay over the course of several years, including expansion of targeted genes from 300 to 471, with additional intronic coverage. We categorized each FGFR alteration described in clinical reports based on the FGFR gene that was altered (FGFR1,2,3,4) and by the category of genomic alteration (fusion/rearrangement, copy number, single-nucleotide variant within protein-coding regions, and likelihood to be pathogenic).
Copy arrays:
Array comparative genomic hybridization (aCGH) was performed on DNA isolated from formalin-fixed paraffin-embedded (FFPE) tissue on the DFCI cohort using either the ThermoFisher Oncoscan CNV assay or the Agilent 1 × 1 M aCGH array, according to the manufacturers’ direction (922719973). All microarrays were analyzed using the Nexus Copy Number Software Package (BioDiscovery, El Segundo, CA) via the FASST2 segmentation with a significant threshold of 1.0E-12. The processed variant calls from our clinical assays were used in this study.
RNA fusion panel
The presence of gene fusions within the DFCI cohort was also assessed using the clinical MGH Snapshot Fusion RNA-based assay that utilizes next generation sequencing from anchored primers within the known gene fusion partner (FGFR1, 3) and was performed on 6 × 5 μm sections of FFPE tissue76. The processed variant calls from our clinical assays were used in this study.
MNP2 (KiTZ) cohort
This cohort was previously described in ref. 22, and includes 425 gliomas from patients <22 years at the time of diagnosis between 2015 and 2019. Tumors were classified by a superfamily class prediction score of >0.9 for any glioma superfamily, irrespective of histology or scores for DNA methylation class families, classes, or subclasses. DNA methylation classes were called used the following classifier (version 12.5): https://www.molecularneuropathology.org/mnp/ > Classifiers > Classifier Versions > Brain classifier version 12.5. The American College of Medical Genetics and Genomics criteria were used to classify mutations as pathogenic/likely pathogenic77. Most analyses were focused on the 30 patients with FGFR alteration. Copy number analysis for patients was performed by visual inspection of copy-number profiles calculated from DNA methylation arrays.
Foundation medicine (FM) cohort
The Foundation Medicine cohort of 502 FGFR-altered patients was derived from a collection of solid tumor clinical cases for which comprehensive genomic profiling had been previously performed3. Selected cases were verified primary CNS tumors harboring alterations in FGFR1-4 known from literature or included in Catalogue Of Somatic Mutations In Cancer (COSMIC) repository, as well as those with likely functional status.
Cell lines
Mouse neural stem cells (mNSC) obtained from the subventricular zone of female CD1 mice39 were cultured as 3D neurospheres in ultra-low adherence flasks and plates (Corning, NY, USA) in tumor stem media (TSM) (500 mL Neurobasal-A (#10888-022), 500 mL DMEM/F12 (#11330-032), 10 mL HEPES Buffer Solution 1 M (#15630-080), 10 mL MEM sodium pyruvate solution 100 mM (#11360-070), 10 mL MEM Non-essential amino acid solution 10 mM (#11140-050), 10 mL GlutaMAX solution (#35050-061), 10 mL Penicillin/Streptomycin (#15140122) and 1X B-27 Supplement Minus Vitamin A (from 50X) (#12587-010) from Invitrogen (MA, USA). When indicated, TSM media was supplemented with 12 ng/mL human-EGF (#78006), 12 ng/mL human-bFGF (#78006), and 2 ug/mL heparin solution 0.2% (#07980) from Stemcell Technologies (Vancouver, Canada). Neurospheres were dissociated using Accutase (#00-4555-56, Invitrogen) every 3–4 days and reseeded as a single-cell suspension with a density of 80–100,000 cells/mL in ULA flasks/plates.
Commercially available H9-derived human neural stem cells (GIBCO, Catalog nos. N7800-100) were immortalized by hTERT virus transduction (CMV-hTERT- Zeo lentivirus: Amsbio, LVP1130-Zeo). They were cultured as an adherent layer (2D) on geltrex-coated (Thermo Fisher, A1413302) tissue culture-treated plates in the TSM media listed above, and accutase was used to detach them from plates.
Cell lines were tested for mycoplasma monthly and STR profiled or validated by immunoblot blot every 3 months.
Model generation
FGFR1 transcript NM_023110.3 was used as a template for the generation of all FGFR1 SVs and SNVs. Similarly, for PTPN11, NM_002834.5 and TACC1 CCDS6109.1 exon 7-13 were used for SNV and SV, respectively. BRAF transcript NM_004333.6 and KIAA1549::BRAF junction exon 15 to 9 were used. FGFR1 N546K – C1638A, FGFR1-ITD, FGFR1::TACC1, and PTPN11 E69K constructs were synthesized into Gateway-compatible entry clones by GenScript with the addition of HA and V5 tags as indicated in the results segment. For the FGFR1-ITD, the following linker was used gatcgcagcctgcgcagcctgtgcagcttttggaaa. Using the Gateway LR Clonase II (Thermo Fisher, 11791020), constructs were cloned into pLX311 (FGFR1s, BRAF, KIAA1549::BRAF, GFP, and HcRed) or pLX307 (PTPN11 and LacZ).
To generate lentivirus, HEK293T cells were transfected with lentiviral expression vectors (10 μg) and packaging plasmids encoding VSVG and PSPAX2 using the lipofectamine 3000 transduction kit (Thermo Fisher, L3000015), media was replaced after 6 h and cell supernatant collected at 24 h for lentivirus concentration using the Lenti-X concentrator (PT4421-2) according to manufacturer’s protocol.
mNSCs were infected using a spinfection protocol (2000 rpm for 2 h at 30 °C). Cells were selected with 0.75 μg/mL puromycin (pLX307) for 3 days or 4 μg/mL blasticidin (pLX311) for 10 days. ihNSCs were infected and selected with 0.5 μg/mL puromycin (pLX307) for 3 days or 2.5 μg/mL blasticidin (pLX311) for 10 days.
Cumulative doubling assays
For cumulative growth assays, cells were seeded a density of 50–66,000 cells/mL (ihNSCs) or 60–100,000 cells/mL (mNSCs) in the presence or absence of FGF and EGF growth factors. Cells were counted every 3–4 days, and when possible, a density of viable cells equal to the initiation of the experiment was reseeded. Doublings were calculated with the formula log2(total viable cells/seeded viable cells) at each passage, and the cumulative results were displayed. These experiments were carried out for 14 days.
Immunoblot/densitometry analysis
Cells for immunoblotting were lysed on ice for 20–30 min in RIPA buffer containing protease and phosphatase inhibitors. Lysates were centrifuged at 13,000 × g for 10 min at 4 °C. Supernatant was collected and protein concentration quantified using the Pierce 660 nm Protein Assay (Thermo Fisher, 22660). Equal amounts of protein for each sample were aliquoted and mixed with 4X LDS Sample loading buffer and 10X NuPAGE Sample Reducing Agent and heated to 70 °C for 10 min. Lysates were loaded and run on NuPAGE Bis-tris 4–12% or NuPAGE Tris-Acetate 3-8% gradient gels. The iBlot transfer system (Life Technologies, IB24001) was used to transfer protein to a PVDF membrane. The membranes were blocked in Advanblock (R-03726-E10) for 1 h at room temperature. Subsequently, membranes were incubated with primary antibodies (listed in Supplementary Data S1) overnight at 4 °C. Membranes were washed 3 × 5 min with 1X TBST and incubated with secondary HRP conjugated species-specific antibodies at room temperature for 1 h, washed again, and then developed using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher, 34578). Image capture was performed using the Fujifilm LAS-3000 Imaging system or the LICOR Odyssey M system. Densitometry analysis was performed using Adobe Photoshop. Full blots are attached in the source file.
In vivo studies
All animal studies were performed according to Dana-Farber Cancer Institute Institutional Animal Care and Use Committee (IACUC)-approved protocols and in full compliance with all of the applicable provisions of the Animal Welfare Act and other Federal Statutes and Regulations relating to research using living vertebrate animals. Animal studies were performed at the Lurie Cancer Imaging Center of the Center for Biomedical Imaging in Oncology, which houses an AAALAC-certified satellite animal facility. All mice (equal females and males) were housed in individually ventilated cages in OptiMICE racks with automatic watering. All mice had unrestricted access to food and water.
Cell injections
The mNSC lines described in Supplementary Fig. S10A were injected stereotactically into the right striatum of 6-week-old female and male NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, The Jackson Laboratory, Bar Harbor, ME). Due to the number of mice injected, for all of the studies, the mice cells were implanted on subsequent days. Briefly, mice were anesthetized with 2% isoflurane mixed with medical air and placed on a stereotactic frame. The skull of the mouse was exposed through a small skin incision, and a small burr hole was made using a 25-gauge needle at the selected stereotactic coordinates. The cells (300,000 cells in 3 µL PBS) were loaded on a 33-gauge Hamilton syringe and injected slowly using the following coordinates: —From Bregma, 0 mm AP, −2 mm ML, −2.5 mm DV. Upon completing injection, the needle was left in place for another minute, then withdrawn slowly to help reduce cell reflux. After closing the scalp with suture and staple, mice were returned to their cages, placed on a warming pad, and visually monitored until full recovery. Mice were then checked daily for signs of distress, including seizures, ataxia, weight loss, and tremors, and euthanized by CO2 asphyxiation as they developed neurological symptoms, including head tilt, seizures, sudden weight loss, loss of balance, and ataxia.
MRI
MRI measurement of tumor volume was performed at the debut of neurological symptoms or around 2 months into the trial if asymptomatic for the study in Fig. 5. For the drug study in Fig. 6, the first MRI was performed 21/22 days post-cell injections and again every subsequent week after until study was completed. MRI was performed using a Bruker BioSpec 7 T/30 cm USR horizontal bore Superconducting Magnet System (Bruker Corp.). This system provides a maximum gradient amplitude of 440 mT/m and slew rate of 3440 T/m/s and uses a 23 mm ID birdcage volume radiofrequency (RF) coil for both RF excitation and receiving. Mice were anesthetized with 1.5% isoflurane mixed with 2 L/min air flow and positioned on the treatment table using the Bruker AutoPac with laser positioning. Body temperature of the mice was maintained at 37 °C using a warm air fan while on the treatment table, and respiration and body temperature were monitored and regulated using the SAII (Sa Instruments) monitoring and gating system, model 1025 T. T2 weighted images of the brain were obtained using a fast spin echo (RARE) sequence with fat suppression. The following parameters were used for image acquisition: repetition time (TR) = 6000 ms, echo time (TE) = 36 ms, field of view (FOV) = 19.2 × 19.2 mm2, matrix size = 192 × 192, spatial resolution = 100 × 100 mm2, slice thickness = 0.5 mm, number of slices = 36, rare factor = 16, number of averages = 8, and total acquisition time 7:30 min. Bruker Paravision 6.0.1 software was used for MRI data acquisition, and tumor volume was determined from MRI images processed using a semiautomatic segmentation analysis software (ClinicalVolumes).
Drug treatment
NSG mice (6 weeks old, male and female) without tumors were used for the 10-day drug tolerability studies. For trametinib (HY-10999, MCE), 1 and 2 mg/kg were tested, and 2 mg/kg was well tolerated. For everolimus (HY-10218, MCE), 5 and 15 mg/kg were tested, and only 5 mg/kg was well tolerated. For infigratinib (HY-13311, MCE), 10, 15, 20, and 30 mg/kg were tested. 15 mg/kg was the only dose well tolerated alone and in combination with the other drugs (Supplementary Fig. S14a, b).
Mice bearing FGFR1::TACC1-altered tumors were treated with (1) Vehicle control (10% DMSO, 40% PEG300, 5% Tween-80, 45% Saline), (2) trametinib (2 mg/kg), (3) everolimus (5 mg/kg), infigratinib (15 mg/kg), (4) trametinib + infigratinib (2 + 15 mg/kg), or 5) everolimus + infigratinib (5 + 15 mg/kg) once daily by oral gavage. Drugs were made in the same solution as the vehicle control and were prepared fresh prior to treatment.
Brain collection, tissue processing, and staining
The mice were euthanized when at least one of the endpoints was reached, including: 15% loss in body weight from peak weight, poor body condition (BCS 2), signs of the animal being in morbid condition, and neurological symptoms. The mouse brains were collected and preserved after euthanasia, either by fixation or flash freezing. The allocation for FFPE and frozen was randomly determined before the experiment began (half and half for the experiment in Fig. 5 and all FFPE for the drug treatment experiments in Fig. 6 and Supplementary Fig. S14. For fixation, murine brains were fixed in 10% formaldehyde for 24 h, then transferred to 70% ETOH until processing. Whole brain coronal sections were placed into cassettes in 70% ETOH and subsequently embedded in paraffin for block generation. Tissues from each paraffin block were cut at 5 μm sections, and routine H&E staining was performed to be evaluated by an expert pathologist and digital pathology. Immunohistochemistry was performed on the Leica Bond III automated staining platform using the Leica Biosystems Refine Detection Kit (Leica; DS9800). FFPE tissue sections were baked for 30 min at 60 °C and deparaffinized (Leica AR9222) prior to staining. Primary antibodies (Supplementary Data S1) with a 30 M citrate antigen retrieval (Leica ER1 AR9961) were incubated for 30 min, visualized via DAB, and counterstained with hematoxylin (Leica DS9800). The slides were rehydrated in graded alcohol and cover-slipped using the HistoreCore Spectra CV mounting medium (Leica 3801733).
Digital neuropathologic analyses
All slides were scanned at 40× magnification using a GT Leica Aperio scanner. An expert neuropathologist confirmed the presence or absence of tumor formation. Tumors were categorized as circumscribed, infiltrative, or mixed based on their border delineation with the surrounding normal brain tissue, as observed from H&E staining by a pathologist. The entire tumor area was annotated as a region of interest for further analysis. AI-based tissue segmentation for vasculature and hemorrhage quantification was performed using DenseNet v2 on the HALO AI image analysis platform (v3.0, Indica Labs, Albuquerque, NM). AI-based quantification of cellularity and IHC marker positivity was carried out using U-Net on Visiopharm image analysis software (v2023.09, Hørsholm, Denmark) for nuclear segmentation with customized apps and specific thresholds. Heatmaps for positive DAB signals were used for the automated selection of hotspot regions with the highest marker positivity for quantification.
Drug response curves
Cells were seeded at a density of 1000 (mNSC) or 2000 (ihNSC) cells/well in three technical replicates in corning 96-well plate (Corning, 3917). The indicated drugs (Supplementary Data S1) were serially diluted before being added to the plate. Cells were incubated for 72 h before viability was assessed by Cell-titer Glo (G7573). Cell titer glo was added per well (1:1), plates were incubated for 10 min at room temperature before readout of luminescence signal on a SpectraMax M5 plate reader. All results were normalized to vehicle control. AUC was calculated using the Broad Institue’s PRISM lab pipeline (https://github.com/cmap/dockerized_mts/tree/master/drc).
Synergy assays
Cells were seeded in 384-well plates (Corning, 3765) at 1000 cells/well followed by drug dispensing (doses in Supplementary Data S1) with the HP D300e Digital Dispenser. Cells were incubated for 72 h before viability was assessed by Cell-Titer Glo (G9242). Cell titer glo was added per well (1:1), plates were incubated for 10 min at room temperature before readout of luminescence signal on the Pherastar plate reader. All results were normalized to vehicle control.
Bulk RNA-seq
mNSC lines
Bulk RNA was extracted from mNSC cells using the Qiagen RNeasy kit (74104) and submitted to the Molecular Biology Core Facilities at Dana-Farber Cancer Institute. Libraries were prepared using Roche Kapa mRNA HyperPrep strand-specific sample preparation kits from 200 ng of purified total RNA according to the manufacturer’s protocol on a Beckman Colter Biomek i7. The finished dsDNA libraries were quantified by Qubit fluorometer and Agilent TapeStation 4200. Uniquely dual-indexed libraries were pooled in an equimolar ratio and shallowly sequenced on an Illumina MiSeq to further evaluate library quality and pool balance. The final pool was sequenced on an Illumina NovaSeq 6000 targeting 40 million 150 bp read pairs per library at the Dana-Farber Cancer Institute Molecular Biology Core Facilities.
Sequenced reads were aligned to the UCSC mm10 reference genome assembly, and gene counts were quantified using STAR (v2.7.3a)78. Differential expression analysis was performed using the R package DESeq2 v1.22.179. RNAseq analysis was performed using the VIPER snakemake pipeline80. Genes with a baseMean (expression) higher than 50, an absolute log2FoldChange higher than 1.5, and a Benjamini and Hochberg corrected p value of less than 0.05 were considered significant.
GSEA with MsigDB
Significant genes were run through the Molecular Signatures Database43. Negative log10(FDR) was calculated for each term, and results were plotted in R using the package ggbarplot.
ihNSC lines
Cell cultures were detached using accutase and collected in TSM, then pelleted by centrifugation and washed twice in ice-cold PBS buffer. RNA was extracted from the cell pellet using the TRI Reagent Protocol (Sigma). In brief, RNA was extracted in TRI reagent, followed by addition of chloroform (no isoamyl alcohol) to aid in phase separation. The aqueous phase (RNA) was transferred into a fresh tube, isopropanol was added to isolate RNA. After incubation with isopropanol, the RNA pellet was washed with 75% ethanol before airdry and resuspension in water. RNA concentration and quality were analyzed with NanoDrop 2000 instrument.
A total of 1 µg of RNA material was used per sample for RNA preparation. RNA was purified from total RNA using poly-T oligo-attached magnetic beads. After fragmentation, the first strand cDNA was synthesized using random hexamer primers, followed by the second strand cDNA synthesis using either dUTP for directional library or dTTP for non-directional library. For the non-directional library, it was ready after end repair, A-tailing, adapter ligation, size selection, amplification, and purification. The library was checked with Qubit and real-time PCR for quantification and bioanalyzer for size distribution detection. Quantified libraries were pooled and sequenced on Illumina platforms, according to effective library concentration and data amount. Sequencing was performed on the Illumina NovaSeq 6000 with 2 × 150 bp read length. Reads were mapped to a reference genome (GRCh38) from multiple databases (NCBI/UCSC/Ensembl) using Hisat2 v2.0.5. featureCounts v1.5.0-p3 was used to count reads, and an FPKM (Fragments per Kilobase of transcript per Million mapped reads) value was calculated per gene. Differentially expressed genes for individual comparisons were determined using the DESeq2 package in R, with the resulting p values adjusted using Benjamini and Hochberg’s approach79. MPAS scores for mNSC and ihNSC models were calculated as previously described in ref. 52. One data point in the FGFR1 N546K mNSC sample was excluded as an outlier for this analysis.
Single-cell RNA-seq
Single-cell RNA-seq was performed on the mNSC lines using the Chromium Next GEM Single Cell 3ʹ Reagent Kits v3.1 (Dual Index) kit as per manufacturer’s instructions. 10,000 cells were loaded on the 10X controller for each sample. Libraries were quantified by Tape station 4200 (Agilent) and submitted to the Molecular Biology Core Facilities at Dana-Farber Cancer Institute. Uniquely indexed libraries were pooled in an equimolar ratio and sequenced on a NovaSeq 6000 S2 flowcell targeting 4 billion reads.
The CellRanger (v6.1.2) pipeline was used to map the sequencing data to the mouse genome (mm10). We then used the Seurat library (4.3.0) to convert the expression data to Seurat objects and perform quality control. First, we filtered for genes that appeared in at least three cells. Consistent with our expectation from mouse models, the fraction of reads that corresponded to mitochondrial RNA was very low (0.32% ± 0.29%); thus, we did not need to filter out cells by mitochondrial content. We filtered out indexes with fewer than 8000 reads to ensure we picked up cells that have one unique index. After filtering of low-quality cells, we obtained 19,009 cells total across the 4 samples. After performing log-normalization (scale.factor = 10,000), we simplified the data set by selecting for the 3000 most highly variable genes. We next performed unsupervised clustering and dimensional reduction via RunPCA, FindNeighbors (30 dimensions), FindClusters (resolution = 0.5), and RunUMAP (30 dimensions) all through the Seurat library to create a two-dimensional visualization of the expression data.
Liquid chromatography tandem mass spectrometry (LC-MS/MS)
A total of 300 µg of protein from each sample was sent to the Barts Cancer Institute Mass Spectrometry Laboratory, Queen Mary University of London. About 150 µg of protein was then used for each, total proteomics and phosphoproteomics analysis. Sample preparation, enrichment of phosphopeptides, LC-MS/MS analysis, and data processing were performed as previously described in ref. 81. Raw data have been deposited on the PRIDE database.
Analyses using publicly available datasets
KiTZ patient tumor-RNA-seq analysis
RNA-sequencing count data from pLGGs harboring FGFR1 or BRAF alterations were obtained and analyzed as previously described4,40,41. Normalization (mean of ratios) of the data and differential gene expression analysis between the BRAF (n = 130) and FGFR1 (n = 16) tumors was performed using the R package DESeq279 with the Wald test. Genes with a baseMean (expression) higher than 50, an absolute log2FoldChange higher than 1.5, and a Benjamini and Hochberg corrected p value of less than 0.05 were considered significant.
Retrieval and processing of bulk RNA-sequencing datasets
Bulk RNA-seq from the Evo-devo project45 and the Brainspan project46 were used for gene expression across brain developmental stages, and GTEx v847 was used for gene expression across adult brain regions.
For the Evo-devo project, raw counts were downloaded and processed using an in-house RNA-seq pipeline as follows. Adapter sequences and the first four nucleotides of each read were removed from the read sets using Trimmomatic82 (v0.32). Reads were scanned from the 5′ end and truncated when the average quality of a four-nucleotide sliding window fell below a threshold (phred33 < 30). Short reads after trimming (<30 base pairs) were discarded. High-quality reads were aligned to the reference genome hg19 (GRCh37) using STAR78 (v2.3.0e) using default parameters. Reads mapping to more than 10 locations (MAPQ < 1) were discarded from downstream analyses. Gene expression levels were estimated by quantifying reads uniquely mapped to exonic regions defined by ensGene annotation set from Ensembl (GRCh37, N = 60,234 genes) using featureCounts83 (v1.4.4). Normalization (mean-of-ratios) and variance-stabilized transformation of the data were performed using DESeq79 (v1.14.1). Brainspan RPKM normalized expression and metadata tables were downloaded and processed as follows: donors with <3 Samples were removed, the provided brain region annotations were grouped into 16 unified regions, and expression data were log-transformed. GTEx v8 (GTEX) TPM normalized expression and metadata tables were downloaded and used directly.
Retrieval and processing of single-cell RNA-sequencing datasets
Single-cell RNA-seq atlases of mouse developmental50,51 and adult84 brain, as well as human developmental49 and adult85,86 brain, were used to query cell type specificity of expression. Datasets were retrieved with cell type labels provided by authors, and gene detection rate (% of cells with expression > 0) per cell type was calculated for visualization purposes.
A single-cell RNA-seq atlas of human fetal organogenesis48 was used to confirm early expression of FGFR3 in human embryo. Raw gene expression matrix and cell type labels from authors were retrieved and processed using Seurat87 (v4.3.0) as follows. Expression values were scaled to 10,000 UMI per cell and log-normalized, as well as z-scored gene-wise. Dimensionality reduction was performed using principal component analysis (PCA) applied to the top 2000 most variant genes. The first 10 principal components were used as input for projection to two dimensions, using uniform manifold approximation and projection (UMAP)88.
Statistics and reproducibility
Statistical analysis was performed using GraphPad Prism (v.10.4.0) for Mac or R (v4.4). Normality tests for determination of a normal distribution and tests of variance were established to determine the appropriate downstream test to perform. To test enrichment in our clinical cohorts and the overlap of gene sets, we performed Fisher’s exact tests. To compare the means of two groups we performed parametric t-tests or Welch’s t-test (does not assume equal SD) or non-parametric Mann–Whitney tests as specified in the text and/or Fig. legend. To compare the mean across multiple groups we performed One-way ANOVA followed by Tukey’s multiple comparison tests, Brown-Forthsythe and Welch’s ANOVA followed by a Dunnett’s T3 multiple comparison tests, or a non-parametric Kruskal–Wallis test. For most of these tests we compared all the means to a vector control/vehicle control condition. The specific information for each Fig. is described in the text and/or Fig. legend. Unless specific otherwise, all error bars shown the mean ± SEM of three independent replicates. P values of <0.05 were considered significant. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The DFCI DNA sequencing data (Oncopanel) from pediatric patients has been previously generated and deposited on dbGAP (Accesion # phs002677.v1.p1 fmethods) and is available to academic collaborators studying cancer and approved by NCI’s Data Access Committee. The length of time to respond to data requests will be as per the Data Usage Committee. There are no limitations on duration of data availability. The DFCI DNA sequencing data (Oncopanel) from adult patients was previously generated and deposited to the AACR GENIE database. The data can be accessed by registered academic users studying cancer and who apply through the AACR Project Genie website (https://www.aacr.org/professionals/research/aacr-project-genie/aacr-project-genie-data/) which also allows for visualization of variant calls in cBioPortal. The DNA sequencing data utilized for this study from Foundation Medicine are derived from clinical samples, and data are available upon request. Due to Health Insurance Portability and Accountability Act requirements, Foundation Medicine is not authorized to share individualized patient genomic data, which contains potentially identifying or sensitive patient information. Foundation Medicine is committed to collaborative data analysis, with well-established and widely utilized mechanisms by which investigators can query our core genomic database of >700,000 de-identified sequenced cancers to obtain aggregated datasets. For more information and mechanisms of access, please contact the corresponding author(s) or the Foundation Medicine, Inc. Data Governance Council at mailto:data.governance.council@foundationmedicine.com. The KiTZ DNA-sequencing (EGAS00001006680) and DNA methylation (GSE2125240) data from 425 patients are publicly available from the KiTZ MNP study.22 Publicly available bulk RNA-seq data from pLGG patient tumors were used from three studies (EGAS00001000381, EGAS00001007072, syn5698493)4,40,41. The KiTZ RNA-sequencing patient cohort information, RNA-sequencing counts, and differential expression data are in Supplementary files. Bulk RNA-seq from mNSCs and ihNSCs and the single-cell RNA-seq from the mNSCs generated in this study have been deposited in GEO under accession number GSE274098. Proteomics data have been deposited to the PRIDE database under the accession number: PXD060246. Source data are provided with this paper. The publicly available Bulk RNA-seq from the Evo-devo project (E-MTAB-8614)45 and the Brainspan project46 were used for gene expression across brain developmental stages, and GTEx v8 (accession no. phs000424.v8)47 was used for gene expression across adult brain regions. Publicly available Single-cell RNA-seq atlases of mouse developmental (GSE210567, GSE118068)50,51 and adult (GSE246717)84 brain, as well as human developmental (EGAS00001007472)49 and adult (RRID:SCR_015820, nemo:dat-3ah9h9x)85,86 brain, were used in addition to a single-cell RNA-seq atlas of human fetal organogenesis (GSE157329)48. All the relevant data are provided in the main manuscript and its supplementary files. Source data are provided with this paper.
References
Ardizzone, A. et al. Role of fibroblast growth factors receptors (FGFRs) in brain tumors, focus on astrocytoma and glioblastoma. Cancers 12, 3825 (2020).
Krook, M. A. et al. Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 124, 880–892 (2021).
Murugesan, K. et al. Pan-tumor landscape of fibroblast growth factor receptor 1-4 genomic alterations. ESMO Open 7, 100641 (2022).
Jones, D. T. et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 45, 927–932 (2013).
Ryall, S. et al. Integrated molecular and clinical analysis of 1000 pediatric low-grade gliomas. Cancer Cell 37, 569–583.e565 (2020).
Zhang, J. et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 45, 602–612 (2013).
Ryall, S. et al. Targeted detection of genetic alterations reveal the prognostic impact of H3K27M and MAPK pathway aberrations in paediatric thalamic glioma. Acta Neuropathol. Commun. 4, 93 (2016).
Mackay, A. et al. Integrated molecular meta-analysis of 1000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32, 520–537.e525 (2017).
Singh, D. et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337, 1231–1235 (2012).
Parker, B. C. et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J. Clin. Investig. 123, 855–865 (2013).
Di Stefano, A. L. et al. Detection, characterization, and inhibition of FGFR-TACC fusions in IDH wild-type glioma. Clin. Cancer Res. 21, 3307–3317 (2015).
Gupta, R. et al. Low-grade glioneuronal tumors with FGFR2 fusion resolve into a single epigenetic group corresponding to ‘Polymorphous low-grade neuroepithelial tumor of the young. Acta Neuropathol. 142, 595–599 (2021).
Palejwala, A. H. et al. Polymorphous low-grade neuroepithelial tumor of the young: rare tumor and review of the literature. Rare Tumors 14, 20363613221083360 (2022).
Tabernero, J. et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 33, 3401–3408 (2015).
Tripathi, A., Li, D. & Pal, S. K. FGFR inhibition: understanding and overcoming resistance. Cancer Discov. 13, 1964–1965 (2023).
Subbiah, V. & Verstovsek, S. Clinical development and management of adverse events associated with FGFR inhibitors. Cell Rep. Med. 4, 101204 (2023).
Bandopadhayay, P. et al. Long-term outcome of 4040 children diagnosed with pediatric low-grade gliomas: an analysis of the Surveillance Epidemiology and End Results (SEER) database. Pediatr. Blood Cancer 61, 1173–1179 (2014).
Johnson, A. et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist 22, 1478–1490 (2017).
van Tilburg, C. M. et al. LOGGIC/FIREFLY-2: a phase 3, randomized trial of tovorafenib vs. chemotherapy in pediatric and young adult patients with newly diagnosed low-grade glioma harboring an activating RAF alteration. BMC Cancer 24, 147 (2024).
Kilburn, L. B. et al. The type II RAF inhibitor tovorafenib in relapsed/refractory pediatric low-grade glioma: the phase 2 FIREFLY-1 trial. Nat. Med. 30, 207–217 (2024).
Bouffet, E. et al. Dabrafenib plus trametinib in pediatric glioma with BRAF V600 mutations. N. Engl. J. Med. 389, 1108–1120 (2023).
Sturm, D. et al. Multiomic neuropathology improves diagnostic accuracy in pediatric neuro-oncology. Nat. Med. 29, 917–926 (2023).
Gessi, M. et al. FGFR1 mutations in Rosette-forming glioneuronal tumors of the fourth ventricle. J. Neuropathol. Exp. Neurol. 73, 580–584 (2014).
Lucas, C. G. et al. Comprehensive analysis of diverse low-grade neuroepithelial tumors with FGFR1 alterations reveals a distinct molecular signature of rosette-forming glioneuronal tumor. Acta Neuropathol. Commun. 8, 151 (2020).
Brady, S. W. et al. Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat. Commun. 11, 5183 (2020).
Engelhardt, S. et al. Frequent FGFR1 hotspot alterations in driver-unknown low-grade glioma and mixed neuronal-glial tumors. J. Cancer Res. Clin. Oncol. 148, 857–866 (2022).
Lazo De La Vega, L. et al. Rare FGFR Oncogenic alterations in sequenced pediatric solid and brain tumors suggest FGFR is a relevant molecular target in childhood cancer. JCO Precis Oncol. 6, e2200390 (2022).
Yang, J. et al. Genomic profiling of circulating tumor DNA from patients with extensive-stage small cell lung cancer identifies potentially actionable alterations. J. Cancer 12, 5099–5105 (2021).
Fina, F. et al. Droplet digital PCR is a powerful technique to demonstrate frequent FGFR1 duplication in dysembryoplastic neuroepithelial tumors. Oncotarget 8, 2104–2113 (2017).
Ryall, S., Tabori, U. & Hawkins, C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol. Commun. 8, 30 (2020).
Qin, A. et al. Detection of known and novel FGFR fusions in non-small cell lung cancer by comprehensive genomic profiling. J. Thorac. Oncol. 14, 54–62 (2019).
Pant, S. et al. Erdafitinib in patients with advanced solid tumours with FGFR alterations (RAGNAR): an international, single-arm, phase 2 study. Lancet Oncol. 24, 925–935 (2023).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Taylor, J. G. et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 119, 3395–3407 (2009).
Levine, K. M., Ding, K., Chen, L. & Oesterreich, S. FGFR4: a promising therapeutic target for breast cancer and other solid tumors. Pharmacol. Ther. 214, 107590 (2020).
Qaddoumi, I. et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 131, 833–845 (2016).
Bale, T. A. FGFR- gene family alterations in low-grade neuroepithelial tumors. Acta Neuropathol. Commun. 8, 21 (2020).
Jones, D. T. W., Bandopadhayay, P. & Jabado, N. The power of human cancer genetics as revealed by low-grade gliomas. Annu. Rev. Genet. 53, 483–503 (2019).
Bandopadhayay, P. et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat. Genet. 48, 273–282 (2016).
Hardin, E. C. et al. LOGGIC core bioclinical data bank: added clinical value of RNA-Seq in an international molecular diagnostic registry for pediatric low-grade glioma patients. Neuro Oncol. 25, 2087–2097 (2023).
Fisher, M. J. et al. Integrated molecular and clinical analysis of low-grade gliomas in children with neurofibromatosis type 1 (NF1). Acta Neuropathol. 141, 605–617 (2021).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Anastasaki, C. et al. Human induced pluripotent stem cell engineering establishes a humanized mouse platform for pediatric low-grade glioma modeling. Acta Neuropathol. Commun. 10, 120 (2022).
Cardoso-Moreira, M. et al. Gene expression across mammalian organ development. Nature 571, 505–509 (2019).
Miller, J. A. et al. Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014).
Consortium, G. T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Xu, Y. et al. A single-cell transcriptome atlas profiles early organogenesis in human embryos. Nat. Cell Biol. 25, 604–615 (2023).
Braun, E. et al. Comprehensive cell atlas of the first-trimester developing human brain. Science 382, eadf1226 (2023).
Jessa, S. et al. K27M in canonical and noncanonical H3 variants occurs in distinct oligodendroglial cell lineages in brain midline gliomas. Nat. Genet. 54, 1865–1880 (2022).
Vladoiu, M. C. et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 572, 67–73 (2019).
Wagle, M. C. et al. A transcriptional MAPK Pathway Activity Score (MPAS) is a clinically relevant biomarker in multiple cancer types. NPJ Precis Oncol. 2, 7 (2018).
Pellegrini, L. Role of heparan sulfate in fibroblast growth factor signalling: a structural view. Curr. Opin. Struct. Biol. 11, 629–634 (2001).
Manoharan, N., Liu, K. X., Mueller, S., Haas-Kogan, D. A. & Bandopadhayay, P. Pediatric low-grade glioma: targeted therapeutics and clinical trials in the molecular era. Neoplasia 36, 100857 (2023).
Krook, M. A. et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-fusion cholangiocarcinoma. Mol. Cancer Ther. 19, 847–857 (2020).
Li, Y. et al. FGFR-inhibitor-mediated dismissal of SWI/SNF complexes from YAP-dependent enhancers induces adaptive therapeutic resistance. Nat. Cell Biol. 23, 1187–1198 (2021).
Lee, A. et al. Erdafitinb in patients with FGFR-altered tumors: Results from the NCI-COG Pediatric MATCH trial arm B (APEC1621B). J. Clin. Oncol. https://doi.org/10.1200/JCO.2023.41.16_suppl.10007 (2023).
Stepien, N. et al. Feasibility and antitumour activity of the FGFR inhibitor erdafitnib in three paediatric CNS tumour patients. Pediatr. Blood Cancer 71, e30836 (2024).
Witt, O. et al. Efficacy and safety of erdafitinib in pediatric patients with advanced solid tumors and FGFR alterations in the phase 2 RAGNAR trial. J. Clin. Oncol. https://doi.org/10.1200/JCO.2024.42.16_suppl.10002 (2024).
Farouk Sait, S. et al. Debio1347, an Oral FGFR inhibitor: results from a single-center study in pediatric patients with recurrent or refractory FGFR-altered gliomas. JCO Precis. Oncol. 5 https://doi.org/10.1200/PO.20.00444 (2021).
Kondyli, M. et al. Trametinib for progressive pediatric low-grade gliomas. J. Neurooncol 140, 435–444 (2018).
Lazow, M. et al. LGG-14. Treatment of two pediatric FGFR-altered low-grade glioneuronal tumors with MEK inhibition. Neuro. Oncol. https://doi.org/10.1093/neuonc/noad073.224 (2023).
Manoharan, N. et al. Trametinib for the treatment of recurrent/progressive pediatric low-grade glioma. J. Neurooncol. 149, 253–262 (2020).
Selt, F. et al. Response to trametinib treatment in progressive pediatric low-grade glioma patients. J. Neurooncol. 149, 499–510 (2020).
Wen, P. Y. et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J. Clin. Oncol. 28, 1963–1972 (2010).
Fangusaro, J. et al. Response assessment in paediatric low-grade glioma: recommendations from the Response Assessment in Pediatric Neuro-Oncology (RAPNO) working group. Lancet Oncol. 21, e305–e316 (2020).
Yvone, G. M. & Breunig, J. J. Pediatric low-grade glioma models: advances and ongoing challenges. Front. Oncol. 13, 1346949 (2023).
Gonzalez-Vega, M. et al. Fibroblast growth factor receptor 1 gene mutation as a potential risk factor for spontaneous intracranial hemorrhage in pediatric low-grade glioma patients. Neurooncol. Adv. 6, vdae074 (2024).
Ishi, Y. et al. Association of the FGFR1 mutation with spontaneous hemorrhage in low-grade gliomas in pediatric and young adult patients. J. Neurosurg. 134, 733–741 (2020).
Siegfried, A. et al. Noonan syndrome, PTPN11 mutations, and brain tumors. A clinical report and review of the literature. Am. J. Med. Genet A. 173, 1061–1065 (2017).
El-Ayadi, M. et al. Occurrence of high-grade glioma in Noonan syndrome: Report of two cases. Pediatr. Blood Cancer 66, e27625 (2019).
Lodi, M. et al. Low-grade gliomas in patients with noonan syndrome: case-based review of the literature. Diagnostics 10, 582 (2020).
Shatara, M. et al. Molecular characterization of gliomas and glioneuronal tumors amid Noonan syndrome: cancer predisposition examined. Front. Oncol. 14, 1453309 (2024).
Khan, S. et al. Neuronal differentiation drives the antitumor activity of mitogen-activated protein kinase kinase (MEK) inhibition in glioblastoma. Neurooncol. Adv. 5, vdad132 (2023).
Cryan, J. B. et al. Clinical multiplexed exome sequencing distinguishes adult oligodendroglial neoplasms from astrocytic and mixed lineage gliomas. Oncotarget 5, 8083–8092 (2014).
Zheng, Z. et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 20, 1479–1484 (2014).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Dobin, A. & Gingeras, T. R. Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinforma. 51, 11 14 11–11 14 19 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Cornwell, M. et al. VIPER: visualization pipeline for RNA-seq, a snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinforma. 19, 135 (2018).
Casado, P. & Cutillas, P. R. A self-validating quantitative mass spectrometry method for assessing the accuracy of high-content phosphoproteomic experiments. Mol. Cell Proteom. 10, M110 003079 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Yao, Z. et al. A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature 624, 317–332 (2023).
Siletti, K. et al. Transcriptomic diversity of cell types across the adult human brain. Science 382, eadd7046 (2023).
Velmeshev, D. et al. Single-cell analysis of prenatal and postnatal human cortical development. Science 382, eadf0834 (2023).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: uniform manifold approximation and projection. J. Open Source Softw. 3, 861 (2018).
Acknowledgements
The authors acknowledge the following funding sources: The Dana-Farber Pediatric Low-Grade Glioma Program (P.B., K.L.L., E.S.F., M.J.E., and S.B.), The Everest Centre for Low-grade Pediatric Brain Tumors which also supports the LOGGIC Core BioClinical DataBank (GN-000707, The Brain Tumor Charity, UK; D.St., O.W., D.T.W.J., T.A.J., L.M.W., and D.Sh.), Team Kermit and the Jared Branfman Sunflowers for Life Fund (P.B., K.L.L., and S.N.C.), Team Brainstorms (P.B. and K.L.L.), Pediatric Brain Tumor Foundation (P.B., K.L.L., E.S.F., and M.J.E.), V Foundation (P.B. and K.L.L.), Team Jack Foundation (P.B. and K.L.L.), Jack’s Drive 55 (P.B. and K.L.L.), Helen Gurley Brown Foundation (P.B.), Damon Runyon-St. Jude Foundation Fellowship (03-24) (AA), The Swedish Childhood Cancer Fund (E.M.), 3000 Miles to the Cure (K.L.L.), Else Kröner Excellence Fellowship (D.St.), The Lady Davis Institute/Dr. Arthur Rosenberg Memorial Fellowship Graduate Scholarship Fund and the James O and Maria Meadows Award from the Faculty of Medicine and Health Sciences at McGill University (B.C.), Alex’s Lemonade Stand POST Program (A.D.L.), the Dietmar Hopp Foundation (KiTZ Clinical Trial Unit 2.0) (O.W.), and the Astro Brain Tumor Fund, UK (T.A.J., L.M.W., and D.Sh.). The MNP2.0 study was supported by the German Childhood Cancer Foundation (DSt 2015.01) Canadian Institutes of Health Research (CIHR) grant PJT-156086 (to C.L.K.). C.L. K. is the recipient of an FRQS Salary Award. We thank the Dana-Farber/Harvard Cancer Center in Boston, MA, for the use of the Specialized Histopathology Core, which provided histology and immunohistochemistry services. We thank the Molecular Biology Core Facilities at the Dana-Farber Cancer Institute for QC and sequencing of bulk and single-cell RNA-seq samples. We thank the CRUK Barts Centre Mass Spectrometry Facility, Queen Mary University of London, for proteomics analysis. We would like to thank Mustafa Kocak from the Broad Institute for help with calculating the AUC values. Finally, we would like to thank and acknowledge the many children and families affected by pediatric gliomas for their generous contributions to this research.
Author information
Authors and Affiliations
Contributions
A.A.A., E.M., S.B., S.O., H.J., E.S.F., M.J.E., Q.D.N., D.T.W.J., K.L.L. and P.B. conceived and designed experiments. A.A.A., E.M., G.A., J.D., S.B., P.C.P., M.M.C., D.N., P.P., C.C.B., S.M., S.W.L., K.Y.C., S.O., A.C., P.R., A.D.L. and L.B. collected data. A.A.A., E.M., D.St., G.A., B.C., P.C.P., R.E.J., J.V., S.M., L.M.W., T.A.J., J.J., J.C., J.S.J., J.W., S.H., S.B., C.K., and Q.D.N. performed analysis. D.St., B.C., J.V., L.M.W., T.A.J., K.M., M.M., L.A.A., S.H.R., C.M.V.T., E.C.H., P.S., F.S., K.K.Y., T.R., S.N.C., K.D.W., S.P., A.P., V.L., S.R., T.A.B., A.A.S., M.T., D.Sh., M.T., N.J., O.W., C.K., S.A., D.T.W.J., and K.L.L. contributed data. A.A.A., E.M., K.L.L. and P.B. wrote the manuscript. All authors reviewed, edited, and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
P.B. serves on paid advisory boards for DayOne Biopharmaceuticals and has served on a paid advisory board for QED Therapeutics. Her lab has received grant funding from Novartis Institute of Biomedical Research. SHR has employment at Labcorp Oncology. C.M.V.T. is on advisory boards for Alexion, Bayer, Novartis, and Roche and has received travel support from Eli Lilly. J.W. is a consultant and equity holder for Soltego. F.S. and D.T.W.J. are co-founders and shareholders of Heidelberg Epignostix GmbH. O.W. is advisory board member or has received research grants from Janssen, Day One Biopharmaceuticals, and Novartis. K.L.L. disclosures: Equity: Travera Inc.; Consulting- Travera Inc., B.M.S., Servier, L.E.K., Integragen, Blaze Bioscience; Research. Research Funding to DFCI: B.M.S. K.M., M.M., L.A.A. are employees of Foundation Medicine, Inc. and stockholders of Roche Holdings AG. E.S.F. is a founder, scientific advisory board (SAB) member, and equity holder of Civetta Therapeutics, Proximity Therapeutics, Neomorph, Inc. (also board of directors), Stelexis Biosciences, Inc., Anvia Therapeutics, Inc., and CPD4, Inc. (also board of directors). He is an equity holder and SAB member for Avilar Therapeutics, Photys Therapeutics, and Ajax Therapeutics, and an equity holder in Lighthorse Therapeutics. E.S.F. is a consultant to Novartis, EcoR1 capital, Odyssey, and Deerfield. The Fischer lab receives or has received research funding from Deerfield, Novartis, Ajax, Interline, Bayer, and Astellas. The Eck lab has received sponsored research support from Novartis, and MJE is a consultant for Syndax Pharmaceuticals. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Apfelbaum, A.A., Morin, E., Sturm, D. et al. A diverse landscape of FGFR alterations and co-mutations suggests potential therapeutic strategies in pediatric low-grade gliomas. Nat Commun 16, 7018 (2025). https://doi.org/10.1038/s41467-025-61820-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61820-z
