
Abstract
Metabolic dysfunction-associated steatotic liver disease is marked by fat accumulation and inflammation, partly due to impaired lipophagy—a cellular process in which lipid droplets are broken down through autophagy. Rubicon, a protein that inhibits this process, worsens the condition by blocking fat breakdown. Small interfering RNA molecules targeting Rubicon show therapeutic potential but face challenges such as instability and off-target effects. Here we show a dual-targeted nanoparticle system designed for efficient delivery of Rubicon-targeting small interfering RNA to liver cells. This system has a core-shell structure that ensures stability in the bloodstream and responsiveness to oxidative stress, commonly found in metabolic dysfunction-associated steatotic liver disease. Once inside the liver cells, the nanoparticles release the RNA molecules, which reduce Rubicon levels, restore lipophagy, and alleviate fatty liver buildup. This strategy offers a flexible platform for targeted gene silencing therapy in liver diseases.
Similar content being viewed by others
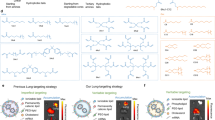


Introduction
Metabolic dysfunction-associated steatotic liver disease presents a complex health challenge intricately linked with hyperlipidemia, progressing along a spectrum from simple steatosis to metabolic dysfunction-associated steatohepatitis (MASH), and may further deteriorate to fibrosis, cirrhosis, hepatocellular carcinoma (HCC), and liver failure in severe cases. Worryingly, over one quarter of the world’s population is affected by MASH, rendering it the most rapidly escalating cause of HCC1. It has been found that the excessive accumulation of lipids, specifically triglycerides, free fatty acids (FFAs), and small amounts of cholesterol within hepatocyte can trigger a cascade of detrimental effects such as lipotoxicity, metabolic dysregulation, and heightened oxidative stress2,3,4. Therefore, accelerating the excessive lipids metabolism represents a promising strategy to impede lipid droplet accumulation and stop the progression of liver diseases.
Autophagy, a vital cellular mechanism, plays a crucial role in maintaining cellular homeostasis5. This process involves the encapsulation of cellular components by double-membraned autophagosomes, which then merge with lysosomes for degradation, thereby recycling materials and generating energy6. Its implications extend across the etiology of numerous diseases, making autophagy a focal point of research interest, particularly regarding its therapeutic potential in regulating lipid metabolism and managing conditions like MASH7. Studies have revealed that suppressing key autophagy-related proteins, Atg7 and Atg5, can significantly reduce intracellular autophagic activity8,9. This suppression results in impaired autophagosome fusion and defective acidification processes, impeding the efficient clearance of lipid droplets within hepatocytes, promoting hepatic steatosis and potentially leading to fibrous changes and oncogenesis10,11. Thus, the strategy with autophagy activation by targeting the core pathophysiological mechanisms of MASH and the related liver diseases emerges as a compelling therapeutic intervention. In the future, enhancing lipid clearance and maintaining cellular homeostasis through autophagy regulation present a promising strategy to block the advancement of these debilitating liver conditions.
Rubicon, a protein known for its RUN domain interaction with Beclin-1 and possession of a cysteine-rich domain, plays a crucial role in the selective autophagy pathway12. Increased Rubicon expression in scenarios of free fatty acid-induced cellular fattening has been consistently observed in mouse MASH models and liver specimens from individuals diagnosed with MASH10. Conversely, interventions that disrupt Rubicon, either genetically or pharmacologically, can promote autophagosome maturation and facilitate fusion with lysosomes, activating the autophagic flux11. This improved lipid metabolism within cells by lipophagy has also been involved in various age-associated diseases treatment13. In the realm of lipid handling or lipophagy—breakdown of lipid droplets via autophagy—Rubicon assumes a critical role, concerning the dismantling and recycling of these droplets with considerable influence10. Dampening the expression or activity of Rubicon within adipocytes increases autophagic activity, accelerating the catabolism of lipid droplets and presenting a promising strategy for alleviating conditions such as MASH12. Therefore, strategic modulation of Rubicon emerges as a frontier in therapy14, offering fresh insights and avenues for tackling diseases characterized by deregulated lipid metabolism10.
Unfortunately, the current landscape lacks small molecule inhibitors tailored to directly target Rubicon, and the therapeutic potential of Rubicon-specific siRNA in MASH remains unclear, primarily hindered by inefficient in vivo RNA delivery methodologies. However, siRNA targeting Rubicon stands as an ideal therapeutic modality due to its precision in mRNA degradation-mediated gene silencing, minimizing off-target occurrences compared to tiny molecule interventions15,16. Yet, the effective delivery of siRNA confronts substantial hurdles: (I) bloodstream stability: unmodified siRNA is prone to rapid degradation in circulation due to both enzymatic actions and hemodynamics. (II) cellular entry dilemma: the inherent negative charge of siRNA impedes cellular endocytosis as it encounters electrostatic repulsion from the similarly charged cell membrane17,18. (III) accumulation and clearance conundrum: although nanoparticles show a propensity to amass in the liver, they are also susceptible to uptake by other organs, and subsequent clearance by phagocytes, limiting the fraction available for targeted liver engagement. (IV) liver-specific precision challenge: even following successful liver accumulation, siRNA delivery platforms must contend with the preferential uptake by non-parenchymal cells, instead of the intended hepatocytes, further complicating targeted intervention. To address these issues, an efficacious siRubicon delivery system must ensure siRNA stability, modify surface charge, control siRNA release, and achieve hepatocyte specificity.
The excessive accumulation of reactive oxygen species (ROS) in liver tissue is a hallmark of MASH development, contributing to the progression of hepatic injury. Consequently, vectors that release payloads in response to high concentrations of ROS would be ideal for the targeted liver delivery of siRubicon19. The asialoglycoprotein receptor (ASGPR), a transmembrane protein abundantly expressed on hepatocyte surfaces, specifically recognizes glycoproteins terminating in N-acetylgalactosamine (GalNAc) in the bloodstream20. Accordingly, GalNAc-modified delivery systems can efficiently target hepatocytes via ASGPR-mediated endocytosis. Here, we demonstrate a pioneering multi-stage targeted nanoparticle system (PPH-PEI/GalNAc@siRNA) that achieves dual targeting of the liver and hepatocytes. This system accumulates in the liver by responding to the high levels of ROS in MASH models, and efficiently targets hepatocytes through modified GalNAc ligands. PPH-PEI/GalNAc@siRNA features a typical core-shell structure, where the positively charged GalNAc-modified PEI25k is first complexed with siRNA to form the inner core PEI25k-GalNAc@siRNA. This core is subsequently coated with mPEG113-b-PLys120-HPBA, forming the outer shell via chemical bonds between the 4-(hydroxymethyl)phenylboronic acid (HPBA) groups on mPEG113-b-PLys120-HPBA and the GalNAc groups on the core. The polymer encapsulated PPH-PEI/GalNAc@siRNA has a negatively charged, PEGylated surface, which helps evade immune clearance during systemic circulation21,22,23. Upon reaching the oxidative microenvironment of fatty liver, the elevated ROS levels trigger the cleavage of HPBA moieties, leading to detachment of the outer mPEG113-b-PLys120-HPBA shell. This process exposes the positively charged PEI25k-GalNAc@siRNA core, allowing GalNAc ligands to engage the highly expressed ASGPR on hepatocytes. Post-internalization, the PEI25k-GalNAc@siRNA complex disassembles in the acidic endosome, liberating siRNA into the cytosol for target interaction5 (Fig. 1).

a Schematic illustration of the multi-stage nanoparticle system, PPH-PEI/GalNAc@siRNA, designed for precise delivery of siRNA to hepatocytes in metabolic dysfunction-associated steatohepatitis (MASH). The nanoparticle comprises a ROS-responsive shell, PEI complexed with GalNAc for hepatocyte targeting, and encapsulated siRNA against Rubicon (siRubicon). b Following retrobulbar intravenous injection, the nanoparticles accumulate in the liver and are specifically activated by ROS in MASH livers. Activation triggers shell disassembly and exposes the PEI25k-GalNAc/siRubicon complex, enabling targeted delivery to hepatocytes via asialoglycoprotein receptor (ASGPR)-mediated endocytosis. c Inside hepatocytes, siRubicon silences Rubicon expression, a key negative regulator of autophagy, thereby enhancing autophagic and lipophagic flux. This leads to increased degradation of lipid droplets via lipophagy, resulting in reduced hepatic steatosis and improved metabolic outcomes in MASH. ROS reactive oxygen species, PEI polyethylenimine, GalNAc N-acetylgalactosamine, siRNA small interfering RNA, ASGPR asialoglycoprotein receptor.
This multi-stage delivery strategy empowers PPH-PEI/GalNAc@siRNA to execute precise siRubicon delivery, efficaciously suppressing target protein expression and inhibiting lipid buildup in hepatocytes through lipophagy. Thus, it constitutes a highly promising gene therapy avenue for siRNA delivery to diseased tissues, particularly in MASH. By addressing siRNA instability and targeting inefficiencies, and exploiting the disease milieu to heighten therapeutic precision and impact, our system exhibits substantial promise for treating MASH and related conditions marked by oxidative stress and lipid imbalance.
Results
Characterization and cellular uptake evaluation of the core-shell nanoparticles
We synthesized core PEI25k-GalNAc@siRNA particles and subsequently coated them with a mPEG113-b-PLys120-HPBA shell, with each synthesis stage validated by 1H NMR to ensure successful material integration (Supplementary Fig. 1). Given the prevalence of Kupffer cells in the liver, we evaluated the phagocytic efficacy of macrophages and hepatocytes toward cationic PEI25k-GalNAc/siRNA complexes at varying mass ratios. When RAW 264.7 macrophages were incubated with PEI25k@Cy5-labeled siRNA complexes (PEI25k-to-siRNA mass ratio of 1), high phagocytic activity was observed, whereas a relatively lower phagocytosis rate was noted for PEI25k-GalNAc@Cy5-labeled siRNA complexes at ratios 1, 2, and 3, potentially due to limited GalNAc recognition sites on macrophages (Fig. 2a, b and Supplementary Fig. 2a, b). Next, we prepared PEI25k-GalNAc@Cy5-labeled siRNA complexes at mass ratios 1, 2, 3, and 5 and exposed them to hepatocytes (Hepa 1–6 cell line), observing a significant decline in uptake beyond a 2:1 ratio, accompanied by cytotoxicity at a 5:1 ratio (Fig. 2c, d and Supplementary Fig. 2c, d). Hence, a 2:1 mass ratio was adopted for core formation. In addition to experiments conducted in established cell lines, we further isolated both liver parenchymal and non-parenchymal cells for endocytosis experiments. The results were consistent with those observed in cell lines, demonstrating that a mass ratio of 2:1 (PEI25k-GalNAc to siRNA) maximized endocytosis efficiency preferentially in hepatocytes (Supplementary Fig. 3a–d).

a representative images IF staining and b FCM analysis of the internalization of various mass ratios of PEI25k-GalNAc to Cy5-labeled siRNA (red) following overnight incubation with RAW 264.7 macrophage cells (n = 3). Scale bar: 10 μm. c IF staining and d FCM analysis of the internalization of different mass ratios of PEI25k-GalNAc to Cy5-labeled siRNA complexes after an overnight incubation with Hepa 1–6 hepatocytes (n = 3). Scale bar: 10 μm. e IF staining and f FCM analysis of the internalization of varied mass ratios of mPEG113-b-PLys120-HPBA with a constant 2:1 mass ratio of PEI25k-GalNAc to Cy5-labeled siRNA, comparing conditions with or without 200 μM H2O2 supplementation in Hepa 1–6 cells. Scale bar: 10 μm. g Size distribution and TEM images showcasing: g PEI25k@siRNA, h PEI25k-GalNAc@siRNA, i PPH-PEI/GalNAc@siRNA, and j PPH-PEI/GalNAc@siRNA after 4 h incubation with 200 μM H2O2 at room temperature. Scale bar for TEM images: 100 nm. k Zeta potential values of PPH-PEI/GalNAc@siRNA under conditions with or without 200 μM H2O2, in conjunction with varying mass ratios of mPEG113-b-PLys120-HPBA to PEI25k-GalNAc@siRNA (n = 3). Error bars indicate means ± SEM. Source data are provided as a Source Data file.
To determine an optimal shell ratio for physiological charge shielding and oxidative stress-responsive release, we varied the mass ratio of mPEG113-b-PLys120-HPBA to PEI25k-GalNAc@siRNA. When co-culturing PPH-PEI/GalNAc@siRNA with hepatocytes under normal or oxidative stress conditions (200 μM H2O2) overnight, an increase in mass ratios of mPEG113-b-PLys120-HPBA to PEI25k-GalNAc@siRNA resulted in decreased endocytic efficiency under normal culturing conditions, indicating the validity of the shielding effect of the shell on nanoparticles (Fig. 2e, f). Under H2O2 culturing condition, we observed an elevation in the phagocytosis rate from 28.8% to 89.2% at a 2:1 shell-to-core mass ratio, demonstrating that this is an appropriate mass ratio for ROS responsiveness (Fig. 2e, f). Consistent with these findings, endocytosis assays using primary parenchymal cells demonstrated that the mPEG113-b-PLys120-HPBA and PEI25k-GalNAc@siRNA complex at a 2:1 mass ratio exhibited enhanced delivery efficiency under oxidative stress conditions (Supplementary Fig. 3e, f).
Dynamic light scattering (DLS) analysis showed PEI25k-GalNAc@siRNA particles possessed a slightly larger diameter than PEI25k@siRNA, averaging around 100 nm, attributed to the inclusion of CDM-GalNAc (Fig. 2g, h). TEM and DLS confirmed shell addition increased particle size to about 190 nm (Fig. 2i), which reduced to ~135 nm under hydrogen peroxide, possibly reflecting the oxidative stress-responsive transformation via electrostatic repulsion (Fig. 2j). Zeta-potential measurements supported this, with a reduction in surface potential upon mPEG113-b-PLys120-HPBA coating but a shift from −0.64 ± 0.31 mV to 0.516 ± 0.245 mV at a 2:1 shell-to-core mass ratio under oxidative conditions (Fig. 2k). Collectively, these results substantiate the rational design and functional effectiveness of the fabricated nanoparticles.
PEI25k-GalNAc@siRNA delivery platforms enhances hepatocytic phagocytic efficiency and selectivity
To elucidate the specific targeting of the synthesized core nanomaterials toward hepatocytes, we implemented a co-culture strategy utilizing transwell inserts with 3 μm pores, arranging macrophages above and hepatocytes beneath. Post-overnight incubation, cells from both chambers underwent immunofluorescence (IF) staining and flow cytometry (FCM) assessment (Fig. 3a). IF data confirmed high phagocytic activity for the materials at mass ratios 1, 2, and 3, except for the hepatotoxicity detected at ratio 5 (Fig. 3b), similar to the results got in Fig. 2c. FCM revealed an equivalent uptake rate between hepatocytes and macrophages at ratio 1 (positive rates: 76.2% versus 73.1%). At ratio 2, a slight uptake advantage in hepatocytes was seen (87.3% versus 76.7%), plausibly attributed to heightened PEI25k-GalNAc interaction with hepatocyte receptors. Conversely, macrophages demonstrated greater phagocytic efficiency at ratio 3 (65.9% versus 81.8%). In parallel, primary parenchymal and non-parenchymal liver cells were isolated and co-cultured in the same way. It revealed that the phagocytic efficiency of non-parenchymal liver cells toward the nanoparticles remained consistently high. In contrast, the uptake efficiency by hepatocytes exhibited an increasing trend as the mass ratio of PEI25k -GalNAc to siRNA approached 2:1 (Supplementary Fig. 4). To validate in vivo selectivity, we employed normothermic machine perfusion (NMP), administering PEI25k@siRNA and PEI25k-GalNAc@siRNA formulations to livers (Fig. 3d). Perfusion was sustained at 3 mL/min under 5–8 mmHg for 6 h, followed by liver retrieval for IF staining (Fig. 3e). Relative to the PEI25k@siRNA cohort, co-localization of hepatocytes with the nanoparticles was significantly elevated in the PEI25k-GalNAc@siRNA group (co-localization positivity: 34.51% ± 6.441% in PEI25k@siRNA group versus 52.62% ± 5.152% in PEI25k-GalNAc@siRNA group) (Fig. 3f). Meanwhile, CD11b positive macrophage and Cy5-labeled siRNA co-localization ratio in PEI25k-GalNAc@siRNA group were comparable with those in PEI25k@siRNA group (PEI25k@siRNA versus PEI25k-GalNAc@siRNA co-localization positivity: 32.18% ± 5.43% versus 26.72% ± 5.79%), emphasizing the exceptional hepatocyte-targeting proficiency of the PEI25k-GalNAc-based siRNA delivery platform (Fig. 3g).

a a co-culture model is established, juxtaposing macrophages with hepatocytes utilizing a transwell system featuring 3-micron pores. b IF and c FCM analysis are employed to evaluate the competitive internalization dynamics of the PEI25k-GalNAc@siRNA complex between hepatocytes and macrophages. Scale bars denote 10 μm. d employing a normothermic machine perfusion setup, 33 μg of the siRNA complex, formulated with a PEI25k-to-siRNA mass ratio of 1:1 and a PEI25k-GalNAc-to-siRNA ratio of 1:2, is introduced into rat livers through a tripartite inlet for extracorporeal perfusion. e 6 h subsequent to perfusion, harvested livers undergo co-staining procedures to visualize hepatocyte/siRNA and macrophage/siRNA associations. Scale bar represents 20 μm. f quantitative assessments detail the co-expression of nanomaterials with either hepatocytes or (g), macrophages (n = 5). Statistical significance demarcated by * p < 0.05 and ** p < 0.01, insignificance was not represented. Error bars indicate means ± SEM. Data were analyzed by two-tailed Student’s t-test. Source data are provided as a Source Data file.
PEI25k-GalNAc@siRubicon therapy mitigates hepatocellular steatosis
To evaluate the efficacy of PEI25k-GalNAc@siRubicon in alleviating hepatocellular steatosis, we initially induced a fatty liver model in vitro with escalating FFA concentrations. This led to an observable rise in lipid droplet accumulation, marked by increase in both number and size (Fig. 4–c). However, CCK8 assays pinpointed a critical viability reduction beyond 500 μM FFA, prompting the selection of 300 μM as the optimal steatosis-inducing dose (Supplementary Fig. 5a). IF staining subsequently disclosed a direct association between steatosis severity and Rubicon expression, peaking at 300 μM FFA (Fig. 4d, e). Western blot analysis corroborated heightened Rubicon and p62 expression in steatotic hepatocytes compared to controls (Fig. 4f, g), implicating steatosis as a barrier to autophagic flux. Next, we screened the siRNA sequences and found that, as determined by WB analysis, the protein expression level in cells transfected with si#2856 was reduced to approximately 30% of that in the control group. Based on this result, we selected si#2856 in the subsequent experiments (Fig. 4h, i). With the aim to explore if Rubicon suppression could boost lipid metabolism by lipophagy, we leveraged PEI25k-GalNAc for siRubicon delivery into hepatocytes. Rapamycin (RAPA) and hydroxychloroquine (CQ) served as positive controls for autophagy stimulation and inhibition, respectively, with optimized concentrations set at 5 μM and 20 μM, respectively (Supplementary Fig. 5b, c). The siRNA concentration was fixed at 30 nM to minimize cell proliferation inhibition (Supplementary Fig. 5d), and a mass ratio below 3 for mPEG113-b-PLys120-HPBA to PEI25k-GalNAc@siRNA was deemed non-disruptive to cell growth (Supplementary Fig. 5e). Following pretreatment with PEI25k-GalNAc@siRubicon, RAPA, or CQ, cells were subjected to 300 μM FFA incubated for 24 h, with the subsequent quantification of free cholesterol (FC) and triglyceride (TG) contents. Compared to the FFA-induced cells, cells receiving PEI25k-GalNAc@siRubicon pre-treatment manifested significantly diminished FC (314.64 mM ± 9.057 mM vs. 457.397 mM ± 10.224 mM in FFA group) and TG levels (4.991 nM ± 1.362 nM vs. 17.295 nM ± 1.493 nM in FFA group), similar to the results got by RAPA treatment (FC: 331.508 mM ± 22.808 mM; TG: 5.694 nM ± 0.275 nM) (Fig. 4k, l). Similarly, IF and Oil Red O staining also depicted attenuated FFA-induced steatosis with RAPA and PEI25k-GalNAc@siRubicon interventions, while worsened post-CQ treatment (Fig. 4m–p). To further validate the regulatory effect of Rubicon on lipid metabolism, primary hepatocytes were treated with 400 μM FFA. Western blot analysis revealed increased expression levels of LC3II and p62, confirming that FFA treatment inhibited autophagic flux (Supplementary Fig. 6a–c). Additionally, Oil Red O staining and quantitative assessments of intracellular TC and TG demonstrated that silencing Rubicon expression significantly reduced intracellular lipid accumulation (Supplementary Fig. 6d–g). This reduction was associated with the restoration of autophagic flux, as indicated by changes in p62 and LC3 expression (Supplementary Fig. 6h–j).

a IF staining visualizes intracellular lipid droplet accumulation in response to varying FFA concentrations. b quantitative assessment of lipid droplet size and c average lipid droplet count per cell under differential concentrations of FFA exposure (n = 7). d IF staining and e quantification of Rubicon in hepatocytes subjected to diverse FFA concentrations (n = 4). f Western blot analysis and g quantification of autophagy markers p62 and Rubicon in hepatocytes treated with 300 μM FFA (n = 3). h Western blot evaluation and i quantification of Rubicon protein levels in hepatocytes after transfection with distinct siRubicon sequences (n = 3). j RT-PCR analysis determining Rubicon gene expression post-transfection with various siRubicon sequences in hepatocytes (n = 4). k Free cholesterol (FC) and l triglyceride (TG) content quantification to gauge the extent of FFA-induced steatosis under alternative treatments (n = 3). m Bodipy staining and n its quantification to analyze the impact of different interventions on FFA-induced hepatic steatosis (n = 3). o Oil Red O staining and p corresponding quantitation to assess the degree of FFA-induced steatosis across disparate treatment regimens (n = 6). Statistical significance is denoted by * p < 0.05, ** p < 0.01, and *** p < 0.001, insignificance was not represented. Error bars indicate means ± SEM in (b, c, e, g, i–l, n, and p). Data were analyzed by oneway ANOVA followed by Tukey’s test. Source data are provided as a Source Data file.
Rubicon silencing promotes lipid metabolism by enhancing autophagic flux
To delve deeper into the hypothesis that Rubicon suppression bolsters lipid metabolism through heightened autophagic flux, we initially harvested proteins from cells subjected to various treatments and conducted Western blot analyses. Our findings illuminated that FFA-induced steatosis provoked a conspicuous elevation in p62 levels alongside a reduction in the LC3B/LC3A ratio, pointing to inhibited autophagic activity. Conversely, downregulating Rubicon dramatically decreased p62 expression and raised the LC3B/LC3A ratio, akin to the effects induced by RAPA. Notably, co-administration with hydroxychloroquine reversed this trend, reinforcing Rubicon’s regulatory function in autophagy modulation (Fig. 5a and Supplementary Fig. 7a). RT-PCR results and p62 IF staining further solidified that Rubicon suppression rejuvenates autophagic flux, thereby expediting fat metabolism (Fig. 5b and Supplementary Fig. 7b, c).

a Western blot assessment of autophagy-linked proteins, LC3 and p62, under diverse treatment regimes. b RT-PCR evaluation of gene expressions implicated in both autophagy and lipid metabolism in response to different treatments (n = 3). c IF visualization and d quantification of Lysotracker/Bodipy co-staining to detect lipophagy status by assessing co-localization of lipid droplets and lysosomes under varying conditions. White arrows highlight co-localization points of Lysotracker and Bodipy (n = 8). e IF detection and f quantification of LC3/Bodipy overlap to evaluate the co-localization of autophagosomes with lipid droplets, indicative of lipid degradation, under different treatments, with white arrows marking LC3-Bodipy co-localization (n = 8). g hepatocytes transfected with mCherry-GFP-LC3B virus and exposed to multiple treatments undergo IF analysis and h quantification to ascertain LC3B autophagic flux dynamics (n = 3). i IF and j quantification of LC3/LAMP1 co-staining to appraise autophagosome-lysosome co-localization, reflecting autophagic activity under varying conditions, where white arrows denote LC3-LAMP1 co-localization sites (n = 3). k TEM analysis provides insights into the state of autophagosomes and lipid droplets within cells, with red arrowheads pinpointing autophagosomes and yellow stars identifying lipid droplets. Statistical significance designated by * p < 0.05, ** p < 0.01, and *** p < 0.001, insignificance was not represented. Error bars indicate means ± SEM in (b, d, f, h, and j). Data were analyzed by oneway ANOVA followed by Tukey’s test. Source data are provided as a Source Data file.
For assessing the interplay between lipid droplets and lysosomes, live-cell imaging with Lysotracker, Bodipy, and Hoechst 33342 stains were conducted, with the subsequent quantification (Fig. 5c). It discloses an augmented colocalization of lysosome and lipid droplets in PEI25k-GalNAc@siRNA and rapamycin-exposed cells, denoting an increase lipid breakdown by lysosomal degradation with Rubicon suppression (Fig. 5d). In concert with these findings, IF staining coupled with quantitative assessment of LC3-Bodipy colocalization served as a robust indicator of autophagosomal lipid degradation. Our results confirmed a marked upregulation in the expression of LC3-Bodipy colocalization events within the PEI25k-GalNAc@siRNA treatment group, further corroborating the notion that Rubicon knockdown accelerates lipid catabolism through enhanced autophagic flux (Fig. 5e, f).
Next, the dual-fluorescent mCherry-GFP-LC3B probe, which discriminates autophagosomes (manifesting as yellow-green) from autolysosomes (appearing red) based on pH differences, were transfected into hepatocytes for tracking the autophagy process. We observed intensified GFP signals in FFA-induced cells while a diminished GFP intensity following PEI25k-GalNAc@siRubicon or rapamycin administration, indicating a promotion on autophagic flux induced by silencing Rubicon (Fig. 5g, h). Likewise, heightened expression of LC3/LAMP1 colocalization could also been observed in PEI25k-GalNAc@siRubicon or rapamycin pre-treatment groups, confirming the importance of Rubicon inhibition in promoting the fusion of autophagosomes and lysosomes, which was vital for autolysosome maturation (Fig. 5i, j). TEM inspections visualized a scarcity of lipid droplets and autophagosomes in normal cells, contrasting with the FFA-triggered scenario marked by pronounced lipid accretion and fewer autolysosomes. Conversely, Rubicon inhibition corresponded with a conspicuous rise in autolysosome numbers (Fig. 5k and Supplementary Fig. 8). To further investigate the potential mechanism by which Rubicon promotes autophagy, we employed Western blotting test to assess the expression levels of key regulatory proteins, including mTOR, p-mTOR, ULK1, p-ULK1, AMPK, and p-AMPK. The results indicated that Rubicon enhanced autophagic flux primarily by modulating the phosphorylation status of mTOR, ULK1, and AMPK (Supplementary Fig. 9).
Rubicon inhibition effectively alleviates FFA-induced ROS damage
To investigate whether Rubicon-mediated autophagic flux recovery could alleviate lipid peroxidation injury, we examined the physiological states of primary hepatocytes under various treatment conditions. TUNEL staining revealed that FFA treatment induced cellular apoptosis, which was mitigated by inhibiting Rubicon expression (Fig. 6a and Supplementary Fig. 10a). This finding was corroborated by Annexin V and PI (AV-PI) flow cytometry analysis, showing that enhancing autophagic flux by Rubicon suppression reduced the FFA-induced hepatocyte apoptosis rate (Fig. 6b and Supplementary Fig. 10b). JC-1 staining demonstrated that FFA treatment caused a marked depolarization of the mitochondrial membrane potential, which was effectively ameliorated by Rubicon expression suppression (Fig. 6c). This improvement correlated with a reduction in intracellular oxidative stress (Fig. 6d, e). Furthermore, RT-PCR analyses demonstrated that promoting autophagic flux via rapamycin or Rubicon inhibition downregulated the gene expression of pro-inflammatory cytokines IL-1β, TNFα, and iNOS (Fig. 6f–h). Additionally, we explored the impact of these nanoparticles on macrophage polarization, revealing that Rubicon inhibition suppressed the expression of M1 macrophage markers under FFA induction (Supplementary Fig. 11a–c). To further examine whether the improved intracellular environment in hepatocytes mediated by Rubicon could influence macrophage polarization state, we established a co-culture model of hepatocytes and macrophages (Supplementary Fig. 11d). Intracellular RT-PCR and cell supernatant ELISA assays showed that oxidative stress reduction within hepatocytes following Rubicon modulation effectively decreased macrophage polarization towards to M1 phenotype (Supplementary Fig. 11e–i).

a TUNEL staining to assess hepatocyte apoptosis under different treatment conditions. b AV-PI flow cytometry analysis of hepatocyte apoptosis under various treatments. c JC-1 staining to evaluate mitochondrial membrane potential in cells. d DCFH-DA assay for intracellular ROS levels and e semi-quantitative analysis (n = 3). f RT-qPCR analysis of IL-1β, g TNF-α, and h iNOS gene expression in cells (n = 3). * p < 0.05, ** p < 0.01, and *** p < 0.001, insignificance was not represented. Error bars indicate means ± SEM in (e–h). Data were analyzed by oneway ANOVA followed by Tukey’s test. Source data are provided as a Source Data file.
PPH-PEI/GalNAc@siRubicon targets hepatocytes in a redox stress-responsive manner
To establish a MASH model, mice were subjected to a high-fat diet (HFD)/methionine-choline deficient (MCD) diet. We first performed histopathological and molecular analyses on liver tissues from mice fed with HFD/MCD for one week for investigating whether PPH-PEI/GalNAc@siRubicon preferentially targets livers undergoing oxidative stress. As expected, this model rapidly induced significant hepatic steatosis and functional impairment (Supplementary Fig. 12), accompanied by a marked upregulation of Rubicon expression (Fig. 7a, b). Further measurement of malondialdehyde (MDA), a biomarker of lipid peroxidation, confirmed the presence of substantial oxidative damage in the livers of these animals (Fig. 7c). Consistent with these findings, RT-qPCR analysis revealed elevated mRNA levels of multiple pro-inflammatory mediators, including TNF-α, IL-6, iNOS, and IFN-γ (Fig. 7d–g), confirming the concept that fatty liver disease is an oxidative stress-associated pathology.

a Western blot analysis of Rubicon expression in livers of normal diet and HFD/MCD diet mice and b semi-quantitative analysis (n = 3). c changes in MDA expression in livers from normal and HFD/MCD diets (n = 6). d RT-qPCR analysis of pro-inflammatory cytokines IL-1β, (e) TNF-α, (f) IL-6, and (g) iNOS gene expression in livers from normal and HFD/MCD diet (n = 7). h relative fluorescence expression of PPH-PEI/GalNAc@Cy5-siRubicon in different organs at 2 and 12 h post-injection in mice fed with normal or HFD/MCD diet. i relative fluorescence expression of PPH-PEI/GalNAc@Cy5-siRubicon in different organs at 2 and 12 h post-injection in MCD-fed mice with or without antioxidant treatment. j Western blot analysis of the efficiency of siRubicon on hepatocytes in HFD/MCD-fed mice and k the relative expression quantification (n = 4). ** p < 0.01, and *** p < 0.001, insignificance was not represented. Error bars indicate means ± SEM in (b–g, and k). Data were analyzed by two-tailed Student’s t-test. Source data are provided as a Source Data file.
To evaluate whether PPH-PEI/GalNAc@siRNA could specifically target livers under oxidative stress conditions, we intravenously administered the nanoparticles to both normal diet and fatty liver–induced mice. Major organs, including heart, liver, lung, and kidney, were collected at 2 and 12 h post-injection for biodistribution analysis. Notably, significantly higher nanoparticle accumulation was observed in the livers of HFD/MCD-fed mice compared to those on a normal diet (Fig. 7h). Importantly, when antioxidant treatment (N-Acetyl-L-cysteine, 200 mg/kg, intraperitoneal injection once a day for three weeks) was co-administered in HFD/MCD-fed mice, nanoparticles accumulation in liver was markedly reduced (Fig. 7i), indicating that the targeting efficiency of our nanocarrier is closely linked to the presence of ROS. To determine whether the nanoparticles preferentially delivered siRubicon to specific liver cell populations, we isolated primary hepatocytes and non-parenchymal liver cells five days after nanoparticle injection and performed Western blot analysis. The results demonstrated that PPH-PEI/GalNAc@siRubicon effectively suppressed Rubicon expression in hepatocytes, while showing minimal effects on Rubicon levels in non-parenchymal cells (Fig. 7j, k), confirming its selective targeting capability toward parenchymal liver cells.
PPH-PEI/GalNAc@siRNA administration effectively inhibits hepatic steatosis
With the established MASH model, samples were harvested at strategic intervals post-induction for comprehensive evaluation. Three days, six days, and nine days subsequent to the primary siRNA nanocomplex injection, hepatic tissues were procured for Western blot analysis and quantitation of Rubicon expression (Supplementary Fig. 13), which revealed sustained, profound suppression of Rubicon over an extended period of 6 days. Accordingly, nanoparticles were injected every 7 days in later experiments to continuously inhibit Rubicon expression in the liver (Fig. 8a). Seven days after nanoparticle injection with HFD/MCD-fed for 2 weeks, we analyzed heart, liver, lung, and kidney tissues for Rubicon expression using RT-PCR assays (Supplementary Fig. 14a–d). The results showed insignificant changes in the heart and kidney but significant reductions in the liver and lung, indicating preferential accumulation in these organs. To assess hepatic steatosis, we performed Oil Red O staining on liver sections (Supplementary Fig. 14e). Although there were no notable differences in inflammatory cell infiltration among the four groups (Supplementary Fig. 14f), mice treated with PPH-PEI/GalNAc@siRubicon exhibited the lowest degree of steatosis (Supplementary Fig. 14g). This group also showed lower levels of ALT and AST, and relatively higher albumin levels (Supplementary Fig. 14h–j), suggesting improved liver function. To ensure the specificity of Rubicon suppression and rule out nonspecific effects from nanoparticle delivery or siRNA transfection, we compared the efficiency of PPH-PEI/GalNAc@siNC (negative control) and PPH-PEI/GalNAc@siRubicon. The data confirmed a strong and specific inhibition of Rubicon by our treatment (Supplementary Fig. 14k, l).

a establishment of a MASH mouse model with nanoparticles treatment. b examinations the levels of TC and c TG levels in liver for evaluating the lipid metabolism status (n = 6). d–f serum biochemical assays for ALT, AST, and albumin in ND (n = 5), PPH-PEI/GalNAc@siNC (n = 7), and PPH-PEI/GalNAc@siRubicon (n = 7) groups, reflecting improvements in liver function. g Oil Red O staining, H&E staining, and Sirius red staining to evaluate the therapeutic efficacy of PPH-PEI/GalNAc@siRubicon. h quantification of the lipid droplet accumulation in the liver in ND (n = 5), PPH-PEI/GalNAc@siNC (n = 6), and PPH-PEI/GalNAc@siRubicon (n = 6) groups, evidencing the degree of steatosis. i assessment of inflammatory cell infiltration in liver tissue for evaluating hepatic inflammation. j semi-quantitative analysis of relative collagen expression, reflecting the degree of fibrosis (n = 6). k IF assay to detect F4/80 and CD86 expression, used as a marker for M1 macrophage polarization. * p < 0.05, ** p < 0.01, and *** p < 0.001, insignificance was not represented. Error bars indicate means ± SEM in (b–f, h–j). Data were analyzed by oneway ANOVA followed by Tukey’s test. Source data are provided as a Source Data file.
To assess the extent of hepatic steatosis, we quantified TC and TG levels in liver tissue with HFD/MCD-fed for 4 weeks. As expected, steatosis induced by HFD/MCD diet led to a significant accumulation of TC and TG in the liver. Notably, treatment with Rubicon inhibition markedly reduced these lipid deposits, confirming its effectiveness in alleviating hepatic steatosis (Fig. 8b, c). Moreover, assessment of liver function via blood samples showed significantly diminished alanine aminotransferase (ALT) and aminotransferase (AST) levels in siRubicon-treated groups compared to untreated group (Fig. 8d, e), while albumin synthesis remained unaltered across all groups (Fig. 8f), affirming its therapeutic potential against MASH. In a bid to further scrutinize hepatic steatosis, Oil Red O staining, HE staining and sirius red staining was carried out (Fig. 8g). Our results demonstrated that suppression of Rubicon expression significantly alleviated hepatic steatosis in fatty liver tissue (Fig. 8h), accompanied by a marked reduction in inflammatory cell infiltration (Fig. 8i). To further evaluate its impact on liver fibrosis, we performed quantitative analyses of collagen deposition in liver tissue (Fig. 8j), as well as mRNA expression levels of TGF-β and vimentin (Supplementary Fig. 15a, b). Western blot analysis of vimentin expression further supported these findings (Supplementary Fig. 15c, d). In addition, we conducted in vitro experiments using FFA-induced mouse hepatic stellate cells to further assess the antifibrotic effects of Rubicon suppression. Consistent with the in vivo results, these cell-based assays also revealed a significant inhibition of fibrotic responses (Supplementary Fig. 15e–g). Collectively, these findings provide compelling evidence that the engineered nanoparticles exert robust therapeutic effects in ameliorating fatty liver pathology and exhibit potential antifibrotic activity.
To further investigate the impact of Rubicon inhibition on hepatic inflammation, we evaluated macrophage polarization and inflammatory cytokine expression in livers with fatty degeneration. Our results revealed a marked upregulation of M1-type macrophages in steatotic livers (Fig. 8k and Supplementary Fig. 16a). Notably, suppression of Rubicon expression via nanoparticle-mediated delivery significantly reduced M1 macrophage infiltration and downregulated the mRNA expression levels of pro-inflammatory cytokines, including TNF-α, IL-1β, iNOS, and IFN-γ (Supplementary Fig. 16b–e). These findings suggest that Rubicon inhibition effectively mitigates chronic inflammatory responses associated with MASH.
Given the close association between hepatic steatosis and metabolic dysregulation, we next assessed glucose and insulin sensitivity in experimental animals. In the HFD/MCD diet-induced model, despite evident hepatic lipid accumulation, mice did not exhibit glucose intolerance (Supplementary Fig. 17a) or insulin resistance (Supplementary Fig. 17b), suggesting that the pathogenic mechanism in this model primarily involves choline deficiency rather than insulin signaling impairment. To better recapitulate metabolic dysfunction in fatty liver disease, we established a more representative model of MASH by feeding mice with HFD for 10 weeks. These HFD-fed mice developed significant glucose intolerance and insulin resistance, which were subsequently ameliorated following nanoparticle-based Rubicon suppression (Supplementary Fig. 17c, d). To comprehensively evaluate the therapeutic potential of Rubicon inhibition in fatty liver disease, we systematically analyzed hepatic inflammation, lipid metabolism, and liver function markers in the HFD-fed model. The results were consistent with those observed in the HFD/MCD model. Suppression of Rubicon expression effectively reduced oxidative stress and hepatic lipid accumulation, and partially restored liver function (Supplementary Fig. 18).
Furthermore, we examined the expression levels of genes and proteins involved in lipid synthesis, transport, and β-oxidation to explore the potential role of Rubicon-regulated autophagy in lipid metabolism. Quantitative analysis showed that siRubicon treatment had minimal effect on the expression of key lipogenic genes such as SCD1 and PPAR-α (Supplementary Fig. 19a), but moderately enhanced lipid transport (Supplementary Fig. 19b). More importantly, Rubicon inhibition promoted lipid β-oxidation, leading to a significant enhancement in lipid clearance from hepatocytes (Supplementary Fig. 19c–e). These findings indicate that Rubicon-mediated autophagy alleviates hepatic lipid accumulation, at least in part, through enhancing lipid transportation and promoting β-oxidation.
Finally, we assessed the in vivo biosafety profile of the nanoparticles. Mice were administered varying doses of the nanoparticle formulation, and major organs (including the heart, liver, lung, and kidney) were examined for histopathological changes. Our results showed that, within a dose range up to 1.5 OD, no significant tissue damage or structural abnormalities were detected (Supplementary Fig. 20a). In addition, blood samples were collected and analyzed for immune cell composition and serum levels of ALT, AST, creatinine, and urea nitrogen (Supplementary Fig. 20b–f). These analyses revealed no marked alterations, indicating that the nanoparticles did not induce systemic toxicity or impair organ function under the tested conditions. Histological evaluation by H&E staining further confirmed the absence of notable pathological changes in the heart, liver, lung, and kidney following repeated administration of the nanoparticles at a therapeutic dose (1 OD) once weekly for three weeks (Supplementary Fig. 20g).
Discussion
Confronting the trifecta of complexities in MASH, comprising lipid accumulation, site-specific oxidative damage, and deteriorated hepatocyte function, this investigation pioneers the design of a groundbreaking multifunctional nanosphere24. Structurally optimized with a sophisticated core-shell configuration, it is meticulously crafted to evade rapid clearance by the bloodstream, selectively accumulate within fatty infiltrated livers, precisely target hepatocytes, and foster lipid autophagy. The key lies in its programmed disassembly under oxidative stress circumstance in fatty liver lesions, resulting in the targeted release of siRubicon-laden positive nanospheres. Internalized by cells, these nanospheres promptly inhibit intracellular Rubicon expression, thereby invigorating the lipid autophagic process and efficaciously mitigating hepatic lipid excess.
In this article, we aim to enhance the autophagic flux within hepatocytes, thereby accelerating the degradation of lipid droplets. This strategy targets the reduction of Rubicon expression in liver cells, with the ultimate goal of mitigating hepatic steatosis. Grounded in the principles of lipid metabolism, our approach seeks to restore energy equilibrium and cellular homeostasis, positioning itself as a promising therapeutic avenue for MASH. In fact, scientists have discovered that lipophagy, standing as a distinctive variant of selective autophagy intracellularly, is vitally important in governing lipid metabolism and maintaining lipid balance. It orchestrates the lysosomal targeting of lipid droplets via a mechanism implicating ubiquitination and the engagement of specialized lipophagy factors and autophagy receptors, thereby intertwining with the regulation of cellular lipid management25. The lipophagic pathway relies on a sophisticated network of autophagy selectivity components and autophagosomes, functioning as vital conduits that interface lipid droplets with the autophagic infrastructure26. These entities navigate a complex protein interplay to facilitate the delivery of lipid droplets to lysosomes, employing either macroautophagic or microautophagic mechanisms. Following this, lysosomal acid lipase initiates the breakdown of cholesterol esters and triglycerides encapsulated within the lipid droplet cores, catalyzing their hydrolysis within the lysosomal milieu27,28,29. The resultant metabolites are then either ejected from the cell or channeled to mitochondria where they undergo fatty acid oxidation, a process that fuels ATP synthesis—the life-sustaining energy currency for both cellular and organismal functions30. The crux of our investigation was to bolster autophagic flux, refining the interface between autophagosomes and lipid droplets to heighten binding efficacy, while accelerating the fusion of these complexes with lysosomes. This strategic approach aimed to optimize the digestive recycling of lipid droplets, thereby enhancing the overall efficiency of this fundamental cellular process.
In the process of autophagy, there are various autophagic proteins involved, among which Rubicon has surfaced as the singular identified negative modulator of cellular autophagy31. Notably, investigations have disclosed elevated Rubicon levels in individuals afflicted with MASH10. On the contrary, genetic ablation of Rubicon in murine models considerably attenuates high-fat-diet-induced hepatic steatosis, implicating its suppressive role in autophagic processes, particularly by impeding autophagosome maturation12. Similar to these results, we found that when subjected to a high-fat milieu, pronounced cellular steatosis alongside with elevated Rubicon expression, evident both in vitro at the cellular level and in vivo at the organ scales, manifest. Upon silencing Rubicon, we observe a notable diminution in hepatic steatosis severity, concomitant with a reduction in the obstruction of autophagic flux. Moreover, our findings highlight that Rubicon’s modulation predominantly enhances the terminal phase of lipophagy—the pivotal fusion event between autophagosomes and lysosomes. Indeed, important insights in last year about the research on the mechanism of the promotion of Rubicon on autophagy have revealed that Rubicon stabilization is markedly amplified by directly with interacting methyltransferase-like protein 3 (METTL3)12, thereby exacerbating the impediment of autophagosome-lysosome fusion and hinders lipid droplet disposal, evidencing the significance of Rubicon-mediated autophagy suppression in MASH’s pathogenesis. Certainly, for a deeper understanding of Rubicon’s mechanism in lipophagy promotion, additional experimental endeavors may be needed in the future. Notably, recent research has proposed that the efficacy of MASH therapy based on Rubicon-targeted may be upon the nutritional milieu; they found that under nutrient-rich conditions, RUBCNL undergoes phosphorylation by MTORC1, leading to its inactivation and lipophagy suppression. Conversely, in nutrient-starved states, dephosphorylation facilitates RUBCNL acetylation, dramatically enhancing the assembly of the homotypic fusion and vacuole protein sorting (HOPS) complex, thereby promoting autophagosome maturation and efficient lipid turnover32. This suggests that for Rubicon based MASH treatment, personalized treatment plans may be developed according to the nutritional status of the patients; moreover, when combined with other lipid drugs, more considerations should be taken into account.
Presently, a multitude of small-molecule candidates for MASH therapy is progressing through preclinical assessments (Madrigal Pharmaceuticals announces FDA approval of rezdiffra™ (resmetirom) for the treatment of patients with noncirrhotic nonalcoholic steatohepatitis with moderate to advanced liver fibrosis, March 14, 2024). By contrast, siRNA delivery, with its inherent targeting precision, can drastically diminish off-target occurrences, thereby curtailing the adverse effects typically associated with small molecules13. This renders targeted siRubicon delivery a compelling alternative therapeutic avenue for MASH. Nonetheless, the swift systemic clearance of unmodified siRNA post-administration underscores the necessity for efficacious nucleic acid delivery vehicles. Considering RNA’s inherent negativity, cationic liposomes emerge as a feasible delivery mode, yet their in vivo safety profile limitations have hindered the broader application of siRNA therapeutics33. Following the pioneering approval of Onpattro, the inaugural siRNA therapy utilizing lipid nanoparticles, preclinical assessments uncovered complications, notably multi-systemic neuropathies attributed to hereditary transthyretin-mediated amyloidosis (hATTR). In recent years, GalNAc-siRNA conjugates have attracted substantial interest from nucleic acid drug developers, owing to their exceptional liver-targeting capabilities and reassuring safety records34. Investigative work has established GalNAc as a specific ligand for the ASGPR, which is profusely expressed on hepatocyte surfaces at around half a million receptors per cell35,36. Following internalization, GalNAc-siRNA complexes migrate from the cell membrane to the cytosol, where they dissociate from ASGPR, enabling the liberated siRNA to silence targeted genes37. Although Polyethylenimine 25 K (PEI25k) boasts high siRNA transfection efficiency and is widely used in nucleic acid delivery, its indiscriminate cellular uptake poses a challenge. Hence, the engineering of PEI25k-GalNAc composite cationic vectors assumes paramount importance, as it aims to augment not only transfection efficacy but also the specificity towards liver cells, thereby addressing a pivotal gap in current delivery strategies.
In addition to achieving precise cellular targeting, delivery systems must also overcome the hurdle of off-target organ retention or premature immune clearance en route to their designated sites. Studies have demonstrated that PEG coatings significantly retard recognition and clearance by the immune system in vivo, thereby enhancing “stealthiness” and extending circulation times38. This stealth characteristic, when coupled with functionally responsive additives, enables PEGylated systems to persist longer in circulation or even home in on specific tissues, spurring the development of numerous core-shell structured nanoparticles. For example, Zhanzhan Zhang and coworkers devised a system featuring a PEI1.8k–HPBA/pDNA core enveloped within a pH and H2O2 dual-responsive mPEG-b-PLys-SGD/CA or mPEG-b-PLys-SGD/SA shell, tailored for efficacious PD-L1 discharge at tumor locales39. Qi Liu and colleagues, meanwhile, constructed a PEI–PBA/pDNA core shielded by a pH-sensitive mPEG113-b-PLys100/DMMA shell, aiming to facilitate protein aggregation and release specifically within the tumor microenvironment40. Expanding upon these precedents, we engineered and established a layer and layer nanoparticles containing a core and a ROS-responsive mPEG113-b-PLys120-HPBA shell layer. This design is purposed to shield the core from unintended accumulation in non-target organs, ensuring that upon arrival at fatty-degenerated liver tissue, the outer shell disintegrates, liberating the core. Thereafter, guided by GalNAc, the exposed core is swiftly endocytosed by hepatocytes, marking a strategic advance in targeted delivery for liver-directed therapies.
This study innovated a dual-shelled nanoparticle architecture fabricated through self-assembly, designed with oxidative stress responsiveness and an explicit focus on hepatocytes within fatty-infiltrated liver tissue. The system excels in conveying siRNA that potently suppresses Rubicon expression within cells, stimulating lipophagy, and thereby ameliorating the pathological manifestations of MASH. Although the PPH-PEI/GalNAc@siRNA system in this study has been shown effective in the treatment of MASH, its long-term safety and application in complex metabolic disease models remain unexplored. Therefore, future studies will further investigate the long-term safety of the PPH-PEI/GalNAc@siRNA system, and its therapeutic potential will be evaluated in other NAFLD models, such as gene-editing models. Moreover, the potential of combining the PPH-PEI/GalNAc@siRNA system with other therapeutic agents will be further investigated.
Methods
Ethics statement
All animal experiments and procedures in this study were in accordance with institutional guidelines on animal welfare and were approved by the Institutional Laboratory Animal Care and Use Committee of Nankai University (Approval number: 2021-SYDWLL-000393). The mice and rats were obtained from Charles River and housed in a specific pathogen-free environment and were maintained on standard chow, a HFD, or a HFD/MCD diet in 12-h light/12-h dark cycles. The ambient temperature and humidity were 23 ± 1.5 °C and 45 ± 15%, respectively. Food and water were provided.
Sex and gender statement
This study included only male mice and male rats due to the focus on sex-specific mechanisms relevant to the investigated pathology. Given the nature of the research, limiting the study to a single sex was considered appropriate to ensure consistency and comparability of results. Further investigations should include both sexes to validate these findings.
Synthesis of PEI25k-GalNAc
The synthesis of PEI25k-GalNAc was achieved by conjugating chondroitin-4-sulfate-derived N-acetylglucosamine (CDM-NAG, Macklin, catalog number 1299292-01-5, China) onto PEI25k. Briefly, 100 mg of PEI25k was first dissolved in sodium bicarbonate solution (pH = 8.5, 100 mM) to reach a concentration of 10 mg/mL, following by the addition of 5.69 mg of CDM-NAG. The reaction solution was stirred at room temperature for 8 h, followed by dialyzed against distilled water and freeze-drying to obtain PEI25k-GalNAc.
Synthesis of mPEG 113 -b-PLys 120-HPBA
For the synthesis of mPEG113-b-PLys120-HPBA, Pinacol boronic ester (PBA-pinacol) was first activated by 4-Nitrophenyl chloroformate (7693-46-1, China). Briefly, 4-(hydroxymethyl) phenylboronic acid pinacol ester (0.8 g, 3.42 mmol) was first dissolved in 50 mL anhydrous dichloromethane, followed by addition of 4-Nitrophenyl chloroformate (1.38 g, 6.84 mmol) and N, N-Diisopropylethylamine (0.53 g, 4.10 mmol) and stirred at room temperature for 12 h. Then the mixture was subjected to rotary distillation and purified by chromatography to obtain HPBA-pinacol-NO2. Poly (ethylene glycol)-b-poly(L-lysine) (mPEG113-b-PLys120) synthesis adhered to previously reported procedure38. Briefly, Lys(Z)-NCA (5.35 g, 18.4 mmol) was dissolved in 30 ml of DMF and polymerized by addition of mPEG113-NH2 (0.61 g, 0.123 mmol) with the terminal primary amino group as the initiator. The reaction mixture was stirred for 3 days at 35 °C under a dry argon atmosphere. Then solvent was evaporated under reduced pressure. The resulting product was dissolved in 15 ml of CHCl3 and then precipitated into excessive diethyl ether to obtain mPEG113-b-PLys120 (Z) (yield 90%). The deprotection of Z group in mPEG113-b-PLys120 (Z) was carried out by addition of HBr (33 wt.% in HOAc, 2 mL) to the solution of mPEG113-b-PLys120 (Z) (2.0 g) in 20 ml CF3COOH for 2 h at 0 °C. After precipitating using cold diethyl ether, the product was re-dissolved in DMF and purified by filtering through a 0.22 µm Millipore filter. The filtrate was precipitated in excessive diethyl ether to remove the residual CF3COOH and obtain mPEG113-b-PLys120 (yield 60%). Then the product was dried at room temperature under vacuum. The mPEG113-b-PLys120-HPBA was then synthesized by a conjugation between the HPBA-pinacol-NO2 and mPEG113-b-PLys120. To a solution of mPEG113-b-PLys120 (100 mg, 0.0052 mmol) in 10 mL sodium bicarbonate buffer (pH 8.5, 50 mM), HPBA-pinacol-NO2 (20.92 mg, 0.052 mmol, dissolved in DMSO) was added and stirred overnight at room temperature. The deprotection of pinacol in mPEG113-b-PLys120-HPBA-pinacol was achieved by dialyzed against HCl solution (0.05 N) for 48 h at room temperature and then lyophilized to afford mPEG113-b-PLys120-HPBA.
Synthesis of PPH-PEI/GalNAc@siRNA
PEI25k-GalNAc and siRNA were combined in equal volumes at varied mass ratios and gently mixed for 15 min at room temperature to facilitate the formation of PEI25k-GalNAc@siRNA nanoparticles. Sequentially, an equivalent volume of mPEG113-b-PLys120-HPBA solution was introduced to the PEI25k-GalNAc/siRNA polyplex mixture at varied mass ratios of mPEG113-b-PLys120-HPBA to siRNA. This secondary mixture was allowed to incubate for an additional 15 min to yield the PPH-PEI/GalNAc@siRNA nanoconjugates.
Characterization and quantification of the nanoparticles
Proton nuclear magnetic resonance spectroscopy (1H-NMR) was employed to analyze the synthesized nanomaterials, ensuring the successful synthesis and integration of diverse components and elemental constituents. To elucidate the particle size distribution and surface charge characteristics, a suite of analyses was conducted, comprising zeta potential measurements, particle sizing, agarose gel electrophoresis retardation tests, and transmission electron microscopy (TEM) imaging. In preparing samples for TEM, they were applied to copper grids for a 60-s adsorption period, surplus unbound material was carefully wicked away, and the grids were then gently dried. Subsequently, 2% phosphotungstic acid was used to stain the samples for 40 s, followed by natural air-drying before proceeding to TEM examination.
Cellular uptake analyses
Hepa 1–6 and RAW 264.7 cells (from mice) were maintained and propagated in Dulbecco’s Modified Eagle Medium (DMEM) or Roswell Park Memorial Institute (RPMI) 1640 medium, respectively, each supplemented with 10% fetal bovine serum (FBS, Gibco). mHSC (mouse derived hepatic stellate cells, Sunncell, China) were cultured in RPMI 1640 medium, supplemented with 10% FBS. To evaluate nanoparticle internalization, a density of 1 × 105 cells/mL was seeded in 12-well plates. Post-overnight incubation, media were refreshed with 1 mL of Opti-MEM™ I Reduced Serum Medium (Invitrogen, catalog number 31985070, USA), incorporating PEI25k-GalNAc@siRNA nanoparticles. To elucidate the cells’ optimal phagocytic efficacy across varying PEI-GalNAc polyplex to siRNA mass ratios, complexes were synthesized at ratios of 1, 2, 3, 5, and 8, with a consistent 30 nM Cy5-labeled siRNA per well. Following an overnight co-incubation with these nanostructures, a subset of cells underwent trypsinization, centrifugation, and preparation for FCM to evaluate Cy5-positive cells. Concurrently, another set was fixed in 4% paraformaldehyde (PFA) to permit actin filament staining through FITC-conjugated phalloidin (Abclonal, RM02836, China), facilitating the evaluation of PEI25k-GalNAc@siRNA nanostructure phagocytosis efficiency. In pursuit of the most advantageous mPEG113-b-PLys120-HPBA to PEI25k-GalNAc@siRNA mass ratio, additional nano-complexes, predicated on a 2:1 PEI25k-GalNAc to siRNA ratio, were engineered at shell-to-core mass ratios of 1, 1.5, 2, and 3. These PPH-PEI/GalNAc@siRNA constructs were subjected to overnight co-culture with Hepa 1–6 cells in Opti-MEM™ I, under both standard conditions and in the presence of 200 μM H2O2, prior to assessment via FCM and IF staining to ascertain the ideal coating-to-core ratio. This comprehensive approach aims to refine our understanding of the parameters governing nanoparticle uptake efficiency in these cell lines.
Targeting efficiency assessment on macrophages and hepatocytes
Hepa 1–6 cells were seeded onto the upper compartment of Transwell inserts (Corning, catalog number 3413, USA) fitted with 3 μm pores, filled with DMEM containing 10% FBS. RAW 264.7 cells were positioned beneath in the lower chamber to establish a co-culture model. To examine the specificity of the materials towards hepatocytes, upon cell attachment, Opti-MEM™ I medium supplemented with PEI25k-GalNAc@siRNA complexes was administered and left to incubate overnight. Subsequently, IF staining and FCM analysis were independently performed on the two cell types to assess targeting selectivity. Furthermore, an in vitro NMP system was utilized, wherein 33 μg of siRNA, complexed either with 66 μg of PEI25k-GalNAc or 33 μg of PEI25k, was infused into the system. Following a 6-h perfusion period at a constant rate of 3 mL/min, samples were gathered, fixed, and readied for examination. This analysis aimed to determine the co-localization of PEI25k-GalNAc@siRNA complexes with hepatic cells, marked by HNF4A (Abcam, ab201460, USA) expression, or with macrophages, marked by CD11b (Biolengend, 101203, USA) expression, thereby elucidating the preferential targeting capabilities of the developed materials within the hepatic microenvironment.
Western blotting protocol
Harvested cells or tissue samples were lysed at a ratio of tissue-to-buffer of 100 mg:1 mL (w/v), and the extracted proteins were quantified using a BCA assay to ensure equal loading. For electrophoretic separation, 30 μg of protein per well was resolved on SDS-polyacrylamide gels, followed by protein transfer onto PVDF membranes. Membranes were blocked with 5% non-fat milk at room temperature for 2 h, then incubated overnight at 4 °C with primary antibodies diluted 1:1000 in 1% non-fat milk: Rabbit polyclonal anti-Rubicon (Abcam, ab156052, USA), rabbit monoclonal anti-p62 (Abclonal, A19700, China), rabbit monoclonal anti-LC3 (Cell Signaling Technology, 12741S, USA), mouse monoclonal anti-α-SMA (Upingbio, YP-Ab-02991, China), rabbit polyclonal anti-AMPK A1 (Upingbio, YP-Ab-14656, China), rabbit polyclonal anti-p-AMPK A1 (Upingbio, YP-Ab-14296, China), rabbit monoclonal anti-ULK1 (Upingbio, YP-Ab-12966, China), rabbit polyclonal anti-p-ULK1 (Upingbio, YP-Ab-14626, China), rabbit polyclonal anti-mTOR (Upingbio, YP-Ab-14867, China), rabbit polyclonal anti-p-mTOR (Upingbio, YP-Ab-14383, China), rabbit monoclonal anti-Vimentin (Huabio, ET1610-39, China), and rabbit polyclonal anti-GAPDH (Abclonal, AC001, China). After extensive washing with PBST thrice for 15 min each, membranes were incubated with a 1:3000 dilution of HRP-conjugated secondary antibody at room temperature for 1 h, with three further PBST washes of 15 min each. Lastly, protein bands were visualized using a chemiluminescence detection system.
RNA extraction and real-time PCR analysis
Cellular or tissue specimens (50 mg tissue in 1 mL TRIzol reagent) were collected and lysed, with subsequent RNA isolation and reverse transcription. The real-time PCR protocol commenced with an initial denaturation step at 105 °C for 5 min, followed by a series of 40 amplification cycles, each including 95 °C for 30 s for denaturation, 60 °C for 30 s for annealing, and 72 °C for 45 s for extension. The primer sequences used were as shown in Supplementary Table 1:
IF staining
With the exception of LysoTracker/Bodipy co-staining that necessitated live-cell labeling, all other cellular or tissue samples underwent IF staining after being fixed in 4% PFA, permeabilized with 0.1% Triton X-100, and blocked with 5% BSA. Primary antibodies, including rabbit polyclonal anti-Rubicon (Abcam, ab156052, USA), rabbit monoclonal anti-p62 (Abclonal, A19700, China), rabbit monoclonal anti-LC3 (Cell Signaling Technology, 12741S, USA), rabbit monoclonal anti-F4/80 (Abcam, ab300421, USA), rabbit monoclonal anti-LAMP1 (Cell Signaling Technology, 9091S, USA), rabbit monoclonal anti-HNF4A (abcam, ab201460, USA), rabbit monoclonal anti-CD86 (Abclonal, A16805, China), were diluted at a 1:200 ratio in 1% BSA and incubated overnight at 4 °C. Fluorescein-conjugated secondary antibodies, Alexa Fluor 488 (Invitrogen, 2420731) and Alexa Fluor 555 (Invitrogen, 2445825), were diluted 1:600 and incubated for 1 h at room temperature, succeeded by nuclear staining with DAPI (Sigma-Aldrich, D9542). For FITC-labelled phalloidin (Abclonal, RM02836, China), a 1:200 dilution in 1% BSA was employed for a 1-h incubation at 37 °C, preceding nuclear staining with DAPI and slide mounting. Co-staining with BODIPY 493/503 (Glpbio, GC42959, China) and Lysotracker (Ye-sen, 40739ES50, China) entailed direct incubation of cells with the dyes at 37 °C for 30 min, with Hoechst 33342 (Beyotime Biotechnology, C1029, China) introduced in the final 5 min for nuclear staining. Fluorescence imaging was promptly executed within 40 min following the staining process to ensure optimal visualization.
Flow cytometry analysis
Cells were harvested from culture dishes and resuspended in staining buffer at a concentration of 1 × 106 cells/mL. We collected approximately 10,000 cells for each sample to ensure robust statistical analysis. Cells were stained with fluorescently labeled nanoparticles or antibodies against specific markers in the dark and washed twice with cold PBS to remove unbound reagents. Flow cytometry was performed using a BD FACSCalibur flow cytometer. Data acquisition and post-acquisition analysis were conducted using FlowJo software. We identified live single cells by excluding debris and doublets through forward scatter (FSC) vs. side scatter (SSC) gating. Specific cell populations (M1 macrophages) were identified based on NOS2 expression levels. Gating strategies included FSC vs. SSC for initial selection, and single-color controls for compensation settings.
Induction of cellular lipid accumulation
To induce lipid accumulation, nanocomplexes were meticulously prepared with variable siRNA concentrations, employing a core composition ratio of PEI25k-GalNAc to siRNA at 2:1 mass ratio. This was complemented by the fabrication of core-shell nanostructures at diverse ratios and testing of a range of FFAs, RAPA, and Q concentrations to gauge their effects on cellular proliferation. Through these assessments, the optimal treatment regimen for the cells was determined. To induce lipid accumulation effectively, we prepared a fFFA inducer by precisely combining sodium oleate (OA) and sodium palmitate (PA) in a 1:2 molar ratio. Both OA and PA were obtained from Sigma-Aldrich (USA), with catalog numbers D7501 and P9767, respectively. This tailored formulation serves as a potent tool for research into lipid metabolism and its impact on cellular processes. Hepa 1–6 cells underwent a 24-h co-culture with varying concentrations of this FFA blend. Subsequently, lipid droplets were visualized through fluorescence staining with BODIPY 493/503, enabling quantitative analysis of droplet size and number across different induction scenarios to pinpoint the most efficacious FFA concentration. Concomitantly, a co-staining strategy involving lipid droplets and Rubicon was implemented to elucidate the interplay between lipid accretion and Rubicon expression patterns. Decisions regarding the final FFA induction concentration, settled at 300 μM based on CCK8 viability assays and lipid droplet fluorescence quantification, further informed the experimental design. Post-induction, cellular harvest took place for Western blot analyses of p62 and Rubicon protein expressions, with statistical validations to strengthen the findings.
Primary hepatocytes culture and induction
Primary hepatic parenchymal and non-parenchymal cells were isolated from the livers of 4- to 6-week-old C57BL/6 mice. Under general anesthesia, a midline abdominal incision was made, and a cannula was inserted into the portal vein for perfusion. The liver was first perfused with Ca²⁺/Mg²−-free HBSS solution containing EGTA to remove residual blood, followed by digestion with collagenase type I dissolved in Ca²⁺/Mg²⁺-supplemented HBSS solution at a flow rate of 5 mL/min until the tissue became soft and easily dissociated. The digested liver was then gently disrupted, and the cell suspension was filtered and centrifuged at 50 × g for 2 min at 4 °C. Non-parenchymal cells were further separated by centrifugation at 600 × g for 10 min under the same conditions. The isolated hepatocytes were seeded onto collagen-coated culture plates and maintained in complete growth medium. Four hours post-seeding, the medium was replaced to remove non-adherent cells. After 24 h of culture, hepatocytes were exposed to 400 μM FFA to induce lipid accumulation.
Therapeutic implications of Rubicon knockdown on cellular lipid accumulation
A panel of distinct siRNA sequences was synthesized (Supplementary Table 2). These siRNAs were encapsulated into nanoparticles with PEI25k-GalNAc at a 1:2 weight ratio and incubated with Hepa 1–6 cells for 24 h. Subsequently, the cells were subjected to WB and RT-PCR analyses to determine the most efficacious siRNA sequence. Autophagic induction was represented by cells pretreated with 5 μM RAPA for 12 h, whereas autophagy inhibition was exemplified by cells pretreated with 20 μM CQ over the same duration. Cells receiving PEI25k-GalNAc-mediated siRubicon transfection constituted the experimental group. Post-induction of lipid accumulation by FFAs, cells were harvested for quantifying triglycerides (TG, Solarbio, BC0625, China) and total cholesterol (TC, Solarbio, BC1895, China) levels utilizing commercial assay kits (Solarbio, BC1895, China) adhering to the supplier’s protocol. Furthermore, lipid droplet formation was evaluated through BODIPY 493/503 fluorescence staining and Oil Red O staining, followed by quantitative assessment. To delve deeper into the consequences of lipid accumulation on macrophage polarization, culture supernatants from Hepa 1–6 cells were combined at a 1:1 ratio with complete RPMI medium and administered to adherent RAW 264.7 macrophages for an additional 24-h co-culture period. Thereafter, co-staining for F4/80 and iNOS was executed to gauge the expression profile indicative of M1 macrophage polarization.
Elucidating the formation of autophagic flux and its interaction with lipid droplet metabolism
Cells from multiple treatment groups were harvested for an extensive analysis suite, including WB, RT-PCR, and IF assays. Hepatocytes were plated to reach 50% confluence overnight and subsequently transduced with Ad-mCherry-GFP-LC3B (Beyotime Biotechnology, C3011, China) at a terminal multiplicity of infection of 20. Twenty-four hours later, the transfection medium was replaced with fresh medium containing 300 μM FFA with or without RAPA and PEI-GalNAc/siRubicon nanoparticles. Cells were harvested 48 h post-transduction for fluorescence-based analysis. To prepare for TEM examination, cells subjected to varied treatments were first detached with trypsin and stabilized in glutaraldehyde. A systematic ethanol gradient (50%–100%) promoted dehydration, followed by a 20-min immersion in pure acetone. Samples were then infiltrated with an acetone-resin mixture, and ultrathin sections, measuring 50–100 nanometers, were sectioned. These slices were stained with 1% lead citrate and 1% uranyl acetate in sequence, air-dried, and ultimately scrutinized under TEM for high-resolution ultrastructure visualization.
Establishment and therapeutic intervention in a murine model of MASH
C57BL/6 male mice aged seven weeks were placed on a HFD/MCD diet to induce MASH. After one week on this regimen, mice were intravenously injected via the tail vein with nanoparticles composed of 33 μg siRNA complexed at mass ratios of 1:1 with PEI25k/siRNA, 1:2 with PEI25k-GalNAc/siRNA, or 1:2 with mPEG113-b-PLys120-HPBA in conjunction with PEI25k-GalNAc/siRNA. Liver tissues were excised 5 days post-injection to assess the silencing efficacy of PEI25k-GalNAc/siRNA nanoparticles against Rubicon. At four weeks, mice were euthanized, and blood was retrieved from the abdominal aorta for serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), and albumin measurements to gauge liver function. Concurrently, livers were carried out Oil Red O staining, HE staining, and Sirius red staining to quantify lipid contents and evaluate inflammatory cell infiltration, elucidating the effect of Rubicon suppression by distinct nanoparticles on fatty metamorphosis. Post-perfusion with heparinized saline, liver specimens were procured for tissue ELISA test to quantify tumor necrosis factor alpha (TNFα; Abclonal, RK00027, China), interferon gamma (IFNγ; Abclonal, RK00019, China), interleukin-6 (IL-6; Abclonal, RK00008, China), and total cholesterol/triglyceride (TC/TG) levels as per kit guidelines. Subsequently, tissues were readied for paraffin embedding or optical cutting temperature freezing, preparatory to IF staining, Oil Red O staining for lipids, periodic acid-Schiff staining for glycogen, and quantitative assessments. Besides, another MASH model was established by feeding the mice with HFD for 10 weeks for glucose detection.
Glucose and insulin tolerance tests
To evaluate systemic glucose homeostasis, glucose tolerance tests were performed after a 14-h fasting period. Mice were intraperitoneally injected with a 20% (w/v) glucose solution at a dose of 1 g/kg body weight. Blood sampling were used for glucose measurement using a calibrated glucometer. Blood glucose levels were recorded at baseline and at 15, 30, 60, 90, and 120 min post-injection. For insulin tolerance tests (ITT), mice were fasted for 3 h Recombinant human insulin was administered intraperitoneally at a dose of 0.75 U/kg body weight. Blood glucose concentrations were measured at baseline and at 15, 30, and 60 min post-injection.
In vivo toxicity study
First, we administered total siRNA doses of 1 OD, 1.2 OD, and 1.5 OD via retro-orbital injection to mice. Forty-eight hours post-injection, mice were euthanized, and blood and major organs were collected to evaluate multi-dose in vivo toxicity. Additionally, we followed a treatment protocol by injecting nanoparticles at a dose of 1 OD weekly for three consecutive weeks. One week after the final injection, blood and tissue samples were collected for safety assessment.
Statistical analysis
The statistical significance of differences between groups was assessed with two-tailed Student’s t test or one-way ANOVA followed by Tukey’s test using GraphPad Prism 9.5.1. A p-value threshold of less than 0.05 denoted statistical significance. In the figures, asterisks indicate levels of significance as follows: ***, p < 0.001; **, p < 0.005; *, p < 0.05; ns (not significant) represents p > 0.5 or where significance was not specified.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that all the data supporting the findings of this study are available within the paper and its supplementary information files. Source data are provided with this paper.
References
Riazi, K. et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 7, 851–861 (2022).
Schuppan, D., Surabattula, R. & Wang, X. Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 68, 238–250 (2018).
Lin, H. Y. et al. The emerging role of microRNAs in NAFLD: highlight of microRNA-29a in modulating oxidative stress, inflammation, and beyond. Cells 9, 1041 (2020).
Su, Q., Kumar, V., Sud, N. & Mahato, R. I. MicroRNAs in the pathogenesis and treatment of progressive liver injury in NAFLD and liver fibrosis. Adv. Drug. Deliv. Rev. 129, 54–63 (2018).
Rozema, D. B. et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl Acad. Sci. USA 104, 12982–12987 (2007).
Mizushima, N. Autophagy: process and function. Genes. Dev. 21, 2861–2873 (2007).
Koga, H., Kaushik, S. & Cuervo, A. M. Altered lipid content inhibits autophagic vesicular fusion. Faseb. J. 24, 3052–3065 (2010).
Inami, Y. et al. Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem. Biophys. Res. Commun. 412, 618–625 (2011).
Park, H. W. et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun. 5, 4834 (2014).
Tanaka, S. et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 64, 1994–2014 (2016).
Singh, R. et al. Autophagy regulates lipid metabolism. Nature 458, 1131–1135 (2009).
Peng, Z. et al. METTL3-m6A-Rubicon axis inhibits autophagy in nonalcoholic fatty liver disease. Mol. Ther. 30, 932–946 (2022).
Barbier-Torres, L. et al. Silencing hepatic MCJ attenuates non-alcoholic fatty liver disease (NAFLD) by increasing mitochondrial fatty acid oxidation. Nat. Commun. 11, 3360 (2020).
Yamamuro, T. et al. Age-dependent loss of adipose Rubicon promotes metabolic disorders via excess autophagy. Nat. Commun. 11, 4150 (2020).
Anderson, M. A., Huang, D. & Roberts, A. Targeting BCL2 for the treatment of lymphoid malignancies. Semin. Hematol. 51, 219–227 (2014).
Davids, M. S. & Letai, A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J. Clin. Oncol. 30, 3127–3135 (2012).
Choi, K. Y. et al. A nanoparticle formula for delivering siRNA or miRNAs to tumor cells in cell culture and in vivo. Nat. Protoc. 9, 1900–1915 (2014).
Pecot, C. V. et al. RNA interference in the clinic: challenges and future directions. Nat. Rev. Cancer 11, 59–67 (2011).
Lv, T. et al. SLC7A11-ROS/αKG-AMPK axis regulates liver inflammation through mitophagy and impairs liver fibrosis and NASH progression. Redox Biol. 72, 103159 (2024).
Nair, J. K. et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 45, 10969–10977 (2017).
Dreaden, E. C. et al. Bimodal tumor-targeting from microenvironment responsive hyaluronan layer-by-layer (LbL) nanoparticles. Acs. Nano. 8, 8374–8382 (2014).
Gu, L., Deng, Z. J., Roy, S. & Hammond, P. T. A Combination RNAi-chemotherapy layer-by-layer nanoparticle for systemic targeting of KRAS/P53 with cisplatin to treat non-small cell lung cancer. Clin. Cancer Res. 23, 7312–7323 (2017).
Lee, K. L. et al. Stealth filaments: Polymer chain length and conformation affect the in vivo fate of PEGylated potato virus X. Acta. Biomater. 19, 166–179 (2015).
Tang, R. et al. Design of hepatic targeted drug delivery systems for natural products: insights into nomenclature revision of nonalcoholic fatty liver disease. Acs. Nano. 15, 17016–17046 (2021).
Ouimet, M. et al. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell. Metab. 13, 655–667 (2011).
Mailler, E. et al. The autophagy protein ATG9A enables lipid mobilization from lipid droplets. Nat. Commun. 12, 6750 (2021).
Robichaud, S. et al. Identification of novel lipid droplet factors that regulate lipophagy and cholesterol efflux in macrophage foam cells. Autophagy 17, 3671–3689 (2021).
Pu, M. et al. ORP8 acts as a lipophagy receptor to mediate lipid droplet turnover. Protein Cell. 14, 653–667 (2023).
Chung, J. et al. The Troyer syndrome protein spartin mediates selective autophagy of lipid droplets. Nat. Cell. Biol. 25, 1101–1110 (2023).
Laval, T. & Ouimet, M. A role for lipophagy in atherosclerosis. Nat. Rev. Cardiol. 20, 431–432 (2023).
Grazide, M. H. et al. Plasma levels of autophagy regulator Rubicon are inversely associated with acute coronary syndrome. Front. Cardiovasc. Med. 10, 1279899 (2024).
Cheng, X. & Sun, Q. RUBCNL/Pacer and RUBCN/Rubicon in regulation of autolysosome formation and lipid metabolism. Autophagy 15, 1120–1121 (2019).
Akinc, A. et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 14, 1084–1087 (2019).
Janas, M. M. et al. The Nonclinical safety profile of GalNAc-conjugated RNAi therapeutics in subacute studies. Toxicol. Pathol. 46, 735–745 (2018).
Springer, A. D. & Dowdy, S. F. GalNAc-siRNA conjugates: leading the way for delivery of RNAi therapeutics. Nucleic Acid. Ther. 28, 109–118 (2018).
Kulkarni, J. A. et al. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 16, 630–643 (2021).
Janas, M. M. et al. Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatotoxicity. Nat. Commun. 9, 723 (2018).
Blanco, E., Shen, H. & Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 33, 941–951 (2015).
Zhang, Z. et al. Dual-locking nanoparticles disrupt the PD-1/PD-L1 pathway for efficient cancer immunotherapy. Adv. Mater. 31, e1905751 (2019).
Liu, Q. et al. Multistage delivery nanoparticle facilitates efficient CRISPR/dCas9 activation and tumor growth suppression in vivo. Adv. Sci. (Weinh.). 6, 1801423 (2018).
Acknowledgements
This research was funded by the National Major Scientific Research Instrument Development Project of China (82127808, Kong DL.), Special Funds of the National Natural Science Foundation of China (82241219, Shen ZY.), National Natural Science Foundation of China (81830060, Kong DL.), National Natural Science Foundation of China (32201073, Lan TT.), the Fundamental Research Funds for the Central Universities, Nankai University (63241463, Kong DL.).
Author information
Authors and Affiliations
Contributions
TT. Lan and QS. Li conceived and designed the study, performed the study, analyzed the data, and wrote the paper. MX. Yu, X. Duan, T. Ming, and S. Li provided crucial help in the experiments. Chunjiong Wang and Yi Zhu conceived the paper. ZY. Shen, DL. Kong and Y. Liu conceived and designed the study and wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lan, T., Li, Q., Yu, M. et al. Dual-targeted siRubicon delivery strategy triggers hepatocellular lipophagy for mitigating liver steatosis. Nat Commun 16, 7455 (2025). https://doi.org/10.1038/s41467-025-61965-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61965-x
