
Abstract
Transcription factors such as Tbx5, Gata4, Mef2c and Pitx2 are required during cardiac development, and in adult cardiac homeostasis. We demonstrate that the gene dosage and modulation of these factors are mediated in vivo by the miR-200 family. Inhibition of a single miR-200 family member within the cluster results in defects of the left ventricle and cardiomyocyte maturation during development. Inhibition of the entire miR-200 family results in a ventricular septal defect and embryonic lethality by embryonic day (E)16.5. Inhibition of each miR-200 family has distinct heart phenotypes in cell specific differentiation and maturation. snRNA-sequencing reveals an immature cardiomyocyte cell state, suggesting reduced differentiation of these cells. The miR-200 family members are critical regulators of early cardiac development through maintaining cardiomyocyte differentiation and maturation. In this report, we identify several transcription factors regulated by miR-200 during heart development, a role for miR-200 in specific heart defects, and an abnormal cardiomyocyte population.
Similar content being viewed by others

Introduction
The field of embryonic cardiac development, historically, has used animal models that recapitulate congenital heart defects (CHDs), observed in human patients1,2,3,4. Many of these animal models have altered expression of what are known as, cardiogenic transcription factors (Tbx5, Nkx2.5, Gata4, Mef2c, Pitx2 etc.), and mutations in these genes are highly associated with CHDs1,2,3,4,5,6,7,8,9. Deleting any of these factors in murine models can alter the expression of the heart gene regulatory network (GRN) and downstream factors of this network2,3,5. In particular, loss of Tbx5 in mice leads to atrial and ventricular septal, ventricular wall, and epicardium defects2. Tbx5 deletion in mice can also cause upper limb abnormalities like what is observed in humans with Holt-Oram syndrome caused by TBX5 genetic mutations2. Using these models, we have come to understand many of the principles, steps and signaling pathways that occur during embryonic cardiac development5,6,7,8,9. Most notably, the study of the murine models and cardiogenic TFs has revealed not only how cardiomyocytes, the contractile cells of the heart, arise but, additionally, how the endocardium, epicardium, and cardiac fibroblasts contribute to the developing heart2,5,10,11,12. Numerous genetic and biochemical studies have provided evidence supporting a highly complex GRN, that drives cardiac development5,6,7,8,9. However, the generation of new animal models to study embryonic cardiac development, the GRN, and CHDs has been limited. Thus, although the contributions of cardiogenic TFs to cardiac development are known, the complex interplay and gene dosage requirements of these TFs and the downstream effects, potentially mediated through microRNAs, have yet to be determined.
The GRN that governs cardiogenesis is comprised of TFs from many different families. In clinics, human genetic mutations in cardiogenic TFs have been known to cause CHD2,5,10,13,14,15,16. Additionally, CHD can occur as both syndromic and non-syndromic2,5,10,13. The phenotypes of each case and mutations vary between patients; however, the first phenotypes observed of CHD persist today2,17. As mentioned, cardiogenesis is a highly regulated process, especially at the transcriptional level18. Over the past two decades, tremendous efforts have provided an understanding of the overly complex GRN and its factors that promote and drive cardiogenesis18. These findings have been corroborated through the introduction of clinical mutations and/or the loss of a TF in mice1,2,3,5,7,8,13,19,20. These mouse models have shown the TF’s which are necessary for cardiac development and the specific roles and mechanisms of the TFs. Further, results have provided the field of cardiac developmental biology with the tools to ask questions about gene regulation during cardiogenesis1,2,3,5,7,8,13,19,20. Bioinformatics have been applied to understand GRNs using TF mutant mice and human samples21,22,23,24,25. However, the role of microRNAs and the miR-200 family controlling GRNs during early murine heart development is unknown.
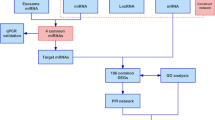
The miR-200 family is subdivided into two functional groups: miR-141/200a (miR-200a group) and miR-200b/c/429 (miR-200c group) (Fig. 1A)26. These miR clusters are located on murine chromosomes 4 and 6 (human 12 and 14)26, with group members found on each chromosome (Fig. 1B). We have shown previously that the miR-200 family members are required for embryonic development of craniofacial tissues27,28,29. In cardiomyocytes (CMs), miR-200 is thought to play a role in CM hypertrophy and hypoxia regulation30,31,32, however studies have only been performed in vitro. The in vivo investigation of miRs has been hindered by the redundancy of miRs, including the miR-200 family33,34. To overcome this barrier, we have designed the Plasmid-based microRNA Inhibitor System (PMIS) to study individual miRs within a cluster in vivo (Supplementary Fig. 1)28,35,36,37,38,39. PMIS interacts with Dicer and the RISC complex to mediate miR inhibition. The PMIS system is efficient, specific, shows no off-target effects and is non-toxic28,35,36. To study the miR-200 family, we generated transgenic mice that inhibit individual groups of the family: PMIS-miR-200a (PMIS-A) and PMIS-miR-200c (PMIS-C)28. PMIS-A and -C mice can be crossed to inhibit the entire miR-200 family (PMIS-AC). These transgenic mice have allowed us to interrogate the molecular mechanisms of miR-200a, miR-200c and the entire family in the context of cardiac development.

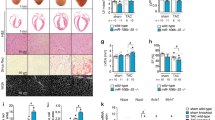
A Seed sequences of the miR-200a and miR-200c subfamilies. B Genomic location of miR-200a (Blue) and miR-200c (Magenta). miRs located on chromosome (Chr) 4 are intragenic and found in the second intron of Ttl10, while miRs located on chromosome 6 are intergenic. C Cardiac expression of miR-200a and miR-200c at E12.5, E14.5, E16.5, and P28. RNA was isolated from wild-type (WT) whole murine hearts. N = 4. (Shown as Ct values). D E14.5 hematoxylin and eosin stained transverse cardiac sections from Wild-Type, PMIS-A, PMIS-C, and PMIS-AC. The arrowhead shows a ventricular septal defect (VSD) and the arrow shows an atrial septal defect (ASD) in PMIS-AC embryos. RA, Right Atrium; LA, Left Atrium; RV, Right Ventricle; LV, Left Ventricle; VS, Ventral Septum. Corresponding anterior/posterior sections are seen in Supplementary Fig. 2. Scale bar = 100 µm. E Compact left ventricle compact myocardium thickness of WT, PMIS-A, PMIS-C, and PMIS-AC. N = 4. F Western Blot for Mef2c, Tbx5, Gata4, and Pitx2. Protein was isolated from E14.5 whole hearts. Lower panel: Quantification of Mef2c, Tbx5, Gata4 and Pitx2 normalized to Gapdh. N = 3. G Functional testing of miR-200a and miR-200c binding to the Mef2c 3’ UTR. Lower panel: identity of miR binding sequences in the Mef2c 3’UTR. N = 5. The statistic test performed was one-way ANOVAwith multiple comparisons. Data are presented as mean values ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.00001.
In this study, we use mouse models that have increased expression of cardiogenic TFs Tbx5, Gata4, Mef2c and Irx1, to study how gene dosage affects cardiac development. The increased combinatorial expression of the TFs due to miR-200 inhibition has not been observed in other models. Altered expression of TFs was achieved in transgenic mice that express an RNA-based inhibitor to the miR-200 family28,35,36,37,38,39, which targets the 3’ UTR of Tbx5, Gata4, Mef2c, and Irx131,40,41. Histological analysis of the mice with reduced miR-200 expression showed phenotypes similar to previous models, and humans with CHD. Single-nuclei multiomics analyses found altered expression of TFs, and resulted in an abnormal cardiomyocyte cell state only in the PMIS-C mice. These cardiomyocytes are marked by the expression of Tbx5. Moreover, these cardiomyocytes have increased expression and more open chromatin around Nppa, a known transcriptional target of Tbx5. Our results from our mouse model provide evidence of the importance of gene dosage during cardiac development. Moreover, these results provide the basis for a major requirement of miRs, specifically the miR-200 family during in vivo cardiac development. Taken together, we found a requirement for miR-200 targeting Tbx5 to allow for cardiac maturation. Our murine models demonstrate the complex interplay of these miRs and TF targets during cardiac development.
Results
The miR-200 family is required during early cardiac development
To determine if miR-200 is a key player in mammalian cardiac development, we profiled miR-200 family expression from embryonic mouse whole heart RNA. Transcripts encoding the miR-200 family members are highly expressed from E12.5 to E16.5, with Ct values between 24.33–26.62 for miR-200a and 24.94–27.25 for miR-200c (Fig. 1C). miR-200 family expression was decreased by P28 (Ct values of 38.25 for miR-200a and 32.26 for miR-200c) (Fig. 1C). The expression profile of the miR-200 family suggests they are active in early heart development.
The function of miR-200 during cardiac development was determined using PMIS-A and PMIS-C mouse models, which significantly inhibit miR-200 expression (Supplementary Fig. 1A, B). These two mouse models allow us to study the impact of individual miR-200 family members. Complete inhibition of the miR-200 family can be achieved by crossing the two mouse lines to yield PMIS-A/C mice. PMIS-A and PMIS-C embryos survive into adulthood, whereas PMIS-A/C embryos are lethal at E16.5, and have severe cardiac abnormalities (Fig. 1D, Supplementary Fig. 2, Supplementary Fig. 3A, B).
Serial sectioning and H&E staining of mouse heart tissue at E12.5, E14.5 and E16.5 revealed unique cardiac phenotypes in PMIS-A, PMIS-C, and PMIS-A/C (Fig. 1D, Supplementary Fig. 2, Supplementary Fig. 3A, B). At E12.5, PMIS-A, PMIS-C, and PMIS-A/C show abnormal morphology of the ventricular chambers (Supplementary Fig. 3A). PMIS-miR-200 heart phenotypes are evident at E14.5 in the ventricle free wall myocardium (Fig. 1D, E, Supplementary Fig. 2). However, the PMIS-A/C compact and trabecular myocardium appear disorganized compared to other hearts (Fig. 1D). PMIS hearts also have abnormal ventricular septal development (Fig. 1D, Supplementary Fig. 2, Supplementary Fig. 3A, B). Serial transverse sections of E14.5 PMIS hearts revealed an atrioventricular septal defect in PMIS-AC (Supplementary Fig. 2D). Additionally, the PMIS hearts have abnormal looping of the heart, based on the positioning of the outflow tract and pulmonary vessels (Supplementary Fig. 2). At E16.5, PMIS-A and PMIS-C retain a non-compacted ventricle free wall myocardium (Supplementary Fig. 3B). The ventricular wall defects observed previously in E14.5 PMIS-A/C embryonic hearts remain at E16.5 and these embryos do not survive beyond this stage (Supplementary Fig. 3B). These defective cardiac phenotypes displayed in the PMIS embryos throughout development demonstrate a role for the miR-200 family in heart development.
In mice, deletion of cardiogenic transcription factors Tbx5, Gata4, Pitx2 and Mef2c can alter gene expression within the cardiac GRN, and downstream factors required for heart development1,2,3,4,5,13,42. Loss of Tbx5 in mice leads to ventricular septal, ventricular wall, and epicardium defects2,21. Tbx5 deletion in mice also causes upper limb abnormalities2. However, overexpression of Tbx5 during cardiogenesis results in malformations and a VSD43,44,45. Additionally, overexpression of other cardiogenic T-Box family members results in cardiovascular phenotypes during development46,47,48,49. Loss of Gata4 is associated with atrioventricular septal defects (AVSD) among other cardiac phenotypes4. Loss of Pitx2 leads to defects in left-right asymmetry of the heart and defects in atrial development3. The PMIS embryos present with defective cardiac phenotypes similar to those observed in mouse models demonstrating disruption of transcription factors known to be targeted by miR-200. These findings further support an essential role for the miR-200 family in cardiac development (Fig. 1D, Supplementary Fig. 2, Supplementary Fig. 3A, B).
Previous reports have confirmed that miR-200 members target the 3’UTRs of Tbx5, Gata4, and Pitx2 in cells31,40,41. To show that miR-200 targets Tbx5, Gata4, Pitx2 and Mef2c in vivo, we performed Western blots, using protein isolated from whole embryonic hearts. Inhibition of the miR-200 family increased the expression of these factors across developmental stages (Fig. 1F, Supplementary Fig. 3C, D). The increased protein expression of the miR-200 targets in PMIS-miR-200 murine cardiac tissues demonstrates a role for miR-200 in heart development.
In addition to these, we identified two binding sites for miR-200a and miR-200c in the 3’ UTR of Mef2c (Fig. 1G). We examined the functionality of these miR-200 sites by cloning the seed sequence site (±25 bp) downstream of a luciferase reporter. The luciferase reporter co-transfected with a plasmid to over express either miR-200a or miR-200c into cells. A significant reduction in luciferase activity occurred for both sites compared to mutant control sites, indicating both sites in the Mef2c 3’UTR are subject to miR-200a and miR-200c regulation.
The miR-200 family members are highly expressed during key stages of embryonic cardiac development. Interestingly, inhibition of the entire miR-200 family results in major cardiac defects, including thinning of the ventricular myocardium, a ventricular septal defect, and embryonic lethality by E16.5, all of which have been observed in ablation of the identified miR-200 targets2,10. Our mouse models provide in vivo confirmation that the miR-200 family modulates the expression of Tbx5, Gata4, Pitx2, and Mef2c. The requirement of the miR-200 family in cardiac development adds to the growing evidence that correct gene dosage is indispensable during development, especially during cardiogenesis.
Inhibition of miR-200 increases TF expression in CMs, leading to dysregulation of TF targets
We performed immunofluorescence (IF) staining of E14.5 apical left ventricle (LV) sections for Tbx5, Gata4, Mef2c, and Nkx2.5 (Fig. 2A). Our results indicate that Tbx5 and Gata4 were significantly over-expressed in PMIS-C and PMIS-AC LV CMs (Fig. 2A, B). The significantly increased expression of Tbx5 in PMIS-C and PMIS-AC is intriguing as previous reports suggest that Tbx5 is lowly expressed in ventricle CMs and is enriched in the atrial CMs at this stage8. This expression offers additional support for the requirement of miR-200c’s targeting Tbx5, and suggests miR-200c may have a role in restricting Tbx5 expression to the atria. Interestingly, Mef2c was significantly increased in all PMIS-miR-200 E14.5 heart CMs (Fig. 2A, B). In addition to the miR-200 targets, the transcription factor Nkx2.5, which is not a direct target of miR-200, was increased significantly in PMIS-A, PMIS-C, and PMIS-AC CMs (Fig. 2A, B). The miR-200 targets have significantly increased expression in apical LV CMs at E14.5, and the miR-200 family specifically regulates Tbx5, Gata4, and Mef2c in LV CMs.

A Immunofluorescent (IF) staining of apical left ventricle sections, at E14.5, for miR-200 family targets Tbx5, Gata4, Mef2c and cardiogenic co-factor Nkx2.5 (green). Sections were co-stained with CM marker (MF20 or cTnT) (Red). TFs are overexpressed in PMIS hearts. Yellow arrows denote CMs that have increased expression of the target TF. Scale bar = 25 μm. B Quantification of Corrected Total Cell Fluorescents (CTCF) of Tbx5, Gata4, Mef2c, and Nkx2.5. N = 12-19. C qPCR for downstream transcriptional targets of miR-200 TFs. RNA was isolated from whole hearts of litter mates used for snMultiOmics experiment. N = 4. The statistic test performed was one-way ANOVA with multiple comparisons. Data are presented as mean values ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.00001.
To evaluate potential downstream effects of increased TF expression, we examined the expression of several transcriptional targets by qPCR (Fig. 2C)5,7,21,44,50. The transcriptional targets Nppa, Myh7, Myh6, Myl7, Myl2 and Bmp10 were significantly increased in PMIS-miR-200 hearts (Fig. 2C). Moreover, miR-200 targeted TFs are known to drive expression of genes required to assemble sarcomeres. IF stain for sarcomere myosin (MF20) and cardiac troponin T (cTnT) were increased in PMIS embryos (Fig. 2A). However, expression of the atrial CM marker and Tbx5 transcriptional target, Nppa at E14.5 in atrial CMs was unchanged in PMIS-miR-200 hearts compared to WT. These results suggest that miR-200 may selectively regulate ventricle CMs, but not atrial CMs.
The observed regulation of Tbx5, Gata4, and Mef2c in PMIS LV CMs is consistent with the ability of the miR-200 family to target these factors in LV CMs. Additionally, the increased expression of Nkx2.5, which is not targeted by miR-200, provides evidence of the downstream changes in PMIS-miR-200 CMs. However, understanding how the increase in Tbx5, Gata4 and Mef2c expression alters the CMs’ transcriptional profiles at a cellular level requires a high-throughput approach.
snRNA-sequencing of E14.5 PMIS hearts reveals changes in transcription profiles of cardiac cell types
To understand the effect of inhibiting miR-200 at a cellular level during cardiac development, we performed single nuclei multiomics sequencing (sn-multiomics) on E14.5 murine cardiac tissue isolated from WT, PMIS-A, PMIS-C, and PMIS-AC embryos, profiling both gene expression and chromatin accessibility simultaneously at the single cell level (Fig. 3). After rigorous quality controls and scVI-based data integration, we carried out Uniform Manifold Approximation and Projection (UMAP) and graph-based cell clustering51 (Fig. 3A). Overall, we captured 12,507 nuclei which belonged to 6 cell type clusters that were assigned based on the top differentially expressed genes (DEGs) (Fig. 3A, B, Supplementary. Data File 1). We detected all major cardiac cell types, including cardiomyocytes (CMs), endocardial cells (EndoCs), epicardial cells (Epi), Endothelium (Endo), fibroblasts (CFs), and immune cells (Fig. 3A, Supplementary. Data File 1). We next quantified the proportion of cell types from each sample (Fig. 3B). CMs were the most prevalent making up ∼29% percent of our dataset. EndoCs/Endo cells (31%), CFs (26%), EPI (9%), and immune cells (3%) (Fig. 3B, Supplementary Dataset 9) accounted for the remainder of the detected nuclei. Differences in the proportions for the CMs, EndoC/Endo, Epi, and FBs cell types were detected amongst the different samples (Fig. 3B, C, Supplementary Dataset 9). Each cell type had cluster-specific DEGs compared to the others (Fig. 3D). Additionally, we detected significant DEGs in nuclei originating from PMIS-miR-200 samples compared to the other samples for each of the cell type clusters (Fig. 3E), indicating inhibiting miR-200 may have functional consequences in cells other than CMs. Iterative subclustering within the major cell types identified 15 unique cell state clusters, including 5 CM populations (Fig. 3F). These results are consistent with recent scRNA-seq studies detailing comparable stages of human and murine ontogeny52,53. High-throughput experiments have revealed cellular heterogeneity25,52,54,55,56, and alteration in TFs can affect the composition of specific cell states. Thus, we sought to determine how inhibiting miR-200 may affect the heterogeneity of CMs.

A UMAP representation of single nuclei transcriptomes from wild-type, PMIS-A, PMIS-C, and PMIS-AC clustered for cell types. Unsupervised clustering identified 9 unique cell clusters. B Fraction of annotated cell clusters from each sample. C UMAP representation of single nuclei transcriptomes from wild-type, PMIS-A, PMIS-C, and PMIS-AC. D Heatmap showing the average expression for the top differentially expressed genes between E14.5 cardiac cell clusters. E Differentially expressed genes for WT v PMIS in cardiac cell clusters. The p-values were obtained using the two-sided Wilcoxon rank-sum test, adjusted for multiple comparisons using the Benjamini-Hochberg procedure. F UMAP representation of single nuclei transcriptomes subclustered for cell type. Unsupervised clustering identified 18 unique cell clusters, including 5 unique CM clusters.
PMIS-miR-200 hearts are enriched for a unique population of CMs
As our previous results found inhibiting miR-200 family members increased expression of target TFs in ventricle CMs (Fig. 1F, Fig. 2A., Supplementary Fig. 3C, D)8, we sought to further understand the cellular changes across PMIS-miR-200 CMs. Gene ontology (GO) enrichment analysis of PMIS-miR-200 CMs showed the top 10 common upregulated terms related to heart, cardiac, and muscle development and differentiation (Supplementary Fig. 4A). Several down-regulated GO terms are related to metabolism and the mitochondria (Supplementary Fig. 4A), processes associated with CM maturation57. Granular sub-clustering of CMs found 5 cell states (CM1, CM2, CM3, CM4, CM5) (Fig. 4A), with each state exhibiting specific signature gene expression (Supplementary Fig. 4D, Supplementary Dataset 10). The fraction of each CM population across samples showed that the CM4 cell state was enriched in PMIS-miR-200 samples (WT- 1.44%, PMIS-A, 16.82%, PMIS-C- 22.37%, PMIS-AC- 13.35%) (Fig. 4B, C). The CM4 cluster exhibits a unique expression profile and DEGs compared to other CM cell states (Fig. 4F, Supplementary Fig. 4B–D, Supplementary Data File 2). Tnni1 shows robust expression across all clusters, confirming CM identity (Fig. 4D). Additionally, Tbx5 and Mef2c were shown to be enriched in PMIS-miR-200 samples across all CMs (Fig. 4E). Based on DEGs from each cluster we annotated the different cell states, including CM1 as ventricle CMs (vCMs) (Hey2, Cacna1c, Ryr2, Gja1), CM2 as proliferating CMs (pCMs) (mKi67) and CM3 as atrial CMs (aCMs) (Pitx2, Tbx3, Myl7)25,54,58,59 (Supplementary Data File 2). The CM4 cluster shows expression of many aCM specific genes (Nppa, Nppb, Myl7, and Myh7) (Fig. 4F)25,58. Additionally, the cluster expresses miR-200 targets Tbx5 and Mef2c (Fig. 4F) (Supplementary Data File 2). Other markers for this PMIS-miR-200 enriched cluster include Sox5 and Fgf12 (Fig. 4F, Supplementary Fig. 4B)60,61,62,63. Moreover, the CM4 cluster also has differential expression of contractile smooth muscle cell markers Tangln, Acta1, and others (Supplementary Fig. 4B, C, E, Supplementary Data File 2)64. Lastly, the CM5 cluster appears to be a more mature CM state as these cells are marked by Tbx20, Tbx2, and Tnnt2 (Supplementary Data File 2)52,65. Analysis of DEGs in CMs of individual PMIS-miR-200 samples vs WT revealed significant DEGs across all 5 CMs clusters (Supplementary Data File 3). Markers of the CM4 cluster, Nppa, Sox5, and Fgf12 show enriched expression in the CM4 cells and minor expression in a subset of CM3 cells (Fig. 4F). As CM3 are likely aCMs, these genes are known to be expressed in aCMs19,58,59,66,67. To validate the in vivo existence of CM4 cells, we stained E14.5 apical LV sections for Nppa and cTnT. PMIS-miR-200 apical LV CMs had increased Nppa expression, which was nearly undetectable in WT. The induced expression at E14.5 of Nppa in LV CMs of PMIS hearts provides supporting evidence that the CM4 cells are present and located in the apical LV. However, the development properties of this CM population must be further investigated.

A UMAP plot of the CM subclustered population at E14.5. B Average percentage of CM cell states selected from 100 cells at random over 10 iterations. PMIS-miR-200 samples have an enrichment of the CM4 population (see boxed region). C UMAP plot of subclustered CMs from each sample. D UMAP feature plot for Tnni1. Higher expression is red. E Violin plots for Tbx5 and Mef2c expression in all identified CMs. PMIS-miR-200 samples show increased expression. F UMAP feature plot for significantly expressed CM4 genes Nppa, Sox5, and Fgf12. UMAP feature plot of Myl9 shows lower expression in CM4. UMAP feature plot of Tbx5 and Mef2c shows these factors are expressed in the CM4 cluster. G Immunofluorescent stain of apical left ventricle sections, at E14.5, for Nppa (Green, Yellow Arrows) and cTnT (Red). Nppa has increased expression in PMIS-miR-200 hearts at E14.5. Four independent experiments were performed with each showing similar results. Scale bar = 25 μm.
The CM4 cell state exhibits immature cell properties
To gain insight into the potential developmental trajectory of the CM populations, we performed pseudotime inference based on RNA velocity (Fig. 5)68. These analyses showed the CM2 cell state appears to transition towards the CM1 and CM3 clusters (Fig. 5A). The CM4 cluster however, did not show a strong likelihood of transitioning any particular CM state. The result could suggest a potential immature cell state (Fig. 5A), as latent time inference clearly shows CM4 as the initiating population. Plotting of the CMs across latent pseudotime revealed that CM4 and CM5 represent terminal cell states in the latent space and we therefore inferred their identity as immature and mature cells, respectively (Fig. 5B), which align with DEGs expressed in the clusters (Supplementary Data File 2). Additionally, these analyses found DEGs across the RNA velocity pseudotime (Fig. 5C). Of note, Tbx5 was found to be expressed at an early pseudotime and correlated with CM4 cells (Fig. 5C)25,52,54. The pseudotime plot of Tbx5 showed increased expression in CM4 and CM3 cells at early time points (Fig. 5D). Expression of Tbx5 is highest in these two populations. As CM3 is most likely aCM59, expression of Tbx5 in these cells was expected67. A similar pattern of expression was found for Mef2c however, this factor is more broadly expressed across all CMs (Fig. 5D)69. Moreover, Tbx5 transcripts in CM4 cells showed an increased ratio of unspliced to spliced RNAs. The presence of more unspliced Tbx5 transcripts suggests the gene is actively being transcribed, and thus, an increase in more immature RNAs (Supplementary Fig. 6A–C). DEGs in CM4 suggested Nppa and Sox5 as markers of these cells (Fig. 4F, Supplementary Data File 2)59,60, therefore we analyzed the expression of CM4 markers across pseudotime. Indeed, Nppa and Sox5 expression were enriched in CM4 cells at early pseudotime (Fig. 5E). Both Nppa and Sox5 have increased expression in CM4 cells compared to more mature CM1 cells. In addition, both Nppa, Sox5 and Fgf12 have an increased ratio of unspliced to spliced, suggesting active transcription of these genes (Supplementary Fig. 5D). These results suggest the CM4 population may be an immature population of CMs activated by miR-200 inhibition, marked by Nppa and Sox5 expression.

A RNA velocity analysis of CM subclusters. B Latent Time of RNA velocity. Purple is time 0, yellow is time 1. C Gene expression dynamics of marker genes in each subcluster ordered along latent time inferred by RNA velocity analysis. Analysis shows that Tbx5 is associated with an earlier latent time and CM4 cluster. D Top: Plot of Tbx5 and Mef2c expression across pseudotime for each subcluster of CM. Both factors show expression in CM4 cells and at earlier pseudotime. Bottom: Violin plot of Tbx5 and Mef2c across CM subclusters. Factors are enriched in the CM4 cluster (boxed regions). E Left: Plot of CM4 markers Nppa and Sox5 expression across pseudotime from each subcluster of CM. Right: Violin plot of Nppa and Sox5 in CM1 and CM4 clusters. CM4 markers are enriched in PMIS-miR-200 samples. F Immunofluorescent stain of apical left ventricle sections, at E14.5, for Sox5 (Green) and Nppa (Red). PMIS-C and PMIS-AC hearts show clear enrichment of cell expression of both Nppa and Sox5 (yellow arrows). Four independent experiments were performed with each showing similar results. Scale bar = 25μm.
To validate Nppa and Sox5 as in vivo markers of CM cells, we stained E14.5 LV apical sections to confirm the presence of CM4 cells. We found the presence of dual Nppa + /Sox5+ cells in PMIS-miR-200 embryos, which appear more frequently in PMIS-C and PMIS-AC hearts (Fig. 5F). The bioinformatic and in vivo results suggest that CM4 has an immature cell transcriptome, and is marked by Tbx5, Nppa, and Sox5 expression. These cells are found in the apical LV of PMIS-miR-200 embryos at E14.5, providing evidence for their existence in vivo.
Analyses of early embryonic CM maturation suggest PMIS-miR-200 CM4 cells are a unique cellular state
To understand and further characterize the predicted immature CMs within the PMIS embryonic heart, we determined if CM4 cells could be found at earlier stages of cardiogenesis. In the LV at E12.5, Nppa and Sox5 expression was observed in WT and PMIS hearts (Supplementary Fig. 6). Across all genotypes, Nppa is expressed in CMs (cTnT + ) within the LV. However, expression of Sox5, in WT and PMIS-A, is found in non-CMs (cTnT-) cells of the LV. Whereas PMIS-C and PMIS-AC CMs show co-expression of Nppa and Sox5 (Supplementary Fig. 6). The result that a subpopulation of WT CMs is not Nppa + /Sox5+ at an earlier developmental time point, suggests an abnormal cellular state is acquired when inhibiting miR-200. To further validate CM4 as an abnormal cell state, we used publicly available scRNA-Seq and snRNA-Seq data from CMs at various developmental time points (Supplementary Fig.7)55. In our model, we identified maturation-correlated genes of CMs from E10.5, E11.5, E12.5, E13.5, and E14.5 CMs, and computed a maturation score (Supplementary Fig. 7, Supplementary Data File 4). The maturation score was calculated based on gene modules classified as “down in development” or “up in development” based on their continuous expression trend from E10.5 to E14.5 (Supplementary Fig. 8, Supplementary Data File 4). Based on this metric, CM clusters CM3 and CM4 had the lowest maturation score (Supplementary Fig. 7B). Additionally, overall PMIS-miR-200 CMs had a lower maturation score versus WT, suggesting a more immature cellular state (Supplementary Fig. 7C). Using the list of genes for maturation scoring (Supplementary Data File 4), we examined genes classified as “down in development”, as these genes have higher expression in immature (i.e. E10.5) CMs versus mature (E14.5) CMs. We compared “down in development” and significantly upregulated PMIS-miR-200 genes to identify a potential set of common markers. These analyses yielded just five genes (Alcam, Crb2, Bex2, Diras2, and Bcar3) (Supplementary Fig. 8A). Of these genes, Alcam has been previously described as a marker of progenitor CMs and is robustly expressed within our dataset (Supplementary Fig. 8B)70. Specifically, Alcam is highly expressed in CM4 cells and increased in PMIS-miR-200 CMs versus WT (Supplementary Fig. 8C). To confirm Alcam expression in CM4 cells, we stained E12.5 and E14.5 hearts for Alcam and Nppa (Supplementary Figs. 9 and 10). At E12.5 and E14.5, WT hearts displayed minimal expression of Alcam (Supplementary Figs. 9 and 10). However, PMIS-miR-200 hearts did express Alcam in CMs within the LV (Supplementary Figs. 9 and 10). Alcam expression was enriched within PMIS-C and PMIS-AC hearts and was co-expressed with Nppa in CMs (cTnT + ) (Supplementary Figs. 9 and 10).
Taken together, the bioinformatic and in vivo results are suggestive that the CM4 cell state is one with unique immature properties. Additionally, the data suggest that this CM4 state is found only when low levels of miR-200 are present in the CM. If the CM4 population was a reversion to an earlier common cell state during normal development, WT hearts would contain Nppa + /Sox5+ and/or Nppa + /Alcam+ CMs at earlier developmental stages. We find that there is little overlap in gene expression between WT cells, and cells of the CM4 cluster found in PMIS-miR-200c mice within the sequencing and processing of the sn-multiomics data. In a non-biased approach, the CM maturity analysis did not show a cell population that had a similar transcriptome to that of our CM4. One would expect if CM4 were indeed a cell-state that is present early in cardiogenesis that the CM4 cell state and WT CMs would share many common genes. We found only a few genes that are common between WT CMs and the CM4 cluster in PMIS-miR-200 hearts.
Chromatin accessibility reveals changes in CM4 genes and Tbx5 transcriptional targets
The cardiogenic TFs regulated by miR-200 have been well documented to respond to epigenetic chromatin modifiers55,58,59,71,72,73,74,75,76,77,78,79,80. We sought to understand if increased expression of TFs by miR-200 inhibition would result in changes to the cellular chromatin landscape. We further leveraged the chromatin accessibility modality in our data measured by ATAC sequencing of murine cardiac tissue derived from WT, PMIS-A, PMIS-C, and PMIS-AC embryos (Fig. 6). After computational processing81, we embedded the cells in an ATAC modality-based UMAP, independent of gene expression51. Clustering showed the same 6 cell types from all major cardiac cell types, mirroring clusters found in analysis based on gene expression. The 5 unique populations of CMs identified in snRNA-Seq (CM1, CM2, CM3, CM4, CM5) are clearly separated in the ATAC UMAP, with enrichment of PMIS samples in the CM4 cluster. To understand the chromatin structure across the CM population, we looked at reads near the locus of genes that mark CM4 cells (Fig. 6A, B, Supplementary Fig. 11, Supplementary Fig. 12). Indeed, CM4 cells had more open chromatin surrounding Nppa (Fig. 6A, B), which was a significant DAR (differentially accessible region) peak between CM clusters (Supplementary File 5) and other CM4 genes Tbx5, Sox5, and Fgf12 (Supplementary Fig. 12A, B). These data suggest the CM4 population has a unique epigenetic structure compared to other CMs. Additionally, the miR-200c targets of Mef2c and Gata4 had increased accessibility in PMIS-C and PMIS-AC samples (Supplementary Fig. 11B, C). Taken together, the epigenetic changes observed in PMIS-miR-200 CM4 cells further support this cluster as an immature cell state. As CM4 marker genes had an increased unspliced to spliced ratio, significantly increased expression in PMIS-miR-200 embryos, and more open chromatin, suggests active transcription of these genes marking CM4. Furthermore, the results showing changes in chromatin access in the PMIS embryos demonstrate that inhibition of miR expression can cause changes at the chromatin level. This notion that inhibition of miR-200 results in epigenetic landscape changes adds a layer of regulation to the functional role of miRs in modulating the expression of mRNA targets.

A Visualization of ATAC-Seq reads of open chromatin around the Nppa locus in each of the CM subclusters. CM4 cluster has more open chromatin compared to other clusters (see arrow). B Visualization of ATAC-Seq reads of open chromatin around the Nppa locus in the CM4 cluster from PMIS-miR-200 samples. The Nppa locus appears more open in PMIS-miR-200 samples compared to WT (see arrows). C Open chromatin of the CM4 cluster was analyzed for enrichment of TF binding motifs. Shown are the top 10 enriched motifs. Motifs are enriched for T-Box family members, with Tbx5 being the most enriched. D UMAP feature blot of RNA expression (Upper) and motif enrichment (chromVAR) (lower) for Tbx5, Mef2c, Gata4, and Nkx2.5. E Heatmap of enriched topics across the CM cell states. CM4 cells are enriched for Topic 9 which contains TBX5 as the topic ranked as a differentially enriched motif (Supplemenatary File 7). F Heatmap of the TFs predicted to bind enriched eRegulons regions based on RNA expression of the TFs. G Nppa-Luciferase activity in AC16 cells stably expressing PMIS-miR-200 or a non-targeting control (PMIS-Scramble) showing activation of the Nppa promoter in PMIS-miR-200 cells. The statistic test performed was one-way ANOVA with multiple comparisons. N = 4. Data are presented as mean values ± SEM.
Based on the previous data, we hypothesized that the increase in miR-200 target TF expression, specifically Tbx5, could result in increased downstream gene expression. We observed a significant increase in Nppa and other Tbx5 transcriptional targets in PMIS-miR-200 embryos compared to WT (Fig. 2B), to support this hypothesis. We identified the significantly DARs in each CM population and performed motif enrichment analyses (Supplementary Fig. 13, Supplementary Data Files 5 and 6)81,82. Each CM population had a unique enrichment of motifs, interestingly the CM4 population showed significant enrichment of multiple T-Box family members, including the Tbx5 motif (Supplementary Fig. 13, Supplementary Data File 6). Along with T-Box members, CM4 had a significant enrichment of GATA, MADS, and NK motifs (Fig. 6C, D, Supplementary Fig. 14, Supplementary Data File 6). However, Tbx5/Tbx5 gene expression and motif enrichment was the only TF that showed a strong correlation to CM4 cells (Fig. 6D, Supplementary Fig. 14).
To further confirm that Tbx5/Tbx5 RNA and motif are correlated in CM4 cells, we performed SCENIC+ analysis83,84. The pipeline leverages both snATAC data to identify TF binding sites in open chromatin regions and snRNA data to identify TFs with high expression in given cell states to infer gene regulatory networks (eRegulons) enriched in different cell states (Fig. 6E, F, Supplementary Fig. 15, Supplementary Data File 7)83,84. Results from SCENIC+ showed the CM4 cell state had greater enrichment for an eRegulon driven by Tbx5 compared to other cell states (Fig. 6E, F, Supplementary Fig. 15). Taken together the increased accessibility near known Tbx5 transcriptional targets (i.e. Nppa), and the results of a high-throughput bioinformatic analysis SCENIC+ are suggestive that Tbx5 plays a role in regulating the transcriptome of the CM4 cell state.
As Tbx5, and other target TFs, are known to drive the expression of Nppa2, this result supports previous results and solidifies the role of Tbx5 as a transcriptional activator during cardiac development58,59,77. Moreover, Tbx5 and Mef2c motifs were enriched in CM4 cells (Fig. 6C, D). Finally, we sought to confirm the bioinformatic results of potential Tbx5 increased transcriptional activity. We transfected AC16 cells, that stably express PMIS-miR-200, with a luciferase reporter plasmid driven by Nppa’s proximal promoter19 which contain Tbx5, Gata4, and Mef2c, binding sites. PMIS-miR-200 AC16 cells were found to have significantly increased luciferase activity compared to controls, suggesting activation of Tbx5 transcriptional activity upon inhibiting miR-200 (Fig. 6G). To understand which motif may be responsible for the increased activity of the Nppa promoter region, we cloned each TF motif in front of a luciferase reporter construct19,85,86,87,88. Results show that the Tbx5, Gata4, and Mef2c motifs had significantly increased luciferase activity in PMIS-A, PMIS-C and PMIS-AC cells (Supplementary Fig. 16). To demonstrate binding specificity each motif was mutated and TF activity was accessed in the luciferase reporter assay (Supplementary Fig. 16). We found that mutation of each motif abrogated TF activity through decreased luciferase activity (Supplementary Fig. 16).
Transcriptional regulation of the miR-200 family in CMs provides insight into the complex mechanisms of miR expression during cardiogenesis
We have shown that members of the miR-200 family are highly expressed during cardiogenesis (Fig. 1C). However, based on our multiomics analysis, we wanted to understand if miR-200 may be regulated across CMs populations. Our previous work found that miR-200 was expressed in cells differentiating from a stem cell niche28, as these miRs target stem cell TFs to promote differentiation. However, their expression pattern in cardiogenesis are not well characterized. As sc/sn RNA-sequencing utilizes a 3’ polyA-based sequence, this excludes miRs from being captured in these experiments. To understand expression of the miR-200 family, we visualized open chromatin in our multiomics experiments (Supplementary Fig. 17). Open chromatin at both the chromosome 4 and chromosome 6 loci (Fig. 1A), were different across cell types within the heart (Supplementary Fig. 17). Specifically, at the chromosome 4 loci (miR-200b, -200a, -429) the DNA was more open in CMs versus other cell types. However, the chromosome 6 loci (miR-200c, -141) was open in endothelial and endocardial cells, but closed elsewhere. The result displays a potential location specific mechanism and expression pattern of the miR-200 family across CMs (Supplementary Fig. 17). To add further evidence of cell type expression of the miR-200 family, we profiled the expression of the 5 family members across E14.5 whole hearts, AC16 cardiomyocytes, C166 endothelial cells, MEC1 epicardial cells, and human cardiac fibroblasts (Supplementary Fig. 18), and found increased expression of miR-200b, miR-200c in AC 16 cardiomyocytes compared to other cell types. We additionally utilized the bioinformatic pipeline of miReact to predict the activity of the miR-200 family across our dataset89. Predictive results from miReact showed a significant increase of miR-200a and miR-200c subfamilies in CMs versus other cell types (Supplementary Fig. 19). Additionally, the program predicted a significantly lower miR-200 activity across PMIS samples (Supplementary Figs. 19 and 20). As our lab has previously described the miR-200 family as a regulator of stemness factors (Sox2, Klf, Bim), we hypothesized that the miR-200 family would be more highly expressed in more mature CMs, such as CM1 and CM5. However, miReact predicted a significantly increased activity in CM1, CM3, and CM4 clusters (Supplementary Fig. 20). The miReact results support an expression pattern of miR-200 within the CM4 cluster, however miR-200 may have a role in other populations of CMs. Additionally, across samples, miR-200 activity was predicted to be significantly decreased in PMIS samples versus WT (Supplementary Fig. 20).
As the bioinformatic results show miR-200 activity in CMs within the heart, we sought to test if expression of the PMIS-miR-200 inhibitors could induce expression of CM4 markers in different cells. We found that in AC16 cells that express PMIS-C and PMIS-AC, they have significantly increased expression of target TFs (Tbx5, Gata4, and Mef2c) and CM4 markers (Nppa, Sox5, and Fgf12) (Supplementary Figs. 21–24). However, PMIS-miR-200 inhibitors did not endogenously increase the expression of these factors in other cell types. This is due to the low expression of the miR-200 family in the other cell types, showing that the miR-200 family is predominantly expressed in CMs.
To gain further insight into the transcriptional regulation of the miR-200 family we tested a potential feedback loop between CM TFs and the miR-200 family. Indeed, over-expression of Tbx5, Gata4, Pitx2, or a combination of these TFs caused increased endogenous miR-200a and miR-200c expression in AC16 cells, providing evidence for a feedback loop (Supplementary Fig. 25). Interestingly, ChIP-Seq data from Akerberg et al. 2019 shows binding of Mef2a, Mef2c, Tbx5, and Nkx2.5 at the chromosome 4 locus but no clear peak at the chromosome 6 locus58 (Supplementary Fig. 26). These results provide evidence of the expression profile of the miR-200 family members during cardiogenesis. Additionally, the results show that the CM4 cell state appears only when miR-200 is inhibited within CMs. Lastly, our results provide insight into the potential regulatory mechanism of miR-200 by various cardiogenic TFs.
Our results show CM4 cells have a unique epigenetic landscape compared to other CMs. Inhibiting miR-200 resulted in CM4 cells with more open chromatin compared to WT, with PMIS-C and PMIS-AC being most enriched. Lastly, motif and functional analysis showed that Tbx5 can be a potential transcriptional driver of the CM4 population, further supporting its role as a major transcriptional regulator during cardiogenesis. Although we have found a potential de novo Tbx5-miR-200c pathway in cardiac development, we hypothesized the PMIS-miR-200 embryos and the CM4 cells may be marked by other TFs.
miR-200c targets Irx1, which is expressed in CM4 cells
The miR-200 family, and more specifically, miR-200c is known to target “stemness” TFs such as Sox2 and Klf428,90. In the heart, miR-200c’s targeting of early cardiogenic TFs Tbx5, Gata4, and Mef2c31,40 complements this miRs function in modulating cell progenitor states. We are also identifying other potential genes regulated by the miR-200 family. We have found miR-200c binds the 3’UTR of Irx1 (Fig. 7A). Supporting miR-200c’s ability to target Irx1, expression of Irx1 is significantly increased in E14.5 PMIS-C and PMIS-AC hearts (Fig. 7B). Previous work has shown Irx1 is expressed during early cardiac development throughout the myocardium and becomes restricted to the ventricular septum CMs by E14.591,92. Staining for Irx1 in apical LV sections showed PMIS-C and PMIS-AC have increased expression of Irx1 in LV CMs (Fig. 7C). Expression of Irx1 was confirmed in septal CMs in wild-type, PMIS-A, PMIS-C, and PMIS-AC embryonic hearts91,92. Thus, miR-200c can specifically target Irx1 in CMs during cardiac development. To understand potential changes at the epigenetic levels, we examined open chromatin reads surrounding the Irx1 locus (Fig. 7D). Open chromatin around Irx1 in CM1 and CM2 were enriched compared to other CM states (Fig. 7D). In addition, changes in chromatin landscape were observed across PMIS samples in CM1, CM2, and CM4 (Supplementary Fig. 27). PMIS-C samples had more open chromatin, especially in CM1 and CM2 cells (Supplementary Fig. 27). Moreover, several regions upstream and downstream of the transcription start site (Fig. 7D), showed increased accessibility suggesting a role for potential cis-regulatory elements in Irx1 (Fig. 7D, Supplementary Fig. 27). Similar results were found for Irx4, which is located near Irx1, and thought to have a similar mechanism of transcriptional regulation93. To understand which CM population expresses Irx1, we analyzed its expression pattern across pseudotime (Fig. 7E). Based on this analysis, Irx1’s expression is correlated with early to middle pseudotime and with CM1 and CM2 populations (Fig. 7E). Finally, to validate the multiomics analysis of Irx1, we stained E14.5 LVs for Irx1, Tbx5, and Nppa. PMIS-C and PMIS-AC CMs show some co-expression of Irx1 with CM4 markers, however Irx1 was found in other CMs. Based on this data, we propose a role for Irx1 in assisting the progression of CM-progenitors toward a more mature state. (Fig. 7F). Further investigation of Irx1, its co-factors, and transcriptional targets may reveal new mechanisms that regulate cardiogenesis.

A Functional testing of the miR-200c regulation site in the Irx1 3’ UTR. N = 4. B qPCR for Irx1 expression in PMIS-miR-200 E14.5 hearts and PMIS transduced AC16 cells. RNA was isolated from whole hearts of litter mates used for snMulti-Omics experiment. N = 3. C Immunofluorescent stain of apical left ventricle sections, at E14.5, for Irx1 (Green) and cTnT (Red). PMIS-C and PMIS-AC hearts show clear enrichment of Irx1 expression (yellow arrowhead). Scale bar = 25 μm. D Visualization of ATAC-Seq reads of open chromatin around the Irx1 locus in the CM4 cluster from each PMIS sample. The Irx1 locus appears more open in PMIS-C and PMIS-AC samples compared to WT. E Gene expression dynamics of marker genes in each subcluster ordered along latent time inferred by RNA velocity analysis. Analysis shows that Tbx5 is associated with an earlier latent time in the CM4 cluster. F Immunofluorescent stain of apical left ventricle sections, at E14.5, for Irx1 (white), Tbx5, and Nppa (red) and cTnT (green). PMIS-C and PMIS-AC hearts show co-localization of Irx1 and Tbx5 and Irx1 and Nppa in CMs (yellow arrowhead). Scale bar = 25 μm. The statistic test performed was one-way ANOVAwith multiple comparisons. Data are presented as mean values ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Discussion
The contribution of miRs to cardiac development is essential94. However, the role and function of a single miR within the broader miR-200 family cluster are not understood. A limited number of miRs and their targets, and the downstream process they modulate have been identified in the context of development95. Our study has identified a layer of regulation of the cardiogenic GRN that drives and promotes cardiac development. The miR-200 family is required during cardiac development to modulate the gene dosage of TFs Tbx5, Gata4, and Mef2c31,40,41. Reduced expression of the family increased expression of targets and lethal developmental defects. Moreover, snMulti-Omic sequencing found inhibition of miR-200 caused transcriptional changes in the developing heart. In CMs, miR-200 promotes maturation through its ability to target Tbx5, Gata4, Mef2c, and Irx1. In PMIS-miR-200 embryos, we found an abnormal population of CMs. Additionally, induced expression of target TFs changed the epigenetic landscape in PMIS-miR-200 hearts, specifically in PMIS-C and PMIS-AC murine embryos. Thus, inhibiting a miR can lead to epigenetic changes demonstrating how essential miRs are to development and the downstream effects of miR suppression. Lastly, our results identified Irx1 as a potential regulator of CM4 cells and mediating maturation progression. Overall, these results provide a unique outlook on the complex nature and requirement miRs have in regulating gene dosage in a developmental context.
miR-1 and miR-133 are part of the same cluster and have roles in cardiac muscle cell development96,97,98. Studies have found miR-1 targets the transcripts of Hand2, Irx5, and Hdac4, while miR-133 targets CyclinD2 and Srf96. This miR cluster has been found to inhibit cardiac growth by regulating their targets96,98. Our group and others have shown that the miR-200 family acts similarly to this cluster with miR-200a targeting Pitx2 and Mef2c41 and miR-200c targeting Tbx5, Gata4 and Mef2c31,40. Interestingly, Srf and Mef2c drive transcription of this cluster adding a feedback loop to further regulate the pathway96. We found a similar result in which expression of Tbx5, Gata4, or Pitx2 in cardiomyocytes increased expression of the miR-200 family, again demonstrating a feedback loop. Other miRs regulate different processes across heart development as well. miR-138 is involved in cardiac patterning by targeting cspg2, miR-126 has a role in endothelial cell angiogenesis, and miR-208 has a role in cardiac maturation and myosin isoform switching95. We demonstrate that inhibition of miR-200 increases the expression of cardiogenic TF targets and downstream pathways that prevent CM maturation. Thus, the seemingly homologous role of miRs in cardiac development, is to decrease the expression of factors that are associated with immature CMs32,99,100,101,102.
Tbx5 is well-known for its association with Holt-Oram syndrome1. Individuals with Holt-Oram have mutations in the TBX5 gene that cause severe congenital heart defects (CHDs) and upper limb abnormalities. Whereas homozygous deletion of Tbx5 in mice caused early embryonic lethality due to major cardiac defects, heterozygous loss of Tbx5 serves as a murine model of human Holt-Oram syndrome2. While increased expression of the TF results in similar phenotypes in cardiac development. Gata4 is one of the earliest transcription factors expressed during cardiac development4. Mutations in Gata4 are known to cause CHD5, including mutation in the 3’ UTR14. Loss of Gata4 in embryos results in failure of the heart to form and early embryonic lethality, making these knockout embryos difficult to study in later stages4. However, familial studies have found that mutations in Gata4 cause both atrial and ventricular septal defects in human patients5. Loss of Mef2c leads to early embryonic lethality, with defective heart looping and dysregulation of cardiac gene expression103. Further investigation has shown a genetic interaction of Tbx5 with Mef2c104. Loss of a single copy of both Tbx5 and Mef2c causes abnormal cardiac development and embryonic lethality21. Taken together, the roles of Tbx5, Gata4 and Mef2c interactions are essential for cardiac development.
We observe similar phenotypes in the PMIS-C and PMIS-A/C embryos which have increased Tbx5 expression. These findings support the idea that there is an exquisite sensitivity of the developing heart to alterations in Tbx5 gene dosage105. Strict maintenance of TBX5 gene dosage is necessary for proper cardiac development to occur in mice as well as in humans where TBX5 haploinsufficiency or duplication is causative for Holt-Oram syndrome106. Gata4 and Mef2c regulate downstream effectors, such as Nppa, Myh7, and Bmp10 in cardiomyocytes. The expression of Nppa in PMIS-miR-200 hearts also marks the immature population of CMs. Furthermore, PMIS-miR-200 CMs have increased chromatin accessibility near genes regulated by Tbx5, Gata4, and Mef2c58,59,77. Motif analysis provided supporting evidence that Tbx5 may initiate these changes because open regions were enriched for T-box binding motifs. As Tbx5, Gata4 and Mef2c are known to interact with chromatin remodelers25,52,54,55,74,79,107,108,109,110,111, and act as pioneer TFs, the change in the epigenetic landscape in PMIS-miR-200 CMs is supported by previous work. Thus, miR-200 targeting of Tbx5, Gata4, and Mef2c is necessary to maintain CM gene expression and chromatin accessibility as development progresses.
In cardiac development, the role of Irx1 appears to be associated with progenitor-like CMs91,92. As our lab and others have defined Irx1 as a regulator of progenitor/stem cell niches112, these results are in line with these reports. However, we do not hypothesize that Irx1+ cells are CM “stem cells,” but rather that Irx1+ CMs happen to be more immature proliferative cells, with Irx1 potentially working to push CMs towards a more mature cell state. Further studies will look to understand the expression of Irx1 in cardiac development, how inhibiting miR-200c can affect Irx1 expression, and the location of Irx1+ CMs in early cardiac development. Further, it would be of benefit to determine if Irx1 has a role during cardiac regeneration. Studies have found that increased Irx1 expression in a disease state is associated with better outcomes, and prevention of heart failure113, suggesting a role for Irx1 in disease. Outside the heart, we have shown that Irx1 functions in regulating the murine incisor and lung stem cell niche112. Additionally, induced Irx1 expression in numerous cancers results in more aggressive and invasive tumors114. Based on Irx1 expression and function, we hypothesize that Irx1 may mark CMs that are less differentiated, immature, and potentially primed for regeneration.
Further experiments will seek to understand the potential of inhibiting the miR-200 family in promoting cardiac regeneration following injury. As PMIS-A and PMIS-C embryos survive into adulthood without developing cancer or toxic metabolic activities, PMIS-miR-200 may have a potential role in regeneration. Interestingly, miR-200 is nearly absent in adult homeostasis, preliminary data indicates that following injury, when CMs revert to an immature state115, miR-200 expression increases. We have identified a unique regulatory loop where, over expression of Tbx5, Gata4, and Pitx2 increases expression of the miR-200 family. Furthermore, we observe open chromatin surrounding the genomic location of the miR-200 family in CM4 cells as well as other CMs. As miR-200 TF targets are known to be activated following injury116,117,118,119 and are associated with regenerative potential, we hypothesize that inhibiting the miR-200 family may mitigate some pathological outcomes following a serious myocardial injury.
This research demonstrates that miR-200 is a critical modulator of gene expression during cardiac development. Our study provides significant evidence of the importance and necessity of the miR-200 family in in vivo cardiac development. These data support and advance the knowledge of previous studies on cardiogenic TFs and provide further evidence of the highly complex GRN that governs cardiac development.
An animal model is required to understand the direct role of each miR in development, proliferation, differentiation, and organ function. One disadvantage of using miR conditional knockout mice to map gene pathways in specific tissues is that they are unable to differentiate the effect of each individual miR within a cluster such as the miR-200 family, and a knockout of both clusters is required to establish a phenotype. We observe that knocking out miRs at later stages of development with tissue-specific Cre drivers has little effect on miR-regulated development. Only by inhibiting miRs very early during development do we observe significant anomalies, demonstrating that miRs regulate early gene networks required for the development of the body and organs. The PMIS system was engineered to be stable, highly specific, with no off-target effects, and non-toxic, as it is not an oligonucleotide. The PMIS transgenic mice express the specific PMIS inhibitor molecule from a Pol III promoter to produce low levels of the transcript. This transcribed RNA molecule is 122nt in length and can physically interact with Dicer and the Ago:RISC complex35. The PMIS-miR transgene can be expressed from a plasmid for use in in vitro studies and therapeutic treatments36. Because the miR-200a/141 group and the miR-200b/200c/429 group are differentiated by one nucleotide, we demonstrate through the mouse models that PMIS-A mice have different heart phenotypes than PMIS-C mice, demonstrating cell-specific differences in these miRs. Thus, we can specifically inhibit miRs with only one nucleotide change in their seed sequence using the highly efficient PMIS system. While we have tried to generate “conditional PMIS-miR mice” due to the Pol III promoter, it is difficult to achieve tissue-specific expression. However, we have found this is not required; (1) as explained, the Cre drivers are expressed after the requirement for specific miRs during development, and (2) the only tissues affected are the tissues that express high levels of the miR, such as the heart with high levels in cardiomyocytes, due to the specificity of the PMIS system towards its target miR. Lastly, we engineered the PMIS system as a potential therapeutic molecule that can be delivered to cells and tissues for regeneration and repair.
Methods
Animals
Mice were housed and handled in accordance with guidelines established by the University of Iowa Institutional Animal Care and Use Committee (IACUC). All experimental techniques were approved by the University of Iowa IACUC guidelines. Mice (C57BL/6) male and female mice and embryos were used in the experiments. Sex does not play a role in phenotypes of the mice. Mice are housed in BSL2 barrier facilities with 12:12 light;dark cycles, at 68–79 degrees F and feed normal hard pellets. Euthanasia was accomplished by anesthesia followed by cervical dislocation. This method is consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association. For developmental studies the mice are euthanized by CO2 asphyxiation and decapitation and the embryos excised from the mother. Embryos are euthanized by rapid cervical dislocation or decapitation.
To construct the miR inhibitor (PMIS) clone vector, we used two BsmBI sites in the most effective inhibitor design. AscI and PmeI sites were inserted between ApaI and XbaI sites before the U6 promoter. A SmaI site was inserted before XhoI after the polIII terminator. This vector is termed pmiRI for plasmid of miR inhibitor. After digestion by BsmBI, pmiRi can be used to clone different miR inhibitors into it after annealing and ligation of different miR binding sites with a central bulge35. To obtain double inhibitor mice, we crossed the PMIS-miR-200a (PMIS-A) and PMIS-miR-200c (PMIS-C) mouse lines to produce PMIS-miR-200a/c (PMIS-AC). E0.5 was designated upon observation of a vaginal plug after mating.
RNA isolation from tissue
RNA was isolated from freshly dissected tissues or cells using Trizol. RNA quality was assessed by gel electrophoresis prior to reverse transcription (RT) applications, 0.5-1 μg of total RNA was used in an RT reaction with the 5x primescript RT kit (Takara Bio). cDNA was diluted 1:5 and 1 μl was used per qPCR reaction. qPCR reactions were normalized to β-actin levels. For miRs, RT was performed using the Qiagen miRScript kit. The resultant cDNA was diluted 1:5 and 1 μl was used per qPCR reaction. qPCR probes for specific genes were provided by Qiagen, as well as the normalization set for murine U6. PCR products were examined by melting curve analysis and the sequences were confirmed. Fold changes was calculated using the 2–ΔΔCT method.
Gene | Forward primer | Reverse primer |
|---|---|---|
PMIS-miR-200a | CTAAACATCGTTACCAATCAGACAGTG | GTCAGCTCTTAGTATTCATGAGATG |
PMIS-miR-200c | CTAATCCATCATTACCCATCAGACAGTATTA | GTCAGCTCTTAGTATTCATGAGATG |
miR-200a | TAACACTGTCTGGTAACGATGATG | |
miR-200b | TAATACTGCCTGGTAATGATGA | |
miR-200c | TAATACTGCCGGGTAATGATGGA | |
miR-141 | TAACACTGTCTGGTAAAGATGG | |
miR429 | TAATACTGTCTGGTAAAACCGT | |
Nppa | GCTTCCAGGCCATATTGGAG | GGGGGCATGACCTCATCTT |
Sox5 | TTTTCCCAACAAGCCTCACTC | TTGCCATCGACTTCCATTGTG |
Fgf12 | GTACCATTGATGGGACCAAGG | ACGCAGTCCTACAGGAATTAGAT |
Myh7 | ACTGTCAACACTAAGAGGGTCA | TTGGATGATTTGATCTTCCAGGG |
Myh6 | GCCCAGTACCTCCGAAAGTC | GCCTTAACATACTCCTCCTTGTC |
Myl2 | ATCGACAAGAATGACCTAAGGGA | ATTTTTCACGTTCACTCGTCCT |
Myl7 | GGCACAACGTGGCTCTTCTAA | TGCAGATGATCCCATCCCTGT |
Bmp10 | ATGGGGTCTCTGGTTCTGC | CAATACCATCTTGCTCCGTGAA |
Gja5 | GGTCCACAAGCACTCCACAG | CTGAATGGTATCGCACCGGAA |
Irx1 (mouse) | ACACCTGACAGCACCACCA | GCAAAAGTAAAAGATGACCCC |
IRX1 (human) | GCTCTTCGGCAGCGACAC | GCTCTGGGGCCTCCTTTG |
Ttll10 | CCTGCATACCAACTGTGAAGT | CACAAAGCGACGCTGTGAC |
β-actin | CTCTTCCAGCCTTCCTTC | ATCTCCTTCTGCATCCTGTC |
Paraffin embedding, sectioning, and staining
After embryo isolation and dissection, embryos were fixed for 30 min at 4 °C in 4% paraformaldehyde (PFA) and dehydrated through an ethanol gradient. Dehydrated tissues were pre-cleared with xylene and incubated with three changes of paraffin, and then embedded into blocks. Paraffin-embedded tissues were sectioned at 7μm and mounted on slides. Prior to staining, slides were de-paraffinized and rehydrated through a reverse ethanol gradient. Hematoxylin and eosin (H&E) staining, was performed to assess tissue morphology27.
Histology and fluorescent immunohistochemistry
Sections to be used for fluorescence immunohistochemistry were rehydrated and treated with 10 mM Sodium Citrate solution for 15 min at a slow boil or microwaved for antigen retrieval28. In brief, sections were rehydrated in xylene and ethanol series and then incubated in Block Buffer (1% BSA, 2.5% goat serum, 1% phosphate-buffered saline with 0.25% Triton X-100, PBST) for 30 min at room temperature, washed, followed by overnight incubation at 4 °C with primary antibody (Ab). The antibodies recognized one of the following proteins: Tbx5 (1:100) (Thermo Fisher), Gata4 (1:100) (Santa Cruz), Mef2c (1:100) (Cell Signaling), Nkx2.5 (1:200) (Abcam), MF20 (1:100) (MF 20 was deposited to the DSHB by Fischman, D.A. (DSHB Hybridoma Product MF 20), cTnT (1:50) (CT3 was deposited to the DSHB by Lin, J.J.-C. (DSHB Hybridoma Product CT3), cTnT (1:500) (Proteintech), Nppa (1:100) (Proteintech), Sox5 (1:25) (PCRP-SOX5-1E3 was deposited to the DSHB by Common Fund – Protein Capture Reagents Program (DSHB Hybridoma Product PCRP-SOX5-1E3), and Irx1 (1:100) (Atlas). After the incubation, the slides were treated with goat anti-mouse Alexa-488 or Alexa-594 and/or goat anti-rabbit Alexa-488, Alexa-555, Alexa-594 or Alexa-647 labeled secondary antibody (Invitrogen) at a concentration of 1:300 for 60 min. Each antibody incubation was followed by 3–6 PBST washes. Nuclei were counterstained with DAPI prior to adding coverslips with mounting media (Vector Laboratories). For subsequent IF primary rabbit Ab was added for overnight incubation at 4 °C. Sections were incubated with secondary Ab for 60 min. Sections were incubated in Blocking Buffer plus 2.5% rabbit serum for 30 min, followed by overnight incubation at 4 °C with primary Ab. Sections were incubated with secondary Ab for 60 min and washed. Nuclei were counterstained with DAPI mounting media.
Cell culture
AC16 cardiomyocytes, MEC1 epicardial cells (Millipore Sigma), C166 endothelial, and 293T cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin and transfected by polyethyleneimine (PEI). HCFs (Sigma) were cultured in Cardiac Fibroblast Growth Media (Sigma). 293 T cells were transfected with Pax2 (psPAX2 was a gift from Didier Trono (Addgene plasmid #12260; http://n2t.net/addgene:12260; RRID:Addgene_12260) and MD2G (pMD2.G was a gift from Didier Trono (Addgene plasmid # 12259; http://n2t.net/addgene:12259; RRID:Addgene_12259) along with one of PMIS-miR-Empty Vector, PMIS-miR-200a, PMIS-miR-200c, or PMIS-miR-200ac to make lentivirus. Media was collected after 48 hours and filtered through 0.45 μm filters. AC16 cells (gift from Dr. James F. Martin) were cultured in lentiviral-containing media in the presence of polybrene. Stable infected cells were selected by puromycin.
In Vitro transfection- Cell lines were cultured prior to and during transfections. Cells were transfected in 100 mm plates with either PMIS plasmids expressing PMIS-miR-Empty Vector, PMIS-miR-200a, PMIS-miR-200c, or PMIS-miR-200ac or plasmid over expression of Tbx5, Gata4, Mef2c, and MTG. After 12 hours media was changed. RNA was collected from cells 3 days post transfection.
Luciferase assays
The TK-Luc plasmid containing 638 bp upstream of the Nppa transcriptional start site, containing a Tbx5 (-100), a Gata4 (-1), and a A/T rich region (Mef2c)(-) has been previously described19. Each transcription factors motif ± 20 bps was cloned individually upstream of the luciferase reporter in the TK-Luc plasmid using BamHI and HDIII sites. Mutant sequences did not contain the motif, but contained identical sequences outside the motif. Various TK-Luc clones, SV-40 β-Gal, and overexpression plasmids with transfected into AC16 cells. Media was changed after 24 hours. At 48 hours, cells were lysed, and luciferase and β-Gal activity was measured. The 3’ UTR of Mef2c or Irx1 which contain a miR-200 seed sequence (±20 bps) were cloned downstream of the Renilla Luciferase reporter in the pPsiCheck2 vector. Mutant sequences did not contain a miR-200 seed sequence. Reporter plasmid and a plasmid overexpressing miR-200a or miR-200c were transfected into 293T cells. At 48 h, cells were lysed, and luciferase activity was using a luminometer120.
All cloned constructs were confirmed by DNA sequencing. All plasmids used for transfection were purified by double banding in cesium chloride.
Forward Oligo | Reverse Oligo | |
|---|---|---|
Mef2c WT | GATCctgacttcatactctaaaaaaaaaataatagctctttcacctgact | AGCTagtcaggtgaaagagctattattttttttttagagtatgaagtcag |
Mef2c Mut | GATCctgacttcatactctaaaaaaaatcacaccgctctttcacctgact | AGCTagtcaggtgaaagagcggtgtgattttttttagagtatgaagtcag |
Gata4 WT | GATCcaagctttgccgaactgataactttaaaagggcat | AGCTatgcccttttaaagttatcagttcggcaaagcttg |
Gata4 Mut | GATCcaagctttgccgaatacgtaactttaaaagggcat | AGCTatgcccttttaaagttacgtattcggcaaagcttg |
Tbx5 WT | GATCctcctctcacacctttgaagtgggggcctcttgaggcaaatcagca | AGCTtgctgatttgcctcaagaggcccccacttcaaaggtgtgagaggag |
Tbx5 Mut | GATCctcctctcacacctttgaagtgggggcctccctaacaaatcagca | AGCTtgctgatttgttagggaggcccccacttcaaaggtgtgagaggag |
Western blot
Immunoblots were performed as previously described28. In brief, tissue or cell lysates were analyzed on SDS-PAGE gels. For embryonic cardiac tissue, 10-15 hearts for each stage and genotype were pooled. Following electrophoresis, the protein was transferred to PVDF membrane (Millipore), immunoblotted, and detected with an HRP-conjugated secondary antibody and ECL reagents (GE Healthcare/Amersham Biosciences). The following polyclonal antibodies were used to detect the proteins: Tbx5 (1:750) (Thermo Fisher), Gata4 (1:1000) (Santa Cruz), Pitx2 (1:2000) (Capra Sciences), Mef2c (1:1000) (Cell Signaling) and Gapdh (1:8000) (Santa Cruz).
Single-nuclei isolation
Isolation of E14.5 single-nuclei from murine hearts was adapted from methods previously described121. Pregnant dams were euthanized by CO2 inhalation and embryos were removed and placed in ice-cold 1x PBS. Hearts were dissected from embryos under a dissection microscope. Hearts were placed in a 1.5 mL tube and rinsed with ice-cold 1x PBS to remove excess blood and placed on ice. The following steps were done at 4 °C. Hearts were rinsed twice in ice-cold PBS and collected by low-speed centrifugation before homogenization in Lysis Buffer (10 mM Tris-HCl (pH7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, 0.1% NP40, 0.1% Digitonin, 1% BSA, and 0.5U/μL RNase inhibitor in Nuclease-free H20). Hearts were pipetted repetitively in Lysis Buffer to dissociate nuclei, and tubes were centrifuged for 60 sec to pellet nuclei. Nuclei were rinsed in wash buffer (10 mM Tris-HCl (pH7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, 5% BSA, and 1U/μL RNase inhibitor in Nuclease-free H20). Nuclei in wash buffer were placed through a pre-wet 40μm cell strainer to remove excess cellular debris. Nuclei were collected in a tabletop centrifuge for 60 sec, and fresh wash buffer was added prior to counting and diluting nuclei for snMulti-omics experiments.
Single-nuclei multiome library preparation
Gene expression using single-nuclei RNA-Seq (snRNA-Seq) and snATAC-Seq sequencing libraries were prepared by the Iowa Institute for Human Genetics (IIHG), Genomics Division Laboratory. Briefly, 10,000 nuclei were targeted for bulk transposition before individual nuclei were encapsulated in oil droplets along with the 10X GEM code beads in the Chip J cartridge (P/N: 1000234) using the 10X Genomics Single Cell iX Chromium Controller. Generation of gel beads in emulsion (GEMs), barcoding, pre-amplification PCR, and ATAC library construction were all performed as recommended by the manufacturer (10X Genomics, Chromium Next GEM Single Cell Multiome + ATAC Reagent Kit, Rev F, P/N:1000283). The gene expression libraries were pooled and sequenced on a NovaSeq 6000 to give at least 20,000 reads per nuclei. The ATAC-Seq libraries were pooled and sequenced on a NovaSeq 6000 to give at least 25,000 reads per nuclei.
Single-nuclei multiomics data processing and integration
All newly snMultiomics datasets underwent processing using a standardized pipeline. The raw sequencing reads were aligned to the mm10 genome build using the 10X Genomics toolkit CellRanger-ARC version 2.0.2 (cellranger-arc count), with default settings. Quality control metrics, generated by CellRanger, were examined for each library. To eliminate background signals originating from ambient transcripts in the RNA modality, the raw RNA UMI count matrices underwent further processing using CellBender {doi.org/10.1038/s41592-023-01943-7} version 0.1.0 (cellbender remove-background). The following parameters were used: “--total-droplets-included = 25000”, “--low-count-threshold = 10”, and “--epochs = 200”. To minimize the loss of valid cell barcodes identified by CellRanger, we also set the “--expected-cells” parameter to 1.5 times the number of nuclei output by CellRanger. The output matrices from CellBender were filtered to include only valid cell barcodes that were also identified by CellRanger.
Additional quality controls were conducted at the single nucleus level for each library. Initially, low-quality nuclei were identified based on predetermined cut-off values: UMI count per nucleus > 200, gene count per nucleus > 150, and mitochondria gene-derived UMI < 25%. For the ATAC modality, we set cut-offs for nucleosome signal <1.2, transcription start site (TSS) enrichment >2 and percent reads in blacklisted regions <5%. All nuclei that failed to meet these criteria were subsequently removed from the datasets. Subsequently, dynamic cut-off values were calculated based on the per-library data distribution to account for inherent heterogeneity across samples. For each library, upper and lower bounds were established at the 75th percentile plus 1.5 times the interquartile range (IQR) and the 25th percentile minus 1.5 times the IQR, respectively, for UMI count, gene count, ATAC fragment count, and the percentage of ATAC fragment in peaks (FRiP) per nucleus. The nuclei fell outside of these bounds and were consequently excluded from the datasets. Next, the remaining nuclei underwent evaluation using the Scrublet tool122 to identify potential doublets, utilizing the following parameters: expected_doublet_rate = 0.15 and call_doublets threshold = 0.25. Around 20% of the nuclei were labeled as potential doublets but were retained in the dataset.
Finally, all samples were integrated, and batch effects were corrected using the probabilistic modeling tool, scVI {doi:10.1038/s41592-018-0229-2}. The 50 dimensions of scVI latent space were subsequently reduced to generate the final global UMAP. We performed an additional round of integration of cardiomyocytes using only the ATAC modality to maximize the resolution provided by the chromatin accessibility independent of RNA expression. Harmony was used for correcting batch effects embedded in the ATAC data.
Clustering and annotation
We applied FindNeighbors function of the Seurat version 4.3.0 package123 to generate the shared nearest-neighbor graph (SNN) using 50 dimensions of the scVI latent space or the harmonized principal components. We defined clusters based on the SNN using Louvain algorithm with an optimized resolution of 1.5. In addition to scVI modeling, we also trained an scANVI model {10.15252/msb.20209620} with previously published mouse cardiac snRNA-seq data sets as a reference. We examined the scANVI-predict cell type identities for each cell cluster. Based on both scANVI-predicted labels and the expression of known cardiac cell type marker genes, we annotated all main clusters.
Subsequent re-clustering of each major cell type was performed using an iterative approach. Within each major cell type, we applied FindNeighbors function using 50 dimensions of the scVI latent space and run FindCluster function with various resolution settings between 0.1 to 1.5. The optimal resolutions were determined by clear separation in the UMAP dimension, and robust identification of >30 significantly differentially expressed genes across subclusters. Subclusters enriched with previously called doublets were examined for the expression of main cell type marker genes and were collectively labeled as ambiguous cells. Only the non-ambiguously annotated nuclei were used for subsequent analyses.
Gene set expression scoring
We used AddModuleScore function provided by the Seurat suite to score each cell in snRNA-seq data for the expression of genes in each gene set. AddModuleScore is an implementation of a scoring approach previously described124. In each cell or spot, this function computes a score equal to the mean expression of genes in the given gene set, subtracted by the average expression of genes in a background gene set. For each gene, the method randomly selects 100 background genes with similar average expressions (computed over all cells).
Detection of differentially expressed genes (DEG)
The detection of signature genes among clusters was performed using the FindAllMarkers function provided by Seurat. Specifically, we specified the return of only upregulated genes with a log2 fold-change cut-off of 0.25 and adjusted P values < 0.01, corrected using the Benjamini-Hochberg method.
Gene ontology and pathway enrichment analysis
GO and pathway were performed using clusterProfiler package (version 4.8.1) for R. We extracted enriched GO terms in the biological process category. Pathway enrichment and the related gene network were determined by the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Database. All enriched terms and pathways were filtered at a threshold of false discovery rate (FDR, Benjamini-Hochberg correction) at <0.05.
RNA velocity inference
RNA velocity is a metric that utilizes the detection of spliced and unspliced transcripts to forecast single-cell development directionality68. To determine the direction in which cells were transitioning along various potential trajectories, we employed the velocyto tool. Specifically, we followed the R implementation velocyto.R (version 0.6), utilizing the CellRanger output BAM file and the GENCODE v38 GTF file as the source to calculate the RNA velocity of individual cells. Subsequently, we visualized the RNA velocities atop the original clustering of the cells using scVelo (version 0.2.5)125. For velocity computation, the “dynamic mode” was used for different cell types. Latent time vector was computed and used to represent cell-cell transition pseudotime.
Mature CM scoring
We used a computational method to measure cardiomyocyte (CM) maturity based on gene expression levels. For unbiased identification of genes indicative of CM maturity, we utilized two sets of single-nuclei RNA sequencing (snRNA-seq) data from murine developing hearts, covering developmental stages E10.5, E11.5, E12.5, E13.5, and E14.5. We integrated data from 12,333 CMs (3,848 C57 CMs and 8485 CD1 CMs) reported by Feng et al.55. We aggregated the normalized expression values of all genes within each age group and computed their mean pseudobulk expression. Next, we calculated the Spearman’s correlation coefficient (SCC) of each gene against the E10.5 to E14.5 timeline. This analysis identified 777 positively correlated genes with SCC = 1 and 1257 negatively correlated genes with SCC = -1. For the current study, we created a single metric representing maturity. For a given CM i, we calculated gene set expression scores Pi and Ni based on the positively and negatively correlated gene sets, respectively (detailed above). Pi and Ni were then transformed into Z-scores based on their distributions within the entire CM population from all samples. Since the Pi score represents the expression of genes upregulated during CM maturation and the Ni score represents those downregulated, we calculated the final maturation score (Mi) for each cell as the difference between the two scores (Mi = Pi - Ni).
miReact
To estimate miR-200a/c activity for each single nucleus, we utilized the miReact software {Sci Rep. 2021 Apr 28;11(1):9170. doi: 10.1038/s41598-021-88480-5}89, with default parameters. miReact leverages motif enrichment analysis to derive miR activity estimates from snRNA-seq data based on well-defined sequence motifs that miRNAs bind to and their typical downregulation of target genes. In this study, we used the seed sequences CAGTGTT for miR-200a and CAGTATT for miR-200c, along with the expression matrix from the snRNA-seq dataset as input for miReact. As a result, per-cell miR-200a and miR-200c activities were calculated and their distributions across different cell types and conditions were compared.
SCENIC +
To run the SCENIC+ pipeline83,84 the count matrix, cell metadata matrix, and gene names from the Seurat analysis were used to create an anndata object, followed by snATAC data preprocessing performed using pycisTopic based on cell state identities annotated from snRNA-seq clustering. Preprocessing steps included generating psuedobulk ATAC-seq profiles for each cell state, calling peaks, creating a consensus peak set, performing QC on the snATAC-seq barcodes, and performing topic modeling to identify sets of co-accessible regions (16 topics were used). Barcode QC for snATAC data was performed by determining TSS enrichment, the number of reads in peaks, the duplicate ratio and the number of unique fragments per barcode, with similar values being used for each of the multiome runs. After pycisTopic preprocessing was complete, enhancer-driven Gene Regulatory Networks (eGRNs) were inferred using the SCENIC+ pipeline. The first step in eGRN inference was to create a Scenic+ object containing both gene expression and chromatin accessibility data as well as the motif enrichment results obtained from pycisTopic analysis, containing only barcodes passing QC standards for both modalities. Then, the original Scenic+ pipeline was followed to identify eGRNs (https://github.com/aertslab/scenicplus/blob/old/docs/pbmc_multiome_tutorial.ipynb). Based on the calculated eGRN enrichment scores, dimensionality reduction was performed to produce a UMAP and tSNE plot, and the original cell states determined by snRNA-seq were projected onto the UMAP. In order to produce a dotplot-heatmap, high quality eRegulons were selected by taking the correlation between transcription factor expression and target region enrichment scores (correlation coefficient>0.5) and the plot was created, with color corresponding to TF expression values and dot size correlating to region enrichment scores. Next, TF cooperativity was inferred by determining the overlap of predicted target regions for TF binding (Top 5 TFs for each cell state).
ChIP-Seq
To determine binding patterns of core cardiac TFs near the genomic loci containing the miR-200 family, embryonic (E12.5) bioChIP-Seq data from Akerberg et al. (2019) were used (GSE124008)58. Briefly,.bw files were input into IGV to visualize TF binding patterns.
Statistical methods
P values were calculated using two tailed, unpaired t tests with a minimum of three biological replicates for each group. Error bars are presented as ± standard error of the mean in every figure. * indicates a P value of <0.05, **<0.01, ***<0.001, ****<0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the findings of this study are available in the article, its Supplementary information and the Source Data file. snRNA-seq dataset has been deposited in the NCBI Gene Expression Omnibus and are accessible through the accession number GSE272154. PMIS constructs, mice and data is available from the corresponding author upon request. Source data are provided with this paper.
References
Bruneau, B. G. et al. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt–Oram syndrome. Dev. Biol. 211, 100–108 (1999).
Bruneau, B. G. et al. A Murine model of Holt-Oram syndrome defines roles of the T-Box transcription factor Tbx5 in cardiogenesis and disease. Cell 106, 709–721 (2001).
Liu, C. et al. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development 129, 5081–5091 (2002).
Molkentin, J. D., Lin, Q., Duncan, S. A. & Olson, E. N. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 11, 1061–1072 (1997).
Garg, V. et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424, 443–447 (2003).
Laforest, B. et al. Atrial fibrillation risk loci interact to modulate Ca2+-dependent atrial rhythm homeostasis. J. Clin. Invest. 129, 4937–4950 (2019).
Maitra, M. et al. Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev. Biol. 326, 368–377 (2009).
Misra, C., Chang, S.-W., Basu, M., Huang, N. & Garg, V. Disruption of myocardial Gata4 and Tbx5 results in defects in cardiomyocyte proliferation and atrioventricular septation. Hum. Mol. Genet. 23, 5025–5035 (2014).
Nadadur, R. D. et al. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci. Transl. Med. 8, 354ra115 (2016).
Ang, Y.-S. et al. Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell 167, 1734–1749 (2016).
Cai, C. L. et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 454, 104–108 (2008).
Diman, N. Y. et al. Tbx5 is required for avian and Mammalian epicardial formation and coronary vasculogenesis. Circ. Res. 115, 834–844 (2014).
Li, Q. Y. et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 15, 21–29 (1997).
Reamon-Buettner, S. M., Cho, S.-H. & Borlak, J. Mutations in the 3’-untranslated region of GATA4 as molecular hotspots for congenital heart disease (CHD). BMC Med. Genet. 8, 38 (2007).
Wang, J., Greene, S. B. & Martin, J. F. BMP signaling in congenital heart disease: new developments and future directions. Birth Defects Res A: Clin. Mol. Teratol. 91, 441–448 (2011).
Zhao, C. M. et al. PITX2 loss-of-function mutation contributes to congenital endocardial cushion defect and Axenfeld-Rieger syndrome. PLoS ONE 10, e0124409 (2015).
Peacock, T. B. Lectures on malformations of the heart. Med Exam. 10, 440–444 (1854).
Buijtendijk, M. F. J., Barnett, P. & van den Hoff, M. J. B. Development of the human heart. Am. J. Med Genet. C: Semin Med Genet. 184, 7–22 (2020).
Ganga, M. et al. PITX2 isoform-specific regulation of atrial natriuretic factor expression: synergism and repression with Nkx2.5. J. Biol. Chem. 278, 22437–22445 (2003).
Liu, C., Liu, W., Lu, M., Brown, N. A. & Martin, J. F. Regulation of left-right asymmetry by thresholds of Pitx2c activity. Development 128, 2039–2048 (2001).
Kathiriya, I. S. et al. Modeling human TBX5 haploinsufficiency predicts regulatory networks for congenital heart disease. Dev. Cell 56, 292–309 (2021).
Holman, A. R. et al. Single-cell multi-modal integrative analyses highlight functional dynamic gene regulatory networks directing human cardiac development. Cell Genomics 4, 100680 (2024).
Gong, W., Koyano-Nakagawa, N., Li, T. & Garry, D. J. Inferring dynamic gene regulatory networks in cardiac differentiation through the integration of multi-dimensional data. BMC Bioinform. 16, 74 (2015).
Hill, J. T., Demarest, B., Gorsi, B., Smith, M. & Yost, H. J. Heart morphogenesis gene regulatory networks revealed by temporal expression analysis. Deveolpment 144, 3487–3498 (2017).
de Soysa, T. Y. et al. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 572, 120–124 (2019).
Humphries, B. & Yang, C. The microRNA-200 family: small molecules with novel roles in cancer development, progression and therapy. Oncotarget 6, 6472–6498 (2015).
Cao, H. J. et al. The Pitx2:miR-200c/141: noggin pathway regulates Bmp signaling and ameloblast differentiation. Development 140, 3348–3359 (2013).
Sweat, M. et al. The miR-200 family is required for ectodermal organ development through the regulation of the epithelial stem cell niche. Stem Cells 39, 761–775 (2021).
Krongbaramee, T. et al. Plasmid encoding microRNA-200c ameliorates periodontitis and systemic inflammation in obese mice. Mol. Thera Nucleic acids 23, 1204–1216 (2021).
Byun, Y. et al. MiR-200c downregulates HIF-1alpha and inhibits migration of lung cancer cells. Cell Mol. Biol. Lett. 24, 28 (2019).
Chen, Z., Zhang, S., Guo, C., Li, J. & Sang, W. Downregulation of miR-200c protects cardiomyocytes from hypoxia-induced apoptosis by targeting GATA-4. Int J. Mol. Med 39, 1589–1596 (2017).
Magenta, A., Ciarapica, R. & Capogrossi, M. C. The Emerging role of miR-200 family in cardiovascular diseases. Circ. Res 120, 1399–1402 (2017).
Chang, H. et al. CRISPR/cas9, a novel genomic tool to knock down microRNA in vitro and in vivo. Sci. Rep. 6, 22312 (2016).
Ventura, A. et al. Cre-lox-regulated conditional RNA interference from transgenes. Proc. Natl Acad. Sci. USA 101, 10380–10385 (2004).
Cao, H. et al. A new plasmid-based microRNA inhibitor system that inhibits microRNA families in transgenic mice and cells: a potential new therapeutic reagent. Gene Ther. 23, 527 (2016).
Eliason, S. et al. Extracellular vesicle expansion of PMIS-miR-210 expression inhibits colorectal tumour growth via apoptosis and an XIST/NME1 regulatory mechanism. Clin. Transl. Med. 12, e1037 (2022).
Hong, L. et al. MicroRNA-200c represses IL-6, IL-8, and CCL-5 expression and enhances osteogenic differentiation. PLoS ONE 11, e0160915 (2016).
Ries, R. J., Yu, W., Holton, N., Cao, H. & Amendt, B. A. Inhibition of the miR-17-92 cluster separates stages of palatogenesis. J. Dent. Res. 96, 1257–1264 (2017).
Sweat, Y. et al. miR-17 acts as a tumor suppressor by negatively regulating the miR-17-92 cluster. Mol. Ther. Nucleic Acids 26, 1148–1158 (2021).
Alzein, M. et al. Differential spatio-temporal regulation of T-box gene expression by microRNAs during cardiac development. J. Cardiovasc. Dev. Dis. 8, 56 (2021).
Sharp, T. et al. A pituitary Homeobox 2 (Pitx2):microRNA-200a-3p:beta-catenin pathway converts mesenchyme cells to amelogenin-expressing dental epithelial cells. J. Biol. Chem. 289, 27327–27341 (2014).
Mechakra, A. et al. A Novel PITX2c Gain-of-Function Mutation, p.Met207Val, in patients with familial atrial fibrillation. Am. J. Cardiol. 123, 787–793 (2019).
Takeuchi, J. K. et al. Tbx5 specifies the left/right ventricles and ventricular septum position during cardiogenesis. Development 130, 5953–5964 (2003).
Liberatore, C. M., Searcy-Schrick, R. D. & Yutzey, K. E. Ventricular expression of tbx5 inhibits normal heart chamber development. Dev. Biol. 223, 169–180 (2000).
Koshiba-Takeuchi, K. et al. Reptilian heart development and the molecular basis of cardiac chamber evolution. Nature 461, 95–98 (2009).
Habets, P. E. M. H. et al. Cooperative action of TBX2 and Nkx2.5 inhibits ANF expression in the atrioventricular canal: implications for cardiac chamber formation. Genes Dev. 16, 1234–1246 (2002).
Shirai, M., Imanaka-Yoshida, K., Schneider, M. D., Schwartz, R. J. & Morisaki, T. T-box 2, a mediator of Bmp-Smad signaling, induced hyaluronan synthase 2 and Tgfbeta2 expression and endocardial cushion formation. Proc. Natl Acad. Sci. USA 106, 18604–18609 (2009).
Christoffels, V. M. et al. T-box transcription factor Tbx2 represses differentiation and formation of the cardiac chambers. Dev. Dyn. 229, 763–770 (2004).
Hoogaars, W. M. H. et al. TBX3 and its splice variant TBX3 + exon 2a are functionally similar. Pigment Cell Melanoma Res. 21, 379–387 (2008).
Schlesinger, J. et al. The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS Genet. 7, e1001313 (2011).
McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: uniform manifold approximation and projection. J. Open Source Softw. 3, 861 (2018).
Cui, Y. et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 26, 1934–1950.e1935 (2019).
Hill, M. C. et al. Integrated multi-omic characterization of congenital heart disease. Nature 608, 181–191 (2022).
Ameen, M. et al. Integrative single-cell analysis of cardiogenesis identifies developmental trajectories and non-coding mutations in congenital heart disease. Cell 185, 4937–4953.e4923 (2022).
Feng, W. et al. Single-cell transcriptomic analysis identifies murine heart molecular features at embryonic and neonatal stages. Nat. Commun. 13, 7960 (2022).
Hesse, J. et al. Single-cell transcriptomics defines heterogeneity of epicardial cells and fibroblasts within the infarcted murine heart. Elife 10, https://doi.org/10.7554/eLife.65921 (2021).
Guo, Y. & Pu, W. T. Cardiomyocyte maturation: new phase in development. Circ. Res 126, 1086–1106 (2020).
Akerberg, B. N. et al. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat. Commun. 10, 4907 (2019).
Sweat, M. E. et al. Tbx5 maintains atrial identity by regulating an atrial enhancer network. Nat. Cardiovasc. Res. 2, 881–898 (2023).
Li, A. et al. Silencing of the Drosophila ortholog of SOX5 in heart leads to cardiac dysfunction as detected by optical coherence tomography. Hum. Mol. Genet. 22, 3798–3806 (2013).
Liu, Y. et al. Spatiotemporal gene coexpression and regulation in mouse cardiomyocytes of early cardiac morphogenesis. J. Am. Heart Assoc. 8, e012941 (2019).
Funakoshi, S. et al. Generation of mature compact ventricular cardiomyocytes from human pluripotent stem cells. Nat. Commun. 12, 3155 (2021).
Yeo, Y. et al. FGF12 (Fibroblast Growth Factor 12) inhibits vascular smooth muscle cell remodeling in pulmonary arterial hypertension. Hypertension 76, 1778–1786 (2020).
Osman, I. et al. YAP1/TEAD1 upregulate platelet-derived growth factor receptor beta to promote vascular smooth muscle cell proliferation and neointima formation. J. Mol. Cell Cardiol. 156, 20–32 (2021).
Mandel, E. M. et al. The BMP pathway acts to directly regulate Tbx20 in the developing heart. Development 137, 1919–1929 (2010).
Chinchilla, A. et al. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ. Cardiovasc. Genet. 110, 958116 (2011).
Postma, A. V. et al. A gain-of-function TBX5 mutation is associated with atypical Holt–Oram Syndrome and paroxysmal atrial fibrillation. Circ. Res. 102, 1433–1442 (2008).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Churko, J. M. et al. Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat. Commun. 9, 4906 (2018).
Gessert, S. et al. DM-GRASP/ALCAM/CD166 is required for cardiac morphogenesis and maintenance of cardiac identity in first heart field derived cells. Dev. Biol. 321, 150–161 (2008).
He, A. et al. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat. Commun. 5, 4907 (2014).
Gonzalez-Teran, B. et al. Transcription factor protein interactomes reveal genetic determinants in heart disease. Cell 185, 794–814.e730 (2022).
Joung, J. et al. A transcription factor atlas of directed differentiation. Cell 186, 209–229.e226 (2023).
Liu, Z. et al. Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature 551, 100–104 (2017).
Paige, S. L., Plonowska, K., Xu, A. & Wu, S. M. Molecular regulation of cardiomyocyte differentiation. Circ. Res. 116, 341–353 (2015).
Rowton, M., Guzzetta, A., Rydeen, A. B. & Moskowitz, I. P. Control of cardiomyocyte differentiation timing by intercellular signaling pathways. Semin Cell Dev. Biol. 118, 94–106 (2021).
Sun, H. et al. Inhibition of p300-HAT results in a reduced histone acetylation and down-regulation of gene expression in cardiac myocytes. Life Sci. 87, 707–714 (2010).
Takeuchi, J. K. et al. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat. Commun. 2, 187 (2011).
Waldron, L. et al. The cardiac TBX5 interactome reveals a chromatin remodeling network essential for cardiac septation. Dev. Cell 36, 262–275 (2016).
Yu, S. J. et al. MicroRNA-200a promotes anoikis resistance and metastasis by targeting YAP1 in human breast cancer. Clin. Cancer Res. 19, 1389–1399 (2013).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
González-Blas, C. B. et al. SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks. Nat. Methods 20, 1355–1367 (2023).
Hiroi, Y. et al. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat. Genet. 28, 276–280 (2001).
Plageman, T. F. Jr & Yutzey, K. E. Differential expression and function of Tbx5 and Tbx20 in cardiac development. J. Biol. Chem. 279, 19026–19034 (2004).
Argentin, S. et al. Developmental stage-specific regulation of atrial natriuretic factor gene transcription in cardiac cells. Mol. Cell Biol. 14, 777–790 (1994).
Morin, S., Charron, F., Robitaille, L. & Nemer, M. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 19, 2046–2055 (2000).
Nielsen, M. M. & Pedersen, J. S. miRNA activity inferred from single cell mRNA expression. Sci. Rep. 11, 9170 (2021).
Zheng, B. et al. miR-200c-SUMOylated KLF4 feedback loop acts as a switch in transcriptional programs that control VSMC proliferation. J. Mol. Cell Cardiol. 82, 201–212 (2015).
Christoffels, V. M., Keijser, A. G. M., Houweling, A. C., Clout, D. E. W. & Moorman, A. F. M. Patterning the Embryonic heart: identification of five mouse iroquois homeobox genes in the developing heart. Dev. Biol. 224, 263–274 (2000).
Houweling, A. C. et al. Gene and cluster-specific expression of the Iroquois family members during mouse development. Mech. Dev. 107, 169–174 (2001).
Kim, K. H., Rosen, A., Bruneau, B. G., Hui, C. C. & Backx, P. H. Iroquois homeodomain transcription factors in heart development and function. Circ. Res. 110, 1513–1524 (2012).
Saxena, A. & Tabin, C. J. miRNA-processing enzyme Dicer is necessary for cardiac outflow tract alignment and chamber septation. Proc. Natl Acad. Sci. USA 107, 87–91 (2010).
Cordes, K. R. & Srivastava, D. MicroRNA regulation of cardiovascular development. Circ. Res. 108, 192872 (2009).
Chen, S. et al. Self-renewal of embryonic stem cells by a small molecule. Proc. Natl Acad. Sci. USA 103, 17266–17271 (2006).
Liu, N. et al. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 22, 3242–3254 (2008).
Zhao, Y. et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 129, 303–317 (2007).
Chen, J. et al. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ. Res. 112, 1557–1566 (2013).
Guzzolino, E. et al. miR-182-5p is an evolutionarily conserved Tbx5 effector that impacts cardiac development and electrical activity in zebrafish. Cell Mol. Life Sci. 77, 3215–3229 (2020).
Saxenaa, A. & Tabin, C. J. miRNA-processing enzyme Dicer is necessary for cardiac out!ow tract alignment and chamber septation. Proc. Natl Acad. Sci. USA 107, 87–91 (2010).
Tao, Y. et al. miR-199a-3p promotes cardiomyocyte proliferation by inhibiting Cd151 expression. Biochem. Biophys. Res. Commun. 516, 28–36 (2019).
Lin, Q., Schwarz, J., Bucana, C. & Olson, E. N. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 276, 1404–1407 (1997).
Ghosh, T. K. et al. Physical interaction between TBX5 and MEF2C is required for early heart development. Mol. Cell Biol. 29, 2205–2218 (2009).
Mori, A. D. et al. Tbx5-dependent rheostatic control of cardiac gene expression and morphogenesis. Dev. Biol. 297, 566–586 (2006).
Basson, C. T. et al. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 15, 30–35 (1997).
George, R. M. et al. Single cell evaluation of endocardial Hand2 gene regulatory networks reveals HAND2-dependent pathways that impact cardiac morphogenesis. Development 150, dev201341 (2023).
Ghosh, T. K., Aparicio-Sánchez, J. J., Buxton, S. & Brook, J. D. HDAC4 and 5 repression of TBX5 is relieved by protein kinase D1. Sci. Rep. 9, 17992 (2019).
Lin, L. et al. Two novel functional mutations in promoter region of SCN3B gene associated with atrial fibrillation. Life 12, 1794 (2022).
Robbe, Z. L. et al. CHD4 is recruited by GATA4 and NKX2-5 to repress noncardiac gene programs in the developing heart. Genes Dev. 36, 468–482 (2022).
Zhang, Q. et al. Super-enhancer-driven lncRNA Snhg7 aggravates cardiac hypertrophy via Tbx5/GLS2/ferroptosis axis. Eur. J. Pharmacol. 953, 175822 (2023).
Yu, W. et al. Irx1 regulates dental outer enamel epithelial and lung alveolar type II epithelial differentiation. Dev. Biol. 429, 44–55 (2017).
Zeng, L., Gu, N., Chen, J., Jin, G. & Zheng, Y. IRX1 hypermethylation promotes heart failure by inhibiting CXCL14 expression. Cell Cycle 18, 3251–3262 (2019).
Guo, X. et al. Homeobox gene IRX1 is a tumor suppressor gene in gastric carcinoma. Oncogene 29, 3908–3920 (2010).
Cui, M. et al. Dynamic transcriptional responses to injury of regenerative and non-regenerative cardiomyocytes revealed by single-nucleus RNA sequencing. Dev. Cell 53, 102–116.e108 (2020).
Tao, G. et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 534, 119–123 (2016).
Malek Mohammadi, M. et al. The transcription factor GATA4 promotes myocardial regeneration in neonatal mice. EMBO Mol. Med. 9, 265–279 (2017).
Inagawa, K. et al. Induction of cardiomyocyte-like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circ. Res. 111, 1147–1156 (2012).
Siatra, P. et al. Return of the Tbx5; lineage-tracing reveals ventricular cardiomyocyte-like precursors in the injured adult mammalian heart. NPJ Regen. Med. 8, 13 (2023).
Cao, H. et al. The Pitx2:miR-200c/141:noggin pathway regulates Bmp signaling and ameloblast differentiation. Development 140, 3348–3359 (2013).
Hill, M. C. et al. A cellular atlas of Pitx2-dependent cardiac development. Development 146, dev180398 (2019).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Tirosh, I. et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016).
Bergen, V., Lange, M., Peidli, S., Wolf, F. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Acknowledgements
The authors thank members of the Amendt laboratory for their expertise and helpful discussions and previous lab members for contributing to the study. We thank the Iowa Institute of Human Genetics (IIHG) Genomics Division for their help with sequencing, and the Flow Cytometry core for cell sorting to establish our stable cell lines. We thank the University of Iowa Cardiovascular Research Center and Cardiovascular Phenotyping Core for advice and support of this project. We also thank the University of Iowa Craniofacial Anomalies Research Center for their support. This work was supported by funding from the University of Iowa to BAA.
Author information
Authors and Affiliations
Contributions
R.L. performed the experimental studies, analyzed data and prepared figures and a draft of the manuscript; Y.Z. performed computational analyses, prepared figures; S.E. performed the experimental studies, analyzed data and managed funds and reagents; K.Z. performed initial studies, analyzed data; A.B. performed initial studies, analyzed data; C.J.H. analyzed data, edited the manuscript, provided critical input on experimental design; R.M.W. analyzed data, edited the manuscript, provided critical input on experimental design; M.S. performed computational analyses, prepared figures, edited the manuscript; X.L. performed computational analyses, prepared figures, edited the manuscript; B.A.A. project development, funding, analyzed data, prepared the manuscript.
Corresponding author
Ethics declarations
Competing interests
Brad A. Amendt is the CSO and founder of NaturemiRI, LLC. All other authors have no disclosures to report.
Peer review
Peer review information
Nature Communications thanks Irfan Kathiriya, and Hiroki Kurihara for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Leonard, R., Zhao, Y., Eliason, S. et al. Single-nuclei multiomics analysis identifies abnormal cardiomyocytes in a murine model of cardiac development. Nat Commun 16, 6947 (2025). https://doi.org/10.1038/s41467-025-62208-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62208-9
