
Abstract
The encapsulation of insulin-secreting cells offers a promising strategy for curative treatment of type 1 diabetes without immunosuppression. However, insufficient oxygen within encapsulation systems remains a major challenge, restricting cell survival, function, and scalability. Here, we report an encapsulation platform combining a miniaturized implantable electrochemical oxygen generator (iEOG) with a scalable, linear cell pouch designed for minimally invasive implantation and retrieval. This system enables continuous oxygen supply via electrolysis of tissue moisture, supporting high-density cell encapsulation (60,000 IEQ/mL). Oxygen generated by our system was stable, controllable, and sufficient to maintain cell viability and function under hypoxic (1% O₂) conditions in vitro. In an allogeneic rat model, the oxygenated system implanted subcutaneously reversed diabetes for up to three months without immunosuppression, while non-oxygenated controls remained hyperglycemic. These findings demonstrate the feasibility of sustained oxygenation to enable functional, high-density islet encapsulation in subcutaneous sites, advancing the development of clinically translatable cell-based therapies.
Similar content being viewed by others

Introduction
Type 1 diabetes (T1D) is a chronic disorder caused by the autoimmune destruction of pancreatic β-cells, which eventually leads to insulin deficiency. Exogenous insulin therapy, the current mainstay to manage this disease, necessitates a stringent and constant effort from patients to carefully manage insulin intake balanced with diet, exercise, sleep, and stress. Despite the significant advances in insulin therapy, it remains non-curative and incapable of maintaining glucose homeostasis, predisposing patients to life-threatening complications including severe hypoglycemia events (SHEs)1,2,3 and irreversible organ damage caused by chronic hyperglycemia4. Cellular therapy has emerged as a potential cure to overcome this challenge given the ability of primary and stem cell-derived insulin-producing cells to inherently respond to blood glucose changes. Multicenter clinical trials of intraportal transplantation of allogenic cadaveric islets in over 1200 patients have consistently demonstrated the superior efficacy and safety of cell therapy, evidenced by physiological glycemic control without exogenous insulin and nearly complete eradication of SHEs5,6. Favorable outcomes of the recent Phase 1/2 clinical trial by Vertex Pharmaceuticals (NCT04786262) further confirmed the feasibility of cell replacement therapy for T1D with the limitation of donor availability being addressed by the sustainable supply of stem cell-derived β cells (SC-β cells). However, the requisite lifelong immunosuppression with deleterious side effects, such as nephrotoxicity, opportunistic infections, and malignancies, remains a formidable obstacle to the widespread clinical application of current cellular therapies7. Thus, developing an immunosuppression-free strategy to protect the transplanted cells is imperative to fully harness the potential of cell therapy for curing T1D.
Over the past few decades, the concept of cell encapsulation has been proposed to circumvent immunosuppression. In principle, enveloping insulin-secreting cells within a semipermeable system could prevent direct contact with the detrimental elements of the immune system while allowing for efficient diffusion of glucose, insulin, nutrients, and metabolic wastes. Among encapsulation strategies, macroencapsulation recently garnered more attraction in the field, especially after SC-β cells became clinically available. As opposed to microencapsulation, cell confinement and the ability to retrieve the whole system of the macroencapsulation devices can address safety and regulatory concerns. Furthermore, the subcutaneous space stands out as an ideal transplantation site given the large capacity, minimally invasive surgical procedure, amenability to frequent monitoring, and safe retrievability. However, despite notable insights from research endeavors, translating this concept into clinical applications has been impeded by persistent and intertwined challenges including insufficient supply of oxygen, foreign body responses (FBR), and inability of maintaining a curative dose of cells functional in a reasonably sized system. Particularly, β-cells have an intrinsically high oxygen consumption rate due to the high metabolic demand for insulin synthesis and secretion. Native pancreatic islets require a disproportionately high proportion of pancreatic blood flow relative to their mass8,9. However, the oxygen supply in encapsulation systems is inadequate due to the lack of direct vascularization of encapsulated cells and relatively low oxygen tension in extrahepatic sites (such as the desirable subcutaneous milieu), leading to hypoxia-induced cell death10,11,12 and irreversibly impaired β-cell function13,14. The oxygen deficit is further exacerbated by the low oxygen permeability of fibrotic tissues surrounding the implants caused by foreign body response against the encapsulating materials15. Moreover, insufficient oxygen supply also limits the packing density of β-cells within the encapsulation system as escalating cell density amplifies the requirement for oxygen owing to the steeper oxygen gradient among cell layers16,17,18,19,20. As a result, low cell densities are typically used to reduce hypoxia-induced cell loss post-implantation, rendering the requisite implant dimensions impractical for delivering a curative dose of β-cells8,21. This limitation in packing density has not always been noted in small animal studies but poses a significant barrier to scaling up for clinical translation.
Enhanced oxygen supply to cell encapsulation systems could potentially address the aforementioned challenges. Integration of oxygen-generating biomaterials (e.g. calcium peroxide16,18,22, sodium percarbonate23,24, and lithium peroxide25) into the encapsulation systems has shown encouraging outcomes in preclinical models. The common downsides of these chemical-based approaches are the short duration of oxygen supply, uncontrolled oxygen generation rate, and undesirable end products16,19. Another approach, by BetaO2 Technologies, tackled these challenges by integrating a refillable oxygen reservoir into a macroencapsulation system, allowing for oxygen replenishment via percutaneous injection ports. Devices supplied daily with supraphysiological levels of oxygen demonstrated long-term diabetes reversal in both an allogeneic rat-to-rat and a xenogeneic rat-to-pig model26,27,28. Nonetheless, the requirement for oxygen replenishing via frequent injections and device bulkiness pose ongoing challenges.
To address this collection of issues, we report here a BioElectronics-Assisted Macroencapsulation (BEAM) system capable of providing a continuous oxygen supply to encapsulated cells via electrolysis of tissue moisture. The components of the implantable electrochemical oxygen generator (iEOG) are assembled in a compact, enclosed design (13 mm diameter x 3.1 mm thick), which is then connected to a linear core-shell cell pouch. Islets are housed within the annular space between an inner gas-permeable silicone tubing and an outer immunoprotective tubing made of a hydrogel-impregnated electrospun membrane. This unique design allows for optimal mass transportation and proximity between cells and the oxygen source. The concentric configuration augments the surface-to-volume ratio and facilitates scalability both longitudinally and radially. The edge-free, round-end cylindrical design of the cell encapsulation pouch eliminates the need for intricate sealing that could lead to sharp edges and rigid connections, which can trigger tissue irritation and fibrosis. The final linear layout of the system simplifies implantation and retrieval through minor incisions using minimally invasive techniques.
We demonstrated consistent and programmable oxygen generation from the system in both benchtop and in vivo contexts, enabling adaptability to various cell types and cell dosages. The beneficial impacts of oxygenation on the viability and secretion of insulin were demonstrated in vitro on insulinoma-1 cell (INS-1) aggregates and human pancreatic islets encapsulated at a high density (60,000 islet equivalents (IEQ)/mL). Additionally, we evaluated the therapeutic potential of the BEAM system after subcutaneous implantation in an allogeneic diabetic rat model. Overall, the BEAM system provides a feasible approach to preserving the viability and function of densely packed insulin-secreting cells in the poorly vascularized but clinically attractive subcutaneous site, thus paving the way for development of a practical cell encapsulation system with realistic dimensions and minimally invasive procedures for immunosuppression-free T1D cell replacement therapy.
Design and fabrication of the BEAM system
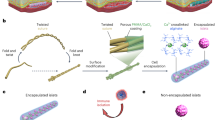
The BEAM system comprises two integral components: an implantable electrochemical oxygen generator engineered to produce a continuous and controllable supply of oxygen through the electrolytic conversion of water and a cell-encapsulating pouch made of alginate-impregnated electrospun fibrous membrane for immunoprotection. Given biocompatibility is a pivotal requisite for the clinical translation of biomedical devices, all the components of the BEAM system were engineered from medical-grade materials. iEOGs were GMP-manufactured by Giner Inc. The electrolyzer components were housed within a medical-grade titanium enclosure with a porous polymeric membrane integrated onto one face, functioning as a window to harvest interstitial water vapor from surrounding tissue for electrolysis. The iEOGs were controlled to a settable current with corresponding voltages from 1.4 to 1.8 V, in the range for the splitting of water into hydrogen and oxygen, which were released through two separate outlets (Fig. 1A). The oxygen outlet delivered gas by convection to an oxygen-permeable tubing traversing the cylindrical cell encapsulation pouch, enabling oxygen diffusion along its length to the encapsulated cells (Fig. 1B, C). An alginate-impregnated fibrous membrane was used as the immuno-protective barrier, which allows for selective diffusion of nutrients, glucose, and insulin while preventing the infiltration of host cells.

The system comprises two integral components: an implantable electrochemical oxygen generator (iEOG) and a core-shell cell encapsulation pouch designed in linear configuration to facilitate minimally invasive implantation, connected by gas-impermeable tubing. A Schematic representation illustrating oxygen generation by electrolytic splitting of tissue moisture. A human sketch is shown to indicate the relative size of a human version system (The power supply needed for electrolysis is not shown). B A representative digital image of a scaled-up BEAM system with a cell encapsulation pouch measuring 15 cm in length and 5 mm in thickness. C Longitudinal section view of the cell encapsulation pouch showing pancreatic islets located in the annular space between an inner silicone tubing and the semipermeable membrane for optimal mass transportation and proximity between cells and the oxygen source. Figure (A) was partly created in BioRender. Pham, T. (2025) https://BioRender.com/k5wqwwr.
Two generations of the iEOG were developed by Giner Inc based on the membrane electrode assembly (MEA) technology as previously described29,30 (Fig. 2A). The MEA consists of a proton exchange membrane that when hydrated serves as the electrolyte, transferring the protons in the balanced electrochemical reaction. The anode and cathode electrocatalysts are pressed on opposite sides of the membrane as a single piece composite. At the surface of the anode, oxygen is evolved and leaves via convection through the oxygen gas port on the anode side (Supplementary Movie 1). At the surface of the cathode, hydrogen is evolved and leaves via convection through the hydrogen gas port on the cathode side. The first version (v1 iEOG) had dimensions of 20 mm in diameter, 4.6 mm in height, and a weight of approximately 4 g. The second version (v2 iEOG) was designed to be smaller in both diameter and thickness by decreasing the MEA area, while maintaining an equivalent oxygen generation capacity. Specifically, the v2 iEOG measures 13 mm in diameter, 3.1 mm in height, and approximately 2 g in weight, which is approximately 25% of the volume and 50% of the weight of the v1 iEOG. Both versions demonstrated precise regulation of oxygen production via modulation of electrical current, allowing for variable oxygen generation rates tailored to meet the metabolic demands of different cells and cell doses, ranging from small numbers ( ~ 1000 IEQ) to clinically relevant doses of islets. The average dose of cadaveric pancreatic islets transplanted into the portal vein during the U.S. CIT-07 trials (NCT00434811) was approximately 800,000 IEQ31. While the optimal dose of islets for extrahepatic transplantation sites has not yet been established, it was predicted that the required dose would range from 300,000 IEQ to 770,000 IEQ for a 70 kg patient32. To evaluate whether the oxygen generation capability of our system could meet the oxygen demand of clinically relevant doses of islets over extended periods, we operated three v1 iEOGs at 10 mA in saline for 11 months, 2 years, and 2.5 years. All three devices exhibited consistent currents close to the setpoint throughout the duration of the study, resulting in a stable oxygen output of approximately 1.94 standard cubic centimeters per hour (scch) (Fig. 2B). This oxygen flow rate is sufficient to support >600,000 human IEQ, assuming an oxygen consumption rate of 2.1 pmol/min/IEQ (Supplementary Table 1). To assess whether the iEOGs could achieve similar oxygen production in vivo, the devices were implanted in immunodeficient nude rats or immunocompetent Sprague-Dawley (SD) rats, operating at 11 mA to simulate expected clinical conditions. After three months of implantation in RNU nude rats, the v1 iEOG generated comparable oxygen levels (2.31 ± 0.10 scch) to those measured pre-implantation (2.25 ± 0.01 scch), which were calculated to be sufficient to support 720,000-740,000 human IEQ (Fig. 2C). Similar results were observed for the v2 iEOG, with oxygen production levels of 2.29 ± 0.03 scch maintained after two months of implantation in nude rats (Fig. 2D). In immunocompetent SD rats, the v2 iEOG also demonstrated stable oxygen generation, with levels measured at 2.44 ± 0.16 scch before implantation and 2.29 ± 0.12 scch one month post-implantation (Fig. 2E). Taken together, these findings suggest the long-term capability of iEOGs to produce sufficient oxygen from interstitial moisture to support clinically relevant doses of human pancreatic islets. Furthermore, histological analysis using Masson’s trichrome staining of tissue surrounding the iEOG in SD rats after one month showed no evidence of adverse tissue reactions and indicated the presence of blood vessels near the water-harvesting window (Fig. 2F, G). We selected the v2 iEOG for further development given the compact design and reduced weight, making it more suitable for implantation.

A A digital image comparing the dimensions of v1 iEOG and v2 iEOG. B Representative 18-h measurements of oxygen flow rate, current, and voltage from a v1 iEOG operated at 10 mA over 2.5 years, collected on day 121, 186, and 879. C Oxygen generation rates of v1 iEOGs operated at 11 mA, measured before and 3 months post-implantation in RNU nude rats (n = 4). Data are presented as mean ± SD. Statistical analysis was performed using paired two-tailed Student’s t-test. D Oxygen generation rates of v2 iEOGs operated at 11 mA, measured 2 months post-implantation in RNU nude rats (n = 8). Data are presented as mean ± SD. E Oxygen generation rates of v2 iEOGs operated at 11 mA, measured before and 1 month after implantation in immunocompetent SD rats (n = 3). Data are presented as mean ± SD. Statistical analysis was performed using paired two-tailed Student’s t test. F A representative stereomicroscope image showing a v2 iEOG implanted in the subcutaneous space of SD rats after 1 month. The skin was removed to exposure the implant. G Masson’s trichrome staining of tissue surrounding the iEOG after 1 month of implantation in SD rats. Arrowheads indicate the presence of blood vessels.
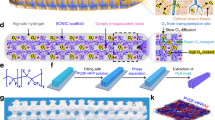
The cell encapsulation pouch was designed to be mechanically robust, scalable, and biocompatible. Mechanical properties, specifically stiffness and elasticity, significantly impact the foreign body responses following implantation33,34,35,36, which is a crucial determinant for the outcomes of a cell encapsulation implant. Ideally, the pouch should be soft to reduce the mechanical mismatch with the surrounding host tissue, which can trigger the activation of inflammatory and fibroblastic cells37,38,39. On the other hand, it also needs to be elastic and durable to maintain its structural integrity during surgical handling and long-term indwelling as intensified fibrotic responses were generally observed in the rough and kinked regions caused by implant deformation40(Supplementary Fig. 2). To meet these requirements, we used an electrospinning technique to prepare a soft, highly porous pouch with round end using medical grade polyether block amide (PEBAX) as a base material (Fig. 3A). Arkema Pebax® thermoplastic elastomer was selected for its inherent softness (77 Shore D) and flexible, rubber-like nature (Flexural modulus of 12). Scanning electron microscopy (SEM) of the membrane revealed a fibrous structure with interconnected pores, which is essential for efficient mass transfer (Fig. 3B, C). The fibers were randomly aligned with sizes ranging from 1.4 to 4 μm (average size: 2.47 ± 0.43 μm) (Fig. 3D); we chose this relatively large fiber sizes to facilitate mass transfer and improve capillary-driven penetration of alginate into the membrane during the hydrogel coating process. The porosity of the membrane was estimated to be 55.12 ± 5.93% (Supplementary Fig. 3). The thickness of the membrane was approximately 70 μm, as determined by SEM. Furthermore, the membrane demonstrated considerable mechanical robustness and elasticity, as indicated by a tensile strength greater than 8 MPa and a fracture strain exceeding 23 (Fig. 3E).

A Schematic illustration for the structure of a cell encapsulation pouch. B Representative digital images showing cell encapsulation pouches in various lengths and diameters, designed to accommodate different cell doses. The inset shows the round end of the pouch. C SEM images of the pouch surface and cross-section. The experiment was repeated independently at least five times with similar results. D Size distribution of PEBAX fibers. E Tensile testing of PEBAX cell encapsulation pouches (n = 3). F Digital images of a nitinol scaffold and a nitinol-reinforced pouch. Both were bent without kink. G Digital images demonstrating the ability of nitinol-reinforced pouches to reverse to their original structural form following twisting deformation. H Representative microscopy image showing the alginate impregnation onto the encapsulation pouch. The experiment was repeated independently at least ten times to prepare for in vivo study with similar results. Figure (E) was partly created in BioRender. Pham, T. (2025) https://BioRender.com/gzlonoy.
One of the challenges in the fabrication of membrane-based encapsulation systems is achieving effective sealing. Conventional sealing methods involving the use of heat, ultrasonic welding, or adhesives can be troublesome due to interference from liquid (media, crosslinking solution) used during the cell loading process. In addition, these methods generally result in rigid and sharp edges, which potentially exacerbate the fibrotic responses. The by-products generated during the application of heat or glue may further contribute to these adverse reactions. Indeed, significantly enhanced fibrotic responses were observed at the heat-sealed ends of membrane-based systems compared to the membranes themselves, regardless of the membranes’ physicochemical properties, across various immunocompetent animal models, including C57BL/6 mice (Supplementary Figs. 4–7), SD rats, and Göttingen minipigs41. The necessity of employing heat sealers, welders, or adhesives also complicates the fabrication process, especially under aseptic conditions. To address these challenges, we designed an edge-free cell pouch with a specific geometry to minimize the area requiring sealing (Fig. 3A). A water-soluble polyethylene glycol (PEG) template was employed to collect the electrospun fibers, thus shaping the cell encapsulation compartment into an edge-free round-ended pouch (Supplementary Fig. 8). A medical-grade polyurethane ring was incorporated into the other open end to serve as a female component in a barbed slide-in connection. This configuration allows the pouch to be sealed by simply sliding the oxygen transportation tubing into the lumen of the cell encapsulation pouch.
Figure 3B shows that the encapsulation pouch can be scaled both radially and longitudinally to accommodate different doses of cells. For long pouches (e.g. human version) which tend to kink, the membrane was reinforced with an elastic, shape-memorable nitinol braided mesh scaffold with a wire thickness of 25.4 μm and pore sizes ranging from 100 to 150 μm (Fig. 3F; Supplementary Fig. 9; Supplementary Movie 2). The reinforced pouches did not kink and were able to revert to their original form after common deformations, such as folding, bending, or twisting, that may occur during the surgical procedure or host movement (Fig. 3G; Supplementary Movie 3). The impact of the nitinol scaffold on mass transfer is expected to be negligible owing to the large pore sizes and thin wire thickness. To impart the immuno-protective property, the fibrous membrane was impregnated with ultrapure alginate (SLG100) using a previously developed method to avoid the detachment of alginate hydrogel after implantation42. This resulted in a hydrogel layer thickness of 30–50 μm, bringing the final thickness of the pouch wall to approximately 100–120 μm (Fig. 3H).
Cell loading and system assembly
A customized cell-loading apparatus, constructed from a perforated polytetrafluoroethylene (PTFE) tube, was developed for cell loading into the BEAM system. PTFE was chosen due to its inertness and hydrophobic properties, which effectively reduce the adhesion of residual alginate solution, thereby minimizing cell loss during encapsulation. To demonstrate the loading procedure, we used INS-1 cell aggregates with a size distribution from 50 to 300 μm as a model to mimic pancreatic islets (Supplementary Fig. 10). These aggregates were gently dispersed in a 2% alginate solution (SLG100) at a density of 60,000 IEQ/mL before being introduced into the cell-loading apparatus. Figure 4A shows the streamlined procedure of cell loading and system sealing. To better visualize the cell loading process, we loaded 150-μm beads (used to simulate cell aggregates) through the loading apparatus, into an encapsulation pouch made of a transparent tube, resulting in a relatively even distribution within the annular space between the loading tool and the pouch wall (Supplementary Fig. 11). When the encapsulation pouch was immersed in a crosslinking solution containing 95 mM calcium chloride and 5 mM barium chloride, the divalent ions diffused through the porous membrane, initiating alginate gelation. Both longitudinal and cross-sectional histology images of the pouches revealed the annular configuration of the aggregate-laden alginate layer with the thickness of 300–500 μm, which is equivalent to about 2–3 layers of cell aggregates (Fig. 4B). This thin hydrogel layer was found to adhere to the fibrous membrane, thus reducing the distance between the cells and the pouch surface to enhance mass transfer. Live/dead staining and histological examination showed no significant changes in aggregate morphology or viability after loading into the BEAM system (Fig. 4B, C).

A A schematic diagram illustrating stepwise procedure for cell loading and pouch sealing in the BEAM system. B Hematoxylin and eosin (H&E) staining of longitudinal and cross-sections of an encapsulation pouch loaded with INS-1 cell aggregates at 60,000 IEQ/mL. The experiment was repeated independently more than three times with similar results. C Bright-field and fluorescence images of the hydrogel layer removed from the encapsulation pouch, containing INS-1 cell aggregates at 60,000 IEQ/mL. Scale bar: 2 mm. D A representative digital image of an oxygen transportation tubing infused with a high flow rate of oxygen. E Representative images depicting the connection between the oxygen transportation tubing and the cell encapsulation pouch. The fibrous membrane was removed for visualization purpose. F The pull-off force measured during the tensile test, indicating the force at which the barb connector is removed from the cell encapsulation pouch (n = 5). Measurements were conducted on 5 devices fabricated across 5 independent production batches. The data are presented as a min to max box-and-whisker plot. The center line represents the median, the bounds of the box indicate the 25th and 75th percentiles, and the whiskers extend to the minimum and maximum values.
Upon crosslinking of the alginate layer, the loading apparatus was withdrawn, allowing for the insertion of the oxygen-transportation tubing. This tubing comprised end-sealed silicone tubing connected to gas-impermeable polyurethane tubing, thereby facilitating oxygen diffusion selectively through the silicone wall to support adjacent cells (Fig. 4D). A barbed connector was integrated onto the gas-impermeable section, designed to fit with the polyurethane ring of the cell encapsulation pouch. Once the oxygen-transportation tubing was fully inserted, the barbed connector provided a tight fit with the ring, effectively sealing the pouch to prevent gas leakage and maintain immunoprotection (Fig. 4E). The connection demonstrated substantial robustness, with the ability to withstand pull forces exceeding 8.5 N (Fig. 4F). The sealed cell encapsulation pouch was finally connected to an iEOG using a custom barb connector before implantation.
Oxygenation by the BEAM system preserved the viability and function of INS-1 aggregates under an in vitro hypoxic condition
The local partial oxygen tension surrounding an encapsulation implant is a critical determinant for the survival and function of encapsulated β-cells, particularly when these cells are loaded at a high density. Unfortunately, post-transplantation oxygen levels in the vicinity of subcutaneous implants have been reported to be critically low ( < 10 mmHg), primarily due to inadequate vascularization at the subcutaneous implantation site and the subsequent formation of a fibrotic capsule43,44,45. Given the inherently high oxygen consumption rate of β-cells, such low oxygen tension is insufficient to support their long-term survival and function. In addition, this also limits the feasibility of loading them at a high density, thus rendering the requisite implant size impractical for clinical applications. We postulated that enhancing the oxygen supply to the implant could improve cell survival and elevate the upper limit of islet cell density. To test this hypothesis, we encapsulated INS-1 832/13 aggregates (2000 IEQ), an insulin-secreting cell line with oxygen consumption rates comparable to that of rat pancreatic islets and higher than that of human islets (Supplementary Table 1), in the BEAM system, resulting in a relatively high loading density (60,000 IEQ/mL). These implants were challenged under 1% O2 cell culture condition ( ~ 7.62 mmHg) for 24 h (Fig. 5A, B). We observed significant cell death in non-oxygenated implants, as evidenced by cell disintegration, shrunken morphology, and live/dead staining (Fig. 5C, D). Histological analysis further revealed a fragmented aggregate structure and cell apoptosis evidenced by weak cytoplasmic staining, nuclear shrinkage, and the formation of apoptotic bodies in the majority of cells (Fig. 5E). Additionally, insulin-positive cells were scarcely detected in these non-oxygenated systems (Fig. 5F). These findings demonstrate a substantial reduction in the viability and function of INS-1 832/13 cells following 24 h of culture under a hypoxic condition. In contrast, cell aggregates in systems supplied with an oxygen generated at an electrical current of 270 μA maintained normal morphology and healthy aggregate structure, with a negligible dead cell number, as indicated by both live/dead staining and histological examination (Fig. 5B–F). Cell aggregates in regions adjacent to silicone tubing (proximal to the oxygen source) and regions near the fibrous membrane (distal to oxygen source) remained highly viable (Supplementary Fig. 12). The oxygen generation rate was approximately 0.016 scch, as estimated using our simulation. In addition, the majority of cells in this group were positive for insulin staining, suggesting that oxygenation by the BEAM system effectively preserved INS-1 832/13 cell aggregate function under a hypoxic culture condition, even at a relatively high cell density.

A A schematic representation of the experimental setup. B Operating electrical current and voltage of iEOG recorded during a 24-h experiment. C Bright-field microscopic images of INS-1 cell aggregates in BEAM systems following a 24-h incubation period under a hypoxic culture condition (1% oxygen), with and without supplemental oxygenation provided by iEOG. D Dual fluorescence staining of INS-1 cell aggregates within BEAM systems following 24-h incubation in 1% oxygen, with and without additional oxygen supply from iEOG. Green: Acridine Orange, Live cells; Red: Ethidium homodimer, Dead cells. E H&E staining of INS-1 cell aggregates before encapsulation and those loaded in BEAM systems subjected to a hypoxic culture condition (1% oxygen) for 24 h, with or without oxygen supplementation from iEOG. Green dashed line represents the interface with the silicone tubing while the red dash line indicates the interface with the fibrous membrane. F Immunofluorescence staining of INS-1 cell aggregates in BEAM systems following 24-h incubation in 1% oxygen, with and without additional oxygen supply from iEOG. Green: Insulin; Blue: Nuclear staining. The experiment in Fig. (C–F) was repeated independently more than six times with similar results. Figure (A) was created in BioRender. Pham, T. (2025) https://BioRender.com/o34farp.
Oxygenation by the BEAM system preserved the viability and function of human islets under an in vitro hypoxic condition
We then assessed the impact of oxygenation provided by the BEAM system on the viability and function of primary human pancreatic islets. Approximately 1800 human IEQ were dispersed in 30 μL of 2% SLG100 solution and subsequently loaded into a BEAM system at a density of 60,000 IEQ/mL and subjected to 1% O2 culture condition (Fig. 6A, B). The BEAM system was operated at 240 μA, corresponding to an oxygen generation rate of approximately 0.009 scch, as estimated by our simulation. After 24 h, pancreatic islets encapsulated in systems without oxygenation exhibited significant cell dissociation and apoptosis (Fig. 6C, D). Immunofluorescence staining revealed a notable decline in cell functionality, as indicated by dominant loss in insulin and glucagon expression compared to those of healthy islets (Fig. 6E). The impaired function was further corroborated by glucose stimulated insulin section (GSIS) assays (Fig. 6F). The levels of insulin secreted from systems cultured under the normoxic condition for 24 h measured 405.6 ± 16.6 μIU/h and 745.9 ± 228.0 μIU/h in the 2.8 mM and 28 mM glucose solutions, respectively. Meanwhile, non-oxygenated systems exhibited significantly lower insulin secretion levels, with 25.5 ± 4.8 μIU/h in a 2.8 mM glucose solution and 99.1 ± 80.9 μIU/h in a 28 mM glucose solution. In contrast, live/dead staining and histological analysis demonstrated that pancreatic islets encapsulated in systems supplied with oxygen remained viable after 72 h of incubation under hypoxic culture condition (Fig. 6C, D). A minor percentage of dead islets was observed at the edges of alginate slabs, likely due to mechanical disruption during sectioning of the hydrogel layer. GSIS and histological evaluations indicated that the function of these islets was comparable to those incubated under normal culture conditions. After 24 h of hypoxic incubation, insulin secretion levels from these oxygenated systems were 330.7 ± 92.0 μIU/h in the 2.8 mM glucose solution and 1,025.5 ± 172.2 μIU/h in the 28 mM glucose solution. Notably, strong insulin and glucagon staining were observed in pancreatic islets of this group after 24 h of incubation (Fig. 6E). These findings suggest that the continuous supply of oxygen by the BEAM system effectively preserved the viability and function of human pancreatic islets encapsulated at a density of 60,000 IEQ/mL.

A A schematic representation of the experimental setup. B Operating electrical current and voltage of iEOG recorded during a 72-h experiment. C Dual fluorescence staining of islets encapsulated in BEAM systems subjected to a 24-h incubation under hypoxic condition (1% oxygen), with and without supplementary oxygenation from iEOGs. Scale bar: 500 μm. Green: Acridine Orange, Live cells; Red: Ethidium homodimer, Dead cells. Dead cells in bottom right panel are thought to be a cutting artifact from processing the annular hydrogel. D H&E staining of encapsulated islets after incubation under a normoxic condition and a hypoxic condition, with and without supplementary oxygenation from iEOG. The experiment in Fig. (C, D) was repeated independently twice with similar results. E Immunofluorescence staining of the encapsulated islets after 24-h incubation under a normoxic and a hypoxic culture condition, with and without supplementary oxygenation from iEOG. Red: Insulin; Green: Glucagon; Blue: Nuclear staining. F Quantification of insulin secretion from islets encapsulated in BEAM systems, following a 24-h incubation under normoxic condition (20% oxygen) (n = 2) and hypoxic condition (1% oxygen), with and without oxygen supplementation from the iEOG (n = 2). Figure (A) was created in BioRender. Pham, T. (2025) https://BioRender.com/o34farp.
Effects of oxygenation by the BEAM system in an allogeneic rat transplantation model
After validating the favorable effect of oxygenation in vitro, we extended our investigation to assess its potential to enhance the viability and function of pancreatic islets in vivo using an allogeneic rat model. Pancreatic islets (6000 IEQ) isolated from Lewis rats were encapsulated in a cell pouch (3.2 mm O.D. x 2 cm length; loading capacity of ~100 μL) of the BEAM system and transplanted into STZ-induced diabetic, immunocompetent SD rats. All rats selected for implantation exhibited blood glucose levels exceeding 450 mg/dL for over 7 consecutive days. The weights of rats before implantation were 418.8 ± 58.5 g, resulting in the islet doses of 14,509.5 ± 1,729.6 IEQ/kg. Owing to the relatively large size of the current electronic controller (3.5 cm × 3.5 cm × 1.2 cm) designed for bench testing, it was not feasible to fully implant the entire BEAM system within the subcutaneous space of rats. Thus, we used an extracorporeal setting, in which electronic components including battery, electronic controller, and iEOG enclosed in a refillable water reservoir, were secured in a pocket worn by the rats (Fig. 7A, B; Supplementary Fig. 13). This setting enables convenient monitoring of iEOG function, water consumption, and battery exchange. For instance, the extracorporeal electronic box allows for the incorporation of a light-emitting diode (LED) to indicate the status of oxygen production for early detection of electrolyzer malfunction or system disconnections, which is crucial in long-term animal study. It is worth noting that for clinical applications, the entire system would be implanted with rechargeable power supply charged weekly via transcutaneous energy transfer (see Supplementary Fig. 14). For the rat study, we designed a customized animal jacket with a small pocket on the back to secure these electronic components. The flank and abdominal area of the jacket were left open to minimize the discomfort or stress on the rats (Supplementary Fig. 15). The use of jackets did not significantly impair the mobility of rats, nor did it cause negative health issues, as evidenced by steady body weight gain over a month (Supplementary Fig. 16). The battery was exchanged every 5 days, and the water reservoir was replenished on a weekly basis. The iEOGs were operated at 280 μA to achieve oxygen generation rates of 0.455 ± 0.015 scc per day (Fig. 7C, D). The cell encapsulation pouch was implanted subcutaneously through a minor skin incision and connected to the iEOG via percutaneous oxygen-impermeable polyurethane tubing. A medical-grade polyester tissue cuff was incorporated at the incision to aid healing and prevent infection. In all 9 animals that underwent transcutaneous implantation, we observed complete wound closure without notable signs of inflammation or adverse reactions at the incision site (Supplementary Fig. 17). Implants without connection to an iEOG (4/4) failed to reverse diabetes, whereas all rats (9/9) received an iEOG-connected implant returned to normoglycemia within 3 days (Fig. 7E–G). Early oxygenation cessation occurred in 3 rats from the first cohort on day 1, day 3, and day 5 because of the damage of the transcutaneous tubing and electronic wire, which were exposed out of the jackets and compromised by the animals (Supplementary Fig. 18; Supplementary Table 2). The implantation location and jacket were then adjusted so these components were secured under the jacket. As a result, the remaining rats with continuous oxygenation maintained normoglycemia for over a month and up to 88 days. Intraperitoneal glucose tolerance test (IPGTT) on day 35 indicated that these rats tolerated glucose comparably to healthy rats, suggesting the normal metabolic function of encapsulated islets (Fig. 7H, I; Supplementary Fig. 19) without significant mass transport limitations. This was further corroborated by the significant increases in serum C-peptide after glucose administration (Supplementary Fig. 20) and the body weight differences between the oxygenated and non-oxygenated groups (Fig. 7G). In contrast, the blood glucose levels of rats implanted with non-oxygenated implants remained higher than 400 mg/dL after 120 min. Oxygenation was accidentally stopped in 2 rats on day 51 and day 72 due to the disconnection of the iEOG with the electronic controller and the cell pouch caused by the rats (Supplementary Fig. 18; Supplementary Table 2). Intriguingly, sharp increases in blood glucose levels were observed right after oxygenation cessation at both early time points (day 1, 3, and 5) and later time points (day 51 and day 72). This suggests that a continuous supply of oxygen may be vital for the long-term function of pancreatic islets in cell encapsulation systems, particularly at high loading density. This is in contrast to a hypothesis in the encapsulation field that short-term oxygenation in the early period may be sufficient.

A A schematic illustration of experimental design. Current, voltage, and battery level were wirelessly spot measured via IR communication. B A digital image demonstrating the BEAM system assembly on a diabetic rat. (1) A pocket on the back of a jacket worn by the rat to secure the iEOG and an electronic box. (2) The oxygen transportation tubing running from iEOG, underneath the jacket to the (3) cell encapsulation pouch implanted in the subcutaneous space of the rat. C Operating current (red) and voltage (blue) of the iEOG recorded during 88-day study from the electronics memory. D Cumulative volume of oxygen generated from the iEOGs used on the rats over a 24-h period (n = 4). E Non-fasting blood glucose levels of rats implanted with a cell encapsulation pouch without oxygen supplementation (n = 4). F Non-fasting blood glucose levels of rats received implantation of a BEAM system (n = 6). G Body weight changes of rats received implants with or without supplemental oxygenation. In F, G, black dots represent NBG levels/body weight changes before implantation while green dots represent NBG levels/body weight changes during continuous oxygenation, and red dots represent NBG levels/body weight changes measured without oxygenation or after the oxygenation cessation. Blue arrows mark the timepoint of implant retrieval. H Intraperitoneal glucose tolerance test conducted on healthy rats (n = 5), diabetic rats (n = 5), rats receiving a BEAM system (n = 4; measured on day 35), and rats receiving a cell encapsulation pouch without oxygen supplementary (n = 4; measured on day 35). Data are presented as mean ± SD. I Corresponding area under the curve for the groups in Fig. (H). Data are presented as mean ± SD. Statistical analyses were performed using one-way ANOVA. p = 0.0725 (diabetic rats vs. without oxygenation), p < 0.001 (with oxygenation vs. without oxygenation), p < 0.001 (heathy rats vs. without oxygenation), p = 0.9964 (heathy rats vs. with oxygenation).
Post-retrieval evaluations further revealed prominent cell death with shrunken morphology in implants without oxygenation and implants with ceased oxygenation, as indicated by both live/dead staining and histology examination (Fig. 8A–D; Supplementary Figs. 21–22). Almost no insulin-positive cells were found in these implants (Fig. 8C). No signs of host cell infiltration were observed in these systems, suggesting the destruction of pancreatic islets was not likely caused by immune cell-mediated rejection. GSIS assay demonstrated low and no significant differences between insulin levels secreted by these cells under a low glucose condition (2.8 mM) and a high glucose condition (16.7 mM), suggesting compromised function of β-cells (Fig. 8E). In contrast, viable islets with healthy morphology and strong insulin/glucagon staining were found in the oxygenated implants on day 32, 40, 45, and 88, suggesting the feasibility of oxygenation to preserve viability and function. GSIS assay on the islets retrieved from these implants showed substantial levels of secreted insulin and the ability to respond to glucose level change. It is worth noting that among these implants, three were retrieved after the blood glucose levels started to increase. We observed a gas bubble partially surrounding the implants in the rats with escalated blood glucose levels (3/6), probably due to the accumulation of excessive oxygen that separated the implants from the surrounding tissue (Supplementary Fig. 23). To confirm this hypothesis, excessive gas was vented through a 23 G needle puncture in a rat, quickly restoring normal blood glucose levels within a few hours and maintaining normoglycemia for an additional 2 weeks until oxygenation accidentally stopped on day 72.

A Stereomicroscopic images showing islet-laden alginate hydrogel layer peeled off from an implant without oxygenation and an implant with oxygenation. B Viability assessment of encapsulated islets by dual fluorescent staining. Green: Acridine Orange, Live cells; Red: Ethidium homodimer, dead cells; Blue: Nuclear staining. C Representative immunofluorescence staining of encapsulated islets in implants without oxygen supplementation and implants with oxygen supplementation. Red: Insulin; Green: Glucagon; Blue: Nuclear staining. D H&E staining of islets before encapsulation, islets in an implant without oxygenation, and islets in an implant with oxygenation. Similar results were observed among the four rats in non-oxygenation groups and the four rats with continuous oxygenation until retrieval. E Ex vivo GSIS of encapsulated islets in implants without oxygen supplementation (n = 3; day 37, 38, and 59) and implants with oxygen supplementation (n = 4; day 32, 40, 45, and 88). Data are presented as individual values (left) and mean ± SD (right). Statistical analysis was performed using paired two-tailed Student’s t test.
Discussion
Macroencapsulation remains a compelling strategy for cell therapy in the treatment of T1D as the use of a permselective membrane could potentially obviate the need for chronic immunosuppression. An increasing body of successful proof-of-concept preclinical studies in mice has demonstrated the potential of this approach to protect encapsulated insulin-secreting cells from immune rejection, thereby enabling long-term glucose homeostasis without the need for immunosuppression46,47,48,49,50,51,52,53,54. However, the translation of these findings to larger animal models, as well as the scaling up of implants for clinical applications, remains a significant challenge. While a few hundred pancreatic islets, loaded within reasonably sized implants at low densities, may be sufficient to reverse diabetes and maintain normoglycemia in mouse models, scaling up these implants for use in larger animals and eventually humans is considerably more complex. The complexity arises from the necessity to pack islets at a high density to achieve an implant size that is practical for implantation. Pancreatic islets have a well-documented reliance on oxygen for their insulin secretion in response to glucose changes. Beta cells can survive under partial oxygen tension levels exceeding 0.1–0.44 mmHg while levels greater than 10 mmHg are necessary to sustain their ability for insulin secretion8,55. Unfortunately, the local oxygen tension within encapsulation systems is significantly below the threshold required for the full function of pancreatic islets when transplanted into the poorly vascularized subcutaneous space43,44,45. This is further exacerbated by the additional mass transport barrier caused by the membrane and the subsequent formation of a fibrotic capsule, which is known to be more severe in larger animals and humans. Results from clinical trials by Viacyte Inc with an immunoprotective device (PEC-Encap™) and an open device (PEC-Direct™) loaded with pancreatic endoderm cells suggest that even with the use of membranes designed to promote vascularization or the membrane with perforations, the devices may fail to achieve sufficient vascularization in both timing and density to support a metabolically adequate functional beta cell mass56. Moreover, increasing cell density further heightens oxygen demand due to the steeper oxygen gradients within cell layers. Thus, in the previously developed cell encapsulation systems, insulin-secreting cells were typically loaded at densities not exceeding 25,000 IEQ/mL (Supplementary Table 3). This would necessitate impractically large encapsulation systems to accommodate a clinically relevant dose of pancreatic islets (300,000 IEQ-770,000 IEQ) at such low densities. For instance, in the clinical trial by ViaCyte (NCT04678557), 12 planar devices with a surface area of approximately 10 cm2 for each were required to achieve a metabolically relevant dose of cells. Computational modeling simulations suggest that a fiber measuring 12.8 m in length with a 1 mm diameter, or a circular slab with a diameter of 11.9 cm and a thickness of 0.5 mm, would be required to deliver 500,000 functional IEQ under 40 mmHg O₂57. Even with microencapsulation, which provides a higher surface-to-volume ratio as compared to macroencapsulation systems, rapid decline in functionality was observed when the loading density was increased to 16,000 IEQ/mL54. Thus, oxygen supplementation is likely needed to realize the clinical translation of cell encapsulation.
Recent advancements in cell encapsulation technology have increasingly focused on the development of encapsulation systems capable of providing oxygenation to preserve viability and function of encapsulated cells16,18,25,58,59. For examples, metal peroxides16,18,22,24,25 have been employed to generate oxygen to enhance cell survival in several encapsulation platforms. However, the inherently short-term oxygen supply in these methods poses a significant barrier to clinical translation, which demands long-term oxygenation. Thus, the need for a method to provide a continuous and sustainable oxygen source remains critical. A recent proof-of-concept study by Krishnan et al. successfully demonstrated the feasibility of using electrolysis-based oxygenation to improve the survival of encapsulated cells in subcutaneous space59. Their device, powered and regulated by resonant inductive coupling, effectively supported a moderate density of pancreatic islets (1000 IEQ/cm2) to correct diabetes in diabetic mice for a month. However, the practicality of resonant inductive coupling in clinical settings for generating an adequate level of oxygen to supply a metabolically relevant dose of cells requires further development and investigation.
In this study, we developed a bioelectronics-assisted encapsulation (BEAM) system incorporating a built-in battery and electronic controller, capable of generating oxygen in a continuously regulated and precise manner. The BEAM system is designed to overcome the limitations of oxygen supply duration associated with chemical-based methods by providing a theoretically unlimited oxygen supply through the electrolysis of tissue moisture, powered by a rechargeable energy source. The oxygen generator, with compact dimensions (13 mm diameter x 3.1 mm thickness) at its full scale, was proven to provide a sustained and adequate oxygen supply to support the upper limits of clinically relevant islet doses (600,000 IEQ-740,000 IEQ) for 11 months to 2.5 years in vitro and three months in vivo. The battery could be designed to be recharged weekly in a wireless fashion via transcutaneous energy transfer (TET), an established technology already in use for FDA cleared rechargeable neurostimulators, thereby reducing the need for patient intervention and compliance requirements. Furthermore, the oxygen generation rate is programmable and precisely controlled, allowing for adaptation to various cell doses and types with differing oxygen consumption rates.
Membrane electrode assembly (MEA) technology was used for the electrolysis of water to facilitate oxygen generation in this system. The MEA comprises a proton exchange membrane that, upon hydration, functions as the electrolyte, enabling proton transfer in the balanced electrochemical reaction. Hydrogen and oxygen evolve on opposite sides of the membrane and are transported through separate gas ports via convection, with less than 1% diffusive crossover, thereby minimizing the risk of spark ignition within the gas phases. The exterior of the iEOG is electrically insulated from the internal components, ensuring the absence of current or voltage hazards upon direct contact. Furthermore, the electrocatalyst-containing electrodes remain isolated from body fluids, as they receive water exclusively in vapor form through a vapor transport membrane, thereby mitigating any potential safety risks.
The linear outline of the BEAM system facilitates minimally invasive implantation and retrieval through a small skin incision. The cell encapsulation compartment is designed as an edge-free, flexible, round-ended cylindrical pouch, which minimizes the risk of enhanced fibrotic reactions typically caused by rigid, sharp edges. A straightforward sequential method for cell loading and system sealing was also developed, streamlining the preparation process, particularly under aseptic conditions. The concentric design of the cell encapsulation pouch, wherein cells are loaded into the thin annular space between the immune-protective membrane and the oxygen source (oxygen-permeable tubing), promotes optimal mass transport and ensures rapid, uniform oxygen diffusion. In addition, this structural configuration is scalable both radially and longitudinally, allowing for loading high islet doses while maintaining proximity between the encapsulated cells and the implant surface as well as the oxygen source.
Our initial results demonstrated that oxygenation provided by the BEAM system successfully preserved the viability and insulin secretion of INS-1 aggregates and human pancreatic islets under a severe hypoxic in vitro condition (1% oxygen or 7.65 mmHg). We then selected rats as the animal model due to the greater thickness of the subcutaneous fat layer, which is more predictive as compared to mouse models when resembling the characteristic of human subcutaneous space48. Additionally, larger animals require higher dose of islets to correct diabetes, making it feasible to load them at a high density. In vivo studies showed that allogeneic islets encapsulated within the BEAM system successfully reversed diabetes and maintained normoglycemia for over one month and up to 88 days, when experiments were prematurely stopped for logistic reasons (i.e. rats compromising the oxygen transport tubing and electrical wire). These results were achieved at a high cell loading density (60,000 IEQ/mL, equivalent to approximately 4200 IEQ/cm²). Meanwhile, islets encapsulated in pouches without oxygenation exhibited significant cell death and predominant loss of function. These findings provide proof-of-concept evidence for the feasibility of using oxygenation to maintain islet viability and function at high cell loading densities within poorly vascularized subcutaneous spaces. The loading density of 60,000 IEQ/mL was selected to achieve a reasonable dose (6000 IEQ) of islets to cure diabetes in rats ( ~ 11,000–15,000 IEQ/kg) using a practically small volume of cell suspension (100 μL). While the packing of 60,000 IEQ/mL or 4200 IEQ/cm2 is among the highest densities reported in literature (Supplementary Table 3), even higher density packing may be possible in clinical application of the BEAM system, especially considering that rat pancreatic islets have a 2–3-fold higher oxygen consumption rate compared to human islets. We estimate that a pouch with an outer diameter of 1 cm and a length of 15 cm would be adequate for encapsulation of approximately 400,000 IEQ at a concentration of 120,000 IEQ/mL (Supplementary Table 4). Alternatively, using seven pouches with an outer diameter of 0.75 cm and a length of 7 cm arranged in parallel could achieve the delivery of 420,000 human IEQ at a density of 60,000 IEQ/mL. The final dimensions of the system including the iEOG and connectors would approximate the size of a credit card (Supplementary Fig. 24). As smaller pancreatic islets (100–150 μm) have been shown to be superior to random-sized pancreatic islets (50–400 μm) in terms of their function57,60,61, the use of smaller sizes may be preferable in preparation of SC-β cell aggregate. We estimate that a system of about half the size of a credit card could be enough to house 500,000 cell aggregates with diameters of 100–125 μm. (Supplementary Fig. 25, 26).
Although the findings from the current design of the BEAM system are promising, several technical challenges must be overcome to realize its potential for clinical translation. Due to the relatively large size of the current electrical controller designed for bench testing, the bioelectronic components could not be fully implanted in rats (a more compact human system has been designed but not implemented). Thus, in this study, we used an extracorporeal setup, wherein, the oxygen generator and electronic components are housed in an external pocket worn by the animal. This setup allows for facile monitoring of iEOG function as well as tracking power and water consumption, which are valuable for the further development of the BEAM system. However, in a clinical context, the entire system—including the cell encapsulation pouch, iEOG, battery, and electrical controller—would need to be fully implantable, with the battery recharged weekly via TET, as is currently used in implantable neurostimulators, left ventricular assist devices and others62,63,64,65,66. We acknowledged that using an extracorporeal system, which relies on transcutaneous tubing and an external jacket to secure the electronic components is troublesome, particularly for long-term studies in flexible animals like rats. In fact, experiments were prematurely terminated in 6 out of 9 rats due to damaged oxygen transportation tubing or rats escaping from jackets (Supplementary Table 2). Furthermore, although the jackets were initially designed to fit comfortably, the rats could outgrow them within a couple of months due to continuous weight gain as they returned to normoglycemia. The use of larger animals, such as pigs, in conjunction with a fully implantable system, is needed to further assess the long-term efficacy of the BEAM system. Second, we envision that in a fully implantable system, it is necessary to develop a strategy to safely dissipate the hydrogen co-product generated during electrolysis from the body. Although hydrogen has been demonstrated to be safe in other contexts67,68,69,70,71, such as in scavenging reactive oxygen species, further investigation is required to assess the long-term impact of hydrogen produced by the BEAM system. A study by Zhu et al. demonstrated that hydrogen can be rapidly cleared from rat tissues within 25 min, even at saturated levels72, supporting the feasibility of safely dissipating hydrogen produced by the iEOG. Our simulations indicate that hydrogen generation occurs at a rate of 0.13 scch (5.8 µmol/h) at 280 µA and 4.95 scch (221 µmol/h) at 11 mA. Given the reported clearance rate, these levels are expected to be safely dissipated through an appropriately designed tissue interface facilitating hydrogen diffusion. One potential strategy to enhance hydrogen clearance involves incorporating a porous membrane at the hydrogen outlet, thereby providing sufficient surface area for controlled diffusion of hydrogen in its soluble phase into the surrounding tissue and circulation.
Third, although the accumulation of excessive oxygen around the cell encapsulation pouch did not appear to adversely affect the encapsulated cells, it resulted in a separation of the system from the surrounding tissue in some cases, thereby hindering mass transport. Interestingly, the oxygen production in this study was measured at approximately 2.43 pmol/IEQ/min, a level comparable to or even lower than reported values for rat pancreatic islets in common culture conditions (Supplementary Table 1). It is noteworthy that over 90% of the encapsulated islets remained viable and functional, exhibiting responses to glucose changes and maintaining the expression of insulin and glucagon, at the time the excessive gas phase was observed. Given that the oxygen consumption rates of beta cells are variable and highly dependent on environmental factors such as glucose and oxygen levels, it is plausible that the total oxygen produced at a constant rate of 2.43 pmol/IEQ/min would exceed the total oxygen demands of these islets under dynamic conditions. Furthermore, as the implant becomes gradually integrated into the subcutaneous space with newly formed vasculature, the oxygen demand may be less compared to the early post-implantation period. In fact, we observed different levels of FBR and vascularization around the cell encapsulation pouches; thus, the oxygen demands of islets in these implants may vary (Supplementary Fig. 27). This challenge could potentially be mitigated by incorporating a built-in sensor (eg. total pressure) within the cell encapsulation pouch, thereby creating a closed-loop system capable of delivering “on-demand” oxygen to the encapsulated cells, as described previously in our patents73,74.
In summary, our study underscores the pivotal importance of adequate oxygenation in encapsulation systems to deliver therapeutic cells. Enhanced oxygen levels efficiently sustained the viability and functionality of encapsulated insulin-secreting cells in both in vitro and in vivo contexts. Notably, continuous oxygen supply allows for the packing of these cells at higher densities without compromising their viability and function. This advancement holds promise for potentially minimizing the required size of encapsulation systems to levels suitable for clinical application, harnessing cell therapy for type 1 diabetes without the need for immunosuppression.
Methods
Preparation of rat INS-1 cell aggregates
INS-1 832/13 cell aggregates were prepared using a commercially available microwell plate (Aggrewell; STEMCELL Technologies). Briefly, 5.9 × 106 INS-1 832/13 cells were added into a well of an AggreWellTM 400 plate, followed by centrifugation at 100 x g for 3 min. After 48-h incubation, the cell aggregates were harvested and cultured in Roswell Park Memorial Institute medium (RPMI-1064 GlutaMAXTM; Gibco) supplemented with fetal bovine serum (FBS; 10% (v/v); Hyclone), HEPES (10 mM; Gibco), sodium pyruvate (1 mM; Gibco), penicillin (100 IU/mL; Gibco), streptomycin (100 µg/mL; Gibco), and β-mercaptoethanol (50 µM; Gibco).
Human pancreatic islets
Human islets (Purity 80–95%) were provided by Dr. Chengyang Liu from the Human Islet Core within the University of Pennsylvania, where the islets were isolated according to the guidelines outlined by the Clinical Islet Transplantation (CIT) consortium protocols. These islets were then transported to our laboratory in cold CIT culture media and cultured for 1–3 days at 37 oC before encapsulation.
Rat pancreatic islet isolation
Rat pancreatic islets were isolated from Lewis rats (200–250 g; Charles River Laboratories) using a previously established enzyme digestion procedure75,76,77. First, 10 mL of LiberaseTM TL (0.15%; Research grade, Roche) in ice-cold Hank’s balanced salt solution (HBSS; Corning) was slowly infused into the pancreas through the pancreatic duct. The pancreas was then incised from associated body parts and incubated at 37 oC for 31 min. Then, 50 mL of ice-cold RPMI-1640 supplemented with 10% FBS was added to terminate the enzymatic digestion. The resulting tissue pellet was washed with RPMI-1640 and filtered through a 450 μm sieve. Islets were purified by a density gradient centrifugation using a Histopaque 1077/RPMI-1640 media gradient. Finally, the islets were manually picked under a light microscope (SZ61, Olympus) to achieve higher purity.
Preparation and assembly of the BEAM system
Manufacture and sterilization of iEOGs
The iEOGs were designed and GMP-manufactured by Giner Inc. Sterilization of the iEOGs was performed using the nitrogen dioxide method (Noxilizer Inc.). Before integration with a cell encapsulation pouch, the operational efficacy of the sterilized iEOGs was evaluated using a highly sensitive mass flow meter (AlicatTM Scientific MW-0.5SSCCM-D). Only iEOGs with oxygen production levels within 85–115% of their theoretical capability were deemed suitable for use in the experimental procedures.
Regulation of oxygen generation
The rate of oxygen generation (Q) is regulated by the modulation of the electrical current (I) applied to the system, as the oxygen flow rate is directly proportional to the current, according to Faraday’s law of electrolysis with adjustments for factors such as temperature, back pressure, and internal losses within the electrolyzer. The equation governing oxygen production is as follows:
Vm is the molar volume of oxygen under standard ambient temperature and pressure (SATP: 25 °C and 1 atm). F is Faraday’s constant derived from the product of the elementary charge (e) and Avogadro’s number (NA). z indicates the number of electrons required to produce one molecule of oxygen, typically 4 electrons per molecule of oxygen. A custom-built electronic controller was developed by Giner Inc. to regulate and maintain a constant current circuit. When the electrolyzer current was set from 0.2 mA to 12 mA, the corresponding voltage required to maintain the set point current was from 1.4 V to 1.8 V, which falls within the expected range for electrolysis.
In vitro assessment of iEOG performance
The long-term in vitro performance of implantable electrochemical oxygen generators (iEOGs) was evaluated in normal saline. Three iEOGs were configured to operate at a current of 10 mA using a custom electronic controller developed by Giner Inc for 11 months, 2 years, and 2.5 years. Current and corresponding voltage were measured every 30 s throughout the study. On a weekly basis, each unit was connected for an 18-h period to a sensitive mass flow meter, calibrated at the factory for oxygen measurement (Whisperlite, Model MW-0.5SSCCM-D, Alicat) to measure the oxygen flow rate and demonstrate it corresponded as expected to the current setpoint.
In vivo assessment of iEOG performance
To assess the in vivo oxygenation capability, sterile iEOGs were implanted into the subcutaneous space of immunodeficient RNU nude rats or immunocompetent SD rats and operated at 11 mA. The flow rates of oxygen generated from these iEOGs were recorded prior to implantation and on the day of explantation. For the experiment on RNU nude rats, the oxygen generation rates were measured from the iEOGs in tissue samples collected post-mortem. For the experiment on SD rats, a flow meter (Whisperlite, Model MW-0.5SSCCM-D, Alicat) was directly connected to the oxygen outlet of the iEOG exposed through the skin.
Preparation of cell encapsulation pouch
The fibrous cell encapsulation pouch was prepared using an electrospinning method. Specifically, medical-grade polyether block amide (Arkema Pebax™) was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at a concentration of 12.5% (w/v) and electrospun at 14 kV through a 23-gauge blunt needle, which served as the spinneret. The polymer solution was fed at a flow rate of 1.5 mL/h, with the spinneret-to-mandrel distance set to 11 cm. To fabricate round-ended, edge-free cell encapsulation pouch, a PEG template of the desired shape was deposited onto a conductive mandrel by injection molding to collect electrospun fibers. For the rat scale pouch, a two-part 3D-printed mold was fabricated using a Formlabs 3 printer (Formlabs Inc.) with Clear V4 photopolymer resin. The mold was assembled onto one end of a steel rod with a diameter of 1.6 mm, and melted PEG was introduced into central cavity within the mold. After allowing the PEG to solidify at room temperature for 10 min, the mold was disassembled, resulting in a 700-μm-thick PEG layer attached to one end of the rod. A 1-cm medical-grade polyurethane ring (Scientific Commodities Inc) was affixed to one end of the PEG template, functioned as a part of the barb connector to seal the pouch. For long pouches, a braided nitinol mesh scaffold (medical grade; Secant Group) with a diameter matching that of the PEG template was first immersed in a 1.5% (w/v) PEBAX solution in HFIP and subsequently positioned over the PEG template. The mandrel was then mounted onto a rotator operating at 450 rpm to facilitate the collection of electrospun fibers for 10 min. Afterward, the mandrel was removed and immersed in distilled water for 30 min to completely dissolve the PEG template. The resulting pouch was washed 5 times with distilled water and then vacuum-dried overnight. Finally, the pouch was sterilized using an Anprolene AN74 ethylene oxide (EO) gas sterilizer (Andersen Sterilizers) following a standard 12-h cycle.
The membrane topology was analyzed using a Gemini 500 scanning electron microscope (Zeiss) at the Cornell Center for Materials Research. Fiber sizes and porosity were measured with ImageJ from 10 frames taken across 3 independent preparation batches. Tensile tests were conducted using an Instron 5965 test system (Instron) and analyzed with Bluehill 3.0 SOP software.
To prepare the alginate-impregnated cell encapsulation pouch, the one-end-open fibrous pouch described above was first submerged in a 2% SLG100 alginate solution (Novamatrix) for 35 min to facilitate the penetration of alginate into the pores of the membrane. Subsequently, a 100 mM barium chloride solution was injected into the pouch through the open end using a 4-inch, 20-gauge blunt needle. The pouch was then held in the alginate solution for 10 seconds to allow the diffusion of barium ions across the fibrous membrane, thereby initiating gelation of the alginate both within and adjacent to the membrane. This process resulted in the formation of a thin alginate hydrogel layer that was securely affixed to the fibrous membrane. The alginate-impregnated pouch was then washed three times with normal saline and stored in a solution containing 95 mM calcium chloride and 5 mM barium chloride until further use.
Cell loading and assembly of BEAM system
A 4-cm section of medical-grade PTFE tubing (0.864 mm I.D. x 1.420 mm O.D.; SAI Infusion Technologies), with one end sealed and approximately 1.5 mm perforations along its length, was utilized as a cell loading apparatus. Pancreatic islets or INS-1 cell aggregates were washed three times with normal saline and subsequently suspended in a 2% SLG100 solution. Specifically, for the rat studies, Lewis rat pancreatic islets (6000 IEQ) were suspended in 100 μL of SLG 100 solution. The prepared cell suspension was pre-loaded into another PTFE tubing, which was then connected to the cell loading apparatus centrally positioned within the cell pouch. The other end of the PTFE tubing was connected to a syringe, which was used to gradually infuse the suspension into the cell compartment through the perforations in the loading apparatus. The outer diameter of the cell-loading device (1.420 mm) was slightly smaller than the inner diameter of the polyurethane ring in the cell encapsulation pouch (1.680 mm) to prevent pressure buildup during infusion of cell suspension. Following this, the cell encapsulation pouch was immersed in a crosslinking solution composed of 95 mM calcium chloride and 5 mM barium chloride for 10 min to facilitate the gelation of the cell-embedded alginate layer. The pouch was then washed 3 times with normal saline. The cell loading apparatus was removed, providing space for the subsequent insertion of oxygen transport tubing.
The oxygen transportation tubing is composed of two distinct segments: an oxygen-permeable segment constructed from USP class VI silicone tubing with an I.D. of 0.635 mm and an outer diameter O.D. of 1.194 mm (Specialty Manufacturing Inc), and an oxygen-impermeable segment fabricated from medical-grade polyurethane tubing with an I.D. of 1.016 mm and an O.D. of 1.379 mm (Scientific Commodities Inc). The length of the oxygen-permeable segment was designed to match the length of the cell-laden section of the encapsulation pouch, which was specifically 2 cm in the version used in rats. To mitigate the risk of kinking post-implantation, a miniature coil spring (D.R. Templeman Co.) was inserted into the oxygen-impermeable segment. Furthermore, a polycarbonate barb connector (Bio-Rad) was incorporated onto the oxygen-impermeable tubing. The cell encapsulation pouch was sealed by simply sliding the oxygen transportation tubing fully into the cell capsules, ensuring that the barb connector fits securely within the polyurethane ring of the cell encapsulation pouch. To assess the durability of the connection, a tensile pull test was conducted. The connector and the polyurethane ring were mounted axially in a holding fixture and tested at a rate of 15 mm per min. The force exerted at the moment of separation between the tubing and the barb was recorded. Finally, the sealed encapsulation pouch was connected to the oxygen outlet of an iEOG. All tubing connections were assembled using a custom metal barb connector (Giner Inc.), and the junction was reinforced using CA-MG Infinity Bond™ cyanoacrylate adhesive (Infinity Bond).
Assessment of cell viability
The viability assessment of pancreatic islets and INS-1 cell aggregates was conducted using a dual fluorescent staining technique. Live cells were stained with acridine orange (AO; Sigma Aldrich), while dead cells were stained with ethidium homodimer-1 (EthD-1, ThermoFisher Scientific), following the provided instructions from the supplier. AO/EthD-1 were diluted in RPMI-1640 medium supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, and 100 IU/mL penicillin and 100 μg/mL streptomycin and incubated with islets or cell aggregates for 1 h. The fluorescent images were captured using a digital inverted fluorescent microscope (InvitrogenTM EVOSTM FL, Fisher Scientific).
Static glucose-stimulated insulin secretion (GSIS)
The functionality of encapsulated pancreatic islets was assessed through a static GSIS assay. Cell encapsulation pouches from three groups—the positive control group (20% oxygen), the negative control group (1% oxygen, without oxygen supplementation), and the experimental group (1% oxygen, with oxygen supplemented from iEOGs)—were subjected to triple rinsing with Krebs-Ringer bicarbonate buffer (KRBB) and preconditioned in a 2.8 mM glucose solution for one hour at 37 oC. Subsequently, these pouches were sequentially exposed to a low glucose solution (2.8 mM), followed by high glucose solutions (16.7 mM or 28 mM), and then returned to a low glucose solution (2.8 mM) under 1% O2 culture condition, with each exposure period lasting one hour. Insulin concentrations in the supernatants were quantified using an enzyme-linked immunosorbent assay (ELISA) kit (Millipore Sigma). For islets encapsulated in alginate segments retrieved from animal models, the total IEQ count in the segments was determined to normalize the insulin secretion levels.
Animal study
Ethics
All animal procedures were approved by the Cornell Institutional Animal Care and Use Committee (IACUC) and supervised by the Cornell Center for Animal Resources and Education (CARE) staffs.
Animal model
Lewis rats (200–400 g; Inotiv Inc) were used as pancreatic islet donors while SD rats (380–420 g; Charles River) were used as recipients. The rats were housed within an environmentally regulated room at the Weill Hall Vivarium Animal Facility, following a standard 12-h dark/light cycle. Daily welfare monitoring was conducted, with specific indicators including marked lethargy, hypothermia, severe dehydration, ataxia, hunched posture, labored breathing, tiptoe or slow ponderous gait, infection at the implant site, and wound dehiscence. Loss of body weight ≥20% compared to baseline and BCS (Body Condition Score) < 2 established as a humane endpoint. Several experiments on iEOG performance were conducted at CBSET (Lexington, MA) on behalf of Giner Inc under IACUC approvals and similar daily monitoring.
Customized neoprene jackets were supplied by Lomir Biomedical Inc. These jackets were made with exposed flanks and an open abdominal area, a deliberate design to prevent discomfort during extended experiments. The backpack segment was seamlessly integrated into the jacket using Velcro, facilitating easy wearing and removal of the jackets. Rats were acclimated to these jackets for a period of 3–14 days before undergoing surgery. Rats wearing jackets were housed individually to prevent potential harm from cage mates. Objects in the cage that could entangle the jackets, such as nest boxes, were removed to avoid incidents.
Diabetes was chemically induced in SD rats by intraperitoneal administration of pharmaceutical-grade streptozotocin (STZ; Zanosar, Sicor Pharmaceutics). Initially, a freshly prepared STZ solution in 100 mM citrate buffer (pH 4.5) was injected into healthy rats (40 mg/kg), followed by a second dose (20 mg/kg) after 5 days. Blood samples were collected daily from the tail vein starting on day 7 after the first STZ dose to confirm diabetes. Rats with at least seven successive blood glucose measurements exceeding 450 mg/dL were considered diabetic and underwent the implantation procedure.
Implantation of cell encapsulation pouch
To prepare for cell encapsulation pouch implantation, anesthesia was induced in rats using 4% isoflurane in oxygen and maintained at 2% throughout the surgery. The fur in the right flank was removed using Nair™ cream (Church & Dwight Company Inc), and the surgical field was asepticized with povidone-iodine cotton swabsticks (Dynarex Corp). Then, a 2-cm skin incision was made above the latissimus dorsi muscle. A 5-cm depth subcutaneous pocket was generated parallelly with the ribs using blunt-tip scissors to make space for the implantation of the BEAM system. The cell encapsulation pouch was then introduced into the pocket. A medical-grade polyester tissue cuff (Surgical MeshTM) was integrated where the oxygen transportation tubing exited from the incision to promote healing and prevent infection. The incision was closed and secured to the transportation tubing using a cruciate suture technique. The transportation tubing was finally connected to an iEOG enclosed in a water-refillable chamber placed in the pocket secured on the back of the rat by a customized jacket. The water was replenished on a weekly basis. The overall weight of the system including internal and external components was under 10% of rat body weight.
Post-implantation assessments
Non-fasting blood glucose (NBG) measurement
NBG levels in rats undergoing implantation were assessed at regular intervals using a Contour Next EZ blood glucose monitoring system (Bayer) following BEAM implantation. Graft failure was defined as NBG levels consistently exceeding 250 mg/dL for three consecutive days.
Intraperitoneal glucose tolerance test (IPGTT)
The metabolic function of transplanted islets in rats was evaluated through an IPGTT conducted on day 30 post-transplantation. After an 18-h fasting period, rats received an intraperitoneal bolus injection of 50% (w/v) dextrose solution (Phoenix Pharmaceutical Inc) at a dosage of 2.0 g/kg. Blood glucose levels were measured at 0, 15, 30, 45, 60, 90, and 120 min following the dextrose injection. Blood samples were collected from the tail vein at 0 and 90 min, allowed to equilibrate at room temperature for 30 min, and then centrifuged at 2000 × g for 10 min at 4 °C to remove clotted material. The serum obtained from the supernatant was stored at –80 °C. Serum C-peptide concentrations were quantified using a Rat C-peptide ELISA kit (ALPCO).
Assessment of iEOG function on animals
An electronic box (3.5 cm × 3.5 cm × 1.2 cm), containing a battery pack and an electronic controller, was developed by Giner Inc. to regulate the electrolysis and thereby control the oxygen production rate. The system was designed to record data on the function of the electrolyzer and battery levels at five-minute intervals. The box was equipped with a built-in infrared (IR) transmitter for communication with an IR receiver connected to a laptop, facilitating touchless monitoring of iEOG functionality from outside the animal cages. In addition, a light-emitting diode (LED) was incorporated into the system to indicate the status of oxygen production, where green indicates normal production rates and orange signals abnormal production rates. The functionality of iEOGs was assessed twice daily, and data from the electronic box were reviewed every four days to detect any malfunctions related to the electrolysis. The battery was recharged every 4–5 days to maintain continuous operation. Oxygen cessation lasting more than 24 h was considered the endpoint of an experiment. In such cases, the cell encapsulation pouches were retrieved within 48 h of the detected oxygen cessation.
Ex vivo assessment of encapsulated pancreatic islets post-implantation
The islet-laden alginate hydrogel layer was carefully removed from the retrieved encapsulation pouch and segmented into three portions for subsequent analyses: viability assessment using AO/EthD-1 staining, functionality assessment via GSIS assay, and histological examination. For histological evaluation, the alginate segments were fixed in 10% neutral-buffered formalin solution (Sigma-Aldrich) for 24 h. To maintain the structural integrity, the hydrogel layer was subsequently embedded in Histogel™ (Thermo Scientific). Samples were then dehydrated, embedded in paraffin (Paraplast, Leica Biosystems), and sectioned at a thickness of 5 µm using a MCT25 manual microtome (Rankin Biomedical). H&E staining was performed utilizing a commercial staining kit (Sigma) in accordance with the manufacturer’s protocol. For immunofluorescence staining, tissue sections were deparaffinized and rehydrated through sequential immersion in xylene, followed by graded ethanol solutions (90%, 75%, 50%, 25%) and distilled water. Antigen retrieval was conducted using a pH 6.0 citrate buffer at boiling temperature, as previously described77. Sections were blocked in PBS containing 1% (w/v) bovine serum albumin (BSA) and 10% (v/v) donkey serum for 2 h at room temperature to prevent non-specific binding. Subsequently, sections were incubated overnight at 4 °C with primary antibodies: rabbit anti-insulin (1:200; ab210560; Abcam) and mouse anti-glucagon (1:200; ab10988; Abcam). Following this, slides were washed five times with PBS and incubated for 1 h at room temperature with secondary antibodies: Alexa Fluor 594-labeled donkey anti-rabbit IgG (1:400; R37119, Invitrogen) and Alexa Fluor 488-labeled donkey anti-mouse IgG (1:400; A-21202, Invitrogen). After five additional washes with PBS, the slides were mounted using Fluoroshield™ containing 4′,6-diamidino-2-phenylindole (DAPI) (Sigma). Immunofluorescence images were captured using an APEXVIEW APX100 microscope (Olympus).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 10. Data are presented as individual values, mean ± standard deviation (SD), standard error of the mean (SEM), or box-and-whisker plots, as specified in the figure captions. The Shapiro-Wilk test and F-test were used to assess the normality and variance equality of the data. Statistical significance was determined at a 95% confidence level, with a p value of less than 0.05 considered significant. For comparisons between two groups, unpaired t tests were conducted for normally distributed data; otherwise, the Mann–Whitney U test was performed. For comparisons involving more than two groups, one-way ANOVA was utilized. All figures were generated using GraphPad Prism 10, with confidence intervals displayed for relevant data points.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analyzed datasets generated during the study are available for research purposes from the corresponding authors on request.
References
Dagogo-Jack, S. Hypoglycemia in type 1 diabetes mellitus: Pathophysiology and prevention. Treat. Endocrinol. 3, 91–103 (2004).
Pedersen-Bjergaard, U. et al. Severe hypoglycaemia in 1076 adult patients with type 1 diabetes: Influence of risk markers and selection. Diab Metab. Res. Rev. 20, 479–486 (2004).
Cooper, M. N., de Klerk, N. H., Jones, T. W. & Davis, E. A. Clinical and demographic risk factors associated with mortality during early adulthood in a population-based cohort of childhood-onset type 1 diabetes. Diabet. Med. 31, 1550–1558 (2014).
Desai, T. & Shea, L. D. Advances in islet encapsulation technologies. Nat. Rev. Drug Discov. 16, 338–350 (2017).
Chetboun, M. et al. Association between primary graft function and 5-year outcomes of islet allogeneic transplantation in type 1 diabetes: a retrospective, multicentre, observational cohort study in 1210 patients from the Collaborative Islet Transplant Registry. Lancet Diab. Endocrinol. 11, 391–401 (2023).
Hering, B. J. et al. Factors associated with favourable 5 year outcomes in islet transplant alone recipients with type 1 diabetes complicated by severe hypoglycaemia in the Collaborative Islet Transplant Registry. Diabetologia 66, 163–173 (2023).
Ryan, E. A., Paty, B. W., Senior, P. A. & Shapiro, A. M. Risks and side effects of islet transplantation. Curr. Diab Rep. 4, 304–309 (2004).
Colton, C. K. Oxygen supply to encapsulated therapeutic cells. Adv. Drug Deliv. Rev. 67-68, 93–110 (2014).
Carlsson, P. O., Palm, F., Andersson, A. & Liss, P. Markedly decreased oxygen tension in transplanted rat pancreatic islets irrespective of the implantation site. Diabetes 50, 489–495 (2001).
Avgoustiniatos, E. S. Oxygen diffusion limitations in pancreatic islet culture and immunoisolation. Thesis (Ph.D.). Massachusetts Institute of Technology (2002).
Avgoustiniatos, E. S. et al. Measurements of the effective diffusion coefficient of oxygen in pancreatic islets. Ind. Eng. Chem. Res. 46, 6157–6163 (2007).
Suzanne, L. A. Eliminating oxygen supply limitations for transplanted microencapsulated islets in the treatment of type 1 diabetes. Thesis (Ph.D.). Massachusetts Institute of Technology (2008).
Dionne, K. E., Colton, C. K. & Yarmush, M. L. Effect of hypoxia on insulin secretion by isolated rat and canine islets of Langerhans. Diabetes 42, 12–21 (1993).
Cantley, J. et al. A preexistent hypoxic gene signature predicts impaired islet graft function and glucose homeostasis. Cell Transpl. 22, 2147–2159 (2013).
Kumosa, L. S. et al. Permeability of subcutaneous tissues surrounding long-term implants to oxygen. Biomaterials 35, 8287–8296 (2014).
Pedraza, E. et al. Preventing hypoxia-induced cell death in beta cells and islets via hydrolytically activated, oxygen-generating biomaterials. Proc. Natl. Acad. Sci. USA 109, 4245–4250 (2012).
Coronel, M. M., Geusz, R. & Stabler, C. L. Mitigating hypoxic stress on pancreatic islets via in situ oxygen generating biomaterial. Biomaterials 129, 139–151 (2017).
Coronel, M. M., Liang, J. P., Li, Y. & Stabler, C. L. Oxygen generating biomaterial improves the function and efficacy of beta cells within a macroencapsulation device. Biomaterials 210, 1–11 (2019).
Gholipourmalekabadi, M. et al. Oxygen-generating biomaterials: A new, viable paradigm for tissue engineering?. Trends Biotechnol. 34, 1010–1021 (2016).
Farris, A. L., Rindone, A. N. & Grayson, W. L. Oxygen delivering biomaterials for tissue engineering. J. Mater. Chem. B 4, 3422–3432 (2016).
de Vos, P. & Marchetti, P. Encapsulation of pancreatic islets for transplantation in diabetes: the untouchable islets. Trends Mol. Med. 8, 363–366 (2002).
Liang, J. P. et al. Engineering a macroporous oxygen-generating scaffold for enhancing islet cell transplantation within an extrahepatic site. Acta Biomater. 130, 268–280 (2021).
Ward, C. L. et al. Oxygen generating biomaterials preserve skeletal muscle homeostasis under hypoxic and ischemic conditions. PLoS One 8, e72485 (2013).
Harrison, B. S. et al. Oxygen producing biomaterials for tissue regeneration. Biomaterials 28, 4628–4634 (2007).
Wang, L.-H. et al. An inverse-breathing encapsulation system for cell delivery. Sci. Adv. 7, eabd5835 (2021).
Colton, C. K. Implantable biohybrid artificial organs. Cell Transpl. 4, 415–436 (1995).
Avgoustiniatos, E. S. & Colton, C. K. Effect of external oxygen mass transfer resistances on viability of immunoisolated tissue. Ann. N. Y Acad. Sci. 831, 145–167 (1997).
Papas, K. K., De Leon, H., Suszynski, T. M. & Johnson, R. C. Oxygenation strategies for encapsulated islet and beta cell transplants. Adv. Drug Deliv. Rev. 139, 139–156 (2019).
Schwenk Melissa, N. & Stone Simon G. Combined electrical lead and gas port terminals and electrolytic gas generator comprising same. U.S. Patent No. 11773496B2 (2023).
Tempelman, L., Stone S. & Papas, K. System for gas treatment of a cell implant. Giner Inc. U.S. Patent No. 10231817B2 (2019).
Ricordi, C. et al. National Institutes of Health-Sponsored Clinical Islet Transplantation Consortium Phase 3 Trial: Manufacture of a Complex Cellular Product at Eight Processing Facilities. Diabetes 65, 3418–3428 (2016).
Owyang, S., Jastrzebska-Perfect, P., Scott, M. & Traverso, G. Re-considering quantity requirements in islet transplantation. Nat. Rev. Bioeng. 1, 382–384 (2023).
Capuani, S., Malgir, G., Chua, C. Y. X. & Grattoni, A. Advanced strategies to thwart foreign body response to implantable devices. Bioeng. Transl. Med. 7, e10300 (2022).
Kämmerling, L. et al. Mitigating the foreign body response through ‘immune-instructive’ biomaterials. J. Immunol. Regenerative Med. 12, 100040 (2021).
Ni, Y. et al. Macrophages modulate stiffness-related foreign body responses through plasma membrane deformation. Proc. Natl. Acad. Sci. 120, e2213837120 (2023).
Carnicer-Lombarte, A., Chen S.-T., Malliaras, G.G., & Barone, D.G. Foreign body reaction to implanted biomaterials and its impact in nerve neuroprosthetics. Front Bioeng. Biotechnol. 9 (2021).
Noskovicova, N. et al. Suppression of the fibrotic encapsulation of silicone implants by inhibiting the mechanical activation of pro-fibrotic TGF-β. Nat. Biomed. Eng. 5, 1437–1456 (2021).
Rolfe, B. et al. The fibrotic response to implanted biomaterials: Implications for tissue engineering. 2011: Citeseer.
Klopfleisch, R. & Jung, F. The pathology of the foreign body reaction against biomaterials. J. Biomed. Mater. Res A 105, 927–940 (2017).
Thanos, C. G., Gaglia J. L., & Pagliuca F. W. Considerations for Successful Encapsulated β-Cell Therapy, in Cell Therapy: Current Status and Future Directions, D. F. Emerich and G. Orive, Editors. 2017, Springer International Publishing: Cham. p. 19-52.
Liu, Q. et al. A Zwitterionic polyurethane nanoporous device with low foreign-body response for islet encapsulation. Adv. Mater. 33, e2102852 (2021).
Liu, W. et al. A safe, fibrosis-mitigating, and scalable encapsulation device supports long-term function of insulin-producing cells. Small 18, e2104899 (2022).
Einstein, S. A. et al. Hypoxia within subcutaneously implanted macroencapsulation devices limits the viability and functionality of densely loaded islets. Front Transpl. 2, 1257029 (2023).
Najdahmadi, A. et al. Non-invasive monitoring of oxygen tension and oxygen transport inside subcutaneous devices after H2S treatment. Cell Transplant. 29, 0963689719893936 (2020).
Mitsugashira, H. et al. Development of a novel method for measuring tissue oxygen pressure to improve the hypoxic condition in subcutaneous islet transplantation. Sci. Rep. 12, 14731 (2022).
An, D. et al. Designing a retrievable and scalable cell encapsulation device for potential treatment of type 1 diabetes. Proc. Natl. Acad. Sci. 115, E263–E272 (2018).
Bose, S. et al. A retrievable implant for the long-term encapsulation and survival of therapeutic xenogeneic cells. Nat. Biomed. Eng. 4, 814–826 (2020).
Grattoni, A. et al. Harnessing cellular therapeutics for type 1 diabetes mellitus: progress, challenges, and the road ahead. Nat. Rev. Endocrinol. 21, 14–30 (2025).
Chendke, G. S. et al. Replenishable prevascularized cell encapsulation devices increase graft survival and function in the subcutaneous space. Bioeng. Transl. Med. 8, e10520 (2023).
Wang, X. et al. A nanofibrous encapsulation device for safe delivery of insulin-producing cells to treat type 1 diabetes. Sci. Transl. Med. 13, eabb4601 (2021).
Nyitray, C. E. et al. Polycaprolactone thin-film micro- and nanoporous cell-encapsulation devices. ACS Nano 9, 5675–5682 (2015).
Veiseh, O. et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nat. Mater. 14, 643–651 (2015).
Vegas, A. J. et al. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat. Biotechnol. 34, 345–352 (2016).
Mukherjee, S. et al. Screening hydrogels for antifibrotic properties by implanting cellularly barcoded alginates in mice and a non-human primate. Nat. Biomed. Eng. 7, 867–886 (2023).
Papas, K. K., Avgoustiniatos, E. S. & Suszynski, T. M. Effect of oxygen supply on the size of implantable islet-containing encapsulation devices. Panminerva Med. 58, 72–77 (2016).
Keymeulen, B. et al. Encapsulated stem cell–derived β cells exert glucose control in patients with type 1 diabetes. Nat. Biotechnol. 42, 1507–1514 (2024).
Ernst, A. U. et al. A predictive computational platform for optimizing the design of bioartificial pancreas devices. Nat. Commun. 13, 6031 (2022).
Lee, I. et al. Electrocatalytic on-site oxygenation for transplanted cell-based-therapies. Nat. Commun. 14, 7019 (2023).
Krishnan, S. R. et al. A wireless, battery-free device enables oxygen generation and immune protection of therapeutic xenotransplants in vivo. Proc. Natl. Acad. Sci. 120, e2311707120 (2023).
Lehmann, R. et al. Superiority of small islets in human islet transplantation. Diabetes 56, 594–603 (2007).
Farhat, B. et al. Small human islets comprised of more β-cells with higher insulin content than large islets. Islets 5, 87–94 (2013).
Pya, Y. et al. First human use of a wireless coplanar energy transfer coupled with a continuous-flow left ventricular assist device. J. Heart Lung Transplant. 38, 339–343 (2019).
Slaughter, M. S. & Myers, T. J. Transcutaneous energy transmission for mechanical circulatory support systems: History, current status, and future prospects. J. Card. Surg. 25, 484–489 (2010).
Dissanayake, T. et al. Transcutaneous Energy Transfer System for Powering Implantable Biomedical Devices. in 13th International Conference on Biomedical Engineering. 2009. Berlin, Heidelberg: Springer Berlin Heidelberg.
Dual, S. A., Cowger, J., Roche, E. & Nayak, A. The future of durable mechanical circulatory support: Emerging technological innovations and considerations to enable evolution of the field. J. Card. Fail. 30, 596–609 (2024).
Campi, T. et al. Wireless power transmission for left ventricular assist devices: advancements, challenges, and future directions. in 2024 IEEE Wireless Power Technology Conference and Expo (WPTCE). 2024.
Ohsawa, I. et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 13, 688–694 (2007).
Ge, L. et al. Molecular hydrogen: a preventive and therapeutic medical gas for various diseases. Oncotarget 8, 102653–102673 (2017).
Wu, Y. et al. Hydrogen Gas from Inflammation Treatment to Cancer Therapy. ACS Nano 13, 8505–8511 (2019).
Saengsin, K., Sittiwangkul, R., Chattipakorn, S. C. & Chattipakorn, N. Hydrogen therapy as a potential therapeutic intervention in heart disease: from the past evidence to future application. Cell. Mol. Life Sci. 80, 174 (2023).
Tamura, T. et al. Efficacy of inhaled hydrogen on neurological outcome following brain ischaemia during post-cardiac arrest care (HYBRID II): A multi-centre, randomised, double-blind, placebo-controlled trial. EClinicalMedicine 58, 101907 (2023).
Zhu, W. et al. Accurate in vivo real-time determination of the hydrogen concentration in different tissues of mice after hydrogen inhalation. Heliyon 8, e10778 (2022).
Simon G. Stone, Linda A. Tempelman & Melissa Schwenk. Self-regulating electrolytic gas generator and implant system comprising the same. U.S. Patent No. 2018135948-A1 (2020).
Simon G. Stone. System and method for controlling oxygen delivery to implanted cells. U.S. Patent No. 12090300B2 (2024).
Pham, T. T. et al. Surface-triggered in situ gelation for tunable conformal hydrogel coating of therapeutic cells and biomedical devices. Adv. Funct. Mater. 31, 2010169 (2021).
Nguyen, T. T. et al. Engineering “cell-particle hybrids” of pancreatic islets and bioadhesive FK506-loaded polymeric microspheres for local immunomodulation in xenogeneic islet transplantation. Biomaterials 221, 119415 (2019).
Tran, P. L. et al. Prolongation of graft survival via layer-by-layer assembly of collagen and immunosuppressive particles on pancreatic islets. Biomaterials 290, 121804 (2022).
Acknowledgements
We acknowledge the contributions of the staff at the Cornell Center for Animal Resources and Education (CARE) for their expertise in husbandry, clinical care, and suggestions regarding improvements to the animal jackets. We extend our thanks to Giner Inc. for supplying iEOGs and electrical instruments and acknowledge contributions by Giner team members including Thomas Regan, Melissa Schwenk, Griffin Marella, Yamini Mohan, and consultant James Littlefield working with authors Dr. Tempelman and Mr. Stone. We thank Kecheng Wang at Cornell University for the assistance in capturing histological images. We extend thanks to Dr. Chengyang Liu at the Human Islet Core, University of Pennsylvania, for providing human pancreatic islets. We also thank the Histology Lab at the Cornell Animal Health Diagnostic Center for their assistance in processing and preparing histological sections. This work was partially supported by the National Institutes of Health (NIH, 1R01DK105967-01A1; R43/R44DK113536), the Hartwell Foundation, Breakthrough T1D (JDRF, 2-IND-2021-1072-I-X), and Novo Nordisk Company.
Author information
Authors and Affiliations
Contributions
T.T.P., P.L.T., L.A.T., and M.M. conceived and designed the project. T.T.P., P.L.T., and A.L. prepared the cell encapsulation pouch. T.T.P. and P.L.T. conducted all in vitro and in vivo cellular studies. P.L.T. performed imaging analysis, ELISA, and tensile test. C.P. measured the oxygen flow rate and captured histological images. S.S. and L.A.T. designed, supervised and/or conducted iEOG in vitro and in vivo studies. T.T.P. and M.M. wrote the manuscript. M.M. and L.A.T. supervised the project. J.A.F. discussed the results and provided suggestions on animal surgery. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
T.T.P., P.L.T., L.A.T., J.A.F., S.S., and M.M. are inventors of the technology described in this paper. T.T.P. and S.G.S. are advisors; L.A.T., J.A.F., and M.M. are co-founders of Persista Bio Inc., a company formed to commercialize the technology.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pham, T.T., Tran, P.L., Tempelman, L.A. et al. A continuously oxygenated macroencapsulation system enables high-density packing and delivery of insulin-secreting cells. Nat Commun 16, 7199 (2025). https://doi.org/10.1038/s41467-025-62271-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62271-2
