
Abstract
The metabolic flexibility of tissues determines the degree and reversibility of organ damage during inflammatory challenges. However, effective treatments for myocardial metabolic dysfunction in septic cardiomyopathy (SCM) are unavailable. Nicotinamide adenine dinucleotide-dependent signaling is fundamental to cellular metabolic homeostasis and inflammatory responses. Here, using male mice models, we reveal that both genetic and pharmacological inhibition of mono-ADP-ribosyl hydrolase MacroD1 which is predominantly enriched in cardiomyocytes alleviates myocardial metabolic impairment, inflammation, dysfunction, and the risk of mortality caused by lipopolysaccharide and cecal ligation and puncture. Mechanistically, MacroD1 selectively modulates the activity of mitochondrial complex I (MCI), which is particularly vulnerable at the early stages of sepsis. Inhibition of MacroD1 preserves MCI activity and bioenergetic reserves of cardiomyocytes by enhancing mono-ADP-ribosylation of Ndufb9 protein, thereby mitigating sepsis-induced myocardial pyroptosis and dysfunction. These preclinical results indicate that MacroD1 dictates cardiac tolerance to sepsis by configuring MCI-coupled bioenergetic reserve and cardiomyocyte pyroptosis.
Similar content being viewed by others


Introduction
Septic cardiomyopathy (SCM) is a common manifestation of sepsis that affects the heart, resulting in myocardial depression. Approximately 40% of sepsis patients admitted to intensive care units are at risk of developing SCM, which complicates hemodynamic evaluation during intensive treatment. The mortality in patients with SCM is up to 70–90% despite adequate management of infection, and even if survived, approximately 5.5% of them are rehospitalized due to heart failure1,2. No consensus guidelines or specific therapies are available for the management of SCM.
The development of SCM involves a complex interplay among microbial toxins, inflammatory mediators, mitochondrial dysfunction, and other factors. Histopathological studies have reported inflammation infiltrates, interstitial and intracellular edema, and intracardiomycocyte lipid accumulation3,4 without significant cardiomyocyte death and sarcomeric structural damage5 in the myocardium of septic patients and animal models. High diffusion of TNF-α expression across different types of cardiac cells and the breakdown of almost all bioenergetic pathways highlight the essence of myocardial inflammometabolic disorders3,6,7. Despite the elevated circulating levels in septic patients, inflammatory cytokines are irrelevant to cardiac dysfunction8. Treatments with TNF-neutralizing antibodies, soluble TNF receptors, Toll-like receptor-4 antibodies, or IL-1 receptor antagonists could not achieve the desired effects8,9,10. In contrast, myocardial bioenergetic status links the intensity of inflammatory insult to contractile working status11. These scenarios support the idea of concurrent management of myocardial metabolism for SCM besides anti-inflammation.
A wide range of myocardial metabolic abnormalities exists in SCM7, including impaired mitochondrial oxidative utilization of fatty acids and lipid accumulation in the myocardium. Glycolysis, glucose oxidation, glycogenolysis, ketone body degradation, and uptake of essential and branched-chain amino acids are also depressed. Mitochondrial dysfunction emerges as a critical mechanism for sepsis-induced multiple organ failure. Functional traits of mitochondria exhibit heterogeneous responses across different types of cells and organs in diseases, including sepsis12. For example, the depressed activity of mitochondrial complex I (MCI), which is most vulnerable to early sepsis13, favors hepatocytes against fulminant endotoxemic injury14 but disfavors other multiple types of cells15. Therefore, identifying cardiomyocyte-specific mediators programming myocardial metabolism against sepsis remains imperative to achieving safe therapy for SCM.
The core roles of nicotinamide adenine dinucleotide (NAD+) in cell or organ homeostasis have been receiving particular attention, i.e., driving circadian physiology of mitochondrial oxidative metabolism16, fueling inflammasome activation17,18, and linking mitochondrial homeostasis to signaling transduction19. Intracellular NAD+ is highly enriched in mitochondria19, where almost all NAD+-consuming signaling reactions occur20. The latest report suggests mitochondria as the sentinel of infections21 and aberrant NAD+ metabolism underlies a root cause of sepsis22. The heart is a mitochondria-enriched organ, and further investigation of cardiac-specific NAD+ signaling pathways for SCM is warranted.
Herein, we profiled and characterized cardiomyocyte-enriched NAD+-signaling reaction mediators linked to myocardial inflammometabolic disorders in sepsis. Mono-ADP-ribosyl hydrolyzing MacroD1 was preferentially expressed in striated muscle cells, including cardiomyocytes, across organs among all NAD+-signaling enzymes known. Its inhibition potentiated myocardial tolerance to sepsis mainly via Ndufb9 mono-ADP-ribosylation (MARylation), which enhanced MCI activity and consequent bioenergetic reserve to withstand NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome activation-coupled cardiomyocyte pyroptosis. MacroD1 blockade protected the heart against sepsis, promising specific prevention of SCM.
Results
Upregulation of cardiomyocyte-enriched MacroD1 links to SCM
To identify NAD+-signaling catalytic enzymes enriched in cardiomyocytes and responsive to septic challenges, we performed cardiac single-cell RNA-sequencing on 10-week-old mice with and without lipopolysaccharide (LPS) injection. Given the higher mortality rate in SCM patients during the first 24 h of sepsis, similarly, prominent cardiac dysfunction at 16–18 h in mice subjected to lipopolysaccharide (LPS) injection23, single-cell sequencing was conducted at 18 h, which enables monitoring of early metabolic and inflammatory responses of cardiomyocytes. The heatmap visualized gene expression profiles of different cell types (Supplementary Fig. 1), complementary to dimensionality reduction analysis with uniform manifold approximation and projection (UMAP) in Fig. 1A. Treatment with LPS alters the proportions of certain cell types with substantial changes observed in endothelial cells, fibroblasts, and immune cells (Fig. 1B, C). Among cell clusters identified, MacroD1 and SIRT3/4/5 are predominantly enriched in ~ 10% of cardiomyocyte compositions, with MacroD1 exhibiting the highest expression levels (Fig. 1D, E and Supplementary Fig. 2). Cardiomyocyte populations contained five distinct subclusters (Supplementary Fig. 3A, B), and LPS treatment induced the differential response of MacroD1 mRNA expression in cardiomyocyte subclusters, increasing in CM1 and decreasing in CM3 (Supplementary Fig. 3C–E). Figure 1F, G, and Supplementary Fig. 4 confirmed the preferential expression of MacroD1 in the heart across major organs.

A UMAP plot illustrating the clustering of distinct cardiac cell populations. B UMAP plot comparing the composition of cardiac cell types between control (CON) and lipopolysaccharide (LPS)-treated groups. C Bar graph representing the relative proportion of each cardiac cell type in the CON and LPS groups. D UMAP plot showing the density of MacroD1-positive cells across various cardiac cell types. E Dot plot displaying the expression levels of MacroD1 across multiple cell types in the CON and LPS groups. F, G Western blotting (WB) examination of MacroD1 protein in major organs of mice (n = 4). H Immunofluorescent staining to show the colocalization of MacroD1 (red) and TOM20 (green for mitochondria) in neonatal rat cardiomyocytes (NRCMs). The cell nucleus was counterstained with DAPI (blue). I WB of MacroD1 expression in cell fractions of NRCMs (Cyto: Cytoplasm without mitochondria; Mito: Mitochondria). J, K WB of MacroD1 expression in hearts of mice 18 h post-intraperitoneal injection of vehicle (Veh) or LPS (10 mg/kg) (n = 4). L–O WB, and quantitative analysis of MacroD1 proteins in NRCMs (L and M) or AC16 cells (N and O) after LPS stimulation (1 μg/ml) in indicated time points (n = 4 cell samples from independent experiments). Data are shown as the mean ± SD. Data analysis was conducted utilizing one-way ANOVA with Dunnett’s multiple comparisons test (G, M, O). Data analysis was conducted using a two-tailed unpaired Student t test (K). Source data are provided as a Source Data file.
We further investigated the subcellular localization and septic response dynamics of MacroD1 in hearts. Immunofluorescent imaging of neonatal rat cardiomyocytes (NRCMs) disclosed its mitochondrial distribution, indicated by overlapped MacroD1- and Tom20-positive signals (Fig. 1H). Analysis of cellular fractions of mouse myocardium confirmed its enrichment in mitochondria (Fig. 1I).
In response to LPS challenges for 18 hours, MacroD1 proteins were upregulated in the myocardium and mitochondrial fractions, whereas its mRNA level was unchanged (Fig. 1J, K and Supplementary Fig. 5A–C). This post-transcriptional regulation of the MacroD1 protein implied a swift cellular adaptation mechanism in response to sepsis. Along the same line, higher LPS concentrations (1 μg/ml) induced a time-dependent upregulation of MacroD1 protein in NRCMs and AC16 human cardiomyocytes instead of its mRNA, whereas lower concentrations (100 ng/ml) had not any effects (Fig. 1L–O and Supplementary Fig. 5D–K). This case suggested that the response of the MacroD1 protein in cardiomyocytes to endotoxin may be relevant to the severity of septic cardiac dysfunction. Overall, the responsive upregulation of MacroD1 protein enriched in cardiomyocyte mitochondria in septic challenge implies a role in cardiac bioenergetics and inflammatory reactions.
Knockout of cardiomyocyte MacroD1 gene enables myocardial tolerance to sepsis
To examine the roles of MacroD1 in SCM, we generated the mouse line with the tamoxifen-induced cardiomyocyte-specific knockout of the MacroD1 gene [MacroD1flox/flox/αMHCMerCreMer (cKO)] (Fig. 2A and Supplementary Fig. 6A–D). cKO mice and littermate controls exhibited comparable viability, cardiac function, and dimensions at baseline. Such conditional gene knockout did not affect MacroD1 proteins in quadriceps muscles compared to 10-week-old MacroD1flox/flox controls (Fig. 2B), highlighting the specificity of the genetic manipulation used.

A Schematic overview of the breeding strategy for creating mice with an inducible, heart-specific MacroD1 knockout. Mice carrying the αMHC-MCM+ MacroD1flox/flox genotype, and upon tamoxifen induction, served as the experimental group (cKO). MacroD1flox/flox littermate mice (Flox) constituted the control cohort. B Western blotting validation of the knockout of MacroD1 in left ventricular tissues and quadriceps muscle from 10-week-old mice. C Kaplan-Meier survival rates analysis of 10-week-old mice subjected to intraperitoneal injection of LPS (10 mg/kg) within 96 h (n = 15). D–F Representative echocardiography (D) and echocardiographic analysis (E, F) in mice 18 h after LPS or saline administration (LVEF: left ventricular ejection fraction; LVFS: left ventricular fractional shortening; n = 7 mice for Flox and cKO; n = 8 mice for Flox + LPS and cKO + LPS). G and H, Serum levels of lactate dehydrogenase (LDH) and cardiac troponin T (cTnT) in Flox and cKO mice injected intraperitoneally with/without LPS for 18 h (n = 5 mice for Flox and cKO; n = 7 mice for Flox + LPS and cKO + LPS). I–K Immunofluorescent staining and quantitative analysis of Myeloperoxidase (MPO; I and J) and α-smooth muscle actin (α-SMA; I and J), and MASSON staining and quantification (I, K) in heart sections. MPO staining was performed from mice 18 h after LPS injection, and α-SMA and MASSON stainings were performed from mice 96 hours after LPS injection (n = 5 for J; n = 6 for K). L Kaplan-Meier survival rates analysis of mice subjected to CLP surgery within 96 hours (n = 15). M–O Representative echocardiography (M) and echocardiographic analysis (N, O) in mice 24 h after CLP or sham surgery (n = 6). P, Q Serum levels of LDH and cTnT in mice 24 h after CLP surgery (n = 5). R, S MPO and α-SMA immunofluorescent staining and quantitative analysis of heart tissues. MPO and α-SMA staining was performed on mice 24 h and 96 h after CLP or sham surgery, respectively (n = 5). Data are shown as the mean ± SD. Data analysis was conducted utilizing one-way ANOVA with Tukey’s multiple comparison test. The log-rank test was employed to analyze survival data. Source data are provided as a Source Data file.
Four days after the LPS challenge, 10-week-old MacroD1flox/flox mice suffered approximately a 25% death rate, but all cKO animals treated with LPS survived (Fig. 2C). Despite comparable cardiac function at baseline, myocardial depression evident in MacroD1flox/flox mice 18 h post-injection of LPS was significantly counteracted by cKO, as demonstrated by echocardiographic evaluation of left ventricular ejection fraction, fractional shortening (Fig. 2D–F), and internal diameters (Supplementary Fig. 7A, B and Table 1). Also, the elevation of circulating LDH and cTnT in MacroD1flox/flox mice under LPS insults was depressed in cKO animals (Fig. 2G, H), implicating restrained myocardial damage.
Consistent with the previous report23, LPS stimulation induced a notable increase in myeloperoxidase (MPO)-positive neutrophils in MacroD1flox/flox hearts (Fig. 2I, J). This response was accompanied by collagen deposition and myofibroblast activation, evidenced by Masson’s trichrome staining and α-smooth muscle actin immunostaining (Fig. 2I–K). In contrast, cKO mitigated these pathological alterations. Notably, cKO did not induce cardiomyocyte apoptosis at baseline or affect LPS-induced apoptosis compared to MacroD1flox/flox controls (Supplementary Fig. 8A, B). Cardiomyocyte size in cKO mice was comparable to MacroD1flox/flox controls and insusceptible to LPS effects (Supplementary Fig. 8C, D). These findings suggest that MacroD1 mediates LPS-induced cardiac inflammation and fibrosis.
Cecal ligation and puncture (CLP) surgery was also performed to mimic bacteremia-induced cardiac dysfunction. As expected, survival rate (Fig. 2L) and cardiac function (Fig. 2M–O; Supplementary Fig. 9A, B and Table 2) were improved in cKO than in MacroD1flox/flox mice. Circulating levels of myocardial damage markers (Fig. 2P, Q) and intensity of myocardial inflammation infiltrate and myofibroblast activation (Fig. 2R, S) were lower in cKO mice, underscoring the importance of MacroD1 in SCM. Thus, the improved signs of myocardial pathology after septic challenges in the cKO mice suggest resistance to myocardial injury.
Preserved bioenergetic metabolism underpins cKO cardioprotection against sepsis
To explore the biological underpinnings for MacroD1 inhibition-afforded myocardial tolerance to sepsis, we conducted cardiac transcriptomics following 18-hour LPS treatment. Principal components analysis (PCA) analysis showcased the clear segregation of replicates, and unsupervised hierarchical clustering showed genotype-associated samples cluster, indicating the reliability of sequencing data (Supplementary Fig. 10A, B). KEGG pathway analysis revealed the significant alterations of genes involved in inflammation, cardiac function, and metabolism, including upregulation of the TNF-α and NF-κB signaling pathways, aligning with a pro-inflammatory profile, which supported our model’s biological relevance. Cardiac dysfunction-related pathways were downregulated in MacroD1flox/flox and cKO hearts following LPS stimulation, such as dilated cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy, and cKO counteracted the depression of the “Citrate Cycle” and “Carbon Metabolism” (Supplementary Fig. 11A–D). Heatmap, gene set enrichment analysis (GSEA), and quantitative PCR analyses confirmed that metabolic genes were downregulated to a lesser extent in cKO hearts (Fig. 3A–D). Strikingly, GDF15, a key mediator of tissue tolerance and metabolic adaptation24, was significantly upregulated in cKO hearts under the LPS challenge (Supplementary Fig. 12A, B). These findings indicate that enhanced cardiac metabolic resilience may underpin the protection of MacroD1 deletion against sepsis-induced cardiac dysfunction.

A Heatmap showing the relative expression of genes involved in the indicated metabolism pathway in the hearts of Flox and cKO mice injected intraperitoneally with/without LPS (10 mg/kg) for 18 h. B, C Gene set enrichment analysis (GSEA) of genes involved in indicated metabolism and oxidative phosphorylation pathways in the hearts of Flox and cKO mice 18 hours after LPS injection. D mRNA expression of genes involved in indicated metabolism pathways in the hearts of mice (n = 4). E, F Representative dihydroethidium (DHE) fluorescent images and quantification of hearts from Flox and cKO mice with and without LPS administration for 18 h (n = 6). G Myocardial ATP contents from Flox and cKO mice 18 hours after saline or LPS injection (n = 6). H Representative transmission electron microscopy images of hearts from mice 18 h after LPS administration. I, J Quantification of cristae number/mitochondria (I; n = 85, 88, 97, and 84 mitochondria were analyzed in the Flox, cKO, Flox + LPS, and cKO + LPS groups, respectively.) and mitochondrial area (J; n = 352, 362, 389, and 384 mitochondria were analyzed in the Flox, cKO, Flox + LPS, and cKO + LPS groups, respectively) from (H). Each group consisted of 5 heart samples from different animals, with five randomly selected electron microscopy fields per sample for analysis of mitochondria. n represents the number of mitochondria. The box boundaries represent the 25th (Q1) and 75th percentiles (Q3) with a central line at the median (50th percentile). The box height spans the interquartile range (IQR = Q3–Q1). Whiskers extend to the lowest/highest data points within Q1–1.5 × IQR and Q3 + 1.5 × IQR bounds, respectively. Minima and maxima correspond to actual extreme values within the whisker-defined range. Data are shown as the mean ± SD for (D–G). Flox: MacroD1flox/flox controls; cKO: MacroD1flox/flox/αMHCMerCreMer mice. Data analysis was conducted utilizing one-way ANOVA with Tukey’s multiple comparison test. Source data are provided as a Source Data file.
We further assessed the effects of MacroD1 on mitochondrial bioenergetics and oxidative stress. MacroD1flox/flox and cKO mice exhibited equivalent DHE fluorescence in hearts under basal conditions. LPS treatment induced a heightened DHE fluorescence in MacroD1flox/flox hearts but a milder increase in cKO, suggesting that MacroD1 inhibition limits cardiac oxidative stress by septic insults (Fig. 3E, F). Accordingly, the content of myocardial ATP was preserved in cKO mice (Fig.3G). Ultrastructural analysis revealed a significant accumulation of lipid droplets in MacroD1flox/flox hearts post-LPS treatment, indicative of compromised lipid catabolism. cKO depressed lipid droplet formation and elevated mitochondrial cristae density with comparable mitochondrial numbers and sizes to those in MacroD1flox/flox hearts, suggesting an enhanced metabolic adaptation to septic stress (Fig. 3H–J and Supplementary Fig. 13A–C). Collectively, the preservation of metabolic pathways, especially mitochondrial oxidative phosphorylation, is tightly linked to cKO-mediated myocardial tolerance to sepsis.
MacroD1 downregulation maintains the bioenergetic process under endotoxemic stress
To confirm the regulation of MacroD1 on mitochondrial functions, we assayed mitochondrial respiration capacity and superoxide production using NRCMs. As shown in Fig. 4A, B, mitochondrial ROS (mitoROS) level was comparable at baseline but depressed under LPS treatment in cardiomyocytes with MacroD1 knockdown (Supplementary Fig. 14A, B). Basal and maximal oxygen consumption rates and ATP-linked respiration of NRCMs depressed by LPS were improved when MacroD1 was downregulated, despite being similar at baseline (Fig. 4C–F). MacroD1 knockdown did not affect the spare respiration of NRCMs with and without LPS treatment (Fig. 4G). In sharp contrast to the scenarios with MacroD1 knockdown, an enhanced mitoROS generation was observed in NRCMs overexpressing MacroD1 in response to LPS insult (Supplementary Fig. 15A–D). Meanwhile, depression of basal and maximal oxygen consumption and ATP-linked and spare respiration deteriorated (Supplementary Fig. 15E–I).

Neonatal rat cardiomyocytes (NRCMs) transfected with MacroD1 SiRNAs (Si-M) or scrambled control (NC) for 36 h and then treated with and without LPS (1 μg/ml) for 18 h to examine mitochondrial function. A, B Representative fluorescence images and quantification of MitoSOX in NRCMs (n = 8). C Seahorse mitochondrial stress test was carried out in NRCMs (n = 8) (OCR: Oxygen consumption rate; Oligo: Oligomycin; Rot/AA: Rotenone/Antimycin). D–G Quantification of basal respiration (D), maximal respiration (E), ATP-linked respiration (F), and spare respiration capacity (G) from C (n = 8). H–K Measurements of oxygen consumption rate and corresponding indexes for seahorse fatty acid oxidation in NRCMs in the presence of bovine serum albumin (BSA) (n = 7) (ETO: Etomoxir, FAO inhibitor). L–O Oxygen consumption rate and corresponding indexes for seahorse fatty acid oxidation in NRCMs in the presence of BSA and palmitate (PAL) (n = 7). Data are shown as the mean ± SD. Data analysis was conducted utilizing one-way ANOVA with Tukey’s multiple comparison test. Source data are provided as a Source Data file.
Whether MacroD1 modulated fatty acid oxidation utilization in mitochondria was also evaluated by substrate-dependent oxygen consumption in NRCMs. In the presence of etomoxir but without palmitoylcarnitine, MacroD1 knockdown did not affect basal, maximal, and acute responsive respiration of NRCMs with LPS treatment compared to negative controls (NC) (Fig. 4H–K). When NRCMs were co-treated by palmitoylcarnitine, MacroD1 knockdown protected against basal and maximal oxygen consumption reduction compared to the NC under LPS treatment (Fig. 4L–O). Hence, MacroD1 downregulation decreases mitochondrial superoxide generation and preserves bioenergetic processes and fatty acid utilization in endotoxemic challenges.
MCI subunit Ndufb9 underpins bioenergetic preservation induced by cKO
We proceeded to figure out the critical mediators for MacroD1 inactivation-dependent bioenergetic metabolism. Using blue native polyacrylamide gel electrophoresis, we resolved that MacroD1flox/flox hearts had mitochondrial membrane protein complexes with comparable levels to cKO ones under basal conditions and an 18-hour LPS challenge (Fig. 5A and Supplementary Fig. 16A, B). Strikingly, cKO selectively elevated MCI activity at baseline and counteracted the depression of MCI and MCV induced by LPS (Fig. 5B–F). Overexpression of MacroD1 increased mitoROS and further depressed MCI activity and ATP production in AC16 human cardiomyocytes treated with LPS. Deletion of the Macro domain in MacroD1, opposite to MacroD1 overexpression, preserved the mitochondrial function of AC16 cells treated with or without LPS, implicating the importance of the Macro domain in MacroD1-dependent mitochondrial function (Fig. 5G–K).

A Blue native gel analysis of the expression of myocardial mitochondrial complexes in mice 18 hafter saline or LPS (10 mg/kg) injection. B–F Mitochondrial complexes activities in the hearts of mice 18 h after saline or LPS injection (n = 5 mice). G Schematic diagram of the sequence of MacroD1 and the MacroD1 mutant protein without the Macro domain (MacroD1 mut). H–K MitoSOX fluorescent staining (H, I; n = 5 cell samples from independent experiments), mitochondrial complex I activity (J; n = 4 cell samples from independent experiments), ATP levels (K; n = 5 cell samples from independent experiments) analysis in AC16 human cardiomyocytes transfected with plasmids of His-MacroD1, Flag-Ndufb9, or His-MacroD1 mut for 36 h and treated with and without LPS (1 μg/ml) for 18 h. L Venn diagram of MacroD1-binding proteins identified by antibody affinity proteomics, complex I-related proteins, and mono-ADP-ribosylated proteins immunocaptured by mono-ADP antibody. M Co-immunoprecipitation (Co-IP) for identifying endogenous interaction of MacroD1 and Ndufb9 in the hearts of mice. N Co-IP of MacroD1 and Ndufb9 in HEK293 cells transfected with His-MacroD1 and/or Flag-Ndufb9 plasmids for 48 hours. O His-MacroD1 or His-MacroD1 mutant with truncation of the Macro domain and Flag-Ndufb9 were co-expressed in HEK293 cells for Co-IP assay. Flox: MacroD1flox/flox controls; cKO: MacroD1flox/flox/αMHCMerCreMer mice. Data are shown as the mean ± SD. Data analysis was conducted utilizing one-way ANOVA with Tukey’s multiple comparison test. Source data are provided as a Source Data file.
Selective effects of MacroD1 on basal MCI activity prompted us to focus on identifying mitochondrial targets that are MARylated, especially for MCI-associated entities. MacroD1 antibody immunoprecipitated 572 proteins compared to IgG-based captures. A mono-ADP ribose antibody-based proteomics identified 261 proteins differentially enriched in cKO compared to MacroD1flox/flox hearts. By cross-referencing with the mouse MitoCarta3.0 inventory25, three proteins related to MCI function were identified (Fig. 5L and Supplementary Data 1), and only Ndufb9 showed MARylation. Physical interaction between MacroD1 and Ndufb9 was verified using respective antibodies- and epitopes-based immunoprecipitation in mouse hearts and HEK293 cells expressing the two recombinants (Fig. 5M, N). Experiments with truncated forms revealed that the Macro domain mediates the binding of MacroD1 to Ndufb9 (Fig. 5O).
Ndufb9 is an accessory assembly factor of MCI proton-pumping module MT-ND5, a rate-limiting step for overall electron transfer in MCI and supercomplex MCI/III/IV assembly26. Its interaction with MT-ND5 is required for this module’s stability. An increased binding of Ndufb9 to MT-ND5 was detected in cKO cardiomyocytes. This physical interaction between Ndufb9 and MT-ND5 was depressed in MacroD1flox/flox hearts by the LPS challenge but maintained in the cKO (Supplementary Fig. 17A, B), which may underlie MCI activity compromise during sepsis and its preservation by MacroD1 blocking.
Ndufb9 MARylation at R173 favors myocardial bioenergetics in sepsis
We further examined whether and how MacroD1 modulates Ndufb9 MARylation and function. At baseline, cKO did not change Ndufb9 protein expression but enhanced its MARylation. Upon LPS challenge, Ndufb9 MARylation was reduced in MacroD1flox/flox, whereas maintained in cKO hearts (Fig. 6A). Ndufb9 MARylation reduced by LPS and preserved by MacroD1 knockdown was reproduced in AC16 cells (Fig. 6B and Supplementary Fig. 18A, B). Overexpression of MacroD1 de-MARylated Ndufb9 compared to MacroD1 knockdown and NC groups, whereas truncated forms of MacroD1 did not (Fig. 6C).

A Co-IP assay for Ndufb9 MARylation in the hearts of mice injected intraperitoneally with and without LPS (10 mg/kg) for 18 h. B Co-IP assay for Ndufb9 MARylation in AC16 cells transfected with MacroD1 siRNA (Si-M) or scrambled control (NC) for 36 h and then treated or untreated with LPS (1 μg/ml) for 18 h. C Co-IP assay for Ndufb9 MARylation in AC16 cells transfected with Si-M, His-MacroD1, or His-MacroD1 mutant plasmids for 48 h. D Mass spectrum identification of the MARylation site of Ndufb9 based on mitochondrial lysates isolated from mice hearts with anti-MARylation antibody. E Sequence conservation display on the MARylation site of Ndufb9 from Homo sapiens, Mus musculus, and Rattus. The red dotted box indicates the MARylation site of Ndufb9. F MARylation site mutation display of the Ndufb9 sequence. The red dotted box indicates the MARylation site mutation (Ndufb9R173A). G Co-IP assay for Ndufb9 MARylation in AC16 cells transfected with Flag-Ndufb9 or Flag-Ndufb9R173A plasmid for 36 h and then treated with/without LPS (1 μg/ml) for 18 h. H Co-IP assay for Ndufb9 MARylation in AC16 cells transfected with the indicated siRNA or plasmids for 48 h. I–L Mitochondrial complex I activity (I; n = 5 cell samples from independent experiments), ATP levels (J; n = 5 cell samples from independent experiments), and MitoSOX fluorescent staining analysis (K, L; n = 5 cell samples from independent experiments) in AC16 cells transfected with indicated siRNA or plasmids for 36 hours and treated with/without LPS (1 μg/ml) for 18 h. The targeted sequence of the Ndufb9 plasmid corresponding to Si-Ndufb9 was synonymously mutated to avoid the interference of Si-Ndufb9 (Flag-SM-Ndufb9). Flox: MacroD1flox/flox controls; cKO: MacroD1flox/flox/αMHCMerCreMer mice. Data are shown as the mean ± SD. Data analysis was conducted utilizing one-way ANOVA with Tukey’s multiple comparison test. Source data are provided as a Source Data file.
Mono-ADP ribose antibody-based affinity proteomics identified MARylation on the arginine residues at position 173 of Ndufb9 (Fig. 6D), which possesses evolutionary conservation among humans, mice, and rats (Fig. 6E). Using a site-directed mutagenesis strategy with alanine in the place of arginine (Fig. 6F) in combination with immunoprecipitation, we found that MARylation of wild-type Ndufb9 proteins rather than mutant (Ndufb9R173A) was reduced in wild-type cells treated with LPS, confirming the site’s responsiveness (Fig. 6G). Opposite to the deMARylation response of wild-type Ndufb9, the mutant’s MARylation was weak regardless of MacroD1 overexpression (Fig. 6H). This evidence suggested R173 in Ndufb9 as a MARylation site responsive to the changes in MacroD1 protein under septic challenge.
To ascertain the function of Ndufb9’s R173 MARylation, we conducted experiments in AC16 cardiomyocytes. While MacroD1 was knocked down, MCI activity and ATP production were unaffected by LPS but depressed upon Ndufb9 downregulation (Fig. 6I, J and Supplementary Fig. 19A, B). Overexpression of a synonymous Ndufb9 mutant (SM-Ndufb9) resistant to interference sequences (Supplementary Fig. 20A, B) reversed the inhibitory effects by Ndufb9 knockdown in the presence of LPS. Still, a mutant with arginine 173 replaced by alanine (SM-Ndufb9R173A) reproduced the phenotype with Ndufb9 knockdown (Fig. 6I, J). In addition, given the role of MCI dysfunction in oxidative stress, we evaluated the effect of Ndufb9 MARylation on mitoROS. Ndufb9 knockdown compromised the resistance of MacroD1 downregulation to LPS-induced mitoROS production, which was rescued by overexpression of SM-Ndufb9 but not SM-Ndufb9R173A (Fig. 6K, L). Thus, R173 MARylation in Ndufb9 is crucial to MacroD1 inhibition-mediated mitochondrial homeostasis against septic insults.
cKO intercepts MCI dysfunction-coupled inflammatory damage of cardiomyocytes
Cellular bioenergetic status programs inflammatory response27, in which the inflammasome is a core player, and a key component NLRP3 mediates caspase-1 activation, gasdermin D (GSDMD) cleavage, the production and release of pro-inflammatory cytokines IL-1β/IL-18, and pyroptotic cell death28. We thus analyzed whether MacroD1-mediated mitochondrial metabolism affected NLRP3 inflammasome activation and pyroptosis of cardiomyocytes in sepsis.
The 18-hour LPS or 24 h CLP challenges induced a comparable increase of serum IL-1β and TNFα in MacroD1flox/flox and cKO mice (Supplementary Fig. 21A–D). However, depressed expression of the apoptosis-associated speck-like protein (ASC), NLRP3, IL1β, IL18, and TNFα mRNAs was observed and validated in cKO hearts exposed to LPS stress or CLP surgery (Fig. 7A–D and Supplementary Fig. 21E–G). Western blotting examinations revealed downregulation of pyroptotic mediators, cleaved caspase-1 and GSDMD, inflammasome components NLRP3 and ASC, and general pro-inflammatory cytokine TNFα proteins (Fig. 7E–H). Levels of ASC and NLRP3 proteins translocated to mitochondria, a critical step for NLRP3 inflammasome activation, were significantly lower in cKO mice (Fig. 7I–K). Immunoprecipitation in the myocardium and proximity ligation assay in isolated adult mice ventricular cardiomyocytes further showed reduced binding of ASC to NLRP3 proteins (Fig. 7L–N). Similar phenomena were reproduced in NRCMs subjected to LPS insult (Fig. 7O–S and Supplementary Fig. 22A–C), highlighting that MacroD1 regulates the priming and activation of NLRP3 inflammasome in cardiomyocytes during septic challenge.

A Heatmap showing the expression of NLRP3 inflammasome pathway-related genes in the hearts of mice injected intraperitoneally with/without LPS (10 mg/kg) for 18 h based on bulk RNA sequence analysis. B–D Expression of IL-1β, IL-18, and TNF-α mRNAs in the hearts of mice with/without LPS administration (n = 5). E–H Expression of NLRP3 inflammasome pathway-related proteins in the heart of mice 18 h after intraperitoneal LPS (10 mg/kg) injection (n = 4). I–K Expression of ASC and NLRP3 proteins in the cytoplasm (Cyto; I and J) and mitochondria (Mito; I and K) isolated from hearts of mice together with LPS administration (n = 4). L Co-IP assay for ASC-NLRP3 interaction in the hearts of mice, together with LPS administration. M, N Proximity Ligation Assay fluorescence detecting interaction between ASC and NLRP3 in isolated ventricular cardiomyocytes from adult mice treated or untreated with LPS (1 μg/ml) for 18 h (n = 60). Red dots show ASC-NLRP3 interaction. Dotted lines outline cardiomyocytes. O–R Expression of NLRP3 pathway-related proteins in neonatal rat cardiomyocytes transfected with Si-MacroD1 (Si-M) or scrambled control (NC) for 36 h and administered ATP (3 mM) 18 h after LPS (1 μg/ml) treatment for 2 h (n = 4). S Representative fluorescence images examining co-localization of ASC (green), NLRP3 (red), and DAPI (blue) staining in NRCMs transfected with Si-MacroD1 (Si-M) or scrambled control (NC) for 36 h and administered ATP (3 mM) 18 h after saline or LPS (1 μg/ml) treatment for 2 h. Flox: MacroD1flox/flox controls; cKO: MacroD1flox/flox/αMHCMerCreMer mice. The three lines in the violin plot represent the third quartile, median, and first quartile, respectively, while other data are shown as mean ± SD. Data analysis was conducted utilizing one-way ANOVA with Tukey’s multiple comparison test (B–D, N). Data analysis was conducted using a two-tailed unpaired Student t test (F, G, J, K, P–R). Source data are provided as a Source Data file.
Moreover, we interrogated whether MCI mediated MacroD1-dependent NLRP3 inflammasome activation. As shown in Supplementary Fig. 23A–E, MCI inhibition with rotenone abolished the protection of MacroD1 knockdown against mitoROS generation, ATP compromise, and increased expression of pro-inflammatory cytokines IL1β and TNFα induced by LPS treatment in NRCMs. Conversely, scavenging of mitoROS produced effects similar to MacroD1 inactivation. Adenovirus-mediated MacroD1 overexpression facilitated mitoROS generation, ATP breakdown, and expression of IL1β and TNFα under LPS stimulation, which was well counteracted by a mitochondria-targeted SOD mimetic mito-TEMPO (Supplementary Fig. 24A–E).
The NAD+/NADH ratio indicates intracellular redox balance. High NAD+ drives a faster metabolic rate, while high NADH leads to a slower one. MacroD1 inhibition significantly resisted LPS-induced reduction in NAD+ contents and NAD+/NADH ratio (Supplementary Fig. 25A, B), consistent with increased activity of MCI in which NADH was oxidized to NAD+ and for fueling bioenergy. MCI subunit Ndufb9 MARylation downregulated by LPS and enhanced by MacroD1 inhibition were NAD+ dependent because intracellular NAD+ depletion by NAMPT inhibitor FK866 abolished such phenomena (Supplementary Fig. 25C). Similar scenarios occurred in regulating MCI activity and inflammasome activation (Supplementary Fig. 25D–F). This evidence implicated that MacroD1 inhibition preserves intracellular redox balance by maintaining NAD+ levels and enhancing NADH oxidation, thereby counteracting LPS-induced metabolic and inflammatory anomalies in cardiomyocytes.
The existence of MacroD1 in the nuclear and cytoplasm of cardiomyocytes (Fig. 1I), albeit in small amounts, implicates a potential biological function. To examine whether non-mitochondrial MacroD1 involves MCI-coupled NLRP3 inflammasome activation, we constructed mutants truncating the mitochondrial targeting sequence in MacroD1 (MacroD1ΔMTS) (Supplementary Fig. 26A, B). Overexpression of MacroD1ΔMTS with a predominant cytoplasmic expression and the scarce in the nuclear in AC16 cardiomyocytes had almost no affection on MCI activity, mitoROS generation, and inflammatory response during LPS treatment, different from MacroD1 overexpression and knockdown (Supplementary Fig. 26C–G). Thus, cardioprotection of MacroD1 blocking against sepsis should most result from its regulatory effects on mitochondria.
Given that MCI inhibition-induced acetyl-CoA accumulation could facilitate mitochondrial protein acetylation and burden Sirt3, the most studied in organ metabolic diseases, including SCM29,30, the MacroD1-MCI signaling axis may involve Sirt3. We found that expression of Sirt3 protein was not susceptible to MacroD1 at baseline (Supplement Fig. 27A). Sirt3 knockdown did not affect Ndufb9 MARylation and MCI activity preserved by MacroD1 blocking (Supplement Fig. 27B, C) while enhanced mitochondrial ROS production and partially weakened anti-oxidative effects mediated by MacroD1 knockdown (Supplementary Fig. 27D, E), suggesting the differential modulation of MacroD1 and Sirt3 on oxidative stress in cardiomyocytes during sepsis. Together, mitochondrial MacroD1 modulates pyroptotic damage of cardiomyocytes via the MCI-mitoROS-NLRP3 inflammasome pathway during sepsis.
Therapeutic potential of MacroD1 blockade for SCM
To explore the potential of targeting MacroD1 to prevent SCM, we adopted the cell-permeable MRS2578 that inhibits MacroD1 activity31,32. In in vitro cultured neonatal mouse cardiomyocytes (NMCMs), Ndufb9 MARylation was significantly increased at baseline and preserved under co-treatment with MRS2578 and LPS (Supplementary Fig. 28A), confirming the effectiveness of the blocking compound. LPS-induced MCI activity compromise and mitoROS generation were depressed by MRS2578 treatment (Supplementary Fig. 28B–D). MRS2578 treatment, similar to MacroD1 knockdown, elevated oxygen consumption response to LPS (Supplementary Fig. 28E–I). Also, it depressed the priming and activation of NLRP3 inflammasome, as evaluated by quantitative PCR assay of IL-1β and TNFα mRNA expression (Supplementary Fig. 28J, K) and Western blotting test of NLRP3, ASC, and TNFα proteins and the cleavage of caspase-1 and GSDMD (Supplementary Fig. 28L–Q).
MRS2578 is also supposed to be a P2Y6 receptor antagonist, and we examined whether this receptor was involved in the Ndufb9/MCI/inflammasome pathway. Using the siRNA-based knockdown in NMCMs, we found that P2Y6 downregulation did not affect Ndufb9 MARylation, mitoROS production, and the expression of IL-1β, TNF-α, NLRP3, and ASC mRNAs in response to LPS insults (Supplement Fig. 29A–H). This case implied that P2Y6 did not mediate inflammometabolic reactions in cardiomyocytes under septic challenges, and the protective effects of MRS2578 should result from inhibition of MacroD1 rather than P2Y6.
We then conducted intraperitoneal injection of MRS2578 in mice, once 8 hours before and after the LPS challenge (Fig. 8A). No death event was observed 96 hours post-injection of LPS in mice receiving the compound, in contrast to an approximately 25% mortality rate in controls (Fig. 8B). Echocardiographic analysis demonstrated that MRS2578 did not affect left ventricular ejection fraction and fraction shortening at baseline and counteracted their depression induced by 18 h LPS stress (Fig. 8C–E and Supplementary Table 3). The restrain of elevated circulating cTnT and LDH and myocardial IL1β and TNFα mRNA expression induced by LPS indicated the prevention of MRS2578 against inflammatory myocardial damage (Fig. 8F, G and Supplementary Fig. 30A, B).

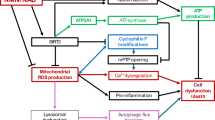
A Schematics for the application of the MacroD1 inhibitor in mice subjected to LPS challenge (MRS: MRS2578; Veh: vehicle; Echo: Echocardiography). B Kaplan-Meier survival analysis of wild-type mice with LPS and MRS2578 or vehicle administration (n = 15). C–E Representative echocardiography (C) and echocardiographic analysis (D and E) in mice (LVEF: left ventricular ejection fraction; LVFS: left ventricular fractional shortening; n = 6). F and G, Serum levels of LDH and cTnT in mice (n = 7). H–K Immunofluorescent analysis of Myeloperoxidase (MPO) (H and I; n = 7), dihydroethidium (DHE) (H and J; n = 7), and Masson’s staining analysis (H and K; n = 6) in the heart of mice. L ATP levels in mice hearts (n = 6). M Schematic illustration displaying the action modality by which MacroD1 modulates heart function in sepsis. Under septic insults, cardiomyocyte MacroD1 inhibits mitochondrial complex I activity via de-monoADP-ribosylating (de-MARylation) Ndufb9, leading to aggravating mitochondrial dysfunction, inflammasome activation-coupled cardiomyocyte pyroptosis, and cardiac dysfunction. In contrast, genetic and pharmacological inhibition of MacroD1 preserves cardiac function by enhancing Ndufb9 MARylation-dependent bioenergetic reserve. Data are shown as the mean ± SD. Data analysis was conducted utilizing one-way ANOVA with Tukey’s multiple comparison test. The log-rank test was employed to analyze survival data. Source data are provided as a Source Data file.
Histopathological analysis with MPO, DHE, and Masson’s stains further revealed alleviation of inflammatory infiltration, oxidative stress, and collagen deposition in the hearts of mice subjected to MRS2578 injection (Fig. 8H–K). Myocardial ATP was preserved (Fig. 8L) when MRS2578 maintained MCI and MCV activity under LPS treatment without affecting other mitochondrial complexes (Supplementary Fig. 31A–E). In addition, despite survivors with normal cardiac function and comparable myocardial oxidative states on day seven post-LPS injection in mice with and without MRS2578 injection, the weak myocardial fibrosis with the application of MRS2578 implied long-term cardiac benefits (Supplementary Fig. 32A–K and Table 4). Overall, the chemical blockade of MacroD1 holds therapeutic potential for preventing SCM (Fig. 8M).
Discussion
Identifying cardiac-specific mediators programming metabolic plasticity is a prerequisite for safely preventing SCM. The current study reveals an essential role of cardiomyocyte-enriched MacroD1 in modulating myocardial tolerance to sepsis. Conditional inactivation of the MacroD1 gene in cardiomyocytes enhances Ndufb9 MARylation, whereby MCI activity and bioenergetics reserve, restraining superoxide reactions, inflammasome activation-coupled myocardial damage, dysfunction, and mortality in sepsis. The pharmacological blockade of MacroD1 recapitulates the cardioprotective benefits of its genetic inactivation and offers a potential approach to preventing SCM.
Metabolic reprogramming of tissues is a crucial response to systemic inflammation and determines the degree and reversibility of organ damage33. Bioenergetic derangement directly links to septic multi-organ dysfunction34,35. Despite NAD+ metabolism being vital to sepsis, anti-inflammatory effects by blocking NAD+ biosynthesis or supplying NAD+ precursors36,37 complicate its roles in SCM. High doses of NAD(H)-loaded nanoparticles38 decreased septic mortality, being a lack of evaluating myocardial damage and function. Herein, we identified that NAD+ underpins the MacroD1/Ndufb9/MCI/inflammasome pathway in cardiomyocytes. In sepsis with myocardial NAD+ starvation, MacroD1 blocking preserves the NAD+/NADH ratio critical for cellular oxidative states and bioenergy, restraining inflammatory damage to cardiomyocytes. Notably, despite its prominent role in the myocardial metabolic disorder, MacroD1 may exert opposing modulation on diabetic cardiomyopathy, in which the PARP1-NAD+-SIRT3 axis is involved, but missing the direct evaluation of mitochondrial protein MARylation39. This discrepancy might partially result from the differential changes in myocardial metabolism patterns, i.e., the breakdown of almost all substrate metabolism in sepsis vs reduced myocardial glucose utilization in diabetes.
Besides effects on mitochondrial bioenergetics, MacroD1 inhibition counteracted sepsis-induced depression in fatty oxidation utilization that provides 60–90% of bioenergetics for normal cardiac function. This accounted for the reduced lipid accumulation in cKO hearts during sepsis. The restrain of dysregulated expression of genes related to lipid, citric cycle, amino acid, and one-carbon metabolism may be partly attributed to the retrograde regulation of MacroD1-inactivated mitochondria on nuclear gene transcription. For example, GDF15 upregulation induced by cKO could program triglyceride metabolism to protect the heart against sepsis24,40 and implicates the roles of MacroD1 in cardio-metabolic disorders.
MCI, a proximal mitochondrial electron transport unit most vulnerable to early sepsis13, dominates ATP production by controlling the formation of mitochondrial supercomplexes containing MCIII and MCIV41 and is identified as a core metabolic effector of MacroD1 inhibition. The enhanced interaction between Ndufb9 and MT-ND5 favors MCI stability42,43 and ensures adequate bioenergetics reserve capacity to support cardiac function. Stable MCI activity may also promote autophagy44 to protect against SCM23. However, direct intervention of MCI is unsuitable for SCM treatment due to its differential effects in various organs14,15. Instead, selective modulation of MacroD1, enriched in cardiomyocytes, promises specific management of myocardial bioenergetics in sepsis.
MCI is also the initiation site of ROS in cardiac mitochondrial dysfunction45. Excessive mitoROS links NLRP3 inflammasome activation to cell pyroptosis, which involves nuclear remodeling of pro-inflammatory cytokine expression and post-translational modifications for inflammasome assembly17,46. MacroD1 modulated mitoROS generation and subsequent activation of the NLRP3 inflammasome via MCI. Its inhibition preserved MCI activity and bioenergetics to limit mitoROS output, NLRP3 inflammasome activation, and cardiomyocyte pyroptosis that drives SCM47,48. By scavenging mitoROS, the MacroD1-MCI-mitoROS-NLRP3 cascade was disrupted. GDF15 upregulated by MacroD1 inhibition should also have anti-inflammatory effects49, although the mechanisms are not yet understood. Of note, the blockade of MacroD1 did not affect serum levels of inflammatory cytokines, including IL1β and TNFα. This case supports the point of no correlation between pro-inflammatory cytokines and SCM8 and that cardiomyocyte inflammatory response may only affect cardiac recruitment of systemic inflammatory cytokines.
Myocardial inflammation is usually characterized by dynamic spatiotemporal crosstalk between cardiac resident cells and the recruited immune cells. Cardiac fibroblasts8,50, endothelial cells51, and immune cells52 participate in myocardial inflammation and affect the contractile activity of cardiomyocytes via paracrine or juxtacrine effects under sepsis. The cardiomyocytes have recently been accepted as an initiating site for myocardial inflammation under nonischemic stresses11. They can produce cytokines that depress the ventricle53 and synchronize immune cell recruitment. Cardiomyocyte-specific regulatory property of MacroD1 on inflammatory response should render a direct target for breaking the vicious cycle of myocardial inflammation and damage in SCM.
Extraordinary mortality of SCM at the onset of sepsis in both patients and animal models underscores early prevention. Despite the importance of oxidative stress in septic organ failure, there have not yet been conclusive benefits for mitochondria-targeted antioxidants. The elusive efficacy may be attributable to indiscriminately blocking ROS across all cell types because oxidative stress initiates survival programming in a given cell type but induces an inflammatory response in other cells54. Targeting MacroD1 may circumvent such a dilemma to enable cardiomyocyte ROS control.
Deciphering mitochondrial MARylation remains challenging, unlike nuclear ADP-ribosylation for genotoxic stress response. On the one hand, MacroD1 is the only ADP-ribose-metabolizing enzyme localized to mitochondria55, while mitochondrial ADP-ribosyltransferases remain unknown56. On the other hand, there is no wide-acknowledged method, although multiple approaches are emerging to characterize MARylation57,58. Using antibodies to immunoprecipitate proteins, combined with mono-ADP ribose antibody-based validations and site-directed mutagenesis, Ndufb9 was identified as a functional effector of MacroD1-mediated inflammometabolic regulation. Even so, the potential contributions of other proteins binding to MacroD1 could not be precluded. In addition, differing from ADP-ribosylation of histidine and lysine residues in muscle mitochondrial Ndufb9 reported59, its arginine residues were MARylated by MacroD1 inhibition. The underlying explanation may be either histidine and lysine residues are inclined to be poly-ADP ribosylated by poly-(ADP-ribose)-polymerases or MARylated by other NAD+-signaling enzymes than MacroD1. Given the effects of mitochondrial ADP-ribosylation on nuclear ADP-ribosylation, MacroD1 may mediate cardiac mitochondrial-nuclear crosstalk.
Mitochondrial MacroD1 holds potential pleiotropic effects on proteins and nucleic acids through its mono-ADP-ribose hydrolyzing activities55. There is currently limited evidence for ADP-ribosylation within mitochondria. Whether MacroD1 performs enzymatic ADPr hydrolyzing of mitochondrial nucleic acids remains a pending issue, despite documented removal of ADPr from phosphorylated double-stranded DNAs by recombinant MacroD1 proteins in vitro experiment55. We cannot preclude the potential modulation of MacroD1 on mitochondrial DNAs and RNAs besides the Ndufb9 protein identified. Nonetheless, comparable expression levels of mitochondrial complexes in cKO and MacroD1flox/flox mice at baseline and septic challenge (Fig. 5A) suggest that MacroD1 does not affect mitochondrial DNAs encoding constitutive proteins of respiratory complexes. Notably, while MRS2578 acts as a MacroD1 inhibitor in our and prior studies31,32, its chemical properties demand cautious interpretation. The compound contains two electrophilic groups that may nonspecifically interact with cellular components, risking off-target effects and complicating data analysis. Although our findings show that MRS2578 mimics genetic MacroD1 inhibition by preserving mitochondrial function and suppressing inflammasome activation, its broad reactivity suggests observed effects may not arise solely from MacroD1 blockade. Future work should prioritize developing MacroD1 inhibitors with enhanced selectivity.
In sum, the potentiation of mitochondrial bioenergetics metabolism plasticity in nonimmune cardiomyocytes is critical for restraining myocardial inflammometabolic disorders and dysfunction in sepsis. Targeting the cardiomyocyte MacroD1-MCI-NLRP3 pathway holds promise for preventing SCM.
Methods
Ethical statement
All animal experiments were approved by the Animal Care Committee of Shanghai Jiaotong University School of Medicine (Protocol No. 2021AW029) and conducted in compliance with the Guide for the Care and Use of Laboratory Animals (National Research Council, US).
Experimental animals
Mice were group-housed (3–5 per cage) in individually ventilated cages (IVC) with corn-cob bedding and environmental enrichment (nestlets, tunnels). Animals were maintained under specific pathogen-free conditions at 22 ± 1 °C with 50 ± 5% humidity under a 12 h light/dark cycle (lights on 07:00), with ad libitum access to autoclaved standard chow (SL0001, Shulaibao Biotech; 24% protein, 5% fat) and acidified water. Daily health monitoring assessed body condition, fur quality, mobility, and signs of distress using a standardized scoring system (1–5 scale).
The mouse strain of cardiac-specific inducible knockout of the MacroD1 gene (cKO) was generated from Cyagen Biosciences (Shanghai, China) by crossing α-myosin heavy chain promoter-driven Cre mice (αMHCMerCreMer) with floxed MacroD1 (MacroD1flox/flox) mice. Tamoxifen (40 mg/kg/d) was dosed intraperitoneally for five consecutive days in 8-week male mice, and after another week, they were employed for ensuing experiments. Genotypes were determined by the polymerase chain reaction (PCR) using tail DNA.
To construct the lipopolysaccharide (LPS)-induced sepsis model, 10-week male mice were intraperitoneally injected with LPS 10 mg/kg for 18, 96, or 168 h to further analysis. An identical volume of saline was given to the control animals.
The mouse model of cecal ligation and perforation (CLP)-induced sepsis was generated as described before60. Briefly, 10-week mice were anesthetized with 3 % isoflurane and exposed to 0.5 L/min oxygen mixed with 1.5 % isoflurane. A 2.5 cm midline incision was done to access the cecum and adjacent intestine. The cecum was ligated with a 4-silk suture at 1 cm from the cecal tip, perforated with a 21-gauge needle, and gently pushed to expel feces from the perforation site. Subsequently, the cecum was reintroduced into the abdominal cavity, and the laparotomy site was sutured with 6-silk sutures. The sham animals were subjected to the same surgical operation, except that the cecum was not ligated and perforated.
Upon completion of the study, mice were deeply anesthetized with sodium pentobarbital (80 mg/kg, i.p.) and euthanized by cervical dislocation; myocardial tissues and serum samples were immediately collected for subsequent biochemical and molecular analyses. Female exclusion was implemented to control for sex hormone influences on cardiac metabolic vulnerability.
Preparation of blood serum
Blood samples were collected via cardiac puncture in the mice under deep anesthesia. A small incision was made in the chest of animals in a supine position to expose the heart. A 23-gauge needle attached to a 1 mL syringe was used to puncture the left ventricle. The blood was allowed to flow into the syringe by gentle aspiration, transferred to microtubes containing EDTA as an anticoagulant, and then centrifuged at 3000 × g for 15 min at 4 °C to separate the serum. The serum was aliquoted into separate tubes and stored at − 80 °C until further analysis.
Quantification of cytokines
The contents of IL-1β, TNF-α, LDH, and cTnT in the supernatants of mouse serum were tested following the manufacturer’s instructions (Mlbio ELISA kit).
Isolation and culture of neonatal rodent cardiomyocytes
Ventricular myocytes were isolated from three-day-old Sprague-Dawley rats (NRCMs) or neonatal mice (NMCMs) using enzymatic dissociation. Briefly, the ventricles of SD rats or mice were detached and cut into small pieces, then digested with 0.75 mg/ml collagenase type II (Worthington) in PBS three times for 15 minutes at 37 °C each time. To stop the digestion, the separated cells were submerged in fetal bovine serum (FBS) and centrifuged at 300 × g for five minutes to form pellets. Subsequently, the pelleted cells were re-suspended and cultivated in Dulbecco’s modified Eagle’s medium (DMEM) with 1% penicillin-streptomycin, 10% FBS, and 1% bromodeoxyuridine for 90 minutes to allow fibroblast adhere to the cell dish. Finally, the floating cardiomyocytes were transferred to a new cell culture dish for further experiments.
Isolation of adult mouse ventricular myocytes (AMCMs)
Adult mice were anesthetized via inhalation of 4% isoflurane followed by cervical dislocation. Hearts were rapidly excised and cannulated via the aorta on a Langendorff perfusion system. Perfusion commenced with calcium-free Tyrode’s solution for 5 min at 37 °C, followed by enzyme solution containing 0.8 mg/ml collagenase type II (Worthington), 0.1 mg/ml trypsin (Sigma), and 0.02 mg/ml protease XIV (Sigma) in Tyrode’s solution. The left ventricle was then dissected, minced, and subjected to gentle trituration in enzyme solution supplemented with 0.2% bovine serum albumin (BSA). The resultant suspension was filtered through a 100 µm mesh and cardiomyocytes sedimented by gravity for 15 min. Cells underwent stepwise calcium reintroduction (0.2 mM, 0.5 mM, 1.2 mM CaCl₂ in BSA-supplemented Tyrode’s solution, 20 minutes per step) before plating on laminin-coated (20 µg/ml) dishes.
Quantitative RT-PCR
Total RNA from tissue and cells was extracted using TRIZOL (TAKARA, Japan), and reversed RNA for cDNA using the TAKARA PrimeScriptTM RT reagent kit (TAKARA, Japan). Real-time PCR was run with TB Green Fast qPCR Mix (TAKARA, Japan) on a Roche LightCycler480 instrument II. The results were defined from the threshold cycle (Ct), and the 2−ΔΔCt method was used to calculate the relative expression levels. The primers used were listed in the Supplementary Tables 5–7.
Echocardiography
A Vevo 3100 System (Visual Sonics) with an MS400 Transducer was used to perform the echocardiographic evaluation in anesthetized (1.5% isoflurane) mice. Left ventricular function was assessed across the middle of the left ventricle using an M-Mode scan.
Transmission electron microscopy
Mice were subjected to transcardial perfusion with 0.1 M PBS (pH 7.4) immediately after sacrifice by intraperitoneally injecting 100 mg/kg pentobarbital sodium. Hearts were extracted, cut into 1-2 mm3 sections, and fixed in the buffer containing 2.5% glutaraldehyde and 0.1 M calcium carbonate at room temperature for 2 h. The sectioned samples were kept at 4 °C overnight, embedded and ultramicrosectioned to obtain 90 nm flakes, and photographed using a Hitachi HT7800 transmission electron microscope (Hitachi, Japan).
Extraction of mitochondria, cytoplasm without mitochondria, and the nucleus
A mitochondria isolation kit was used to separate mitochondrial and cytosolic fractions of ventricle tissues using differential centrifugation according to the manufacturer’s instructions (Beyotime, C3606). Nucleus fraction was extracted by nuclear protein extraction kit (Beyotime, P0027). The protein concentrations were determined using an assay kit (BioRad, Hercules, CA).
Blue Native PAGE (BN-PAGE) assay
The expression of mitochondrial complexes was detected using the Blue Native PAGE (BN-PAGE) assay following the manufacturer’s instructions (Zhongke Ruitai Technology, RTD6136). Briefly, mitochondria was extracted from heart tissue and the protein concentration was determined. Then, 200 μg of mitochondria were dissolved in a mixture of 5 μl 4 × BN/CN-PAGE protein sample buffer and 15 μl 10% digitonin solution. The resulting mixture was placed on ice for 20 minutes, followed by centrifugation at 4 °C 20000 × g for 10 min. Next, 7.5 μl of the supernatant was taken and mixed with 2.5 μl 5% G-250 dye and loaded onto a 3–12% PAGE Bis-Tris gel, which was run according to the manufacturer’s protocols. Finally, the gel was subjected to Coomassie brilliant blue staining to detect the expression of mitochondrial complexes.
Mitochondrial complexes activity assays
Mitochondrial complexes activity was detected according to the manufacturer’s experimental protocol (Abbkine, KTB1850 for mitochondrial complex I activity analysis; KTB1860 for mitochondrial complex II; KTB1870 for mitochondrial complex III; KTB1880 for mitochondrial complex IV; KTB1890 for mitochondrial complex V).
Immunohistochemistry
Following collection, hearts were fixed in 10% formalin or implanted in OCT (SAKURA) as needed. Immunohistochemistry was performed as previously described61. Antibodies used for immunostaining were as followed: α-SMA (1:100) and MPO (1:100). After DAPI counterstaining, the tissue slides were microphotographed.
For Masson’s staining, the hearts of mice were extracted, fixed with formaldehyde, embedded in paraffin, and sectioned at 8 μm thickness. The sections were stained using regular protocols23 and imaged with a DM5000B microscope (Leica).
The fluorescence intensity of MPO- and α-SMA-staining images acquired with consistent microscope settings was measured using ImageJ software. The fluorescence intensity of samples in experimental groups was normalized to intact flox controls. In addition, Masson’s Trichrome staining of collagen deposition was quantified by measuring the blue-stained collagen fibers with ImageJ to evaluate cardiac fibrosis. The results were expressed as a percentage of the total tissue area.
Proximity ligation assay (PLA)
The proximity ligation assay was performed in triplicate with the Duolink proximity ligation assay kit (Millipore Sigma DUO92101). AMCMs were plated onto confocal dishes, fixed with 4% formaldehyde for 20 min, permeabilized by 0.3% Triton X-100 for 10 min, and blocked with Duolink blocking buffer for 30 min. After incubation with primary antibodies anti-NLRP3 (1:100) and anti-ASC (1:50) at 4 °C overnight, AMCMs were sequentially washed with buffer A twice for 5 min at room temperature, incubated with the plus and minus assay probes for 1 h at 37 °C, washed twice with Duolink buffer A and then incubated with 1 × ligation buffer containing ligase for 30 minutes at 37 °C. Following washing with buffer A twice for 5 minutes at room temperature and incubation by 1 × DuoLink Amplification Buffer with polymerase for 100 min at 37 °C, AMCMs were washed twice with Duolink wash buffer B, mounted with Duolink proximity ligation assay mounting medium and 4’,6-diamidino-2-phenylindole (DAPI). The speckles were imaged using a Leica SP8 confocal microscope.
Gene silencing and overexpression of cardiomyocytes
Cardiomyocytes were isolated and cultured in DMEM/F12 medium containing 10% FBS and 0.1 mM BrdU for 36 hours. Following the transfection with MacroD1 siRNA, P2Y6 SiRNA or NC, recombinant adenovirus vectors carrying the MacroD1 gene (Adv-MacroD1, OED) or the GFP gene (Adv-GFP, Vect) at the multiplicity-of-infection of 50 for 36 hours, cardiomyocytes were treated with indicated reagents and then used to further examinations. siRNAs and adenovirus were obtained from Generay Biotech (Shanghai, China) and WZ Bioscience (Shandong, China), respectively. The sequence of siRNAs was listed in the Supplementary Table 8.
Transfection of siRNA or/and Plasmids for HEK293 cells or AC16 cells
HEK293 or AC16 cells were transfected with indicated siRNAs or plasmids using Lipofectamine 3000 (Thermo Fisher, L3000008) in line with the manufacturer’s instructions for 36 hours. Then, the cells were subjected to the corresponding treatment, followed by protein extraction of the cells or other experiments. The plasmids and siRNA employed in the experiment included siRNA MacroD1 (Si-M), siRNA Ndufb9 (Si-Ndufb9), siRNA Sirt3 (Si-Sirt3), His-tagged MacroD1 plasmid, His-tagged MacroD1 with truncated N-terminus (MacroD1△MTS) plasmid, Flag-tagged Ndufb9 plasmid, His-tagged MacroD1 with truncated Macro domain (MacroD1 mut) plasmid, Flag-tagged Ndufb9R173A mutant plasmid, Flag-tagged SM-Ndufb9 plasmid, and Flag-tagged SM-Ndufb9R173A mutant plasmid. Plasmids and siRNAs were constructed and supplied by Hanheng Biotechnology (Shanghai, China) and Generay Biotech (Shanghai, China), respectively. The sequences of siRNAs were listed in the Supplementary Table 8.
Immunocytochemistry
The isolated NRCMs were seeded at an adequate density on confocal dishes. After washing three times with PBS, the cells were treated with 4% formaldehyde buffer at room temperature for 30 minutes. Next, the cells were rinsed three times using PBS and permeabilized with PBS containing 1% Triton100 ×. Following the three times rinsing with PBS, cells were blocked by 3% BSA for 30 minutes at room temperature and incubated with primary antibodies overnight at 4 °C. Nuclei were counterstained with DAPI after incubation with secondary antibodies at room temperature for another hour. The antibodies included ASC (1:50), NLRP3 (1:100), MacroD1 (1:100), TOM20 (1:100).
Cell bioenergetic profiling
Oxygen consumption rate (OCR) was measured using a Seahorse XFe96 cell efflux analyzer (Agilent Technologies, USA). Briefly, 3 × 104 cells per well were plated in assay microplates for 24 h followed by related treatments. After the culture medium was replaced by Seahorse XF DMEM buffer supplementing with 10 mM glucose, 2 mM glutamine, and 1 mM pyruvate, cells were cultured for 1 h in a CO2-free incubator at 37 °C. OCR was detected in real-time when the plate was sequentially injected with 1.5 μM oligomycin, 0.5 μM FCCP, and 0.5 μM Rot/AA.
The Seahorse XF Palmitate Oxidation Stress Kit (Agilent Technologies, USA) was used to evaluate fatty acid oxidation (FAO) metabolism following the manufacturer’s instructions. The FAO test was conducted over two days with two pretreatment steps. NRCMs were plated at a density of 3 × 104 cells per well for 24 h. The medium was then replaced by substrate-limited growth medium, i.e., DMEM supplemented with 0.5 mM glucose, 1.0 mM GlutaMAX, 0.5 mM carnitine, and 1% FBS, and cells were incubated for 24 h. After removing the substrate-limited growth medium, cells were incubated with substrate-limited analysis medium (Seahorse XF DMEM medium containing 2 mM glucose and 0.5 mM carnitine) for another 45 min in a CO2-free incubator. Before the assay, the analysis medium was refreshed and added BSA or palmitate-BSA. OCR was measured in real-time while the sequential injection of 4 µM etomoxir, 1.5 μM oligomycin, 0.5 μM FCCP, and 0.5 μM Rotenone/Antimycin (Rot/AA).
Assay of ROS and ATP contents
According to the manufacturer’s instructions, NRCMs or AC16 cardiomyocytes were cultured in a medium containing 5 μM MitoSOX (MCE, HY-D1055) and 1 × Hoechst 33342 (Beyotime, C1028) for 30 min at 37 °C to detect mitochondrial ROS levels.
In frozen sections of rat hearts, the OCT was removed by washing with PBS. To prepare the Dihydroethidium (DHE; Invitrogen) (5 µM) dye, the original solution was diluted with PBS solution. The tissue was incubated with the diluted DHE dye solution for 30 min in the dark. After then, the slides were washed with PBS three times to stop the staining process. Nuclei were counterstained with DAPI for 10 min and washed with PBS. The anti-fluorescence quenching solution was applied to the tissue before being covered with a coverslip.
The ATP contents of tissues or cells were quantified using ATP Assay Kit (Beyotime, S0026). Briefly, 20 mg of fresh heart tissue was homogenized in normal saline, centrifuged at 3000 × g/min for 10 min, and the supernatant was collected. Tests were conducted separately by the manufacturer’s instructions.
NAD+ assay
To evaluate the levels of NAD+ within cells, the NAD+/NADH Assay Kit (Beyotime, S0175) was employed according to the guidelines provided by the manufacturer.
Co-immunoprecipitation
On detecting mono-ADP-ribosylation assay, the cells were cultured under different treatments in a 10 cm dish and harvested with precooled PBS on ice. Cells were pelleted by 500 × g centrifugation at 4 °C for 5 min, lysed, and boiled for 5 min in 200 μl immunoprecipitation (IP) buffer (Beyotime, P0013) containing 250 nM ADP-HPD (Sigma-Aldrich, A0627), 2.5 μM Olaparib (McdChem Express, HY-10162), and 1 × protease inhibitor cocktail (Epizyme Biotech, GRF101). For heart tissue, 80 mg heart tissue was mechanically homogenized for 60 s at 60 hz and boiled for 5 min after adding 400 μl IP buffer containing 250 nM ADP-HPD, 2.5 μM Olaparib, and 1 × protease inhibitor cocktail. The lysates of cells or heart tissues were sonicated on ice and centrifuged for 10 min at 12,000 × g before the supernatant was transferred to a fresh tube. A BCA protein assay kit (Beyotime, P0009) was used to determine the protein concentration, and the lysate was diluted with LPS containing 250 nM ADP-HPD, 2.5 μM Olaparib, and 1 × protease inhibitor cocktail. 400 μg protein of lysates containing 4 μg anti-Ndufb9 antibody was shaken in a 1.5 ml tube for 8 hours at 4 °C. Then, 20 μl prewashed protein A/G magnetic beads (Med Chem Express, HY-K0202) were transferred to the sample, incubated, and shaken for 2 h at room temperature. The beads were separated by a magnetic rack and washed three times with PBS. The homologous IgG was used as a negative control, and 20% lysate as an input control. Finally, the beads were boiled in 1% SDS loading buffer, electrophoresed, western transferred, and blotted with the indicated antibodies.
To conduct co-immunoprecipitation, cells or heart tissues were lysed in IP lysis buffer containing a protease inhibitor cocktail and incubated with the indicated antibodies on a rotator overnight at 4 °C. The protein A/G magnetic beads were added and incubated for 2 h at 4 °C. The beads were then separated by a magnetic rack and washed three times with PBS, boiled in 1% SDS loading buffer, electrophoresed, western transferred, and blotted with the indicated antibodies. The antibodies used in the co-immunoprecipitation experiments included MacroD1, Ndufb9, Flag, His, ASC, NLRP3, and MT-ND5.
Mass spectrometry analysis
As for mass spectrometry analysis for mono-ADP-ribosylation of mitochondrial protein, mitochondrial fractions of Flox or cKO mice hearts (n = 5 for each group) were extracted and pooled according to the manufacturer’s instructions (Beyotime, C3606). Briefly, mitochondria samples were lysed and boiled for 5 min in 250 μl IP buffer containing 250 nM ADP-HPD, 2.5 μM Olaparib, and 1 × protease inhibitor cocktail, and ultrasonicated on ice. The lysates were centrifuged for 10 min at 12,000 × g before the supernatant was transferred to a fresh tube. Next, the lysates were diluted with IP buffer containing 250 nM ADP-HPD, 2.5 μM Olaparib, and 1 × protease inhibitor cocktail. Then, the lysates were digested with sequencing-grade trypsin (Promega, USA), purified, and lyophilized to get peptides. The lyophilized peptides were dissolved in 1 × AP buffer (50 mM TRIS pH 8.0, 50 mM NaCl, 1 mM MgCl2, and 250 μM DTT) and incubated with 4 μg anti-mono-ADPr antibody (antibodies specifically recognizing mono-ADP-ribosylated proteins) for 4 to 6 h. After that, 20 μl protein A/G magnetic beads prewashed by PBS were added to the peptide mixture and incubated for 2 h in a head-over-tail mixer. After magnetic beads were separated by a magnetic rack and washed with cold PBS and MilliQ water, the ADP-ribosylated peptide was eluted with 2% formic acid and further treatment for mass spectrometry.
To identify MacroD1-interacted proteins, the mitochondrial fractions of hearts from wild-type mice (n = 5 for each group) were extracted and pooled according to the manufacturer’s instructions and then lysed with 250 μl immunoprecipitation (IP) lysis buffer and centrifuged at 12,000 × g for 15 min to collect the supernatant. The lysate was incubated with the anti-MacroD1 or IgG antibody at 4 °C overnight and with protein A/G magnetic beads for another 8 h. Bead-antibody-antigen complexes were collected using a magnetic separator and washed twice with IP lysis buffer. The precipitated protein samples were placed in − 20 °C and sent to the Institute of Botany, Chinese Academy of Sciences, for further processing.
Protein digestion was conducted using the filter-aided sample preparation (FASP) method with slight modifications. In brief, the total of protein from each group was added to 200 μL of UA buffer (8 M urea and 50 mM Tris−HCl, pH 8.0), loaded onto a 10 kDa ultrafiltration centrifuge tube (Sartorius), and centrifuged at 14,000 × g for 30 min. This step was repeated twice. Subsequently, the samples were subjected to reductive alkylation by adding 100 μL of UA buffer containing 10 mM Tris (2-carboxyethyl) phosphine (TCEP) and 20 mM 2-chloroacetamide (CAA) with continuous shaking for 30 min. Another 300 μL of UA buffer was added, and the samples were centrifuged at 14,000 × g for 30 min. After this step was repeated twice, 300 μL of 50 mM NH4HCO3 solution was added, followed by centrifugation at 14,000 × g for 20 min. This step was repeated twice. Next, 2 μL of trypsin solution (1 μg of trypsin in 100 μL of 100 mM NH4HCO3) was added, and the samples were shaken at 37 °C for 12 h. The collection tube was replaced, and the digests were collected by centrifugation at 14,000 × g for 20 min. The filtrate was collected, and a 0.1% trifluoroacetic acid solution was added, followed by desalting with C18 cartridges (Thermo Scientific). The peptide concentration was determined using the Pierce Quantitative Colorimetric Peptide Assay kit (Thermo Scientific)
Experiments were performed on an Orbitrap HF-X mass spectrometer that was coupled to Easy nLC (Thermo Fisher Scientific). The peptide mixture was loaded onto a the C18-reversed phase column (25 cm long, 75 μm inner diameter) packed in-house with RP-C18 1.9 μm resin in buffer A (0.1% Formic acid in HPLC-grade water) and separated with a linear gradient of buffer B (0.1% Formic acid in 80% acetonitrile) at a flow rate of 300 nl/min. MS data was acquired using a data-dependent top20 method dynamically choosing the most abundant precursor ions from the survey scan (375–1500 m/z) for HCD fragmentation. Determination of the target value is based on predictive Automatic Gain Control (pAGC). Dynamic exclusion duration was 45 s. Survey scans were acquired at a resolution of 60,000 at m/z 200, and resolution for HCD spectra was set to 15,000 at m/z 200. Normalized collision energy was 28 eV.
Sequence database searching and data analysis for ADP-ribosylated peptides: the MS data were analyzed using Byonic software (Protein Metrics, Cupertino, CA, USA). Peptide sequences (and hence protein identity) were determined by matching the UniProt Mus musculus Proteome database (UP000000589, https://www.uniprot.org/proteomes/UP000000589). An initial search was set at a precursor mass window of 10 ppm. The search followed an enzymatic cleavage rule of Trypsin and allowed maximal two missed cleavage sites and a mass tolerance of 0.02 Da for fragment ions. Carbamidomethylation of cysteines was defined as a fixed modification, while protein N-terminal acetylation, methionine oxidation and ADP-ribose modification of amino acids (cysteine, glutamic acid, asparagine, arginine, serine, lysine) were defined as variable modifications for database searching. The cutoff of global false discovery rate (FDR) for peptide and protein identification was set to 0.01. The screening for significantly differentially expressed proteins was conducted based on the criterion that proteins enriched in the cKO group exhibited a fold-change ratio of at least 1.5 compared to the Flox group.
Sequence database searching and data analysis for MacroD1-interacted peptides: the MS data were analyzed using Thermo proteome Discoverer2.5.0.400 software. MS data were searched against the Peptide sequences (and hence protein identity) were determined by matching the UniProt Mus musculus Proteome database (UP000000589, https://www.uniprot.org/proteomes/UP000000589). An initial search was set at a precursor mass window of 10 ppm. The search followed an enzymatic cleavage rule of Trypsin and allowed maximal two missed cleavage sites and a mass tolerance of 0.02 Da for fragment ions. Carbamidomethylation of cysteines was defined as a fixed modification, while protein N-terminal acetylation and methionine oxidation were defined as variable modifications for database searching. The cutoff of global false discovery rate (FDR) for peptide and protein identification was set to 0.01. Screening for significantly differentially expressed proteins was conducted based on the criteria that the expression level of proteins enriched by the MacroD1 antibody was more than 1.5 times higher than that of the corresponding proteins enriched by the IgG.
RNA sequencing and analysis
RNA sequencing analysis was conducted by Paiseno Biotechnology Co., Ltd. Briefly, Total RNAs were extracted from the left ventricular tissues of mice for RNA sequencing. An Agilent Bioanalyzer was employed to evaluate the quality of RNA. Sequencing libraries were generated using the TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA, USA). High-throughput sequencing was performed using the NovaSeq 6000 Sequencing System (Illumina). Raw reads from each library were mapped to the mouse genome (GRCm39).
The DESeq (v.1.24.0 R package), with the criteria of |Log2FC (fold change)| > 1 and adj. p-value < 0.05, was used to identify the differentially expressed genes (DEGs) in WT and cKO samples. KEGG pathway enrichment analysis and Gene Set Enrichment Analysis (GSEA) performed with clusterProfiler R package (4.7.1.3). The Bioconductor package pheatmap (v.1.0.12) was used to create heat maps.
Cardiac single-cell RNA sequencing (scRNA-Seq)
APExBIO Company executed the cardiac tissue processing and sequencing analyses. Specifically, upon sample acquisition, the fresh cardiac tissues were rapidly frozen in liquid nitrogen for 1 h, followed by storage at − 80 °C. A nuclei suspension was prepared using the Nuclei Isolation Kit (BioYou). The nuclei concentration was quantified using the Countess II Automated Cell Counter, and subsequently adjusted to a range of 700–1200 nuclei/μL. The single-cell suspensions were introduced into the MobiNova-100 platform. For library preparation, the MobiCube Single-Cell 3′ RNA-seq Kit v1.0 (MobiDrop) was employed, and sequencing was performed on the Illumina NovaSeq 6000 Systems.
The resultant FASTQ files were aligned to the genome using MobiVision v3.2, leveraging a customized GRCm38 reference genome. Cells of inferior quality, defined by gene counts fewer than 300 or exceeding 5000, or mitochondrial read proportions surpassing 12%, were eliminated using Seurat v.4.3.0 in R. The high-quality samples were normalized with SCTransform before integration. After integration, the dataset was scaled, and UMAP dimensionality reduction was conducted utilizing the foremost 15 principal components. Unsupervised Louvain clustering was applied to a shared nearest neighbor graph, with the resolution parameter at 0.4. Differential expression analysis was performed using FindAllMarkers (Seurat) to identify DEGs within each cluster. The cell-type identity of each cluster was manually annotated based on the expression of canonical markers and corroborated with existing literature. Visualizations of gene expression were generated using Seurat’s DoHeatmap, DotPlot, and VlnPlot functions, in the form of heatmaps, dot plots, and violin plots.
Western blotting
The proteins of cells or tissue samples were extracted for Western blotting examination. Briefly, RIPA or IP lysis buffer was used to extract the total protein from the cultured cells or tissue, and a BCA Protein Assay Kit (Beyotime, P0009) was used to measure the protein concentrations. The proteins were boiled in a denatured loading buffer containing sodium dodecyl sulfate (SDS) (EpiZyme, LT101) and then loaded onto a 10% or 12% polyacrylamide gel. Following electrophoresis, the gels were transferred onto nitrocellulose transfer membranes. The transfer membranes were blocked with 5% fat-free dry milk for 1 h to minimize background staining and then incubated with the indicated primary antibodies and HRP-conjugated secondary antibodies. Western blotting procedure was carried out using an ECL Western Blotting Detection Kit. Details of antibodies used in immunofluorescence, western blotting, and Co-immunoprecipitation are available in the Source data.
Statistics and reproducibility
The results were expressed as mean ± SD. Statistical analysis was conducted using GraphPad Prism (Version 8.4.3, San Diego, CA, USA). A two-tailed unpaired Student’s ttest was applied to compare the two groups. When more than two groups were compared, one-way ANOVA and subsequent Tukey’s post hoc tests or Dunnett’s multiple comparisons tests were employed for homogeneous variances. The Kaplan-Meier method was used to calculate the survival rate, and the log-rank (Mantel-Cox) test evaluated the survival curve difference between groups. A p-value less than 0.05 was statistically significant. All experimental data derive from multiple biological replicates or independently repeated trial. Specific replication details for each assay are provided in the corresponding figure legends. To ensure reproducibility, a minimum of three distinct biological samples were utilized across all tests, with all collected data included in the final analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data associated with this study are in the paper or the Supplementary Materials. The raw and analyzed RNA-sequencing and cardiac single-cell sequencing data produced in this study have been deposited in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) with the accession numbers: GSE253829 and GSE285720. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository62,63 with the dataset identifiers PXD059021 and PXD059022. Source data are provided in this paper.
References
Prescott, H. C. & Angus, D. C. Enhancing recovery from sepsis: A review. JAMA 319, 62–75 (2018).
Zhang, K. et al. TREM2(hi) resident macrophages protect the septic heart by maintaining cardiomyocyte homeostasis. Nat. Metab. 5, 129–146 (2023).
Celes, M. R., Prado, C. M. & Rossi, M. A. Sepsis: going to the heart of the matter. Pathobiology 80, 70–86 (2013).
Vasques-Novoa, F. et al. Myocardial Edema: An overlooked mechanism of septic cardiomyopathy?. Shock 53, 616–619 (2020).
Crouser, E. A rare glimpse behind the mask of sepsis-induced organ failures provides hope for an eventual cure. Am. J. Respir. Crit. Care Med. 187, 460–462 (2013).
Richardson, P. et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 93, 841–842 (1996).
Kawaguchi, S. & Okada, M. Cardiac Metabolism in Sepsis. Metabolites 11, https://doi.org/10.3390/metabo11120846 (2021).
Landesberg, G. et al. Myocardial dysfunction in severe sepsis and septic shock: No correlation with inflammatory cytokines in real-life clinical setting. Chest 148, 93–102 (2015).
Clark, M. A. et al. Effect of a chimeric antibody to tumor necrosis factor-alpha on cytokine and physiologic responses in patients with severe sepsis-a randomized, clinical trial. Crit. Care Med. 26, 1650–1659 (1998).
Opal, S. M. et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 309, 1154–1162 (2013).
Suetomi, T., Miyamoto, S. & Brown, J. H. Inflammation in nonischemic heart disease: initiation by cardiomyocyte CaMKII and NLRP3 inflammasome signaling. Am. J. Physiol. Heart Circ. Physiol. 317, H877–H890 (2019).
Ngo, J., Osto, C., Villalobos, F., & Shirihai, O. S. Mitochondrial heterogeneity in metabolic diseases. Biology 10, https://doi.org/10.3390/biology10090927 (2021).
Vanasco, V. et al. Cardiac mitochondrial biogenesis in endotoxemia is not accompanied by mitochondrial function recovery. Free Radic. Biol. Med. 77, 1–9 (2014).
Ai, Q. et al. Rotenone, a mitochondrial respiratory complex I inhibitor, ameliorates lipopolysaccharide/D-galactosamine-induced fulminant hepatitis in mice. Int. Immunopharmacol. 21, 200–207 (2014).
Li, N. et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 278, 8516–8525 (2003).
Peek, C. B. et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342, 1243417 (2013).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Misawa, T. et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 14, 454–460 (2013).
Katsyuba, E., Romani, M., Hofer, D. & Auwerx, J. NAD(+) homeostasis in health and disease. Nat. Metab. 2, 9–31 (2020).
Covarrubias, A. J., Perrone, R., Grozio, A. & Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 22, 119–141 (2021).
Brokatzky, D. & Hacker, G. Mitochondria: intracellular sentinels of infections. Med. Microbiol. Immunol. 211, 161–172 (2022).
Suchard, M. S. & Savulescu, D. M. Nicotinamide pathways as the root cause of sepsis - an evolutionary perspective on macrophage energetic shifts. FEBS J. 289, 955–964 (2022).
Sun, Y. et al. Beclin-1-dependent autophagy protects the heart during sepsis. Circulation 138, 2247–2262 (2018).
Luan, H. H. et al. GDF15 Is an inflammation-induced central mediator of tissue tolerance. Cell 178, 1231–1244.e1211 (2019).
Rath, S. et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547 (2021).
Sazanov, L. A. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 16, 375–388 (2015).
Liu, T. F. et al. Fueling the flame: bioenergy couples metabolism and inflammation. J. Leukoc. Biol. 92, 499–507 (2012).
Ogura, Y., Sutterwala, F. S. & Flavell, R. A. The inflammasome: first line of the immune response to cell stress. Cell 126, 659–662 (2006).
Chen, W. W., Freinkman, E., Wang, T., Birsoy, K. & Sabatini, D. M. Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell 166, 1324–1337 (2016).
Wagner, G. R. & Payne, R. M. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 288, 29036–29045 (2013).
Haikarainen, T., Maksimainen, M. M., Obaji, E. & Lehtio, L. Development of an inhibitor screening assay for mono-ADP-ribosyl hydrolyzing macrodomains using alphaScreen technology. SLAS Discov. 23, 255–263 (2018).
Mattila, S. et al. Macrodomain binding compound MRS 2578 inhibits Alphavirus replication. Antimicrob. Agents Chemother. 65, e0139821 (2021).
Weis, S. et al. Metabolic adaptation establishes disease tolerance to sepsis. Cell 169, 1263–1275 (2017).
Brealey, D. et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360, 219–223 (2002).
Vico, T. A. et al. Mitochondrial bioenergetics links inflammation and cardiac contractility in endotoxemia. Basic Res. Cardiol. 114, 38 (2019).
Van Gool, F. et al. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat. Med. 15, 206–210 (2009).
Gerner, R. R. et al. NAD metabolism fuels human and mouse intestinal inflammation. Gut 67, 1813–1823 (2018).
Ye, M. et al. NAD(H)-loaded nanoparticles for efficient sepsis therapy via modulating immune and vascular homeostasis. Nat. Nanotechnol. 17, 880–890 (2022).
Liu, Y. T. et al. Macrod1 suppresses diabetic cardiomyopathy via regulating PARP1-NAD(+)-SIRT3 pathway. Acta Pharmacol. Sin. 45, 1175–1188 (2024).
Chung, H. K. et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol. 216, 149–165 (2017).
Vercellino, I. & Sazanov, L. A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 23, 141–161 (2022).
Stroud, D. A. et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 538, 123–126 (2016).
Wu, M., Gu, J., Guo, R., Huang, Y. & Yang, M. Structure of mammalian respiratory supercomplex I(1)III(2)IV(1). Cell 167, 1598–1609 (2016).
Thomas, H. E. et al. Mitochondrial complex I activity is required for maximal autophagy. Cell Rep. 24, 2404–2417 (2018).
Kushnareva, Y., Murphy, A. N. & Andreyev, A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 368, 545–553 (2002).
Tschopp, J. & Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production?. Nat. Rev. Immunol. 10, 210–215 (2010).
Li, N. et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 24, 101215 (2019).
Fujimura, K. et al. NLRP3 inflammasome-driven IL-1beta and IL-18 contribute to lipopolysaccharide-induced septic cardiomyopathy. J. Mol. Cell Cardiol. 180, 58–68 (2023).
Deng, Y. et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 128, 232–245 (2021).
Zhang, W. et al. Carbon monoxide releasing molecule-3 improves myocardial function in mice with sepsis by inhibiting NLRP3 inflammasome activation in cardiac fibroblasts. Basic Res. Cardiol. 112, 16 (2017).
Kang, W. et al. Neuregulin‑1 protects cardiac function in septic rats through multiple targets based on endothelial cells. Int. J. Mol. Med. 44, 1255–1266 (2019).
Kong, C. et al. ICA69 aggravates ferroptosis causing septic cardiac dysfunction via STING trafficking. Cell Death Discov. 8, 187 (2022).
Comstock, K. L. et al. LPS-induced TNF-alpha release from and apoptosis in rat cardiomyocytes: obligatory role for CD14 in mediating the LPS response. J. Mol. Cell Cardiol. 30, 2761–2775 (1998).
Martindale, J. L. & Holbrook, N. J. Cellular response to oxidative stress: signaling for suicide and survival. J. Cell Physiol. 192, 1–15 (2002).
Agnew, T. et al. MacroD1 is a promiscuous ADP-ribosyl hydrolase localized to mitochondria. Front. Microbiol. 9, 20 (2018).
Hopp A. K., Gruter P. & Hottiger M. O. Regulation of glucose metabolism by NAD(+) and ADP-ribosylation. Cells 8, https://doi.org/10.3390/cells8080890 (2019).
Vivelo, C. A. & Leung, A. K. Proteomics approaches to identify mono-(ADP-ribosyl)ated and poly(ADP-ribosyl)ated proteins. Proteomics 15, 203–217 (2015).
Hopp, A. K. & Hottiger, M. O. Uncovering the invisible: Mono-ADP-ribosylation moved into the spotlight. Cells 10, https://doi.org/10.3390/cells10030680 (2021).
Hopp, A. K. et al. Mitochondrial NAD(+) controls nuclear ARTD1-induced ADP-ribosylation. Mol. Cell 81, 340–354.e345 (2021).
Lewis, A. J. et al. Use of biotelemetry to define physiology-based deterioration thresholds in a murine cecal ligation and puncture model of sepsis. Crit. Care Med. 44, e420–e431 (2016).
Li, Y. et al. Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol. 38, 101771 (2021).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–d1217 (2019).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 50, D1522–D1527 (2022).
Acknowledgements
We thank Prof. Jiang Hong and Liqun Zhao at the Department of Intensive Medicine, Shanghai General Hospital, Shanghai Jiaotong University School of Medicine, for their constructive comments, MS. Shanshan Wang and Lianyan Jing at the Core Facility Center of CAS Center for Excellence in Molecular Plant Sciences, for their assistance in proteomic assays. This work was supported by the National Original Exploration Program of China (82150001 to J.L.), the National Key Research & Development Program of China (2018YFC1312504 to J.L.), the National Natural Science Foundation of China (82070414, 81670360, 81870293 to J.L.; 82000376 to F.L.; 82000375 to Q.Z.), and the Popularization & Management Optimization Program of Diagnosis and Treatment Technique of Shanghai (SHDC22023241 to H.X.).
Author information
Authors and Affiliations
Contributions
J.L. and S.L. designed the study. X.C. and T.Y. executed in vitro experiments and part of the in vivo experiments. D.Z. performed some in vivo experiments with the help from M.Y. F.L. and X.C. performed all statistical analyses. H.X. analyzed single-cell sequencing and tissue RNA-seq. X.C., J.L., and S.L. wrote the manuscript. All authors thoroughly discussed the results and provided comments on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Paul Delgado Olguin, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, X., Yuan, T., Zheng, D. et al. Cardiomyocyte mitochondrial mono-ADP-ribosylation dictates cardiac tolerance to sepsis by configuring bioenergetic reserve in male mice. Nat Commun 16, 8119 (2025). https://doi.org/10.1038/s41467-025-62384-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62384-8
