
Abstract
Optimizing both on-target and off-target potencies is essential for developing effective and selective small-molecule therapeutics. Free energy calculations offer rapid potency predictions, usually within hours and with experimental accuracy and thus enables efficient identification of promising compounds for synthesis, accelerating early-stage drug discovery campaigns. While free energy predictions are routinely applied to individual proteins, here, we present a free energy framework for efficiently achieving kinome-wide selectivity that led to the discovery of selective Wee1 kinase inhibitors. Ligand-based relative binding free energy calculations rapidly identified multiple novel potent chemical scaffolds. Subsequent protein residue mutation free energy calculations that modified the Wee1 gatekeeper residue, significantly reduced their off-target liabilities across the kinome. Thus, with judicious use of this gatekeeper residue selectivity handle, applying this computational strategy streamlined the optimization of both on-target and off-target potencies, offering a roadmap to expedite drug discovery timelines by decreasing unanticipated off-target toxicities.
Similar content being viewed by others


Introduction
Potency optimization is a crucial component of drug discovery campaigns. Without sufficient on-target potency, modulation of the therapeutic target will be inadequate to achieve the desired efficacy. Without sufficient on-target selectivity, off-target binding and the potential for in vivo safety liabilities will make it very challenging to successfully progress a given molecule through the discovery process. Achieving selectivity for inhibitors that bind to conserved regions in a gene family is particularly challenging, including kinome-wide selectivity for inhibitors targeting the ATP-binding pocket of a given kinase1,2,3.
Free energy simulations are the backbone of many structure-based drug discovery programs to accurately and efficiently predict the potency of design ideas2,3. In ligand relative binding free energy simulations (L-RB-FEP+), as depicted in Fig. 1a, a reference compound with measured binding affinity to the protein target is alchemically perturbed to a design idea both in solvent and in the context of the protein binding site. The free energy of binding of a design idea relative to the reference compound is computed from the thermodynamic cycle based on these alchemical simulations. To ensure adequate selectivity profiles, the binding affinity of a design idea is often predicted from the alchemical simulations performed in the context of the corresponding off-target binding pockets. Thus, a typical computational workflow, as shown in Fig. 1b, may first predict the potency of design ideas through simulations in the on-target protein and then progress promising designs for potency profiling in the off-target protein. For drug discovery programs where several off-targets are of concern, profiling in multiple distinct off-target protein structures can be performed in a cascading fashion.

a Thermodynamic cycle leveraged by ligand-based relative binding free energy calculations (L-RB-FEP+) to estimate the binding affinity of a design idea (orange) (ΔGexptdesign) relative to experimental binding affinity of reference compound (green) (ΔGexptreference) in the crystallographic structure of AZD1775 (compound 1) within the Wee1 binding pocket (gray surface representation; PDB ID 5V5Y47). Arrows A and B represent the alchemical perturbation between the reference and design idea in solvent (ΔGFEPsolvent) and the binding pocket, (ΔGFEPcomplex), respectively, while arrows 1 and 2 represent experimental binding affinities. b Sample schematic of an L-RB-FEP+ profiling cascade to predict potency and selectivity against three off-targets such that only the most promising designs need to be promoted to subsequent stages.
The accuracy and utility of the L-RB-FEP+ approach as a computational binding affinity assay have been validated extensively, generating predictions for small molecule binding within 1.0 kcal/mol (6–8-fold) of experimental values on average. This error encapsulates contributions from the force field, approximations from finite sampling times, and uncertainties associated with the experimental data4,5,6.
For kinase inhibitors, the high degree of conservation of the ATP-binding site can often lead to a significant number of off-targets being inhibited along with the target of interest, in which case, the physics-based computational approach utilizing individual protein structures for scores of kinases becomes intractable. QSAR7,8, Free-Wilson9, and ML-based10,11,12,13 computational approaches have been proposed for predicting kinome-wide polypharmacology. However, these models are trained using sparse datasets; even with the large numbers of compounds in the respective training sets, relatively few compounds have binding affinity measurements for more than a dozen kinases, and none of the models have been trained across the human kinome. These models are also limited by the chemical diversity represented in the respective training sets and, thus, the predictions may not extrapolate reliably into unexplored chemical space in active drug discovery programs.
Extensive coverage of chemical space is especially important in the context of hit-to-lead and lead optimization phases, where diverse functional groups and core changes are explored routinely. Thus, we propose a physics-based approach to approximate the binding of compounds to off-target kinases by simulating ligand binding in the presence of protein point mutations of the on-target. Point mutations are specifically chosen as selectivity handle residues that contribute to the uniqueness of the binding pocket across the kinome. The differential impact on ligand binding due to crucial sequence changes in the ATP-binding pockets between the on-target and off-target kinases can be modeled using protein residue mutation free energy calculations (PRM-FEP+), which follow a similar thermodynamic cycle as small-molecule relative binding free energy simulations. In L-RB-FEP+ simulations, the ligand is perturbed alchemically in the presence of a specific protein sequence, while in PRM-FEP+ simulations protein residues are perturbed alchemically in the presence of a specific ligand, as depicted in Fig. 2a, b illustrates the corresponding in silico profiling funnel that relies on PRM-FEP+ simulations for modeling selectivity. Protein residue mutation free energy calculations have been used previously for estimating protein-protein binding affinity14,15 and protein thermostability16 as well as predicting effects of resistance mutations on ligand binding17,18. A recent analysis of protein residue mutation FEP+ calculations for over 200 single-point mutations in protein-protein complexes confirmed that the relative binding free energy changes for most protein mutations were correctly predicted within ~1 kcal/mol19, which is sufficiently accurate to positively impact decision-making in drug discovery projects. Herein, we report what is to our knowledge the first successful utilization of protein residue mutation free energy calculations to enable the discovery of highly selective kinase inhibitors, as exemplified by perturbing the Wee1 Asn gatekeeper residue (i.e., selectivity handle) to design novel Wee1 inhibitors with optimal kinome-wide selectivity.

a Thermodynamic cycle leveraged by protein residue mutation free energy calculations (PRM-FEP+) calculations to estimate the binding affinity of a design idea (gray, surface rendering) bound to the wild-type sequence (ribbon + green CPK) (ΔGexptWT) compared to the mutant sequence (ribbon + orange CPK) (ΔGexptmutant) of the Wee1 binding pocket (PDB ID: 5V5Y47). Arrows A and B represent the alchemical perturbation between the wild-type and mutant sequence in solvent (ΔGFEPsolvent) and the binding pocket (ΔGFEPcomplex), respectively, while arrows 1 and 2 represent experimental binding affinities. b Sample schematic of profiling cascade combining ligand-based relative binding free energy calculations (L-RB-FEP+) and protein residue mutation free energy calculations (PRM-FEP+) to identify potent and selective inhibitors efficiently.
With an arsenal of computational strategies available, including active-learning FEP+20, machine-learning models and de novo design workflows21,22,23, our Wee1 program design goals were to discover a potential best-in-class, highly selective molecule containing a new scaffold and balanced drug-like properties, while minimizing drug-drug interactions and off-target binding liabilities. As depicted in Fig. 3, Wee1 is a serine/threonine kinase that is the gatekeeper of the G2-M cell cycle checkpoint. In the presence of errors or damage during DNA replication, cell cycle checkpoint proteins, including ATR, CHK1, and Wee1, and DNA repair machinery work in concert to delay cell cycle progression until sufficient repair has been achieved. Healthy cells repair damaged DNA during G1 arrest, prior to DNA replication in the S phase. However, cancer cells usually have a deficient G1-S checkpoint and rely on later (S and G2-M) checkpoints for DNA repair before mitosis. Wee1 inhibition in cancer cells abrogates the G2-M checkpoint, releases cell cycle brakes, and results in premature catastrophic mitosis and tumor cell apoptosis, all of which make Wee1 an attractive target for oncology drug development.

a Schematic of Wee1 in cell cycle regulation. The cell cycle is composed of four major phases, including G1 (gap 1), S (DNA synthesis), G2 (gap 2) and M (mitosis). Wee1 is a gatekeeper of the G2-M cell cycle checkpoint through phosphorylation of CDK1/2. Inhibition of Wee1 forces cells into unscheduled mitosis and culminates in premature cell death. b Binding pocket representation of AZD1775 bound to Wee1 (PDB ID: 5V5Y47; generated in Maestro (Schrödinger Release 2024-2: Maestro, Schrödinger, LLC, New York, NY, 2024.) c 2D schematic of AZD1775 scaffold with highlighted R1 group occupying the hydrophobic pocket around the gatekeeper (GK) residue, R2 group located under the p-loop and the R3 group protruding into the solvent-exposed region.
While several Wee1 inhibitors have entered clinical development for the treatment of solid tumors, there are currently no approved Wee1 inhibitors. AstraZeneca’s Wee1 inhibitor, AZD1775 (Compound 1), licensed from Merck & Co was studied in multiple Phase 2 clinical trials over a period of nine years before being discontinued due to a narrow therapeutic window mainly driven by combined myelosuppression and gastrointestinal toxicity. Off-target Inhibition of PLK124,25 has been hypothesized to contribute to the observed AZD1775 toxicities. This hypothesis is currently being tested in the ongoing clinical trials of PLK family-sparing Wee1 inhibitors from Debio (Debio 012326,27), Impact (IMP706828) and Aprea (APR-105129). Zentalis is currently investigating its Wee1 inhibitor, ZN-c3, in multiple trials30,31,32,33.
In this communication, we summarize rapid advances during the early stage of developing a novel Wee1 inhibitor. The drug discovery campaign started from the crystallographic structure of AZD1775 (Fig. 3b, c), first, to identify novel chemical series with promising potency against Wee1 and reduced potency against PLK1 using the L-RB-FEP+ and, second, to efficiently optimize the selectivity profile of each series across the kinome by using focused PRM-FEP+.
Results and discussion
Rapid identification of new potent series by free energy calculations
Through the use of large-scale AutoDesigner23 enumerations and hand-drawn ideation, 6.7 billion design ideas covering a wide range of chemical space were explored at the onset of the program. The design ideas were prioritized using physicochemical property filters, docking, as well as machine-learning models. In total, 9000 design ideas were profiled with L-RB-FEP+ within the Wee1 binding pocket to identify those designs that were predicted to be potent against Wee1. Compounds that looked promising in the Wee1 L-RB-FEP+ model were also profiled by L-RB-FEP+ in a crystallographic structure of the PLK1 binding pocket to ensure that the design was predicted to lose potency against the off-target compared to our reference compounds. Through this hierarchical profiling strategy, within 7 months, 80 compounds were prioritized for synthesis and multiple novel series were identified with nanomolar affinity against Wee1 and enhanced selectivity (up to 1000-fold) over PLK1. Among the 80 compounds 7 originated from AutoDesigner workflows and the rest were hand-drawn de novo designs. We recently illustrated the efficiency of this hit-identification strategy in the discovery of a Wee1 inhibitor with a novel 5,5-core34.
Protein residue mutation-free energy calculations recapitulate kinome selectivity patterns
Representative compounds (compounds 2–5 in Fig. 4a) from our novel subseries were profiled in Eurofins’ DiscoverX scanMAX panel of 403 wild-type human kinases. These experiments revealed that while the compounds did not inhibit PLK1, they were hitting many other proteins in the kinome (see Fig. 4a) compared to known Wee1 inhibitors. In our experience, issues of selectivity at this stage of compound optimization could be addressed by adding one or several problematic off-targets to the explicit L-RB-FEP+ modeling cascade to ensure that binding to these off-targets would be avoided in subsequent rounds of synthesis. However, the sheer number of kinases inhibited by the identified leads rendered this approach intractable. Thus, a more sequence-based analysis of the scanMAX panel results was undertaken.

a scanMAX plots for ligands in the retrospective analysis which includes representatives from our novel 5,6-cores (blue shaded) and 6,6-cores (purple shaded), as well as representatives from three known literature scaffolds (gray shaded) of Wee1 inhibitors (pyrazolopyrimidinone core, compound 6; pyrimidine-based tricyclic core45,60, compound 7; and pyrimidopyrimidinone core26, compound 8). For full-size scanMAX plots see Supplementary Information, Fig. SI-1. b Propensity of experimentally observed hits in the scanMAX panel according to gatekeeper (GK) family correlates to PRM-FEP+ pKi predictions for each of the seven ligands bound to mutant Wee1 binding pockets; i.e., each ligand modeled while perturbing from the WT Wee1 binding pocket to mutant binding pockets containing each of the five common gatekeeper mutations in turn (Asn376 → Thr, Phe, Met, Leu, and Val). scanMAX % kinases hit reflects the percentage of kinases with a given GK residue among the scanMAX kinases that have % Ctrl <10, with a minimum of 1% where \({{{\mathrm{\%}}}}{{{\mathrm{Ctrl}}}}{{=}}\frac{{{{\mathrm{test}}}}\; {{{\mathrm{compound}}}}\; {{{\mathrm{signal}}}}{{{\mathrm{-}}}}{{{\mathrm{positive}}}}\; {{{\mathrm{control}}}}\; {{{\mathrm{signal}}}}}{{{{\mathrm{negative}}}}\; {{{\mathrm{control}}}}\; {{{\mathrm{signal}}}}{{{\mathrm{-}}}}{{{\mathrm{positive}}}}\; {{{\mathrm{control}}}}\; {{{\mathrm{signal}}}}}\times 100\%\). Gatekeeper residue assignments across scanMAX kinases, raw scanMAX results and PRM-FEP+ predictions are provided as a Source Data Excel file.
Our novel chemical series included both 5,6-cores (compounds 2 and 3) and 6,6-cores (compounds 4 and 5) and the two scaffolds differed in which kinases they hit: the 6,6-cores more frequently hit kinases with threonine (Thr) and valine (Val) gatekeeper (GK) residues whereas the 5,6-cores hit kinases regardless of gatekeeper residue, including phenylalanine (Phe) and leucine (Leu). Since Wee1 and Wee2 are rare among the kinome with asparagine (Asn) as the gatekeeper residue, protein residue mutation FEP+ simulations were undertaken to estimate the change in binding affinity of a ligand in the presence of single-point mutations from the Asn gatekeeper (i.e., selectivity handle) in the Wee1 binding pocket to other amino acids. With these PRM-FEP+ simulations perturbations to each of the five most common gatekeepers for kinases with approved small-molecule kinase inhibitors, i.e., threonine, phenylalanine, methionine, leucine and valine35, were evaluated. Figure 2a illustrates the thermodynamic cycle used to predict the potency change for a given ligand migrating from Wee1 into other kinase gatekeeper families, where in this context, a gatekeeper family is composed of kinases with a given gatekeeper residue. Thus, for each ligand, pKi’s were predicted for each of the five mutant binding pockets, where each prediction represented the binding propensity to a distinct kinase gatekeeper family.
Binding affinity predictions from PRM-FEP+ calculations for our four compounds, as well as representatives from three known literature scaffolds (compounds 6–8 in Fig. 4a) that were evaluated in scanMAX panels, strongly correlated with the percentage of kinases in the panel that had the corresponding gatekeeper residue and were hit experimentally as depicted in Fig. 4b. These PRM-FEP+ calculations could accurately differentiate between promiscuous ligands (representatives from the 5,6-cores (compounds 2 and 3)), ligands targeting Asn, Thr and Val kinase families (representatives from the 6,6-cores (compounds 4 and 5) and selective Wee1 inhibitors (known literature compounds). The PRM-FEP+ predictions and percentage of kinases hit in scanMAX by gatekeeper are included in the Supplementary Information (Table SI-1). Prevalent binding of a compound to a given gatekeeper family was defined as >10% of members in a gatekeeper family hit in the scanMAX panel and having predicted pKi >8 for the corresponding gatekeeper point mutation in the PRM-FEP+ calculation. With this classification, PRM-FEP+ calculations for five-point mutations modeled across these 7 scaffolds have 91% accuracy, with only 1 false positive and 2 false negatives. This retrospective analysis indicated that the PRM-FEP+ strategy, though it only probes the identity of the gatekeeper residue, is able to capture the likelihood of a compound binding to another gatekeeper family of kinases. Further, this computational approach could suggest which gatekeeper families would need to be targeted for an improved selectivity profile for each scaffold, and a given compound could be classified as “clean” when PRM-FEP+ simulations for all five of the GK perturbations yield predicted pKi’s less than 8. To our knowledge, this is the first application of protein residue mutation FEP+ simulations to estimate kinome-wide selectivity.
Efficient optimization of kinome-wide selectivity by protein residue mutation free energy calculations
With the quantitative retrospective performance of this protein residue mutation FEP+ strategy, the focus turned to prospective optimization of series selectivity. During a three-month design and modeling campaign, more than 6000 compounds were profiled computationally. L-RB-FEP+ was used to estimate on-target Wee1 potency and, for each promising design, five PRM-FEP+ simulations were used to calculate the design’s selectivity. In each of the series, chemical matter was identified that was predicted to be potent and improve selectivity. During these three months, 42 designs were prioritized for synthesis to validate this physics-based approach prospectively, and 22 of these compounds met the project potency and selectivity goals of maintaining single-to-double-digit nanomolar potency on Wee1 as achieved for the early hits while improving kinome selectivity profile to comparable or better than AZD1775. To experimentally validate prospective PRM-FEP+ results, a subpanel of 20 kinases was assembled to cover positive and negative controls for kinases hit or spared within the initial novel series representatives. Kinases were selected for the subpanel based on diversity of location on the traditional kinome tree (see Supplementary Information, Fig. SI-2), distribution of gatekeeper residues, availability of the corresponding binding assays, and availability of crystallographic structures. In the rest of this communication, we present the evolution of three series that were efficiently optimized through this modeling campaign and prospective computational profiling.
Optimization of pyrrolopyrimidine core series
The selectivity profiles of the 5,6-cores were efficiently optimized using protein residue mutation FEP+ calculations and results for key compounds in the series are summarized in Table 1. The initial pyrrolopyrimidine representative, compound 2, that was identified in the original Wee1/PLK1 L-RB-FEP+ profiling cascade, though it did not hit PLK1, hit a relatively even distribution of kinases in the scanMAX panel, i.e., % Control <10 for 14% of the 72 kinases with Thr gatekeepers, 16% of the 149 kinases with Met gatekeepers, and 19% of the 58 kinases with Phe gatekeepers. The corresponding retrospective PRM-FEP+ panel estimates concurred that most of the gatekeeper perturbations would be well-tolerated for compound 2; specifically, mutations from the WT to Phe and Met mutations would lead to increased potency, mutations to Thr and Leu would result in comparable binding affinity, whereas a mutation to Val would lead to ~1 log unit loss of potency. With uncertainties in the PRM-FEP+ simulations on the order of 1 kcal/mol19, a predicted loss of potency greater than 2 kcal/mol for each mutation was targeted in order to dial out off-target liabilities.
While awaiting kinome panel profiling for the representative compounds, other variations of the core from nitrogen-walks around the 5-membered ring were prioritized for synthesis based on Wee1 L-RB-FEP+ potency predictions. From among these compounds, compound 9, with the transposed pyrrolopyrimidine core had measured Wee1 IC50 of 7.8 nM, and was predicted by PRM-FEP+ simulations to be significantly more selective compared to compound 2. Indeed, compound 9 inhibited less than half of the subpanel kinases that were inhibited by compound 2. Given the promise of this pyrrolopyrimidine core containing the nitrile near the gatekeeper residue, the chloro-thiophene directed towards the backpocket and the phenyl piperazine towards solvent, different R-group variations were explored. Within the subsequent two rounds of ideation, in silico profiling and synthesis around this promising core, compound 10 containing an unsubstituted thiophene and pyrazolopiperidine emerged as a clear front-runner and was comparable to AZD1775 with sub-nanomolar potency against Wee1 and similar scanMAX selectivity scores.
Optimization of 6,6-core series
This promising protein residue mutation FEP+ strategy was also applied successfully to optimize the selectivity of the 6,6-cores. In the scanMAX panel, the 6,6-core series representative, compound 4, proved to be promiscuous across the kinome exhibiting % Control <10 for 82% of the kinases with Thr gatekeepers, 30% of the kinases with Met gatekeepers and 9% of the kinases with Phe gatekeepers. Compound 4 showed >100-fold selectivity in only half of the subpanel kinases (see Supplementary Information, Table SI-2, for more details) and was the starting point from which the PRM-FEP+ strategy was employed to identify chemical variations that would be selective for Wee1. Table 2 summarizes the key compounds in the optimization of the selectivity profiles for this 6,6-scaffold.
In the first round of profiling variations of compound 4’s core, compound 11 was predicted in FEP+ to lose some potency against Wee1 yet have significantly fewer selectivity liabilities with more than 2 log units predicted loss of potency in PRM-FEP+ with gatekeeper perturbations from Asn to Thr, Met, Phe, and Leu. On-target L-RB-FEP+ and off-target PRM-FEP+ predictions were realized as compound 11 had measured potency of 22 nM and, remarkably, had more than 100-fold loss of potency in each of the 20 kinases in the subpanel compared to Wee1.
The next round of optimization of this core included variations of the solvent-exposed group and minor modifications of the core substituents. From among these designs, compound 12 was selected for synthesis based on the combined L-RB-FEP+ and PRM-FEP+ strategy and was confirmed experimentally to maintain the exquisite selectivity profile in the kinase subpanel while improving the measured Wee1 potency to 1 nM. Furthermore, compound 12 when profiled in the scanMAX panel hit only 3 kinases with % Control <10: ABL1, MAP3K4 and Wee1. The measured potency in our in-house ABL1 assay was 1.9 µM, suggesting a false positive in the scanMAX panel.
Discovery of a novel potent scaffold while maintaining kinome selectivity
Another common need in drug discovery programs is the ability to explore unexplored chemical space to optimize scaffolds for various properties while maintaining a favorable on-target potency and selectivity profile against off-targets. Here, the applicability of the developed L-RB-FEP+ and PRM-FEP+ workflow for predicting potency and kinome selectivity was investigated for de novo core designs inspired by compound 8, a representative of the pyrimidopyrimidinone-based Wee1 inhibitors from Debiopharm26,36. Compound 8 had a measured potency of 0.6 nM, and the scanMAX panel confirmed that only 8 kinases were hit with less than 10% control remaining.
Series of compounds with tricyclic cores based on compound 8 were designed and evaluated in the combined L-RB-FEP+ and PRM-FEP+ workflow and Table 3 summarizes the key compounds discovered in the process. The prototype design, compound 13, was prioritized for synthesis and, indeed, had a measured potency of 42 nM for Wee1 and maintained >100-fold loss of potency in each of the 20 kinases in the subpanel compared to Wee1. The second round of optimization around the tricyclic scaffold focused on improving on-target potency while continuing to demonstrate selectivity. Through FEP+ profiling of hand-drawn designs as well as designs from an AutoDesigner enumeration, 19 compounds were prioritized for synthesis, and each one improved the Wee1 potency to between 1 and 15 nM, with nine compounds exhibiting Wee1 IC50s < 5 nM. Several compounds were profiled in the kinase subpanel and, as illustrated by compound 14 in Table 3, selectivity windows for all kinases in the subpanel were excellent. Compound 14 was also selective in the full scanMAX panel compared to the initial 6,6-cores with only 14 kinases hit with % Control <10, thereby confirming successful and efficient identification of a novel potent and selective scaffold (see Supplementary Information, Table SI-3, for more details).
Cellular activity and ADME properties profiling
To demonstrate cellular on-target activity for identified selective Wee1 inhibitors we have evaluated the molecular effects of the compounds in cancer cell lines, specifically the target engagement of Wee1 measured by phosphorylation of Wee1’s substrate CDK1 (CDC2) at Tyr15, and inhibition of cell viability in A427 (non-small cell lung cancer) and OVCAR3 (ovarian cancer) cells. All these compounds demonstrate the inhibition of Wee1 signaling in cells and anti-tumor activity in vitro. The data including the benchmark compounds AZD1775 and Compound 8 is provided in Supplementary Information, Table SI-5. In addition, identified Wee1-selective inhibitors were profiled in a subset of ADME assays and data is summarized in Supplementary Information, Table SI-6.
Generalizing selectivity optimization strategy across kinases
While optimization of Wee1 inhibitors was achieved from modeling perturbations of the gatekeeper Asn residue in the kinase ATP pocket, this physics-based strategy is applicable whenever a sufficiently high-quality model of the on-target is available, including a validated binding mode of the chemical matter, and where a selectivity handle can be identified for the on-target. In this context, a selectivity handle is any residue or combination of residues in the vicinity of the binding pocket that differ between the on-target and off-targets that contributes to selective on-target binding. Various approaches have been reported to propose selectivity handles based on sequence variability and/or structural differences. For example, Huang et al.37 presented a network analysis of kinase ATP-binding pockets to estimate the potential for selective inhibition of each kinase and the resulting network could be used to infer key residues to target for designing selective inhibitors for a given target. In another approach, Wang et al.38 developed a Human Kinase Pocket database (HKPocket) including 1717 pockets from 255 kinases. From the resulting 91 pocket clusters sequence conservation, critical interactions, and pocket hydrophobicity could guide the identification of selectivity handle residues. Zhang et al.39 identified structurally-defined binary units, that is, pairs of residues in close proximity to one another within kinase binding pockets. Their analysis of binary units across the kinome demonstrated that all 495 kinases contain binary units that are shared by less than seven other kinases and 331 kinases have at least a single unique binary unit that may distinguish it from all other kinases. Thus, these rare or unique binary units, as visualized in their Kinase Drug Selectivity software, could become the two selectivity handle residues proposed to perturb in the PRM-FEP+ simulations to efficiently optimize the on-target selectivity of a chemical series or ensure its selectivity will be maintained as other properties are optimized.
This physics-based approach could also be extended beyond kinases to protein families where ligand binding pockets within the protein family have significant sequence and structural homology, including, protease active sites40 intracellular domains within GPCRs41 and the DNA-binding domain of nuclear hormone receptors, which contain conserved zinc finger motifs42.
Structural insights
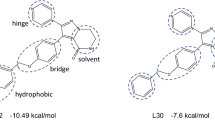
Crystallographic structures obtained for each of the three subseries confirm overall binding modes that are very similar to those observed for AZD1775 (1) bound to Wee1 (PDB ID: 5V5Y in Fig. 3b), i.e., two H-bond interactions with main-chain atoms of Cys379 in the hinge region, one H-bond acceptor interacting with the terminal NH2 group of the gatekeeper Asn376 sidechain, an R1 group occupying the hydrophobic pocket around the GK residue, an R2 group under the p-loop and an R3 group protruding into the solvent-exposed pocket.
The central fused rings of each scaffold are coplanar with those of AZD1775, have the same hinge binding motif and similar π-stacking interactions with Phe433. However, each series achieves the H-bond with Asn376 by different means: the 5,6-core represented by compound 10 replaces the carbonyl with a nitrile; the 6,6-core represented by compound 11 utilizes the exocyclic amide carbonyl, while the tricyclic core represented by compound 14 employs a lactam carbonyl.
Other research groups have proposed structural rationales in the back hydrophobic pocket for potency and selectivity profiles of Wee1 inhibitors. Palmer et al.43 explored the SAR around the phenyl ring occupying this pocket, though they could not design compounds that significantly improved the selectivity of the compounds for Wee1 over c-Src. Matheson et al.44 proposed modulating the electronic character of the R1 group to impact the strength of the hydrogen bond between the pyrazolidinone carbonyl and Asn376. Tong et al.45 in their analysis of AbbVie pyrimidine-based tricyclic Wee1 inhibitors suggested that the hydrogen bond with the gatekeeper residue, Asn376, promotes selectivity since human kinases rarely have H-bond donor residues in the gatekeeper position (<0.5% across 500 kinases). Guler et al.46 targeted features of the selectivity pocket around the gatekeeper to enhance the selectivity of Wee1 inhibitors over PLK1.
Within our chemical series, the thiophene of compound 10 occupies the same region in the backpocket as the allyl group in AZD1775. The backpocket di-Cl phenyl group of compound 14 overlays almost exactly with that in the crystallographic structure of PD-166285 (15, PDB ID: 5VC546,47, Fig. 5d), laying perpendicular to the core and forming a halogen bond with the backbone NH of Ala326. The di-Cl phenyl group of compound 11 has a slightly different orientation in the pocket and halogen bonds with the backbone of Asn376 instead. As can be seen in the overlay of compounds 10, 11, and 14 with AZD1775 in Fig. 5e, none of the representative compounds in our scaffolds occupy the region under the p-loop to the same extent as the decorated phenyl group in AZD1775. While potency can be enhanced with larger groups at this R2 position, they were not necessary in these series for selectivity gains.

a Compound 10 (PDB ID: 9D0R), b compound 11 (PDB ID: 9D0Q), c compound 14 (PDB ID: 9D0S), d PD-166285 (compound 15; PDB ID: 5VC547), and e overlay of compounds 10, 11, and 14 (green) with AZD1775 (orange) in the Wee1 binding pocket (select residues for 5V5Y displayed in cyan ribbons and gray surface). All ligand interaction diagrams generated in Maestro (Schrödinger Release 2024-2: Maestro, Schrödinger, LLC, New York, NY, 2024).
In their SAR analysis of Wee1 inhibitors, Tong et al.45 noted that a monocyclic aromatic group attached to the core is able to fit through the narrow channel towards the solvent and that bulkier groups are tolerated within the solvent-exposed pocket itself. Consistent with this trend, in each of our scaffolds, a planar ring (i.e., phenyl or pyrazole) at R3 is used to exit through the solvent channel while a bulkier group with more 3-dimensional character occupies the solvent-exposed pocket. However, the geometry of the core influences the exit vector. For example, the piperazines of compound 11 and AZD1775 occupy a similar region in the solvent pocket while the R3 vectors of compounds 10 and 14 are at an ~30° angle relative to AZD1775, leaving the corresponding piperidines of compounds 10 and 14 displaced by ~5 Å relative to AZD1775.
Physics-based insights beyond qualitative SAR
While understanding the SAR in the various parts of the binding pocket is a significant help in designing potent and selective inhibitors for a given target, rigorous physics-based strategies can identify non-intuitive activity cliffs and non-additive SAR across regions of the binding pocket. Rather than the presence or absence of hydrogen bonding to Asn376 being the primary criterion for predicting selectivity, PRM-FEP+ calculations account for highly complex compensatory effects, including the desolvation of the ligand to enter the binding pocket, the loss of entropy upon ligand binding, as well as the interplay among all of the protein-ligand interactions within the pocket. For example, AZD1775 and the matched pair in which the allyl moiety is replaced with a methyl group (compound 16) both maintain the pyrazole carbonyl interaction with Asn376. While AZD1775 is about 10-fold more potent than compound 16, it is also more selective in the kinase subpanel as summarized in Table 4.
Protein residue mutation FEP+ predictions efficiently drove the selectivity optimization campaign by providing estimates of potency losses that went beyond qualitative rationalizations. For some point mutations in the presence of AZD1775 and compound 16 could be rationalized based on steric bulk; for example, replacing Asn at the gatekeeper position with Phe results in a predicted loss of potency (ΔpKi −2.1) due to the bulkier Phe sidechain clashing with the allyl group in AZD1775 while this bulkier Phe is well accommodated by the compact methyl group in compound 16 (ΔpKi 0.3). However, the magnitude of the predicted potency losses were not consistent for each of the five point mutations assessed and could not be accounted for by simplistic differences in hydrogen bonding patterns or steric bulk and even counting interactions throughout the simulation trajectory itself does not correlate with predicted free energy losses or gains.
Early on in the program, we identified diverse R1 replacements of the allyl group on AZD1775 that would maintain potency and not bind to PLK1. PRM-FEP+ calculations were then used to rapidly identify candidates that were likely to be selective across the kinome. Compound 17 is one such compound which was predicted to be a weak binder for kinases with Phe and Leu gatekeepers with ~3 pKi loss of potency and only be a modest binder to kinases with Thr and Met gatekeepers with pKi losses of 1–1.5 log units. Compound 17, indeed, across the subpanel of 20 kinases demonstrated a marked improvement in selectivity compared to compound 16 and has a comparable profile to AZD1775 (see Supplementary Information, Table SI-4, for more details).
Summary
Chemical series exploration and compound prioritization in drug discovery campaigns are streamlined using free energy calculations to predict on-target potency accurately and efficiently. Routine experimental screening of large kinase panels to identify selective compounds would be time-consuming and cost-prohibitive. While focused in vitro mutagenesis experiments to survey gatekeeper point mutations of the wild-type sequence would reduce the expense of the binding assays, the investment of resources to synthesize enough compounds, often through many DMTA cycles, remains in order to identify selective candidates. Rigorous physics-based simulations can identify selective compounds to guide synthesis decisions. However, relative binding FEP+ (L-RB-FEP+) to evaluate selectivity can become intractable when many off-targets must be simulated individually, given the need for high-resolution structural models of each off-target and high computational resources. Here, we demonstrate how PRM-FEP+ can be leveraged to rapidly tune the selectivity profile against a family of off-targets and exemplify how strategic use of PRM-FEP+ enabled the transformation of diverse scaffolds from promiscuous to Wee1-selective inhibitors and, for another series, ensured the maintenance of a favorable selectivity profile. In this work, the gatekeeper residue in the kinase ATP pocket was used as a selectivity handle and was systematically perturbed from the Wee1 Asn to other common gatekeeper residues to model the binding propensity of design ideas across the broader kinome. This approach, however, is not limited to modeling gatekeeper differences or to kinases, but can be applied whenever there are more off-targets than can be modeled individually and where interactions within on-target and the problematic off-targets can be differentiated by a single or small number of point mutations in the vicinity of the binding pocket. With reliable physics-based strategies for modeling on-target potency and dialing out off-target liabilities, drug discovery campaigns can become more efficient by rapidly identifying promising development candidates.
Methods
FEP+ calculations
All simulations were performed using the OPLS4 force field6 with the FEP/REST sampling method within the Desmond GPU MD simulation package4. Most compounds were profiled using Schrödinger Release 2019-3: Maestro, Schrödinger, LLC, New York, NY, 2019. Compound 10 was profiled using Schrödinger Release 2019-4, and Compounds 12 and 14 were profiled using Schrödinger Release 2020-1. The Force Field builder was used to obtain customized torsional parameters for the small molecules. Input structures were solvated with a 5 Å buffer of simple point charge water in a rectangular box and equilibrated through a series of minimization and restrained MD phases. Each equilibrated structure was then subjected to a 10 ns FEP/REST simulation. The FEP/REST phase was performed in the isothermal-isobaric ensemble with a Berendsen thermostat and barostat. The equations of motion were integrated using a reversible reference system propagator algorithm scheme with an inner time step of 2.0 fs and an outer time step of 6.0 fs. The change in Gibbs free energy, ΔG, was computed from the difference between the bound and unbound simulations, giving the relative binding affinity (ΔΔG). For Wee1 simulations, the publicly available crystallographic structure with AZD1775 (PDB ID: 5V5Y47) bound was used, while for PLK1 simulations, a proprietary crystallographic structure with AZD1775 bound was used (PDB ID: 9D0P).
For small-molecule perturbations, by default, 12 lambda windows were used in a 10 ns FEP/REST schedule to perturb from a reference compound to a design idea. For initial profiling of designs in large libraries, short triaging simulations of 2 ns were utilized. For charged perturbations and for larger perturbations, up to 36 lambda windows and simulation lengths up to 25 ns were used to ensure adequate simulation convergence. The perturbations for the bound simulations were performed in the context of the Wee1 or PLK1 binding pocket with wild-type sequence, and the unbound simulations were performed in a simulated water box.
For all protein residue perturbations, 12 lambda windows were used in the 10 ns FEP/REST schedule to perturb residue 376 in Wee1 from the wild-type Asn residue to either Thr, Phe, Met, Val, or Ile and residue 376 was included in the REST hot region. The perturbations for the bound simulations were performed in the presence of a small-molecule and the unbound simulations were modeled using apo structures.
General information for compound availability and synthesis
AZD1775 was acquired through commercial sources. For the other synthesized compounds detailed experimental procedures and characterization data are available in the Supplementary Information.
Wee1 kinase biochemical assay
The Wee1 kinase domain was obtained from Carna (catalog #05-177). Poly-(Lys, Tyr 4:1) hydrobromide was used as a substrate (Sigma-Aldrich) to assess the activity of Wee1. ADP production was measured with the ADP-Glo Kinase Assay kit (Promega). The kinase reaction was conducted in a buffer of 40 mM Tris-HCl, 30 mM MgCl2 supplemented with 0.1 mg/mL bovine serum albumin, and 2 mM DDT. The final reaction mix contained 1 nM Wee1 enzyme, 15 µM ATP, and 2 ng/mL Poly-(Lys, Tyr 4:1) hydrobromide substrate, with a reaction time of 4 h at 25 °C. The Envision plate reader was used to measure the ADP-Glo signal. Per cent inhibition was calculated by Eq. 1.
where Ssample is the signal of compounds, SHighCtrl the signal of the high control (DMSO) and SLowCtrl the signal of the low control (positive inhibition at saturating concentration).
PLK1 kinase biochemical assay
The full-length PLK1 kinase was obtained from Carna (catalog #05-157). Dephosphorylated Native Cow Casein protein was used as a substrate (Abcam) to assess the activity of PLK1. ADP production was measured with the ADP-Glo Kinase Assay kit (Promega). The kinase reaction was conducted in a buffer of 40 mM Tris-HCl, 20 mM MgCl2 supplemented with 0.1 mg/mL bovine serum albumin, and 2 mM DDT. The final reaction mix contained 10 nM PLK1 enzyme, 3 µM ATP, and 5 µM Native Cow Casein protein substrate, with a reaction time of 2 h at 25 °C. The Envision plate reader was used to measure the ADP-Glo signal. Percent inhibition was calculated from Eq. 1.
Cellular assays
Details of the experimental protocols for the cellular target engagement assay (phosphorylated CDC2) and cell viability assay are provided in Supplementary Information.
Subpanel kinase selectivity assays
ADP-Glo assays at either two-dose titration or full-dose titration were performed for ABL, PDGFRb, CDK9, FLT3, HIPK2, TRKA, AKT1, MAP4K2, AXL, MERTK, RET, and SLK.
MSA (mobility shift assays) at either two-dose titration or full dose titration were performed for ERK2, KIT, CLK4, DYRK1a, JAK2, NEK2, IRAK4, and CSNK1E. Peptide substrates, reagents and reaction times are indicated in Table 5. Reactions were performed with 1.5 µM peptide substrate in a buffer containing 50 mM HEPES at pH 7.5 supplemented with 10 mM MgCl2, 0.05% Brij-35, 2 mM DTT, 0.05% BSA, 1 mM EGTA. Reactions were incubated for the time indicated in Table 5 at 25 °C before terminating the reaction and reading the signal on EZ Reader (PerkinElmer). All assays were performed at ATP Km.
Binding affinity and kinase selectivity
Binding affinity (KdELECT) and kinase selectivity (scanMAX) assays were performed at Eurofins DiscoverX Corporation using KINOMEscan platform, which utilizes an active site-directed competition binding assay to measure interactions between test compounds and selected human kinases, without the need for ATP. Compounds were screened at 500 nM or 1 μM in the scanMAX assay covering 468 human kinases. For selected kinase hits, compounds were followed up with dose–response to determine the binding affinity using KdELECT assay. Protocols are briefly described below; please see vendor’s website for additional assay description: https://www.eurofinsdiscovery.com/catalog/scanmax-kinase-panel-kinomescan-discoverx/87-0001-1000.
KINOMEscan protocol description
Most kinases were produced from kinase-tagged T7 phage strains grown in an Escherichia coli host derived from the BL21 strain. E. coli were grown to log-phase before infection with T7 phage (multiplicity of infection = 0.4), and incubation with shaking at 32 °C for 90–150 min. Cells were lysed and lysates centrifuged and filtered to remove cell debris. The remaining kinases were produced in HEK-293 cells and subsequently tagged with DNA for qPCR detection.
Streptavidin-coated magnetic beads were treated with biotinylated small-molecule ligands for 30 min at room temperature to generate affinity resins for kinase assays. Ligand-bound beads were then blocked with excess biotin to remove unbound ligand and reduce non-specific binding and washed with a blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT). Binding reactions included kinases, ligand-bound beads, and test compounds in 1× binding buffer (20% SeaBlock, 0.17× PBS, 0.05% Tween 20, 6 mM DTT).
Test compounds for percentage inhibition assays were prepared as 100× stocks in 100% DMSO, whereas for Kd assays, they were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point threefold serial dilution of each compound. Prepared compounds were directly diluted into the assays. All reactions were carried out in polypropylene 384-well plates in a final volume of 0.02 mL incubated at room temperature with shaking for 1 hour followed by a wash in a buffer containing 1× PBS and 0.05% Tween 20. The beads were resuspended in an elution buffer containing 1× PBS, 0.05% Tween 20, and 0.5 μM non-biotinylated affinity ligand, and incubated at room temperature with shaking for 30 min. Kinase concentration in the eluate was measured using qPCR.
Determination of percentage inhibition and Kd
In case of percentage inhibition assays, test compounds were screened at a single concentration of 500 or 1000 nM, and the percentage inhibition of a kinase was calculated as follows:
where the negative control is DMSO (0% inhibition) and the positive control is the control compound (100% inhibition).
Kd’s were calculated with a standard dose–response curve using the Hill equation:
The Hill Slope was set to −1, and curves were fitted using a non-linear least square fit with the Levenberg–Marquardt algorithm.
Crystallographic structure determination
The PLK1 crystallographic construct was produced and crystallized, and the structure was determined as described48. Data was collected at a temperature of 100° K with radiation at a wavelength of 0.99990 Å at Swiss Light Source beamline X10SA. The model was initially refined with the program REFMAC49. Manual model building and refinement were completed using Maestro and PrimeX50.
The Wee1 crystallization construct was produced as described51. Crystals of the Wee1 complex with compounds 10 or 14 were obtained using the sitting-drop vapor diffusion method. Wee1 at a concentration of 6.9 mg/ml (50 mM Tris/HCl, 150 mM NaCl, 1 mM TCEP, pH 8.0) was pre-incubated with 1.0 mM (4.7-fold molar excess) compound (150.0 mM in DMSO) for 1 h. 1 µL of the protein solution was then mixed with 1 µL of reservoir solution (0.10 M Hepes/NaOH pH 6.9, 3.4 M NaCl) and equilibrated at 12 °C over 0.6 ml of reservoir solution. Diffraction data were collected a temperature of 100° K at the BESSY synchrotron radiation source beamline 14.1 using the Pilatus3 S 6 M detector and radiation with a wavelength of 0.91840 Å. Data was integrated using MOSFLM52, with merging and scaling using SCALA53.
Crystals of the Wee1 complex with compound 11 were obtained using the sitting-drop vapor diffusion method. Wee1 at a concentration of 6.9 mg/ml (50 mM Tris/HCl, 150 mM NaCl, 1 mM TCEP, pH 8.0) was pre-incubated with 1 mM (4.7-fold molar excess) of compound 11 (150 mM in DMSO) for 1 h. 0.2 µL of the protein solution was then mixed with 0.02 µL of a seed stock and 0.18 µL of reservoir solution (0.1 M bis-tris-propane pH 7.0, 2.8 M sodium acetate pH 7.0) and equilibrated at 20 °C over 0.2 mL of reservoir solution. Diffraction data was collected at a temperature of 100° K at the Diamond Light Source beamline i04-1 using the Pilatus 6M-F detector and radiation with a wavelength of 0.91210 Å. Data were integrated using Dials54, and merged and scaled using Aimless55.
All three Wee1 structures were determined by molecular replacement with PHASER56 in the CCP4 program suite57 utilizing PDB code 2IN658 as the search model. The model was initially refined with the program REFMAC49. Manual model building and refinement were completed using Maestro and PrimeX50.
The coordinates and structure factors for all four structures have been deposited in the Protein Data Bank. Additional information is presented in the Supplementary Information, Table SI-7 and Figs. SI-3 and SI-4.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The crystallographic structure data generated in this study have been deposited in the PDB under accession codes 9D0P, 9D0Q, 9D0R, and 9D0S. Initial and final configurations for the ligand FEP+ and protein residue mutation FEP+ simulations are available at Zenodo59. Supplementary Information contains the kinase gatekeeper assignments (Supplementary Dataset 1), raw ScanMAX results (Supplementary Dataset 2, Fig. SI-1), location of subpanel kinases on a phylogenetic tree of the human kinome (Fig. SI-2), more detailed simulation results and ScanMAX results (Tables SI-1 through SI-4), cellular (Table SI-5), ADME data (Table SI-6) and experimental procedures including characterization data for proprietary compounds (Table SI-7 and Figs. SI-3 and SI-4). Source data are provided with this paper.
References
Davis, M. I. et al. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 (2011).
Armacost, K. A. & Thompson, D. C. Free energy methods in drug discovery: current state and future directions. (American Chemical Society (ACS), Washington, DC, 2021).
Adediwura, V. A., Koirala, K., Do, H. N., Wang, J. & Miao, Y. Understanding the impact of binding free energy and kinetics calculations in modern drug discovery. Expert Opin. Drug Discov. 19, 671–682 (2024).
Wang, L. et al. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J. Am. Chem. Soc. 137, 2695–2703 (2015).
Ross, G. A. et al. The maximal and current accuracy of rigorous protein-ligand binding free energy calculations. Commun. Chem. 6, 222 (2023).
Lu, C. et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 17, 4291–4300 (2021).
Kothiwale, S., Borza, C., Pozzi, A. & Meiler, J. Quantitative structure-activity relationship modeling of kinase selectivity profiles. Molecules 22, 1576 (2017).
Sheinerman, F. B., Giraud, E. & Laoui, A. High affinity targets of protein kinase inhibitors have similar residues at the positions energetically important for binding. J. Mol. Biol. 352, 1134–1156 (2005).
Sciabola, S., Stanton, R. V., Johnson, T. L. & Xi, H. Application of Free–Wilson selectivity analysis for combinatorial library design. Chem. Lib. Des. 685, 91–109 (2011).
Merget, B., Turk, S., Eid, S., Rippmann, F. & Fulle, S. Profiling prediction of kinase inhibitors: toward the virtual assay. J. Med. Chem. 60, 474–485 (2017).
Li, X. et al. Deep learning enhancing kinome-wide polypharmacology profiling: model construction and experiment validation. J. Med. Chem. 63, 8723–8737 (2019).
Wu, J. et al. Large-scale comparison of machine learning methods for profiling prediction of kinase inhibitors. J. Cheminf. 16, 13 (2024).
Schifferstein, J., Bernatavicius, A. & Janssen, A. P. A. Docking-informed machine learning for kinome-wide affinity prediction. J. Chem. Inf. Model. 64, 9196–9204 (2024).
Clark, A. J. et al. Free energy perturbation calculation of relative binding free energy between broadly neutralizing antibodies and the gp120 glycoprotein of HIV-1. J. Mol. Biol. 429, 930–947 (2017).
Clark, A. J. et al. Relative binding affinity prediction of charge-changing sequence mutations with fep in protein-protein interfaces. J. Mol. Biol. 431, 1481–1493 (2019).
Duan, J., Lupyan, D. & Wang, L. Improving the accuracy of protein thermostability predictions for single point mutations. Biophys. J. 119, 115–127 (2020).
Smith, M. B. K. et al. Monte Carlo calculations on HIV-1 reverse transcriptase complexed with the non-nucleoside inhibitor 8-Cl TIBO: contribution of the L100I and Y181C variants to protein stability and biological activity. Protein Eng. Des. Sel. 13, 413–421 (2000).
Hauser, K. et al. Predicting resistance of clinical Abl mutations to targeted kinase inhibitors using alchemical free-energy calculations. Commun. Biol. 1, 70 (2018).
Sampson, J. M. et al. Robust prediction of relative binding energies for protein-protein complex mutations using free energy perturbation calculations. J. Mol. Biol. 436, 168640 (2024).
Knight, J. L., Leswing, K., Bos, P. H. & Wang, L. Impacting drug discovery projects with large-scale enumerations, machine learning strategies, and free-energy predictions. In Free Energy Methods in Drug Discovery: Current State and Future Directions, 205–226 (American Chemical Society, Washington, DC, 2021).
Konze, K. D. et al. Reaction-based enumeration, active learning, and free energy calculations to rapidly explore synthetically tractable chemical space and optimize potency of cyclin-dependent kinase 2 inhibitors. J. Chem. Inf. Model. 59, 3782–3793 (2019).
Ghanakota, P. et al. Combining cloud-based free-energy calculations, synthetically aware enumerations, and goal-directed generative machine learning for rapid large-scale chemical exploration and optimization. J. Chem. Inf. Model. 60, 4311–4325 (2020).
Bos, P. H. et al. AutoDesigner, a design algorithm for rapidly exploring large chemical space for lead optimization: application to the design and synthesis of d-amino acid oxidase inhibitors. J. Chem. Inf. Model. 62, 1905–1915 (2022).
Matheson, C. J. et al. A WEE1 inhibitor analog of AZD1775 maintains synergy with cisplatin and demonstrates reduced single-agent cytotoxicity in medulloblastoma cells. ACS Chem. Biol. 11, 921–930 (2016).
Wright, G. et al. Dual targeting of WEE1 and PLK1 by AZD1775 elicits single agent cellular anticancer activity. ACS Chem. Biol. 12, 1883–1892 (2017).
Hewitt, P., Burkamp, F., Wilkinson, A., Miel, H. & O’dowd, C. Pyrimidopyrimidinones useful as Wee-1 kinase inhibitors. World Patent WO2018162932A1, (2018).
Papadopoulos, K. P. et al. Results of a phase 1, dose-finding study of Debio 0123 as monotherapy in adult patients with advanced solid tumors: safety, pharmacokinetic, and preliminary antitumor activity data. J. Clin. Oncol. 42, 3120 (2024).
Cai, S. X. et al. Abstract 3091: discovery and development of a potent and highly selective WEE1 inhibitor IMP7068. Cancer Res. 83, 3091 (2023).
US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/study/NCT06260514 (2024).
Meric-Bernstam, F. et al. Abstract CT029: Safety and clinical activity of single-agent ZN-c3, an oral WEE1 inhibitor, in a phase 1 trial in subjects with recurrent or advanced uterine serous carcinoma (USC). Cancer Res 82, CT029 (2022).
Huang, P. Q. et al. Discovery of ZN-c3, a highly potent and selective Wee1 inhibitor undergoing evaluation in clinical trials for the treatment of cancer. J. Med. Chem. 64, 13004–13024 (2021).
Liu, J. F. et al. Phase II study of the WEE1 inhibitor adavosertib in recurrent uterine serous carcinoma. J. Clin. Oncol. 39, 1531–1539 (2021).
Tolcher, A. et al. Abstract CT016: clinical activity of single-agent ZN-c3, an oral WEE1 inhibitor, in a phase 1 dose-escalation trial in patients with advanced solid tumors. Cancer Res 81, CT016 (2021).
Bos, P. et al. AutoDesigner - core design, a de novo design algorithm for chemical scaffolds: application to the design and synthesis of novel selective Wee1 inhibitors. J. Chem. Inf. Model. 64, 7513–7524 (2024).
Zhou, Y., Xiang, S., Yang, F. & Lu, X. Targeting gatekeeper mutations for kinase drug discovery. J. Med. Chem. 65, 15540–15558 (2022).
Harrison, T. et al. Pyrimidopyrimidinones useful as Wee-1 kinase inhibitors. World Patent WO2015092431A1 (2015).
Huang, D., Zhou, T., Lafleur, K., Nevado, C. & Caflisch, A. Kinase selectivity potential for inhibitors targeting the ATP binding site: a network analysis. Bioinformatics 26, 198–204 (2010).
Wang, H. et al. HKPocket: human kinase pocket database for drug design. BMC Bioinformatics 20, 617 (2019).
Zhang, M., Liu, Y., Jang, H. & Nussinov, R. Strategy toward kinase-selective drug discovery. J. Chem. Theory Comput. 19, 1615–1628 (2023).
Ćwilichowska, N., Świderska, K. W., Dobrzyń, A., Drąg, M. & Poręba, M. Diagnostic and therapeutic potential of protease inhibition. Mol. Asp. Med. 88, 101144 (2022).
Yang, D. et al. G protein-coupled receptors: structure- and function-based drug discovery. Signal Transduct. Target. Ther. 6, 7 (2021).
Helsen, C. et al. Structural basis for nuclear hormone receptor DNA binding. Mol. Cell. Endocrinol. 348, 411–417 (2012).
Palmer, B. D. et al. Structure-activity relationships for 2-anilino-6-phenylpyrido[2,3-d]pyrimidin-7(8H)-ones as inhibitors of the cellular checkpoint kinase Wee1. Bioorg. Med. Chem. Lett. 15, 1931–1935 (2005).
Matheson, C. J., Casalvieri, K. A., Backos, D. S. & Reigan, P. Development of potent pyrazolopyrimidinone-based WEE1 inhibitors with limited single-agent cytotoxicity for cancer therapy. ChemMedChem 13, 1681–1694 (2018).
Tong, Y. et al. Pyrimidine-based tricyclic molecules as potent and orally efficacious inhibitors of Wee1 kinase. ACS Med. Chem. Lett. 6, 58–62 (2015).
Guler, S. et al. Selective Wee1 inhibitors led to antitumor activity and correlated with myelosuppression. ACS Med. Chem. Lett. 14, 566–576 (2023).
Zhu, J.-Y. et al. Structural basis of Wee kinases functionality and inactivation by diverse small molecule inhibitors. J. Med. Chem. 60, 7863–7875 (2017).
Rudolph, D. et al. BI 6727, a polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 15, 3094–3102 (2009).
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 (1997).
Bell, J. A. et al. In International Tables for Crystallography, (ed. E. Arnold, D. M. H. & M. G. R.) vol. F, 534–538 (New York: Wiley, 2012).
Squire, C. J., Dickson, J. M., Ivanovic, I. & Baker, E. N. Structure and inhibition of the human cell cycle checkpoint kinase, Wee1A kinase: an atypical tyrosine kinase with a key role in CDK1 regulation. Structure 13, 541–550 (2005).
Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 (2011).
Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 (2006).
Winter, G. et al. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. D Struct. Biol. 74, 85–97 (2018).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Agirre, J. et al. The CCP4 suite: integrative software for macromolecular crystallography. Acta Crystallogr. D Struct. Biol. 79, 449–461 (2023).
Smaill, J. B. et al. Synthesis and structure-activity relationships of N-6 substituted analogues of 9-hydroxy-4-phenylpyrrolo[3,4-c]carbazole-1,3(2H,6H)-diones as inhibitors of Wee1 and Chk1 checkpoint kinases. Eur. J. Med. Chem. 43, 1276–1296 (2008).
Bos, P. et al. Harnessing free energy calculations for kinome-wide selectivity in drug discovery campaigns with a Wee1 case study (v1.0.0). Zenodo. https://doi.org/10.5281/zenodo.15882242 (2025).
Tong, Y., Penning, T. D., Florjancic, A. S., Miyashiro, J. & Woods, K. W. Tricyclic inhibitors of kinases. US Patent US20120220572A1 (2012).
Acknowledgements
The authors thank Howook Hwang for python development work for the kinase gatekeeper annotations, Alfred Cervantes and James Evans for assistance with the images, Laura Madden for strategic discussions, our partners in medicinal chemistry, biology and crystallography and our Schrödinger colleagues and leadership for logistical support and fruitful discussions.
Author information
Authors and Affiliations
Contributions
A.J.C. conceived of using protein residue mutation FEP for broad kinome profiling and designed compounds. A.J.C. and J.L.K. performed and analyzed FEP and protein residue mutation FEP simulations and identified top compounds for synthesis. J.W., A.P., and A.I.G. designed compounds, identified compounds for synthesis and oversaw all syntheses. P.H.B. and S.B. designed compounds through AutoDesigner enumerations and identified top compounds for synthesis. J.A.B. oversaw the crystallography and refinement to prepare structures for use in FEP simulations. S. Silvergleid and W.Y. compiled the subpanel of kinases and oversaw assay development. S. Silvergleid, W.Y., F.G., and S. Sun oversaw all in vitro experiments. K.A. and R.A. facilitated the computational and experimental work of the team. J.L.K., P.H.B., J.W., and A.I.G. outlined the manuscript, and J.L.K. wrote the first draft. All authors contributed to the paper editing and preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Knight, J.L., Clark, A.J., Wang, J. et al. Harnessing free energy calculations for kinome-wide selectivity in drug discovery campaigns with a Wee1 case study. Nat Commun 16, 7962 (2025). https://doi.org/10.1038/s41467-025-62722-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62722-w
