
Abstract
Difluoromethylene moiety has gained widespread applications in pharmaceuticals, agrochemicals, and materials owing to its augmented lipophilicity and being bioisosteric to ethereal oxygen. Possessing two orthogonal reactivity modes for bridging an electrophile and a radical acceptor to give gem-difluorides (R1-CF2-R2), the efficient difluoromethylene radical anion synthon (diFRAS) has been long sought after. In this work, we successfully utilize the readily available difluoromethyl phenyl sulfone (PhSO2CF2H) to couple with electrophiles and radical acceptors, thereby enabling PhSO2CF2H to serve as a novel diFRAS in organic synthesis. The generation of radicals (•CF2R) via visible light-promoted homolytic cleavage of C−S bonds in (phenylsulfonyl)difluoromethylated derivatives (PhSO2CF2R) is the linchpin in the diFRAS strategy to construct gem-difluorides (R1-CF2-R2) with structural complexity.
Similar content being viewed by others
Introduction
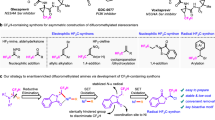
The incorporation of fluorine into pharmaceutical candidates often leads to improvements in metabolic stability, lipophilicity, and augmented biological activity1,2. Among fluorinated motifs, difluoromethylene has been known to serve as a bioisostere of ethereal oxygen and is increasingly applied in medicinal chemistry3,4,5,6. The conventional route to gem-difluorides (R1-CF2-R2) is the direct fluorination (C−F bond formation), which necessitates pre-functionalized molecular frameworks and often meets limitations due to functional group incompatibility7,8,9. Synthetic chemists aspire to connect two distinct components with the difluoromethylene moiety, fabricating difluorides in a modular fashion (Fig. 1a). Difluorocarbene is an equivalent of bipolar difluoromethylene unit, and its tandem reactions are among the most extensively employed in difluoromethylene synthon strategies. Reactions involving difluorocarbene typically proceed via interception by a nucleophile (Nu−), yielding a difluoroalkyl carbanion (Nu−CF2−). Subsequently, the carbanion attacks an electrophile (E+), leading to the construction of a gem-difluoride (Nu−CF2−E)10,11,12. However, this difluorocarbene protocol is limited by the potential reaction between the nucleophile (Nu−) and electrophile (E+). On the other hand, synthetic protocols with difluoromethylene diradical13,14,15 and dianion16,17,18,19 synthons also have drawbacks such as the singular reactivity profile (Fig. 1b).

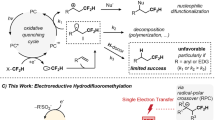
a Synthetic route to gem-difluorides and its bioisostere. b Previously developed difluoromethylene synthon. c Challenges for difluoromethylene radical anion synthon (diFRAS). d This work: PhSO2CF2H as diFRAS for modular synthesis of gem-difluorides with chemical complexity. FG functional group, LG leaving group, PG protecting group.
Bearing two orthogonal reactivity modes, difluoromethylene radical anion synthon (diFRAS, •CF2−) has significant advantages in the modular synthesis of gem-difluorides20. However, in spite of the benefits of the diFRAS, the intrinsic dilemma between its two activation modes poses a considerable obstacle (Fig. 1c). For the initial introduction of an electrophile to the potential diFRAS, the generation of carbanion is often challenging; even if the difluoroalkyl carbanion is successfully generated, its nucleophilicity is low in the absence of an auxiliary group owing to the negative fluorine effect21. Initiating the polar reaction encounters a dilemma between carbanion stability and radical reactivity: on one hand, for highly reactive radical precursors, such as halides and sulfonium salts, their corresponding unstable carbanions have a propensity to decompose into difluorocarbene22,23,24, impeding the introduction of electrophiles. On the other hand, regarding the leaving groups (such as arylthio, arylphosphoryl, and nitro groups) which are compatible with carbanion generation, the corresponding radical reactions are usually difficult to be initiated15,20,25,26,27,28 (Fig. 1c). Previous work pertaining to the diFRAS-involved radical reaction has been only limited to intramolecular cyclizations20,29,30,31,32,33,34. The diFRAS-enabled double intermolecular processes involving the annexation of two additional molecules to forge difluorides (R1-CF2-R2) still remains a challenging task.
In 2016, we reported the first example of sulfone-enabled radical fluoroalkylation via S−C bond cleavage of difluoromethyl benzothiazolyl sulfone (BTSO2CF2H, 1b)35. Although the radical fluoroalkylation with 1b has found different applications35,36,37,38,39,40,41,42,43,44,45, the instability of the corresponding carbanion of 1b (BTSO2CF2−)22 precludes 1b from serving as an efficient diFRAS. On the other hand, we have found that difluoromethyl phenyl sulfone (PhSO2CF2H, 1a), a readily available and easy-to-handle compound46, can serve as a robust nucleophilic difluoroalkylation reagent47,48,49 for various electrophiles such as alkyl halides50,51, disulfides18, aldehydes, ketones52,53, and imine54,55. The obtained products (PhSO2CF2R) constitute a library of broad range of fluoroalkyl sulfones, and therefore, we envisioned that 1a could be developed as a privileged diFRAS if we can tackle the challenge of the homolytic cleavage of the S−C bond of PhSO2−CF2R to generate difluoroalkyl radical species (•CF2R). Herein, we reported a visible light-promoted desulfonylalkylation of the phenyl sulfones to generate difluoroalkyl radicals. Merging nucleophilic (phenylsulfonyl)difluoromethylation of electrophiles with a sequential radical coupling with radical acceptors, we designed a strategy for modular synthesis of gem-difluorides using PhSO2CF2H as a diFRAS that introduces two distinct components and unlocks great chemical complexity (Fig. 1d).
Results
Reaction optimization
In light of the radical-anion-promoted desulfonylation and the potential risk of uncontrolled overreduction of fluoroalkyl radical by harsh reductants (e.g., Na-Hg, Mg-HOAc)56,57,58, robust yet mild conditions are crucial for the radical cleavage of the PhSO2–CF2R bond59. Photocatalysis is a powerful tool in radical chemistry60,61,62, and most processes in which sulfones break to generate radicals revolve around photocatalysis24,35,36,37,38,39,40,41,42,43,45,63,64. Using vinylphenyldimethylsilane (2a) as a radical acceptor, we explored the hydrofluoroalkylation with functionalized phenyl sulfones (1c) obtained from the nucleophilic addition of propionaldehyde with PhSO2CF2H52 (Table 1). BTSO2CF2H (1b), a widely-used fluoroalkyl radical precursor, and PhSO2CF2H (1a) were also tested under the series of conditions. BTSO2CF2H showed a moderate to good reactivity in various conditions (Table 1, entries 1-6). Meanwhile, phenyl sulfones 1a and 1c were hardly activated, and either no target product or only a small amount of the target product was obtained. Inspired by radical defluorination of trifluoromethylarenes65,66, potassium formate was applied as a reductant to react with phenyl sulfones, affording the products in moderate yields67,68,69 (Table 1, entry 7). Subsequently, it was discovered that when 2-naphthalenethiol (2-NpSH) was used as a hydrogen atom transfer (HAT) catalyst, a moderate yield could still be observed without additional expensive photocatalysts70 (Table 1, entries 8,9). Using DMSO as solvent resulted in better yields (Table 1, entry 10). During our investigation, the catalytic activity of arenethiol as both photocatalyst and HAT catalyst was reported by Shang71,72 and Molander73,74. The catalytic performance of other arenethiols was evaluated, and all showed lower activity compared to 2-NpSH (Table 1, entry 11; see Supplementary Information, Table S1 for performance of additional arenethiols). Finally, when blue light was replaced with the most widely used white light, it gave the products in the highest yield (Table 1, entry 12). Hence, 2-NpSH/HCO2K in DMSO under white light irradiation was identified as the optimal condition.
Substrate scope
The robustness of the optimized reaction conditions was illustrated across the substrate scope (Fig. 2). Initially, evaluation was conducted on the sulfone derivatives that were obtained through the nucleophilic addition of PhSO2CF2H (1a)47,48,49. The sulfones derived from a wide range of cyclic (3a, 3b), linear (3c), sterically hindered (3d), and aryl aldehydes (3e), as well as ketone (3f), showed compatibility in our methodology. This reaction was also applicable to sulfones obtained through cascade cyclization (3g) (see Supplementary Information, Fig. S4 and Table S3) and Michael addition (3h). Imine-derived sulfones enabled the synthesis of β-amino-α,α-difluorides (3i-3l). Amino and silyl groups were tolerated in the method (3m, 3n). Notably, naturally occurring bioactive motifs could be coupled to either radical or anion ends of diFRAS. The incorporation of radical linkers like quinine (3o), and electrophilic linkers, for instance, pregnenolone (3p), 5a-cholestan-3-one (3q), estrone (3r) and fenofibrate (3s) into diFRAS was demonstrated. Because of steric hindrance, several of their corresponding radicals preferentially abstract hydrogen atoms in HAT catalysts rather than react with alkenes.

Reaction conditions: 2 (0.5 mmol, 1.0 equiv), 1 (2.0 equiv), 2-NpSH (20 mol%), HCO2K (3.0 equiv), DMSO (4 mL), white LED, room temperature, 18 h. All yields are isolated yields. a1 (0.5 mmol, 1.0 equiv), 2 (2.0 equiv).
A comprehensive evaluation has been conducted on sulfone derivatives (derived from substitutions involving PhSO2CF2H)18,50,51,75, showcasing their potential applications in organic synthesis (Fig. 3). The coupling proceeded effectively for sulfones adorned with a distal hydroxyl group derived from cyclic sulfates (4a–4c). Linear difluoroalkyl sulfones, obtained from the substitution reactions between PhSO2CF2H and alkyl halides, were also compatible with this protocol (4d–4i). Arylthio-modified sulfones derived from disulfides were likewise converted into SCF2-containing species (4j–4l), underscoring the broad functional group tolerance of this approach. The scope of radical acceptors featuring alkene moieties was also explored with various functional groups such as sulfonyl (4m), amino (4n), hydroxy (4o,4p), and carboxylic acid (4q). The methodology can be extended to drug derivatives, including alibendol (4r), ibuprofen (4s), tarenflurbil (4t), and paclitaxel (4u). The inherent chemical versatility of hydroxyl groups facilitates further derivatization of γ-hydroxyl-α,α-difluoroalkyl phenyl sulfones, underscoring their potential for structural modification. A peptide-embellished difluoroalkyl moiety was integrated into a paclitaxel derivative, linking two bioactive motifs via diFRAS (4v). To further substantiate the synthetic utility, we synthesized a bioisostere of pitolisant (4w), an FDA-approved drug for the treatment of narcolepsy76, wherein the oxygen was replaced by a difluoromethylene group.

Reaction conditions: 2 (0.5 mmol, 1.0 equiv), 1 (2.0 equiv), 2-NpSH (20 mol%), HCO2K (3.0 equiv), DMSO (4 mL), white LED, room temperature, 18 h. All yields are isolated yields. aK2CO3 (1.0 equiv) as an additive. bThe reaction was conducted on 0.2 mmol scale.
Apart from employing alkenes as radical acceptors, we delved deeper into investigating the feasibility of our method with different types of radical acceptors (Fig. 4). Our research efforts were centered around the construction of CF2−S structure, which not only holds critical importance in pharmaceutical chemistry but also garners substantial interest among synthetic chemists77,78,79,80,81,82,83,84. With DBU as a base in lieu of a reductive formate, we expanded the scope of radical acceptors to include arenethiols (5a-5e). Sulfones 1-4a underwent reactions with various thiophenols to produce a series of thioethers (5f–5o) with the additional photocatalyst Ir(ppy)3 under blue LED. In addition to electrically neutral arenethiols, both electron-rich and electron-deficient ones proved to be effective coupling partners in this reaction. Ortho- and meta-substituted and multi-substituted arenethiols exhibited compatibility under the reaction conditions (5k, 5m–5o). Remarkably, Ar-CF3 was stable under the conditions (5l–5n), experiencing minimal defluorination. Furthermore, the conversion of 2-PySH (5p), a prevalent structural motif in numerous bioactive molecules85, was successfully conducted.

Reaction conditions: 2 (0.5 mmol, 1.0 equiv), 1 (2.0 equiv), DBU (2.0 equiv), DMSO (4 mL), white LED, room temperature, 18 h. All yields are isolated yields. a1 (0.5 mmol, 1.0 equiv), 2 (2.0 equiv). bfac-Ir(ppy)3 (0.2 mol%) as an additive, blue LED instead of white LED. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
In scenarios without radical acceptors, the generation of difluoromethyl-containing products can be achieved via hydrodesulfonylation process from sulfone derivatives. Conventionally, this transformation was accomplished under acidic conditions employing highly reactive metals such as sodium amalgam and magnesium56,57,58. Herein, our tactic offers a moderate and efficient method for converting a (phenylsulfonyl)difluoromethyl group to a difluoromethyl group (Figs. 5 and 6a–i).

Reaction conditions: 1 (0.5 mmol), 2-naphthalenethiol (10 mol%), HCO2K (3.0 equiv), DMSO (4 mL), white LED, room temperature, 18 h. All yields are isolated yields.

Reaction conditions: 2 (0.5 mmol, 1.0 equiv), 1 (2.0 equiv), 2-NpSH (20 mol%), HCO2K (3.0 equiv), DMSO (4 mL), white LED, room temperature, 18 h. All yields are isolated yields. aAdditional H2O (0.5 mL) was added. bFollowing conditions for construction of CF2-S bond in Fig. 4.
Besides a range of structurally diverse fluoroalkyl sulfones, the parent sulfone PhSO2CF2H (1a) also proved to be suitable for the reaction conditions (Fig. 6). The hydrodifluoromethylation of alkenes demonstrated commendable functional group tolerance (7a–7m), as observed with the sulfone derivatives (Figs. 2 and 3). Difluoromethylation of arenethiol (7n) could be achieved following the procedure outlined in Fig. 4. Late-stage modification of a range of pharmaceutical compounds, namely tarenflurbil (7o), oxaprozin (7p), ibuprofen (7q), mefenamic acid (7r) and alibendol (7s), and naturally occurring molecules, such as allylestrenol (7t), boldenone undecylenate (7u) and caryophyllene oxide (7v), was also found to be fruitful (Fig. 6).
Synthetic applications
When sabinene, bearing a radical clock, was subjected to the reactions, the cyclopropane ring-opening product (8a) was isolated with a 5:1 selectivity for the 6-membered over the 5-membered ring (Fig. 7a). The methodology also lightened a route to the synthesis of saturated rings via radical cyclization of unconjugated dienes. Octahydropentalene (8b) was constructed following the tactic from cycloocta-1,5-diene (Fig. 7b). Of particular note, sunlight, the most ubiquitous and cost-free light source, proved highly effective in promoting the transformation (Fig. 7c). The practicality of this method was further confirmed through a gram-scale synthesis of gem-difluorides in a good yield (Fig. 7d).

a Radical clock reaction. b Radical cyclization of unconjugated dienes. c Sunshine-induced difluoroalkylation. d Gram-scale reaction. e UV-vis absorption spectra of species in the transformation and oxidative quenching cycle of 2-napthalenethiolate. f Proposed mechanism.
Mechanistic investigation
The UV-vis absorption spectra unambiguously show that 2-napthlanenethiolate exhibits absorption onset wavelength at 487 nm, which falls within the visible light range. Additionally, it indicates that the photoexcitation of 2-NpS− is more facile compared to reported arenethiolates71,72,73,74, as the latter necessitate irradiation at shorter wavelengths for excitation. Additionally, although p-MeOC6H4SH showed low activity under 450 nm blue LED irradiation (Table 1, entry 11), the thiol effectively catalyzed the reaction under 420 nm violet LED irradiation in 79% yield (see supplementary information, Table S5, entry 3). The negligible hypsochromic shift observed between iv and v (as shown in Fig. 7e) supports that the thiolate directly participates in photoexcitation without undergoing electron donor-acceptor (EDA) complexation. Based on the spectra and electrochemical data (Ep/2(2-NpS•/2-NpS−) = 0.09 V, see supplementary information, Fig. S13), strong reducibility of the excited thiolate was confirmed (Ecal(2-NpS•/[2-NpS−]*) = −2.49 V)86. The low quantum yield (Φ = 0.20) does not strongly support a chain process (see Supplementary Information 2.4.4). In reference to previous reports65,66,71, we propose a thiol-catalyzed reductive radical mechanism (Fig. 7f). Upon photoexcitation, thiolate (2-NpS−) transitions to its excited state [2-NpS−]*. This strong reducing agent [2-NpS−]* readily undergoes a single electron transfer (SET) to a sulfone, resulting in the generation of a fluoroalkyl radical and a thiyl radical (2-NpS•). The thiyl radical can then undergo a tandem reaction with formate, yielding a carbon dioxide radical anion (CO2•−) and regenerating the thiolate. Alternatively, the thiyl radical (2-NpS•) can also undergo SET process with the carbon dioxide radical anion (CO2•−), leading to the regeneration of the thiolate (2-NpS−). Meanwhile, the alkene synchronously reacts with the fluoroalkyl radical, forming an adduct intermediate. As a hydrogen atom transfer (HAT) catalyst, the thiol (2-NpSH) facilitates the transfer of a hydrogen atom to the adduct intermediate, culminating in the formation of gem-difluorides (3,4). Besides alkenes, the fluoroalkyl radical may be converted to ArSCF2R (5) with a thiyl radical (see supplementary information 2.4.3 for more details). Additionally, the fluoroalkyl radical can be directly hydrogenated to generate RCF2H (6).
Discussion
In conclusion, we discover PhSO2CF2H (1a), a readily available reagent, as a remarkably versatile difluoromethylene radical anion synthon (diFRAS, •CF2−), which serves as an efficient -CF2- bridge between an electrophile and a radical acceptor. Naturally occurring molecules and pharmaceutically relevant molecules can be late-stage functionalized by either radical or anion end of the diFRAS (•CF2−. This synthetic protocol not only offers a powerful and practical tool for the assembly of a wide range of structurally diverse gem-difluorides (R1-CF2-R2), it also provides new insights into the privileged chemical reactivities of fluoroalkyl sulfones.
Methods
General procedure for incorporation of an alkene with sulfone 1
To a dry Schlenk tube were added 2-Naphthalenethiol (0.1 mmol, 20 mol%) and potassium formate (1.5 mmol, 3.0 equiv), alkene (0.5 mmol, 1.0 equiv), sulfone 1 (1.0 mmol, 2.0 equiv) and dry DMSO (4.0 mL) under argon atmosphere. The mixture was stirred under irradiation with 10 W white LED for 18 h at room temperature. After the reaction was complete, water and brine were added and the mixture was extracted with diethyl ether three times. The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel to afford the product.
General procedure for incorporation of an arenethiol with sulfone 1
To a dry Schlenk tube were added arenethiol (0.5 mmol, 1.0 equiv), sulfone 1 (1.0 mmol, 2.0 equiv), fac-Ir(ppy)3 (0.001 mmol, 0.2 mol%), DBU (1.0 mmol, 2.0 equiv) and dry DMSO (4.0 mL) under argon atmosphere. The mixture was stirred under irradiation with 10 W white LED for 18 h at room temperature. After the reaction was complete, water and brine were added and the mixture was extracted with diethyl ether three times. The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel to afford the product.
General procedure for hydrodesulfonylation of sulfone 1
To a dry Schlenk tube were added 2-Naphthalenethiol (0.05 mmol, 10 mol%) and potassium formate (1.5 mmol, 3.0 equiv), sulfone 1 (0.5 mmol, 1.0 equiv), and dry DMSO (4.0 mL) under argon atmosphere. The mixture was stirred under irradiation with 10 W white LED for 18 h at room temperature. After the reaction was complete, water and brine were added and the mixture was extracted with diethyl ether three times. The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel to afford the product.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Crystallographic data for the structures of sulfone 1i (precursor of 3g) reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 2452837. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Experimental details and the spectroscopic data of the corresponding compounds are provided in the Supplementary Information. All data supporting the study are available from the corresponding author on request.
References
Meanwell, N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 61, 5822–5880 (2018).
Ogawa, Y., Tokunaga, E., Kobayashi, O., Hirai, K. & Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. iScience 23, 101467 (2020).
Blackburn, G. M., England, D. A. & Kolkmann, F. Monofluoro- and difluoro-methylenebisphosphonic acids: isopolar analogues of pyrophosphoric acid. J. Chem. Soc. Chem. Commun. 15, 930–932 (1981).
Blackburn, G. M., Kent, D. E. & Kolkmann, F. The synthesis and metal binding characteristics of novel, isopolar phosphonate analogues of nucleotides. J. Chem. Soc. Perkin Trans. 1, 1119–1125 (1984).
Xu, Y. et al. Structure−activity relationships of fluorinated lysophosphatidic acid analogues. J. Med. Chem. 48, 3319–3327 (2005).
O’Hagan, D., Wang, Y., Skibinski, M. & Slawin, A. M. Z. Influence of the difluoromethylene group (CF2) on the conformation and properties of selected organic compounds. Pure Appl. Chem. 84, 1587–1595 (2012).
Middleton, W. J. New fluorinating reagents. Dialkylaminosulfur fluorides. J. Org. Chem. 40, 574–578 (1975).
Lal, G. S., Pez, G. P., Pesaresi, R. J., Prozonic, F. M. & Cheng, H. Bis(2-methoxyethyl)aminosulfur trifluoride: a new broad-spectrum deoxofluorinating agent with enhanced thermal stability. J. Org. Chem. 64, 7048–7054 (1999).
Beaulieu, F. et al. Aminodifluorosulfinium tetrafluoroborate salts as stable and crystalline deoxofluorinating reagents. Org. Lett. 11, 5050–5053 (2009).
Xie, Q. & Hu, J. A journey of the development of privileged difluorocarbene reagents TMSCF2X (X = Br, F, Cl) for organic synthesis. Acc. Chem. Res. 57, 693–713 (2024).
Dilman, A. D. & Levin, V. V. Difluorocarbene as a building block for consecutive bond-forming reactions. Acc. Chem. Res. 51, 1272–1280 (2018).
Zeng, X., Li, Y., Min, Q.-Q., Xue, X.-S. & Zhang, X. Copper-catalysed difluorocarbene transfer enables modular synthesis. Nat. Chem. 15, 1064–1073 (2023).
Kostromitin, V. S., Sorokin, A. O., Levin, V. & Dilman, A. D. Aminals as powerful XAT-reagents: activation of fluorinated alkyl chlorides. Chem. Sci. 14, 3229–3234 (2023).
Zhang, Z.-Q. et al. Programmable synthesis of difluorinated hydrocarbons from alkenes through a photocatalytic linchpin strategy. Chem. Sci. 14, 11546–11553 (2023).
Reutrakul, V., Thongpaisanwong, T., Tuchinda, P., Kuhakarn, C. & Pohmakotr, M. Difluorophenylsulfanylmethyl radical and difluoromethylene diradical synthons: gem-difluoromethylene building block. J. Org. Chem. 69, 6913–6915 (2004).
Beier, P., Alexandrova, A. V., Zibinsky, M. & Surya Prakash, G. K. Nucleophilic difluoromethylation and difluoromethylenation of aldehydes and ketones using diethyl difluoromethylphosphonate. Tetrahedron 64, 10977–10985 (2008).
Kosobokov, M. D. et al. Geminal silicon/zinc reagent as an equivalent of difluoromethylene bis-carbanion. Org. Lett. 16, 1438–1441 (2014).
Prakash, G. K. S., Hu, J., Mathew, T. & Olah, G. A. Difluoromethyl phenyl sulfone as a selective difluoromethylene dianion equivalent: one-pot stereoselective synthesis of anti-2,2-difluoropropane-1,3-diols. Angew. Chem. Int. Ed. 42, 5216–5219 (2003).
Trifonov, A. L. & Dilman, A. D. Synthesis of difluoroalkylated heteroarenes via difluorocarbene. Org. Lett. 23, 6977–6981 (2021).
Li, Y. & Hu, J. Stereoselective difluoromethylenation using Me3SiCF2SPh: synthesis of chiral 2,4-disubstituted 3,3-difluoropyrrolidines. Angew. Chem. Int. Ed. 46, 2489–2492 (2007).
Zhang, W., Ni, C. & Hu, J. Selective fluoroalkylation of organic compounds by tackling the “negative fluorine effect”. Top. Curr. Chem. 308, 25–44 (2012).
Prakash, G. K. S., Ni, C., Wang, F., Hu, J. & Olah, G. A. From difluoromethyl 2-pyridyl sulfone to difluorinated sulfonates: a protocol for nucleophilic difluoro(sulfonato)methylation. Angew. Chem. Int. Ed. 50, 2559–2563 (2011).
Sap, J. B. I. et al. [18F]Difluorocarbene for positron emission tomography. Nature 606, 102–108 (2022).
Wei, Z., Zheng, W., Wan, X. & Hu, J. Copper-catalyzed enantioselective difluoromethylation-alkynylation of olefins by solving the dilemma between acidities and reduction potentials of difluoromethylating agents. Angew. Chem. Int. Ed. 62, e202308816 (2023).
Korth, H.-G., Sustmann, R., Dupuis, J. & Giese, B. Zum Mechanismus der Denitrierung aliphatischer Nitro-Verbindungen mit Trialkylstannyl-Radikalen. Eine ESR-kinetische Untersuchung. Chem. Ber. 120, 1197–1202 (1987).
Kashihara, M. et al. Catalytic generation of radicals from nitroalkanes. Synlett 34, 1482–1486 (2022).
Zhang, J.-Q. et al. Conversion of triphenylphosphine oxide to organophosphorus via selective cleavage of C-P, O-P, and C-H bonds with sodium. Commun. Chem. 3, 1 (2020).
Zhang, J.-Q. et al. Selective C–P(O) bond cleavage of organophosphine oxides by sodium. J. Org. Chem. 85, 14166–14173 (2020).
Keereewan, S. et al. Diastereoselective addition of PhSCF2SiMe3 to chiral N-tert-butanesulfinyl ketimines derived from isatins: synthesis of enantioenriched gem-difluoromethylenated spiro-pyrrolidinyl and spiro-piperidinyl oxindoles. J. Org. Chem. 87, 15963–15985 (2022).
Korvorapun, K. et al. Stereoselective nucleophilic addition of PhSCF2SiMe3 to chiral cyclic nitrones: asymmetric synthesis of gem-difluoromethylenated polyhydroxypyrrolizidines and -indolizidines. Chem. Asian J. 10, 948–968 (2015).
Masusai, C. et al. Synthesis of gem-difluoromethylenated polycyclic cage compounds. J. Org. Chem. 80, 1577–1592 (2015).
Peewasan, K. et al. α-Difluoro-α-phenylsulfanyl-α-trimethylsilylmethane as “·CF2−” synthetic building block for the preparation of gem-difluoromethylenated cyclopentanols. J. Fluor. Chem. 135, 367–372 (2012).
Punirun, T., Soorukram, D., Kuhakarn, C., Reutrakul, V. & Pohmakotr, M. Oxidative difluoromethylation of tetrahydroisoquinolines using TMSCF2SPh: synthesis of fluorinated pyrrolo[2,1-a]isoquinolines and benzo[a]quinolizidines. J. Org. Chem. 83, 765–782 (2018).
Thaharn, W. et al. Asymmetric synthesis of gem-difluoromethylenated linear triquinanes via cascade gem-difluoroalkyl radical cyclization. J. Org. Chem. 80, 816–827 (2015).
Rong, J. et al. Radical fluoroalkylation of isocyanides with fluorinated sulfones by visible-light photoredox catalysis. Angew. Chem. Int. Ed. 55, 2743–2747 (2016).
Fu, W. et al. Visible-light-mediated radical oxydifluoromethylation of olefinic amides for the synthesis of CF2H-containing heterocycles. Chem. Comm. 52, 13413–13416 (2016).
Rong, J., Wang Yunze, N. C., Cuiwen, K., Yucheng, G. & Jinbo, H. Radical fluoroalkylation of aryl alkenes with fluorinated sulfones by visible-light photoredox catalysis. Acta Chim. Sin. 75, 105–109 (2017).
Zou, G. & Wang, X. Visible-light induced di- and trifluoromethylation of N-benzamides with fluorinated sulfones for the synthesis of CF2H/CF3-containing isoquinolinediones. Org. Biomol. Chem. 15, 8748–8754 (2017).
Trump, L. et al. Late-stage 18F-difluoromethyl labeling of N-heteroaromatics with high molar activity for PET imaging. Angew. Chem. Int. Ed. 58, 13149–13154 (2019).
Zhu, M. et al. Visible-light-induced radical di- and trifluoromethylation of β, γ-unsaturated oximes: synthesis of di- and trifluoromethylated isoxazolines. Eur. J. Org. Chem. 2019, 1614–1619 (2019).
Zhu, M., You, Q. & Li, R. Synthesis of CF2H-containing oxindoles via photoredox-catalyzed radical difluoromethylation and cyclization of N-arylacrylamides. J. Fluor. Chem. 228, 109391 (2019).
Bao, K., Wei, J., Yan, H. & Sheng, R. Visible-light promoted three-component tandem reaction to synthesize difluoromethylated oxazolidin-2-imine. RSC Adv. 10, 25947–25951 (2020).
Zhang, B. & Wang, J. Acyldifluoromethylation enabled by NHC-photoredox cocatalysis. Org. Lett. 24, 3721–3725 (2022).
Zhou, X., Ni, C., Deng, L. & Hu, J. Electrochemical reduction of fluoroalkyl sulfones for radical fluoroalkylation of alkenes. Chem. Commun. 57, 8750–8753 (2021).
Mao, E., Prieto Kullmer, C. N., Sakai, H. A. & MacMillan, D. W. C. Direct bioisostere replacement enabled by metallaphotoredox deoxydifluoromethylation. J. Am. Chem. Soc. 146, 5067–5073 (2024).
Hu, J., Liu, R. & Liu, A. Preparation method of difluoromethyl sulfone compound. China patent CN 112574076A (2021).
Hu, J. Nucleophilic, radical, and electrophilic (phenylsulfonyl)difluoromethylations. J. Fluor. Chem. 130, 1130–1139 (2009).
Ni, C., Hu, M. & Hu, J. Good partnership between sulfur and fluorine: sulfur-based fluorination and fluoroalkylation reagents for organic synthesis. Chem. Rev. 115, 765–825 (2015).
Jia, R., Wang, X. & Hu, J. Recent advance in synthetic applications of difluoromethyl phenyl sulfone and its derivatives. Tetrahedron Lett. 75, 153182 (2021).
Prakash, G. K. S., Hu, J., Wang, Y. & Olah, G. A. Nucleophilic difluoromethylation of primary alkyl halides using difluoromethyl phenyl sulfone as a difluoromethyl anion equivalent. Org. Lett. 6, 4315–4317 (2004).
Prakash, G. K. S., Hu, J., Wang, Y. & Olah, G. A. Difluoromethyl phenyl sulfone, a difluoromethylidene equivalent: use in the synthesis of 1,1-difluoro-1-alkenes. Angew. Chem. Int. Ed. 43, 5203–5206 (2004).
Prakash, G. K. S., Hu, J., Wang, Y. & Olah, G. A. Convenient synthesis of difluoromethyl alcohols from both enolizable and non-enolizable carbonyl compounds with difluoromethyl phenyl sulfone. Eur. J. Org. Chem. 2005, 2218–2223 (2005).
Hu, M., Gao, B., Ni, C., Zhang, L. & Hu, J. Nucleophilic difluoro(phenylsulfonyl)methylation of carbonyls with PhSO2CF2H reagent in the presence of in situ generated substoichiometric amount of base. J. Fluor. Chem. 155, 52–58 (2013).
Li, Y. & Hu, J. Facile synthesis of chiral α-difluoromethyl amines from N-(tert-butylsulfinyl)aldimines. Angew. Chem. Int. Ed. 44, 5882–5886 (2005).
Liu, J., Li, Y. & Hu, J. Stereoselective synthesis of di- and monofluoromethylated vicinal ethylenediamines with di- and monofluoromethyl sulfones. J. Org. Chem. 72, 3119–3121 (2007).
Sabol, J. S. & McCarthy, J. R. A new route to 1,1-difluoro olefins: application to the synthesis of 2′-deoxy-2′-difluoromethylene nucleosides. Tetrahedron Lett. 33, 3101–3104 (1992).
Stahly, G. P. Nucleophilic addition of difluoromethyl phenyl sulfone to aldehydes and various transformations of the resulting alcohols. J. Fluor. Chem. 43, 53–66 (1989).
Ni, C. & Hu, J. Nucleophilic difluoromethylation of carbonyl compounds using TMSCF2SO2Ph and Mg0-mediated desulfonylation. Tetrahedron Lett. 46, 8273–8277 (2005).
Kim, S. et al. Radical hydrodifluoromethylation of unsaturated C−C bonds via an electroreductively triggered two-pronged approach. Commun. Chem. 5, 96 (2022).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322–5363 (2013).
Romero, N. A. & Nicewicz, D. A. Organic photoredox catalysis. Chem. Rev. 116, 10075–10166 (2016).
Wei, Z., Lou, Z., Ni, C., Zhang, W. & Hu, J. Visible-light-promoted S-trifluoromethylation of thiophenols with trifluoromethyl phenyl sulfone. Chem. Commun. 58, 10024–10027 (2022).
Zhang, F.-X., Lin, J.-H. & Xiao, J.-C. Difluoromethylsulfonyl imidazolium salt for difluoromethylation of alkenes. Org. Lett. 24, 7611–7616 (2022).
Crespi, S. & Fagnoni, M. Generation of alkyl radicals: from the tyranny of tin to the photon democracy. Chem. Rev. 120, 9790–9833 (2020).
Wang, H. & Jui, N. T. Catalytic defluoroalkylation of trifluoromethylaromatics with unactivated alkenes. J. Am. Chem. Soc. 140, 163–166 (2018).
Vogt, D. B., Seath, C. P., Wang, H. & Jui, N. T. Selective C–F functionalization of unactivated trifluoromethylarenes. J. Am. Chem. Soc. 141, 13203–13211 (2019).
Hendy, C. M., Smith, G. C., Xu, Z., Lian, T. & Jui, N. T. Radical chain reduction via carbon dioxide radical anion (CO2•–). J. Am. Chem. Soc. 143, 8987–8992 (2021).
Xiao, W., Zhang, J. & Wu, J. Recent advances in reactions involving carbon dioxide radical anion. ACS Catal. 13, 15991–16011 (2023).
Shen, Q., Cao, K., Wen, X. & Li, J. Formate salts: the rediscovery of their radical reaction under light irradiation opens new avenues in organic synthesis. Adv. Synth. Catal. 366, 4274–4293 (2024).
Jia, R. Radical Fluoroalkylation Based on Fluoroalkylsulfones. Master’s Thesis, Zhengzhou University (2021).
Liu, C., Li, K. & Shang, R. Arenethiolate as a dual function catalyst for Photocatalytic defluoroalkylation and hydrodefluorination of trifluoromethyls. ACS Catal. 12, 4103–4109 (2022).
Liu, C., Shen, N. & Shang, R. Photocatalytic defluoroalkylation of trifluoroacetates with alkenes using 4-(acetamido)thiophenol. Synthesis 55, 1401–1409 (2023).
Shreiber, S. T. et al. Visible-light-induced C–F bond activation for the difluoroalkylation of indoles. Org. Lett. 24, 8542–8546 (2022).
Matsuo, B. et al. Transition metal-free photochemical C–F activation for the preparation of difluorinated-oxindole derivatives. Chem. Sci. 14, 2379–2385 (2023).
Ni, C., Liu, J., Zhang, L. & Hu, J. A remarkably efficient fluoroalkylation of cyclic sulfates and sulfamidates with PhSO2CF2H: facile entry into β-difluoromethylated or β-difluoromethylenated alcohols and amines. Angew. Chem. Int. Ed. 46, 786–789 (2007).
Syed, Y. Y. Pitolisant: first global approval. Drugs 76, 1313–1318 (2016).
Jiang, H. et al. A facile preparation of 2-bromodifluoromethyl benzo-1,3-diazoles and its application in the synthesis of gem-difluoromethylene linked aryl ether compounds. J. Fluor. Chem. 133, 167–170 (2012).
Xiong, H.-Y., Pannecoucke, X. & Besset, T. Recent advances in the synthesis of SCF2H- and SCF2FG-containing molecules. Chem. Eur. J. 22, 16734–16749 (2016).
Orsi, D. L., Easley, B. J., Lick, A. M. & Altman, R. A. Base catalysis enables access to α,α-difluoroalkylthioethers. Org. Lett. 19, 1570–1573 (2017).
Geri, J. B., Wade Wolfe, M. M. & Szymczak, N. K. The difluoromethyl group as a masked nucleophile: a Lewis acid/base approach. J. Am. Chem. Soc. 140, 9404–9408 (2018).
Brigham, C. E., Malapit, C. A., Lalloo, N. & Sanford, M. S. Nickel-catalyzed decarbonylative synthesis of fluoroalkyl thioethers. ACS Catal. 10, 8315–8320 (2020).
Santos, L. et al. Deprotonative functionalization of the difluoromethyl group. Org. Lett. 22, 8741–8745 (2020).
Zubkov, M. O. et al. A novel photoredox-active group for the generation of fluorinated radicals from difluorostyrenes. Chem. Sci. 11, 737–741 (2020).
Xu, J. et al. Construction of C–X (X = S, O, Se) bonds via Lewis acid-promoted functionalization of trifluoromethylarenes. ACS Catal. 13, 7339–7346 (2023).
De, S. et al. Pyridine: the scaffolds with significant clinical diversity. RSC Adv. 12, 15385–15406 (2022).
Buzzetti, L., Crisenza, G. E. M. & Melchiorre, P. MechaniStic Studies In Photocatalysis. Angew. Chem. Int. Ed. 58, 3730–3747 (2019).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFF0701700, J.H.), the National Natural Science Foundation of China (22261132514, J.H.; 22271299, C.N.), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0590000, J.H.).
Author information
Authors and Affiliations
Contributions
J.H. conceived the concept. S.S., R.J., X.Z. and J.R. performed the experiments. S.S., R.J., X.Z. Z.W. and C.N. contributed to the analysis and interpretation of the data. S.S., X.Z., and J.H. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Takashi Koike, Rui Shang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, S., Jia, R., Zhou, X. et al. Modular synthesis of CF2-containing compounds with PhSO2CF2H reagent through difluoromethylene radical anion synthon strategy. Nat Commun 16, 7731 (2025). https://doi.org/10.1038/s41467-025-62834-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62834-3
