
Abstract
Organs collaborate to maintain metabolic homeostasis in mammals. Spatial metabolomics makes strides in profiling the metabolic landscape, yet can not directly inspect the metabolic crosstalk between tissues. Here, we introduce an approach to comprehensively trace the metabolic fate of 13C-nutrients within the body and present a robust computational tool, MSITracer, to deep-probe metabolic activity in a spatial manner. By discerning spatial distribution differences between isotopically labeled metabolites from ambient mass spectrometry imaging-based isotope tracing data, this approach empowers us to characterize fatty acid metabolic crosstalk between the liver and heart, as well as glutamine metabolic exchange across the kidney, liver, and brain. Moreover, we disclose that tumor burden significantly influences the host’s hexosamine biosynthesis pathway, and that the glucose-derived glutamine released from the lung as a potential source for tumor glutamate synthesis. The developed approach facilitates the systematic characterization of metabolic activity in situ and the interpretation of tissue metabolic communications in living organisms.
Similar content being viewed by others


Introduction
Spatial omics is becoming a powerful tool for profiling cellular heterogeneity, complex tissue architectures and dynamic molecular changes during development and disease1,2,3 and has been widely heralded as a new frontier in the biomedical and life sciences fields4. Remarkably, mass spectrometry imaging (MSI) can be used to simultaneously locate and visualize thousands of molecules, such as metabolites, peptides, glycans, and drugs, within the spatial context of various tissues, providing multidimensional information that is essential for spatial omics investigations. MSI-driven spatial metabolomics is widely used to decipher metabolic mechanisms that underlie diverse physiological and pathological processes5. However, the metabolite levels measured by MSI can only reflect molecular relative abundances and may not imply metabolic pathway activity, as these levels are the net product of multiple metabolic reactions, which constantly synthesize and consume metabolites. This limitation hinders research to clarify the dynamic role of metabolites and metabolic rewiring across heterogeneous tissues in health and disease.
Isotope tracing allows for the direct interrogation of metabolic pathway activity6. Generally, isotopically labeled tracers are introduced into living systems, such as cultured cells7, model animals8 and patients9. Mass spectrometry can then be used to detect downstream metabolites and identify heavy atom (e.g., 2H, 13C, 15N) enriched isotopologues10. By tracking isotope incorporation and distinctive labeling patterns, this technique provides an extra dimension to infer metabolite interconversion and estimate biochemical pathway activities11. In particular, the integration of stable isotope tracing and MSI has proven to be highly beneficial for investigating metabolic activity in heterogeneous tissues12 and spatially mapping the flux of fatty acid synthesis and elongation in gliomas and healthy brain tissue13. Nevertheless, current spatial isotope labeling analyses predominantly focus on a few specific metabolites and targeted pathways, making it challenging to identify metabolites derived from nonclassical or unexpected reactions in situ.
To date, a collection of software solutions has been developed to broaden the coverage of the labeled metabolites, including MetTracer14, X13CMS15, NTFD16, and others17,18,19, which are tailored for liquid chromatography coupled with mass spectrometry (LC‒MS) or gas chromatography coupled with mass spectrometry (GC‒MS) techniques. Due to the absence of separation and enrichment steps in MSI and the relatively low abundance of metabolite isotopologues, identification of labeled metabolites from complex MSI datasets in an automated and unbiased manner is difficult. While Blanc et al. proposed a variation of the Kendrick mass defect concept to detect 13C-enriched molecules with lower technical demand20, this method is more applicable to lipids with a greater number of carbons and cannot provide information on labeling fractions. Therefore, a tool that can comprehensively identify labeled metabolites for MSI and automatically calculate labeling fractions for spatial isotope tracing is urgently needed.
In living organisms, individual organs coordinate and communicate to acquire essential metabolites, thereby maintaining metabolic homeostasis21,22. One of the key examples is the Nobel Prize discovery of the “Cori cycle”, which involves glucose–lactate shuttling between skeletal muscle and the liver23. Increasing evidence suggests that systemic metabolic imbalances occur within an organism during disease states. In certain circumstances, such as cancer, tumor cells may interact with peripheral tissues to reallocate energy resources to sustain their proliferation24. For instance, alanine cycling between melanoma and healthy liver tissue has been revealed using a zebrafish model25. Similarly, researchers found that glutamate and glutathione synthesis in patient-derived tumor xenografts could be largely fueled by host liver-derived glutamine26. These efforts underscore that inter-tissue crosstalk is paramount in the context of organismal development and has the potential to pinpoint new metabolic vulnerabilities for therapeutic intervention.
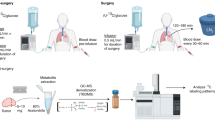
Here, we propose a framework that enables deep probing of the metabolic fate of nutrients and explores metabolic communication in living organisms (Fig. 1a). Specifically, a robust computational tool, MSITracer, which leverages the metabolic profile provided by stable isotope tracing and laboratory-built, highly sensitive ambient airflow-assisted desorption electrospray ionization (AFADESI)-MSI27 was introduced for spatial tracing metabolic network within tissues. We demonstrate that effective quantification of the metabolite fingerprint in situ can be achieved, yielding ample molecular insights towards distribution differences between native metabolites and corresponding isotopologues from whole-body animals to microregions of tissues. Our results show interactions between the liver and heart in fatty acid metabolism. This approach further allows us to characterize the global metabolome and lipidome alterations induced by tumor burden and to unravel the lung provides glucose-derived glutamine to the tumor via the circulation, thereby promoting glutamate metabolism within the tumor.

a Overview of the spatial isotope deep tracing framework using LC‒MS/MS and AFADESI-MSI. Created in BioRender. Li, X. (https://BioRender.com/sakosq5). b High-coverage identification of labeled metabolites in the mass range (m/z 70−1200) under three LC conditions following U-13C glucose infusion. c Numbers of labeled metabolite ions and isotopologues after U-13C glucose and U-13C glutamine infusion. The isotopologues counted refer to those with labeling fractions greater than zero across all three samples. d Labeling profile of the metabolite ion m/z 385.2226 across serum and organs. e Classification of the labeled metabolites in various matrices following infusion of U-13C glucose (left) and U-13C glutamine (right). Metabolite ions detected under different LC‒MS conditions were counted once. f The distributions of shared and unique labeled metabolites across the body. Each column represents the number of labeled metabolites identified in the specific matrix, which are displayed as black solid dots in the lower section of the plot. Source data are provided as a Source Data file.
Results
In vivo stable-isotope deep tracing
The first objective of this study was to establish a workflow to deeply track the metabolic fate of nutrients across various organs in vivo. Serum and nine organs, including the brain, liver, kidney, heart, spleen, lung, pancreas, muscle, and brown adipose tissue (BAT), were collected from mice following intrajugular vein infusion of U-13C glucose and U-13C glutamine, which are commonly used tracers. To maximize the detection of labeled metabolites using LC‒MS/MS, the metabolome and lipidome were extracted and analyzed separately for each sample. Polar metabolites were separated using hydrophilic interaction chromatography (HILIC), while other metabolites and lipids were separated using two different reversed-phase chromatography (RP) systems. LC‒MS/MS was performed for all samples in both positive and negative ion modes. MetDNA228 and MetID29 were used to generate annotations of signals in unlabeled samples, and MetTracer14 software was used to extract all possible isotopologues and quantify the labeling fraction. All potential labeled features were manually curated to exclude false positives.
After the isotopic steady state was reached in mice (Supplementary Fig. 1a–d), we determined and identified a large number of isotopically enriched metabolites utilizing the developed tracking workflow. The labeled extent of compounds varied between 0.02 and 0.69 across the ten biological matrices (Fig. 1b, and Supplementary Fig. 2a). Specifically, 1274 labeled metabolites and 3227 isotopologues from 41 metabolic pathways were tracked in the U-13C glucose infusion samples (Fig. 1c, and Supplementary Fig. 2b, 2c), while 462 labeled metabolites and 1018 isotopologues, covering 36 metabolic pathways, were identified using the U-13C glutamine tracer (Fig. 1c, and Supplementary Fig. 2d). Compared to prior work30, we identified a higher number of labeled metabolites from in vivo infusion experiments, and these values increased by nearly threefold after U-13C glucose infusion (Supplementary Fig. 2e). For metabolites that were previously unreported, we assigned chemical formulas based on their accurate mass, natural isotope distribution, and MS/MS spectra. Surprisingly, after U-13C glucose infusion, three metabolites showed higher labeling fraction than those of intermediates related to glycolysis and the TCA cycle (Supplementary Figs. 2f, 3a–c). Metabolite ion corresponding to the molecular formula C20H33O7 (m/z 385.2226) was highly labeled in the serum, liver, kidney, lung, heart, and BAT tissues (Fig. 1d). Further elaboration on the structure and function of the compounds exceeds the scope of this study, but we included the data here as a resource that provides a comprehensive overview of the metabolic landscape following the administration of glucose and glutamine (Supplementary Data 1).
Considering compartmentalized metabolic profiles across various organs, we next compared the inter-tissue differences in the number and class distribution of labeled metabolites. The results demonstrated that the liver contained the most considerable amount of 13C isotopologues, while the muscle contained the least for both U-13C nutrients (Fig. 1e). Moreover, the brain and pancreas were the second-largest metabolic organs that incorporated 13C labels after U-13C glucose and U-13C glutamine infusion, respectively. This discrepancy in enrichment is consistent with the knowledge that the brain primarily uses glucose for energy, and the pancreas is sensitive to glutamine. Moreover, the accumulation of labeled metabolites was various among organs, as the liver was enriched mostly in lipids; the brain and kidney were enriched in lipids and comparable amounts of amino acids. Further cross-organ analysis revealed that nearly three-quarters of the labeled metabolites were uniquely detected in only one matrix after infusion with U-13C glucose (Fig. 1f), suggesting that heterogeneous metabolic properties occurred among the organs. Based on this collective evidence, deep stable isotope tracing enables the comprehensive mapping of labeled metabolites in vivo and the exploration of unknown metabolic processes, paving the way for the characterization of metabolic activity in organs.
MSITracer: a tool for spatial isotope tracing
Based on the labeled metabolites identified by LC‒MS/MS, we developed a tool called MSITracer, which was tailored for MSI datasets to achieve spatial isotope tracing. Studies by us and other researchers revealed that the types and abundances of adduct ions between LC‒MS and MSI systems vary31,32; thus, an MSI-specific database was introduced for U-13C glucose and U-13C glutamine infusion experiments. To improve the detection coverage, the database encompasses both experimentally determined and theoretically predicted adduct ions. Accordingly, it comprised 2394 metabolite ions and 65,303 corresponding isotopologues following the administration of U-13C glucose and 928 metabolite ions with 27,458 corresponding isotopologues after the administration of U-13C glutamine (Supplementary Data 2).
Building on this comprehensive database, MSITracer functions through the following major steps: (1) extracting the intensity of targeted isotopologues; (2) selecting ions with sufficient imaging signals, and (3) quantifying labeling patterns and fractions (Fig. 2a). Briefly, in the first step, the tool automatically performs isotopologues matching through comparing measured and theoretical m/z values within 5 ppm error range. The corresponding signal intensity for each ion is also recorded. Subsequently, unlabeled isotopologue groups are removed according to the set threshold of 12C-metabolite ion intensities and the isotopologue intensity ratios between labeled and unlabeled samples. Finally, after natural isotope abundance correction, a file containing compound names, molecular formulas, and isotopologue ion intensities is generated, as well as a file containing corrected labeled fractions. Supplementary Fig. 4 illustrates the ability of MSITracer to achieve efficient and accurate analysis for spatial isotope tracing. Uridine can be detected in both [M+Cl]– and [M − H]– forms via AFADESI-MSI. However, [M+Cl]– adduct ions exhibit higher intensity across multiple organs in AFADESI-MSI compared to LC‒MS, resulting in better imaging of the isotopologue ions (such as M5). This highlights the importance and necessity of database extension, as additional examples provided in Supplementary Figs. 5a and 5b. Moreover, two consecutive kidney sections were ground and subjected to MSI and LC‒MS analyses, consistent metabolite isotope labeling patterns were observed for five representative metabolites involved in the TCA cycle, glycolysis, and amino acid metabolism, indicating the quantitative accuracy of MSITracer (Supplementary Fig. 5c).

a Schematic diagram of the MSITracer workflow. b AFADESI-MS images of native glutamate and 13C-enriched isotopologues in whole-body mice after two 13C-nutrient infusions. The organs of interest are delineated with dashed lines in different colors. Intensity shown as relative abundance (normalized to 100%). c, d AFADESI-MS images of native glutamate and representative 13C-isotopologues in the kidney (c) and brain (d). For each figure, from left to right, the images correspond to the unlabeled, U-13C glucose-infused, and U-13C glutamine-infused samples. Statistics were calculated using one-way ANOVA and post hoc Tukey’s correction, based on three replicates, each with two representative regions of interest (ROI) selected from each slice (mean ± SD). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Adjusted p values for comparisons reaching statistical significance are provided in Supplementary Table 2. Signal intensity is shown in ion counts. Oc, outer cortex; Ic, inner cortex; Me, medulla; Pe, pelvis; AM, amygdala; HY, hypothalamus; TH, thalamus; IC, internal capsule; HP, hippocampus; CC, corpus callosum; CTX, cerebral cortex. Source data are provided as a Source Data file.
Given the advantages of MSITracer, the tool facilitates the comprehensive quantitative mapping of metabolic activity from whole-body animals to heterogeneous tissues. According to the whole-body MSI analysis, the infusion of U-13C glucose led to the predominant distribution of labeled glutamate within the brain, whereas following U-13C glutamine infusion, labeled glutamate was primarily observed in the pancreas (Fig. 2b, and Supplementary Fig. 6a). Additionally, in heterogeneous tissues (such as the kidney), the labeled fraction of glutamate isotopologue M2 was highest in the medulla (Me), with a labeled fraction of 0.12 following U-13C glucose infusion; furthermore, the isotopologue M5 was most intense in the inner cortex (Ic), at 0.15 following U-13C glutamine infusion (Fig. 2c, Supplementary Fig. 6b). Similar region-specific differences in nutrient utilization were also detected in the brain (Figs. 2d, and Supplementary Fig. 6c–d).
Characterization of organ-specific metabolic activity
Next, we sought to directly visualize and explore the metabolic activity of individual organs with the MSITracer. When U-13C glucose enters the blood, the isotope distribution of glucose M6 provides a readout of glucose uptake from circulation, and isotopologue M3 serves as an indicator of gluconeogenic activity within tissues (Supplementary Fig. 7a). Compared with native glucose (M0), which was prominently distributed in the liver (Fig. 3a1), isotopologues M6 and M3 were notably enriched in the lung and kidney, respectively, with normalized labeling of 0.43 in the lung and 0.10 in the kidney (Fig. 3a2, a3). In terms of U-13C glutamine infusion, the native form (M0) exhibited high intensity in the brain, heart, and pancreas (Fig. 3b1), and the highest normalized labeling (1.09) of uniformly labeled ions (M5) was observed in the pancreas (Fig. 3b2). This result further confirms the tendency of the pancreas to utilize glutamine.
The labeled characteristics of downstream metabolites originating from the two U-13C nutrients were also elucidated (Supplementary Fig. 8). Besides isotopologue M2, highly enriched M4 form of malate, succinate, aspartate, and N-acetyl aspartate (NAA) were dominant in the brain following U-13C glucose infusion (Fig. 3c1-c4), suggesting that glucose metabolism was robust and the TCA cycle occurred at least twice within this organ. Moreover, the oxidation product gluconate M3 was intensely labeled in the liver regardless of whether U-13C glucose or glutamine was infused, highlighting the significance of the liver in glucose generation and oxidative metabolism (Fig. 3c5, and Supplementary Fig. 7b). Unexpectedly, lactate labeling was widely enriched throughout the body, with a greater abundance in the brain (Fig. 3c6). For all labeled metabolite ions discussed above, no interfering signal was detected in the mice that did not receive the infusions (Supplementary Figs. 7c–f, 6d, and 4e).
To further validate the organ-specific metabolic activity, we analyzed the dynamic metabolic labeling profiles by sampling at six time points following U-13C glucose infusion: 10, 30, 60, 120, 180, and 240 min. Over time, the mean enrichment (ME) of labeled metabolites exhibited a progressive increase. In particular, the accumulated ME of labeled lactate was similar across the five major organs, while for TCA intermediates, such as succinate, it was highest in the brain (Fig. 3d). A first-order kinetic equation was then employed to estimate labeling rates in those organs33. The decay rate (k) of each metabolite was determined and the curve fitting was assessed by the correlation coefficient (r), with r > 0.80 indicating a good fit. Using succinate as an example, the k value was calculated to be 0.0011 min−1, with an r value of 0.991 (Supplementary Fig. 9a). These results demonstrate that, unlike lactate, TCA-related metabolites turnover most rapidly in brain tissue (Fig. 3e, Supplementary Fig. 9b).

a AFADESI-MS images of glucose M0, M6, and M3 following U-13C glucose infusion. The heatmap shows normalized labeling across various organs. Signal intensity is shown in ion counts. BAT, brown adipose tissue; Bra, brain; Kid, kidney; Liv, liver; Lun, lung; Pan, pancreas. b AFADESI-MS images of glutamine M0, M5, and M2 following U-13C glutamine infusion. The heatmap shows normalized labeling across various organs. Signal intensity is shown in ion counts. c Representative AFADESI-MS images of organ-specific labeled metabolites following U-13C glucose infusion. The organs of interest are delineated with dashed lines in different colors. Signal intensity is shown in ion counts. NAA, N-acetyl aspartate. d Time-course mean enrichment of lactate and succinate in the brain, lung, heart, kidney and liver. e Exponential fitting of the unlabeled fraction of lactate and succinate in the five organs. Data are presented as mean ± SD (n = 3 biological replicates). Source data are provided as a Source Data file. f Notable spatial distribution differences among the native aspartate and corresponding isotopologues following U-13C-glutamine infusion. Signal intensity is shown in ion counts. g Schematic diagram of 13C-atom translations from U-13C-glutamine. Colored circles denote 13C carbons, while white circles represent 12C carbons. Red circles indicate labeling in the first turn of the TCA cycle, whereas blue circles indicate labeling in the second turn of the TCA cycle. h Illustration of the glutamine-glucose metabolic coordination between kidney, liver and brain. Created in BioRender. Li, X. (https://BioRender.com/gc0ge5m).
After U-13C glutamine infusion, one of the intriguing findings is the notable spatial distribution differences among various forms of isotopologues for certain metabolites. For example, higher-order labeling isotopologues (e.g., aspartate M4, glutamine M5, glutamate M5, and succinate M4) were enriched in the spleen, pancreas or liver, whereas their M2 isotopologues appeared in the brain (Fig. 3f, 3b3, 2b, Supplementary Fig. 10a). Given the vigorous TCA activity in the brain, we suspected that the enrichment of M2 metabolites indicates active 13C-glucose oxidation occurring (Fig. 3g). Additionally, the predominant enrichment of glucose M3 in the kidney and liver, as detected by AFADESI-MSI and LC − MS (Supplementary Fig. 10b, 10c), points to enhanced gluconeogenic activity in these two organs. This is further supported by the elevated expression of glycolysis-related enzymes, including fructose-1,6-bisphosphatase (FBPase), glucose-6-phosphatase (G6Pase), and phosphoenolpyruvate carboxykinase (PEPCK), in the liver and kidney compared to other organs (Supplementary Fig. 10d). Coupled with the higher levels of serum glucose M3 (Supplementary Fig. 10c), these results suggest that the newly synthesized glucose is entering the systemic circulation. Therefore, following 13C-glutamine infusion, the glutamate M2 and M1 observed in the brain are primarily derived from glucose produced via gluconeogenesis in the kidney and liver (Fig. 3h). Taken together, spatially resolved metabolic tracing supported the quantitative comparison of metabolic activities across tissues and offers valuable clues for discovering inter-organ metabolic communication.
Deciphering inter-organ metabolic communication
Drawing from this concept, we investigated the differences in spatial distribution between M0 and Mn (n is the total number of carbons in the metabolite). Interestingly, we observed significant differences in the spatial patterns of nascent fatty acids (FAs) compared to those of preexisting FAs. For instance, 12C-palmitic acid (M0) was predominantly localized in the brain, while its isotopologues (M1 to M6) were most abundant in BAT (Fig. 4a, Supplementary Fig. 11a). A similar spatial fingerprint was observed for myristic acid (Supplementary Fig. 11b). As glucose can be converted into acetyl-CoA to provide two-carbon units that support the de novo synthesis of FAs (Fig. 4b), this data provides evidence that BAT is a major producer of FAs.

a AFADESI-MS images of palmitic acid with a heatmap representing the signal intensity across organs (left) and images of palmitic acid M6 with a heatmap representing the ME across organs (right). Signal intensity is shown in ion counts. Bra, brain; Liv, liver; Kid, kidney; Hea, heart; Spleen, spl; Lun, lung; Pan, pancreas; BAT, brown adipose tissue; Int, intensity. b Schematic illustrating the incorporation of carbon from U-13C-glucose into palmitic acid. Light green circles denote 13C carbons, while white circles represent 12C carbons. CAR, carnitine; CoA, coenzyme A. c AFADESI-MS images of acetylcarnitine M2. Signal intensity is shown in ion counts. d Normalized labeling fraction of acetylcarnitine M2 across organs, as measured by LC‒MS. Data are presented as mean ± SD (n = 3 biological replicates). e Normalized labeling fraction of palmitoylcarnitine isotopologues in the BAT and brain. Data are presented as mean ± SD (n = 3 biological replicates). f Normalized labeling fraction of hexanoylcarnitine M2 in BAT, brain, heart and serum. Data are presented as mean ± SD (n = 3 biological replicates). g Labeling profile of palmitic acid in BAT, brain and serum. Data are presented as mean ± SD (n = 3 biological replicates). Source data are provided as a Source Data file. h Representative lipid profiles of TG (48:1) in BAT, brain, liver and serum. The color in the heatmap represents labeled fraction (LF) after logarithmic transformation. i Distinct labeling profile of representative lipids between the brain and liver. Lipids in the brain can incorporate more 13C carbons (up to M7), whereas lipids in the liver can only be labeled with M3, as measured by LC‒MS. j The potential 13C-carbon allocation of labeled TG (48:1). k Illustration of the proposed metabolic crosstalk model, created in BioRender. Li, X. (https://BioRender.com/plw7j67).
We further analyzed the in vivo oxidative metabolism of FAs, which can provide energy by converting FAs into even-chain carnitine species (Fig. 4b). AFADESI-MS images and LC‒MS data revealed that the acetylcarnitine M2 exhibited the highest ion intensity and normalized labeling in the heart (Fig. 4c, d). Since acetylcarnitine can be used to buffer excess acetyl-coenzyme A, we checked the labeling patterns of longer-chain carnitines using LC‒MS data. As a result, labeled palmitoylcarnitine were exclusively detected in BAT and the brain (Fig. 4e), while hexanoylcarnitine was highly labeled in the BAT, brain and heart, with no detectable labeling in the serum (Fig. 4f). These results suggest that the heart utilizes both circulating fatty acids and glucose simultaneously for energy production, and fatty acid oxidation is more pronounced in the brain and BAT compared to the heart.
As no labeled free FAs were detected in the heart, we were curious about their origin. The most likely source is free FAs in the circulation. Yet, despite the high ion intensity of FAs in the serum, no labeled forms were detected via LC‒MS analysis (Fig. 4g). We then examined alternative lipid carriers, with triglycerides (TGs) emerging as the primary candidates. We detected labeled TG (48:1) in BAT, brain, liver, and serum but with distinct profiles across these samples (Fig. 4h). In BAT, 13C-labeling extends to isotopologue M10, while in the brain, it reaches M7. In contrast, the liver and serum exhibited only M3-labeled TG. Other classes of lipids, such as diacylglycerol (DG, 34:0) and lysophosphatidylcholine (LPC, 16:0), exhibited similar distribution patterns (Fig. 4i).
Theoretically, triglyceride molecules can be labeled from two different sources: the glycerol backbone and the acyl chain (Fig. 4j). The isotopologue spectra of the highly abundant and highly labeled TG (48:1) suggest that the labeling patterns observed in the brain and BAT result from a combination of both labeling pathways, while the liver predominantly utilizes the labeled glycerol backbone for lipid synthesis. As the isotope distribution pattern of lipids in the serum aligns with that in the liver, the blood could selectively transport lipids synthesized in the liver to other organs, such as the heart, for energy. Therefore, the following metabolic exchange model may exist between organs: BAT and the brain utilize glucose for de novo synthesis of FAs and simultaneously oxidizes the FAs for metabolism within tissues. Moreover, the liver synthesizes lipids using the glycerol backbone generated from glucose oxidation and transports them to other organs for interorgan energy exchange, thereby maintaining overall lipid homeostasis (Fig. 4k).
To validate the proposed metabolic communications, we assessed the expression of key enzymes involved in the aforementioned process. Acetyl-CoA carboxylase 1 (ACC1) and fatty acid synthase (FASN) are two key enzymes in the fatty acid synthesis. Results from the immunohistochemistry (IHC) and quantitative proteomics assays demonstrated that the two enzymes were most abundant in BAT, with the lowest expression in the heart (Supplementary Fig. 11c-11e), further supporting BAT’s central role in synthesizing FAs. Diacylglycerol O-acyltransferase 2 (DGAT2), recognized as the rate-limiting enzyme in triglyceride biosynthesis with greater substrate affinity than DGAT134, is more highly expressed in BAT and the liver (Supplementary Fig. 11f), consistent with the triglyceride synthesis observed in this study. Moreover, the lipoprotein lipase (LPL) and carnitine O-palmitoyltransferase 1b (CPT1b) are responsible for the hydrolysis of triglycerides and the β-oxidation of long-chain FAs, respectively35. In BAT and the heart, the elevated expression of LPL and CPT1b underscores their dependence on fatty acids as a primary energy source (Supplementary Fig. 11g, 11h). This is further supported by the uniquely enhanced levels of uncoupling protein 1 (UCP1) in BAT, highlighting its role in non-shivering thermogenesis by converting energy into heat36 (Supplementary Fig. 11i). By comparison, the heart exhibits greater levels of ATP synthase F1 subunit alpha (ATP5F1A) and ATP5F1B, reflecting its reliance on ATP production for contractile activity37 (Supplementary Fig. 11j, 11k).
Systemic loss of metabolic coordination in tumor-bearing mice
A disturbance in metabolic homeostasis is a defining characteristic of cancer. Yet, the impact of tumors on host metabolism at the system level is not fully understood. Here, we infused U-13C glucose into HepG2 tumor-bearing mice and employed LC‒MS and AFADESI-MSI for untargeted stable-isotope tracing metabolomics to map metabolic changes across whole-body mice (Fig. 5a). We identified 231 13C-labeled metabolites and 1360 13C-labeled isotopologues in positive mode, as well as 264 13C-labeled metabolites and 1096 13C-labeled isotopologues in negative mode using LC‒MS/MS in tumors (Supplementary Fig. 12a). Most of these labeled ions were lipids, suggesting that autonomous FA oxidation and synthesis processes occurred within the tumor to support its proliferation (Supplementary Fig. 12b, 12c). Moreover, MSITracer annotated the labeled metabolites in the tumor within 5 min and allowed us to identify heterogeneous distribution of labeled metabolites across various microregions (Fig. 5b, Supplementary Fig. 12d). For example, labeled glucose M6 and its derivative, glucosyl-glycerol M3, were predominantly distributed in the necrotic and stromal regions, with no presence in the proliferating zone (Fig. 5b, c). In contrast, lactate was distributed throughout the tumor, with higher concentrations in the necrotic areas. Other metabolites involved in the TCA cycle (e.g., succinate), FA oxidation (e.g., acetylcarnitine), and glutathione metabolism (e.g., glutathione), were predominantly distributed in the proliferative region (Fig. 5b, c). These results indicate that tumor exhibits extensive glucose metabolism.

a Scheme of in vivo deep U-13C glucose tracing in tumor-bearing mice. Created in BioRender. Li, X. (https://BioRender.com/rq6a86p). b H&E-stained image of a tumor section and ×100 magnified H&E-stained images of tumor microregions derived from a HepG2 subcutaneous xenograft mouse infused with U-13C-glucose; scale bar = 2 mm for the whole tissue section, scale bar = 20 μm for the magnified images (left). Images are representative of at least three independent experiments. T1, proliferating areas; T2, necrotic areas; T3, stromal areas. AFADESI-MS images of the isotopologues of glucose, lactate, succinate, acetylcarnitine, glucosyl-glycerol, proline, malate and glutathione across the tumor (right). Intensity shown as relative abundance (normalized to 100%). c Normalized labeling fraction of representative metabolites in the tumor proliferation zone, stroma, and necrotic region, as calculated by MSITracer. Statistics were calculated using one-way ANOVA and post hoc Tukey’s correction. Data are mean ± SD (n = 9; 3 biological replicates, each with 3 independent regions selected from each slice). *p < 0.05, **p < 0.01, and ***p < 0.001. Adjusted p values for comparisons reaching statistical significance are provided in Supplementary Table 2. d AFADESI-MS images of glutamate and glutamine isotopologues M2, M3, and M4 in tumors following U-13C glucose infusion. Intensity shown as relative abundance (normalized to 100%). e, Labeling fingerprints of representative metabolites in tumors, as calculated by MSITracer. Data are mean ± SD (n = 9; 3 biological replicates, each with 3 independent regions selected from each slice). f Heatmap of the differences in the normalized labeling of UDP-GIcNAc isotopologues across organs between normal and tumor-bearing mice. Each data point represents the means of normalized labeling from three biological replicates. Source data are provided as a Source Data file. g Illustration of the impact of tumor burden on the HBP pathway in host organs and the glutamine communication between the host and the tumor. Created in BioRender. Li, X. (https://BioRender.com/yewga37).
When examining the labeling patterns in tumors using MSITracer, we observed that the 13C-fingerprint of glutamate was distinct from that of other metabolites (Fig. 5d, Supplementary Fig. 13a). Specifically, the labeled fraction of glutamate isotopologues was higher compared to other TCA cycle intermediates (Fig. 5e). Using LC‒MS, we further discovered a progressive decrease in 13C enrichment among metabolites extracted from tumors (Supplementary Fig. 12e), while the labeled fraction of glutamate M2 was higher than that of its anticipated precursor, α-ketoglutarate (α-KG) M2 (Supplementary Fig. 12f). Glutamate and α-KG are theoretically in isotopic equilibrium, allowing glutamate enrichment measurements to serve as a proxy for α-KG enrichment in certain cases38. The discrepancy in labeling between these two metabolites suggested an additional 13C source may exist to facilitate glutamate synthesis. Moreover, glutamate can be converted to glutamate semialdehyde, which subsequently leads to the formation of proline and arginine39. The labeled fractions of proline M2 and arginine M1 in tumors were approximately 0.02 and 0.01, respectively, confirming the presence of active glutamate metabolism (Supplementary Fig. 12g).
We next sought to determine the carbon source that could complement the metabolic demand for glutamate in tumors. Glutamine can be swiftly converted to glutamate by glutaminase (GLS), thereby facilitating TCA cycle anaplerosis40. After infusing 13C-glucose into tumor-bearing mice, glutamate and glutamine predominantly accumulated in brain (Supplementary Fig. 13b, c), and the labeled fractions of these two metabolites were linearly correlated, with an R2 of 0.984 (Supplementary Fig. 13d). Nevertheless, this correlation decreased in tumor, with an R2 of 0.919, suggesting that additional sources of glutamine may contribute to the pool (Supplementary Fig. 13e). At the protein level, we noticed that the expression of GLS and glutamine transporters responsible for shuttling glutamine into the cytosol (SLC1A5, SLC38A1) were significantly elevated in tumors compared to livers of the tumor-bearing mice (Supplementary Fig. 14a–c). In contrast, the expression of glutamine synthetase (GS) in tumors was much lower than that in livers (Supplementary Fig. 14d), suggesting that the tumors may rely more on importing glutamine from the extracellular environment rather than synthesizing it internally. Subsequently, we used LC‒MS to examine the labeling of glutamine in the serum and other tissues. The highest labeled fractions of glutamine M2 and M4 were detected in the brain, consistent with the data obtained by MSITracer. Additionally, the isotopologue profiles in the lung and serum were the most closely matched (Supplementary Fig. 14e). Based on their isotopologue similarities, we infer that glutamine released from the lung may serve as a source of glutamine for tumors (Supplementary Fig. 14f).
Given that N-acetyl-aspartyl-glutamate (NAAG) was recently identified as a glutamate reservoir41, we explored the possibility that metabolites containing glutamyl or glutamate groups could fuel glutamate synthesis in tumors. Through in-depth in vivo isotope tracing, five labeled metabolites were detected with this characteristic including NAAG, N-acetyl-glutamine, glutamylalanine, glutamylglutamate, and glutamylglutamine. All of them exhibited higher labeling in the brain, indicating robust glutamate synthesis and utilization (Supplementary Fig. 14g). However, the labeling profiles in serum and tumor differed significantly. For example, no labeled NAAG was detected in the serum, and NAAG labeling in the tumor was significantly lower than in the brain. These results suggest that the contribution of circulating intermediates containing glutamate groups to glutamate synthesis is likely insignificant.
Another question of interest was what metabolic alterations occurred in the host in response to the metabolic burden imposed by the tumor. Utilizing LC‒MS based deep isotope tracing, our results demonstrated that UDP-N-acetyl-glucosamine (UDP-GIcNAc) showed the most significant reduction in labeling extent across organs. For example, in the lung of normal mice, UDP-GIcNAc was labeled to isotopologue M8, indicating incorporation from glucose M6 and acetyl-CoA M2 molecules (Fig. 5f, and Supplementary Fig. 15a). However, in tumor-bearing mice, no labeled UDP-GIcNAc was detected. UDP-GIcNAc, the end product of the hexosamine biosynthetic pathway (HBP) (Supplementary Fig. 15b), plays a crucial role in protein N-glycosylation and O-linked N-acetylglucosamine modification42. The shift in labeling fraction of other key metabolites in this pathway, such as N-acetylglucosamine-6-phosphate (GlcNAc-6-P) (Supplementary Fig. 15c), further highlights that tumor burden exerts a considerable impact on host HBP metabolism. In conclusion, these results indicated that the tumor burden significantly impacted the HBP pathway in host organs, with the lungs sustaining the tumor’s metabolic demands through glucose-derived glutamine communication (Fig. 5g).
Discussion
In the field of spatial isotope tracing, the lack of methods to comprehensively identify labeled metabolites causes significant challenges. In this study, we introduced a framework to track labeled metabolites spanning from polar metabolites to lipids for both LC‒MS and MSI. Although several efforts have been made to automatically access LC‒MS based isotope tracing spectra, and we attempted to avoid reinventing the wheel, our study has several distinctive advantages in comparison with previous publications. First, a thoroughly optimized workflow that encompasses LC‒MS/MS data collection conditions, peak detection parameters, metabolite identification tools, and isotopologue feature tracing algorithms was developed (see “Methods”). This workflow has significantly improved the identification coverage of labeled metabolites and can be applied to any given 13C-labeled tracer. As an added value, the largest available resource of labeled metabolites in mammals (after the in vivo infusion of U-13C glucose and U-13C glutamine) was provided, complementing and substantially improving the knowledge accumulated by conventional metabolomics studies. This workflow enabled us to observe significant differences among various organs in the uptake and utilization of the two nutrients that are most essential for cell metabolism, emphasizing the brain’s metabolic preference for glucose and the pancreas’s sensitivity towards glutamine. Notably, many previously unreported metabolites were identified, three of which were labeled more greatly than those of the TCA cycle-related intermediates. Future studies are needed to elucidate the biological mechanisms underlying the rapid transformation of these metabolites in vivo.
Second, a computational tool was introduced for spatial isotope tracing that can achieve the untargeted identification of labeled metabolites and qualify the labeled fraction, offering streamlined and automated MSI data analysis. For LC‒MS data, isotope labeling signals can be identified by comparing the signal intensity of naturally occurring isotope profiles with that of heavy atom-enriched profiles. However, conducting this kind of calculation per pixel in complex MSI spectra can be convoluted and computationally intensive. Hence, the main advancement in MSITracer resulted from the integration of a thorough database that contained potentially labeled signals. As we have fully considered the difference in the adduct ions between MSI and LC‒MS, this strategy enhances annotation coverage and reduces the number of false positives through two intensity filtering functions (see “Methods”). While we only demonstrated the efficiency of the in-house-developed AFADESI-MSI system, MSITracer is designed to accommodate all types of MSI data and supports the input of tabular forms. The tool is also accessible without a requirement for advanced computational capabilities and professional bioinformatics knowledge. Moreover, MSITracer can directly export information such as metabolite names, molecular formulas, adduct forms, and labeled fractions and allow users to customize databases and thresholds for each step as needed. In the case of multiple labeled nutrients being introduced, this approach is also effective for delineating differences in nutrient utilization among various regions. For example, distinct labeling patterns were detected in the kidney and brain after two U-13C nutrient infusions (Fig. 2c, d), which may reflect the expression of relevant transporters and enzymes.
Furthermore, a strategy is proposed to elucidate inter-tissue metabolic communication by examining spatial distribution differences between isotopically labeled metabolites. In particular, MSITracer enables intuitively and visually uncovers clues in three dimensions: original and newly assimilated metabolites (M0 and Mn), various labeled isotopologues of the same metabolite (M1 and Mn), and different newly synthesized metabolites within the same pathway (Mx and My). Since it is generally assumed that the spatial distribution of metabolites and their isotopologues is uniform, this unexpected labeling pattern prompted us to use LC‒MS for further analysis. Accordingly, we characterized the turnover of glutamine between the kidneys, liver, and brain, and conversion of glucose-derived lipids between the liver and heart. In these scenarios, MSI and LC‒MS functions act as flashlight and magnifying glass, enabling the identification of metabolic exchanges and their underlying causes. Meanwhile, we can also effectively identify the spatial distribution differences within heterogeneous tissues, such as native glutamate and glutathione, and their corresponding isotopologues (Figs. 2c, d and Supplementary Fig. 6e). This phenomenon sheds light on metabolic crosstalk at the mircoreigon level of tissue.
Cancer metabolism is regulated by cell-intrinsic factors, interactions within the tumor microenvironment, and overall metabolic homeostasis43. While numerous studies have focused on the first two aspects, the impact of a tumor on distant tissues remains poorly understood. Naser, Jackstadt, et al. proposed a tumor-liver alanine cycle using a zebrafish model and highlighted some organs within an organism that are likely to reprogram their metabolism to complement the tumor25. Our results further support this idea in a mouse xenograft model, tumors can exploit lung-derived glutamine to meet their high metabolic demands. While the inter-organ glutamine trafficking has been proposed before44,45, it has been directly demonstrated using MSI and 13C-isotope tracing in this study. We showed that the lung of host mice, rather than the muscle, make a greater contribution to the tumor’s glutamine supply, despite both being primary producers in healthy mice. Our data also suggested a system-wide loss of metabolic coordination across various organs in the tumor-bearing mice, which was uniquely evidenced by the downregulation of the HBP throughout the entire body. It has been reported that 3%-5% of glucose participates in the HBP and leads to the production of UDP-GIcNAc, an important regulator of cell signaling and protein glycosylation46. This observation indicates that tumors typically downregulate certain physiological tasks to conserve energy, exemplified by the reduction in the rate of proteins synthesis observed in the pancreas within pancreatic cancer models8. Therefore, we argue that although the organism undergoes metabolic reprogramming to adapt to tumor proliferation, the tumor exerts irreversible effects on host metabolism. These changes may contribute to cancer-associated cachexia and hijacking systemic metabolic alterations holds the potential to benefit patients.
The technical limitations of the study include that the number of labeled metabolites that can be visualized by MSI is smaller than that measurable by LC‒MS and that the signal for higher-order labeling (e.g., M4 in citrate and malate) is poor in MSI. Since varying sample efficiencies occur between these two techniques, integrating LC‒MS and MSI is highly encouraged to elucidate critical metabolic interactions. Secondly, unlike LC − MS, which can differentiate metabolites based on retention times, the current MSITracer cannot distinguish isomeric molecular species. The presence of the same molecular weights may lead to multiple tentative annotations. Integrating orthogonal techniques, such as ion mobility MS47, presents a promising solution to this challenge. Moreover, we employed ex vivo rapid freezing of tissues to terminate enzymatic activity during sample collection. Since this approach may not entirely prevent post-mortem degradation of metabolites, studies examining metabolites that rapidly change after death can consider alternative organ sampling methods, such as in-situ focused microwave irradiation48, to effectively halt metabolic activities. Finally, the spatial resolution of the AFADESI platform applied in this study was approximately 100 micrometers. The efficiency of MSITracer in single-cell analysis has yet to be validated. As higher spatial resolution MSI techniques continue to evolve49, we anticipate that this tool will improve our ability to investigate cellular interactions and elucidate dynamics within the tissue microenvironment.
Methods
Ethics
All animal experiments were conducted with the approval of the Animal Ethical Committee at the Institute of Materia Medica, Chinese Academy of Medical Science, and Peking Union Medical College (grant no. 00008359).
Chemicals and reagents
HPLC-grade acetonitrile (ACN), methanol (MeOH), isopropanol (IPA), and methyl tertiary butyl ether (MTBE) were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Ammonium acetate (AmAc) was purchased from Sigma‒Aldrich (St. Louis, MO. USA). Ammonium hydroxide (NH4OH) was purchased from Fisher Chemical (Fair Lawn, NJ, USA). Formic acid was purchased from J&K Scientific (Shanghai, China). U-13C glucose and U-13C glutamine were purchased from Cambridge Isotopes laboratories (MA, USA). Pure water was purchased from Wahaha Co., Ltd. (Hangzhou, China). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin–streptomycin, 0.25% trypsin-ethylenediaminetetraacetic acid, and phosphate-buffered saline (PBS) were purchased from Gibco (Life Technologies, Carlsbad, CA). Other materials used for cell culture were purchased from Corning (North Carolina, NY, USA) unless otherwise noted.
Mouse xenografts
Male BALB/c nude mice weighing 16 − 19 g and aged 4–5 weeks were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and housed under specific pathogen-free conditions with a constant room temperature of 20−24 °C, 35 − 55% air humidity, and a 12-hour light/dark cycle. Sex was not considered in this study as our primary focus was the development of the spatial isotope deep tracing method and the investigation of tumor metabolic crosstalk.
Human hepatocarcinoma (HepG2) cells were originally purchased from ATCC. The cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin at 37 °C under 5% CO2. To establish a subcutaneous xenograft tumor model, exponentially growing cells were harvested and suspended in PBS. A total of 3.5 × 106 HepG2 cells were inoculated into the right flank of each mouse. To ensure a homogeneous tumor size, when the tumor reached approximately 1 cm3 in size, the tumor masses were resected, minced into pieces of approximately 2 mm3 and transplanted subcutaneously into the next cohort of experimental nude mice. The tumor volume was measured every 2 days and calculated using the formula V = length × width2 × 0.5.
In vivo 13C labeling experiments
The BALB/c nude mice were randomly divided into noninfusion or infusion groups. Catheters were inserted into the jugular vein of mice under anaesthesia. U-13C glucose and U-13C glutamine were infused into conscious, free-moving animals for 3 h at constant rates of 80 and 30 nmol/min/g, respectively. For the dynamic 13C labeling experiments, the infusion groups received U-13C glucose at a rate of 80 nmol/min/g for six time points, namely, 10, 30, 60, 120, 180, and 240 min, with no infusion group used as a control. For the flank hepatocellular carcinoma mouse model, U-13C glucose infusions were initiated when the tumors reached 1.0-1.2 cm in diameter. The infusion rate was set at 80 nmol/min/g for conscious, free-moving animals over a period of 3 hours.
For whole-body animal (WBA) MSI analysis, the samples were collected as follows: After the infusion, blood was collected from the orbital sinus, and the mouse was euthanized. The whole-body mouse was then quickly snap-frozen in liquid nitrogen within 30 s to halt metabolic processes. The samples were subsequently stored at −80 °C until section preparation.
For MSI and LC–MS analysis of individual organs, the samples were collected as follows: After the infusion, blood was collected from the orbital sinus, and the mouse was euthanized. Nine organs (liver, kidney, spleen, pancreas, heart, lung, brain, brown adipose tissue, and muscle) were sequentially harvested and quickly snap-frozen in liquid nitrogen to halt metabolic processes and ensure comparability between different groups. The samples were stored at −80 °C until the preparation of tissue section for MSI analysis and tissue homogenates for LC–MS analysis.
Metabolite extraction from serum and tissues
For metabolomics analysis, 500 μL of extraction solvent (ACN: MeOH: H2O = 2:2:1, v/v/v) was added to 100 μL of serum or 25 mg of tissue sample. The mixture was vortexed for 30 s, followed by homogenization and sonication for 5 min in an ice-water bath; this process was repeated 3 times. Then, the mixture was incubated for 1 h before centrifugation at 15000 × g for 15 min at 4 °C. An aliquot of the supernatant was used for the LC‒MS assay. For lipidomic analysis, 480 μL of extraction solution (MTBE: MeOH = 5: 1, v/v) was sequentially added to 200 μL of water. Apart from the supernatant collection, all other procedures were the same as those described above. After solution layering, the supernatant was transferred and vacuum-dried. Finally, the samples were resolubilized in ACN/IPA/H2O (65:30:5, v/v/v) containing 5 mM AmAc prior to analysis.
Tissue slide preparation
For the tissue slide samples, frozen brains, kidneys, tumors, and WBA were preserved intact at −80 °C. For brain, kidney and tumor analyses, 15 µm-thick sections were collected on a cryostat (CM1860 UV, Leica Microsystems, Wetzlar, Germany) and thaw-mounted onto Superfrost Plus microscope slides (Thermo Scientific, USA) for AFADESI-MSI. Prior to analysis, the sections were desiccated under vacuum for 30 min. Consecutive tissue sections were collected for hematoxylin and eosin staining.
Each WBA sample was soaked in a metal mold container filled with 3% carboxymethyl cellulose sodium (CMC-Na) gel, and 25 µm thick sections were collected using a cryomacrotome (CM3600, Leica Microsystems, Wetzlar, Germany) and mounted onto an epoxy-gel-coated slide. Drying was carried out for 2 h at −20 °C and then for 2 h at room temperature before analysis. Optical images of the WBA sections were obtained by a Microtek scanner (Shanghai, China).
LC‒MS analysis
Metabolomics and lipidomics data were acquired using a UHPLC system (ACQUITY, Waters) coupled to a Q-Orbitrap mass spectrometer (Q Exactive, Thermo Scientific). For metabolome analysis, a Waters BEH Amide column (2.1 mm × 100 mm, 1.7 μm) and a Waters HSS T3 column (2.1 mm × 100 mm, 1.8 μm) were used for LC separation in HILIC and RP modes, respectively. For lipidome analysis, a Waters BEH C8 column (2.1 mm × 100 mm, 1.7 μm) was utilized. In HILIC mode, the mobile phase consisted of water containing 25 mmol/L AmAc, 25 mmol/L NH4OH (A) and ACN (B) in both positive and negative ion modes. The gradient program was set as follows: 0–0.5 min, 5% A; 0.5–7.0 min, 5% A-35% A; 7.0–8.0 min, 35%–60% A; 8.0–9.0 min, 60% A; and 9.0–12.0 min, 5% A. The flow rate was 0.5 mL/min, and the sample injection volume was 5 μL. In RP mode, the mobile phase consisted of water containing 0.1% formic acid (A) and ACN (B) for both positive and negative ion modes. The gradient program was set as follows: 0–1.5 min, 98% A; 1.5–15 min, 98-0% A; 15–22 min, 0% A; 22–22.1 min, 0-98% A; 22.1–27 min, 98% A. The flow rate was 0.25 mL/min, and the sample injection volume was 5 μL. For lipid analysis, the mobile phase consisted of ACN/H2O (60:40, v/v) (A) and IPA/ACN (90:10, v/v) (B), both containing 10 mM AmAc, for positive and negative ion modes. The gradient program was set as follows: 0–1.5 min, 68% A; 1.5–15.5 min, 15% A; 15.5-15.6 min, 3% A; 15.6–18 min, 3% A; 18–18.1 min, 68% A; and 18.1–20 min, 68% A. The flow rate was 0.26 mL/min, and the sample injection volume was 5 μL.
The HESI source parameters were as follows: spray voltage, 3.6 kV (positive) and −3.2 kV (negative); capillary temperature, 400 °C (for HILIC mode) or 350 °C (for RP mode and lipid analysis); sheath gas, 25 arb; and aux gas, 20 arb. For all the samples, full-scan LC‒MS data were acquired. For quality control samples, an additional ddMS2 scan was applied to obtain MS/MS spectra. The detailed parameters were as follows: full scan, resolution, 140000; AGC target, 1e6; maximum injection time, 100 ms; scan range, 70 − 1000 Da (for HILIC and RP mode) or 200 − 1200 Da (for lipid analysis); ddMS2 scan, orbitrap resolution, 70000; AGC target, 1e5; maximum injection time, 60 ms; scan range, 50 − 1200 Da; top N setting, 15; isolation width, 1.0 m/z; collision energy mode, stepped; collision energy type, normalized; HCD collision energies (%), 20, 30, 40 (for HILIC and RP mode) or 25, 35, 45 (for lipid analysis).
AFADESI-MSI experiments
The AFADESI-MSI platform mainly comprises a home-built AFADESI source, an ESI sprayer, a sample carrier platform, a 3D translational stage, a height-adjustable desk, and an electricity control module50. The AFADESI source was constructed with a stainless-steel transport tube (i.d. 3 mm, o.d. 4 mm, length 50 cm) and a vacuum pump that produces the extracting gas for remote ion transport, with a flow rate of 45 L/min. The spray solution was ACN/H2O (80:20, v/v), and was delivered to the sprayer at a flow rate of 10 μL/min for WBA samples and 5 μL/min for tissue samples. The spray gas was N2, and the pressure was set at 0.7 MPa. The spray voltage was set at 7.0 kV in positive ion mode and −7.0 kV in negative ion mode. The sprayer and glass slide were mounted on a 3D translational stage to allow their positioning relative to the transport tube with submillimeter precision. Before data collection, mass calibration was conducted in both positive and negative ion modes via electrospray ionization, using the respective calibration solution (Pierce, Thermo Scientific). The mass error was maintained below 3 ppm within the m/z range of 70–1000. For WBA sample analysis, the 3D translational stage moved continuously at a horizontal speed of 0.35 mm/s with a vertical step size of 0.50 mm. For tissue sample analysis, the 3D translational stage moved continuously at a horizontal speed of 0.10 mm/s with a vertical step size of 0.15 mm. Accordingly, the time required for MSI of each whole-body mouse, brain, kidney, and tumor section is approximately 140 min, 130 min, 100 min, and 120 min, respectively. The pixel sizes for AFADESI-MSI are 100 μm × 76 μm for a tissue and 500 μm × 270 μm for a whole-body animal. The mass spectrometer was operated in full scan mode. The capillary temperature was set at 350 °C, and S lens voltage was set at 55 V. The maximum injection time was 200 ms, and AGC target was 3 × 106. The resolution was set at 140,000, and the scan range was m/z 70−1000.
Comprehensive isotope tracing workflow using the LC‒MS technique
After the infusion of two U-13C tracers, metabolomics and lipidomics data were collected from nine organs and serum in both positive and negative ion modes, yielding a total of 60 datasets. Each dataset was processed according to the following procedure. ProteoWizard (version 3.0.22143) was used to convert the raw MS data (.raw) files to the.mzXML format (for full scan mode) and.mgf (for ddMS2 mode) format. The R package AutoTuner (version 1.4.0) was utilized to select dataset-specific parameters to ensure reliable data processing (Supplementary Table 3)51. Then, these optimized key values were used to group mzXML data files from noninfusion samples for peak detection, retention time correction, and peak alignment using the R package XCMS (version 3.12.0)52. For datasets acquired under HILIC mode, the resulting MS1 peak table and MS2 files were input into the R package metID (version 1.2.19)29 and MetDNA2 (version 1.4.1; http://metdna.zhulab.cn/)28 for metabolite annotation, with the liquid chromatography set to “HILIC”. For datasets acquired under the RP mode, metID was used to perform metabolite annotation using public databases embedded in its library. According to the definition of the metabolomics standards initiative, the confidence of metabolite annotation was assigned to level 2 (public MS2 database) and level 3 (public MS1 database). The generated metabolite annotation tables were further filtered and modified to meet the data formatting requirements of the R package MetTracer (version 1.0.4); these modifications included removing unreliable adduct forms, eliminating isotope annotations, adding molecular formula information, etc. Finally, default parameters were applied to globally track labeled metabolites within the dataset14.
Manual curation of U-13C labeled metabolites
To improve the accuracy of identifying labeled metabolites and reduce false positives, we further manually checked the results outputted by MetTracer using the following criteria in the raw files: (1) for the unlabeled sample, the extracted ion chromatogram at the given m/z exhibited a well-defined peak shape; (2) the retention time difference between isotopic peaks should be less than 0.1 min and consistent with the results recorded in the table; (3) for labeled samples, the calculated labeling fraction should derive from the corresponding isotopic peaks rather than nearby ion interferences; and (4) ions identified as M0 should not be isotopic peaks.
Categorization of metabolites
We used the Human Metabolome Database (HMDB) (www.hmdb.ca) to classify the metabolites into seven categories: energy, carbohydrate, nucleotide, amino acid, peptide, lipid, and others.
MSI data processing
In the AFADESI-MSI experiment, the tissue sample is scanned line by line, with each line’s data saved as a raw file. These raw files were converted to.cdf format using Xcalibur (Thermo Fisher Scientific, version 2.2) and then imported into MassImager software53. By typing the physical length and height of the tissue sample, the scan count and sampling time can be checked automatically by the MassImager (version 1.0) upon completing line sequence creation. Peak picking was conducted by identifying local maxima with a slope threshold of 100, determined based on the peak shape. To reduce background interference, a non-tissue background area was selected, and background subtraction was performed using a proportional coefficient (k = 1). The deduction formula is: Adjusted intensity = Original intensity- k × Background average intensity. The m/z tolerance for background subtraction is set to 0.001 Da. Data normalization was performed on the entire data matrix to further reduce the mass intensity variant in each mass pixel. After selecting the regions of interest (ROIs), the m/z and peak intensity list were exported as text files and imported into MarkerView software (Sciex, version 1.2.1) for peak alignment (mass tolerance: 5 ppm; minimum required response: 100).
The workflow of MSITracer
MSITracer features a high-coverage labeled metabolite database constructed from in vivo 13C-tracing experiments. Since the chemical formulas were known for each labeled metabolite from the LC‒MS results, we first calculated the theoretical m/z values of all 12C-metabolites with potential adduct ions, including [M + H]+, [M + Na]+, [M + K]+, [M + NH4]+, and [M + H − H2O]+ in positive ion mode and [M − H]−, [M + Cl]− and [M − H − H2O]− in negative ion mode (Supplementary Table 1). Subsequently, the m/z values of the 13C-isotopologues corresponding to each metabolite were calculated based on a mass difference of 1.0033548378. Users can also flexibly define and customize ion types in the database file (Supplementary Data 2).
Using this database, the MSITracer package processes data in three major steps: (1) Extracting the intensity of the targeted isotopologues. After inputting the average mass spectra data, MSITracer automatically matched the intensities between the experimentally measured m/z and the targeted isotopologues list with a tolerance of 5 ppm for each dataset. If multiple peaks are present in the specified window, MSITracer will record the most intensive signal. (2) Selecting ions with sufficient imaging signals. For the generated table, if the intensity of the 12C-metabolite ion was less than 100 in more than half of the samples, this ion and its corresponding isotopologue group were removed. This threshold of 100 counts can be customized by the users according to the instrument sensitivity. For the remaining isotopologue groups, ratio analysis was performed by dividing the average ion intensities of the isotopologues (M2 and M3) in the labeled sample group by the corresponding average ion intensities of the isotopologues in the unlabeled sample group. For the two ratios obtained, if one of them exceeded 10, the isotopologue group was retained, indicating sufficient labeled signals following 13C-nutrient infusion. The selection of M2 and M3 instead of M1 for the ratio calculations was because nutrients commonly engage in downstream metabolism via two or three carbon units. Additionally, M1 might be susceptible to greater interference from natural isotopes. The threshold of 10 for ratio analysis was empirical but adjustable based on the type of tracer administered and the degree of isotope enrichment. Finally, intermediate files for each step and tables containing the names, molecular formulas, and ion intensities of the labeled metabolites were generated as outputs. This data filtering strategy greatly reduced data complexity and improved the accuracy of labeled metabolite identification. (3) Quantification of labeling patterns and fractions. The R package AccuCor (version 0.3.0; https://github.com/XiaoyangSu/AccuCor)54 was further integrated to perform 13C natural isotope abundance correction and provide the mass isotopologue distribution table. This result was output as another file.
Definition of labeled extent, mean enrichment and normalized labeling
Following the infusion of the 13C-labeled tracer, the labeled extent (LE) represents the overall 13C enrichment of one metabolite and can be calculated by Eq. 116 as follows:
where IMi is the peak intensity of isotopologue Mi, LM0 denotes the labeled fraction of M0 and N represents the carbon count of the metabolite.
The mean enrichment (ME) reflects the average carbon atom labeling of one metabolite in a heterogeneous region and can be calculated by Eq. 214 as follows:
where LMi is the labeled fraction of isotopologue Mi and N represents the carbon count of the metabolite.
Normalized labeling was calculated by dividing the labeled fraction by the relative enrichment of the 13C tracer (glucose or glutamine) in circulation55.
Immunohistochemistry
Formalin-fixed paraffin-embedded tissue blocks were cut into 4 μm slices and were mounted on polylysine-charged glass slides. Endogenous peroxidase activity was blocked by exposure to 3.0% H2O2 for 30 min. Antigen retrieval was performed in a citrate buffer (pH 6.0) at 120 °C for 10 min. Sections were then incubated at 4 °C overnight with FASN (C20G5) rabbit mAb (#3180, CST, 1:50 dil) and ACC1 monoclonal antibody (#67373-1-Ig, Proteintech, 1:500 dil). After washing, the sections were further incubated with the goat anti-rabbit IgG (H + L) secondary antibody, HRP (#EF0002, Sparkjade, 1:200 dil) and goat anti-mouse secondary antibody, HRP (#EF0001, Sparkjade, 1:200 dil) for 1 h at room temperature, respectively. Sections incubated with isotype and concentration matched immunoglobulins without primary antibodies were used as isotype controls. Peroxidase activity was visualized with the DAB working solution (#CW0125M, CWBIO), and brown coloration of tissues represented positive staining. Images were captured using a Pannoramic MIDI scanner (3DHISTECH, Budapest, Hungary).
Quantitative proteomics
Tissue samples were retrieved from the −80 °C freezer and ground into powder using liquid nitrogen. An appropriate amount of the powder was then transferred to 1.5 mL centrifuge tubes, followed by the addition of lysis buffer (containing 8 M urea, 1 mM PMSF, and 2 mM EDTA). Ultrasonic lysis was performed on ice for 5 min. The samples were then centrifuged at 15000 × g and 4 °C for 10 min to collect the supernatant. Finally, protein concentration was determined using a BCA assay kit. Based on the determined protein concentration, 100 μg of protein solution was taken, and the volume was adjusted to 200 μL with 8 M urea. The sample was then reduced with 5 mM DTT at 37 °C for 45 min, followed by alkylation with 11 mM iodoacetamide (IAM) in the dark at room temperature for 15 min. Afterward, 800 μL of 25 mM ammonium bicarbonate solution and 2 μL of Trypsin (Promega, V5280) were added, and the mixture was incubated overnight at 37 °C for digestion. The pH of the digested peptides was then adjusted to 2 − 3 using 20% TFA, followed by desalting with C18 resin (Millipore, Billerica, MA). Finally, the peptide concentration was measured using a Pierce™ Quantitative Peptide Assay Kit with standards (Thermo Fisher Scientific).
All data were acquired using a Vanquish Neo UHPLC system coupled with an Orbitrap Astral mass spectrometer (Thermo Fisher Scientific). Mobile phase A consisted of 0.1% formic acid in water, while mobile phase B was ACN containing 0.1% formic acid. A trap-and-elute dual-column setup was used, with a PepMap Neo Trap Cartridge (300 μm × 5 mm, 5 μm) serving as the trapping column, and an Easy-Spray™ PepMap™ Neo UHPLC column (150 μm × 15 cm, 2 μm) as the analytical column. The column temperature was maintained at 55 °C, with an injection volume of 200 ng and a flow rate of 2.5 μL/min. The effective gradient lasted 6.9 min, and the total runtime was 8 min. Mass spectrometry was conducted in positive ion mode. MS1 spectra were acquired with a precursor ion scan range of 380-980 m/z, a mass resolution of 240000 at 200 m/z, a normalized AGC target of 500%, and a maximum injection time (IT) of 5 ms. MS2 spectra were acquired using DIA mode with 299 scan windows, an isolation window of 2 Th, an HCD collision energy of 25%, a normalized AGC target of 500%, and a maximum IT of 3 ms.
MS raw data were analyzed using DIA-NN (v1.8.1) with a library-free method. The uniprotkb_proteome_UP000000589_mouse_54910_20240528.fasta database (containing 54910 sequences) was used to generate a spectra library through deep learning algorithms based on neural networks. The Match Between Runs (MBR) option was employed to create a spectral library from the DIA data, which was then reanalyzed using this library. The false discovery rate for the search results was controlled to < 1% at both the protein and precursor ion levels, and the remaining identifications were used for further quantification analysis.
In the DIA-NN search settings, the maximum number of missed cleavages was set to 1, and only one variable modification per peptide was allowed. N-terminal methionine excision and cysteine carbamidomethylation were set as fixed modifications, while methionine oxidation (Ox[M]) was selected as a variable modification. Peptide length was restricted to 6–35 amino acids, and precursor charge range was set to 2–5. Protein inference was performed based on protein names from the FASTA file, and a minimum of one unique peptide was required for protein identification. All other parameters were left at their default values.
Statistical analysis
The statistical significance of the difference between two groups was analyzed using an unpaired two-tailed Student’s t test. P values were corrected for multiple testing using the Benjamini‒Hochberg procedure. For multiple comparisons, statistics were calculated using one-way ANOVA and post hoc Tukey’s test. The data are expressed as the means ± SDs, and differences were considered statistically significant at *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Both GraphPad Prism (version 9.0.0) and R (version 4.2.0) were used to conduct the tests. The exact sample sizes and statistical tests used are indicated in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The LC‒MS raw files of metabolomics data generated in this study have been deposited in the Metabolomics Workbench56 with study ID ST004031 (https://doi.org/10.21228/M8HR85) and OMIX database of the National Genomics Data Center under accession number OMIX006225. The AFADESI-MSI data were uploaded to METASPACE and are available at https://metaspace2020.org/project/li-2024. The proteomics data have been deposited in the Integrated Proteome Resources under project number IPX0009979001. The remaining data are available within the supplementary material of this article. Source data are provided with this paper.
Code availability
MSITracer is available for non-commercial use as an R package at (https://github.com/xinzhul/MSITracer) alongside clear example code vignettes. A version of the code has been archived on Zenodo with the: (https://doi.org/10.5281/zenodo.15760474).
References
Moffitt, J. R., Lundberg, E. & Heyn, H. The emerging landscape of spatial profiling technologies. Nat. Rev. Genet. 23, 741–759 (2022).
Yuan, Z. et al. SEAM is a spatial single nuclear metabolomics method for dissecting tissue microenvironment. Nat. Methods 18, 1223–1232 (2021).
Zhang, H. et al. Single-cell lipidomics enabled by dual-polarity ionization and ion mobility-mass spectrometry imaging. Nat. Commun. 14, 5185 (2023).
Bressan, D., Battistoni, G. & Hannon, G. J. The dawn of spatial omics. Science 381, eabq4964 (2023).
Alexandrov, T. Spatial metabolomics: from a niche field towards a driver of innovation. Nat. Metab. 5, 1443–1445 (2023).
Jang, C., Chen, L. & Rabinowitz, J. D. Metabolomics and isotope tracing. Cell 173, 822–837 (2018).
Grankvist, N. et al. Profiling the metabolism of human cells by deep 13C labeling. Cell Chem. Biol. 25, 1419–1427 (2018).
Bartman, C. R. et al. Slow TCA flux and ATP production in primary solid tumours but not metastases. Nature 614, 349–357 (2023).
Johnston, K. et al. Isotope tracing reveals glycolysis and oxidative metabolism in childhood tumors of multiple histologies. Med. 2, 395–410.e4 (2021).
Lehmann, W. D. A timeline of stable isotopes and mass spectrometry in the life sciences. Mass Spectrom. Rev. 36, 58–85 (2017).
Yuan, M. et al. Ex vivo and in vivo stable isotope labelling of central carbon metabolism and related pathways with analysis by LC‒MS/MS. Nat. Protoc. 14, 313–330 (2019).
Wang, L. et al. Spatially resolved isotope tracing reveals tissue metabolic activity. Nat. Methods 19, 223–230 (2022).
Schwaiger-Haber, M. et al. Using mass spectrometry imaging to map fluxes quantitatively in the tumor ecosystem. Nat. Commun. 14, 2876 (2023).
Wang, R. et al. Global stable-isotope tracing metabolomics reveals system-wide metabolic alternations in aging Drosophila. Nat. Commun. 13, 3518 (2022).
Llufrio, E. M., Cho, K. & Patti, G. J. Systems-level analysis of isotopic labeling in untargeted metabolomic data by X(13)CMS. Nat. Protoc. 14, 1970–1990 (2019).
Hiller, K., Metallo, C. M., Kelleher, J. K. & Stephanopoulos, G. Nontargeted elucidation of metabolic pathways using stable-isotope tracers and mass spectrometry. Anal. Chem. 82, 6621–6628 (2010).
Capellades, J. et al. geoRge: A computational tool to detect the presence of stable isotope labeling in LC/MS-based untargeted metabolomics. Anal. Chem. 88, 621–628 (2016).
Agrawal, S. et al. El-MAVEN: a fast, robust, and user-friendly mass spectrometry data processing engine for metabolomics. Methods Mol. Biol. 1978, 301–321 (2019).
Chen, Q. et al. Global determination of reaction rates and lipid turnover kinetics in Mus musculus. Cell Metab. 35, 711–721.e4 (2023).
Blanc, L. et al. Kendrick mass defect variation to decipher isotopic labeling in brain metastases studied by mass spectrometry imaging. Anal. Chem. 93, 16314–16319 (2021).
Argiles, J. M., Stemmler, B., Lopez-Soriano, F. J. & Busquets, S. Inter-tissue communication in cancer cachexia. Nat. Rev. Endocrinol. 15, 9–20 (2018).
Jang, C. et al. Metabolite exchange between mammalian organs quantified in pigs. Cell Metab. 30, 594–606.e3 (2019).
Cori, G. T. & Cori, C. F. Glucose-6-phosphatase of the liver in glycogen storage disease. J. Biol. Chem. 199, 661–667 (1952).
Schmidt, S. F., Rohm, M., Herzig, S. & Berriel Diaz, M. Cancer cachexia: more than skeletal muscle wasting. Trends Cancer 4, 849–860 (2018).
Naser, F. J. et al. Isotope tracing in adult zebrafish reveals alanine cycling between melanoma and liver. Cell Metab. 33, 1493–1504.e5 (2021).
Sun, R. C. et al. Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nat. Commun. 8, 1646 (2017).
Sun, C. et al. Spatially resolved metabolomics to discover tumor-associated metabolic alterations. Proc. Natl. Acad. Sci. USA. 116, 52–57 (2019).
Zhou, Z. et al. Metabolite annotation from knowns to unknowns through knowledge-guided multi-layer metabolic networking. Nat. Commun. 13, 6656 (2022).
Shen, X. et al. metID: a R package for automatable compound annotation for LC‒MS-based data. Bioinformatics 38, 568–569 (2022).
Lopes, M. et al. Metabolomics atlas of oral 13C-glucose tolerance test in mice. Cell Rep. 37, 109833 (2021).
Zhu, Y. et al. An organ-specific metabolite annotation approach for ambient mass spectrometry imaging reveals spatial metabolic alterations of a whole mouse body. Anal. Chem. 94, 7286–7294 (2022).
Li, X. et al. Database-driven spatially resolved lipidomics highlights heterogeneous metabolic alterations in type 2 diabetic mice. Anal. Chem. 95, 18691–18696 (2023).
Shi, X. et al. Comprehensive isotopic targeted mass spectrometry: reliable metabolic flux analysis with broad coverage. Anal. Chem. 92, 11728–11738 (2020).
Yen, C. L. et al. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 49, 2283–2301 (2008).
Tracey, T. J. et al. Neuronal lipid metabolism: multiple pathways driving functional outcomes in health and disease. Front. Mol. Neurosci. 11, 10 (2018).
Marlatt, K. L. & Ravussin, E. Brown adipose tissue: an update on recent findings. Curr. Obes. Rep. 6, 389–396 (2017).
Croston, T. L. et al. Evaluation of the cardiolipin biosynthetic pathway and its interactions in the diabetic heart. Life Sci. 93, 313–322 (2013).
Hubbard, B. T. et al. Q-Flux: a method to assess hepatic mitochondrial succinate dehydrogenase, methylmalonyl-CoA mutase, and glutaminase fluxes in vivo. Cell Metab. 35, 212–226.e214 (2023).
Wang, Y. et al. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat. Commun. 10, 201 (2019).
Cluntun, A. A., Lukey, M. J., Cerione, R. A. & Locasale, J. W. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer 3, 169–180 (2017).
Hoang, G. et al. Uncovering metabolic reservoir cycles in MYC-transformed lymphoma B cells using stable isotope resolved metabolomics. Anal Biochem 632, 114206 (2021).
Lam, C., Low, J.-Y., Tran, P. T. & Wang, H. The hexosamine biosynthetic pathway and cancer: Current knowledge and future therapeutic strategies. Cancer Lett. 503, 11–18 (2021).
Elia, I. & Haigis, M. C. Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat. Metab. 3, 21–32 (2021).
Hensley, C. T., Wasti, A. T. & DeBerardinis, R. J. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest 123, 3678–3684 (2013).
Erez, A. & Kolodkin-Gal, I. From prokaryotes to cancer: glutamine flux in multicellular units. Trends Endocrinol. Metab. 28, 637–644 (2017).
Itano, N. & Iwamoto, S. Dysregulation of hexosamine biosynthetic pathway wiring metabolic signaling circuits in cancer. Biochim. Biophys. Acta, Gen. Subj. 1867, 130250 (2023).
Luo, M. et al. A mass spectrum-oriented computational method for ion mobility-resolved untargeted metabolomics. Nat. Commun. 14, 1813 (2023).
Sugiura, Y., Honda, K., Kajimura, M. & Suematsu, M. Visualization and quantification of cerebral metabolic fluxes of glucose in awake mice. Proteomics 14, 829–838 (2014).
Zhang, H., Delafield, D. G. & Li, L. Mass spectrometry imaging: the rise of spatially resolved single-cell omics. Nat. Methods 20, 327–330 (2023).
He, J. et al. A sensitive and wide coverage ambient mass spectrometry imaging method for functional metabolites based molecular histology. Adv. Sci. (Weinh.) 5, 1800250 (2018).
McLean, C. & Kujawinski, E. B. Autotuner: high fidelity and robust parameter selection for metabolomics data processing. Anal. Chem. 92, 5724–5732 (2020).
Smith, C. A., Want, E. J., O’Maille, G., Abagyan, R. & Siuzdak, G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787 (2006).
He, J. et al. MassImager: A software for interactive and in-depth analysis of mass spectrometry imaging data. Anal. Chim. Acta 1015, 50–57 (2018).
Su, X., Lu, W. & Rabinowitz, J. D. Metabolite spectral accuracy on Orbitrap. Anal. Chem. 89, 5940–5948 (2017).
Hui, S. et al. Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118 (2017).
Sud, M. et al. MetabolomicsWorkbench: an international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 44, D463–D470 (2016).
Acknowledgements
We would like to thank Prof. Jun Tu (Jiangxi University of Chinese Medicine) for valuable discussions and insightful advice. This research was supported by the National Natural Science Foundation of China (Grant Nos. 21927808, Z. A.; 82293684, R. Z., and 82374158, Q. Z.), the Chinese Academy of Medical Science (CAMS) Innovation Fund for Medical Sciences (CIFMS, Grant No. 2021-I2M-1-028, R. Z.).
Author information
Authors and Affiliations
Contributions
Z.A., R.Z., Q.Z. and X.L. (Xinzhu Li) conceptualized the study. Q.Z., and X.L. (Xinzhu Li) designed the experiments. X.L. (Xinzhu Li) and Y.Z. conceived and developed MSITracer. Q.Z., X.L. (Xinzhu Li), and T.L. performed the stable isotope tracing experiments. X. L. (Xinzhu Li) and Q. Z. carried out all the remaining experiments. H.Z., T.T., T.L. and F.L. assisted with the biological validation experiments. X.T. assisted in mice experiments and code development. S.Z. helped with the LC‒MS data acquisition and manually checking the labeling of metabolites. L.W., F.L., and X.L. (Xin Li) helped with the MSI experiments. C.S. performed the H&E staining and provided fruitful suggestions. X.L. (Xinzhu Li) analyzed the data and completed the figures. X.L. (Xinzhu Li) and Q.Z. wrote the manuscript. R.Z. and Z.A. revised the paper. Z.A., R.Z. and Q.Z. supervised and guided the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zheng-Jiang Zhu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, X., Zhu, Y., Li, T. et al. Spatial isotope deep tracing deciphers inter-tissue metabolic crosstalk. Nat Commun 16, 7934 (2025). https://doi.org/10.1038/s41467-025-63243-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63243-2
