
Abstract
In multiple sclerosis (MS), chronic compartmentalized inflammation is thought to drive relentless clinical deterioration. Here, we investigate the link between unresolved parenchymal inflammation and cellular senescence in MS progression. Single-cell transcriptomic analysis of human brain tissue reveals an accumulation of senescent-like glial cells in diseased white matter, especially in chronic active lesions, and to a lesser extent in the cortex. Spatial transcriptomics show gradients of senescence-like signatures extending from lesion cores to periplaque regions, alongside rewired cellular networks. Experimental induction of senescence in MS hiPSC-derived neural organoids demonstrates that microglia are especially vulnerable to inflammation-induced senescence, which can be partially rescued by CNS-penetrant anti-inflammatory drugs. At the patient level (n = 466), increased 3T MRI-estimated brain-age is observed, especially in individuals with more than four chronic active lesions. These findings suggest that chronic inflammation might accelerate senescence-like processes, potentially contributing to disease progression, and that its modulation might help limit further propagation.
Similar content being viewed by others


Introduction
Multiple sclerosis (MS) is a lifelong and disabling disease of the central nervous system (CNS) in which waves of peripheral immune attacks on the CNS are accompanied by compartmentalized inflammatory and neurodegenerative processes, leading to cumulative disability1. While existing disease-modifying treatments effectively control peripheral immune responses and mitigate relapse-associated inflammatory activity, they demonstrate limited efficacy in halting disease progression that occurs independently of relapses (referred to as PIRA or silent progression)2. Growing evidence supports the role of unresolved smoldering CNS inflammation, as occurs at the leading edge of chronic active MS lesions (observable in vivo as paramagnetic rim lesions or PRL on MRI)3,4, in relentless clinical progression5,6,7. Through single-nucleus RNA sequencing (snRNA-seq) blueprinting, we recently highlighted the coexistence at the chronic active lesion edge of self-sustaining glia-mediated pathological processes as well as, to a lesser extent, resident lymphocyte-driven inflammation8.
In this work, we asked whether cellular senescence9, a cellular state featuring permanent cell-cycle arrest and inflammatory secretome (senescence-associated secretory phenotype or SASP), is among the relevant self-propagating pathological processes disrupting tissue homeostasis and its reparative capacity in chronic active lesions, as well as in other MS brain tissue. Using single-cell transcriptomic data from human autopsy brains (including snRNA-seq and spatial transcriptomics), we identified subsets of senescent-like cells with cell-type and regional specificity in the brains of relatively young individuals with progressive MS. Strategic brain tissue sampling enabled the investigation of white matter (WM), cortex, and different lesion pathological stages, separately. To dissect some of the drivers of this cellular program in MS, we developed an inflamed human-induced pluripotent stem cell (hiPSC)-derived organoid model to experimentally study and modulate inflammation-induced cellular senescence-like processes in vitro.
Finally, in the absence of definite in vivo biomarkers indicative of cellular senescence in the living brains of MS patients, we applied the MRI-estimated brain age paradigm. This macroscopic approach provides a global estimation of the brain’s structural integrity, extending beyond brain volume. Since we identified senescent-like cells in chronic active lesions, we specifically investigated whether, at the patient level, MS patients with a high burden of MRI-visible chronic active lesions (PRL) also exhibit an increased MRI-estimated brain age and the corresponding likelihood of disease progression.
Results
Senescent-like cells show regional and cell-type vulnerability in the MS brain
We investigated the regional and cell-type specificity of senescent-like cells in the MS brain. Following comprehensive histological characterization of autopsy tissue blocks, we performed snRNAseq on 43 sampling areas from 32 snap-frozen brain autopsy tissue, using a cold 3-mm punch-biopsy tool that enabled strategic sampling of WM and cortex separately, as well as different pathological MS lesion stages (Fig. 1a–d, samples are listed in Supplementary Data 1). Of these 43 samples, 20 WM samples were included in our prior single-cell transcriptomic study8.

a Study design for punch-based sampling of brain tissues (cortex and WM at different locations and pathological stages, i.e., myelinated cortex, demyelinated cortex, periplaque WM, lesion edge, and lesion core) from relatively young progressive MS cases (n = 30) and non-neurological controls (n = 13). Created in BioRender. Absinta, M. (2025) https://BioRender.com/d0hxq2g. b Representative image of punch-based sampling of CAL edge, periplaque NAWM, and myelinated cortex (see Supplementary Data 1 for a detailed description of each sample and identification number). c Number of tissue samples for each location (control cortex, MS myelinated and demyelinated cortex, control WM) and pathological stages. d Representative multiplex immunostaining showing the pathological staging of MS tissue based on myelination status, presence of neurons, and characterization of the myeloid infiltrate (TMEM119+ for surveilling microglia and MHCII+ for activated antigen-presenting microglia). Dotted lines correspond to the lesion edge in CA and CI lesions. Scale bar 50 μm. e snRNA-seq clustering of 197,912 nuclei by cell type, labelled based on known lineage markers, and visualized as UMAP plot. Each dot corresponds to a single nucleus and each colour to a cell-type cluster. f Dot plot depicting selected differentially expressed genes for each cluster and associated cluster labelling. Dot size corresponds to the percentage of nuclei expressing the gene in each cluster, and the colour represents the average expression level. g Averaged percentage of cell populations in the different microenvironments. snRNA-seq single-nucleus RNA sequencing, WM white matter, NAWM normal appearing white matter, MS multiple sclerosis, CA chronic active, CAL chronic active lesion, UMAP uniform manifold approximation and projection, CI chronic inactive, VAS vascular cells, OPC oligoprecursor cells, AST astrocytes, LYM lymphocytes, OLIGO oligodendrocytes, NEU neurons.
To minimize the confounding effects of normal brain aging on the identification of senescent-like cells, we selected relatively young progressive MS cases and non-neurological controls for the analysis. From 15 progressive MS cases (women 7, mean age 51.9, range 35–69 years), we processed 13 samples from the cortex (6 myelinated vs. 7 demyelinated) and 17 from the WM (at different pathological lesion stages, including chronic active lesions, chronic inactive lesions, and periplaque normal appearing WM [NAWM]). An additional 6 samples of cortex and 7 samples of WM were obtained from 8 age-matched non-neurological controls (women 4, mean age 54.9, range 45–60 years). Comprehensive demographic and clinical data of the autopsy cases, detailed information about each sample, and quality control metrics for each snRNA-seq dataset are provided in Supplementary Data 1.
In total, 197,912 nuclei (average of 4602 nuclei per sample) were processed for unsupervised clustering, which identified 14 cellular clusters, rendered as uniform manifold approximation and projection (UMAP) plots (Fig. 1e). The bioinformatic analysis is fully detailed in the “Methods” section. The cluster annotation was performed based on the expression of well-established lineage marker genes (Fig. 1f). The top 100 differentially expressed genes by cell type are shown in Supplementary Data 2. Cell-type proportions by location and pathological stage are shown in Fig. 1g. As expected, and validating our sampling strategy, neurons accounted for approximately 50% of nuclei in the cortex but less than 10% in the WM. Oligodendrocytes constituted 70% of nuclei in NAWM, 1.8% in demyelinated WM lesions, 8% in demyelinated MS cortex, and 30% in myelinated cortex, on average. The highest percentage of microglia and lymphocytes was observed at the chronic active lesion edge (19% and 1.4%, respectively).
Following the recent international consensus guidelines for the multi-omics identification of senescent cells10 (Fig. 2a), we first applied the SenMayo gene set11 to our snRNAseq dataset (Fig. 2b). The SenMayo gene set11 is a panel of 125 genes previously validated in both human and mouse across aged tissue types, including the CNS. As expected, in our control brain tissue, the SenMayo gene signature scores correlated with chronological age (higher scores in older non-neurologically affected people, Pearson correlation coefficient r = 0.26, p = 0.009, Supplementary Fig. 1a). Figure 2b shows the SenMayo gene signature scores for each nucleus, projected on UMAPs based on disease status (MS vs. non-neurological controls) and sample location (WM vs cortex). Following the binarization of nuclei into SenMayo-defined senescent vs. non-senescent cells, we observed an increase of senescent-like cells in the WM of MS patients (average 13% in MS WM vs. 8% in control WM and 7% in MS cortex vs. 10% in control cortex; two-way ANOVA p = 0.05, Fig. 2c).

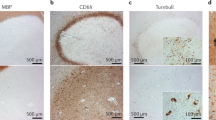
a Schematic representation of the consensus-based strategy used for senescent cell identification. Cell cycle arrest was evaluated by scoring cell cycle-related gene signatures using the senescence index tool (SIT), followed by protein validation of p16INK4a levels in MS tissues. Cell cycle arrest and SASP were assessed by scoring SenMayo gene signature. DNA damage response and nuclear reorganization were validated by identifying TBP53 and nuclear lamin B1 protein levels in MS tissues. Created in BioRender. Absinta, M. (2025) https://BioRender.com/d0hxq2g. b UMAP showing mapping SenMayo gene signature scores in the cortex and white matter from MS cases vs. controls. c Quantification (%, mean ± SEM) of SenMayo-informed senescent cells by location (white matter and cortex) in MS tissues vs. non-neurological controls (n = 43 biological samples, two-way ANOVA, p = 0.05, post-hoc multiple comparison analysis * p = 0.028). d Random Forest on the percentage of SenMayo-defined senescent cells highlighting the CNS cell type, pathological stage, and subject age as the most relevant factors. e Heatmap showing the percentage of SenMayo-defined senescent cells by CNS cell type and pathological stage (linear mixed model with fixed effects [sex, age, and pathological lesion stage] by each CNS cell type, * p < 0.05, **p < 0.01). f Correlation between SenMayo- and SIT-defined senescent-like cells (Pearson correlation coefficient r = 0.47, p < 0.001, equation y = 0.6278*x + 7.077). g A representative example of the CA lesion edge. Within the CA lesion edge, most cells (both microglia and endothelia) are positive for the senescence marker p16INK4a (arrows). Separate channels are shown to facilitate the visualization of different markers. Dotted lines indicate the lesion edge. h A representative example of the lesion core. Within the lesion core, the arrows indicate astrocytes positive for the senescence marker p16INK4a. i A representative example of p16INK4a+ ependymal cells. j Violin plot showing the quantification of p16INK4a+ cells (%) on human brain tissue by pathological condition from eighteen 5-μm paraffin-embedded tissue sections (ANOVA p < 0.0001; Tukey’s multiple comparisons * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001). The violin plot shows the median (black line), quartiles (dotted lines), as well as the minima and maxima. snRNA-seq single-nucleus RNA sequencing, UMAP uniform manifold approximation and projection, MS multiple sclerosis, CA chronic active, CI chronic inactive, NAWM normal appearing white matter, OPC oligoprecursor cells, SASP senescence-associated secretory phenotype.
To determine which clinical and pathological variables most effectively explain the number of senescent-like cells in the snRNAseq dataset, we applied a random forest model, which identified CNS cell type, lesion pathological stage, and subject age, as significant prioritized variables (Fig. 2d). Supporting this finding, a two-way ANOVA test (main effect p < 0.05) found a significant interaction between CNS cell type (row effect p < 0.0001) and pathological lesion stage (column effect p = 0.018). Overall, these results indicate a regional and CNS cell-type-specific distribution of SenMayo-defined senescent-like cells in the MS brains compared to controls.
Within the MS brain tissue, microglia, endothelia, and astrocytes were the most prominent cells featuring senescence-related signatures (Fig. 2b, e). For each CNS cell type, a linear model with fixed effects (sex, age, and pathological lesion stage) was applied to assess the distribution of SenMayo-defined senescent-like cells in tissue. Among all locations and pathological stages, senescent-like microglia cells were significantly enriched in chronic active lesions (p = 0.008) and chronic inactive lesions (linear model R2 = 0.49, p = 0.042). Similarly, senescent-like astrocytes were significantly enriched in chronic active lesions (linear model R2 = 0.26, p = 0.008), as were senescent-like vascular cells in chronic inactive lesions (linear model R2 = 0.30, p = 0.02). We obtained similar results by re-analyzing two other independent snRNAseq MS datasets12,13 (Supplementary Fig. 1b–d).
Of note, in MS, WM had higher percentages of senescent-like cells compared to cortex (Fig. 2c). Among the 13 cortical MS samples examined, only one demyelinated cortical MS sample (from a woman in her 40 s) showed a net increase in the percentage of endothelial, astrocytic, and microglial senescent-like cells (66%, 44%, and 34%, respectively). This suggests that although less frequent in our dataset, glial cells in the cortex can also be affected by cellular senescence-associated processes, particularly following inflammatory demyelinating events. Differential gene expression and pathway analysis of neuronal transcriptome profiles within the different microenvironment (myelinated vs. demyelinated MS cortex vs. control cortex) are discussed in the Supplementary Material and Supplementary Data 3.
Validation using senescence index tool and multiplex immunostaining
Since the SenMayo signature is enriched in inflammatory genes (part of the senescence-associated inflammatory secretome), as an additional analysis, we implemented a recently developed senescence index tool (SIT)14 that identifies senescent cells based on a smaller set of established senescence genes (CDKN2A, CDKN2B, IL23A, CDKN1A, CDKN1B, SERPINE1) combined with cell cycle arrest signatures (Fig. 2a). In our dataset, the SenMayo- and SIT-defined senescent-like cells were correlated at the gene expression level (Pearson correlation coefficient r = 0.47, p < 0.001; Fig. 2f).
Our transcriptomic data were validated by multiplex immunofluorescence detecting consensus-based senescence markers10 p16INK4a (indicative of cell cycle inhibition), p53 binding protein (TP53BP1, indicative of DNA damage response), and loss of nuclear lamin B1 (indicative of nuclear reorganization). Consistent with our transcriptomic results, MS brains displayed a higher percentage of p16INK4a+ cells compared to control brain tissues (ANOVA p < 0.0001, Fig. 2j), specifically within areas associated with microglia-mediated smoldering inflammation (chronic active lesion edge; Fig. 2g), prior demyelinated regions (lesion core; Fig. 2h) in both white and gray matter, and at the ependymal barrier lining the brain’s ventricular system (Fig. 2i). These areas also showed a trend toward decreased of nuclear lamin B1 in MHCII+ microglia cells (Supplementary Fig. 2).
Immune cell subclustering identified MIMS-foamy and dendritic cells as more prone to cellular senescence-like processes in MS brain tissue
To determine which subset of immune cells was more prone to cellular senescence, we performed an unsupervised subclustering of the immune cells, encompassing 12,599 nuclei (Fig. 3a). The subclustering identified 11 cellular clusters, visualized as a UMAP plot (Fig. 3b). Cluster annotation was performed based on the gene expression module scores of immune cell clusters as described and validated in Absinta et al.8 (Fig. 3b).

a Immune cell subset based on snRNAseq. b Immune cell subclustering (UMAP plots) and annotation based on Absinta et al.8 Heatmap displaying gene module scores between subclusters and Absinta et al.8. c Percentage of the different immune cell subclustering by location and pathological stage. d Heatmap showing the expression of genes included in the SenMayo signature in microglia by tissue location and pathological stage. e Quantification (%) of SenMayo-defined senescent cells in each immune subclustered population demonstrating the presence of subsets of senescent-like cells in each subcluster. f Representative multiplex immunostaining showing the co-existence of p16INK4a positive and negative IBA1+, MHCII+, and CD68+ microglia at the chronic active lesion edge. g Correlation between the SenMayo signature score and the GO negative regulation of autophagy signature score by cell type (n = 43 samples, Pearson correlation coefficient r = 0.48, p < 0.0001, equation y = 0.3555*x + 0.0747). Each dot in the scatter plot represents the average of the scores per sample, cell type, and signature. h LFB-PAS staining showing lipofuscin-laden microglia cells at the chronic active lesion edge (arrows, magnified views). DC dendritic cells, MG microglia, MIMS microglia inflamed in MS, CAL chronic active lesion, PV M0 perivascular macrophages, WM white matter, CX cortex, LFB-PAS Luxol fast blue-periodic acid Schiff.
As expected, a high percentage of surveilling microglia (both homeostatic and stressed) was observed in control brain tissue (approximately 83% in both the control WM and cortex), whereas this percentage was lower in MS brain tissue (ranging between 40% and 60% of nuclei in the MS WM, and 60%–70% in the MS cortex, Fig. 3c). The highest percentage of microglia inflamed in MS (MIMS) was observed at the edge of chronic active lesions (18% of nuclei). Dendritic cells accounted for approximately 5% of nuclei in both the control WM and cortex, but increased to about 30% in and around WM lesions, 10% in the MS myelinated cortex, and 20% in the demyelinated MS cortex. The highest percentage of perivascular macrophages was found in the lesion core (11% of nuclei). Lymphocytes (both T cells and plasmablasts) comprised approximately 6% to 8% of nuclei in and around WM lesions (compared to 2% in the control WM) but constituted only 3% in the MS cortex (compared to 0.9% in the control cortex)
Figure 3d shows the average expression of the SenMayo genes in the microglia cluster (including all the states) in the different microenvironments. Among genes of the SenMayo signature, microglia at the chronic active lesion edge were enriched in genes involved in cytokine signaling (IL7, IL32, IL15, IL18, IL6ST, CCL5, CCL20, CXCR2, CXCL12), TNF signaling (TNFRSF1A, TNFRSF1B, TNFRSF10C, TNFRSF11B), extracellular matrix remodeling (MMP2, MMP9, MMP14), and osteopontin signaling (SPP1); whereas microglia in the cortex enriched in genes involved in the IGF1 pathway (IGF1, IGFBP2, IGFBP4, IGFBP5, IGFBP6, IGFBP7, Fig. 3d).
For each immune cell subcluster, we identified the SenMayo-defined senescent cells (Fig. 3e). The clusters with the highest number of senescent-like cells were MIMS-foamy (33% of nuclei), dendritic cells (30% of nuclei), and lymphocytes (27%), while the lowest was observed in perivascular macrophages (10% of nuclei). Approximately 18% of the homeostatic microglia exhibited the transcriptional signature of senescent cells, indicating that the senescent-like program is prevalent across different microglial states but is more prominent in some of them (Fig. 3e). Validating these transcriptomic results, in Fig. 3f, the co-existence of p16INK4a-senescent and non-senescent IBA1+ MCHII+ CD68+ microglia cells is shown at the chronic active lesion edge.
Since MIMS-foamy, an inflammatory microglial transcriptional state characterized by increased expression of phagocytosis and lipid processing genes8, showed a high percentage of senescent-like cells, we investigated the correlation between cellular senescence and autophagy signatures. In senescent-like microglia (regardless of the cellular state), we observed a correlation between SenMayo signature scores and reduced autophagy signature scores15 (Pearson correlation coefficient r = 0.48, p < 0.0001 [Fig. 3g]). This finding potentially indicates a reduced capacity of senescent-like microglia to process phagocytosed myelin and cellular debris, a task that is especially critical in areas of smoldering demyelination (chronic active lesion edge). In tissue, this was also consistent with the presence of lipofuscin-enriched microglia at the chronic active lesion edge (Fig. 3h)16,17.
Subclustering of the other cell types identified endothelial cells and astrocytic subsets as more prone to cellular senescence-like processes in MS brain tissue
Subclustering analysis of the vascular cells and astrocytes is presented in Fig. 4 and described in detail in the Supplementary Material. Through this analysis, we identified specific cellular subsets exhibiting transcriptional features of cellular senescence. Notably, among vascular cells in the MS brain, approximately one-third of venous endothelial cells were classified as senescent-like cells (Fig. 4a–e). This finding is particularly significant, as the postcapillary venule serves as a critical site of interaction between CNS-infiltrating lymphocytes and the endothelium. Subclustering analysis identified also subsets of astrocytes inflamed in MS (AIMS), reactive astrocytes, and the recently described ciliated astrocytes18 featuring a cellular senescence-associated transcriptional program. These astrocytic states were especially abundant within WM lesions (ranging 11–15% of astrocytes) compared to the MS cortex (less than 0.5% of astrocytes) (Fig. 4f–j).

a Vascular cell subset based on snRNAseq initial clustering. b UMAP of vascular subclusters and annotation based on Yang et al.62 Heatmap displaying gene module scores between subclusters and Yang et al.62. c Percentage of the vascular cell populations by location and pathological stage. d Quantification (%) of SenMayo-defined senescent-like cells in each vascular subclustered population demonstrating the presence of subsets of senescent cells in each subcluster. e Representative multiplex immunostaining showing the co-existence of p16INK4a+ and p16INK4a− CD31+ vascular cells at the chronic active edge. f UMAP of astrocyte subtypes and annotation by cortex and white matter based on Absinta et al.8 and Lerma-Martin et al.18. g Dot plot of averaged z-transformed gene expression of marker genes for each astrocyte subtype. h Percentage of the astrocyte subclustered populations by location and pathological stage. i Quantification (%) of SenMayo-defined senescent cells in each astrocyte subclusters. j Representative multiplex immunostaining showing the co-existence of p16INK4a+ and p16INK4a− Vimentin+ inflammatory astrocytes at the chronic active lesion edge. Cl cluster, WM white matter, CX cortex, AIMS astrocytes inflamed in MS.
Spatial transcriptomic mapping of MS brain tissue
To resolve transcriptomic cellular senescence signatures within the spatial microenvironment of MS brain tissue, we processed and characterized twelve 6.5 mm² tissue blocks using 10× Visium technology (Fig. 5a). The MS brain tissue comprised various lesion pathological stages: active lesion, chronic active lesion, chronic inactive lesion, remyelinated lesion, cortex, and apparently non-lesional (non-demyelinated) tissue (Supplementary Fig. 3). Demographic and clinical features are detailed in Supplementary Data 4. A total of 45,355 55-µm spots (with an average of 3778 spots per section) were analyzed, containing an average of 600 UMI counts per spot. An average of 12,797 unique genes per Visium slide were detected (Supplementary Data 4).

a Overview of the study design. Multimodal data were imputed in each spot, including gene expression, definition of microenvironments/niches (unsupervised and/or manual clustering), snRNA-seq informed spot’s cell-type deconvolution, and ligand-receptor pair-based cell communication. Created in BioRender. Absinta, M. (2025) https://BioRender.com/d0hxq2g. b Pie chart showing the percentage of spots by anatomo-pathological niches (total number of spots = 45,335; Supplementary Data 4 for a detailed description of each sample and identification number). c Violin plots showing the number of gene counts by niches for each location and pathological stage. d Heatmap showing the deconvoluted cell-type proportion by niches (asterisks identify conditions with more than 15% of total deconvoluted cells). e Representative example of the comprehensive spatial transcriptomic analysis of a CAL (woman with progressive MS in her 40 s). By histology, CAL are demyelinated lesions with an inflammatory edge of MHCII+ myeloid cells. A 6.5-mm2 of tissue (white square) was processed for spatial transcriptomics using the 10× Visium platform. Both manual and unsupervised clustering are shown. The unsupervised clustering identified transcriptional microenvironments (CAL edge, core, WM periplaque, and cortex; red spots) overlaid onto the LFB-stained tissue. These microenvironments are in line with the pathological tissue staging. f Enhanced spatial gene expression of SPP1, CHIT1, FTL, APOE, and C1QB that are the among top differentially expressed genes at the CAL edge. g Spatially-resolved microglia subclustering (red spots) after deconvolution of the snRNAseq dataset from Absinta et al.8 are overlaid onto the LFB-stained tissue. The previously described MIMS-iron and MIMS-foamy are microglia clusters located at the CAL edge. Stressed microglia are sparsely seen in the CAL core, edge, and periplaque. Homeostatic microglia are rarely located in the periplaque WM only. WM white matter, CAL chronic active lesion, CI chronic inactive lesion, LFB-PAS luxol fast blue-periodic acid-Schiff.
For each spatial spot, multimodal data were imputed at (xi, yj), including gene expression counts, microenvironments (spatial niches, identified through both manual annotation and BayesSpace19 unsupervised clustering), cell-type deconvolution using our processed snRNAseq data (SPOTlight)20, and spot-level cell-cell communication analysis based on ligand-receptor (LR) pairs (Fig. 5a). The bioinformatic analysis is fully detailed in the “Methods” section and dataset integration shown in Supplementary Fig. 4.
Across the entire spatial dataset, 52% of all spots were classified as NAWM, 23% as demyelinated lesion core, 11% as cortex, 1% as active lesion, 2% as CAL edge, 7% as CI lesion edge, and 3% as remyelinated lesion (Fig. 5b). Only 2% of all spots were excluded due to tissue folding. The distribution of gene counts per microenvironment shown in Fig. 5c is in line with prior publications using the same technology18,21.
The cell-type deconvolution results aligned with the anticipated composition based on the anatomical-pathological region (Fig. 5d, individual slides shown in the Supplementary Fig. 3). A high proportion of microglia (18% of all spots) and lymphocytes (8%) were observed at the chronic active lesion edge and within the active lesion. Oligodendrocytes accounted for approximately 40% of the NAWM and 30% of remyelinated lesion. The demyelinated lesion cores were primarily composed of astrocytes (20% of the spots) and vascular cells (27% of the spots), whereas oligodendrocytes represented less than 10%. OPCs constituted between 3% and 5% of the spots, regardless of the microenvironment. As expected, neurons accounted for 37% of the cortical spots (Fig. 5d).
Notably, the microenvironment of the chronic active lesion edge (Fig. 5e) exhibited a distinct transcriptional profile compared to other pathological stages, marked by elevated expression of microglial genes (SPP1, CHIT1, FTL, APOE, C1QB) [Fig. 5f]. Validating our prior work8, immune cell deconvolution revealed the presence of microglia inflamed in MS (MIMS)-foamy at the sharp edge of the chronic active lesion (consistent with their role in myelin phagocytosis), while MIMS-iron microglia were observed to extend into the periplaque (consistent with their involvement in antigen presentation), indicating their distinct roles within chronic active lesions (Fig. 5g). Stressed microglia were prevalent in both the normal appearing tissue and lesion core, whereas homeostatic microglia were notably scarce in this dataset and sparsely located in the normal appearing tissue.
Spatial transcriptomic analysis reveals centrifugal propagation of the cellular senescence signatures
Following the comprehensive analysis of the spatial transcriptomic dataset, we spatially mapped cellular senescence-related genes and signatures. In Fig. 6a, representative Visium slides highlighted in different pathological stages the distribution of inflammatory-related genes, including SPP1 and C3, as well as senescence-related genes, including SERPINA3, HMGB1, IGFBP5, and CDKN1A.

a Representative expression of inflammatory (SPP1, C3) and senescence-associated genes (SERPINA3, HMGB1, IGFBP5, and CDKN1A) in different MS pathological tissue (i.e., active lesion, CAL, chronic inactive lesion, remyelinated lesion, and non-lesional MS tissue). b Gradients of senescence-associated astrocyte signature (55 genes)22 was observed extending from the lesional areas toward the periplaque in Visium slides with CAL and chronic inactive lesions. c, d Spatial differential expression analysis of genes along a linear trajectory identified non-random gene gradients (SPARKX p < 0.01). Error bands of line plots indicate the confidence interval (level 0.95). Cellular senescence-related genes and SenMayo genelist exhibited non-random gradients not only within lesional areas but also in the surrounding periplaque in chronic active lesion slide V01, whereas such pattern was not observed in the active lesion slide V05. e SenMayo genelists spatial gradient for Visium slide, including a remyelinated lesion. Error bands of line plots indicate the confidence interval (level 0.95). Remyelination was assessed by both lipid staining (LFB) and myelin PLP immunostaining. The asterisk indicates the postcapillary central venule. CAL chronic active lesion, MBP myelin basic protein, LFB luxol fast blue, WM white matter, PLP myelin proteolipid protein.
To test the hypothesis that cellular senescence extends beyond focal demyelination, we performed two complementary bioinformatic approaches. In the first one, the brain tissue was manually segmented in concentric gradients and cellular senescence signatures applied across all Visium slides (Fig. 6b). Using this approach, we identified gradients of senescence signatures22 highest in the lesion core and gradually decreasing toward the WM periplaque in both chronic active and chronic inactive lesions (Fig. 6b). Interestingly, this senescence-like gradient was less evident in remyelinated and missing in the slides with normal appearing (non-lesional) tissue. Senescence signatures were seen within the active lesion, but not in the periplaque region. Overall, also in the independent spatial transcriptomics dataset, the MS cortex did not exhibit an increase in the cellular senescence signatures.
In the second approach, we leveraged a recently published method23 that enables spatial differential expression analysis of genes along a linear trajectory in tissue (SPATA2). For each Visium slide, we identified significant genes exhibiting non-random tissue gradients (p < 0.01, FDR < 0.05). Figure 6c presents a representative example of a slide with a chronic active lesion. Consistent with the findings from the previous analysis, we observed that many cellular senescence-related genes, including NUPR1, HMGB1, and IGFBP5, followed a gradual descending pattern (p < 0.01). The same pattern was observed when the entire SenMayo gene signature was applied. For comparison and reference, Fig. 6c also highlights microenvironment-specific genes such as SV2A (a synaptic gene showing an ascending late pattern with highest gene expression in the cortex), CHIT1 (a microglial gene exhibiting a peak pattern with highest gene expression at the chronic active lesion edge), MBP (ascending gradually pattern, corresponding to myelin gene distribution, with low expression in the demyelinated lesion core and higher expression in the periplaque white matter), and GFAP (descending linear pattern, reflecting the distribution of astrocyte genes, which are more highly expressed within the lesion core than in the rest of the tissue slide). Additional representative examples of the spatial distribution of the SenMayo gene signature in tissue are shown in Fig. 6d, e. In the active lesion (Fig. 6d), the SenMayo signature was increased within the lesion core but not yet in the periplaque white matter. In contrast, in the remyelinated lesion, a more gradual descending pattern is observed (Fig. 6e).
Overall, these findings support the concept that MS tissue pathology extends beyond focal demyelinated areas. They also suggest that cellular senescence-like processes might occur early in lesion development, in line with recent findings in a marmoset experimental autoimmune encephalomyelitis (EAE) model24, but might contribute to the centrifugal propagation of tissue dysfunction only in later pathological stages.
Inflammation-induced senescent-like cells rewire cellular networks in MS tissue
To assess whether and how senescent-like cells can rewire the cellular network in MS tissue, we performed a differential cell-cell communication analysis employing both snRNAseq and spatial transcriptomic data (workflow shown in Fig. 7a). For this analysis, we focused on the cell types (i.e., microglia, endothelia, and astrocytes) we found in the MS tissue most vulnerable to the cellular senescence program. Overall, using CellChat25 on the snRNAseq dataset, senescent-like cells showed more signaling interactions with other cell types than non-senescent cells (Fig. 7b for microglia, Supplementary Fig. 5 for vascular cells and astrocytes, respectively), confirming their influence in shaping the surrounding microenvironment.

a Workflow schematic of the cell-cell communication analysis using both snRNAseq and spatial transcriptomic datasets (CellChat, NICHES tools). Created in BioRender. Absinta, M. (2025) https://BioRender.com/d0hxq2g. b Circle plot representing the comparison between the cellular communication (ligand-receptors [LR] pairs) between senescent-like microglia and other cell populations vs. non-senescent microglia and other cell populations using CellChat on snRNAseq data. Red lines indicate increased number of interaction ligand-receptors pairs, while blue line reduced number of interactions. c Chord diagram showing detailed communication through upregulated pathways and providing insights into the autocrine- vs. paracrine-acting pathways for senescent microglia using CellChat. Significant upregulation was defined by an interaction with p < 0.05 and logFC for ligand expression >0.2. Colours correspond to the different cell-types. d Spatially mapping of prioritized ligand-receptor pairs involving senescent-like microglia (SPP1-CD44, TREM2-APOE, HLADB1-CD4, NAMPT-ITGAV5, PSAP-GPR37) within the Visium spot and their relative quantification (mean ± SD) by location and pathological stage (n = 7 locations/pathological stages from 12 Visium slides, ANOVA p < 0.0001 for all pairs, except PSAP-GPR37, post-hoc multiple comparison analysis *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). Representative icons with ligand-receptors pairs created in BioRender. Absinta, M. (2025) https://BioRender.com/d0hxq2g.
When comparing their cell-cell communication, senescent-like microglia exhibited significantly more interactions with astrocytes, vascular cells, lymphocytes, and oligodendrocytes (Fig. 7b). Among the top upregulated pathways in the senescent-like vs. non-senescent microglia were APOE, C3, SPP1, NAMPT, IL16, LAIR1, CD39, MHCII, ADGRE, and GRN (Fig. 7c). After prioritizing LRs pairs, we spatially mapped and quantified cell-cell communication at the spot level using NICHES26, leveraging cell-type deconvolution and the intrinsic spatial interactions among cells within the same 55-µm Visium spot. Figure 7d presents representative examples of prioritized LR pairs on Visium slides, including lesions at different pathological stages. Across the entire spatial dataset, significantly higher LRs pairs average scores per spot (e.g., SPP1-CD44) were found, especially in the active and chronic active lesions.
Similarly, senescent-like astrocytes showed significantly more interactions with the other cell types, principally vascular cells, OPCs, and microglia. Among the top upregulated intercellular genes and pathways in the senescent astrocytes were ANGPTL4, VEFGA, complement C3, as well as FGF signaling (Supplementary Fig. 5a, b). Supplementary Fig. 5c demonstrates spot-level C3-ITGB2 (astrocyte-immune cells) signaling quantified in the spatial transcriptomic dataset, which was found to be significantly upregulated in those spatial spots belonging to the active and chronic active lesions.
Concerning the vascular cells, senescent-like endothelia also showed significantly more interactions with the other cell types, mostly astrocytes but also lymphoid and myeloid cells (Supplementary Fig. 5d), potentially suggesting some interference with the regulation of the blood-brain barrier. Among the top upregulated intercellular pathways in senescent-like endothelia were macrophage migration inhibitory factor (MIF), bone morphogenetic protein 6 (BMP6), tight-junction and/or adhesion-related molecules (CDH5, ESAM, ICAM, NECTIN; Supplementary Fig. 5e). Supplementary Fig. 5f exhibits spot-level MIF-CXCR4 (endothelia-immune cells) and BMP6-BMPR1B (endothelia-astrocytes) signaling quantified in the spatial transcriptomic dataset. MIF-CXCR4 signaling is relevant for endothelia-immune cell recruitment and interaction at the blood-brain barrier, while BMP6 signaling has been implying in astrogliosis and blockade of OPC differentiation27.
MS-inflamed CSF and selected inflammatory cytokines increase the number of senescent-like cells in hiPSC-derived glia-enriched brain organoids
To investigate potential drivers of inflammation-induced cellular senescence observed in MS tissue, we implemented a hiPSC-derived organoid model that we recently developed28. Within an 8-week timeframe, glia-enriched organoids consist of an intricate network of CNS-like cell types (Fig. 8a), including astrocytes, oligodendrocyte lineage cells (both OPC and myelinating oligodendrocytes, as observed through electron microscopy, Fig. 8b), neurons (both excitatory and inhibitory), and microglia; see Fagiani et al.28 for detailed description of the organoid model. Oligodendrogenesis was specifically fostered by overexpressing the SOX10 transcription factor in a subset of neural precursor cells (NPCs) within the first 2 weeks of differentiation. After 8 weeks of organoid differentiation, single-cell transcriptional profiles showed higher resemblance to adult rather than fetal human brain28.

a Immunostaining of NeuN+ and MAP2+ neurons, MBP+ oligodendrocytes, SOX10-eGFP+ OPCs, GFAP+ astrocytes, and MHCII+ microglia out of DAPI in whole cryosections from 8 weeks-old submillimetric glia-enriched organoids (30×). Scale bar: 100 μm. b Trasmission electron microscopy showing myelinated axons (red arrows) within the glia-enriched organoid. Scale bar: 500 nm. c Representative images and quantification of SA-β-galactosidase activity along the organoid differentiation (n = 3 MS hiPSC-lines; at least 25 glia-enriched organoids per condition; one-way ANOVA p < 0.0001, Tukey’s post-hoc multiple comparison analysis * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001). The violin plots show the median (red line), quartiles (dotted lines), as well as the minima and maxima; each dot corresponds to one glia-enriched organoid. d Representative images of untreated organoids (left) vs. organoids exposed to 10% CSF (right) for 24 h (top, > 100 glia-enriched organoids analyzed per experimental condition) and 6 days (bottom, > 40 analyzed organoids per experimental condition) and relative quantification of SA-β-gal staining area (n = 3 MS hiPSC-lines, t-test *** p = 0.001; **** p < 0.0001). The violin plots show the median (black line), quartiles (dotted lines), as well as the minima and maxima; each dot corresponds to one glia-enriched organoid. e Single-cell RNA-seq clustering of 118,706 cells, visualized as UMAP plot, from 8-week-old glia-enriched organoids (from 3 MS hiPSC-lines). Each dot corresponds to a single cell and each colour to a cell cluster. f Averaged percentage of each cell population by experimental condition. g Dot plot of averaged z-transformed gene expression of marker genes for each cluster. h Box plot showing the percentage of SIT-defined senescent cells (mean ± SEM; n = 3 MS hiPSC-lines; ANOVA p = 0.028, Kruskal–Wallis post-hoc multiple comparison analysis *p = 0.011). Increased number of senescent-like cells is seen after stimulation with MS CSF or inflammatory cytokines. i Heatmap showing the averaged percentage of SIT-defined senescent cells in the different cell-types by experimental condition (two-way ANOVA p = 0.05, row effect p = 0.012, column effect p = 0.015, post-hoc multiple comparison analysis *p < 0.05). j Volcano plot reports gene expression changes in SIT-defined senescent vs. non-senescent microglia from glia-enriched organoids. W8 week 8, MS multiple sclerosis, SEM standard error of the mean, CSF cerebrospinal fluid, NEU neurons, ASTRO astrocytes, OPC oligodendrocyte precursor cells, OL oligodendrocytes, GLIA_IMM immature glia, PROG cycling progenitors, MG hiPSC-derived microglia, SA-β-gal senescence-associated β-galactosidase activity, MBP myelin basic protein, SIT senescence index tool.
To assess whether glia-enriched organoids are suitable for studying cellular senescence in vitro, senescence associated β-galactosidase (SA-β-gal, an enzyme that becomes highly active in senescent cells at pH 6.0, but not in proliferating or quiescent cells) staining was performed at different time points, revealing a progressive increase in SA-β-gal staining area, with a peak at week 16 (Fig. 8c). To assess whether a complex MS-relevant inflammatory stimulus can trigger cellular senescence, we exposed glia-enriched organoids to the 10% supernatant CSF of untreated MS patients selected for having chronic active lesions (PRL) by 7T MRI. As expected, the cytokine/chemokine profile of the MS CSF was enriched in inflammatory mediators (available in Fagiani et al.28). To assess both short and long-term effect of inflamed MS-CSF onto the organoid culture, SA-β-gal staining was performed on sequential cryosections at different levels of the organoid volume. Compared to control conditions, SA-β-gal staining was increased both 24 h (p < 0.0001) and 6 days (p < 0.001) after CSF exposure (Fig. 8d), suggesting that MS CSF contains inflammatory mediators that may induce cellular senescence in the organoids.
Next, to understand which cell types were prominently affected by CSF-triggered cellular senescence, we performed scRNA-seq of glia-enriched organoids, using 3 hiPSC lines from MS patients (2 women and 1 man, all in their 30 s at the time of skin biopsy) (Fig. 8e). To also determine which category of mediators might be responsible for inducing cellular senescence, we exposed glia-enriched organoids not only to the inflamed MS CSF but also to: (1) CSF from 3 non-neurological controls (obtained during spinal anesthesia due to knee surgery); (2) a cocktail of inflammatory cytokines prioritized in our prior snRNAseq autopsy study8 (IFNγ, TNFα, IL1β, C1q); and (3) human control immunoglobulins G (IgG), which are uniquely present in the CSF of MS patients as oligoclonal bands (a widely used diagnostic biomarker in MS). After standard preprocessing, scRNA-sequencing of organoids yielded a total of 118,706 single cells (Supplementary Data 5 and Fig. 8e) generated from 3 independent stem cell lines. The transcriptomic expression of a total of 23,391 distinct genes was studied. Unsupervised clustering identified 7 clusters, annotated using known lineage marker genes (Fig. 8f, g, top 100 differentially expressed gene by cell type in Supplementary Data 6) and revealing distinct populations of neurons, astrocytes, oligodendrocytes, OPC, and microglia (Fig. 8f, g). Overall, exposure to inflammatory cytokines and MS-inflamed CSF, but not to immunoglobulins, was associated with ~threefold increase of senescent-like cells by SIT scoring (ANOVA p = 0.026, Fig. 8h). Of note, CSF from non-neurological controls did not significantly increase the percentage of senescent-like cells.
Among the different organoid cell types, microglia were the most affected by inflammation-induced cellular senescence, followed by astrocytes (Fig. 8h, i), partially recapitulating in vitro our transcriptomic findings from autopsy brain MS cases. Overall, when compared to non-senescent organoid microglia (by SIT scoring), senescent-like organoid microglia enriched not only in cell cycle arrest genes (CDKN1A, NFKB1, LMNB1, CDKN1B, UBC, CDKN2A, CDKN2B), but also in inflammatory pathways, including cytokine signaling (CXCL10, CCL22, IL32, CCL5, IL12B, IL15, IL6), IFNγ signaling (IRF1, IRF4, GBP1, GBP4, PTPN1), antigen presentation (B2M, HLA-A, HLA-B, HLA-F, TAP1), NFκB signaling pathway (NFKB2, NFKB1, BIRC3, BCL2A1, NFKBIA, CD40) (volcano plot in Fig. 8h and pathway analysis in Supplementary Fig. 6a). We also performed a principal component analysis (PCA) of the pseudobulk scRNA from microglia exposed to d ifferent stimuli. As shown in Fig. 9a, in the PCA space, microglia treated with MS-CSF and inflammatory cytokines clustered separately from those in the untreated condition, as well as from those exposed to CSF from non-neurological controls and immunoglobulins. Notably, despite the changes in gene expression, the different stimuli did not significantly affect the phagocytosis capacity of microglia cells at 24 h (Supplementary Fig. 6b).

a Principal component analysis of microglia pseudobulk RNA from scRNAseq glia-enriched organoid data and pathway analysis performed on the top 50 genes of PC1 and PC2, respectively. Each dot corresponds to one microglia sample. Organoids’ microglia samples exposed to MS CSF and inflammatory cytokines are clustering separately from untreated microglia as well as microglia samples exposed to control CSF and human immunoglobulins G. b Graphical representation of the experimental design for drug treatment in organoids exposed to 10% CSF for 6 h and, then, co-treated with 1,5 μm dasatinib, 50 μm ibudilast, 100 μm α-lipoic acid, and 1 μm tolebrutinib for additional 18 h. Created in BioRender. Absinta, M. (2025) https://BioRender.com/kl03zc0. c, d Quantification of SA-β-gal and MBP+ staining area, respectively (n = 3 MS hiPSC-lines; at least 30 glia-enriched organoids analyzed per experimental condition, respectively; mixed-effect analysis with treatment as fixed effect p < 0.0001; Tukey’s post-hoc multiple comparison analysis * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001). The violin plots show the median (red line), quartiles (dotted lines), as well as the minima and maxima; each dot corresponds to one glia-enriched organoid. e Graphical abstract summarizing the results from the hiPSC-derived glia-enriched organoid experiments. The X represents conditions that are not inducing or that are reducing cellular senescence-like processes in the model. Created in BioRender. Absinta, M. (2025) https://BioRender.com/kl03zc0. PC principal component, MS multiple sclerosis, CSF cerebrospinal fluid, SA-β-gal senescence-associated β-galactosidase activity, MBP myelin basic protein.
In inflamed glia-enriched organoids, pharmacological modulation of inflammation reduces cellular senescence
To evaluate whether pharmacological modulation of CNS-compartmentalized inflammatory processes may counteract cellular senescence, glia-enriched organoids were exposed to the 10% supernatant CSF of untreated MS patients for 6 h and then co-treated with the following brain-penetrant drugs for additional 18 h: (1) 50 μM ibudilast, (2) 100 μM R-α-lipoic acid, and (3) 1 μM tolebrutinib (Fig. 9b). Lower concentrations of drugs (1:10) were also tested (Supplementary Fig. 6c, d). These drugs were shortlisted based on their potential effect on glia-mediated smoldering inflammation, based on our prior in silico microglia-drug interaction analysis from human snRNAseq data8 and/or prior preclinical and clinical studies supporting their scientific rationale. Additionally, dasatinib (1.5 μM) was included as a positive control due to its well-known senolytic properties. No significant differences in glia-enriched organoids’ cell viability were observed after inflammatory stimuli and treatment vs. control condition (Supplementary Fig. 6c).
After 24 h post-CSF treatment, we observed a significant reduction in SA-β-gal staining area in organoids treated with ibudilast (p < 0.0001), R-α-lipoic acid (p < 0.0001), and tolebrutinib (p < 0.0001), compared to CSF-exposed organoids (Fig. 9c). This effect was maintained after 6 days. A dose effect was seen for all drugs that showed no effect with 1:10 of the dose, except for tolebrutinib, that also maintained its effect at 1:10 of the dose (0.1 μM). These results indicate that upstream pharmacological inhibition of inflammatory processes might hinder the onset of cellular senescence. Of note, among the different drugs, tolebrutinib was also able to rescue CSF-induced oligodendrocyte toxicity (MBP+ staining area ANOVA p < 0.0001, Fig. 9d). A graphical abstract summarizing the main results obtained using the hiPSC-derived organoid model is shown in Fig. 9e.
Older brain biological age in chronic active multiple sclerosis by both transcriptome and in vivo MRI
In the absence of definite in vivo biomarkers indicative of cellular senescence in the living brains of MS patients, we applied a more global assessment using the brain age paradigm applied to both in vivo MRI data and tissue transcriptomic data.
We first estimated the biological age of human autopsy samples of MS lesions classified by their pathological stage. After retrieving publicly available snRNAseq datasets8,12,13,29 of both MS (n = 44 samples, chronological age range 34–58 years) and non-neurological control brain samples (n = 21, chronological age range 34–82 years), we first constructed a normative time-series reference using RAPToR, a bioinformatic tool designed to estimate the biological age of a tissue sample by its transcriptome profile (Fig. 10a). On average, MS tissue samples showed an older estimated biological age (mean age gap 21.3 years, SD 12.2, p < 0.0001). Relative to non-neurological control brain tissue, a clear gradient of biological-chronological age gap was seen starting from the normal appearing MS white matter (NAWM, 8.9 year-gap) to those regions affected by prior (lesion core, 34.8 year-gap) and ongoing inflammation (chronic active and active lesions, 27.3 and 20.3 year-gap, respectively) [ANOVA Kruskal–Wallis p < 0.0001; Fig. 10c].

a Graphical representation of the estimation of the brain biological age by transcriptome or MRI. After obtaining a normative time series for each modality, human brain MS samples or MRI scans were staged for biological vs. chronological age. See “Methods” for additional detail. Created in BioRender. Absinta, M. (2025) https://BioRender.com/d0hxq2g. b Representative 3T MRI case of a man in his ‘50 with MS. Several chronic active lesions (box and magnification) can be seen by MRI as T2-FLAIR-positive hyperintense lesions with a paramagnetic rim (PRL) on susceptibility-based MRI sequences. Scale bars: 10 mm. c Bar plot showing the brain age gap (mean ± SD, years) by transcriptome in brain samples from non-neurological controls (n = 21 samples) and MS (n = 44 samples at different pathological stage). Among all conditions, chronic active lesion edge and core showed higher brain biological age (ANOVA Kruskal–Wallis p < 0.0001, Dunn’s multiple comparison ** p < 0.01, **** p < 0.0001). d Bar plot showing the brain age gap (mean ± SD, years) by MRI analysis in non-neurological controls (n = 22), MS-mimics (n = 44), and MS patients (n = 466). Higher brain age gap is seen in MS patients, especially in those with more than 3 chronic active lesions by MRI (PRL, one-way ANOVA p < 0.0001, Tukey’s post-hoc multiple comparisons * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). e Scatter plot of chronological vs. brain age (years), by MRI, for each individual (n = 532). One dot equals one individual. MS patients were classified based on the PRL number and color-coded. Brain-age trajectories confirmed prior data as seen in (c). f Reverse Kaplan–Meier curves of the probability of the disability milestone EDSS of ≥4 for both chronological (top, comparison of survival curves using log-rank Mantel–Cox test p = 0.0028, median survival 63 years-old for PRL 0, 61 years-old for PRL 1–3, 58 years-old for PRL > 4, respectively) and biological MRI-brain age (bottom, comparison of survival curves using log-rank Mantel–Cox test p = 0.5 n.s., median survival 73.3 years-old for PRL 0, 71.5 years-old for PRL 1–3, 73.7 years-old for PRL > 4, respectively). snRNA-seq single-nucleus RNA sequencing, MS multiple sclerosis, NAWM normal appearing white matter, PRL paramagnetic rim lesions, EDSS Expanded Disability Status Scale.
To parallel and expand these findings and to assess whether chronic compartmentalized inflammation, as seen in chronic active lesions (paramagnetic rim lesions or PRL, in vivo imaging biomarker), can influence in vivo the estimated brain-age, we aggregated and analyzed a multicenter cross-sectional clinical-MRI dataset, in which the brain-age of 528 adults was predicted by applying a previously validated machine learning algorithm30 to 3T MRI brain images (Fig. 10a). Of these, 466 individuals were diagnosed with MS and 44 with MS mimics (neuromyelitis optica spectrum disorder, CNS vasculitis, HIV and HTLV infection with white matter brain lesions); 22 were healthy controls. Demographic, clinical, and MRI data are detailed in Supplementary Data 7. Overall, MS cases had higher mean brain-age gap (9.0 ± 9.4 years), defined as the subject chronological age subtracted from the predicted brain-age, compared to both age-matched healthy controls (−2.1 ± 6.2 years) and MS-mimics (2.5 ± 6.7 years; ANOVA p < 0.0001). When MS cases were stratified according to the number of chronic active lesions (brain PRL), higher brain-age gap was seen in those with ≥4 PRL (14.6 ± 10.1 years) than those with 1–3 PRL (9.1 ± 9.1 years) and no PRL (6.2 ± 8.1 years; ANOVA p < 0.0001; Fig. 1d,e). In the MS subset, a multivariable linear mixed model, including clinical center, PRL category, chronological age (cutoff = 55 years), and white matter lesion load as covariates, confirmed that brain-age gap (dependent variable) was higher in patients with ≥4 PRL (estimate 4.5 years; p = 4.8 × 10−5), high lesion load (estimate 0.5 year/mL; p = 2 × 10−16), and older chronological age (estimate −3.9 years; p = 0.0009). No significant center-specific effect influenced the results of the multivariable analysis.
To explore the role of brain atrophy in the estimation of MRI-based brain-age, brain volume was also included in a second multivariable linear mixed model, which accounted for clinical center, sex, and brain volume as fixed variables, along with PRL category and chronological age (cutoff = 55 years) as covariates. In this model, an increased brain-age gap was associated with having PRL ≥4 (estimate 9.25 years, p < 0.001), as well as with the interaction between PRL category and chronological age category (estimate −5.5 years, p = 0.02), indicating a higher brain-age gap in younger patients (less than 55 years old) with PRL ≥4. This confirms that MRI-estimated brain-age does not necessarily reflect brain volume only. The relevance of our findings relate to that fact that an increased brain-age gap was observed in young patients who, despite not yet exhibiting brain atrophy, showed signs of chronic CNS inflammation (evident as PRL).
In line with prior studies3,4,31, in this dataset, MS patients with ≥4 PRL had also significantly higher disability and severity scores, including Expanded Disability Status Scale (EDSS), MS severity score (MSSS), and age-related MSSS (ANOVA p < 0.05, respectively; Supplementary Data 7). On the basis of these data, we constructed survival curves to estimate the median chronological age and the median brain-age, respectively, to reach the disability milestone of EDSS 4 (ambulation limited to 1 kilometer). The median chronological age to EDSS 4 was 64 and 65 years in cases with PRL 0 or PRL 1–3, respectively, compared to 56 years in those with ≥4 PRL (Mantel–Cox test p = 0.028; Fig. 10f). When the median brain-age to reach EDSS 4 was evaluated, there were no significant differences among the survival curves (median brain-age 74, 74, and 71 years, respectively; Mantel–Cox test p = 0.5 n.s.).
Altogether, these results suggest that the MS brain shows both transcriptomic and MRI features normally associated with an aged brain. In vivo, substantial chronic parenchymal brain inflammation (≥4 PRL) is significantly and specifically associated with early aging (MRI-predicted brain-age) of the MS brain (vs. MS-mimicking conditions) and anticipates by a decade the risk of fixed disability and clinical progression.
Discussion
In our quest to understand—and potentially target—the mechanisms driving self-sustaining compartmentalized inflammation in MS, we investigated the presence and role of cellular senescence-like processes in MS brain tissue. The accumulation of senescent cells, which undergo stable cell cycle arrest and produce a distinct senescence-associated secretory phenotype (SASP), has been reported in age-related diseases, where they actively promote low-grade tissue inflammation and further disruption of tissue homeostasis9. Senescent growth arrest requires coordinated action of the p53/p21 and p16INK4A/retinoblastoma pathways. Key features of senescent cells include prolonged DNA damage signaling, increase in senescence-associated β-galactosidase activity, and nuclear loss of lamin B132 or HMGB133.
A recent international consensus statement highlighted the risks of relying on one single biomarker to identify cellular senescent cells. Therefore, we leveraged advanced single-cell genomics techniques (both snRNAseq and spatial transcriptomics) and histopathological validation for multiple hallmarks10,34 to investigate cellular senescence in MS brains with high-resolution granularity. By applying validated senescence signatures11,14,22 to our expanded snRNAseq human dataset, we identified subsets of senescent-like cells with cell-type and regional specificity in the brains of relatively young individuals with progressive MS (mean age 51 years). Among different CNS cell types, specific subpopulations of microglia, endothelia, and astrocytes showed transcriptional signatures of cellular senescence, particularly in WM areas affected by smoldering inflammation. Importantly, while these cellular subsets exhibit an overall inflammatory secretome (SASP), our data showed that they do not fully overlap with specific disease-associated inflammatory cellular states (i.e., only 30% of MIMS-foamy were senescent).
In terms of CNS regionality, we discovered that, in MS, senescent-like cells are prevalent in the WM than in cortical regions, potentially indicating a differential response of the brain tissue to the inflammatory injury. In line with this finding, major morphological and transcriptional differences were recently highlighted for glial cells in different microenvironments in MS35,36. For example, in the WM, higher and prolonged microglia inflammatory responses to injury were seen when compared to the affected (demyelinated) cortical environment36. In our study, along the lesion pathological stage trajectory, the presence of senescent-like cells was especially pronounced at the edges of chronic active lesions, indicating a potential association between inflammation-driven senescence and lesion pathology. In this regard, in chronic active lesions, the mechanisms underlying MS treatment-refractory chronic inflammation occurring behind a partially closed BBB are not fully elucidated and not properly targeted. We previously suggested the coexistence of self-sustaining, glia-mediated pathological processes and, to a lesser extent, resident lymphocyte-driven inflammation8. Our data support the idea that cellular senescence-like processes might be among those self-autonomous glial processes in MS tissue.
After identifying senescence cell-type specificity and their patterned spatial localization, a critical question remained about whether cellular senescence is a beneficial or detrimental response to injury in the context of MS tissue. To address this question, we opted for three different approaches. The first approach was indirect, evaluating the presence of a centrifugal gradient of cellular senescence transcriptional signatures in the human MS brain tissue starting from demyelinated plaques, using spatial transcriptomic analysis. Gradients of senescence-like signatures extending from lesion cores to the surrounding periplaque were observed in older lesions (i.e., chronic active and inactive lesions), but not in the new actively demyelinating ones, indicating that cellular senescence might play a detrimental role by participating in chronic non-remitting inflammation, thereby contributing to tissue dysfunction. In line with our evidence, macroscopic gradients of tissue pathology were identified using in vivo MRI, focusing mostly on the CSF-gradient interfaces and diffusible molecules37,38. Thus, we suggest that intrinsic pathological processes (e.g., cellular senescence) might also contribute to the interpretation of these data. The second approach was to assess how the presence of senescent-like cells can rewire the cellular networks in tissue. By differential snRNAseq- and spatial transcriptomics-informed cell-cell communication analysis, we demonstrated increased intercellular signaling, mostly mediated by increased senescent cell-related ligands, in MS tissue, confirming that senescent-like cells can strongly affect cell communication and critically shape the surrounding microenvironment.
The third approach was to investigate the detrimental vs. beneficial role of cellular senescence in MS employing a hiPSC-derived 3D organoid model. Lately, hiPSC-derived models have been increasingly used to study neuroinflammation in neurodegenerative diseases39. One published study40 and one preprint41 have investigated cellular senescence in NPCs from MS hiPSCs. Since the presence and the role of NPCs in adult tissue are controversial, we decided to investigate cellular senescence in a more complex MS hiPSC-derived organoid model consisting of differentiated CNS cell types (astrocytes, neurons, oligodendroglia, and microglia). In our study, we found that inflammation-induced by a complex MS-relevant stimulus, in this case, inflamed CSF from untreated MS patients (as well as a cocktail of MS-associated inflammatory cytokines, but not immunoglobulins), triggered cellular senescence-like processes, suggesting inflammation’s role in driving this process. This result led us to hypothesize that inflammation-driven senescence-like events may occur early in MS pathology. Over time, these senescent-like cells may contribute to non-relapsing, compartmentalized inflammation by impairing critical cellular functions (such as inefficient debris removal from senescent-like microglia) and by secreting SASP. SASP can negatively impact the functionality of neighboring CNS cells through the secretion of proinflammatory growth factors, cytokines, and chemokines, thereby promoting proinflammatory cellular phenotypes that lessen tissue regenerative capacity.
Building on our previous investigation, which showed that microglia act as the initial responders to inflamed MS CSF in our glia-enriched organoid system28, we pursued a therapeutic strategy by pharmacologically manipulating upstream microglia-mediated inflammation to counteract accelerated cellular senescence-like processes and its role in low-grade neuroinflammation. Specifically, we selected drugs known to potentially affect MS-specific neurodegenerative glial populations. These included: (1) ibudilast, a phosphodiesterase type 4 and macrophage MIF inhibitor, previously tested in a phase 2 clinical trial where it reduced brain atrophy, though not new lesion accrual, and identified in our in silico microglia-drug interaction analysis using human snRNAseq data8; (2) R-α-lipoic acid for its antioxidant and anti-inflammatory effects (with an ongoing clinical trial, NCT03161028); and (3) tolebrutinib, a brain-penetrant BTK inhibitor known to modulate both B cell and microglia activity. In our in vitro model, tolebrutinib was also able to significantly reduce CSF-driven MBP+ oligodendrocyte toxicity. Of note, tolebrutinib was recently reported to delay the time to 6-month confirmed disability progression by 31% in a phase 3 clinical trial of non-relapsing secondary progressive MS, compared to placebo42. A recent post-hoc analysis of this trial showed that this protective effect was significantly seen in those patients with PRL compared to those without43. Our results demonstrated that these drugs partially halted inflammation-driven cellular senescence-like processes and that our glia-enriched organoids might serve as a relevant platform for studying drug effects on human CNS-like tissue, potentially complementing animal preclinical testing and aiding in prioritizing drugs for phase 2 clinical trials. As reference, these results were also compared with the organoid treatment with dasatinib, which was included as a positive control due to its well-known senolytic properties. In a recent animal study, depletion of senescent-like cells using senolytics was shown to reduce the severity of EAE symptoms44.
Finally, in the absence of definitive in vivo biomarkers indicative of cellular senescence in the living brains of MS patients, we applied the MRI-estimated brain age paradigm. Adding to prior evidence30,45,46,47,48, our in vivo MRI data (MRI-based brain-age) indicated advanced biological age of the MS brain in strict relation to chronic inflammation. Our findings showed that the presence of chronic active lesions (seen on MRI as PRL) was associated with higher MRI-estimated brain-age gap in MS patients compared to those without PRL. These results align with the observed clinical outcomes, with patients harboring PRL exhibiting higher disability scores and reaching these milestones at a comparatively younger chronological age, emphasizing the clinical relevance of both smoldering inflammation in MS progression. Our data from more than 400 people with MS also identified the critical “brain-age” (range of 71–74 years) at which patients exponentially accumulate disability.
Our study has some limitations, including a potentially controversial use of terminology given the topic’s intrinsic complexity. We used the term “aging” primarily when referring to results derived from in vivo brain-age imaging data. In contrast, our single-cell genomic human studies and hiPSC modelling specifically focused on cellular features and transcriptomic profiles reminiscent of inflammation-induced cellular senescence, which is not synonymous with aging. Moreover, the disentanglement of chronic inflammation from cellular senescence-associated pathways might be incomplete, considering the SenMayo gene signature includes inflammatory genes as part of the SASP. Our results, however, have been confirmed and validated using the SIT bioinformatic tool, which defines senescent cells based on cell cycle arrest and a small gene list (CDKN2A, CDKN2B, IL23A, CDKN1A, CDKN1B, SERPINE1). Beyond that, our bioinformatics approaches combined both unsupervised and supervised methods chosen depending on the research question and bioinformatic tool availability. For instance, alongside unsupervised analysis, we also used SPATA223 to analyze spatial transcriptomics data along user-defined, manually drawn trajectories within limited tissue areas and on individual slices only, without aggregating the data. This manual process might have introduced bias, potentially limiting the generalizability of our findings. Our in vitro experiments on a patient-derived neural organoid model relied on a limited number of MS hiPSC lines (n = 3), which might limit the generalization of our results. Although axonal myelination was observed, the poor myelin compaction and lack of typical brain cytoarchitecture represent significant limitations of the neural organoid model. Finally, the survival of hiPSC-derived microglia once engrafted within the neural organoid spans only a few weeks, which restricts the possibility of chronic inflammatory stimulation of this system. Further development of this patient-derived cellular model, as well as validation of our results, would be desirable in future work.
Overall, our results suggest that in MS, the immediate response to brain injury might trigger detrimental, long-lasting effects. During the initial stages of demyelination and tissue injury, the activation of senescence-associated pathways might hinder the further proliferation of microglia and other glial cell types, particularly in the presence of a lymphocyte-driven inflammatory environment. In line with this, a recent paper reported astrocytic and ependymal senescence before the onset of MRI-visible demyelination in a marmoset EAE model24. However, long-term senescence could contribute to sustained low-grade neuroinflammation, impaired repair processes, and the progressive spread of tissue damage over time, exacerbating the progression of MS pathology (schematic interpretative framework in Supplementary Fig. 7). In line, a recent EAE mouse study demonstrated that senescent-like microglia limit remyelination through the SASP49. These findings suggest potential clinical implications, pointing to chronic inflammation and cellular senescence pathways as areas of future interest for possibly slowing MS progression.
Methods
Ethical compliance for human tissue studies
This research complies with all relevant ethical regulations and has been approved by the ethical committee of IRCSS San Raffaele Scientific Institute, Milan, Italy (IRB 45/INT/2021 and 137/2021). The human brain samples were obtained from the Netherlands Brain Bank, Netherlands Institute for Neuroscience, Amsterdam (open access: www.brainbank.nl), the NINDS/NIH, US (16-N-0055) and the Rocky Mountain Tissue Bank, US (https://www.rmtb.org/). All Material has been collected from donors for or from whom a written informed consent for a brain autopsy and the use of the material and clinical information for research purposes had been obtained by the NBB, NIH, and Rocky Mountain Tissue Bank, respectively.
Human single-nucleus RNA sequencing
Brain tissue
A total of 32 frozen brain tissue blocks were provided by the Netherlands Brain Bank, the Rocky Mountain Brain Bank, and the NINDS/NIH (15 patients with progressive MS and 8 age- and sex-matched non-affected, non-dementia controls). When available, a mirror paraffin-embedding block was obtained and processed for immunostaining tissue characterization and validation. For tissue selection, initial pathological staging and characterization of MS lesions was provided directly by the tissue banks but detailed characterization was then performed in our lab prior the execution of single-cell genomics experiments. Post-mortem interval was limited to 12 h to ensure tissue quality. Brain tissue sampling was performed using a cold 3-mm punch-biopsy tool allowing multiple sampling of a tissue block: a total of 43 samples were obtained from the 32 frozen brain tissue blocks. Details regarding the brain sample location and pathological stage as well as the associated clinical features of the autopsy cases are reported in Supplementary Data 1.
Nuclei extraction and snRNAseq
Nuclei extraction from snap-frozen human brains was performed according to the following protocol8. On wet ice, the frozen, dissected tissue was lysed using a Dounce homogenizer (10 strokes with the loose pestle followed by 20 strokes with the tight pestle) in 500 μL of freshly made cold lysis buffer (low-sucrose buffer, Triton ×-100 20%, RNase inhibitor 0.8 U/μL). The homogenate was filtered through a 40-μm cell strainer for nuclei extraction with 4 mL of low-sucrose buffer (0.64 M sucrose, HEPES pH8, CaCl2, MgAc, EDTA, DTT, and nuclease-free water). Mechanical homogenation was then performed for 10 s at 1000 rmp (VWR® 200 Homogenizer). To remove cellular and myelin debris, a sucrose gradient (12 mL of high-sucrose buffer (2 M sucrose, HEPES pH 8, MgAc, DTT, nuclease-free water) underneath the low-sucrose buffer) was centrifuged at 3200 × g for 30 min at 4 °C, and the supernatant was discarded. Nuclei were resuspended in 1 mL resuspension solution (1× PBS, 5% BSA and RNase inhibitor 0.2 U/μL) and centrifuged for 12 min at 3200 × g. Nuclei were again resuspended in 50 μL cold resuspension solution. Nuclei were counted using trypan blue and a haemocytometer under bright field. Only round and dark-blue stained nuclei were considered of good quality and included in the final count. A yield of 700–1200 nuclei per μL per sample was targeted. For droplet-based scRNA-seq, libraries were prepared using the Chromium Single Cell 3ʹ v3, according to the manufacturer’s protocol (10× Genomics) without modifications, targeting 5000 cells per sample. Using the Illumina Novaseq platform, all samples were sequenced at 50,000 reads per cells. Preparation of cDNA libraries and sequencing support were performed at the Center for Omics Sciences (San Raffaele Scientific Hospital, Milan).
Bioinformatic analysis
Downstream analysis utilized the computational resources of the research cluster of San Raffaele Scientific Hospital, Milan. CellRanger software (v.6.1.2; 10× Genomics) was used for library demultiplexing, reads processing, and unique molecular identifier (UMI) counts. Human transcriptome refdata-gex-GRCh38-2020-A was used as reference. Single samples were processed with SoupX50 using default parameters. Filtered matrices were loaded in Seurat (v 4.3) using the Read10× function with a cut-off value of 200 unique molecular identifiers (UMIs) and min.cells parameter of 20. The filters for mitochondrial reads and the range of the total number of features was tailored for specific samples to remove low-quality barcodes. Single samples were also processed with DoubletFinder51 to identify and remove doublets. Counts matrices from individual samples were merged into a single one using the merge function. After normalization, the top 4000 highly variable features we selected. Data were scaled, regressing out the contribution from the proportion of mitochondrial counts, the total counts, the cell cycle scores, the tissue of origin and the source facility. Integration was performed using Harmony52 using the individual sample id as grouping variable. The entire dataset was projected onto two-dimensional space using the RunUMAP function, retaining the top 30 principal components. The FindNeighbors function run on the first 30 principal components to construct the k-nearest neighbors graph based on the Euclidean distance in PCA space, and the FindClusters function applies the Louvain algorithm to iteratively group nuclei (resolution = 0.2). The assignment of cell type identity to clusters was based on known cell lineage markers, as well as comparison with previously published and reported snRNA-seq studies. For each cluster, we extracted the average gene expression for all identified genes as well as the top 100 differentially expressed genes comparing each cluster against all the others. A differentially expressed gene was defined as a gene significantly expressed (p adjusted for multiple comparison, p < 0.05) in ≥25% of cell populations and showing, on average, >0.25-fold difference (log scale) between groups of cells.
Subclustering analysis
To identify more granular cell states, we performed subclustering analysis on the previously annotated immune, astrocyte, and vascular cell populations. Each of these populations was isolated from the main dataset and re-clustered independently, using the same unsupervised algorithm and downstream analysis pipeline as in the initial clustering.
Mapping cellular senescence gene signatures
To quantify the senescent cells in the human snRNAseq dataset, we first calculated a senescence score per nucleus by applying the function AddModuleScore in Seurat and by using as geneset the SenMayo signature11. The quantification of the senescent cells was performed by counting the nuclei with a module score greater than the imposed threshold. Briefly, the distribution of the senescent score was grouped by cluster per tissue type (WM vs. cortex) and lesion pathological stage condition. Since distinct CNS cell types had different senescence score distributions independently from the patient group of origin (non-neurological controls vs. MS), for counting purposes, we defined an arbitrary, tailored threshold of senescence for each CNS cell type based on the top 10% of nuclei with the highest senescence scores in the control cortex. For each CNS cell type, MS nuclei with a senescence score greater than the threshold were labelled as senescent. The comparison between MS and healthy control per CNS cell type was therefore expressed in terms of fold-change increase or decrease relative to the control cortex.
Senescence index tool (SIT)
To quantify the number of senescent-like cells in tissue, the Senescence Index tool (SIT) was used following the developers’ guidelines14. Essentially, the algorithm relies on typical senescent cell features: the increased expression of established senescent-associated genes and cell cycle arrest. The workflow comprises four key steps: (1) defining the Senescence Signature (SS): each cell initially underwent scoring based on the expression of established senescent genes (CDKN2A, CDKN2B, IL23A, CDKN1A, CDKN1B, SERPINE1); (2) Defining Cell Cycle Arrest Signatures (K or R or WP): cells were scored using three distinct cell cycle signatures—KEGG CELL CYCLE M7963 (K), REACTOME CELL CYCLE M543 (R), and WP CELL CYCLE M39650 (WP)—via ss-GSVA. Cell cycle arrest signatures were established as the inverse of scores for each signature; (3) Establishing Cell Senescence Scores (SS_K, SS_R, SS_WP): these scores represented the summation of the senescence signature SS and the cell cycle arrest signatures (K or R or WP); and (4) Identifying senescent cells: the senescence scores SS_K, SS_WP, and SS_R were divided based on quartiles. Cells with a senescence score within the top quartile for each score were categorized as senescent-like cells.
Spatial transcriptomics
Brain tissue
Frozen brain tissue blocks from patients with progressive MS were provided by the Netherlands Brain Bank. Post-mortem interval was limited to 12 h to ensure tissue quality. Details regarding the brain sample location and pathological stage as well as the associated clinical features of the autopsy cases are reported in Supplementary Data 4.
Tissue processing and Visium data generation
Frozen samples were embedded in OCT and cryosectioned at −20 °C (LEICA CM1950). 10-mm-thick sections were placed on chilled Visium Tissue Optimization Slides (catalog no. 3000394, 10× Genomics) and Visium Spatial Gene Expression Slides (catalog no. 2000233, 10× Genomics), and captured firmly by warming the back of the slide. Tissue sections were then stained according to Visium Spatial Tissue Optimization User Guide (catalog no. CG000238 Rev A, 10× Genomics) or with 1% Sudan Black (199664-25 G, Sigma-Aldrich) in ethylene glycol and Nuclear Fast Red (NFR, ab246831, Abcam) as described in Lin et al.24 Based on Visium Spatial Tissue Optimization User Guide (catalog no. CG000238 Rev A, 10× Genomics), tissue was permeabilized for 18 min. Brightfield histology 4× tiling imaging were taken with a Nikon Eclipse Ci 1195 microscope. Stitched images were exported as.tiff files for downstream analysis. cDNA libraries were prepared according to the Visium Spatial Gene Expression User Guide (https://assets.ctfassets.net/an68im79xiti/3pyXucRaiKWcscXy3cmRHL/a1ba41c77cbf60366202805ead8f64d7/CG000239_VisiumSpatialGeneExpression_UserGuide_Rev_F.pdf). cDNA libraries were loaded at 300 pM and sequenced on a NovaSeq 6000 System (Illumina) using a NovaSeq S4 Reagent Kit (200 cycles, catalog no. 20027466, Illumina), at a sequencing depth of approximately 250–400 10 × 106 read-pairs per sample. Sequencing was performed using the following read protocol: read 1: 28 cycles; i7 index read: 10 cycles; i5 index read: 10 cycles; and read 2: 91 cycles.
Bioinformatic analysis
Imaging and sequencing data were processed using SpaceRanger v.21.1 to generate spatially resolved counts from 4992 spots/slide. Following automatic image thresholding in SpaceRanger, each tissue slice was manually inspected for quality, segmentation accuracy, and correspondence between read density and tissue histology. Seurat v4.3 have been used for spot normalization and regression. High-variable genes and top 30 PCs have been used for UMAP construction. Marker genes per cluster have been identified using the FindAllMarkers function with the Wilcoxon rank-sum test using default cutoffs implemented in Seurat. For spatially-aware unsupervised clustering, we utilized BayesSpace19. This algorithm integrates both transcriptional similarity and spatial proximity to refine cluster definitions and enhance the identification of tissue domains. We also relied on a supervised approach to define ROIs of centrifugal gradients around MS lesions. The ROIs have been used to measure average gene expression for the aging gradient analysis. Cell-type spot deconvolution has been performed with SPOTlight20 using default parameters and annotated snRNASeq brain dataset8. The AddModules function, implemented in Seurat, was used to score each spot for specific senescence signatures.
Seurat spatial transcriptomic dataset integration
The integration of all the Visium slide datasets was performed following the recommendation provided by 10× Genomics [https://www.10xgenomics.com/analysis-guides/correcting-batch-effects-in-visium-data]. First, individual slides were merged into a single object, then normalized and scaled using the SCTransform function from the Seurat package53. The nCount_Spatial and percent_mt covariates were regressed out. Harmony52 was used to correct for batch effects, with the single slide ID as the grouping variable. Finally, the integrated object obtained was used for downstream analysis.
Spatially enhanced gene expression resolution
Enhancement of the resolution of gene expression was achieved using BayesSpace19. For each gene, a model using the PC vectors of each spot was trained to predict the spot-level gene expression, and the fitted model was used to predict subspot expression from the subspot PCs. The algorithm is implemented in the enhanceFeatures() function. enhanceFeatures() has been used on a subset of genes of interest to improve the quality of the depiction of the expression on the tissue slice. Pre-enhanced values have been used for the regular bioinformatic analysis.
Spatial differential expression analysis of genes along a linear trajectory
The selection of top gene features along a gradient was performed using the SPATA2 package23. Briefly, pre-processed spatial Seurat objects were collected and converted into a SPATA object using the asSPATA2 function. For each Visium slide, a linear trajectory was manually drawn to intersect the lesion edge perpendicularly. From the object, only spatially variable features were shortlisted using the runSPARKX function. Only features with a SPARKX p < 0.01 were retained. Features were then scored along the user-defined drawn trajectory using the spatialTrajectoryScreening function. Features with a false discovery rate (FDR) below 0.05 were considered significant. The smallest mean absolute error (MAE) was used to rank the best trend type for each feature.
Inference of cell-to-cell communication
To infer cell-cell communication, two independent objects were generated from the snRNAseq dataset: one containing senescent-like microglia (according to SenMayo classification) along with all other cell populations at the chronic active lesion edge, and the other containing non-senescent microglia with all other cell populations at the chronic active lesion edge. Cell-cell interactions, based on the expression of known LR pairs in different cell types, were inferred using CellChat (v.2.1.0)25. Briefly, we loaded the normalized counts into CellChat and applied the preprocessing functions “identifyOverExpressedGenes” and “identifyOverExpressedInteractions” with standard parameters. For the database, we selected the “Secreted Signaling” pathways and used the pre-compiled human “Protein-Protein Interactions” as a priori network information. For the main analysis, the core functions “computeCommunProb”, “computeCommunProbPathway”, and “aggregateNet” were applied using standard parameters with fixed randomization seeds. Finally, to determine the senders and receivers in the network, the function “netAnalysis_computeCentrality” was applied to the “netP” data slot. The two processed objects were then merged using “mergeCellChat”. The functions “computeNetSimilarityPairwise”, “netEmbedding”, and “netClustering” were applied to group signaling pairs based on structural similarity. Finally, we identified overexpressed signaling using “identifyOverExpressedGenes”. Significant upregulation was determined if the interaction was significant (p < 0.05) and the logFC of the ligand expression was greater than 0.2. The same analysis was performed to evaluate differential cell-cell communication between senescent and non-senescent vascular cells and astrocytes, respectively.
To perform spatial cell-cell communication analysis on Visium spatial transcriptomics data, the NICHES package26 was used. Briefly, after collecting the pre-processed spatial Seurat objects and the LR database (the same one used for the CellChat analysis), the LR database was converted into a format compatible with NICHES. As recommended in the vignette (https://msraredon.github.io/NICHES/articles/01%20NICHES%20Spatial.html), we imputed gene expression data using the RunALRA function implemented in SeuratWrappers (https://github.com/satijalab/seurat-wrappers). Then, the NICHES pipeline was run with the custom LR database. The NeighborhoodToCell slot was selected and processed to scale the data, perform dimensionality reduction (PCA and UMAP), and clustering. The final object was used for downstream analysis and visualization.
Human multiplex immunofluorescence imaging
Multiplex immunofluorescence protocol
Multiplex fluorescence immunostaining was performed on 5-µm-thick paraffin sections using single or iterative rounds of sequential immunostaining with antibody panels (rat anti-GFAP [Invitrogen, 13-0300, 1:100], rat anti-MBP [Millipore, MAB386,1:50], mouse anti-HLA-DP, DQ, DR [Dako, M0775,1:100], rabbit anti-NeuN [ABCAM, ab104225, 1:500], chicken anti-IBA1 [Cederlane Labs, 234006(SY), 1:100], mouse anti-MAP2 [Millipore, MAB 3418, 1:500], mouse anti-CDKN2A/p16INK4a [Abcam, ab54210, 1:100], guinea pig IgG anti-CD31 [Cederlane Labs, 351004(SY), 1:100], mouse anti-CD68 [Agilent/DAKO, M087601-2, 1:100], rabbit anti-TMEM119 [Sigma-Aldrich, HPA051870, 1:100], chicken anti-Vimentin [Millipore Sigma, AB5773, 1:100], rabbit anti-TPPP [Abcam, ab92305, 1:100], rabbit anti-lamin B1 [Abcam, ab133741, 1:100], mouse anti-53BP1 [Invitrogen, MA5-26830, 1:100]. Each round of antibody staining was imaged by fluorescence microscopy, after which antibodies were stripped and the tissue re-stained with a different antibody panel, as described in Absinta et al.8 In brief, after deparaffinization and antigen retrieval (a 10-min heat-mediated step in 10 mM Tris-EDTA buffer pH 9.0 using an 800 W microwave set at 100% power), sections were incubated with Human BD Fc Blocking Solution (BD Biosciences) to saturate endogenous Fc receptors, and then in True Black Reagent (Biotium) to quench intrinsic tissue autofluorescence. The sections were incubated for 1 h at room temperature using a cocktail of immunocompatible primary antibodies, then rinsed with PBS supplemented with 1 mg ml−1 BSA and incubated with a cocktail of the appropriately cross-adsorbed secondary fluorescent antibodies. After washing, sections were counterstained using DAPI (Thermo Fisher Scientific) for visualization of cell nuclei. Slides were then cover-slipped using Immu-Mount medium (Thermo Fisher Scientific). Briefly, each round of antibody staining was imaged by ImageXpress Confocal MC system (Molecular Devices, San Jose, CA, USA) with a PlanApoLambda 20×/0.75 for 4 channels: DAPI (nuclei), 488, Cy3, and Cy5. A grid with multiple images were collected for individual tissue in order to cover the entire section. The segmentation was performed using the software ArivisVision4D (ZeissAG, Germany).
After imaging, slides were incubated with 1 M Tris buffer (pH = 7.5) in order to remove disaccharides. Then, tissue-bound primary and secondary antibodies were stripped off the slides after a 30-min incubation at 56 °C in stripping buffer containing 10% SDS (1:10) and 0.5 M Tris HCl (pH = 6,8) (1:16), and 2-mercaptoethanol (1:250) diluted in distilled water. Then, the slides were washed 3 times with 1× PBS, followed by a 1-min additional heat-mediated antigen-retrieval step in Tris-EDTA buffer. The above processing cycle, beginning with re-blocking of tissues in Human BD Fc Blocking solution, was repeated, and the same sections then incubated using the antibody mixtures.
Image analysis and quantification
Pseudocoloured, coregistered images acquired from all the rounds of antibody staining and imaging were overlaid as individual layers to create multicoloured merged composites. Segmentation of the tissue in region of interest was performed manually based on the anatomopathological tissue composition. On the DAPI staining, individual nuclei were identified with CellPose alghoritm54. Each nucleus had a unique identifier and the size-enlarged mask associated to the nuclei were then filtered as “positive” based on the value of mean intensity of the specific channel. The position (X and Y) and the number of positive cells over the total number of nuclei for each area were then calculated and exported for statistical analyses.
hiPSC-derived glial-enriched organoids
Human subjects
Permission to generate hiPSCs from human skin fibroblasts was granted by the ethical committee of the San Raffaele Scientific Institute (Milan, Italy) (BancaINSpe - INSPE1178). Fibroblasts were isolated from skin biopsies from 3 individuals with a diagnosis of relapsing-remitting MS (2 women, 1 man). Collection of CSF by lumbar puncture from untreated MS cases (2 women, 2 men) with chronic active MS lesions by 7T MRI was approved by the National Institute of Health (Bethesda, US) (protocol number: 89-N-0045; principal investigator: Daniel S. Reich) and shared under a materials transfer agreement. CSF collection by lumbar puncture from 3 non-neurological controls undergoing spinal anesthesia for knee surgery (1 woman, 2 men) was granted by the ethical committee of the San Raffaele Scientific Institute (Milan, Italy) (BancaINSpe–INSPE1178). Written consent was obtained from all subjects.
Neural organoid generation
hiPSC-derived NPCs with the SOX10-eGFP-expressing lentivirus were generated as described by Fagiani et al.28 To generate self-organizing forebrain SOX10-eGFP organoids, NPCs were expanded in Matrigel-coated flasks in N2B27 medium containing equal parts of Neurobasal (Invitrogen) and DMEM-F12 medium (Invitrogen) with 1:100 B27 supplement lacking vitamin A (Invitrogen), 1:200 N2 supplement (Invitrogen), 1% penicillin/streptomycin/glutamine, supplemented with 3 μm CHIR, 1 μM SAG, 150 μM ascorbic acid. At 100% confluence, NPCs were enzymatically detached and counted. 2,5 × 106 cells per well were plated in 3 mL of Reinhardt complete medium in non-treated 6 well-plates. Cells were grown in Reinhardt complete medium for 24 h under constant gyratory shaking (88 rpm). Subsequently, medium was changed to glial induction medium (100% DMEM-F12, 1:100 B27 lacking vitamin A, 1:200 N2, 1% Penicillin/Streptomycin, and 1% L-Glutaminase), supplemented with 10 ng/mL triiodo-L-thyronine (T3), 10 ng/mL rhPDGF, 10 ng/mL rhNT3, 1 μM SAG, 10 ng/mL μM IGF-1, 200 μM ascorbic acid, and 1 μg/mL doxycycline from day1 to day 3. From day 5 till the end of the differentiation protocol, medium was replaced with glial differentiation medium (GDM) (100% DMEM-F12, 1:100 B27 lacking vitamin A, 1:200 N2, 1% Penicillin/Streptomycin, and 1% L-Glutaminase), supplemented with 60 ng/mL T3, 10 ng/mL rhNT3, 10 ng/mL IGF-1, 200 μM ascorbic acid, and 100 μM cyclic AMP analog (dbcAMP) to promote OPC proliferation and maturation. Doxycycline at the final concentration of 1 μg/mL was added to GDM medium from day 5 until day 14. On day 14, 0.01 μg/mL human recombinant GDNF and BDNF were added to the culture medium till the end of the differentiation protocol. Half of the medium was changed three times a week. Cultures were maintained at 37 °C, 5% CO2 under constant gyratory shaking (88 rpm) for up to 8 weeks.
hiMicroglia differentiation
hiPSCs were first differentiated into hematopoietic progenitors’ cells using STEMdiffTM Hematopoietic Kit (Catalog #05310) and then, STEMdiffTM Microglia Differentiation Kit (Catalog #100-0019), and STEMdiffTM Microglia Maturation Kit (Catalog #100-0020) to generate microglia, according to the manufacturer’s instructions. FACS analysis was performed to check each maturation stage, according to the manufacturer’s instructions. At day 38 of differentiation, microglia were added to 7-week-old organoids (to have 10% microglia into the neuron-glia network, 200,000 cells/well of a 6-well-plate were plated). Organoids were harvested at different time points for downstream analysis.
CSF, cytokines, and drug treatments
Organoids from 3 different cell lines integrating hiPSC-derived microglia were exposed to: (1) 10% CSF from 3 non-neurological healthy controls for 24 h as negative control; (2) 10% CSF from a pool of 5 MS CSF from MS patients with 7T chronic active lesions for 24 and 48 h; (3) to an inflammatory cytokine cocktail (30 ng/mL TNFα [Peprotech, #300-01A-100 μg], 10 ng/mL IL1β [Peprotech, #200-01B-10 μg], 10 ng/mL IFNγ β [Peprotech, #300-02-100 μg], and 400 ng/mL C1q [Mybiosource, #MBS147305) for 24 h; (4) immunoglobulins IgG (Invitrogen, #02-7102) at the concentration of 8 μL/mL for 24 h. For drug treatments, after 6 h-treatment with a pool of 6 CSF from MS patients, organoids were treated with vehicle (i.e., DMSO) and selected drugs at two concentrations: 1,5 and 0,15 μM dasatinib (Sigma-Aldrich, SML2589); 50 and 5 μM ibudilast (MedChemExpress, Cat. No.: HY-B0763); 1 and 0,1 μM tolebrutinib (MedChemExpress, HY-109192); 100 and 10 μM α−lipoic acid (MedChemExpress, CAS No. 1200-22-2) for 18 h.
Immunofluorescent staining
Organoids were fixed in 4% paraformaldehyde (PFA) for 1 h at room temperature (RT) and then, washed in 1× PBS (3 times) and transferred to 30% sucrose solution overnight. Subsequently, they were embedded in OCT and stored at −80 °C. For immunofluorescence analysis, 15-μm-thick sections were cut using a Leica cryostat. Cryosections were air dried for at least 40 min and then, rinsed twice with 1× PBS for 5 min. Nonspecific binding sites were blocked by incubation in the blocking solution (1× PBS containing 0.1% Triton ×-100 (Sigma-Aldrich, ×-100), 10% serum) for 1 h at RT. Cryosections were incubated at 4 °C for 72 h with primary antibodies diluted in blocking solution in a humidity chamber. The following primary antibodies were used for immunofluorescence: anti-GFAP (rabbit, 1:500, Agilent DAKO, Z0334); anti-MBP (rat, 1:50, Millipore, MAB386); anti-MHCII (HLA-DR, DQ, DP, mouse, 1:100, Dako, M0775); anti-NeuN (rabbit, 1:500, ABCAM, ab104225); anti-MAP2 (mouse, 1:500, Millipore, MAB 3418); anti-GFP (rabbit, 1:500, Life Technologies, A-11122). For double labeling, some primary antibodies were incubated simultaneously. After incubation with the primary antibodies, cryosections were washed three times for 5 min with 1× PBS containing 0.1% Triton ×-100. Then, cryosections were incubated for 2 h at RT in the dark with appropriate fluorophore-conjugated secondary antibodies (Alexa Fluor 488, Alexa Fluor 546, and Alexa Fluor 633), diluted in 1× PBS containing 1% serum and 0.1% Triton ×-100. In all immunofluorescence staining, nuclei were stained with DAPI (1:25,000; Roche) in 1× PBS for 2 min. Sections were mounted with Dako fluorescent mounting medium and imaged on Olympus FluoVIEW FV3000RS confocal microscope. Images were processed in ImageJ (Fiji Version 2.0.0).
Transmission electronic microscopy
Organoids were incubated with Karnovsky’s fixative (2% paraformaldehyde and 2.5% glutaraldehyde). Then, the organoids were washed in 0.1 M cacodylate buffer. Post-fixed in 2% osmium, 1.5% potassium ferrocyanide for 1 h on ice, rinsed, dehydrated, and embedded in Epon resin (Electron Microscopy Science). Ultrathin sections were mounted onto slot grids for viewing using Talos 120 C (Fei) microscope, images were acquired with a 4k × 4k Ceta CMOS camera (Thermo Fisher Scientific).
Senescence associated-β-galactosidase staining
In organoids, the presence of SA-β-gal activity was detected using the Senescence SA-β-gal staining kit (Catalog # 9860, Cell Signaling), according to the manufacturer’s instructions. Briefly, 15-μm-thick cryosections were air dried for at least 40 min and then, rinsed twice with 1× PBS for 5 min, fixed with 1× Fixative solution (provided by the kit) for 15 min at RT, and finally washed with 1× PBS. Then, sections were incubated with SA-β-gal staining solution (pH = 6) at 37 °C for 24 h in a dry incubator (no CO2). Sections were mounted with Dako fluorescent mounting medium and imaged on a digital brightfield slide scanning (20×) for downstream quantification. For each experimental condition, SA-β-gal staining area was quantified in at least 30 organoids using ImageJ (Fiji Version 2.0.0).
Immunohistochemistry
Organoids were fixed in 4% PFA for 1 h at RT and then, washed in 1× PBS (3 times) and transferred to 30% sucrose solution overnight. Subsequently, they were embedded in OCT and stored at −80 °C. 15-μm-thick sections were cut using a Leica cryostat. Cryosections were air dried for at least 40 min and then, rinsed twice with 1× PBS for 5 min and treated for endogenous peroxidase inactivation with 3% hydrogen peroxidase (H2O2) solution in 1× PBS for 15 min at RT and, then, washed with 1× PBS (2 times). Nonspecific binding sites were blocked by incubation in the blocking solution (1× PBS containing 0.1% Triton ×-100 (Sigma-Aldrich, ×-100), 10% serum) for 1 h at RT. Cryosections were incubated with anti-MBP primary antibody (rat, 1:50, Millipore, MAB386) diluted in blocking solution in a humidity chamber at 4 °C for 72 h. After incubation with the primary antibody, cryosections were washed for 5 min with 1× PBS (3 times). Then, cryosections were incubated for 1 h at RT with appropriate goat anti-rat IgG (H + L) biotinylated secondary antibody (Vector, BA9400), diluted in 1× PBS containing 1% serum. Cryosections were washed for 5 min with 1× PBS (3 times), followed by incubation for 45 min at RT with a solution containing Reagent A and Reagent B (VECTASTAIN® Elite® ABC-HRP Kit, Peroxidase (Standard), Vector, PX-6100). Cryosections were washed for 5 min with 1× PBS (3 times). Finally, cryosections were developed with the DAB solution (Liquid DAB+ Substrate Chromogen system, Dako, K3468) for a few seconds, followed by rinsing in PBS. Sections were mounted with Dako fluorescent mounting medium and imaged on a digital brightfield slide scanning (20×) for downstream quantification. For each experimental condition, MBP staining area was quantified in at least 30 organoids using ImageJ (Fiji Version 2.0.0).
Organoid viability assay
Organoid viability was assessed using the CellTiter-Glo® 3D Cell Viability Assay (Promega), which is based on the quantitation of adenosine triphosphate present as marker of metabolically active cells. Briefly, 5 organoids per condition (treated with the selected inflammatory stimulus and/or pharmacological compounds) were added to experimental wells of opaque-walled 96-well plates in 100 µL of culture medium. The plate was then equilibrated to RT for 30 min. An equal volume of CellTiter-Glo® 3D Reagent (100 µL) was added to each well, and the contents were mixed vigorously for 5 min to induce cell lysis. The plate was then incubated at RT for an additional 25 min to stabilize the luminescent signal. Luminescence was recorded using a BertholdTech Mithras LB 940 (integration time: 0.5 s per well). tert-Butyl hydroperoxide (concentration of 500 mM for 30 min) was used as positive control to trigger cell death. Experiments were run in triplicate per condition.
Phagocytosis assay
hiPSC-derived microglia cell lines derived from 4 different human donors (n = 4 people with MS) were cultured on MaES-precoated 96-well tissue culture plates at a density of 100,000 cells/well and treated with (1) 10% CSF from a pool of CSF from 5 MS patients, (2) an inflammatory cytokine cocktail (3 ng/mL TNFα, 1 ng/mL IL1β, 1 ng/mL IFNγ, and 40 ng/mL C1q), and (3) immunoglobulins IgG at the concentration of 8 μL/mL. After 24 h of treatments, the phagocytic capacity of microglia was assessed using pHrodo™ Green Zymosan Bioparticles™ Conjugate for Phagocytosis (P35365, Thermo Fischer Scientific). pHrodo™ Green Zymosan Bioparticles™ were added to the cells at a concentration 100 μg/mL in microglia medium and incubated for 1 h at 37 °C. Cells were, then, harvested and processed for flow cytometry (BD LSRFortessa™ Cell Analyzer). For each sample, 50,000 events were recorded. Viability was assessed with Zombie Aqua Fixable Viability Kit (Biolegend). Cell data were analyzed with FlowJo software (Tree Star).
Single-cell RNA-sequencing
For the enzymatic cellular dissociation, 30 organoids, for each experiment and for each donor, were first washed in Hank’s balanced salt solution (HBSS) and then, incubated with a freshly prepared dissociation solution (HBSS, papain and DNAase I) for 45 min at 37 °C on an orbital shaker (90 rpm), and gently triturated using a P1000 pipette. Cells were incubated again for 15 min at 37 °C on an orbital shaker (90 rpm) centrifuged and gently triturated using a P1000 pipette. After the single cell dissociation, the cell suspension was mixed (1:1) with 10 mg/mL ovomucoid protease inhibitor solution and centrifuged at 300 × g for 5 min. The pellet was resuspended in 1× PBS containing BSA (1:50). Cells were counted using trypan blue and a haemocytometer under bright field. A yield of 700–1200 cells per μL per sample was targeted. For each condition, cell viability was assessed using Luna II Automated Cell Counter. Samples with more than 80% viable cells were processed for downstream analysis. For droplet-based scRNA-seq, libraries were prepared using the Chromium Single Cell 3ʹ v3, according to the manufacturer’s protocol (10× Genomics) without modifications, targeting 10,000 cells per sample. Using the Illumina Novaseq platform, all samples were sequenced at 50,000 reads per cells. Preparation of cDNA libraries and sequencing support were performed at the Center for Omics Sciences (San Raffaele Scientific Hospital, Milan).
Bioinformatic analysis
CellRanger software (v.6.1.2; 10× Genomics) was implemented for library demultiplexing, barcode processing, fastq file generation, gene alignment to the human genome (refdata-gex-GRCh38-2020-A), and unique molecular identifier (UMI) counts. Filtered matrices were loaded in Seurat (v 4.1.1). The analysis pipeline included data quality control (outlier nuclei and artifact detection), nuclei filtering (mitochondrial contamination, very low and high library size), normalization, scaling, doublets removal (DoubletFinder), detection of highly variable genes, dimensional reduction (using PCA) and clustering (heatmaps and UMAP plots), identification of differentially expressed genes (clusters biomarkers), assignment of cell type identity to clusters (based on known cell lineage markers and comparison with data from literature). For each cluster, we extracted the average gene expression for all identified genes as well as the top 100 differentially expressed genes comparing each cluster against all the others. A differentially expressed gene was defined as a gene significantly expressed (p adjusted for multiple comparison, p < 0.05) in ≥25% of cell populations and showing, on average, >0.25-fold difference (log scale) between groups of cells. Aging/senescence gene signatures and SIT algorithm were applied as described above.
Donor deconvolution
Donor deconvolution was performed to assign each cell from the pooled single-cell RNA-seq library to one of the three hiPSC-line donors. Raw sequencing data were first processed using CellRanger (v.6.1.2, 10× Genomics). The resulting BAM file was then provided as input to cellsnp-lite (Mode 1a) to pile up expressed alleles at single-nucleotide polymorphism (SNP) locations55. Finally, the SNP profiles were used by Vireo to demultiplex the data, clustering cells and assigning each to its donor of origin56.
Brain-age prediction from human snRNAseq samples
The biological age of brain tissue was estimated from transcriptional profiles using the Real Age Prediction from Transcriptome staging on Reference (RAPToR) tool57. Briefly, the workflow includes two main steps: (1) reference gene expression time-series are interpolated to build a near-continuous, high-temporal-resolution reference; (2) the correlation profile of each sample against the reference is defined. The output is the estimated biological age defined as the timing of the correlation peak. To construct the reference, we retrieved snRNAseq data8,12,13,29 from samples of control human brain spanning six adult decades (n = 21 samples, age range 34–82 years, average 55.1, SD 14.0). Gene expression matrices were aggregated to sample-wise pseudobulks; the gene counts were then normalized to transcripts per million. MS brain samples were retrieved from published MS snRNAseq papers (n = 44 samples, age range 35–58 years, average 47.6, SD 6.9)8,12,13. Finally, all the MS samples were staged following RAPToR’s recommended workflow without modifications. Scripts used for data preprocessing, reference building, and sample staging are shared at https://github.com/AbsintaLab/.
Clinical-MRI multicenter study
Study participants
Between March 2012 and March 2023, imaging and clinical cross-sectional data were prospectively collected under institutional review board-approved protocols in adults from 3 academic research hospitals, after written, informed consent: the NIH Clinical Center (Bethesda, MD, USA; IRB 89-N-0045, principal investigator Daniel S. Reich), the Johns Hopkins University Hospital (Baltimore, MD, USA; IRB CIR00106571, principal investigator Peter Calabresi), and the Saint-Luc University Hospitals (Brussels, Belgium, IRB BA03200000104, principal investigator Pietro Maggi). These protocols allow exploratory analysis and pooling of collected data. Clinical data were obtained from experienced MS clinicians. Only adult subjects (older than 18 years-old) with availability of a high-resolution susceptibility-based 3T MRI scan were included in the study. The enrolled subjects were classified according to their diagnosis: (1) patients with a diagnosis of MS according to the 2017 McDonald criteria, (2) patients with a diagnosis of inflammatory or infectious diseases affecting the CNS (here termed “MS-mimics”), and (3) subjects without neurological and psychiatric conditions (here termed “healthy controls”).
MRI acquisition and analysis
MRI studies were performed on 4 MRI 3-tesla scanners: 2 Philips Intera or Ingenia scanners (Philips Medical Systems, The Netherlands), 1 Siemens Skyra scanner (Siemens AG, Germany) and 1 General Electric Signa Premier (GE Healthcare, Waukesha, Wisconsin, USA). On all scanners, submillimeter isotropic 3D T2*-weighted segmented echo-planar-imaging (EPI; TR, 64 milliseconds; TE, 35 milliseconds; FA, 10°; echo-train length, 15; AT, 5:46; 256 sagittal slices; 0.65-mm isotropic voxels; voxel volume, 0.27 µL)58,59, providing both magnitude and phase images, was acquired before or during intravenous injection of a single dose (0.1 mmol/kg) of gadobutrol (Gadavist; Bayer Healthcare, Leverkusen, Germany). The same 3D T2*-EPI sequence was adapted and optimized to the different MRI scanners with minimal parameter modification59. Additional routine MRI images acquired for clinical use, including 3D T2-FLAIR images and 3D precontrast T1-weighted MPRAGE sequences, were not standardized across the scanners, and implemented for T2-lesion and brain volumes, as previously described59.
Machine learning MRI-based brain-age estimation
Brain-predicted age (hereafter just termed brain-age) calculation implemented 3D FLAIR and 3D T1-weighted brain images as input and followed a previously established protocol without modifications (https://github.com/james-cole/brainageR). Briefly, after segmentation and normalization using SPM12, images were loaded in R using RNifti package and vectorized. The brain-age values were then predicted using a trained model in kernlab (435 principal components). As suggested by the tool developers, adjustment of brain-age was applied to remove age-related variance (output subtracted of 3.33 and then divided by 0.91)30. The brain-age was subtracted from the chronological age and implemented for downstream analysis. A positive difference indicates that the brain is predicted by the machine learning algorithm to be older than their chronological age.
PRL assessment
In each individual, the number of supratentorial chronic non-enhancing PRL on unwrapped filtered phase images was determined by consensus of 2 raters blinded to all patient information and according to the NAIMS guidelines for PRL identification60. A chronic non-enhancing MS lesion was defined as a PRL if it demonstrated a paramagnetic rim on unwrapped filtered phase images, and an isointense/slightly paramagnetic core when compared to the extralesional WM58,59. Specific rim features include: (1) colocalization with the edge of an MS lesion on T2-FLAIR or T1-weighted images; (2) visibility on at least two planes for 3D images; and (3) coverage of at least 2/3 of the lesion WM edge on the slice of maximum visibility. MS patients were classified into PRL status (0, 1–3 and ≥ 4 PRL) based on prior evidence of clinical relevance for disability and clinical progression risk4.
Statistical analysis
Demographic, clinical, and MRI differences were assessed with ANOVA and Tukey’s post-hoc multiple-comparison test, or with Fisher’s exact or chi-squared test, as appropriate. The association between brain-age, brain-age difference with chronological age, log-lesion volume, presence of PRL was tested in a multivariable linear mixed model with random participant effect.
Statistical analysis and data visualization
Statistical analysis used two-tailed parametric or non-parametric t-tests for two-group comparisons, and Fisher’s exact test, one-way ANOVA, and two-way ANOVA with corresponding post hoc tests for multiple group comparisons, multivariable linear mixed and random forest models, when appropriate. Data distributions are presented as box or violin plots (with individual data points when n < 10). p-values are labeled as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Gene expression values in dot plots and heat maps were averaged, mean-centered, and z-score-scaled. Dot size indicates the percentage of nuclei in the cluster where the gene was detected. Among the nuclei in which the gene was detected, the expression level was mean-centered and scaled. For aggregated gene lists, a relative color scheme was used to indicate expression levels. Volcano plots were generated by plotting the log₂(fold change) between two cell populations on the x-axis and the negative log₁₀ of the adjusted p-value on the y-axis (only genes with adjusted p < 0.05 were included). See the dedicated paragraph for details on the snRNA-seq bioinformatic analysis. Differentially expressed genes were defined as those significantly expressed (adjusted p < 0.05 for multiple comparisons) in ≥25% of cell populations, with an average log-fold difference >0.25 between groups of nuclei. Additional statistical analyses were performed using GraphPad Prism (GraphPad Software v9.0). Statistical methods were not used to recalculate or predetermine sample sizes.
Graphical objects in Figs. 1a, 2a, 5a, 7a, d, 9b, 10a as well as in Supplementary Figs. 5c, f and 7 were created with BioRender.com (https://BioRender.com/d0hxq2g).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Single-cell transcriptomic data generated in this study were deposited in Gene Expression Omnibus (GEO) and are publicly available under accession number GSE277435, GSE277430, and GSE280341. This research paper re-analyzes existing, publicly available genomic datasets, including GSE180759, GSE118257, SRA: PRJNA544731, GSE147528. Source data are provided as a Source Data file. Source data are provided with this paper.
Code availability
Bioinformatic codes were posted in GitHub (https://github.com/AbsintaLab) and Zenodo (https://doi.org/10.5281/zenodo.16569909)61. Additional information is available from the lead contact upon request.
References
Kuhlmann, T. et al. Multiple sclerosis progression: time for a new mechanism-driven framework. Lancet Neurol. 22, 78–88 (2023).
Kappos, L. et al. Contribution of relapse-independent progression vs relapse-associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA Neurol. 77, 1132–1140 (2020).
Absinta, M. et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J. Clin. Invest. 126, 2597–2609 (2016).
Absinta, M. et al. Association of chronic active multiple sclerosis lesions with disability in vivo. JAMA Neurol. 76, 1474–1483 (2019).
Absinta, M., Lassmann, H. & Trapp, B. D. Mechanisms underlying progression in multiple sclerosis. Curr. Opin. Neurol. 33, 277–285 (2020).
Maggi, P. et al. B cell depletion therapy does not resolve chronic active multiple sclerosis lesions. EBioMedicine 94, 104701 (2023).
Cagol, A. et al. Association of spinal cord atrophy and brain paramagnetic rim lesions with progression independent of relapse activity in people with MS. Neurology 102, e207768 (2024).
Absinta, M. et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 597, 709–714 (2021).
Baker, D. J. & Petersen, R. C. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J. Clin. Invest. 128, 1208–1216 (2018).
Suryadevara, V. et al. SenNet recommendations for detecting senescent cells in different tissues. Nat. Rev. Mol. Cell Biol. 25, 1001–1023 (2024).
Saul, D. et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat. Commun. 13, 4827 (2022).
Jäkel, S. et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543–547 (2019).
Schirmer, L. et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573, 75–82 (2019).
Troiani, M. et al. Single-cell transcriptomics identifies Mcl-1 as a target for senolytic therapy in cancer. Nat. Commun. 13, 2177 (2022).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Papadopoulos, D. et al. Accelerated cellular senescence in progressive multiple sclerosis: a histopathological study. Ann. Neurol. 97, 1074–1087 (2025).
Safaiyan, S. et al. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 19, 995–998 (2016).
Lerma-Martin, C. et al. Cell type mapping reveals tissue niches and interactions in subcortical multiple sclerosis lesions. Nat. Neurosci. 27, 2354–2365 (2024).
Zhao, E. et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 39, 1375–1384 (2021).
Elosua-Bayes, M., Nieto, P., Mereu, E., Gut, I. & Heyn, H. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 49, e50 (2021).
Alsema, A. M. et al. Spatially resolved gene signatures of white matter lesion progression in multiple sclerosis. Nat. Neurosci. 27, 2341–2353 (2024).
Ximerakis, M. et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 22, 1696–1708 (2019).
Kueckelhaus, J. et al. Inferring histology-associated gene expression gradients in spatial transcriptomic studies. Nat. Commun. 15, 7280 (2024).
Lin, J.P. et al. 4D marmoset brain map reveals MRI and molecular signatures for onset of multiple sclerosis-like lesions. Science 387, eadp6325 (2025).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Raredon, M. S. B. et al. Comprehensive visualization of cell-cell interactions in single-cell and spatial transcriptomics with NICHE. Bioinformatics 39, btac77 (2023).
Petersen, M. A. et al. Fibrinogen activates BMP signaling in oligodendrocyte progenitor cells and inhibits remyelination after vascular damage. Neuron 96, 1003–1012.e7 (2017).
Fagiani, F. et al. A glia-enriched stem cell 3D model of the human brain mimics the glial-immune neurodegenerative phenotypes of multiple sclerosis. Cell Rep. Med. 5, 101680 (2024).
Leng, K. et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 24, 276–287 (2021).
Cole, J. H. et al. Longitudinal assessment of multiple sclerosis with the brain-age paradigm. Ann. Neurol. 88, 93–105 (2020).
Altokhis, A. I. et al. Longitudinal clinical study of patients with iron rim lesions in multiple sclerosis. Mult. Scler. 28, 2202–2211 (2022).
Freund, A., Laberge, R.-M., Demaria, M. & Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 23, 2066–2075 (2012).
Davalos, A. R. et al. p53-dependent release of alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 201, 613–629 (2013).
Gil, J. The challenge of identifying senescent cells. Nat. Cell Biol. 25, 1554–1556 (2023).
van der Poel, M. et al. Transcriptional profiling of human microglia reveals grey-white matter heterogeneity and multiple sclerosis-associated changes. Nat. Commun. 10, 1139 (2019).
Chomyk, A. et al. Transcript profiles of microglia/macrophage cells at the borders of chronic active and subpial gray matter lesions in multiple sclerosis. Ann. Neurol. 95, 907–916 (2024).
Magliozzi, R. et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol. 68, 477–493 (2010).
Magliozzi, R. et al. Ependymal-in’ gradient of thalamic damage in progressive multiple sclerosis. Ann. Neurol. 92, 670–685 (2022).
Summers, R. A., Fagiani, F., Rowitch, D. H., Absinta, M. & Reich, D. S. Novel human iPSC models of neuroinflammation in neurodegenerative disease and regenerative medicine. Trends Immunol. 45, 799–813 (2024).
Nicaise, A. M. et al. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA 116, 9030–9039 (2019).
Park, B. et al. Integrative single-cell analysis of neural stem/progenitor cells reveals epigenetically dysregulated interferon response in progressive multiple sclerosis. Preprint at bioRxiv. https://doi.org/10.1101/2024.02.09.579648 (2024).
Fox, R. J. et al. Tolebrutinib in Nonrelapsing Secondary Progressive Multiple Sclerosis. N. Engl. J. Med. 392, 1883–1892 (2025).
Oh, J. et al. Paramagnetic rim lesions as prognostic and predictive biomarker in the Tolebrutinib Phase 3 trials for disability outcomes. Oral presentation at ACTRIMS Forum Annual Meeting (2025).
Drake, S. S. et al. Senolytic treatment diminishes microglia and decreases severity of experimental autoimmune encephalomyelitis. J. Neuroinflammation 21, 283 (2024).
Kaufmann, T. et al. Common brain disorders are associated with heritable patterns of apparent aging of the brain. Nat. Neurosci. 23, 295 (2020).
Romme, C. J. A. et al. Analysis and visualization of the effect of multiple sclerosis on biological brain age. Front. Neurol. 15, 1423485 (2024).
Høgestøl, E. A. et al. Cross-sectional and longitudinal MRI brain scans reveal accelerated brain aging in multiple sclerosis. Front. Neurol. 10, 450 (2019).
Brier, M. R. et al. Brain age’ predicts disability accumulation in multiple sclerosis. Ann. Clin. Transl. Neurol. 10, 990–1001 (2023).
Gross, P. S. et al. Senescent-like microglia limit remyelination through the senescence associated secretory phenotype. Nat. Commun. 16, 2283 (2025).
Young, M. D. & Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 9, giaa151 (2020).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e4 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Stringer, C., Wang, T., Michaelos, M. & Pachitariu, M. Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106 (2021).
Huang, X. & Huang, Y. Cellsnp-lite: an efficient tool for genotyping single cells. Bioinformatics 37, 4569–4571 (2021).
Huang, Y., McCarthy, D. J. & Stegle, O. Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biol. 20, 273 (2019).
Bulteau, R. & Francesconi, M. Real age prediction from the transcriptome with RAPToR. Nat. Methods 19, 969–975 (2022).
Absinta, M. et al. Identification of chronic active multiple sclerosis lesions on 3T MRI. Ajnr. Am. J. Neuroradiol. 39, 1233–1238 (2018).
Maggi, P. et al. Paramagnetic rim lesions are specific to multiple sclerosis: an international multicenter 3T MRI study. Ann. Neurol. 88, 1034–1042 (2020).
Bagnato, F. et al. Imaging chronic active lesions in multiple sclerosis: a consensus statement. Brain 147, 2913–2933 (2024).
Absinta, M. MS brain senescence (v1.0.0). Zenodo. https://doi.org/10.5281/zenodo.16569909 (2025).
Yang, A. C. et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603, 885–892 (2022).
Acknowledgements
M.A. was supported by Cariplo Foundation (2019-1677), FRRB Early Career Award (1750327), NMSS (RFA-2412-44156), and FISM–Fondazione Italiana Sclerosi Multipla co-financed with the “5 per mille” public funding (2023/R-Single/015). F.F. was supported by a research fellowship from FISM - Fondazione Italiana Sclerosi Multipla co-financed with the “5 per mille” public funding (2023/BR/004), and a starting grant from the BraYn association (https://www.braynassociation.com). P.M. was supported by Fondation Charcot Stichting Research Fund 2023, Fund for Scientific Research (F.R.S., FNRS 40008331), Cliniques universitaires Saint-Luc “Fonds de Recherche Clinique”. P.A.C. was supported by NIH R01NS082347. P.P.B. was supported by FISM - Fondazione Italiana Sclerosi Multipla co-financed with the “5 per mille” public funding (2022/R-Single/026). G.V.M. was supported by the International Progressive MS Alliance (PA-1604-08492 BRAVEinMS). D.S.R. was supported by Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, and Intramural Research Program of NINDS, NIH. This research was supported in part by the Intramural Research Program of the National Institutes of Health (NIH). The contributions of the NIH authors are considered Works of the United States Government. The findings and conclusions presented in this paper are those of the authors and do not necessarily reflect the views of the NIH or the U.S. Department of Health and Human Services. We thank Dr Francesca Giannese, Dr Lorenza Moronetti and Dr Caterina Oneto of the Center for Omics Sciences (IRCCS San Raffaele Scientific Institute, Milan) for help with the 10× Genomics experiments and sequencing. We thank Dr Maira Gironi for recruiting patients for skin biopsy; Dr Giacomo Sferruzza, Prof Giovanni Landoni, and Dr Antonella Crescenti for CSF collection from non-neurological controls. We thank Prof. Tanja Kuhlmann (Münster University, Germany) for help with NPC differentiation. We thank Dr Valeria Berno (ALEMBIC Core facility, IRCCS San Raffaele Scientific Institute) and Dr Fabio Pasqualini (Histology Core facility, IRCCS Humanitas Research Institute) for help with light imaging. We thank Amleto Fiocchi (IRCCS San Raffaele Scientific Institute) for help with brightfield slide scanning. We thank Dr Chiara Camisaschi and Dr Ilaria Montali for their assistance with flow cytometry (Cytometry Core facility, IRCCS Humanitas Research Institute, Milan). This work also implemented the Research Cluster of IRCCS San Raffaele Scientific Institute for the computational analysis.
Author information
Authors and Affiliations
Contributions
F.F. performed experiments on human postmortem brain tissues and hiPSC-derived cultures. F.F. and J.P.L. performed the Visium experiments. E.P. analyzed single-cell genomic data. M.S.M. analyzed spatial transcriptomic data. D.M. performed part of the multiplex immunostaining. G.M., P.P.B., G.G., F.R., E.B., and C.P. helped with the experiments using hiPSC-derived cultures. M.A., F.F., and E.P. analyzed the biological data. M.A., D.S.R., P.M., C.V.B., and P.A.C. supplied MRI scans and analyzed the MRI data. M.A., F.F., and E.P. wrote the manuscript with input from all authors. M.A., G.M., P.P.B., P.A.C., D.S.R., and P.M. provided intellectual contribution to the manuscript. M.A. contributed with experimental design and supervision of the project.
Corresponding author
Ethics declarations
Competing interests
P.A.C. received research funding from Genentech and consulting honoraria for serving on advisory boards for Idorsia Spolia Therapeutics and Lilly. P.M. received consulting and/or speaker honoraria from Sanofi, Biogen, and Merck. D.S.R. received research funding from Abata Therapeutics and Sanofi. M.A. received consulting and/or speaker honoraria from GSK, Sanofi, Biogen, Immunic Therapeutics, Viatris, and Abata Therapeutics. All other authors report no relevant declaration of interest.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fagiani, F., Pedrini, E., Martire, M.S. et al. Spatially-restricted inflammation-induced senescent-like glia in multiple sclerosis and patient-derived organoids. Nat Commun 16, 8477 (2025). https://doi.org/10.1038/s41467-025-63371-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63371-9
